







Języki
Strony
Prawny


Dane nr 7
Analiza danych w programach Analiza danych w programach bioinformatycznychbioinformatycznych
Panek PaulinaPauch JoannaPawłowska UrszulaPawłowski CyprianPryczynicz Ewa
GRUPA II

Dane nr 7numer osobnika
ojcowie
matki
waga markery

PROGRAMY
Programy użyte do analizy naszych danych:Programy użyte do analizy naszych danych: GDAGDA QTL ExpressQTL Express SASSAS EMLDEMLD GOLDGOLD PHASEPHASE

GDA
Struktura pliku wejściowego
10 loci
Allele

HIPOTEZY
Program ten sprawdza nam postawioną Program ten sprawdza nam postawioną przez nas HIPOTEZĘ:przez nas HIPOTEZĘ:
H0: dane locus jest w równowadze H-WH0: dane locus jest w równowadze H-W
H1: dane locus nie jest w równowadze H-WH1: dane locus nie jest w równowadze H-W Lmax przyjmujemy 0,01Lmax przyjmujemy 0,01 jeżeli Lmax<Lt to przyjmujemy H0jeżeli Lmax<Lt to przyjmujemy H0 jeżeli Lmax>Lt to przyjmujemy H1jeżeli Lmax>Lt to przyjmujemy H1

WYNIKI
Locus 1 jest w równowadze H-W
Locus 8 jest w równowadze H-W
Kombinacja locus 2 do locus 6 nie jest w równowadze H-W
Kombinacja locus 4 do locus 6 jest w równowadze H-W

AnalizaAnalizazaburzeńzaburzeń
Najczęściej występujące allele
Loci wzięte pod uwagę
----->Współczynnik D przyjmuje wartości
-0,25>D>0,25 --------->Jeżeli D=0 to dane
loci jest w równowadze
Wartość testu
Wartość oszacowana
odchylenie

QTL ExpressQTL Express
Half-sib Analysis

PLIKIPLIKI
Do tego programu trzeba było stworzyć Do tego programu trzeba było stworzyć 3 pliki wejściowe:3 pliki wejściowe:
- - Genotype File Genotype File
- Map File- Map File - Phenotype File - Phenotype File
Plik z wymyślonymi odległościami markerów
na chromosomie

WYNIKIWYNIKI

WYNIKIWYNIKI
Mamy 2 rodziny, ponieważ mamy 2 ojców

WYNIKIWYNIKI
Najbardziej prawdopodobna lokalizacja QTL
100cM
Wartość testu F Wartość testu LRT

WYNIKIWYNIKI
Największego skupienia QTL
należy spodziewać się
w pozycji 100cM

SASSAS
Struktura pliku wejściowego


WYNIKIWYNIKI
Możliwe są 4 haplotypy:11,12,21,22
W kombinacji loci 4 5 utworzyły się następujące haplotpypy 11, 21, 22
W count mamy podana ich liczebność
a w percent ich procentową frekwencję

WYNIKIWYNIKIrekombinowane
nierekombinowane
rekombinacje

EMLDPlik wejściowy – rozszerzenie .dat
55 10Liczba osobnikówLiczba markerów
Poszczególne allele
Liczba porządkowa osobników (1-55)

HapFreq plik wynikowy1 2 3
1 – markery
2 – sprzężenia pomiędzy poszczególnymi markerami
3 – wielkość próby (ilość osobników)

Plik wynikowy - LD
Wartości nierównowagi sprzężeń pomiędzy markerami

Statystyka DStatystyka D
Markery w największej nierównowadze sprzężeń
Markery będące najbliżej równowagi sprzężeń
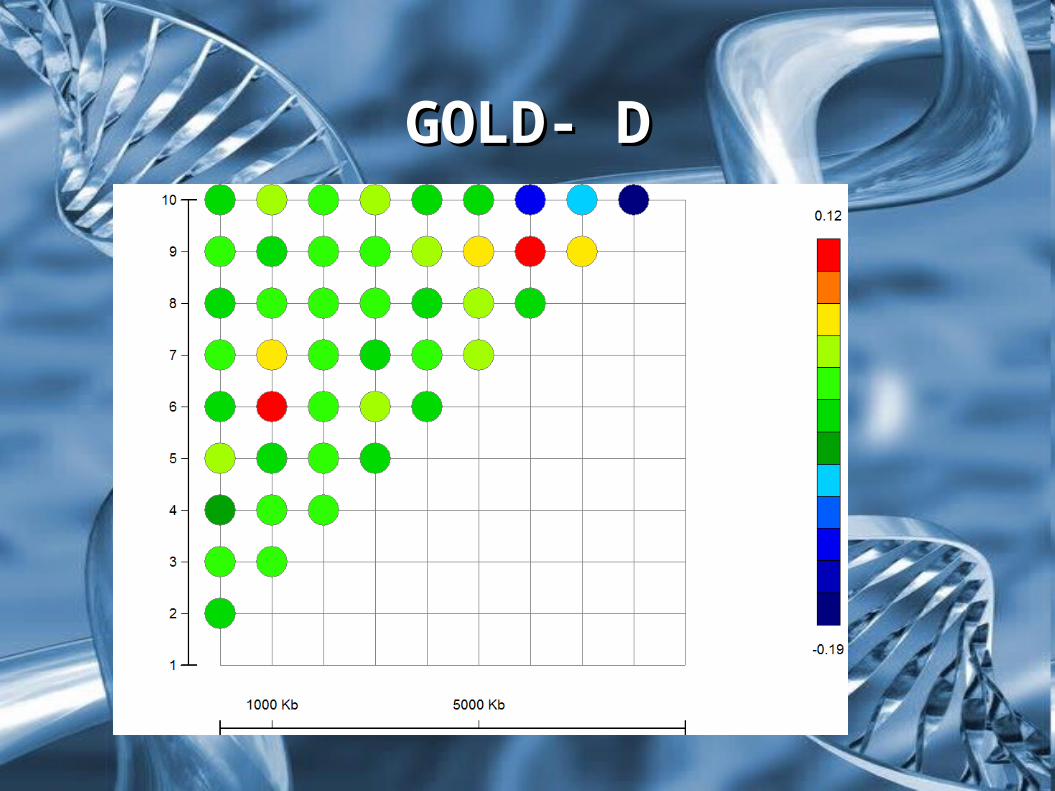
GOLD- DGOLD- D

Statystyka D’Statystyka D’
Nierównowaga sprzężeń
Nierównowaga sprzężeń
Najbliżej równowagi sprzężeń

GOLD – D’GOLD – D’

Statystyka r2
Markery najbliżej równowagi sprzężeń
Markery w pełnej nierównowadze sprzężeń

GOLD – rGOLD – r22

PHASEPHASEIle osobnikówIle markerówS- Snipy
Przekonwertowane dane:11 112, 21 H22 2

PHASE – uruchomienie z wiersza poleceń
-n : nie został podane numery osobników
-f2 : zaznaczamy, że chcemy pracować na pliku, który wcześniej przekonwertowaliśmy1 1112, 21 H22 2
100 : ile powtórzeń ma program wykonać

Pliki wynikowe :
Plik o rozszerzeniu .out_freqs : - zawarte w nim są frekwencje haplotypów
dla całej populacji, oraz podana jest częstość ich występowania dla całej populacji.
Plik o rozszerzeniu .out_pairs : - podanie w nim są haplotypy, których
wystąpienie jest możliwe u danego osobnika.

.out_freqs
1. Ile programutworzył haplotypów dla danej populacji
2. Jakie to haplotypy
3. Frekwencje poszczególnych haplotypów.
4. Błąd, jaki program wykonał podczas obliczania.
1 432

.out_pairs.out_pairs
Numery Numery osobnikówosobników

Dziękujemy za uwagęDziękujemy za uwagę
Top Related