
WYKŁAD 3 - home.agh.edu.plhome.agh.edu.pl/~graboska/doc/FPP-WIRT-wyklad_3.pdf · Jeśli proces (...
44
WYKŁAD 3
-
Upload
phungthuan -
Category
Documents
-
view
213 -
download
0
Transcript of WYKŁAD 3 - home.agh.edu.plhome.agh.edu.pl/~graboska/doc/FPP-WIRT-wyklad_3.pdf · Jeśli proces (...

WYKŁAD 3

Termodynamika a Kinetyka chemina W rozważaniach termodynaminych występuje pojęcie powinowactwa termodynaminego danego procesu (np. reakcji cheminej). Powinowactwo chemine można wyrazić za pomocą zmiany entalpii swobodnej (∆G(T,p) = Lmax uż.), towayszącej pejściu danego układu ze stanu poątkowego do końcowego.
éophile de Donder (1872-1957)
De Donder wprowadził do termodynamiki – jako miarę powinowactwa cheminego reakcji cheminej, zachodzącej py T, p =const wielkość:
A = -∑ νi μi = - ∆G gdzie: νi - wółynnik stechiometryny substancji „i” μi – potencjał cheminy
Pykładowo dla reakcji syntezy wody: 2H2 + O2 = 2H2O dla wodoru νH = -2, dla tlenu νO = 1, dla wody νH2O = 2.
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Jeśli proces ( np. reakcja chemina) zachodzi bez obecności pracy użytenej (Luż.
= 0) to wg De Dondera zachodzi związek:
• gdy A > 0 ( yli ∆G < 0) oznaa możliwość zachodzenia samoutnie ( jeśli pozwolą na to opory kinetyne) reakcji z lewa na prawo (tj. substraty → produkty) • gdy A = 0 (yli ∆G = 0) określa stan równowagi
Kinetyka chemina a termodynamika
A ≥ 0
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Pomiędzy powinowactwem cheminym procesu a jego szybkością
(T,p = const) zachodzi związek:
gdzie v – szybkość procesu Ze wzoru wynika, że A i v muszą mieć ten sam znak. Pykładowo: - jeśli A > 0, to v > 0, to reakcja pebiega „ → ” - jeśli A < 0, to v < 0, to reakcja pebiega w peciwną stronę „ ← “ - jeśli A v = 0, to układ znajduje się w stanie równowagi
termodynaminej.
Kinetyka chemina a termodynamika
A v ≥ 0,
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH
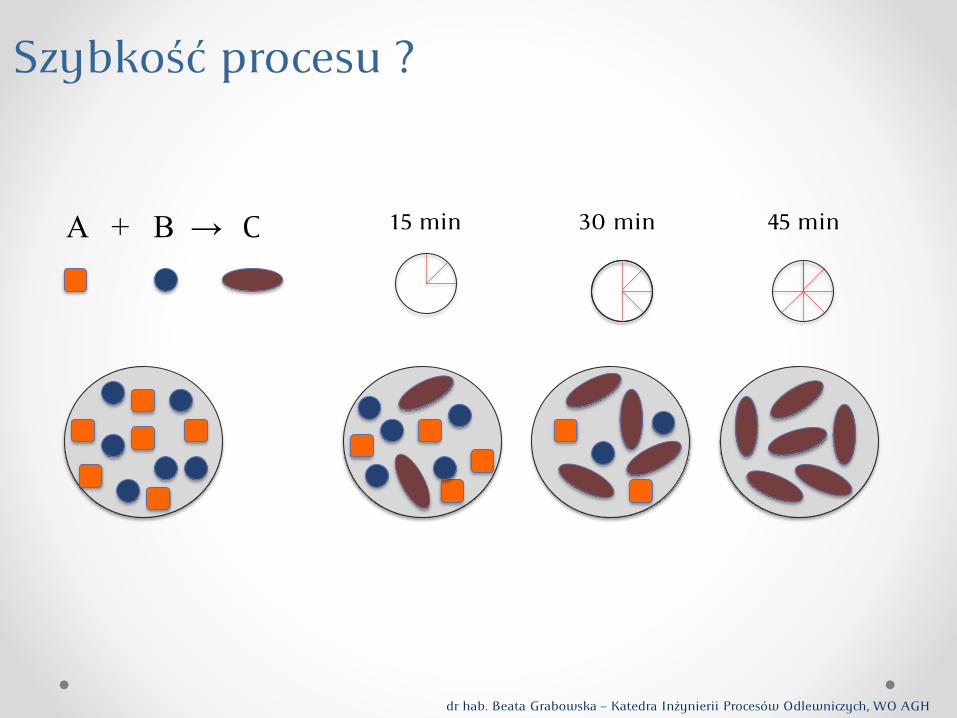
A + B → C
Szybkość procesu ?
15 min 30 min 45 min
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Szybkość procesu ?
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH
http://www.geekweek.pl
https://pl.wikipedia.org
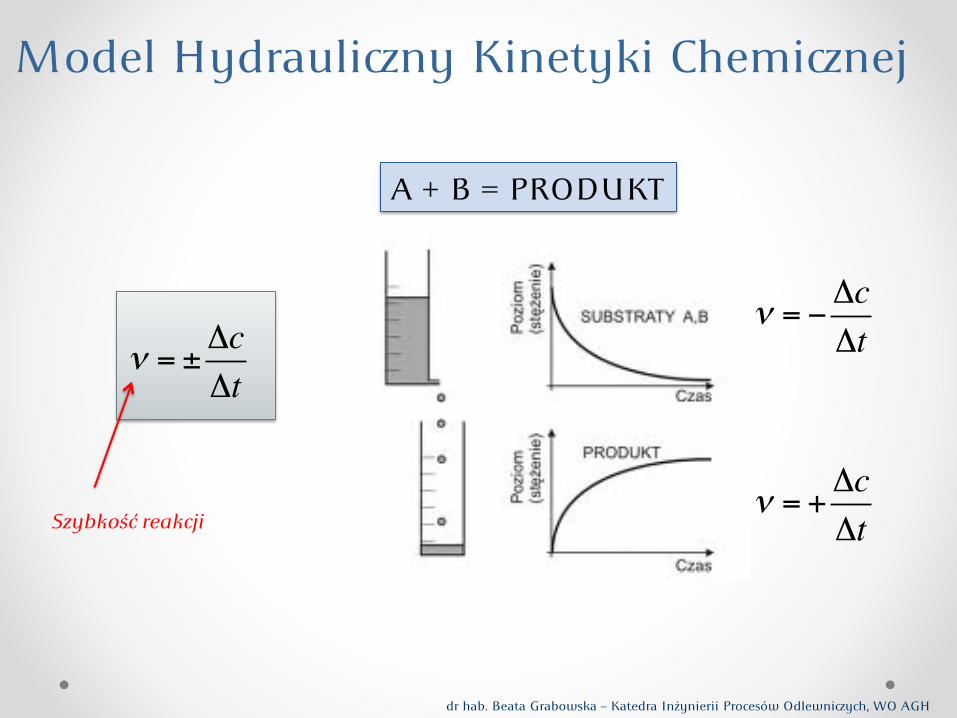
! = ±!c!t
Model Hydrauliny Kinetyki Cheminej
A + B = PRODUKT
! = !"c"t
! = +!c!tSzybkość reakcji
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

SZYBKOŚĆ REAKCJI homogeninej zachodzącej w stałej objętości:
dtdc1v i
iν=
v - szybko!" reakcji [mol! dm-3!s-1], [mol! dm-3!min-1], [mol! dm-3!h-1],
ic - chwilowe st#$enie i-tego reagenta [mol! dm-3],
i! - wspó%czynnik stechiometryczny i-tego reagenta,
„+” - produkt, „-„ - substrat t - czas [s-1], [min-1], [h-1],
Kinetyka chemina
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Kinetyka reakcji homogeninych (jednorodnych)
zachodzących w obrębie jednej fazy
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Kinetyka reakcji homogeninych • Określona jest najęściej zmianą stężenia któregoś z reagentów w jednostce
asu.
• Zależność szybkości reakcji cheminej w układzie homogeninym od stężenia wyraża kinetyne prawo działania mas
prawo Guldberga – Waage’go (1864):
szybkość reakcji nieodwracalnej w stałej temperatue, w danym momencie asu, jest proporcjonalna do chwilowego stężenia (ściślej aktywności) substratów w odpowiednich potęgach
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Kinetyka reakcji homogeninych
W pypadku gdy rozpatrujemy szybkość reakcji vr tylko w kierunku twoenia się produktów:
aA + bB + cC + … → produkty Wówas prawo działania mas wyraża równanie: gdzie: cA, cB, cC - stężenia chwilowe substratów, nA, nB, nC – wyznaone wykładniki potęgowe py odpowiednich stężeniach, k – stała szybkości reakcji zależna od temperatury, a także od obecności i
rodzaju katalizatora
vr = k.cAnA k.cB
nB k.cCnC
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH
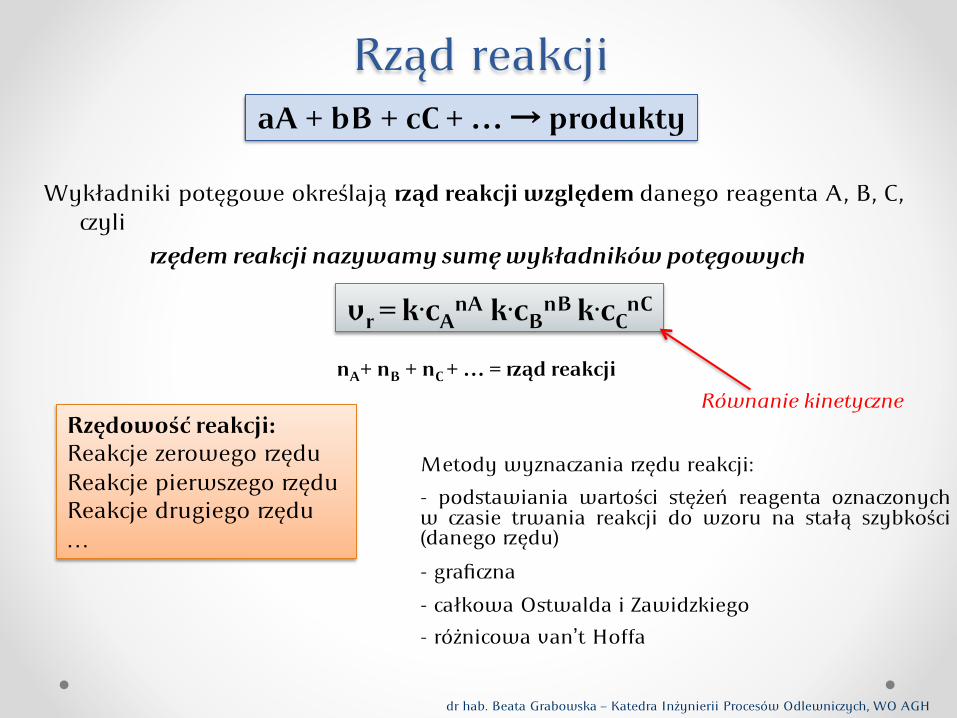
Rząd reakcji
Wykładniki potęgowe określają ąd reakcji względem danego reagenta A, B, C, yli
ędem reakcji nazywamy sumę wykładników potęgowych
nA+ nB + nC + … = ąd reakcji
Metody wyznaania ędu reakcji:
- podstawiania waości stężeń reagenta oznaonych w asie trwania reakcji do wzoru na stałą szybkości (danego ędu)
- grafina - całkowa Ostwalda i Zawidzkiego - różnicowa van’t Hoffa
vr = k.cAnA k.cB
nB k.cCnC
aA + bB + cC + … → produkty
Rzędowość reakcji: Reakcje zerowego ędu Reakcje pierwszego ędu Reakcje drugiego ędu …
Równanie kinetyne
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Równanie reakcji a równanie kinetyne
v = k cAcB H2 + I2 = 2HI
H2 + Br2 = 2HBr v =k1 H2[ ] Br2[ ]1/2
1+ k2HBr[ ]Br2[ ]
v = k c2np. 2HI = H2 + I2
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

(1) 2 N2O5 → 4 NO2 + O2 (w fazie gazowej lub w roztwoe)
v = ! 12d N2O5[ ]
dt= k N2O5[ ] n = 1
(2) (C2H5)3N + C2H5I → (C2H5)4NI (w roztwoe)
v = !d C2H5I[ ]
dt= k C2H5( )3
N"# $% C2H5I[ ]n = 2
(3) CO + Cl2 → COCl2
v =d COCl2[ ]
dt= k CO[ ] Cl2[ ]
32
n = 5/2
Równanie reakcji a równanie kinetyne
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

v = k c0 = kv
c
v
t
co - pocz!tkowe st"#enie substratu , [mol!dm-3], [mg dm-3], [mg cm-3],
c - st"#enie substratu po czasie t, [mol!dm-3], [mg dm-3], [mg cm3],
!dcdt= k
dc = !k dt
dc = ! kt=0
t
"c!
c
" dt
c-c! = !k t
c = c! ! k tc co
t
α tg ! = a = -kt0.5 =
c!2k
k = c! - ct
k[ ] = [mol dm-3 h-1], [mol dm-3 min-1], [mol dm-3 s-1]
Kinetyka reakcji zerowego ędu Szybkość reakcji nie zmienia się w asie
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Pykłady reakcji zerowego ędu
np. reakcje fotochemine
a b s o r p c j a k w a n t u p r o m i e n i o w a n i a elektromagnetynego (fotonu) o odpowiedniej energii niezbędnej do pejścia ze stanu kwantowego o niższej energii na stan kwantowy o wyższej lub powstanie wolnych rodników
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH
E’ (stan wzbudzony)
E0 (stan podstawowy) wzb
udze
nie
emis
ja

Równanie kinetyne dla reakcji jednoąstekowej:
A → produkty
Chwilowa szybkość reakcji (vr) jest wyrażona pochodną stężenia substratu względem asu dcA/dt.
Kinetyka reakcji pierwszego ędu
! r = !dcAdt
= kcA
szybkość reakcji wprost proporcjonalna do jednego łonu stężeniowego
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Kinetyka reakcji pierwszego ędu
Ogólnie szybkość reakcji homogeninych określona jest pochodną ze stężenia któregoś ze substratów albo któregoś z produktów reakcji. Znak „+" odnosi się do pypadku, kiedy substancja „i” jest produktem, znak „-” , kiedy substancja „i" jest substratem. Stężenie substancji reagujących po asie t wynosi:
gdzie: - stężenie poątkowe substancji (tj. dla t = 0), cA - stężenie chwilowe substancji A po asie t, py ym cA = - x , gdzie x
oznaa ubytek stężenia substancji A po asie t: kt
xaa
=−
ln
! = ±dcdt
ln acA= kt
aa
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Zależność ln cA = f (t) ma charakter liniowy
ln c
ln co
t
α
tg ! = a = -k
t
c co
ln c = ln co – kt
c = c!e!k t
n = 1
t0.5 =ln 2
k
Zależność cA = f (t) ma charakter kywej wykładniej (gdy t rośnie, cA oraz vA maleje)
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Pykłady reakcji pierwszego ędu
np. reakcje promieniotwórego rozpadu
np. reakcje terminego rozkładu
np. reakcje estryfikacji w rozcieńonych roztworach
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH
α

v = k cAcB
v = k c2
Kinetyka reakcji drugiego ędu
szybkość reakcji wprost proporcjonalna do dwóch łonów stężeniowych
szybkość reakcji wprost proporcjonalna do jednego z łonów stężeniowych w kwadracie
lub np. H2 + I2 = 2HI
np. 2HI = H2 + I2
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

c = c!1+ k t c!
t0.5 =1
k c!k = 1
tc! ! c( )c! "c
k[ ] = [dm3 mol-1 h-1], [dm3 mol-1 min-1], [dm3 mol-1 s-1]
tg ! = a = k
t
c1
οc1
α
1c=
1c!+ k t
!dcdt= k c2
dcc2 = !k dt
1c2 dc = ! k
t=0
t
"c!
c
" dt
!1c+
1c!= !k t
v = k c2
co - pocz!tkowe st"#enie substratu , [mol!dm-3], [mg dm-3], [mg cm-3], c - st"#enie substratu po czasie t, [mol!dm3], [mg dm-3], [mg cm-3],
n = 2
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Okres połowinej pemiany
Ø inaej - połówkowy as reakcji Ø charakteryzuje się kinetyny charakter procesu, jest tzw. okres połowinej pemiany (t 1/2 lub t0.5 ) Ø jest to as konieny na to, aby pereagowała połowa substratu Ø dla reakcji pierwszego ędu: yli:
W pypadku reakcji pierwszego ędu okres połowinej pemiany nie zależy od stężenia poątkowego a, natomiast dla reakcji ędu n , w pypadku ogólnej postaci Równania kinetynego: okres ten jest odwrotnie proporcjonalny do an-1 :
12/11~−na
t
t1/2 = t0.5 = (ln 2)/ k
k = (ln 2)/t1/2
! r = !dcdt
= kcn
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Ø odwracalne
Ø równoległe
Ø następe
Kinetyka reakcji złożonych
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Reakcje odwracalne Ściśle biorąc wszystkie reakcje są odwracalne (kinetynie), a w stanie równowagi szybkość reakcji w prawo jest równa szybkości reakcji w lewo. Dla reakcji odwracalnej I -go ędu ogólne równanie chemine:
Natomiast równanie kinetyne w postaci różnikowej:
Reakcje równoległe
Jeśli substraty w danych warunkach mogą reagować na kilka osobów, twoąc różne rodzaje produktów:
k1 B A k2 C
to zakładając, że reakcja A →B i A → C są pierwszego ędu, otymamy następujące równanie różnikowe określające szybkość pemiany cheminej substancji A w produkty (A i C):
BAk
k
⎯⎯←
⎯→⎯
2
1
BAA ckckdtdc
21 −=
BAA ckckdtdc
21 +=−
Pykłady: nitrowanie toluenu, nitrowanie fenolu, chlorowanie fenolu.
Pykłady: proces estryfikacji, synteza amoniaku.
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Reakcje następe
Rozpatmy ogólną reakcję: pebiegającą w stadiach: Załóżmy: każde stadium stanowi reakcję pierwszego ędu. Substancja B twoy się w wyniku rozpadu substancji A i sama się rozpada na substancję C . Równanie różnikowe na szybkość: W pypadku reakcji złożonej z kilku etapów następych, znanie różniących się szybkością, etap najwolniejszy będzie określał szybkość wypadkową reakcji złożonej (np. A→ B). Jeśli np. k1 << k2 stała szybkości k reakcji wypadkowej (A → C) będzie równa praktynie k1. Oznaa to, że najwolniejszym etapem reakcji wypadkowej będzie etap pierwszy (A → B); etap ten będzie stanowił etap kontrolujący szybkość reakcji sumarynej (wypadkowej).
CA k⎯→⎯
BA k⎯→⎯ 1 B k2! "! C
CBA kk ⎯→⎯⎯→⎯ 21
BAB ckckdtdc
21 −=
Pykłady: pebiegające w dwóch etapach zmydlanie szawianu dietylowegowodorotlenkiem sodowym, reakcje łańcuchowe, procesy wchłaniania i eliminacji leków z ustroju.
c
t
A
B
C
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Kinetyka reakcji heterogeninych
Pykłady procesów heterogeninych: Ø procesy związane z odgazowywaniem metali, Ø procesy rozpuszania ciał stałych w cieach, Ø procesy wysokotemperaturowej korozji metali w agresywnych gazach, Ø krystalizacja ciey lub kondensacja pary, Ø pemiany fazowe zachodzące podas obróbki terminej metali i stopów.
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Kinetyka reakcji heterogeninych
Najbardziej charakterystyną cechą reakcji heterogeninych jest ich złożony mechanizm, obejmujący niekiedy 5 etapów:
1. proces tranou substratów (pez konwekcję lub dyfuzję) na miejsce
reakcji, którą jest z reguły granica faz (ciało stałe - gaz, ciało stałe - cie, cie - gaz itp.),
2. adsorpcji substratów na granicy faz, 3. właściwa reakcja na granicy faz, 4. desorpcji produktów na granicy faz, 5. proces tranou produktów z miejsca reakcji w głąb graniących ze sobą
faz.
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Kinetyka reakcji heterogeninych
Etapem kontrolującym ogólną szybkość procesu jest etap najwolniejszy, stąd wyróżniamy:
Ø kontrolę kinetyną procesu (w niskich temperaturach), Ø kontrolę dyfuzyjną procesu (wpływ mieszania), Ø kontrola mieszana procesu kinetyno-dyfuzyjna (szybkość dyfuzji i
szybkość właściwej reakcji cheminej są zbliżone),
Szybkość procesów w układach wielofazowych zależy zwykle w znanym stopniu od wielkości powiechni podziału faz (tj. powiechni zetknięcia faz), na której zachodzi reakcja.
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Kinetyka rozkładu ciał stałych
Rozkład ciał stałych pod wpływem temperatury mozna pedstawićkilkoma schematynymi zapisami, le z punktu widzenia niniejszej pracy najwazniejsza jest postać:
AB(s) → C(s) + D(g)
Ø Szybkość reakcji pechodzi pez maksimum Ø Szybkość reakcji jest maksymalna w chwili jej rozpoęcia Ø Istotną rolę odgrywają makro- i mikrodefekty struktury
Rozkład węglanu wapnia: CaCO3(s) → CaO(s) + CO2(g)
Szybkość reakcji rozkładu węglanów jest proporcjonalna do stopnia odstępstwa od stanu równowagi wyrażanego różnicą pomiędzy prężnością rozkładową pr
CO2, a aktualnie panującym ciśnieniem ąstkowym pCO2, w danej temperatue oraz do wielkości powiechni fazy reagującej:
v = krS(prCO2 + pCO2)
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Czynniki wpływające na szybkość reakcji
Ø rodzaj reagentów (typ reakcji) Ø stężenie reagentów Ø ciśnienie gazowych reagentów Ø temperatura Ø katalizator Ø promieniowanie elektromagnetyne Ø rodzaj rozpuszalnika Ø stopień rozdrobnienia substratów
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Wpływ temperatury na szybkość reakcji
Reguła van 't Hoffa
W pypadku reakcji homogeninych
ęsto stwierdza się podwojenie (w pybliżeniu) szybkości reakcji py
każdym wzroście temperatury o 10°C.
Jacobus Henricus van 't Hoff 1852-1911
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Wpływ temperatury na szybkość reakcji
Zależność stałej szybkości reakcji, a zatem szybkości reakcji od temperatury, określona jest równaniem Arrheniusa (1889 r.): gdzie: A - stała, tzw. wółynnik ęstości, Ea - energia aktywacji.
Równanie Arrheniusa można stosować z powodzeniem do wielu reakcji w fazie gazowej, do reakcji zachodzących w roztworach skondensowanych, a także do reakcji heterogeninych.
Po zlogarytmowaniu:
k = A exp (- Ea / RT )
ln k = ln A - Ea / RT
Svante August Arrhenius 1859-1927
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH
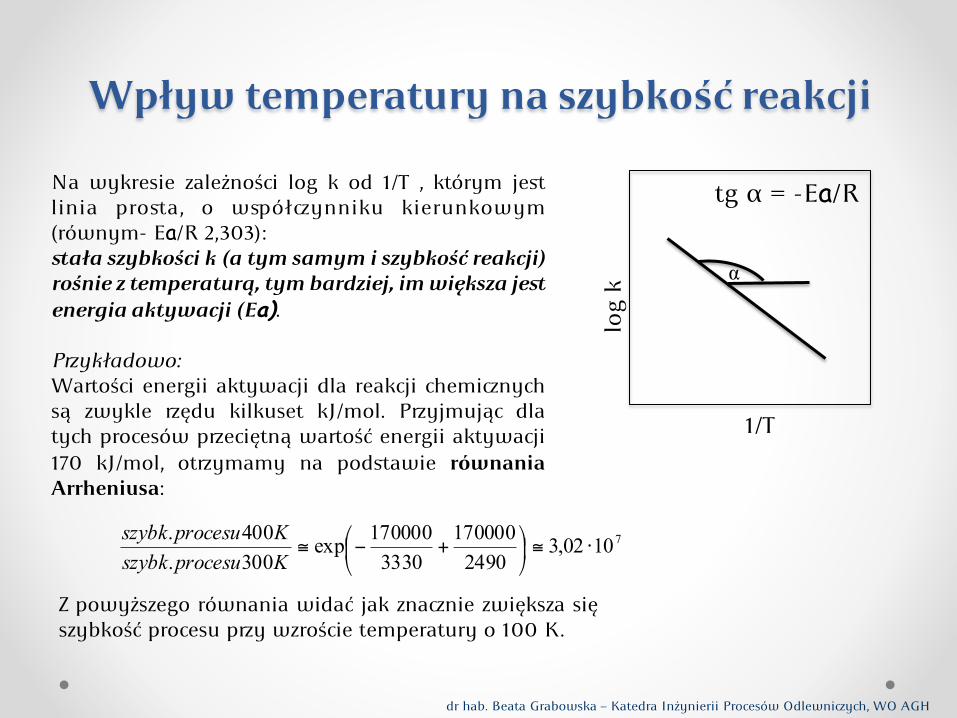
Wpływ temperatury na szybkość reakcji
Na wykresie zależności log k od 1/T , którym jest linia prosta, o wółynniku kierunkowym (równym- Ea/R 2,303): stała szybkości k (a tym samym i szybkość reakcji) rośnie z temperaturą, tym bardziej, im większa jest energia aktywacji (Ea). Pykładowo: Waości energii aktywacji dla reakcji cheminych są zwykle ędu kilkuset kJ/mol. Pyjmując dla tych procesów peciętną waość energii aktywacji 170 kJ/mol, otymamy na podstawie równania Arrheniusa:
71002,32490170000
3330170000exp
300.400.
⋅≅⎟⎠
⎞⎜⎝
⎛ +−≅KprocesuszybkKprocesuszybk
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH
α
log
k
1/T
tg α = -Ea/R
Z powyższego równania widać jak znanie zwiększa się szybkość procesu py wzroście temperatury o 100 K.
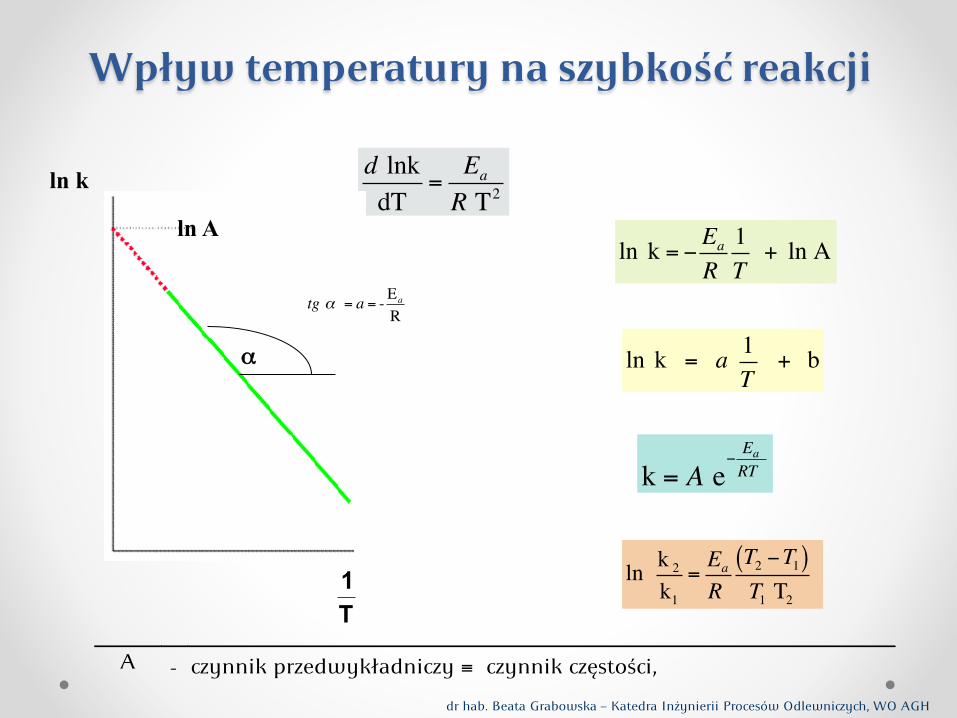
d lnkdT
=Ea
R T2ln k
T1
ln A
α
tg ! = a = - Ea
R
A - czynnik przedwyk!adniczy ! czynnik cz"sto#ci,
k = A e!EaRT
ln k 2
k1
=Ea
R T2 !T1( )T1 T2
ln k = ! Ea
R 1T
+ ln A
ln k = a 1T
+ b
Wpływ temperatury na szybkość reakcji
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

Teoria zdeeń aktywnych Na podstawie teorii zdeeń aktywnych liba zdeeń aktywnych „z" (a tym samym szybkość reakcji w danej temperatue) można obliyć, biorąc pod uwagę, że reagują ze sobą tylko te ąsteki, które py zdeeniu maja energię równą lub większą od pewnej waości minimalnej, zwanej energią aktywacji Ea: z = z0 exp (- E / RT ) gdzie: z0 - ogólna liba zdeeń. Zakładając, że szybkość reakcji, a ściślej biorąc stała szybkości reakcji k, jest określona pez libę zdeeń aktywnych z , otymamy: k = z exp (- Ea / RT )
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH
a

Teoria stanu pośredniego Ø inaej teoria bezwzględnych szybkości reakcji lub teoria
kompleksu aktywnego,
Ø opiera się na ogólnym założeniu, że kiedy dwie ąsteki reagujące, mające energie równą lub większą od energii aktywacji, zdeą się ze sobą wówas twoy się kompleks aktywny, który rozpada się samoutnie na ostateny produkt reakcji.
Ø teoria kompleksu aktywnego pozwala obliyć stałą szybkości reakcji k , a tym samym szybkość reakcji.
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH
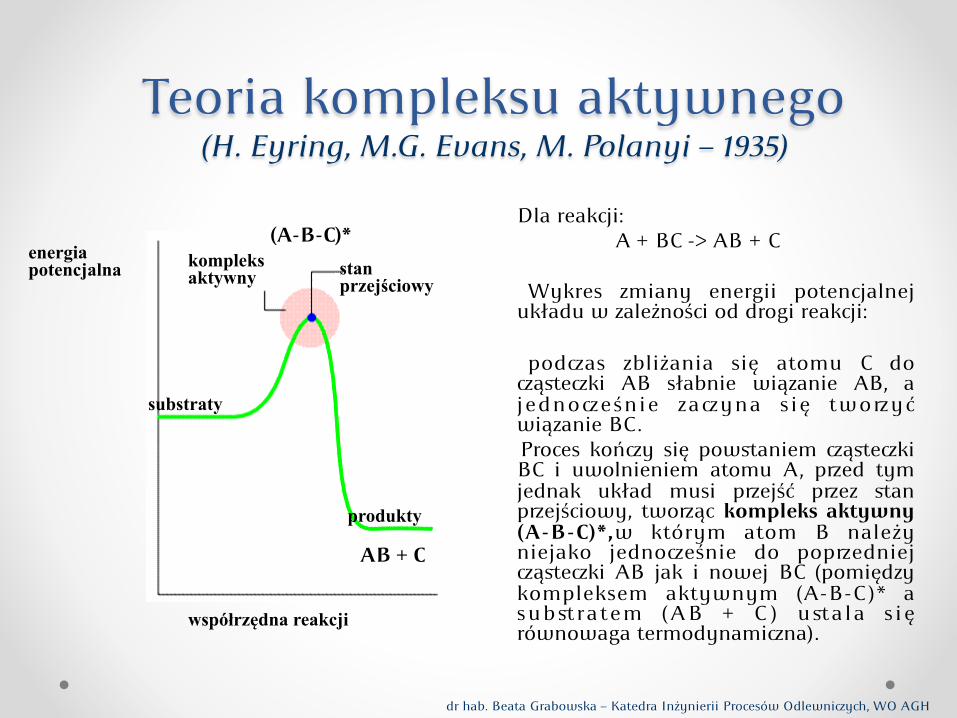
Teoria kompleksu aktywnego (H. Eyring, M.G. Evans, M. Polanyi – 1935)
Dla reakcji: A + BC -> AB + C
Wykres zmiany energii potencjalnej układu w zależności od drogi reakcji:
podas zbliżania się atomu C do ąsteki AB słabnie wiązanie AB, a jednoeśnie zayna się twoyć wiązanie BC. Proces końy się powstaniem ąsteki BC i uwolnieniem atomu A, ped tym jednak układ musi pejść pez stan pejściowy, twoąc kompleks aktywny (A-B-C)*,w którym atom B należy niejako jednoeśnie do popedniej ąsteki AB jak i nowej BC (pomiędzy kompleksem aktywnym (A-B-C )* a subst ratem (AB + C ) ustala s ię równowaga termodynamina).
A + BC
energia potencjalna
współrzędna reakcji
substraty
produkty
kompleks aktywny stan
przejściowy
(A-B-C)*
AB + C
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH
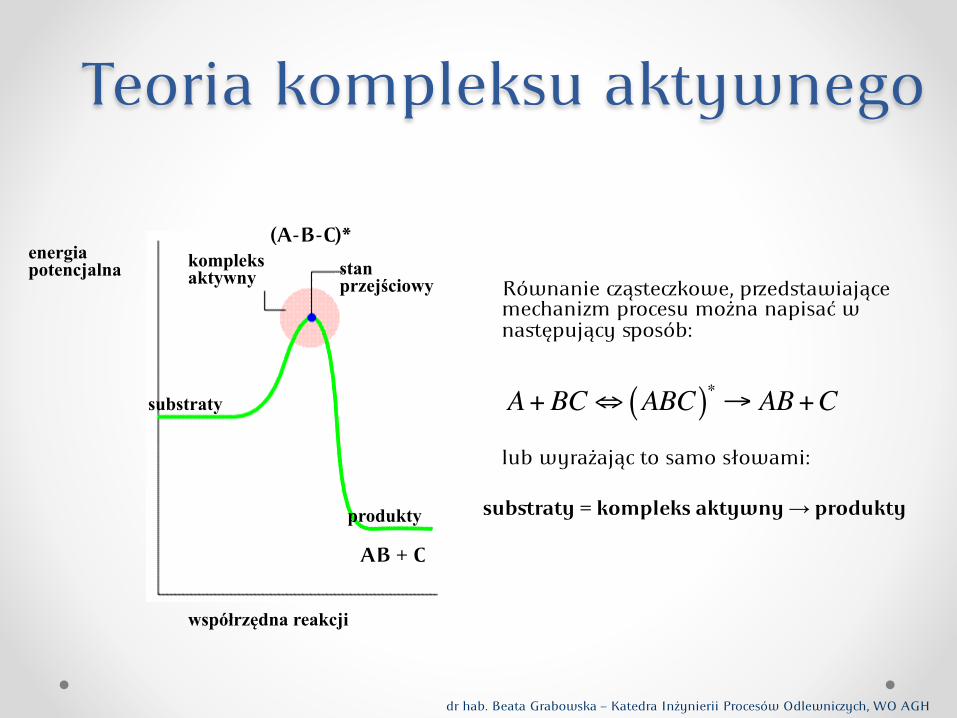
Teoria kompleksu aktywnego
Równanie ąstekowe, pedstawiające mechanizm procesu można napisać w następujący osób:
lub wyrażając to samo słowami:
substraty = kompleks aktywny → produkty
A+BC! ABC( )" # AB+C
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH
energia potencjalna
współrzędna reakcji
substraty
produkty
kompleks aktywny stan
przejściowy
(A-B-C)*
AB + C

Na podstawie teorii stanu pośredniego zależność stałej szybkości reakcji k od temperatury można wyrazić następującym równaniem (stanowiącym odpowiednik prawa Arrheniusa) :
k = A exp (- Ea / RT ) [ x (kT/h) exp (∆S* / R)] exp (-∆H* / RT) yli: Ea = ∆H* gdzie: x - wółynnik transmisji, który określa prawdopodobieństwo rozpadu
kompleksu aktywnego na ąsteki produktów, k - stała Boltzmana, h - stała Plancka, ∆S* i ∆H* - pyrost odpowiednio entropii i entalpii w procesie twoenia się kompleksu aktywnego z substratów.
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH
Teoria kompleksu aktywnego
Wpływ katalizatora …
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH
Pykłady reakcji katalizowanych … Rodzaje katalizatorów …
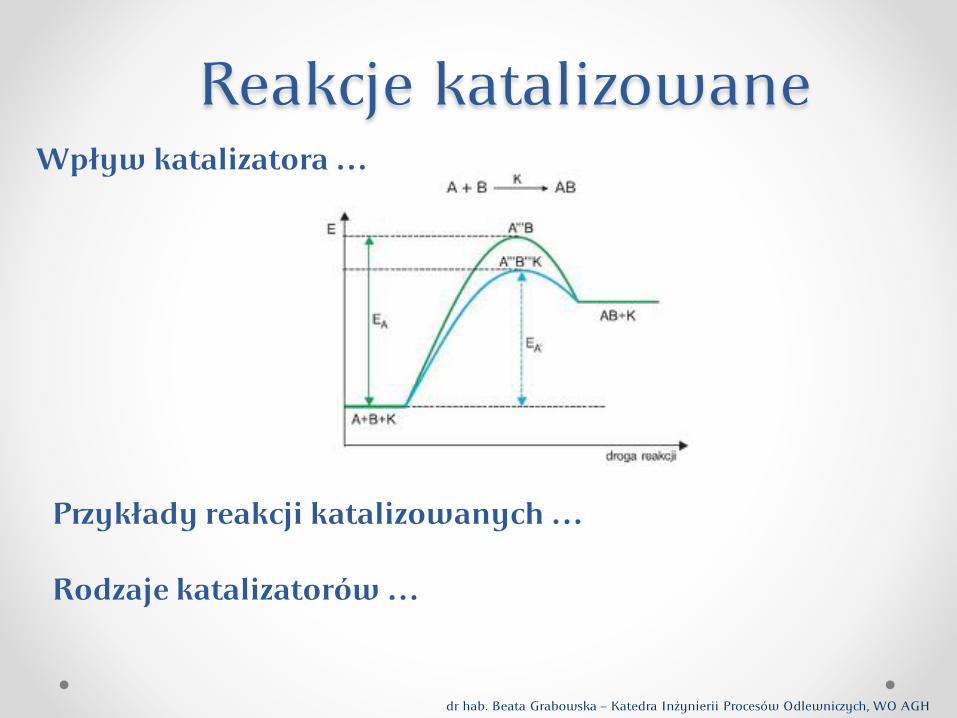
Reakcje katalizowane
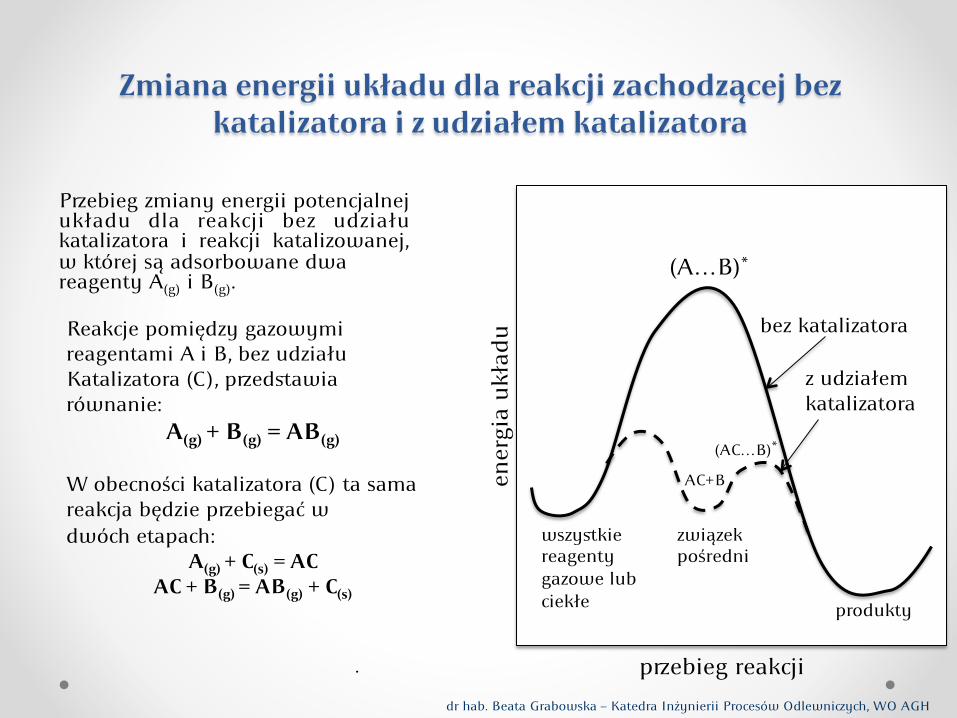
Zmiana energii układu dla reakcji zachodzącej bez katalizatora i z udziałem katalizatora
Reakcje pomiędzy gazowymi reagentami A i B, bez udziału Katalizatora (C), pedstawia równanie:
A(g) + B(g) = AB(g) W obecności katalizatora (C) ta sama reakcja będzie pebiegać w dwóch etapach:
A(g) + C(s) = AC AC + B(g) = AB(g) + C(s)
.
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH
((
(A…B)*
bez katalizatora
z udziałem katalizatora
wszystkie reagenty gazowe lub ciekłe produkty
związek pośredni
AC+B
(AC…B)*
ener
gia
ukła
du
pebieg reakcji
Pebieg zmiany energii potencjalnej układu dla reakcji bez udziału katalizatora i reakcji katalizowanej, w której są adsorbowane dwa reagenty A(g) i B(g).

[1] A. Staronka. Chemia fizyna. Wyd. AGH 1994 (i dalsze wydania). [2] K. Pigoń, Z. Ruziewi: Chemia Fizyna, PWN, Warszawa 2014. [3] E. Brian Smith: Basic chemical thermodynamics, Imperial College Press, 2014. [4] S. K. Bose, S. Roy: Principles of Metallurgical ermodynamics, Universites Press 2014. [5] Strona internetowa: http://home.agh.edu.pl/~graboska/ [6] Fotografie: https://pl.wikipedia.org
Literatura
dr hab. Beata Grabowska – Katedra Inżynierii Procesów Odlewniych, WO AGH

koniec











![OCENA ZAGROśENIA I SYSTEM OCHRONY PRZED ...holmes.iigw.pl/~llewicki/HomepageLL/ZIOPP/wyklad_3.pdfLiczba ludno ści ogółem [mln] Wska źnik zaludnienia [l.osób / km 2] Ludno ść](https://static.fdocuments.pl/doc/165x107/60b0c42257acd75ed85668b4/ocena-zagroenia-i-system-ochrony-przed-llewickihomepagellzioppwyklad3pdf.jpg)






