
Wydzial Chemii praca magisterska - chemia.uj.edu.plzcht/mgrfiles/2008-wlodarczyk.pdf · praca...
53
Uniwersytet Jagiello´ nski Wydzial Chemii praca magisterska Implementacja metody TDDFT uwzgl , edniaj , aca w przybli ˙ zony spos´ob wplyw konfiguracji podw´ojnie wzbudzonych na energie wzbudze´ n Radoslaw Stanislaw Wlodarczyk Praca wykonana w Zakladzie Metod Obliczeniowych Chemii pod kierunkiem dr Grzegorza Mazura 2008
Transcript of Wydzial Chemii praca magisterska - chemia.uj.edu.plzcht/mgrfiles/2008-wlodarczyk.pdf · praca...

Uniwersytet Jagiellonski
Wydzia l Chemii
praca magisterska
Implementacja metody TDDFTuwzgl edniaj aca w przyblizony sposob wp lyw
konfiguracji podwojnie wzbudzonych naenergie wzbudzen
Rados law Stanis law W lodarczyk
Praca wykonana w Zak ladzie Metod Obliczeniowych Chemiipod kierunkiem dr Grzegorza Mazura
2008

Serdecznie dzi ekuj e Panu dr Grzegorzowi Mazurowi
za olbrzymi a wyrozumia losc, nieocenion a pomoc,
a przede wszystkim za zarazenie zapa lem do pracy naukowej.
Dzi ekuj e takze Panu dr hab. Jackowi Korchowcowi
za udost epnienie komputera do przeprowadzenia obliczen.

Spis tresci
Rozdzia l 1. Wstep . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
Rozdzia l 2. TDDFT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
2.1. Podstawy formalizmu . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
2.2. J adro korelacyjno-wymienne . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
2.3. Wybrane warianty TDDFT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
2.4. Dressed TDDFT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
2.5. Ograniczenia Dressed TDDFT . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
Rozdzia l 3. Implementacja metody TDDFT . . . . . . . . . . . . . . . . . . 19
3.1. Implementacja TDA-TDDFT . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
3.2. Implementacja RPA-TDDFT . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
3.3. Automatyczna generacja kodu . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
3.4. Wydajnosc . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
3.5. Implementacja Dressed TDA-TDDFT . . . . . . . . . . . . . . . . . . . . . . . 30
Rozdzia l 4. Obliczenia dla wybranych polienow liniowych . . . . . . . . . 31
4.1. Wst ep . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
4.2. Obliczenia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
4.3. Wyniki i dyskusja . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
Rozdzia l 5. Podsumowanie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
Bibliografia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43
Dodatek A. Eliminacja wspolnych podwyrazen . . . . . . . . . . . . . . . . 46
Streszczenie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
Abstract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51
1

Rozdzia l 1
Wstep
Opis w lasciwosci elektronowych stanow wzbudzonych stanowi istotny czynnik roz-
woju wielu dzia low wspo lczesnej chemii i biochemii. Nieodzowne jest posiadanie do-
k ladnych, a zarazem praktycznie stosowalnych, modeli i metod obliczeniowych, umoz-
liwiaj acych badania teoretyczne oraz interpretacj e danych doswiadczalnych.
Mozliwosc badania stanow wzbudzonych daj a niektore z metod obliczeniowych
opartych na funkcji falowej [1]. Uwzgl ednienie korelacji kulombowskiej jest jednak
bardzo kosztowne i wykonywanie obliczen staje si e utrudnione bardzo szybko ze wzro-
stem uk ladu. Poszukiwanie bardziej wydajnych metod sta lo si e koniecznosci a wraz
z nadejsciem ery badan duzych uk ladow biologicznych.
W 1964 roku Hohenberg i Kohn [2] dowiedli, ze do pe lnego opisu stanu podsta-
wowego wystarczy znajomosc rozk ladu g estosci elektronowej w tym stanie. Ponadto
sformu lowali zasad e wariacyjn a wskazuj ac a kierunek poszukiwan optymalnej g estosci
stanu podstawowego. Ich twierdzenia stanowi a podstaw e rozwoju metod obliczenio-
wych opartych na g estosci elektronowej.
Rok pozniej Kohn i Sham [3] zaproponowali praktyczny schemat obliczeniowy
w ramach Density Functional Theory (DFT). Pozwala on szybko przeprowadzac dosc
dok ladne obliczenia dla stanow podstawowych.1 Efektywnie umozliwia to obliczenia
niektorych energii wzbudzen, jednak metoda ich wyznaczania, tak zwana ∆-DFT, jest
rzadko stosowana.
1 stan podstawowy oznacza energetycznie najnizej lez acy stan o danej symetrii
2

Sukces DFT dla stanow podstawowych zach eci l do poszukiwania pokrewnych me-
tod opisuj acych stany wzbudzone. Mi edzy innymi Runge oraz Gross rozwazali, oparty
na tej metodzie, zalezny od czasu opis eksperymentow rozproszeniowych. Przeprowa-
dzona przez nich proba generalizacji DFT zaowocowa la prze lomow a publikacj a z 1984
roku [4]. Zawarte w niej twierdzenie jest odpowiednikiem twierdzenia Hohenberga-
-Kohna dla zaleznego od czasu rownania Schrodingera [5].
Bazuj aca na twierdzenu Runge-Grossa metoda Time-Dependent Density Functio-
nal Theory (TDDFT) wyprowadzona zosta la przez zastosowanie rachunku zaburzen,
przy wykorzystaniu formalizmu Kohna-Shama. Pozwala ona niskim kosztem oblicze-
niowym uzyskac opis stanow wzbudzonych przy dobrym uwzgl ednieniu korelacji elek-
tronowej. Dzi eki temu stosowac j a mozna nawet do stosunkowo duzych uk ladow.
Niniejsza praca ma dwa cele. Pierwszym z nich jest opracowanie wydajnej imple-
mentacji TDDFT oraz rozszerzenia TDDFT, metody Dressed TDDFT (DTDDFT).
Drugim celem jest zbadanie stosowalnosci i dok ladnosci DTDDFT dla wybranych
zwi azkow chemicznych.

Rozdzia l 2
TDDFT
Time-Dependent DFT oparte jest na rachunku zaburzen zaleznych od czasu. Zabu-
rzenie dzia laj ace na dowolny uk lad wywo luje jego reakcj e, a zachodz ace zmiany mozna
opisac za pomoc a funkcji odpowiedzi. W szczegolnosci, gdy zjawisko to dotyczy za-
burzenia g estosci elektronowej uk ladu znajduj acego si e w stanie podstawowym, miar a
zaburzenia jest funkcja odpowiedzi g estosci stanu podstawowego.
Stosowany w formalizmie TDDFT rachunek zaburzen moze byc w ogolnosci rozwi-
ni ety do dowolnego rz edu. Akronim TDDFT utozsamiany jest jednak z teori a opart a
na rachunku zaburzen pierwszego rz edu. Przedstawione w niniejszej pracy podstawy
teoretyczne tej metody bazuj a na pracach E.K.U. Grossa et al [5–7].
2.1. Podstawy formalizmu
Celem przytoczonego wyprowadzenia jest uzyskanie wyrazenia na energie wzbu-
dzen Ω rzeczywistego uk ladu. Przyjmuj e, ze przejscie uk ladu do stanu wzbudzonego
moze zachodzic w efekcie oddzia lywania z zaburzeniem zewn etrznym, np. w postaci fali
elektromagnetycznej. Zaburzenie to wyraza si e poprzez zmian e potencja lu zewn etrz-
nego, w jakim znajduje si e uk lad. Funkcja odpowiedzi g estosci elektronowej stanu
podstawowego na zmian e potencja lu zewn etrznego jest zdefiniowana jako
χσσ′(r, t, r′, t′) =δρσ(r, t)
δV extσ′ (r′, t′)
(2.1)
4

2.1. Podstawy formalizmu
gdzie δV extσ′ (r′, t′) oznacza infinitezymaln a zmian e potencja lu zewn etrznego, natomiast
δρσ(r, t) to odpowiedz g estosci na zaburzenie. Poniewaz energie wzbudzen uk ladu dane
s a przez bieguny χσσ′(r, t, r′, t′), dlatego tez g lownym celem staje si e badanie ci ag losci
tej funkcji.
By otrzymac rozwi azanie oparte na DFT, nalezy rozwazyc uk lad cz astek Kohna-Sha-
ma (KS), ktorych g estosc reprodukuje g estosc stanu podstawowego. Z definicji cz astki
te znajduj a si e w efektywnym potencjale Kohna-Shama V KSσ′ , ktory zdefiniowany jest
jako
V KSσ′ (r, t) = V ext
σ′ (r, t) + V H(r, t) + V xcσ′ (r, t) (2.2)
gdzie V extσ′ to potencja l zewn etrzny pochodz acy od dodatniego ladunku j ader atomo-
wych, V H jest potencja lem odpychania cz astka-cz astka, a potencja l korelacyjno-wy-
mienny V xcσ′ odpowiada za wszystkie pozosta le oddzia lywania wielocia lowe.
Korzystaj ac z regu ly lancucha dla pochodnych funkcjonalnych [8], row. (2.1) mozna
przekszta lcic do postaci
χσσ′(r, t, r′, t′) =δρσ(r, t)
δV extσ′ (r′, t′)
=∑σ′′
∫∫δρσ(r, t)
δV KSσ′′ (r′′, t′′)
δV KSσ′′ (r′′, t′′)
δV extσ′ (r′, t′)
dr′′dt′′ (2.3)
Wyst epuj acy w powyzszym rownaniu element δρ(r, t)/δV KSσ′′ (r′′, t′′) opisuje liniow a
odpowiedz g estosci na infinitezymaln a zmian e efektywnego potencja lu Kohna-Shama
χsσσ′′(r, t, r′′, t′′) =δρσ(r, t)
δV KSσ′′ (r′′, t′′)
(2.4)
Jest to definicja funkcji liniowej odpowiedzi g estosci nieoddzia luj acych cz astek Kohna-
-Shama.
Uwzgl ednienie zaleznosci (2.2) pozwala jawnie zapisac drugi z cz lonow wyst epuj a-
cych pod ca lk a w row. (2.3) jako
δV KSσ′′ (r′′, t′′)
δV extσ′ (r′, t′)
=δV ext
σ′′ (r′′, t′′)
δV extσ′ (r′, t′)
+δV H(r′′, t′′)
δV extσ′ (r′, t′)
+δV xc
σ′′ (r′′, t′′)
δV extσ′ (r′, t′)
(2.5)
Wykonanie w otrzymanym rownaniu rozniczkowania funkcjonalnego, przy uwzgl ednie-
niu zaleznosci (2.1), prowadzi do
δV KSσ′′ (r′′, t′′)
δV extσ′ (r′, t′)
= δσ′′σ′δ(r′′ − r′)δ(t′′ − t′) +
+∑σ′′′
∫∫ [δ(t′′ − t′′′)|r′′ − r′′′|
+δV xc
σ′′ (r′′, t′′)
δρσ′′′(r′′′, t′′′)
]χσ′′′σ′(r′′′, t′′′, r′, t′) dr′′′dt′′′ (2.6)
5

2.1. Podstawy formalizmu
Rownanie na funkcj e odpowiedzi uk ladu na zaburzenie zewn etrzne (2.3) przyjmuje
ostateczn a, scis l a w ramach rozwazania odpowiedzi liniowej, samouzgodnion a postac
po wstawieniu row. (2.6) oraz (2.4)
χσσ′(r, t, r′, t′) = χsσσ′(r, t, r′, t′) +
+∑σ′′σ′′′
∫∫∫∫χsσσ′′(r, t, r′′, t′′)
[δ(t′′ − t′′′)|r′′ − r′′′|
+ fxcσ′′σ′′′(r′′, t′′, r′′′, t′′′)
]×
× χσ′′′σ′(r′′′, t′′′, r′, t′) dr′′dt′′dr′′′dt′′′ (2.7)
gdzie wprowadzone j adro korelacyjno-wymienne
fxcσ′′σ′′′(r′′, t′′, r′′′, t′′′) =δV xc
σ′′ (r′′, t′′)
δρσ′′′(r′′′, t′′′)(2.8)
jest zdefiniowane jako pochodna funkcjonalna potencja lu korelacyjno-wymiennego wzgl e-
dem g estosci elektronowej.
Wymnozenie obu stron rownania (2.7) przez δV extσ′ (r′, t′), a nast epnie wysumowanie
po σ′ i wyca lkowanie po zmiennej r′, prowadzi do
∑σ′
∫χσσ′(r, t, r′, t′) δV ext
σ′ (r′, t′) dr′ =
=
[∑σ′
∫χsσσ′(r, t, r′, t′) δV ext
σ′ (r′, t′) dr′
]+
+∑σ′′σ′′′
∫∫∫∫χsσσ′′(r, t, r′′, t′′)
[δ(t′′ − t′′′)|r′′ − r′′′|
+ fxcσ′′σ′′′(r′′, t′′, r′′′, t′′′)
]×
×
[∑σ′
∫χσ′′′σ′(r′′′, t′′′, r′, t′)δV ext
σ′ (r′, t′)dr′
]dr′′dt′′dr′′′dt′′′ (2.9)
Wyst epuj acy w tym rownaniu cz lon
∑σ′
∫χσσ′(r, t, r′, t′) δV ext
σ′ (r′, t′) dr′ =
=∑σ′
∫δρσ(r, t)
δV extσ′ (r′, t′)
δV extσ′ (r′, t′) dr′ ≡ δρσ(r, t) (2.10)
jest definicj a rozniczki g estosci jako funkcjona lu potencja lu zewn etrznego.
6

2.1. Podstawy formalizmu
Rownanie (2.9) przyjmuje zatem postac
δρσ(r, t) =
[∑σ′
∫χsσσ′(r, t, r′, t′) δV ext
σ′ (r′, t′) dr′
]+
+∑σ′σ′′
∫∫∫∫χsσσ′(r, t, r′, t′)
[δ(t′ − t′′)|r′ − r′′|
+ fxcσ′σ′′(r′, t′, r′′, t′′)
]×
× δρσ′′(r′′, t)dr′dt′dr′′dt′′ (2.11)
Wykonanie transformacji Fouriera pozwala zapisac
δρσ(r, ω) =
[∑σ′
∫χsσσ′(r, r′, ω) δV ext
σ′ (r′, ω) dr′
]+
+∑σ′σ′′
∫∫χsσσ′(r, r′, ω)
[1
|r′ − r′′|+ fxcσ′σ′′(r′, r′′, ω)
]δρσ′′(r′′, ω) dr′dr′′ (2.12)
Rownanie to wi aze odpowiedz g estosci rzeczywistego uk ladu δρ z funkcj a odpowiedzi
uk ladu cz astek Kohna-Shama χs. Jest to istotne, gdyz bieguny funkcji odpowiedzi χs
s a znane.
Dalsze przekszta lcenie rownania (2.12) pozwala otrzymac ca lkow a postac rownania
na liniow a odpowiedz g estosci.
∑σ′
∫χsσσ′(r, r′, ω) δV ext
σ′ (r′, ω) dr′ =
=∑σ′′
∫ [δσσ′′δ(r − r′′) −
∑σ′
∫χsσσ′(r, r′, ω)×
×(
1
|r′ − r′′|+ fxcσ′σ′′(r′, r′′, ω)
)dr′]δρσ′′(r′′, ω) dr′′ (2.13)
Funkcj e odpowiedzi g estosci cz astek Kohna-Shama χs mozna przedstawic w bazie
niezaburzonych orbitali KS φ. Niech indeksy r, s, t, u oznaczaj a dowolne orbitale KS,
i oraz j zaj ete orbitale KS, natomiast a i b wirtualne orbitale KS. Jezeli ωrsσ = εrσ−εsσoznacza roznic e energii orbitalnych, a nr oraz ns to odpowiadaj ace liczby obsadzen,
wtedy
χsσσ′(r, r′, ω) = δσσ′
∑sr
(nsσ − nrσ)φ∗sσ(r)φrσ(r)φsσ(r′)φ∗rσ(r′)
ω − ωrsσ + ıη(2.14)
Z rownania na χsσσ′(r, r′, ω) wynika, ze opisywana funkcja odpowiedzi uk ladu cz astek
KS ma bieguny dla ω = ωrsσ.
Energie wzbudzen rzeczywistego uk ladu Ω s a w ogolnosci rozne od energii wzbudzen
7

2.1. Podstawy formalizmu
uk ladu cz astek Kohna-Shama (Ω 6= ωrsσ). Zatem dla rzeczywistych energii wzbudzen
ω → Ω lewa strona rownania (2.13) pozostaje okreslona. Z drugiej strony, funkcja li-
niowej odpowiedzi g estosci rzeczywistego uk ladu, a wi ec takze δρσ′′(r′′, ω), ma bieguny
dla energii wzbudzen ω = Ω. Dlatego operator ca lkowy, wyst epuj acy po prawej stronie
rownania (2.13)
∑σ′′
∫ [δσσ′′δ(r − r′′) −
∑σ′
∫χsσσ′(r, r′, ω)×
×(
1
|r′ − r′′|+ fxcσ′σ′′(r′, r′′, ω)
)dr′]dr′′ (2.15)
dzia laj acy na δρσ′′(r′′, ω), nie moze byc odwracalny dla ω → Ω. Inaczej mowi ac, war-
tosci w lasne tego operatora musz a si e zerowac dla ω = Ω. Wystarczy zatem znalezc ω
dla ktorych spe lniony jest warunek
∑σ′′
∫ [δσσ′′δ(r − r′′) −
∑σ′
∫χsσσ′(r, r′, ω)×
×(
1
|r′ − r′′|+ fxcσ′σ′′(r′, r′′, ω)
)dr′]ξσ′′(r′′, ω) dr′′ = 0 (2.16)
lub inaczej
∑σ′σ′′
∫∫χsσσ′(r, r′, ω)
(1
|r′ − r′′|+ fxcσ′σ′′(r′, r′′, ω)
)ξσ′′(r′′, ω) dr′dr′′ =
= ξσ(r, ω) (2.17)
Otrzymane rownanie ca lkowe jest bardzo trudne to rozwi azania. By uzyskac rozwi a-
zanie w bazie niezaburzonych orbitali Kohna-Shama, nalezy wstawic jawn a postac
χsσσ′(r, r′, ω) z row. (2.14). Otrzymuje si e wtedy zaleznosc
∑sr
(nsσ − nrσ)φ∗sσ(r)φrσ(r)
ω − ωrsσ + ıηζrsσ(ω) = ξσ(r, ω) (2.18)
gdzie
ζrsσ(ω) =
=∑σ′′
∫∫φ∗rσ(r′)φsσ(r′)
(1
|r′ − r′′|+ fxcσσ′′(r′, r′′, ω)
)ξσ′′(r′′, ω) dr′dr′′ (2.19)
8

2.1. Podstawy formalizmu
Wstawienie ξσ(r, ω) z rownania (2.18) do (2.19) daje postac rowania macierzowego
∑σ′
∑ut
Mrsσtuσ′(ω)
ω − ωtuσ′ + ıηζtuσ′(ω) = ζrsσ(ω) (2.20)
gdzie
Mrsσtuσ′(ω) = (nuσ′ − ntσ′)×
×∫∫
φ∗rσ(r′)φsσ(r′)
(1
|r′ − r′′|+ fxcσσ′(r′, r′′, ω)
)φtσ′(r′′)φ∗uσ′(r′′)dr′dr′′ (2.21)
W ten sposob skomplikowane rownanie ca lkowe (2.17) przekszta lcone zosta lo do formy
rownania macierzowego.
Rownanie (2.20) da si e przeformu lowac do postaci zagadnienia w lasnego na energie
wzbudzen Ω. Jezeli ω = Ω, to
∑σ′
∑ut
Mrsσtuσ′(Ω)
Ω− ωtuσ′ + ıηζtuσ′(Ω) = ζrsσ(Ω) (2.22)
Wstawienie nowej zmiennej βtuσ′ = ζtuσ′(Ω)/(Ω− ωtuσ′)∑σ′
∑ut
Mrsσtuσ′(Ω)βtuσ′ = (Ω− ωrsσ)βrsσ (2.23)
i proste przekszta lcenie doprowadzaj a do ostatecznej postaci uogolnionego zagadnienia
w lasnego ∑σ′
∑ut
(Mrsσtuσ′(Ω) + δsu δrt δσσ′ ωrsσ)βtuσ′ = Ωβrsσ (2.24)
Rozwi azanie otrzymanego zagadnienia jest przeprowadzane w praktyce przy ograni-
czeniu rozwini ecia zastosowanego w row. (2.14) do sumy skonczonej.
Istnieje wygodniejsza postac rozwi azania (2.24). Aby j a uzyskac, nalezy w rownaniu
(2.12) skorzystac z odpowiedzi g estosci wyrazonej przez [5]
δρσ(r, ω) =∑ia
[γiaσ(ω)φiσ(r)φ∗aσ(r) + γaiσ(ω)φaσ(r)φ∗iσ(r)] (2.25)
9

2.1. Podstawy formalizmu
By zachowac przejrzystosc wyprowadzenia rozwazam poszczegolne elementy row-
nania (2.12)
δρσ(r, ω) =
[∑σ′
∫χsσσ′(r, r′, ω) δV ext
σ′ (r′, ω) dr′
]+
+∑σ′σ′′
∫∫χsσσ′(r, r′, ω)
[1
|r′ − r′′|+ fxcσ′σ′′(r′, r′′, ω)
]δρσ′′(r′′, ω) dr′dr′′
Wstawienie jawnej postaci funkcji odpowiedzi g estosci cz astek Kohna-Shama
χsσσ′(r, r′, ω) do pierwszego z elementow sumy wyst epuj acej po prawej stronie prowadzi
do
∑σ′
∫χsσσ′(r, r′, ω) δV ext
σ′ (r′, ω) dr′ =
=∑σ′
∫δσσ′
∑sr
(nsσ − nrσ)φ∗sσ(r)φrσ(r)φsσ(r′)φ∗rσ(r′)
ω − ωrsσ + ıηδV ext
σ′ (r′, ω) dr′ =
=∑sr
(nsσ − nrσ)φ∗sσ(r)φrσ(r)
ω − ωrsσ + ıηδV ext
rsσ (2.26)
gdzie
δV extrsσ =
∫φ∗rσ(r′)δV ext
σ (r′, ω)φsσ(r′)dr′ (2.27)
Dok ladnie w ten sam sposob przekszta lcam drugi element sumy
∑σ′σ′′
∫∫χsσσ′(r, r′, ω)
[1
|r′ − r′′|+ fxcσ′σ′′(r′, r′′, ω)
]δρσ′′(r′′, ω) dr′dr′′ =
=∑σ′′
∑sr
(nsσ − nrσ)φ∗sσ(r)φrσ(r)
ω − ωrsσ + ıη
∫∫φsσ(r′)φ∗rσ(r′) ×
×[
1
|r′ − r′′|+ fxcσσ′′(r′, r′′, ω)
]δρσ′′(r′′, ω) dr′dr′′ (2.28)
W otrzymanym rownaniu w miejsce δρσ′′(r′′, ω) nalezy dokonac podstawienia korzy-
staj ac z row. (2.25)
∑σ′σ′′
∫∫χsσσ′(r, r′, ω)
[1
|r′ − r′′|+ fxcσ′σ′′(r′, r′′, ω)
]δρσ′′(r′′, ω) dr′dr′′ =
=∑sr
(nsσ − nrσ)φ∗sσ(r)φrσ(r)
ω − ωrsσ + ıη
∑jb
∑σ′′
Krsσbjσ′′γbjσ′′(ω) +Krsσjbσ′′γjbσ′′(ω) (2.29)
10

2.1. Podstawy formalizmu
gdzie element macierzy Krsσbjσ′′ , zwanej macierz a sprz egaj aca, jest zdefiniowany ana-
logicznie do Mrsσtuσ′ z row. (2.21)
Krsσbjσ′′ =
=
∫∫φ∗rσ(r′)φsσ(r′)
[1
|r′ − r′′|+ fxcσσ′′(r′, r′′, ω)
]φbσ′′(r′′)φ∗jσ′′(r′′)dr′dr′′ (2.30)
Zatem rownanie (2.12) przyjmuje postac
∑ia
[γiaσ(ω)φiσ(r)φ∗aσ(r) + γaiσ(ω)φaσ(r)φ∗iσ(r)] =
=∑sr
(nsσ − nrσ)φ∗sσ(r)φrσ(r)
ω − ωrsσ + ıη×
×(δV ext
rsσ +∑jb
∑σ′′
Krsσbjσ′′γbjσ′′(ω) +Krsσjbσ′′γjbσ′′(ω)
)(2.31)
Podwojna suma∑
sr, wyst epuj aca po prawej stronie rownania, rozci aga si e po
parze dowolnych orbitali Kohna-Shama. Orbitale te mog a byc zaj ete b adz wirtualne.
Oznacza to, ze w tej sumie zawarte s a cztery kombinacje grup orbitali: zaj ety-zaj ety,
zaj ety-wirtualny, wirtualny-zaj ety oraz wirtualny-wirtualny. Ze wzgl edu na cz lon (nsσ−nrσ) niezerowy wk lad daj a tylko kombinacje zaj ety-wirtualny oraz wirtualny-zaj ety.
Rownanie (2.31) mozna zatem przepisac nast epuj aco
∑ia
[γiaσ(ω)φiσ(r)φ∗aσ(r) + γaiσ(ω)φaσ(r)φ∗iσ(r)] =
=∑ia
(niσ − naσ)φ∗iσ(r)φaσ(r)
ω − ωaiσ + ıη×
×(δV ext
aiσ +∑jb
∑σ′′
Kaiσbjσ′′γbjσ′′(ω) +Kaiσjbσ′′γjbσ′′(ω)
)+
+∑ia
(naσ − niσ)φ∗aσ(r)φiσ(r)
ω − ωiaσ + ıη×
×(δV ext
iaσ +∑jb
∑σ′′
Kiaσbjσ′′γbjσ′′(ω) +Kiaσjbσ′′γjbσ′′(ω)
)(2.32)
11

2.1. Podstawy formalizmu
Jest to suma dwoch sprz ezonych wzgl edem siebie rownan
∑ia
γiaσ(ω)φiσ(r)φ∗aσ(r) =
=∑ia
(naσ − niσ)φ∗aσ(r)φiσ(r)
ω − ωiaσ + ıη×
×(δV ext
iaσ +∑jb
∑σ′′
Kiaσbjσ′′γbjσ′′(ω) +Kiaσjbσ′′γjbσ′′(ω)
)(2.33)
∑ia
γaiσ(ω)φaσ(r)φ∗iσ(r) =
=∑ia
(niσ − naσ)φ∗iσ(r)φaσ(r)
ω − ωaiσ + ıη×
×(δV ext
aiσ +∑jb
∑σ′′
Kaiσbjσ′′γbjσ′′(ω) +Kaiσjbσ′′γjbσ′′(ω)
)(2.34)
ktore po prostych przekszta lceniach daj a
−∑ia
φ∗aσ(r)φiσ(r)
ω + ωaiσ + ıηδV ext
iaσ =
=∑ia
φ∗aσ(r)φiσ(r)
ω + ωaiσ + ıη×
×(∑
jb
∑σ′′
(δijδabδσσ′′ωaiσ +Kiaσjbσ′′ + ω
)γjbσ′′(ω) +Kiaσbjσ′′γbjσ′′(ω)
)(2.35)
−∑ia
φ∗iσ(r)φaσ(r)
ω − ωaiσ + ıηδV ext
aiσ =
=∑ia
φ∗iσ(r)φaσ(r)
ω − ωaiσ + ıη×
×(∑
jb
∑σ′′
(δijδabδσσ′′ωaiσ +Kaiσbjσ′′ − ω
)γbjσ′′(ω) +Kaiσjbσ′′γjbσ′′(ω)
)(2.36)
12

2.2. J adro korelacyjno-wymienne
Rownosci te s a spe lnione, gdy dla kazdej kombinacji ia
− δV extiaσ =
=∑jb
∑σ′′
(δijδabδσσ′′ωaiσ +Kiaσjbσ′′ + ω
)γjbσ′′(ω) +Kiaσbjσ′′γbjσ′′(ω) (2.37)
− δV extaiσ =
=∑jb
∑σ′′
(δijδabδσσ′′ωaiσ +Kaiσbjσ′′ − ω
)γbjσ′′(ω) +Kaiσjbσ′′γjbσ′′(ω) (2.38)
Otrzymane rownania mozna przedstawic w postaci macierzowej [9][(A B
B∗ A∗
)− ω
(1 0
0 −1
)](X
Y
)=
(−δV−δV ∗
)(2.39)
gdzie
Aaiσbjσ′ = δijδabδσσ′ωaiσ +Kaiσbjσ′
Baiσbjσ′ = Kaiσjbσ′
Xbjσ′ = γbjσ′
Ybjσ′ = γjbσ′
W granicy nieskonczenie ma lego zaburzenia otrzymujemy zatem(A B
B∗ A∗
)(X
Y
)= ω
(1 0
0 −1
)(X
Y
)(2.40)
Ograniczenie si e do rzeczywistych orbitali Kohna-Shama pozwala ostatecznie zapi-
sac (A B
B A
)(X
Y
)= ω
(1 0
0 −1
)(X
Y
)(2.41)
Takie sformu lowanie uogolnionego zagadnienia w lasnego nazywane jest postaci a ma-
cierzow a Casidy [10]. Energie wzbudzen dane s a przez wartosci w lasne, natomiast
struktura stanow wzbudzonych opisana jest przez wektory w lasne.
2.2. Jadro korelacyjno-wymienne
Zdefiniowane w rownaniu (2.8) j adro korelacyjno-wymienne jest skomplikowanym
funkcjona lem g estosci. Znajomosc dok ladnej postaci fxc pozwoli laby na odnalezienie
dok ladnych energii wzbudzen ω. Jednak wyst epuj aca zaleznosc fxc od cz estotliwosci
13

2.3. Wybrane warianty TDDFT
stanowi powazne utrudnienie w rozwi azaniu rownania (2.41). Zastosowanie przyblize-
nia adiabatycznego [11]
fAxc(r, r′) ≈ fxc(r, r
′, ω) (2.42)
umozliwia rozprz egni ecie rownania (2.41) i otrzymanie w efekcie zagadnienia w lasnego.
Ponadto w praktyce stosowane jest przyblizenie lokalne, w ktorym dodatkowo za-
k ladamy
fxc(r, ω) ≈ δ(r − r′)fxc(r, r′, ω) (2.43)
Zastosowanie obu przyblizen rownoczesnie oznacza, ze j adro korelacyjno-wymienne
jest zalezne jedynie od g estosci elektronowej i, na poziomie GGA, gradientu g estosci
w danym punkcie przestrzeni. Przyblizenie to jest cz esto okreslane mianem Adiabatic
Local Density Approximation (ALDA).
2.3. Wybrane warianty TDDFT
Postac macierzowa zagadnienia w lasnego opisania rownaniem (2.41) jest okreslana
mianem wariantu Random Phase Approximation (RPA), czy tez sformu lowaniem ma-
cierzowym Casidy. To w lasnie rownanie w tym kszta lcie jest standardowo stosowane do
obliczen stanow wzbudzonych metod a TDDFT. Poniewaz wi ekszosc obliczen TDDFT
wykonywanych jest w przyblizeniu RPA, wi ec okreslenie “metoda TDDFT” rozumiane
jest jako“metoda RPA-TDDFT”. Dla uproszczenia, dalsze rozwazania w tym rozdziale
prowadzone s a dla uk ladow zamkni etopow lokowych.
Rozwazaj ac pojedyncze, izolowane wzbudzenie z orbitalu zaj etego i na wirtualny
orbital a, uzyskujemy przyblizenie Small Matrix Approximation (SMA) [7]. Energia
wzbudzenia w tym przyblizeniu wyraza si e wzorem
ωSMA =√ωai (ωai + 2〈ai|fAHXC |ai〉) (2.44)
Mozliwe jest takze zaniedbanie sprz egania konfiguracji pojedynczo wzbudzonych
poprzez konfiguracje wzbudzone podwojnie. Taki wariant przyblizenia nazywany jest
Tamm-Dancoff Approximation TDDFT (TDA-TDDFT) [12]. Wtedy w rownaniu (2.41)
zaniedbywana jest macierz B, a przez to ca le zagadnienie w lasne zostaje uproszczone
do postaci
AX = ωX (2.45)
Rozwazanie w ramach TDA-TDDFT pojedynczego izolowanego wzbudzenia z or-
bitalu zaj etego i na wirtualny orbital a nazywane jest przyblizeniem Single-Pole Ap-
proximation (SPA). Energia wzbudzenia opisanego w ramach tego przyblizenia wyraza
14

2.4. Dressed TDDFT
si e wzorem
ωSPA = ωai + 2〈ai|fH |ai〉+ 2〈ai|fAXC(ωq)|ai〉 (2.46)
Do tego samego rezultatu prowadzi ograniczenie si e do pierwszego elementu rozwini e-
cia w szereg Taylora wyrazenia na energi e w przyblizeniu SMA. Szczego lowa analiza
stosowalnosci przyblizenia SPA opisana jest w publikacjach [13,14].
Sposrod opisanych wariantow TDDFT, w praktyce obliczeniowej stosowane s a
RPA-TDDFT oraz TDA-TDDFT. Warianty SMA oraz SPA uzywane s a przede wszyst-
kim do rozwazan modelowych.
2.4. Dressed TDDFT
Istotnym ograniczeniem metody TDDFT jest nieprawid lowy opis stanow zawiera-
jacych domieszk e konfiguracji podwojnie wzbudzonych [15]. Jednym z proponowanych
rozszerzen poprawiajacych ten stan rzeczy jest Dressed TDDFT. W niniejszej pracy
przedstawione s a jedynie robocze rownania tej metody, ktorej podstawy zosta ly opisane
w publikacji [15].
Rozwazmy prosty uk lad modelowy, ktorego baz e stanowi a dwa niezdegenerowa-
ne stany oznaczane przez Φq oraz ΦD. Hamiltonian takiego uk ladu zdefiniowany jest
nast epuj aco
H =
(Hqq HqD
H∗qD HDD
)(2.47)
Diagonalizacja prowadzi do uzyskania energii stanow Ψ1 oraz Ψ2
Ψ1 = mΦq +√
1−m2ΦD (2.48)
Ψ2 =√
1−m2Φq −mΦD (2.49)
gdzie 0 < m < 1 jest wspo lczynnikiem okreslaj acym stopien mieszania konfiguracji.
Przekszta lcaj ac zagadnienie w lasne dane przez (2.47) otrzymujemy rownanie
(Hqq − E)(HDD − E) = |HqD|2 (2.50)
ktore umozliwia sformu lowanie diagonalizacji hamiltonianu w postaci samouzgodnio-
nej. Zaleznie od postaci rownania oraz od startowej energii otrzymuje si e w procesie
iteracyjnym jedno z dwoch rozwi azan: o nizszej lub wyzszej energii. Nizsza energia jest
wyznaczana przez iteracyjne rozwi azanie rownania
E = Hqq +|HqD|2
E −HDD
(2.51)
15

2.4. Dressed TDDFT
pod warunkiem, ze proces iteracji rozpoczniemy od wartosci E < HDD.
W przypadku braku oddzia lywania mi edzy stanami Φq i ΦD rownanie (2.51) uprasz-
cza si e do postaci E = Hqq. W miar e wzrostu oddzia lywania mi edzy stanami coraz
istotniejszy staje si e element |HqD|2. Id ac dalej t a sciezk a rownanie (2.51) mozna zinter-
pretowac jako wyrazenie na energi e stanu Φq poprawion a o oddzia lywanie ze stanem
ΦD. Im wi eksze sprz ezenie mi edzy stanami bazy, tym wi eksza wartosc |HqD|2, a co
za tym idzie tym wi ekszy wp lyw poprawki. Zeruje si e ona przy braku oddzia lywan
pomi edzy stanami bazy. Jest to rozwi azanie scis le.
Dotychczasowe rozwazania s a prawdziwe dla dowolnych dwoch stanow, w szcze-
golnosci dla dwoch dowolnych konfiguracji. Niech Φq oznacza konfiguracj e pojedynczo
wzbudzon a, ΦD konfiguracj e dwukrotnie wzbudzon a. Wtedy cz lon hamiltonianu HqD
oznacza sprz ezenie mi edzy tymi konfiguracjami.
Na potrzeby rozwazan dotycz acych stanow wzbudzonych wygodnie jest odnosic
wszelkie wartosci do energii stanu podstawowego. Niech ω oznacza energi e wzbudzenia
ze stanu podstawowego do stanu wzbudzonego. Wtedy E = H00 + ω, a wzor (2.51)
mozna przekszta lcic do
ω = (Hqq −H00) +|HqD|2
ω − (HDD −H00)(2.52)
Wariant SPA-TDDFT daje dok ladne wyniki obliczen w przypadku braku oddzia ly-
wan mi edzy konfiguracjami wzbudzonymi pojedynczo. Rozwazany model moze byc za-
tem zastosowany do SPA-TDDFT bez dalszych przyblizen. Otrzymana energia wzbu-
dzenia w takim uk ladzie wynosi, zgodnie ze wzorem (2.46)
ωSPA = Hqq −H00 = ωq + 2〈q|fAHXC |q〉 (2.53)
Na podkreslenie zas luguje, iz na wartosc energii wzbudzenia (2.53) nie wp lywa obec-
nosc konfiguracji podwojnie wzbudzonej. A posteriori mozna podj ac prob e uwzgl ednie-
nia sprz ezenia konfiguracji wzbudzonej pojedynczo Φq z konfiguracj a ΦD wzbudzon a
podwojnie. Wstawienie (2.53) do rownania (2.52) daje
ω = ωq + 2〈q|fAHXC |q〉+|HqD|2
ω − (HDD −H00)(2.54)
Na tak wyrazon a energi e wzbudzenia ω sk ladaj a si e trzy wielkosci: energia konfi-
guracji pojedynczo wzbudzonej ωq, cz lon zalezny od adiabatycznego j adra korelacyj-
no-wymiennego fAHXC oraz zalezna od cz estotliwosci poprawka wynikaj ac a z oddzia ly-
wania konfiguracji wzbudzonej pojedynczo z konfiguracj a podwojnie wzbudzon a.
16

2.4. Dressed TDDFT
J adro korelacyjno-wymienne wyznaczna roznic e pomi edzy energi a wzbudzenia uk la-
du cz astek Kohna-Shama a energi a wzbudzenia uk ladu rzeczywistego (rozdz. 2.1).
W przypadku stanow zawieraj acych domieszk e konfiguracji podwojnie wzbudzonej,
adiabatyczne j adro korelacyjno-wymienne nie wystarcza i konieczne staje si e zdefinio-
wanie poprawionego fHXC
2〈q|fHXC(ω)|q〉 = 2〈q|fAHXC |q〉+|HqD|2
ω − (HDD −H00)(2.55)
gdzie 〈q|fHXC(ω)|q〉 oznacza zmodyfikowane, zalezne od cz estostliwosci (nieadiaba-
tyczne), j adro korelacyjno-wymienne.
Rownanie (2.54) przybiera zatem ostateczn a postac
ω = ωq + 2〈q|fHXC(ω)|q〉 (2.56)
W ten sposob uwzgl edniono oddzia lywanie konfiguracji wzbudzonej podwojnie na ener-
gie wzbudzenia uzyskan a metod a SPA.
Przedstawione powyzej podejscie ma ograniczon a stosowalnosc, ze wzgl edu na za-
lozone przyblizenie SPA-TDDFT, ktorego uzywa si e jedynie do rozwazan modelowych.
W przeciwienstwie do niego, wariant TDA-TDDFT uzywany jest do obliczen na real-
nych uk ladach. Dlatego tez warto rozwazyc aplikacj e poprawki do tego wariantu.
Niech
|q〉 =∑i
ci|qi〉 (2.57)
b edzie stanem wzbudzonym, w przyblizeniu TDA-TDDFT danym przez kombinacj e
liniow a konfiguracji pojedynczo wzbudzonych. Uwzgl ednienie oddzia lywania konfigu-
racji pojedynczo wzbudzonych |qi〉 z konfiguracj a podwojnie wzbudzon a |qD〉 da po-
prawion a energi e stanu |q〉
ω = ωq +∑ij
〈qi|H|qD〉〈qD|H|qj〉ω − ωq
(2.58)
Identyfikacja odpowiadaj acych sobie elementow macierzy j adra korelacyjno-wy-
miennego oraz elementow macierzy rozwazanego hamiltonianiu, prowadzi do wyrazenia
na nieadiabatyczne j adro korelacyjno-wymienne w przyblizeniu Dressed TDA-TDDFT
(DTDA-TDDFT)
2〈qi|fHXC(ω)|qj〉 = 2〈qi|fAHXC |qj〉+〈qi|H|qD〉〈qD|H|qj〉
ω − ωq(2.59)
17

2.5. Ograniczenia Dressed TDDFT
Wariant TDA daje pe lne informacje o energiach wzbudzen z ograniczeniem do
uwzgl edniania jedynie konfiguracji wzbudzonych pojedynczo. Wariant RPA uwzgl ednia
ponadto sprz ezenia pomi edzy konfiguracjami wzbudzonymi pojedynczo za posrednic-
twem odpowiednich konfiguracji wzbudzonych podwojnie (elementy macierzy B za-
gadnienia w lasnego (2.41)). Bezposrednie uwzgl ednienie proponowanej przez Maitr e
et al. [15] poprawki prowdzi loby do przeszacowania wp lywu konfiguracji wzbudzonych
podwojnie. Formalnie przyczyn a tego jest, pojawiaj aca si e w wariancie RPA-TDDFT,
nieliniowa zaleznosc energii wzbudzen od j adra korelacyjno-wymiennego. Jest to za-
sadnicza roznica w stosunku do wariantow SPA oraz TDA. Dlatego nie jest mozliwe
zastosowanie w wariancie RPA poprawki w rozwazanej postaci.
Problem ten latwo mozna zilustrowac analizuj ac przyblizenie SMA-TDDFT. Ana-
logiczne w stosunku do poprzednich przypadkow post epowanie, a wi ec wstawienie
rownania na energi e (2.44)
ωSMA = Hqq −H00 =√ωq (ωq + 2〈q|fAHXC |q〉)
do row. (2.52)
ω =√ωq (ωq + 2〈q|fHXC |q〉) +
|HqD|2
ω − (HDD −H00)(2.60)
nie pozwala na uzyskanie poprawki do funkcjona lu korelacyjno-wymiennego w rozwa-
zanej postaci.

Rozdzia l 3
Implementacja metody TDDFT
Programistyczna cz esc niniejszej pracy, a wi ec implementacja metody TDDFT,
zosta la wykonana jako rozszerzenie pakietu niedoida [16]. Projekt niedoida, rozwijany
w Zak ladzie Metod Obliczeniowych Chemii UJ, ma na celu utworzeniu samodzielnego
pakietu bibliotek umozliwiaj acych wykonywanie obliczen kwantowomechanicznych.
3.1. Implementacja TDA-TDDFT
Prac e programistyczn a rozpocz eto od implementacji przyblizenia Tamma-Dancoffa
opisanego w rozdziale (2.3). Jest to wariant stosowany do praktycznych obliczen, przy
czym rozwi azywane zagadnienie w lasne ma stosunkowo prost a postac
AX = ωX
Formalnie macierz Aai,bj jest czteroindeksowa, co oznacza rozmiar rz edu N2occN
2virt,
gdzie Nocc i Nvirt oznaczaj a odpowiednio: liczb e orbitali zaj etych i wirtualnych. Ze
wzgl edu na duzy rozmiar, zarowno jawne wyliczenie elementow macierzy, jak i tym
bardziej jej diagonalizacja, s a w praktyce wykonalne tylko dla niewielkich uk ladow.
Wyznaczenie wszystkich wartosci w lasnych nie jest jednak konieczne, gdyz w obli-
czeniach TDDFT liczy si e stosunkowo niewiele najnizszych energii wzbudzen. Potrze-
ba zatem metody, ktora jest w stanie wyznaczyc pewn a liczb e najnizszych wartosci
19

3.1. Implementacja TDA-TDDFT
w lasnych bez koniecznosci przechowywania wszystkich elementow macierzy. Takimi
mozliwosciami charakteryzuj a si e algorytmy iteracyjne.
Kluczow a cech a iteracyjnych metod poszukiwania wartosci w lasnych jest fakt, ze
nie wymagaj a znajomosci macierzy, a potrzebuj a jedynie mozliwosci obliczenia jej
iloczynu z pewnym wektor probnym, czyli
σai =∑bj
Aai,bjXbj (3.1)
gdzie Xbj jest wektorem probnym. Przez wstawienie definicji A z row. (2.39)
Aai,bj = δijδab(εa − εi) + (ai|jb) + (ai|fxc|jb)− cHF (ab|ji)
otrzymuje si e iloczyn σai zapisany w bazie orbitali molekularnych
σai = (εa − εi)Xai +∑bj
((ai|bj) + (ai|fxc|bj)− cHF (ab|ji))Xbj (3.2)
Taka postac σai jest niewygodna do prowadzenia obliczen. Do wyliczenia kazdej
ca lki dwuelektronowej konieczne jest wyznaczenie ca lek dwuelektronowych w bazie
AO, a nast epnie ich transformacja do bazy MO. Taka transformacja limituje szyb-
kosc obliczen, gdyz jej z lozonosc jest rz edu N5, gdzie N jest liczb a orbitali. Dalsze
przekszta lcenia sumy
∑bj
((ai|bj) + (ai|fxc|bj)− cHF (ab|ji))Xbj =
=∑bj
∑µν
∑κλ
CµaCνiCκbCλj((µν|κλ) + (µν|fxc|κλ)− cHF (µκ|λν))Xbj =
=∑µν
∑κλ
CµaCνi((µν|κλ) + (µν|fxc|κλ)− cHF (µκ|λν))∑bj
CκbCλjXbj (3.3)
umozliwiaj a jednak zwi ekszenie efektywnosci obliczen przez zastosowanie innego algo-
rytmu.
Wygodne jest wprowadzenie macierzy pseudog estosci, ktorej element zdefiniowany
jest analogicznie do elementu macierzy g estosci:
Pκλ =∑bj
CκbCλjXbj (3.4)
20

3.1. Implementacja TDA-TDDFT
Wtedy
∑bj
((ai|bj) + (ai|fxc|bj)− cHF (ab|ji))Xbj =
=∑µν
∑κλ
CµaCνi((µν|κλ)− cHF (µκ|λν) + (µν|fxc|κλ))Pκλ =
=∑µν
CµaCνi∑κλ
((µν|κλ)− cHF (µκ|λν))Pκλ + (µ|fxc ρ|ν) ≡
≡∑µν
CµaCνiFµν (3.5)
Wprowadzony element ρ jest zdefiniowany analogicznie do g estosci
ρ = Tr(PS) (3.6)
gdzie S jest macierz a ca lek nak ladania, a P to poprzednio wprowadzona macierz pseu-
dog estosci. Analogicznie, ρ to pseudog estosc, a F to pseudo macierz Focka, nazywana
tak ze wzgl edu na postac bardzo zblizon a do macierzy operatora Focka.
Ostatecznie
σai = (εa − εi)Xai +∑µν
CµiCνaFµν (3.7)
Zastosowanie pseudo macierzy Focka umozliwia efektywne wykonywanie obliczen, gdyz
transformacja tak liczonych ca lek do bazy orbitali molekularnych charakteryzuje si e
z lozonosci a N3.
Kulombowska i wymienna cz esci macierzy F jest obliczana w sposob analogiczny
do tych cz esci macierzy Focka. Dlatego tez uzywany jest istniej acy kod, stosowany
w trakcie samouzgodnionego wyznaczania postaci orbitali molekularnych. Drobnych
zmian wymaga jedynie uwzgl ednienie niesymetrycznosci macierzy pseudog estosci P
wprowadzonej w miejsce symetrycznej macierzy g estosci P . Wymaga to nieznacznych
modyfikacji obliczen macierzy wymiennej. Modyfikacje te zosta ly juz wczesniej wyko-
nane na potrzeby zaimplementowanych obliczen CIS.
Istotnie nowym elementem programu jest obliczanie elementow macierzowych j adra
korelacyjno-wymiennego. Analogicznie jak w przypadku macierzy potencja lu korela-
cyjno-wymiennego s a one obliczane przez ca lkowanie numeryczne. Wykorzystywana
jest kwadratura1 uzyta do ca lkowania potencja lu korelacyjno-wymiennego w uprzednio
wykonanych obliczeniach SCF.
Analogicznie jak w CIS, diagonalizacja macierzy sprz egaj acej w TDA-TDDFT wy-
1 wykonywana w fizycznej, trojwymiarowej przestrzeni; jest iloczynem odpowiednich kwadraturjednowymiarowych
21

3.2. Implementacja RPA-TDDFT
konywana jest iteracyjnym algorytmem Davidsona-Liu [17]. Kluczow a cech a tej me-
tody jest mozliwosc rownoczesnego obliczania zadanej liczby najmniejszych wartosci
w lasnych i odpowiadaj acych im wektorow w lasnych.
Wst epny wybor wektorow startowych odbywa si e na podstawie energii diagonal-
nych. W ustalonej w ten sposob podprzestrzeni przeprowadzana jest wst epna diagona-
lizacja. Wektory w lasne odpowiadaj ace najmniejszym wartosciom w lasnym uzywane
s a nast epnie jako wektory startowe dla procedury Davidsona-Liu.
Opracowuj ac implementacj e TDA-TDDFT wykorzysta lem duze podobienstwo tej
metody do CIS. Efektem jest jednolita implementacja obu metod, efektywnie wykorzy-
stuj aca wszystkie ich podobienstwa poprzez identyfikacj e wspolnego zestawu abstrakcji
i eliminacj e nadmiarowosci z kodu.
3.2. Implementacja RPA-TDDFT
Wariant RPA wymaga rozwi azania niehermitowskiego zagadnienia w lasnego przed-
stawionego w row. (2.41)(A B
B A
)(X
Y
)= ω
(1 0
0 −1
)(X
Y
)
Rozwi azanie go bezposrednio wymaga loby istotnej modyfikacji procedury diagonali-
zacji, gdyz standardowy algorytm Davidsona-Liu przeznaczony jest do diagonalizacji
macierzy symetrycznych.
Jednym z rozwi azan jest symetryzacja zagadniena w lasnego przy pomocy transfor-
macji unitarnej
U =1√2
(1 1
−1 1
)Po takiej tranformacji zagadnienie w lasne przyjmuje postac symetryczn a
ΩF = ω2F (3.8)
gdzie
Ω = −S− 12 (A+B)S−
12
F = S12 (X + Y )
S = −(A−B)−1
22

3.3. Automatyczna generacja kodu
To podejscie ma ograniczenia zwi azane z koniecznosci a obliczenia S−12 . Dla funk-
cjona low niehybrydowych (A−B) jest macierz a diagonaln a
(A−B)ai,bj = δijδab(εa − εi)
wi ec
S− 1
2ai,bj = δijδab(εi − εa)
12 (3.9)
Jednak w przypadku funkcjona low hybrydowych (A − B) zawiera cz lony pozadiago-
nalne
(A−B)ai,bj = δijδab(εa − εi)− cHF ((ab|ji)− (aj|bi)) (3.10)
co w praktyce uniemozliwia obliczenie S−12 . Oznacza to, ze dla funkcjona low hybrydo-
wych konieczne jest zastosowanie algorytmu diagonalizacji dla macierzy niesymetrycz-
nych. Zagadnienia tego nie podj eto w niniejszej pracy.
Zsymetryzowane rownania RPA-TDDFT s a bardzo podobne do rownan TDA-DFT,
dlatego wykorzystywany jest kod implementuj acy przyblizenie TDA. Wszystkie roznice
pomi edzy wariantami da si e uwzgl ednic przez prost a modyfikacj e macierzy pseudog e-
stosci i elementow diagonalnych Ω.
3.3. Automatyczna generacja kodu
Do przeprowadzenia obliczen metod a TDDFT konieczne jest obliczenie j adra ko-
relacyjno-wymiennego zdefiniowanego w row. (2.8). Wymaga to policzenia wartosci
drugich pochodnych energii wzgl edem g estosci elektronowej i gradientu g estosci oraz
odpowiednich pochodnych mieszanych (tab. 3.1). Liczba funkcjona low stosowanych
do obliczen jest duza, a ich postac jest zazwyczaj z lozona, wi ec wyprowadzenie wzo-
row na analityczn a postac pochodnych staje si e czasoch lonne. Pozniejsze przenoszenie
wzorow do kodu zrod lowego dodatkowo stwarza mozliwosc pope lnienienia trudnych do
zdiagnozowania b l edow.
Tabela 3.1. Liczba unikalnych funkcjonalnych pochodnych funkcjona lu korelacyjno-wymien-nego koniecznych do wyliczenia dla roznych typow obliczen
Typ obliczen Liczba pochodnychLDA GGA
stan podstawowy (DFT) 1(∂Exc∂ρσ
)3(∂Exc∂ρσ
, ∂Exc∂γστ
)stan wzbudzony (TDDFT) 2
(∂2Exc∂ρσ∂ρτ
)9(∂2Exc∂ρσ∂ρτ
, ∂2Exc∂ρσ∂γστ
, . . .)
razem 3 12
23

3.3. Automatyczna generacja kodu
Problemy te mozna rozwi azac przez zastosowanie programu do automatycznej ge-
neracji kodu. Sposrod istniej acych implementacji dost epne s a jedynie: zmodyfikowana
wersja dfauto [18], zaimplementowana na potrzeby “Density Functional Repository”,
oraz pakiet cclib [19].
Dfauto oparta jest na komputerowym systemie obliczen symbolicznych Maple [20].
Niestety, implementacja ta nie daje mozliwosci samodzielnego definiowania kolejnych
funkcjona low. Ponadto wydaje si e zawierac b l edy uniemozliwiaj ace uzyskanie popraw-
nych wynikow dla funkcjona low gradientowych.
Cclib jest nowym projektem i znajduje si e w fazie rozwoju. Pakiet ten zbudowany
zosta l na bazie systemu algebry komputerowej Maxima [21]. Maxima charakteryzuje
si e duz a z lozonosci a obs lugi, co utrudnia poszerzanie projektu o nowe funkcjona ly.
Ponadto nie jest do konca jasne z jakich powodow autorzy programu do automatycz-
nej generacji kodu do l aczyli do niego szereg funkcjona low wyprowadzonych r ecznie.
Dodatkowy brak jasnych uregulowan prawnych dotycz acych licencji, na ktorej oparta
jest dystrybucja pakietu cclib, sk loni l do poszukiwania innego rozwi azania.
Aby miec pe ln a swobod e w implementowaniu potrzebnych funkcjona low, podj eto
prob e wykonania w lasnego narz edzia do automatycznej generacji kodu. Rdzen takiego
pakietu stanowi system algebry komputerowej (CAS), ktorego zadaniem jest wyzna-
czanie analitycznej postaci pochodnych funkcjona low. Sposrod bezp latnie dost epnych
programow CAS najwi eksze mozliwosci daje pakiet Maxima. Jego minusem jest dosc
duza z lozonosc obs lugi. Niestety Maxima posiada mozliwosc generowania kodu wyni-
kowego jedynie w j ezyku Fortran. Stwarza to dodatkowy k lopot, gdyz programistyczna
cz esc pracy magisterskiej realizowana jest w j ezyku C++.
Alternatyw e stanowi system Yacas [22]. Pakiet ten jest mniej skomplikowany zarow-
no w budowie jak i w obs ludze. Ponadto Yacas doskonale wpasowuje si e w bibliotek e
niedoida dzi eki wbudowanej generacji kodu wynikowego bezposrednio w j ezyku C++.
Te zalety zadecydowa ly, pakiet Yacas zosta l wybrany do roli systemu CAS.
Niestety, procedura generacji kodu wbudowana wpakiet Yacas jest nieadekwatna do
tak z lozonego problemu. Kompilacja otrzymywanego kodu C++ jest zbyt czasoch lon-
na. Rozmiar otrzymywanych wyrazen przekracza mozliwosci optymalizacji uzywanych
kompilatorow, a otrzymywany kod wynikowy jest bardzo nieefektywny.
Zwi ekszenie efektywnosci kodu uzyskano na drodze eliminacji wspolnych podwy-
razen (CSE). Procedur e rozpoczyna wst epna transformacja z lozonego wyrazenia do
ci agu prostszych wyrazen. Podwyrazenia wyst epuj ace w takim ci agu wi ecej niz jeden
raz s a z niego wyci agane, a nast epnie wyliczane jednokrotnie. Tak przetransformowany
kod daje w krotszym czasie wynik identyczny wzgl edem kodu pierwotnego. Przedsta-
24

3.3. Automatyczna generacja kodu
wiona procedura wykonywana jest rekurencyjnie, gdyz powsta le wyrazenie rowniez
moze zawierac powtarzaj ace si e podwyrazenia.
Proces ten jest przeprowadzany standardowo w trakcie kompilacji, jednak kompi-
latory stosuj a algorytm dla ma lo z lozonych wyrazen. Dlatego tez konieczna sta lo si e
rozszerzenie generatora kodu o funkcj e eliminuj ac a wspolne podwyrazenia jeszcze przed
dostarczeniem z lozonego kodu do kompilatora. Procedura ta ma miejsce na poziomie
wyrazen wygenerowanych w Yacasie.
Szkic algorytmu
Powtarzaj
— Znajdz w wyrazeniu E wszystkie proste podwyrazenia i zachowaj w L
— Usun z L podwyrazenia wyst epuj ace tylko raz
— Jezeli L pusta, wyjdz z p etli
— Skompresuj L (usun kopie)
— Przyporz adkuj unikalne identyfikatory elementom L, zachowaj pary w S
— Zamien w E podwyrazenia z S na odpowiadaj ace im identyfikatory
Wygeneruj instrukcje przypisania dla par nalez acych do S
Wygeneruj kod dla wyrazenia E
Zastosowanie powyzszego algorytmu w znacz acy sposob zwi ekszy lo efektywnosc
generatora kodu. Otrzymywane pliki implementuj ace funkcjona ly s a do ponad pi eciu
razy mniejsze, a sama struktura kodu jest znacznie uproszczona. Dzi eki temu czas
kompilacji zosta l skrocony dramatycznie.
Korzystaj ac z opisanego mechanizmu generacji kodu zaimplementowano do tej pory
nast epuj ace funkcjona ly:
korelacyjne
— VWN3 [23], VWN5 [23], VWN5RPA [23], LYP [24], PBE [25], PW91 [26],
PW92 [27]
wymienne
— Slater [28, 29], Becke88 [30], Becke3 [31], PW91 [26], mPW91 [32], PBE [25],
revPBE [33]
korelacyjno-wymienne
— kombinacje powyzszych, w szczegolnosci:
SVWN, SLYP, BLYP, PW91 [26], PBE [25], B3LYP [31], PBE0 [34]
Przyk lad procedury generacji kodu na podstawie jednego z najprostszych funkcjo-
na low, funkcjona lu wymiennego PBE [25]
EPBEX =
∑σ
ρσeXFX (3.11)
25

3.3. Automatyczna generacja kodu
gdzie
eX = −3kF4π
(3.12)
FX = 1 + κ− κ
1 + µs2
κ
(3.13)
s =
√γσσ
2kFρσ(3.14)
kF = (3π2ρσ)13 . (3.15)
Na poziomie przyblizenia GGA (Generalized Gradient Approximation) [35] funkcjo-
na l korelacyjno-wymienny jest funkcj a g estosci spinowej ρσ i niezmiennika gradientu
g estosci spinowej
γστ = ∇ρσ∇ρτ (3.16)
gdzie σ, τ oznaczaj a spin.
W opartym na Yacasie systemie generacji kodu definicja funkcjona lu korelacyj-
no-wymiennego PBE (3.11) jest zapisywana nast epuj aco
mu := 0.2195149727645171;
kF(rho) := (3 * Pi^2 * rho)^(1/3);
eX(rho) := -3/4 * kF(rho) / Pi;
S(rho,gamma) :=
1/2 * Sqrt(gamma) / (kF(rho) * rho + Tiny);
FX(s, kp) := 1 + kp - kp / (1 + (mu*s^2)/kp);
XPBE(rhoa, rhob, gammaaa, gammaab, gammabb) := [
Local(kp);
kp := 0.804;
rhoa * eX(2*rhoa) * FX(S(2*rhoa, 4*gammaaa), kp) +
rhob * eX(2*rhob) * FX(S(2*rhob, 4*gammabb), kp);
];
Uruchomienie procedury generacji kodu nast epuje w trakcie kompilacji pakietu nie-
doida. Dla kazdego zdefiniowanego funkcjona lu generowane s a odpowiednie pochodne,
ktore wraz z funkcjona lem zostaj a automatycznie poddane procesowi eliminacji wspol-
nych podwyrazen, a nast epnie zapisane w postaci kodu C++ przy pomocy wbudowa-
nego generatora kodu Yacasa.
26

3.4. Wydajnosc
3.4. Wydajnosc
Formalnie obliczenie iloczynu macierzy sprz egaj acej i wektora probnego (row. (3.1))
skaluje si e szesciennie, ze wzgl edu na z lozonosc obliczeniow a transformacji z bazy or-
bitali atomowych do bazy orbitali molekularnych. W praktycznych zastosowaniach
skalowanie jest lepsze. Wynika to z faktu, ze wspo lczynnik przy cz lonie szesciennym
jest bardzo ma ly i tego rz edu skalowanie pojawi si e dopiero dla bardzo duzych uk ladow.
Dla wykonywanych w praktyce obliczen TDDFT wi ekszosc czasu zajmuj a trzy
elementy: obliczenie j adra korelacyjno-wymiennego, j adra kulombowskiego oraz cz esci
wymiany Focka. Obliczenia j adra kulombowskiego oraz cz esci fockowskiej charaktery-
zuj a si e z lozonosci a tego samego rz edu, inn a niz dla j adra korelacyjno-wymiennego.
Optymalizacja procedury ca lkowania j adra korelacyjno-wymiennego zosta la wyko-
nana przez analogi e do odpowiedniego kodu implementuj acego ca lkowanie potencja lu
korelacyjno-wymiennego. Kluczowym dla wydajnosci obliczen czynnikiem jest prescre-
ening2 na etapie konstrukcji kwadratury. Dzi eki wykorzystaniu algorytmu z pracy [36],
proces ca lkowania skaluje si e liniowo ze wzrostem rozmiaru uk ladu. Ponadto prescre-
ening na poziomie bloku punktow i na poziomie pojedynczych punktow kwadratury,
jakkolwiek nie zmienia postaci skalowania, znacznie skraca czas prowadzonych obliczen.
Obliczenia j adra kulombowskiego i, w przypadku zastosowania funkcjona lu hy-
brydowego, cz esci fockowskiej charakteryzuj a si e z lozonosci a identyczn a jak generacja
macierzy Focka. Dzi eki zastosowaniu prescreeningu opartego na nierownosci Schwartza,
w granicy duzych uk ladow mozna oczekiwac skalowania kwadratowego. W praktyce,
juz dla niezbyt duzych uk ladow mozna oczekiwac z lozonosci rz edu O(N2.3), gdzie N
jest liczb a orbitali.
Skalowanie metody TDDFT sprawdzono przeprowadzaj ac obliczenia dla szeregu
polienow3 i oligotiofenow. Wszystkie obliczenia wykonano przy uzyciu funkcjona lu
PBE0, uzywaj ac uprzednio zoptymalizowanych geometrii stanu podstawowego. Obli-
czenia dla polienow przeprowadzono w bazie 6-311G**, natomiast dla oligotiofenow
w bazie 6-31G**. Dane o rozmiarzach badanych uk ladow przedstawione s a w tab. 3.2
i tab. 3.3. Porownanie czasu pojedynczej iteracji metody Davidsona dla obliczen pierw-
szego stanu wzbudzonego przedstawiono na rys. 3.1 i rys. 3.2.
Ze wzgl edu na coraz powszechniejsze stosowanie klastrow i komputerow wieloproce-
sorowych, istotne znaczenie dla szybkosci dzia lania programu ma zrownoleglenie kodu.
Zaimplementowany algorytm obliczen zosta l zrownoleglony przy uzyciu modelu Single
2 Prescreening to ogolne okreslenie metody zwi ekszania wydajnosci algorytmow obliczeniowych.Polega ono na oszacowaniu z gory obliczanej wartosci i pomini eciu obliczen, jezeli oszacowana wartoscjest mniejsza od pewnej za lozonej wartosci progowej. Prescreening oczywiscie ma sens jedynie wtedy,gdy dost epna jest metoda szacowania znacz aco tansza obliczeniowo od pe lnego algorytmu.
3 Dok ladna charakterystyka badanych w niniejszej pracy polienow jest przedstawiona w rozdz. 4.
27

3.4. Wydajnosc
Tabela 3.2. Podstawowe informacje o rozmiarze obliczen dla szeregu polienow w bazie6-311G**
Cz asteczkaLiczba
at. w egla atomow pow lok orbitali
butadien 4 10 44 108heksatrien 6 14 62 156oktatetraen 8 18 80 204dekapentaen 10 22 98 252
0
10
20
30
40
50
60
70
80
4 5 6 7 8 9 10
Cza
sob
licz
en(j
edn.
um
owna)
Rozmiar uk ladu (liczba atomow w egla)
J adro korelacyjno-wymienne
3
3
3
3
3J adro kulombowskie i wymiana Focka
++
+
+
+Razem
2
2
2
22
Rysunek 3.1. Czas pojedynczej iteracji metody Davidsona dla obliczen pierwszego stanuwzbudzonego dla szeregu polienow. Obliczenia PBE0 w bazie 6-311G**.
Tabela 3.3. Podstawowe informacje o rozmiarze obliczen dla szeregu oligotiofenow w bazie6-31G**
Cz asteczkaLiczba
pierscieni atomow pow lok orbitali
tiofen 1 9 33 94bitiofen 2 16 60 178tertiofen 3 23 87 262quatertiofen 4 30 114 346quintetiofen 5 37 141 430seksitiofen 6 44 168 514
28
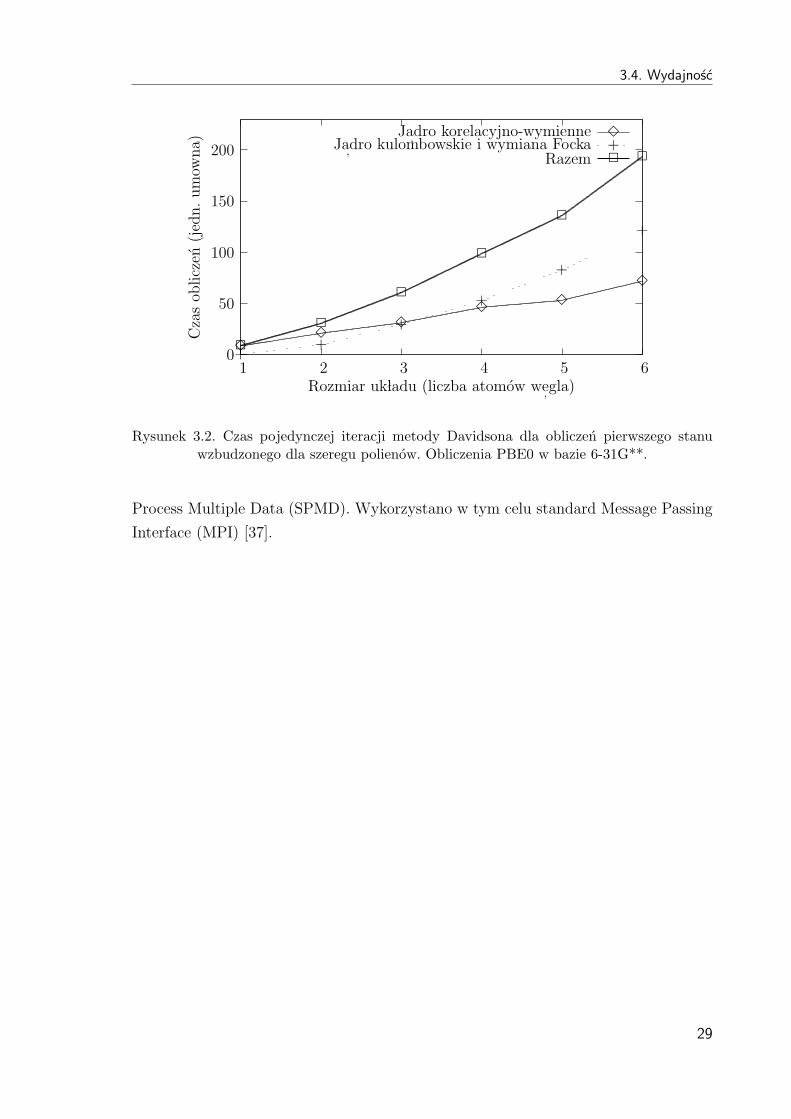
3.4. Wydajnosc
0
50
100
150
200
1 2 3 4 5 6
Cza
sob
licz
en(j
edn.
um
owna)
Rozmiar uk ladu (liczba atomow w egla)
J adro korelacyjno-wymienne
33
33
3
3
3J adro kulombowskie i wymiana Focka
++
+
+
+
+
+Razem
2
2
2
2
2
22
Rysunek 3.2. Czas pojedynczej iteracji metody Davidsona dla obliczen pierwszego stanuwzbudzonego dla szeregu polienow. Obliczenia PBE0 w bazie 6-31G**.
Process Multiple Data (SPMD). Wykorzystano w tym celu standard Message Passing
Interface (MPI) [37].
29

3.5. Implementacja Dressed TDA-TDDFT
Implementacja metody Dressed TDA-TDDFT zosta la wykonana jako modyfikacja
metody TDA-TDDFT. Zaimplementowany algorytm jest nast epuj acy.
Najpierw przeprowadzane s a standardowe obliczenia TDA-TDDFT. Nast epnie wy-
szukiwane s a konfiguracje podwojnie wzbudzone o energii zblizonej do obliczonych
energii wzbudzen. Brane pod uwag e s a tylko te konfiguracje podwojnie wzbudzone,
ktore, zgodnie z regu lami Slatera-Condona [38], mog a oddzia lywac z danym wzbu-
dzeniem. Energie konfiguracji podwojnie wzbudzonych szacowane s a z odpowiednich
energii orbitalnych. Elementy poprawki wyrazone s a przez ca lki zgodnie z regu lami
Slatera-Condona. Wartosci ca lek obliczane s a przy uzyciu orbitali Kohna-Shama. War-
tosc ω jest szacowana jako energia odpowiedniego wzbudzenia.
W nast epnym kroku przeprowadzane s a ponowne obliczenia TDA-TDDFT, przy
czym tym razem uzywaj ac zmodyfikowanego j adra korelacyjno-wymiennego. Uzyski-
wane energie wzbudzen s a uzywane do szacowania wartosci ω w kolejnym kroku. Po-
wyzsza procedura wykonywana jest iteracyjnie, az do uzbieznienia wszystkich energii
wzbudzen.
W celu przyspieszenia obliczen wyszukiwanie konfiguracji podwojnie wzbudzo-
nych jest wykonywane tylko raz na pocz atku obliczen. Wyniki wyszukiwania s a za-
pami etywane i uzywane we wszystkich nast epnych krokach. Ponadto wektory wzbu-
dzen z poprzedniego kroku s a wykorzystywane jako wektory startowe dla procedury
Davidsona-Liu w kolejnym kroku.

Rozdzia l 4
Obliczenia dla wybranych polienow
liniowych
4.1. Wstep
Lancuchy polienowe wchodz a jako chromofory w sk lad wielu zwi azkow o znaczeniu
biologicznym, takich jak: pigmenty, witaminy czy uk lady antenowe. Ze wzgl edu na rol e
tych cz asteczek, istotne staje si e zagadnienie fotofizyki polienow [39], w tym poznanie
kwantowomechanicznego opisu najnizszych singletowych stanow wzbudzonych, 11Bu
oraz 21Ag. Dlatego tez opis wzbudzen elektronowych krotkich lancuchow polienowych
od lat stanowi cel wielu opracowan eksperymentalnych, teoretycznych i obliczeniowych
[39–55].
Z lozona struktura wzbudzen elektronowych i niewielki rozmiar cz asteczek czyni a
z krotkich lancuchow polienowych doskona ly obiekt do testowania nowych metod ob-
liczeniowych.
W niniejszej pracy skoncentrowano si e na badaniu energii wertykalnych
wzbudzen elektronowych s-trans-buta-1,3-dienu (butadienu),1 2(s-trans),4(s-trans)--
heksa-1,3(E),5-trienu (heksatrienu), 2(s-trans),6(s-trans)-okta-1,3(E),5(E),7-tetraenu
(oktatetraenu) oraz 2(s-trans),8(s-trans)-deka-1,3(E),5(E),7(E),9-pentaenu (dekapen-
taenu). Wzory strukturalne tych cz asteczek przedstawione s a na rys. 4.1.
1 W nawiasach podane s a skrocone nazwy, uzywane w dalszej cz esci tekstu.
31

4.1. Wst ep
Rysunek 4.1. Wzory strukturalne wybranych polienow
32

4.1. Wst ep
Dla wszystkich tych cz asteczek symetria stanu podstawowego to Ag, natomiast
dwa najnizsze singletowe stany wzbudzone maj a symetri e Bu oraz Ag. Stan 11Bu
ma charakter jonowy [56] i zdominowany jest przez konfiguracj e pojedynczo wzbu-
dzon a (HOMO) → (LUMO). Kowalencyjny stan 21Ag zawiera dominuj acy wk lad
podwojnie wzbudzonej konfiguracji (HOMO)2 → (LUMO)2 [53]. Przejsciem dozwo-
lonym jest 11Ag → 11Bu, natomiast wzbudzenie 11Ag → 21Ag jest zabrononie przez
symetri e.
Przez lata kontrowersje budzi lo ustalenie samej kolejnosci stanow 11Bu i 21Ag.
Pocz atkowo uwazano, ze najnizej lezacym singletowym stanem wzbudzonym polie-
now jest 11Bu. Eksperymenty przeprowadzone w latach siedemdziesi atych ubieg lego
wieku wskaza ly jednak na znaczn a roznic e w zdolnosci do luminescencji pomi edzy
heksa-1,3,5-trienem (brak fluorescencji) [41], a okta-1,3,5,7-tetraenem (obecnosc fluore-
scencji) [57]. Zjawisko to zasugerowa lo, ze w szeregu homologicznym polienow zmianie
ulega kolejnosc najnizszych stanow wzbudzonych [58,59]. Stanowisko to by lo stopniowo
potwierdzane przez eksperymenty i wyliczenia teoretyczne [41,43,52–55,57–60].
Rowniez silnie skorelowane metody oparte na funkcji falowej, jak Active Space with
Second-order Perturbation Theory (CASPT2) [54,60] czy Multireference Møller-Plesset
(MRMP) [53], potwierdzi ly zmian e kolejnosci stanow 11Bu i 21Ag wyst epuj ac a od
pewnej d lugosci lancucha polienowego. Dla butadienu najnizej lez acym stanem wzbu-
dzonym jest 11Bu, dla heksatrienu stany te maj a bardzo zblizone energie, a pocz awszy
od oktatetraenu nast epuje inwersja i najnizszym energetycznie stanem wzbudzonym
staje si e 21Ag. Otrzymywane tymi metodami wyniki s a na tyle dok ladne, ze mozna
je traktowac jako referencyjne w przypadku braku wiarygodnych danych eksperymen-
talnych. Doswiadczalne energie wzbudzen wertykalnych oraz wybrane wyniki obliczen
przedstawiono w tab. 4.1.
W przypadku MRMP jedynie dla cz asteczki butadienu zosta la uzyta odpowiednio
duza baza i spora przestrzen aktywna. Rownolegle zbadany zosta l wp lyw doboru ba-
zy i przestrzeni aktywnej na otrzymywane wyniki. Obliczenia dla wi ekszych uk ladow
zosta ly wykonane w mniejszych bazach prawdopodobnie ze wzgl edu na zbyt wyso-
kie koszty obliczeniowe. Uzyskane tak wyniki s a mniej dok ladne niz dla butadienu.
Uwzgl ednienie korekty na efekt bazy, a wi ec pewnego rodzaju ekstrapolacja wynikow,
pozwoli lo na lepsz a reprodukcj e danych doswiadczalnych. Ekstrapolowane wyniki obli-
czen MPRP szeregu homologicznego polienow z dosc dobr a dok ladnosci a reprodukuj a
energie wzbudzen wertykalnych wyznaczone eksperymentalnie oraz obliczone CASPT2.
Badania wykonane roznymi metodami Coupled-Cluster (CC) [55] potwierdzaj a ko-
lejnosc stanow i zachodz ac a inwersj e. Nie daj a jednak energii zblizonych do ekspery-
mentalnych ze wzgl edu na koniecznosc stosowania zbyt ma lych baz.
33

4.1. Wst ep
Tabela 4.1. Wyniki referencyjne
Wzbudzenie CASSCFa CASPT2b MRMPc ekstr. MRMPd Eksperyment
Butadien11Bu 7.73 6.23 6.21 – 5.92e
21Ag 6.67 6.27 6.31 – –
Heksatrien11Bu 7.06 5.01 5.37 5.10 5.13f
21Ag 5.64 5.20 5.34 5.09 5.21g
Oktatetraen11Bu 6.62 4.42 4.81 4.66 4.41h
21Ag 5.16 4.38 4.72 4.47 –
Dekapentaen11Bu 6.37 – 3.97 4.05 4.02i
21Ag 4.32 – 3.95 3.65 3.48j
a [53]b [54,60]c [53]d [53]e [49]f [45,46]g [61]h [50]i [47]j [47]
34

4.1. Wst ep
Sposrod silnie skorelowanych metod opartych na funkcji falowej w chwili obecnej
znane s a wyniki CASPT2, MRMP i CC. Obliczenia CC dla wszystkich cz asteczek
uda lo si e przeprowadzic w nieadekwatnie ma lych bazach, dok ladne obliczenia CAS-
PT2 dost epne s a tylko do oktatetraenu w l acznie, natomiast dobre wyniki MRMP dla
wi ekszych cz asteczek otrzymano dopiero po ekstrapolacji wynikow.
Ograniczenia tych metod wynikaj a ze z lego skalowania kosztow obliczeniowych ze
wzrostem uk ladu. Wyd luzenie lancucha zwi eksza nie tylko liczb e kombinowanych orbi-
tali atomowych czy tez korelowanych elektronow. Powi ekszenie poduk ladu π-elektro-
nowego wymusza zwi ekszenie przestrzeni aktywnej dla przejsc π → π∗. Pomniejszenie
bazy staje si e zatem koniecznosci a. Powoduje jednak spadek jakosci wynikow. Duzo
lepiej skaluj a si e obliczenia TDDFT.
Jednak dla cz asteczek polienow TDDFT nie reprodukuje dobrze energii stanow
11Bu i 21Ag. Energia 11Bu jest systematycznie zanizana, natomiast energia 21Ag sys-
tematycznie zawyzana (tab. 4.2). Nie jest obserwowana inwersja stanow wraz ze wzro-
stem d lugosci lancucha [39,62].
Stan 11Bu ma charakter jonowy, w duzym stopniu podobny do stanu z przeniesie-
niem ladunku (Charge-Transfer, CT). TDDFT nie opisuje dobrze stanow o charakterze
Charge-Transfer i w pe lnym TDDFT (wariant RPA) energia takich stanow jest zaniza-
na. Zastosowanie przyblizenia TDA-TDDFT w duzym stopniu poprawia otrzymywane
wyniki [56,62]. W niniejszym opracowaniu wszystkie obliczenia TDDFT wykonywane
s a w wariancie TDA.
Obliczenia CASSCF dla wszystkich badanych polienow potwierdzi ly znacz acy udzia l
konfiguracji dwukrotnie wzbudzonej (HOMO)2 → (LUMO)2 (1b2g → 2a2u) w funkcji
falowej stanu 21Ag [53]. Niestety, TDDFT nie opisuje dobrze stanow zawieraj acych
znacz acy wk lad konfiguracji dwukrotnie wzbudzonych (rozdz. 2.4) [15]. Dla badanych
cz asteczek konfiguracja (HOMO)2 → (LUMO)2 ma wyzsz a energi e, niz konfigura-
cje pojedynczo wzbudzone wchodz ace w sk lad stanu 21Ag (tab. 4.3). Na tej podsta-
wie mozna przypuszczac, ze uwzgl ednienie oddzia lywania mi edzy tymi konfiguracjami
efektywnie obnizy loby energi e stanu.
Przeprowadzenie obliczen wariantem TDA-TDDFT poprawia energi e stanu 11Bu.
Uzyskanie bardziej dok ladnego wyniku dla stanu 21Ag wymaga polepszenia opisu sta-
now zawieraj acych znaczny wk lad dwukrotnie wzbudzonych konfiguracji.
Stan 21Ag uzyskany w TDDFT to kombinacja z lozona g lownie z dwoch konfiguracji
pojedynczo wzbudzonych: 1bg → 2bg oraz 1au → 2au. Tymczasem, jak by lo to juz
wspomniane, obliczenia CASSCF wskazuj a na istotny udzia l konfiguracji dwukrotnie
wzbudzonej 1b2g → 2a2u.
Grupa Cave’a [63] na dwukonfiguracyjnym uk ladzie 1bg → 2bg i 1au → 2au spraw-
35

4.1. Wst ep
Tabela 4.2. B l edy systematyczne TDDFT. Porownanie wertykalnych energii wzbudzen11Ag → 11Bu i 11Ag → 21Ag (eV) otrzymanych metod a B3LYP/TDDFT z wartosciami
eksperymentalnymi.
WzbudzenieB3LYPa
EksperymentTDA RPA
Butadienb
11Bu 5.90 5.59 5.92c
21Ag 6.48 6.46 –
Heksatriend
11Bu 5.06 4.64 5.13e
21Ag 5.66 5.65 5.21f
Oktatetraeng
11Bu 4.39 3.98 4.41h
21Ag 4.83 4.83 –
Dekapentaeni
11Bu 3.90 3.52 4.02j
21Ag 4.22 4.21 3.48k
a [62]b baza 6-311(2+,2+)G**c [49]d baza 6-31++G**e [45,46]f [61]g baza 6-31++G**h [50]i baza 6-31++G**j [47]k [47]
Tabela 4.3. Energie diagonalne najnizej lez acych podwojnie wzbudzonych konfiguracji orazenergie stanow 21Ag otrzymane metod a TDA-TDDFT. Wzystkie wyniki w eV.
Cz asteczkaB3LYP/6-311++G** PBE0/6-311++G**
(HOMO)2 → (LUMO)2 21Ag (HOMO)2 → (LUMO)2 21Ag
butadien 10.93 6.56 11.76 6.77heksatrien 8.83 5.69 9.57 5.90oktatetraen 7.52 4.86 8.19 5.06dekapentaena 6.67 4.29 7.29 4.48
a baza 6-311G**
36

4.2. Obliczenia
dzi la oddzia lywanie z konfiguracj a dwukrotnie wzbudzon a 1b2g → 2a2u. Do oszacowania
oddzia lywania mi edzy orbitalami Kohna-Shama wzi ete zosta ly odpowiednie elemen-
ty macierzy hamiltonianu metody Restricted Hartree-Fock (RHF). Konsekwentnie,
energi e konfiguracji podwojnie wzbudzonej zaczerpni eto z energii orbitalnych orbitali
RHF. Wyniki dla butadienu i heksatrienu uzyskane tak przyblizon a metod a okaza ly
si e nadzwyczaj trafne i zach eci ly do wykonania pe lnej implementacji metody.
4.2. Obliczenia
Obliczenia TDA-TDDFT oraz DTDA-TDDFT dla cz asteczek: butadienu, heksa-
trienu, oktatetraenu oraz dekapentaenu przeprowadzono przy uzyciu hybrydowych
funkcjona low B3LYP oraz PBE0, w bazach 6-311++G** i 6-311G**.
Funkcjona l PBE0 zastosowano ze wzgl edu na bardzo dobry opis stanow wzbudzo-
nych niewielkich cz asteczek [34, 64]. Funkcjona lu B3LYP uzyto w celu sprawdzenia
zaleznosci wynikow od zastosowanego funkcjona lu.
Baza 6-311++G** dla ma lych cz asteczek organicznych stanowiu praktycznie limit
bazy dla funkcjona lu PBE0 [64]. Baz e 6-311G** wybrano w celu sprawdzenia stabil-
nosci metody wzgl edem stosowanej bazy funkcyjnej.
Wszystkie obliczenia wykonano dla geometrii zoptymalizowanych przy zastosowa-
niu metody B3LYP/6-31G**. Obliczenia TDDFT wykonano przy uzyciu pakietu nie-
doida [16], poszerzonego o implementacj e TDDFT uwzgl edniaj ac a wp lyw konfiguracji
wzbudzonych podwojnie. Szczego lowy opis implementacji zosta l zawarty w rozdz. 3.
Samouzgodniona procedura obliczen DTDA-TDDFT, zgodnie z oczekiwaniami,
jest szybko zbiezna. We wszystkich przypadkach wystarczy lo zaledwie kilka iteracji
aby uzyskac zbieznosc na poziomie 10−3 eV. Przebieg samouzgodnienia zilustrowano
w tab. 4.4 i 4.5.
37

4.2. Obliczenia
Tabela 4.4. Analiza zbieznosci DTDA-TDDFT dla cz asteczki butadienu(PBE0/6-311++G**)
Iteracja Energia stanu 21Ag (eV) Zmiana energii (eV)
0 6.77195 –
1 6.29538 -0.476572 6.33953 +0.044153 6.33579 -0.003744 6.33611 +0.00032
Tabela 4.5. Analiza zbieznosci DTDA-TDDFT dla cz asteczki heksatrienu(PBE0/6-311++G**)
Iteracja Energia stanu 21Ag (eV) Zmiana energii (eV)
0 5.90104 –
1 5.09251 -0.808532 5.23950 +0.146993 5.21688 -0.022624 5.22046 +0.003585 5.21990 -0.00056
38

4.3. Wyniki i dyskusja
Obliczenia dla wszystkich cz asteczek z wyj atkiem dekapentaenu przeprowadzono
w bazie 6-311++G**. W przypadku dekapentaenu w bazie orbitali atomowych wyst e-
puj a silne zaleznosci liniowe. Ich usuni ecie jest mozliwe i stanowi jedno z zagadnien
przewidzianych do realizacji w niedalekiej przysz losci. Jednak w obecnej wersji progra-
mu niemozliwe jest uzbieznienie obliczen SCF dekapentaenu w bazie 6-311++G**, co
z kolei nie daje mozliwosci przeprowadzenia obliczen TDDFT. Przy czym, jakkolwiek
dla butadienu roznica wynikow otrzymanych przy i bez uzycia funkcji dyfuzyjnych
wynosi nawet 0.5 eV, to dla oktatetraenu spada juz ponizej 0.15 eV. Swiadczy to o co-
raz szybszym wysycaniu bazy wraz ze wzrostem rozmiaru cz asteczki. Mozna zatem
oczekiwac, ze energie wertykalnych wzbudzen otrzymane w bazie 6-311G** s a bardzo
bliskie rezultatom dla bazy 6-311++G**. Rezultaty przeprowadzonych obliczen zosta ly
zebrane w tabeli 4.6.
Przeprowadzenie obliczen DTDA-TDDFT, zgodnie z oczekiwaniami, nie wp lywa
na energi e stanu 11Bu. Natomiast uwzgl ednienie poprawek od konfiguracji dwukrotnie
wzbudzonych zmienia energi e stanu 21Ag o od -0.4 eV do -0.8 eV. Ponadto wykonanie
obliczen DTDA-TDDFT zaciera istniej ac a dla TDA-TDDFT zaleznosc energii stanu
21Ag od zastosowanej bazy, co swiadczy o stabilnosci DTDA-TDDFT wzgl edem zmia-
ny bazy.
Energie doswiadczalne (obliczenia CASPT2 gdy brak danych doswiadczalnych) s a
bardzo dobrze reprodukowane. Stan 11Bu o charakterze jonowym jest prawid lowo opi-
sywany dzi eki zastosowaniu przyblizenia TDA-TDDFT. Uwzgl ednienie oddzia lywania
konfiguracji pojedynczo wzbudzonych z konfiguracjami podwojnie wzbudzonymi po-
prawia opis stanu 21Ag. Porownanie wynikow DTDA-TDDFT z rezultatami uzyska-
nymi przy zastosowaniu innych metod (tab. 4.1) zamieszczono w tabeli 4.7
Otrzymane wyniki potwierdzi ly skutecznosc zaimplementowanej metody DTDA-
-TDDFT dla nisko lez acych stanow wzbudzonych polienow liniowych.

4.3. Wyniki i dyskusja
Tabela 4.6. Energie wzbudzen wertykalnych (eV) nisko lez acych stanow singletowychwybranych polienow. Obliczenia TDA-TDDFT oraz DTDA-TDDFT wykonano w bazie6-311++G** (wyniki w nawiasie w bazie 6-311G**) przy uzyciu pakietu niedoida. Geometri e
uprzednio zoptymalizowano metod a B3LYP/6-31G**.
WzbudzenieB3LYP PBE0
TDA DTDA TDA DTDA
Butadien11Bu 6.02 (6.44) 6.02 (6.44) 6.13 (6.50) 6.13 (6.50)21Ag 6.56 (7.03) 6.06 (6.16) 6.77 (7.23) 6.34 (6.46)
Heksatrien11Bu 5.08 (5.30) 5.08 (5.30) 5.16 (5.35) 5.16 (5.35)21Ag 5.69 (5.84) 4.92 (4.91) 5.90 (6.05) 5.22 (5.22)
Oktatetraen11Bu 4.41 (4.55) 4.41(4.55) 4.48 (4.60) 4.48 (4.60)21Ag 4.86 (4.94) 4.07(4.06) 5.06 (5.13) 4.36 (4.36)
Dekapentaena
11Bu – (4.01) – (4.01) – (4.06) – (4.06)21Ag – (4.29) – (3.48) – (4.48) – (3.77)
a Obliczen dla bazy 6-311++G** nie uda lo si e uzbieznic ze wzgl edu na silne zaleznosci liniowew bazie orbitali atomowych
Tabela 4.7. Roznice (eV) pomi edzy wynikami DTDA-TDDFT (PBE0/6-311++G**),a: TDA-TDDFT, CASPT2, ekstrapolowanym MRMP oraz danymi eksperymentalnymi
(tab. 4.1). W nawiasach odpowiednie wartosci dla B3LYP/6-311++G**
Wzbudzenie ∆DFT ∆CASPT2 ∆MRMP ∆Eksp.
Butadien11Bu 0.00 ( 0.00) -0.10 (-0.21) -0.08 (-0.19)a 0.21 (0.10)21Ag -0.43 (-0.50) 0.07 (-0.21) 0.03 (-0.25)b – (–)
Heksatrien11Bu 0.00 ( 0.00) 0.15 ( 0.07) 0.06 (-0.02) 0.03 (-0.05)21Ag -0.68 (-0.77) 0.02 (-0.28) 0.13 (-0.17) 0.01 (-0.29)
Oktatetraen11Bu 0.00 ( 0.00) 0.06 (-0.01) -0.18 (-0.25) 0.07 (0.00)21Ag -0.70 (-0.79) -0.02 (-0.31) -0.11 (-0.40) – (–)
Dekapentaenc
11Bu 0.00 ( 0.00) – (–) 0.01 (-0.04) 0.04 (-0.01)21Ag -0.71 (-0.81) – (–) 0.12 (-0.17) 0.29 ( 0.00)
a MRMP bez ekstrapolacjib MRMP bez ekstrapolacjic Rezultaty dla bazy 6-311G**
40

Rozdzia l 5
Podsumowanie
Niniejsza praca mia la dwa zasadnicze cele. Pierwszym z nich by lo opracowanie
wydajnej implementacji TDDFT oraz rozszerzenia TDDFT, metody Dressed TDDFT
(DTDDFT). Drugim celem by lo zbadanie stosowalnosci i dok ladnosci obliczen metod a
DTDDFT dla wybranych polienow.
W ramach pracy wykonano implementacj e metody TDDFT (czyli wariantu RPA-
-TDDFT) i TDA-TDDFT dla uk ladow zamkni etopow lokowych. Dla wariantu TDA
mozliwe s a obliczenia przy uzyciu hybrydowych j ader korelacyjno-wymiennych, nato-
miast dla wariantu RPA ograniczono si e jedynie do j ader LDA i GGA. Zastosowanie
podejscia obiektowego i wykorzystanie istniej acego kodu pozwoli lo na stworzenie czy-
telnej, latwej do rozszerzania i wydajnej implementacji. Algorytm obliczen zosta l zrow-
noleglony przy wykorzystaniu modelu Single Process Multiple Data (SPMD). Wprowa-
dzenie automatycznej generacji kodu znacz aco u latwi lo i przyspieszy lo implementacj e
nowych funkcjona low korelacyjno-wymiennych na potrzeby obliczen DFT i TDDFT.
Ponadto w ramach pracy zaimplementowano rozszerzenie metody TDA-TDDFT, Dres-
sed TDA-TDDFT (DTDA-TDDFT), maj acej na celu poprawienie dok ladnosci obliczen
energii stanow o duzym udziale konfiguracji podwojnie wzbudzonych.
Realizuj ac drugi cel pracy, wykonano obliczenia TDA-TDDFT i DTDA-TDDFT
energii nisko lez acych elektronowych stanow wzbudzonych w cz asteczkach s-trans-
-buta-1,3-dienu (butadienu), 2(s-trans),4(s-trans)-heksa-1,3(E),5-trienu (heksatrienu),
2(s-trans),6(s-trans)-okta-1,3(E),5(E),7-tetraenu (oktatetraenu) oraz 2(s-trans),8(s-
41

-trans)-deka-1,3(E),5(E),7(E),9-pentaenu (dekapentaenu). W przeciwienstwie do
RPA-TDDFT i TDA-TDDFT, DTDA-TDDFT poprawnie reprodukuje kolejnosc
wzbudzen we wszystkich badanych uk ladach. Wartosci energii wzbudzen otrzyma-
nych z obliczen DTDA-TDDFT s a bardzo zblizone do wynikow CASPT2, przy czym
DTDA-TDDFT charakteryzuje si e znacznie mniejszym kosztem obliczeniowym. Otrzy-
mane wyniki potwierdzaj a, ze DTDA-TDDFT znacz aco poprawia dok ladnosc obliczen
energii stanow o duzym udziale konfiguracji podwojnie wzbudzonych. Wykonane ob-
liczenia potwierdzi ly tez, ze, w ramach TDDFT, opis nisko lez acych stanow wzbudzo-
nych w polienach wymaga uwzgl ednienia konfiguracji podwojnie wzbudzonych.

Bibliografia
[1] Helgaker, T.; Jorgensen, P. and Olsen, J., Molecular Electronic Structure Theory, John
Wiley and Sons, ltd, 2000.
[2] Hohenberg, P. and Kohn, W., Phys Rev, 1964, 136(3B), 864–871.
[3] Kohn, W. and Sham, L. J., Phys Rev A, 1965, 140(4), 1133 – 1138.
[4] Runge, E. and Gross, E. K. U., Phys Rev Lett, 1984, 52, 997.
[5] Marques, M. and Gross, E., Ann Rev Phys Chem, 2004, 55, 427.
[6] Petersilka, M.; Gossmann, U. J. and Gross, E. K. U., Phys Rev Lett, 1996, 76, 1212–1215.
[7] Grabo, T.; Petersilka, M. and Gross, E., J Mol Struc (Theochem), 2000, 501–502,
353–367.
[8] Nalewajski, R. F., Podstawy i Metody Chemii Kwantowej, Wydawnictwo Naukowe
PWN, Warszawa, 2001.
[9] Stratmann, R. E.; Scuseria, G. E. and Frisch, M. J., J Chem Phys, 1998, 109, 8218.
[10] Casida, M. In Recent developments and applications in density functional theory, Semi-
nario, J., Ed.; Elsevier, Amsterdam, 1996.
[11] Bauernschmitt, R. and Ahlrichs, R., Chem Phys Lett, 1996, 256, 454.
[12] Hirata, S. and Head-Gordon, M., Chem Phys Lett, 1999, 314, 291–299.
[13] Gonze, X. and Scheffler, M., Phys Rev Lett, 1999, 82, 4416.
[14] Appel, H.; Gross, E. K. U. and Burke, K., Phys Rev Lett, 2003, 90(4), 043005.
[15] Maitra, N. T.; Zhang, F.; Cave, R. J. and Burke, K., J Chem Phys, 2004, 120(13), 5932.
[16] Mazur, G.; Makowski, M.; Piskorz, W.; Cwiklik, L.; Sterzel, M.; Radon, M.;
Jagoda-Cwiklik, B.; Kulig, W. and B lazewicz, D., Niedoida 0.3, 2007.
[17] Leininger, M. L.; Sherrill, C. D.; Allen, W. D. and III, H. F. S., J Comp Chem, 2001,
22(13), 1574–1589.
[18] Strange, R.; Manby, F. and Knowles, P., Comp Phys Comm, 2001, 136, 310.
[19] Sa lek, P. and Hesselmann, A., J Comp Chem, 2007, 28(16), 2569–2575.
[20] Maple http://www.maplesoft.com/, ?
[21] Maxima http://maxima.sourceforge.net/, ?
43

Bibliografia
[22] Yacas http://yacas.sourceforge.net/, ?
[23] Vosko, S.; Wilk, L. and Nusair, M., Can J Phys, 1980, 58, 1200.
[24] Lee, C.; Yang, W. and Parr, R. G., Phys Rev B, 1988, 37(2), 785.
[25] Perdew, J. P.; Burke, K. and Ernzerhof, M., Phys Rev Lett, 1996, 77, 3865.
[26] Perdew, J. P.; Chevary, J. A.; Vosko, S. H.; Jackson, K. A.; Pederson, M. R.; Singh,
D. J. and Fiolhais, C., Phys Rev B, 1992, 46(11), 6671.
[27] Perdew, J. P. and Wang, Y., Phys Rev B, 1992, 45(23), 13244.
[28] Dirac, P. A. M., Proceedings of the Cambridge Philosophical Society, 1930, 26, 376.
[29] Bloch, F., Zeitschrift fur Physik, 1929, 57, 545.
[30] Becke, A. D., Phys Rev A, 1988, 38(6), 3098–3100.
[31] Becke, A. D., J Chem Phys, 1993, 98, 5648.
[32] Adamo, C. and Barone, V., J Chem Phys, 1998, 108, 664.
[33] Zhang, Y. and Yang, W., Phys Rev Lett, 1998, 80, 890.
[34] Adamo, C. and Barone, V., J Chem Phys, 1999, 110, 6158–6170.
[35] Langreth, D. C. and Perdew, J. P., Phys Rev B, 1980, 21, 5469.
[36] Perez-Jorda, J. M. and Yang, W., Chem Phys Lett, 1995, 241, 469–476.
[37] MPI-2: Extensions to the message-passing interface
http://www-unix.mcs.anl.gov/mpi/mpi-standard/mpi-report-2.0/mpi2-report.htm,
?
[38] Piela, L., Idee Chemii Kwantowej, Wydawnictwo Naukowe PWN, Warszawa, 2003.
[39] Catalan, J. and de Paz, J. L. G., J Chem Phys, 2006, 124, 034306.
[40] Mulliken, R. S., J Chem Phys, 1939, 7, 364.
[41] Gavin, R. M.; Risemberg, S. and Rice, S. A., J Chem Phys, 1973, 58, 3160.
[42] Mosher, O. A.; Flicker, W. M. and Kuppermann, A., J Chem Phys, 1973.
[43] R. M. Gavin, J. and Rice, S. A., J Chem Phys, 1974, 60, 3231.
[44] McDarmid, R., J Chem Phys, 1976, 64, 514.
[45] Flicker, W. M.; Mosher, O. A. and Kuppermann, A., Chem Phys Lett, 1977, 45, 492.
[46] Kuppermann, A.; Flicker, W. M. and Mosher, O. A., Chem Rev, 1979, 79, 77–96.
[47] D’Amico, K. L.; Manos, C. and Christensen, R. L., J Am Chem Soc, 1980, 102(6), 1777.
[48] Doering, J. P. and McDiarmid, R., J Chem Phys, 1980, 73, 3617.
[49] Doering, J. P. and McDiarmid, R., J Chem Phys, 1981, 75, 2477.
[50] Heimbrook, L. A.; Kohler, B. E. and Levy, I. J., J Chem Phys, 1984, 81, 1592.
[51] Leopold, D. G.; Pendley, R. D.; Roebber, J. L.; Hemley, R. J. and Vaida, V., J Chem
Phys, 1984, 81, 1592.
[52] Beljonne, D.; Shuai, Z.; Serrano-Andres, L. and Bredas, J. L., Chem Phys Lett, 1997,
279, 1.
[53] Nakayama, K.; Nakano, H. and Hirao, K., Int J Quantum Chem, 1998, 66(2), 157–175.
[54] Serrano-Andres, L.; Merchan, M. and Nebot-Gil, I., J Chem Phys, 1993, 98(4), 3151.
44

Bibliografia
[55] Cronstrand, P.; Christiansen, O.; Norman, P. and Agren, H., Phys Chem Chem Phys,
2001, 3(13), 2567.
[56] Wang, Y.-L. and Wu, G.-S., Int J Quantum Chem, 2008, 108, 430–439.
[57] R. M. Gavin, J.; Weisman, C.; McVey, J. K. and Rice, S. A., J Chem Phys, 1978, 68,
522.
[58] Hudson, B. S. and Kohler, B. E., Chem Phys Lett, 1972, 14(3), 299.
[59] Hudson, B. and Kohler, B. E., Ann Rev Phys Chem, 1974, 25, 437.
[60] Serrano-Andres, L.; Lindh, R.; Roos, B. O. and Merchan, M., J Phys Chem, 1993, 97,
9360.
[61] Fujii, T.; Kamata, A.; Shimizu, M.; Adachi, Y. and Maeda, S., Chem Phys Lett, 1985,
115, 369–372.
[62] Hsu, C.-P.; Hirata, S. and Head-Gordon, M., J Phys Chem A, 2001, 105(2), 451–458.
[63] Cave, R. J.; Zhang, F.; Maitra, N. T. and Burke, K., Chem Phys Lett, 2004, 389, 39–42.
[64] Ciofini, I. and Adamo, C., J Phys Chem, 2007, 111, 5549–5556.

Dodatek A
Eliminacja wspolnych podwyrazen
Ponizej przedstawiono implementacj e w systemie algebry symbolicznej yacas algo-
rytmu eliminacji wspolnych podwyrazen (CSE) stworzon a na potrzeby automatycznej
generacji kodu s luz acego do obliczania wartosci funkcjona low korelacyjno-wymiennych
i wartosci ich pochodnych.
// For an expression e return a list of all simple subexpressions
Function("fcs", e)
[
Local(r);
r := ;
If(Not IsAtom(e),
[
Local(p, l);
l := Listify(e);
If(ExpressionDepth(e)=2,//<4,
[
r := l;
],
46

Eliminacja wspolnych podwyrazen
[
ForEach(p, l) [
Local(x);
x := fcs(p);
If(x != ,
[
r := Concat(r, x);
]);
];
]);
]);
r;
];
// For a list of (listified) expressions return list of unique
// expressions which are present more then once in the original
// list
Function("rcs", cs)
[
Local(r);
r := ;
ForEach(s, cs) [
If(Count(cs, s) > 1,
[
Push(r, s);
]);
];
RemoveDuplicates(r);
];
// For a list of (listified) expressions return list of
// pairs (unique_id, expression)
Function("bcs", l)
[
Local(r);
r := ;
ForEach(i, l) [
47

Eliminacja wspolnych podwyrazen
Push(r, UniqueConstant(), i);
];
r;
];
// Given a list of pairs (identifier, listified_subexpression), replace
// all occurences of subexpression in expression e with the corresponding
// identifier
Function("scs", sl, e)
[
Local(r);
r := e;
ForEach(i, sl) [
r := Subst(UnList(i[2]), i[1])r;
];
r;
];
// Generate list of assignements from the list of pairs
// (identifier, listified_expression)
Function("acs", sl)
[
Local(r);
r := ;
ForEach(i, sl) [
Local(a);
a := :=, i[1], UnList(i[2]) ;
Push(r, UnList(a));
];
r;
];
// Generate list of strings with declarations and initializations
// representing the assignements in al
Function("dcs", al)
[
Local(r);
48

r := ;
ForEach(i, al) [
Push(r, "const double " : CForm(i) : ";");
];
r;
];
// For an expression e:
// - find all simple subexpressions present more then once
// - generate unique ids for for each of them
// - substitute them with the ids
// - return substituded list of substitutions and substituted id
Function("gcs", e)
[
Local(re);
re := e;
Local(csl);
csl := ;
Local(t);
t := ;
Until(Length(t) = 0) [
t := bcs(rcs(fcs(re)));
If(Length(t) != 0,
[
re := scs(t, re);
csl := Concat(csl, t);
]);
];
re, csl ;
];

Streszczenie
Niniejsza praca mia la dwa zasadnicze cele. Pierwszym z nich by lo opracowanie
wydajnej implementacji TDDFT oraz rozszerzenia TDDFT, metody Dressed TDDFT
(DTDDFT). Drugim celem by lo zbadanie stosowalnosci i dok ladnosci obliczen metod a
DTDDFT dla wybranych polienow liniowych.
W ramach pracy wykonana zosta la czytelna, latwa do rozszerzania i wydajna im-
plementacja metody RPA-TDDFT oraz TDA-TDDFT dla uk ladow zamkni etopow lo-
kowych. Wprowadzono takze mechanizm automatycznej generacji kodu funkcjona low
korelacyjno-wymiennych. Algorytm obliczen zosta l zrownoleglony. Ponadto, w ramach
pracy zaimplementowano metod e DTDA-TDDFT, maj ac a na celu poprawienie dok lad-
nosci obliczen energii stanow o duzym udziale konfiguracji podwojnie wzbudzonych.
Realizuj ac drugi cel pracy, wykonano obliczenia TDA-TDDFT i DTDA-TDDFT
energii nisko lez acych elektronowych stanow wzbudzonych w cz asteczkach butadie-
nu, heksatrienu, oktatetraenu oraz dekapentaenu. Pokazano, ze w przeciwienstwie do
RPA-TDDFT i TDA-TDDFT, DTDA-TDDFT poprawnie reprodukuje kolejnosc ni-
sko lez acych wzbudzen we wszystkich badanych uk ladach. Wartosci energii wzbudzen
otrzymanych z obliczen DTDA-TDDFT s a bardzo zblizone do wynikow CASPT2, przy
czym DTDA-TDDFT charakteryzuje si e znacznie mniejszym kosztem obliczeniowym.
S lowa kluczowe
stany wzbudzone, TDDFT, polieny liniowe
50

Abstract
The aim of the thesis was twofold. The first of the objectives was to design an
efficient implementation of the TDDFT method and its extension, the Dressed TDDFT
(DTDDFT) method. The other objective was to study the applicability and accuracy
of DTDDFT for selected linear polyenes.
A readable, easily extensible and efficient computational scheme of the RPA-TDDFT
and TDA-TDDFT methods for closed-shell systems was designed and implemented.
A facility for automatic code generation for the purpose of the exchange-correlation
kernel calculations was implemented. Additionally, the DTDDFT method was imple-
mented. The method aims at improving the accuracy of excitation energy calculations
for states having significant admixture of doubly-excited configurations.
Moreover, TDA-TDDFT and DTDA-TDDFT calculations for low-lying excited sta-
tes in butadiene, hexatriene, octatetraene and decapentaene were performed. It was
shown that, contrary to RPA-TDDFT and TDA-TDDFT, DTDA-TDDFT correctly
reproduces low-lying excited states ordering in all studied systems. Excitation energy
values obtained from the DTDA-TDDFT calculations are very close to their CASPT2
counterparts, at the significantly lower computational cost of the DTDA-TDDFT me-
thod.
Key words
excited states, TDDFT, linear polyenes
51


















