
Wykres a) ilustruje zale no pr no ci par składników A i B ... · odst ępstwa od prawa...
10
Destylacja i rektyfikacja Destylacja to metoda rozdzialu i oczyszczania mieszanin substancji cieklych. W najprostszym przypadku polega na doprowadzeniu cieczy do wrzenia i skropleniu powstalej pary w chlodnicy. W sytuacji, gdy w ruchu jest tylko jedna faza, czyli para, mamy do czynienia z destylacją prostą. Jeśli w przeciwprądzie do par przemieszczać się będzie część skroplonych par (orosienie, flegma) splywające do naczynia z destylowaną cieczą będziemy mieli zjawisko deflegmacji lub rektyfikacji. Jeśli dwie ciecze A i B rozpuszczają się nawzajem w dowolnym stosunku zachodzi wówczas zjawisko nazywane prawem Raoulta, które mówi, Ŝe pręŜność pary danego skladnika cieklej mieszaniny nad roztworem jest proporcjonalna do ulamka molowego tego skladnika w roztworze: p A = p o x A gdzie p 0 - pręŜność par skladnika A nad czystym skladnikiem, x A - ulamek molowy skladnika A w roztworze, p A - pręŜność pary skladnika A nad roztworem. PoniewaŜ proces krzepnięcia i wrzenia jest silnie powiązany z pręŜnością par nad krzepnącą lub wrzącą cieczą, stąd pochodna od prawa Raoulta zasada zmian temperatury wrzenia i krzepnięcia. Regula ta w praktyce sluŜyla do wyznaczania masy cząsteczkowej nieznanej substancji metodą ebulioskopową (wrzenie) lub krioskopową (krzepnięcie) poprzez badanie wielkości zmian temperatury wrzenia (krzepnięcia) po rozpuszczeniu znanej ilości nieznanej substancji w rozpuszczalniku o znanej stalej ebulioskopowej (krioskopowej). Prawo Raoulta jest sluszne dla roztworów idealnych (brak oddzialywań międzycząsteczkowych) a dla roztworów rzeczywistych stosuje się w zasadzie tylko w przypadku roztworów rozcieńczonych. Dla wielu przypadków, szczególnie gdy mamy do czynienia z roztworami stęŜonymi (kilkuprocentowymi) i o silnych oddzialywaniach (np. etanol-woda) występują silne odstępstwa od prawa (azeotropia). Jeśli mieszanina dwuskladnikowa spelnia bez odchyleń prawo Raoulta mamy przypadek roztworu doskonalego. Taka sytuacja zachodzi w nielicznych przypadkach, gdy mamy roztwory substancji z szeregów homologicznych, np.: benzen – toluen, metanol – etanol. Wykres a) ilustruje zaleŜność pręŜności par skladników A i B od skladu tej mieszaniny przy stalej temperaturze (izoterma). Na wykresie b) przedstawiono zaleŜność między temperaturą cieczy i pary w zaleŜności od skladu mieszaniny przy stalym ciśnieniu (izobara).
-
Upload
nguyendang -
Category
Documents
-
view
214 -
download
0
Transcript of Wykres a) ilustruje zale no pr no ci par składników A i B ... · odst ępstwa od prawa...

Destylacja i rektyfikacja Destylacja to metoda rozdziału i oczyszczania mieszanin substancji ciekłych. W najprostszym przypadku polega na doprowadzeniu cieczy do wrzenia i skropleniu powstałej pary w chłodnicy. W sytuacji, gdy w ruchu jest tylko jedna faza, czyli para, mamy do czynienia z destylacją prostą. Jeśli w przeciwprądzie do par przemieszczać się będzie część skroplonych par (orosienie, flegma) spływające do naczynia z destylowaną cieczą będziemy mieli zjawisko deflegmacji lub rektyfikacji. Jeśli dwie ciecze A i B rozpuszczają się nawzajem w dowolnym stosunku zachodzi wówczas zjawisko nazywane prawem Raoulta, które mówi, Ŝe pręŜność pary danego składnika ciekłej mieszaniny nad roztworem jest proporcjonalna do ułamka molowego tego składnika w roztworze:
pA = poxA
gdzie p0 - pręŜność par składnika A nad czystym składnikiem, xA - ułamek molowy składnika A w roztworze, pA - pręŜność pary składnika A nad roztworem. PoniewaŜ proces krzepnięcia i wrzenia jest silnie powiązany z pręŜnością par nad krzepnącą lub wrzącą cieczą, stąd pochodna od prawa Raoulta zasada zmian temperatury wrzenia i krzepnięcia. Reguła ta w praktyce słuŜyła do wyznaczania masy cząsteczkowej nieznanej substancji metodą ebulioskopową (wrzenie) lub krioskopową (krzepnięcie) poprzez badanie wielkości zmian temperatury wrzenia (krzepnięcia) po rozpuszczeniu znanej ilości nieznanej substancji w rozpuszczalniku o znanej stałej ebulioskopowej (krioskopowej). Prawo Raoulta jest słuszne dla roztworów idealnych (brak oddziaływań międzycząsteczkowych) a dla roztworów rzeczywistych stosuje się w zasadzie tylko w przypadku roztworów rozcieńczonych. Dla wielu przypadków, szczególnie gdy mamy do czynienia z roztworami stęŜonymi (kilkuprocentowymi) i o silnych oddziaływaniach (np. etanol-woda) występują silne odstępstwa od prawa (azeotropia). Jeśli mieszanina dwuskładnikowa spełnia bez odchyleń prawo Raoulta mamy przypadek roztworu doskonałego. Taka sytuacja zachodzi w nielicznych przypadkach, gdy mamy roztwory substancji z szeregów homologicznych, np.: benzen – toluen, metanol – etanol.
Wykres a) ilustruje zaleŜność pręŜności par składników A i B od składu tej mieszaniny przy stałej temperaturze (izoterma). Na wykresie b) przedstawiono zaleŜność między temperaturą cieczy i pary w zaleŜności od składu mieszaniny przy stałym ciśnieniu (izobara).
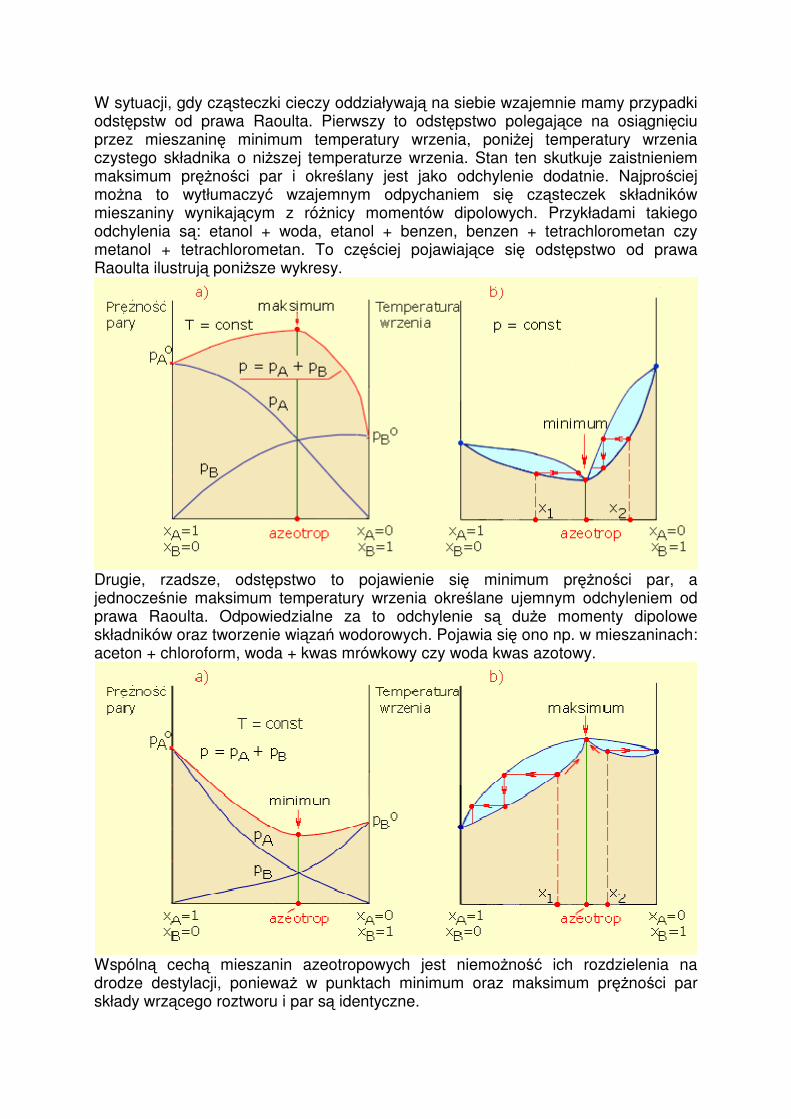
W sytuacji, gdy cząsteczki cieczy oddziaływają na siebie wzajemnie mamy przypadki odstępstw od prawa Raoulta. Pierwszy to odstępstwo polegające na osiągnięciu przez mieszaninę minimum temperatury wrzenia, poniŜej temperatury wrzenia czystego składnika o niŜszej temperaturze wrzenia. Stan ten skutkuje zaistnieniem maksimum pręŜności par i określany jest jako odchylenie dodatnie. Najprościej moŜna to wytłumaczyć wzajemnym odpychaniem się cząsteczek składników mieszaniny wynikającym z róŜnicy momentów dipolowych. Przykładami takiego odchylenia są: etanol + woda, etanol + benzen, benzen + tetrachlorometan czy metanol + tetrachlorometan. To częściej pojawiające się odstępstwo od prawa Raoulta ilustrują poniŜsze wykresy.
Drugie, rzadsze, odstępstwo to pojawienie się minimum pręŜności par, a jednocześnie maksimum temperatury wrzenia określane ujemnym odchyleniem od prawa Raoulta. Odpowiedzialne za to odchylenie są duŜe momenty dipolowe składników oraz tworzenie wiązań wodorowych. Pojawia się ono np. w mieszaninach: aceton + chloroform, woda + kwas mrówkowy czy woda kwas azotowy.
Wspólną cechą mieszanin azeotropowych jest niemoŜność ich rozdzielenia na drodze destylacji, poniewaŜ w punktach minimum oraz maksimum pręŜności par składy wrzącego roztworu i par są identyczne.

PoniewaŜ rozdział azeotropu na drodze destylacji nie jest moŜliwy, aby go dokonać naleŜy posłuŜyć się innymi technikami. Pierwsza to wprowadzenie do mieszaniny trzeciego składnika, który spowoduje zmianę właściwości rozdzielanej mieszaniny. Sposób ten wykorzystywano w przeszłości przy otrzymywaniu bezwodnego etanolu. Do rektyfikatu (96% v/v) dodawany był benzen i przeprowadzano destylację frakcyjną. W pierwszej kolejności oddestylowywał azeotrop trójskładnikowy (etanol-woda-benzen), następnie etanol z benzenem, a na końcu czysty (99,8% v/v) etanol. Metoda ta ma znaczenie historyczne, poniewaŜ z racji na toksyczność benzenu została zaniechana. Obecnie jako czynnika tworzącego azeotrop trójskładnikowy uŜywa się cykloheksanu. Inna metoda umoŜliwiająca rozdział azeotropu to zmiana ciśnienia, pod jakim prowadzi się destylację. Ilustruje to przykład:
Nowymi metodami stosowanymi w rozdziale azeotropów, wprowadzonymi pod koniec XX w. są perwaporacja i permeacja. Polegają na wykorzystaniu membran polimerowych w celu oddzielenia jednego ze składników azeotropu na drodze dyfuzji przez porowatą przegrodę. DESTYLACJA PROSTA I Z DEFLEGMACJĄ Destylację prostą przeprowadza się w aparaturze zbudowanej z kotła destylacyjnego (1) zaopatrzonego na szczycie w hełm (3), którego zadaniem jest niedopuszczenie do przedostania się do chłodnicy drobinek cieczy porwanych przez parę. Ogrzewanie dokonywane jest za pomocą wbudowanej węŜownicy zasilanej parą wodną lub innym medium grzejnym (2). Pary skraplane są w chłodnicy (4), skąd destylat spływa do odbieralników (5).
Podobnie przebiega destylacja z deflegmacją, z tym Ŝe między kotłem destylacyjnym a chłodnicą umieszczany jest deflegmator. Jego zadaniem jest wzbogacanie

destylatu w składnik bardziej lotny. Część par wędrujących przez deflegmator w górę ulega skropleniu, oddając ciepło kondensacji mieszaninie wędrującej do góry. Tą drogą para ulega wzbogaceniu w składnik bardziej lotny, dzięki czemu rozdział mieszaniny jest
dokładniejszy.
Deflegmator – przekrój poprzeczny DESTYLACJA Z PARĄ WODNĄ Przy wydzielaniu niewykorzystanych reagentów, izolacji produktów reakcji oraz izolacji olejków eterycznych stosowana jest destylacja z parą wodną. Rządzi nią prawo Daltona, które mówi, Ŝe ciśnienie wywierane przez mieszaninę par jest równe sumie ciśnień wywieranych przez składniki mieszaniny, gdyby kaŜdy z nich był umieszczany osobno w tych samych warunkach objętości i temperatury, jest ono zatem sumą ciśnień cząstkowych.
Tak więc by przeprowadzić destylację z parą wodną wydzielana substancja nie moŜe mieszać się z wodą i musi wykazywać pewną, nawet niewielką pręŜność par w temperaturze wrzenia wody. Aparat do destylacji zbudowany jest z kotła destylacyjnego (1) doprowadzeniem pary do wnętrza przez bełkotkę (2). Stosuje się tu ogrzewanie bezpośrednie, gdyŜ para wodna jednocześnie słuŜy do doprowadzenia zawartości kotła do temperatury wrzenia oraz przenosi destylowaną substancję do chłodnicy (3). Destylat trafia do flaszki florentyńskiej, z której woda jest lewarowana na zewnątrz, a produkt, lŜejszy od niej, pozostaje we wnętrzu naczynia.

Aparatura do destylacji z parą wodną DESTYLACJA MOLEKULARNA Aby poddać destylacji substancje wraŜliwe na rozkład w wysokiej temperaturze stosujemy destylację pod zmniejszonym ciśnieniem lub destylację molekularną, z tym, Ŝe destylacja pod zmniejszonym ciśnieniem znalazła zastosowanie dla substancji o niewielkiej masie cząsteczkowej, zaś destylacja molekularna w przypadku substancji o duŜej masie cząsteczkowej. Wielkość przemysłowych aparatów do destylacji próŜniowej ogranicza konieczność sprostania wielkim siłom, jakie po obniŜeniu ciśnienia wywiera na ich powierzchnię ciśnienie atmosferyczne. Konstrukcja aparatury jest podobna do aparatów stosowanych przy destylacji prostej, z tym, Ŝe zapewniona musi być ich większa wytrzymałość. Korzyścią jest mniejsze zapotrzebowanie na energię potrzebną do destylacji. Konieczne jest jednak staranne uszczelnienie aparatury i dostarczenie próŜni o odpowiedniej jakości. Proces tem prowadzony jest przy ciśnieniach od 100 do 0,01 mmHg. Z kolei destylacja molekularna wymaga jeszcze wyŜszej próŜni rzędu 0,01 do 0,0001 mmHg. W przypadku destylacji w warunkach wysokiej próŜni lotności względne związków chemicznych zaleŜą nie tylko od stosunku pręŜności par nasyconych składników destylowanej mieszaniny, lecz takŜe od pierwiastka ze stosunku mas molowych rozdzielanych związków chemicznych, pod warunkiem, Ŝe nie występują między nimi wiązania wodorowe i zbyt silne oddziaływania międzycząsteczkowe. Przy tak niskich ciśnieniach droga swobodna cząsteczek ulega znacznemu wydłuŜeniu i pozwala na pokonanie odległości między powierzchnią destylowanej cieczy a powierzchnią

kondensacyjną bez zderzeń z innymi cząsteczkami. Przykładowe przekroje urządzeń do destylacji molekularnej pokazuje poniŜsza ilustracja.
W procesie destylacji molekularnej istotną rolę odgrywa właściwe przygotowanie cieczy poddawanej destylacji. Musi ona być odgazowana i pozbawiona resztek rozpuszczalników, co mogło by zaburzyć przebieg procesu. Tak przygotowana ciecz wprowadzana jest pod postacią cienkiego filmu na rozgrzane do temperatury 150 - 300°C walce. Po odparowaniu substancja pokonuje drogę ok. 2 cm i dociera do ochłodzonej powierzchni, na której się skrapla. PoniŜsze ilustracje przedstawiają urządzenia do destylacji molekularnej pracujące w pozycji pionowej, w których destylowana substancja spływa w dół po ogrzewanych walcach.
REKTYFIKACJA

Jeśli mamy rozdzielić mieszaninę kilku cieczy o zbliŜonych temperaturach wrzenia konieczne jest dokonanie wielokrotnego odparowania i skroplenia. W ten sposób przebiega rektyfikacja. Na kaŜdej z półek kolumny rektyfikacyjnej zachodzi proces destylacji prostej. Czynnikiem odpowiedzialnym za ogrzewanie są przemieszczające się w górę pary rozdzielanej mieszaniny, które kondensując na kolejnych półkach przekazują znajdującej się tam cieczy ciepło skraplania doprowadzają ją do wrzenia. Konstrukcja kolumny rektyfikacyjnej umoŜliwia wymianę ciepła i masy pomiędzy wędrującymi ku górze parami a spływającą w dół cieczą. W ten sposób pary wzbogacane są w składnik bardziej lotny, a spływająca do kotła destylacyjnego ciecz w składnik o mniejszej lotności. Pary dochodzące do szczytu kolumny są skraplane. Warunkiem przeprowadzenia rektyfikacji jest zawracana części kondensatu z powrotem do kolumny. Oznacza to, Ŝe jedynie część kondensatu odbierana jest jako destylat.
Zasada działania kolumny dzwonowej Na szczycie kolumny ulokowana jest głowica, która odpowiada za skraplanie par i ich rozdział na dwa strumienie: destylat i odciek. Zawracanie części destylatu spełnia w rektyfikacji zasadniczą rolę. Ilość destylatu powracająca do kolumny wpływa na parametry procesu. Zwiększenie odcieku z jednej strony zwiększa rozdzielczość kolumny, z drugiej podnosi koszty procesu i moŜe powodować trudności techniczne, np. zalanie kolumny. Kolumny rektyfikacyjne mogą mieć róŜnorodną budowę. NajwyŜszą sprawnością wyróŜniają się kolumny dzwonowe oraz kapkowe, będące modyfikacją kolumn dzwonowych. W tych rodzajach kolumn dna są pełne, a przepływ par zachodzi w obrębie dzwonów lub kapek, ciecz przepływa zaś przez rury przelewowe. PoniewaŜ utrzymanie cieczy na półkach nie zaleŜy od prędkości przepływu pary moŜliwy jest stosunkowo długi kontakt pary z cieczą, dzięki czemu istnieje moŜliwość ustalenia się równowagi ciecz-para i dokładny rozdział.

Kolumny rektyfikacyjne półkowe
Kolejny typ to kolumny sitowe, których półki zbudowane są z materiału perforowanego. Otwory mają średnicę tak dobraną, by przemieszczająca się ku górze para równowaŜyła ciśnienie hydrostatyczne cieczy utrzymując ją na półce.
KaŜda półka ma otwór przelewowy, który pozwala cieczy spływać na niŜszy poziom. Warunkiem poprawnego działania tego typu kolumny jest doskonałe
wypoziomowanie półek, by warstwa cieczy miała na całej powierzchni jednakową grubość. Sprawność tych kolumn w porównaniu z dzwonowymi jest niŜsza, poniewaŜ
ciecz na półkach utrzymywana jest przez pary wędrujące ku górze.
Oprócz kolumn wymienionych rodzajów uŜywane są równieŜ kolumny z wypełnieniem, w których za kontakt par z cieczą odpowiadają umieszczone wewnątrz

kształtki o rozwiniętej powierzchni. W tym rodzaju kolumn nie ma półek, a całą objętość kolumny zajmują elementy wypełnienia. Wykonuje się je z róŜnorodnych, odpornych chemicznie materiałów, np. ze stali, ceramiki i szkła. Wypełnienie moŜe mieć róŜne kształty, a ilustrują to poniŜsze zdjęcia:
Pierścienie zwijane z perforowanej taśmy stalowej
Pierścienie Rashiga wykonane z ceramiki
Szklane pierścienie Rashiga Ceramiczne siodełka Berla
Wypełnienie strukturalne Kolumny z wypełnieniem są prostsze w konstrukcji i tańsze od kolumn półkowych, lecz ich sprawność jest mniejsza. Wykorzystuje się je do rektyfikacji pod ciśnieniem atmosferycznym, ale dobrze sprawują się przy pracy pod zmniejszonym ciśnieniem, poniewaŜ stawiają znacznie mniejszy opór od kolumn półkowych.
Przykłady kolumn z wypełnieniem
Proces rektyfikacji moŜe być prowadzony metodami okresową lub ciągłą. W metodzie okresowej po napełnieniu kotła destylacyjnego rozdzielaną mieszaniną prowadzi się proces rektyfikacji do momentu wyczerpania się pozyskiwanego składnika. W kotle pozostaje ciecz wyczerpana. Przy rektyfikacji ciągłej poddawana rozdziałowi mieszanina (surówka) wprowadzana jest na półkę, na której skład cieczy jest identyczny z rozdzielaną. Zasilająca mieszanina jest ogrzana do temperatury wrzenia przed wprowadzeniem na kolumnę. Ilustruje to poniŜszy schemat.













![3 ISKY.DEPO No.2 No.a No.4 No.5 No.6 No.7 No.8 No.9 No. 10 ... · 3 ISKY.DEPO No.2 No.a No.4 No.5 No.6 No.7 No.8 No.9 No. 10 No. 11 No. 12 No. 13 No. 14 Y276-0004 Baa c] : 00) TEL](https://static.fdocuments.pl/doc/165x107/5f9d923ad67aba75cc50432e/3-iskydepo-no2-noa-no4-no5-no6-no7-no8-no9-no-10-3-iskydepo-no2.jpg)





