Presentazione di PowerPoint - MIT - Massachusetts Institute of...
53
1 LE (MALATTIE) NEOPLASIE MIELOPROLIFERATIVE CRONICHE PHILADELPHIA-negative parte I
Transcript of Presentazione di PowerPoint - MIT - Massachusetts Institute of...

1
LE (MALATTIE) NEOPLASIEMIELOPROLIFERATIVE
CRONICHE PHILADELPHIA-negative
parte I

Malattie (Neoplasie) Mieloproliferative Croniche
Malattie che originano dalla trasformazione neoplastica di una cellula staminale emopoietica pluripotente che genera un clone
con crescita non regolata nel midollo osseo e in sedi extramidollari.
Si manifestano prevalentementea carico di una o più linee delle cellule del sangue e si
accompagnano ad alterazioni del microambiente midollare

Myeloproliferative NeoplasmsMyeloproliferative Neoplasms
CML “Classical”CNL
CEL-NOSMastocytosis
MPN-u
Polycythemia Vera
Essential Thrombocythemia
Primary Myelofibrosis
BCR-ABL1
Reviewed in Vannucchi et al. CA Cancer J Clin. 2009; 59(3):171-91
• L. cronica neutrofili• L. cronica-eosinofili*• Mastocitosi• Forme inclassificabiliWHO-2008

4
Emopoiesi policlonale Emopoiesi monoclonale
XA
XA XB ZIGOTE
XA XB XA XB EMBRIONEXA XB
XA XXBB XA XB XA XB
Lyonizzazione
XA XA
XB XBXA
XA
Cellula uovoCellula uovo SpermatozooSpermatozooXB
Predominanza “clonale” vs normale emopoiesi policlonale

AA 2010-11 Lezioni di Ematologia prof A.M. Vannucchi
5
Emopoiesi
B linfocitiT linfocitiNK
DC
GranulocitiMacrofagi
PiastrineEmazie
Staminali
Progenitori
MEP

6
• Sono malattie relativamente poco frequenti: 5 / 100.000,
ma tra le più comuni neoplasie ematologiche
• Sono malattie sporadiche, ma esistono «famiglie» con una
elevata incidenza (autosomica dominante?)
• Esiste certamente una predisposizione familiare: il rischio
è circa 7 volte superiore rispetto alla popolazione
normale per chi ha un familiare affetto da una MPN
•Aplotipo di predisposizione «46/1» sul cr 9, ed altri su Myb e
TERT
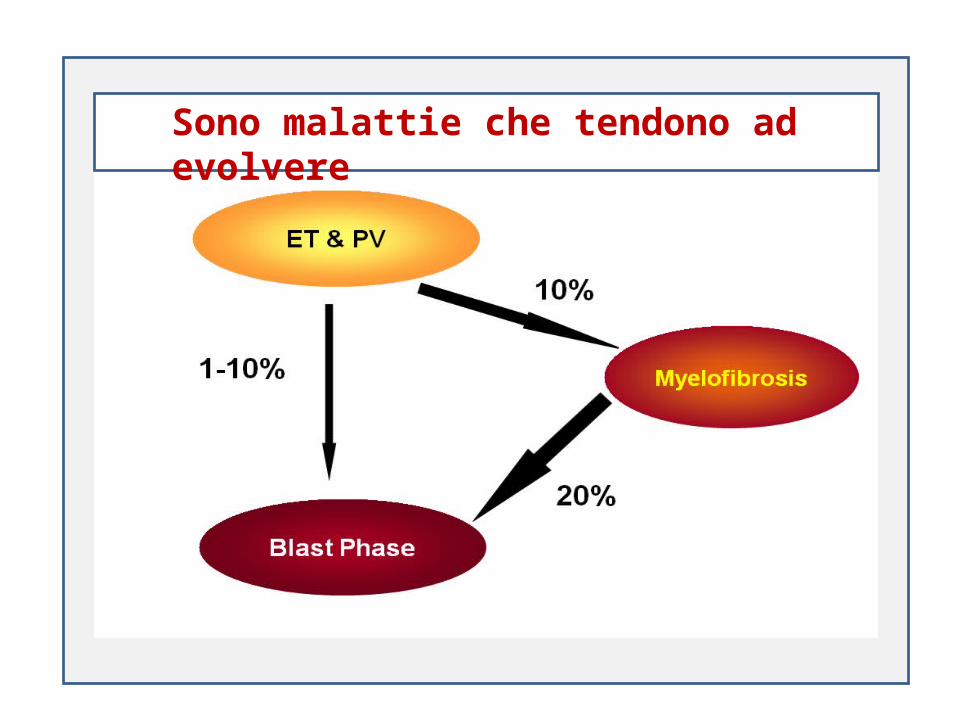
SSono malattie che tendono ad evolvere

Policitemia Vera Trombocitemia Essenziale Mielofibrosiprimaria e secondaria
JAK2 ESONE12
Altre (rare)
JAK2 V617F
MPL W515
NESSUNA
CALR mutazioni
PROFILO MUTAZIONALE

9
La mutazione JAK2V617F
Una G-T mutazione a livello del nucleotide 1849 (nell’ esone 14) porta alla sostituzione di valina con fenilalanina in posizione 617 (V617F) Il codone mutato V617F si trova nel dominio pseudochinasico
Ulteriori mutazioni/delezioni/inserzioni sono state dimostrate a livellodell’ esone 12

10
Mutazioni di MPL
• MPL codifica per il recettore della trombopoietina
• Il gene si trova sul crom 1• Le mutazioni sono localizzate
al codone 515, e determinano una instabilità del recettore stesso che si attiva in assenza del ligando
• Rare mutazioni al codone 505 in forme familiari di piastrinosi

AA 2010-11 Lezioni di Ematologia prof A.M. Vannucchi
11
Kaushansky K. N Engl J Med 2006;354:2034-2045
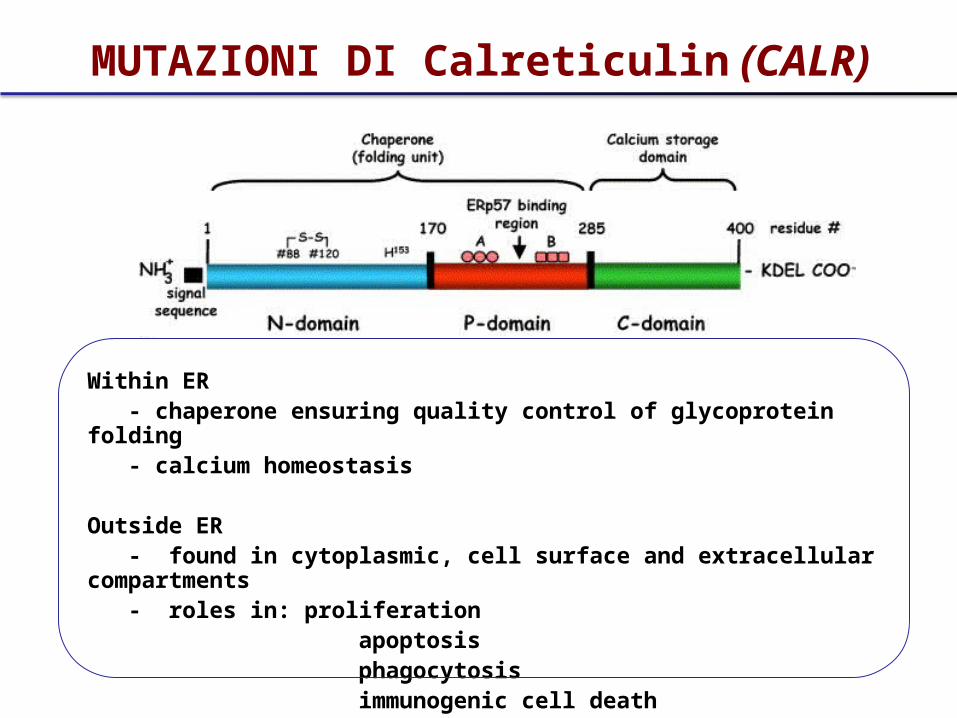
Within ER - chaperone ensuring quality control of glycoprotein folding - calcium homeostasis
Outside ER - found in cytoplasmic, cell surface and extracellular compartments - roles in: proliferation apoptosis phagocytosis immunogenic cell death
MUTAZIONI DI Calreticulin (CALR)

MUTAZIONI DI Calreticulin (CALR)
• Le mutazioni di CALR sono tutte localizzate nell’esone 9, coinvolto nel legame del Ca2+ e nella localizzazione della proteina nel RE
• Oltre 50 tipi descritti.• Delezione 52 bp e inserzione di 9 bp le più comuni• Determinano tutte un frameshift, e quindi viene
codificata una proteina con una porzione C-terminale anomala
• Sono espresse in un progenitore multipotente
AA 2010-11 Lezioni di Ematologia prof A.M. Vannucchi
14
Vannucchi et al. CA Cancer J Clin. 2009; 59(3):171-91
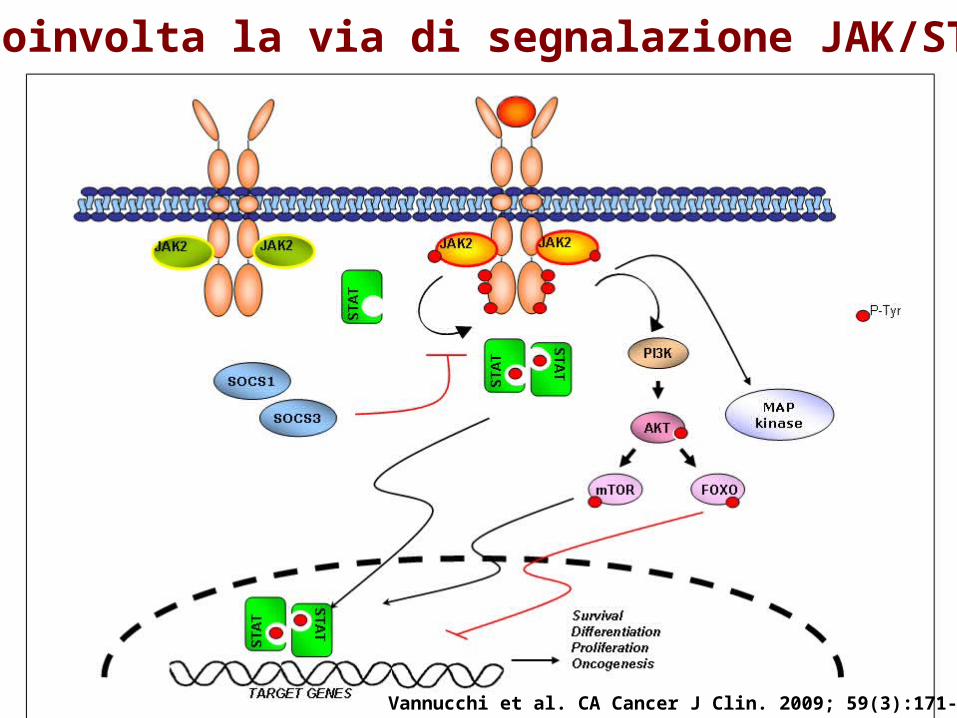
Coinvolta la via di segnalazione JAK/STAT

EEC= Colonie eritroidi endogene («spontanee»)
• Crescono in assenza dello stimolo proliferativo legato all’azione dell’Eritropoietina (tipiche della PV, ma presenti anche in ET e PMF più raramente).
• Fenomeno simile anche a carico dei progenitori mieloidi e megacariocitari

16
POLICITEMIA VERA:diagnosi

Policitemia Vera (PV)Malattia mieloproliferativa cronica caratterizzata da una eccessiva e afinalistica produzione di globuli rossi ( talvolta anche di globuli bianchi e piastrine) Nella maggioranza dei casi si trova la mutazione di JAK2 V617F, o exon 12
TerminologiaEmatocrito: E’ la percentuale di sangue che è occupata da globuli rossi Emoglobina: si misura in grammi per 100 ml di sangue interoGlobuli rossi . Sono espressi per microlitro di sangue interoPlasma: E’ la parte non corpuscolata del sangue
Va tenuta distinta dalle forme reattive (JAK2!)

18
ERITROCITOSI
assoluta relativa (vera) (spuria)
• ERITROCITOSI PRIMARIA Congenita Acquisita (=PV)
ERITROCITOSI SECONDARIADa secrezione appropriata di EpoDa secrezione inappropriata di Epo
• EMOCONCENTRAZIONE secondaria a disidratazione: diarrea, sudorazione profusa, vomito, diuretici, etanolo)
• IPERTENSIONE• PRE-ECLAMPSIA• FEOCROMOCITOMA

19
ERITROCITOSI SECONDARIA- I
DA SECREZIONE APPROPRIATA DI ERITROPOIETINA
• Congenite:– Emoglobine ad alta affinità O2 (aut. dominanti)– Metemoglobinemia ereditaria – Deficit di 2,3-DPG (aut. recessiva)
• Acquisite:– Anomalie cardiovascolari con shunt destro-sinistro– Anomalie dei vasi polmonari – Soggiorno a grandi altezze– Malattie polmonari croniche (BPCO, enfisema, fibrotorace,...)– S. della ipoventilazione incluse le apnee notturne– Cardiopatie acquisite con scompenso cronico– Carbossiemoglobinemia (eritrocitosi dei fumatori)– Alterazioni neurologiche (disfunzioni centri respiratori)– HbM (farmaci, tossici)– Cobalto

20
DA SECREZIONE INAPPROPRIATA DI ERITROPOIETINA• Congenite:
– Forma Cuvash ed altre da mutazioni del gene VHL (aut. recessiva)• Acquisite:
– Cisti renali (rene policistico)– Leiomioma uterino– Feocromocitoma – Carcinoma renale– Meningioma– Carcinoma epatocellulare – Carcinoma renale, polmonare, ovarico– Emangioblastoma cerebellare
• Altre:– Dopo trapianto di rene– Doping con Epo– Trattamento con androgeni
ERITROCITOSI SECONDARIA - II

Come si effettua la diagnosi (I)
E’ fondamentale eseguire il corretto iter diagnostico:
confermare la diagnosi secondo i criteri WHO, escludendo le possibili cause di eritrocitosi secondaria
distinguere accuratamente la PV dalle altre forme di neoplasie mieloproliferative croniche

Come si effettua la diagnosi (II)
1. EMOCROMO e striscio di sangue periferico:- incremento del numero di globuli rossi, del valore dell’ematocrito e della emoglobina;-nell’80-85% dei casi: incremento del numero di globuli bianchi (“leucocitosi”); neutrofilia, ma non cellule mieloidi immature-incremento del numero delle piastrine (“piastrinosi” o “trombocitosi”) nel 70-80% dei casi

Come si effettua la diagnosi (III)
2. Ricerca della mutazione genetica JAK2V617F:-presente in almeno il 95% dei casi di PV; nel 2-3% mutazioni più rare a carico dell’esone 12 del gene JAK2-JAK2V617F non è specifica della PV: si dimostra nel 60% circa dei casi di TE o di PMF-nessuna delle forme secondarie di eritrocitosi presenta queste alterazioni
3. Dosaggio dell’ERITROPOIETINA (EPO):ormone che regola la produzione dei globuli rossia livello del midollo osseo: livelli MOLTO RIDOTTI

Come si effettua la diagnosi (IV)
• Biopsia osteomidollare e aspirato midollare: dimostrazione delle tipiche alterazioni : aumento della cellularità
e proliferazione trilineare, senza alterazioni morfologiche dei magacariociti né fibrosi
fornisce informazioni molto utili per una corretta diagnosiconsente la distinzione dalle altre forme di neoplasie
mieloproliferative cronicheANCHE SE non è sempre necessario eseguire l’esame per
porre la diagnosi

25

Diagnosi: criteri WHO (2008) • Criteri maggiori
• Emoglobina superiore a 18.5 g/dL nel maschio e a 16.5 g/dL nella femmina (o altra dimostrazione di incremento della massa eritrocitaria)
• Presenza della mutazione JAK2617V>F o di altre mutazioni funzionalmente simili (mutazioni dell’esone 12 del gene JAK2)
1. Criteri minori2. Biopsia del midollo osseo che mostra ipercellularità per l’età con
proliferazione trilineare (panmielosi) con iperplasia sia della serie eritroide che di quella granulocitaria e megacariocitaria
3. Eritropoietina sierica inferiore al normale range di riferimento.4. Formazione di colonie eritroidi endogene in vitro (EEC)
Per la diagnosi sono richiesti: entrambi i criteri maggiori +1 minore
oppureil primo criterio maggiore + 2 criteri minori

27
TROMBOCITEMIA
ESSENZIALE:
diagnosi

28
TROMBOCITOSI:limite superiore normale delle piastrine 450x109/L
SPURIACrioglobulineBatteriemieBlasti leucemiciEritroblasti
VERAPrimitivaSecondaria (o reattiva)
da produzioneda alterata distribuzione

29
CAUSE DI TROMBOCITOSI REATTIVA
• Anemia Fe-carenziale• Anemie emolitiche• Post-emorragiche• In corso di malattie
infiammatorie croniche• Collagenopatie • Neoplasie• Infezioni• Correzione di carenza di
vit. B12/ ac folico
• Dopo interventi chirurgici• Forme “rebound”• Dopo abuso di etanolo• Malattie renali croniche
• Post-splenectomia / agenesia splenica
• Sforzo fisico intenso• Adrenalina

• Trombocitemia Essenziale• Policitemia Vera• Mielofibrosi Primaria• Leucemia Mieloide cronica• Sindrome del(5q)• Anemia refrattaria con
sideroblasti ad anello e trombocitosi (RARS-T)
• Mutazioni del recettore della trombopoietina (MPL),[S505N]
• Mutazioni del gene della trombopoietina
TROMBOCITOSI PRIMARIE
acquisite congenite

31
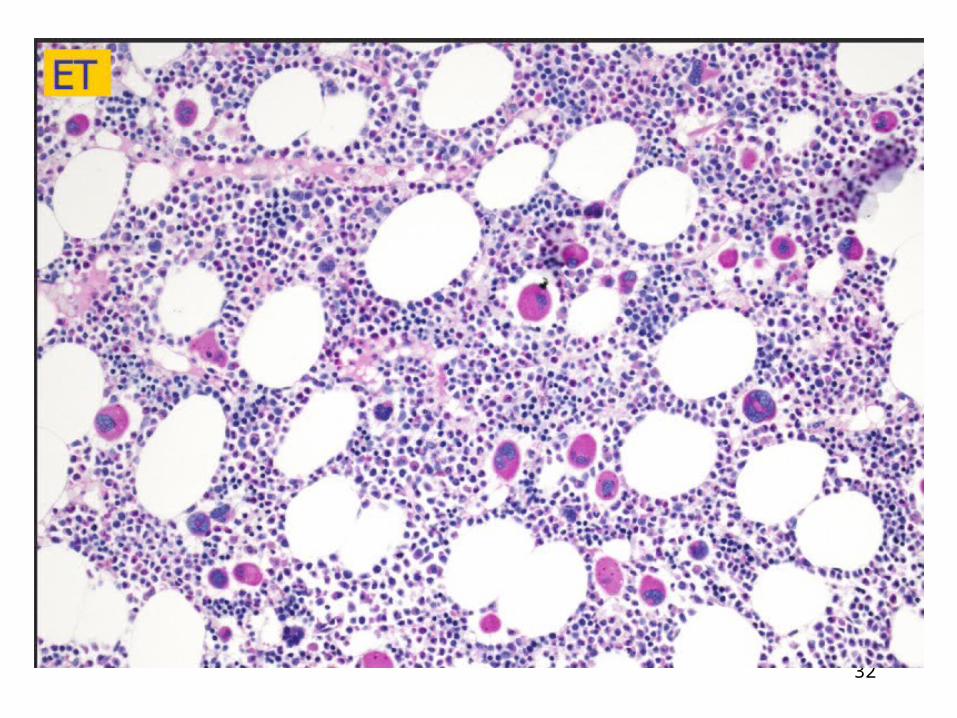
32

33
Essential Thrombocythemia
Major criteria
1. Platelet count ≥ 450 x 109/L2. Megakaryocyte proliferation with large and mature morphology. No or little granulocyte or erythroid proliferation.3. Not meeting WHO criteria for CML, PV, PMF, MDS or other myeloid neoplasm4. Demonstration of JAK2V617F or other clonal marker (will include CALR) or no evidence of reactive thrombocytosis
Minor criteria
Diagnostic combinations All four criteria must be met
WHO 2008 DIAGNOSTIC CRITERIA

PV & ET
Presentazione clinica
34

35
Anomalie di laboratorio: piastrinosi, leucocitosi, assente/moderato aumento LDH.Esame obiettivo: indifferente, modesta splenomegaliaSintomatologia:
Asintomatica (>2/3 dei casi)Manifestazioni emorragiche ( 5%)
Manifestazioni trombotiche ( 15%)
TROMBOCITEMIA ESSENZIALE
Scoperta quasi sempre per caso in corso di esami di routine, più raramente al tempo d’oggi in occasione di un evento trombotico o emorragico

PV: Come si manifesta (I)
• Scoperta per caso (esami del sangue eseguiti per altri motivi o per check-up di routine) alterazione dei parametri ematologici
• Comparsa di sintomi generici:
- rossore al volto, arrossamento delle congiuntive, spesso accompagnati da sensazione di calore diffuso
- sensazione di stanchezza e di debolezza generale (“astenia”)- episodi ricorrenti di cefalea- alterazioni transitorie della vista, come lampi luminosi (“scotomi”) o zone scure
del campo visivo- alterazioni dell’udito (“acufeni”), come rumori o fischi
spesso attribuibili all’aumento della viscosità ematica con alterazioni a carico del microcircolo(sintomi riconducibili agli organi colpiti)

PV: Come si manifesta (II)
• Altri sintomi a carico del microcircolo:
- disturbi di sensibilità, formicolii (“parestesie”), in particolare alle estremità (polpastrelli delle dita, piedi)
- episodi di arrossamento intenso, bruciore e dolore, spesso urente, alle estremità (“eritromelalgia”)
- vertigini
- riscontro di ipertensione arteriosa

A. Burning pain and redwarm congestion of the big and little toes, sole of forefoot and lateral edge of right foot.
B. Burning aching pain and motlled red-blue ischemic discoloration of the sole of the right foot.
C and D Erythromelalgia of the big second and third toes before (C) and after (D) treatment with aspirin.
Eritromelalgia
Crisi doloroseacute che colpiscono la parte distale
delle estremità che si associano a rossore e gonfiore

PV: Come si manifesta (III)• Altre manifestazioni cliniche:
- Prurito, localizzato agli arti o al tronco oppure diffuso, spesso scatenato dal bagno o dalla doccia (”acquagenico”)
- sensazione di ingombro e di pesantezza all’addome, con possibili disturbi digestivi, senso di sazietà precoce o alterazioni dell’alvo (per aumento del volume della milza, “splenomegalia”)
- dolori ossei, muscolari, osteoarticolari
- gastrite
- “Sintomi costituzionali”(generali o sistemici): sudorazioni profuse (specie nelle ore notturne) febbricola ricorrente senza causa infettiva dimagrimento (perdita del 10% del peso corporeo entro circa 6
mesi)
- Complicanze dell’iperuricemia: episodi di gotta, calcoli renali di acido urico (“nefrolitiasi”)

40
Thrombosis-free Survival in PV and ET Thrombosis-free Survival in PV and ET
Time (months)
PVn=397
CE=59 (14.9%)
ETn=637
CE=73 (11.5%)
0.70
0.75
0.80
0.85
0.90
0.95
1.00
0 360 720 1080 1440 1800 2160 2520 2880 3240 3600
Prob
abili
ty o
f thr
ombo
sis-
free
sur
viva
l
PVn=397
CE=59 (14.9%)
ETn=637
CE=73 (11.5%)Cause of death %
Fatal thrombosis 41
Hemorrhages 4
AML/MF 13
Other cancers 20
Other cause 22
3.5 decessi/100 persone/anno (=2.1 volte superiore alla popolazione italiana standardizzata per sesso ed età)

complicanza vascolare maggiore:
-emorragie maggiori (es. tratto gastrointestinale, genitourinario, cerebrale).
- trombosi arteriose maggiori: es. infarto del miocardio, trombosi dei vasi cerebrali;
- trombosi venose maggiori: es. trombosi venose profonde, embolia polmonare, trombosi in distretti inusuali come quelle addominali (v. porta, v. mesenterica o v. splenica) e a carico della vena retinica;
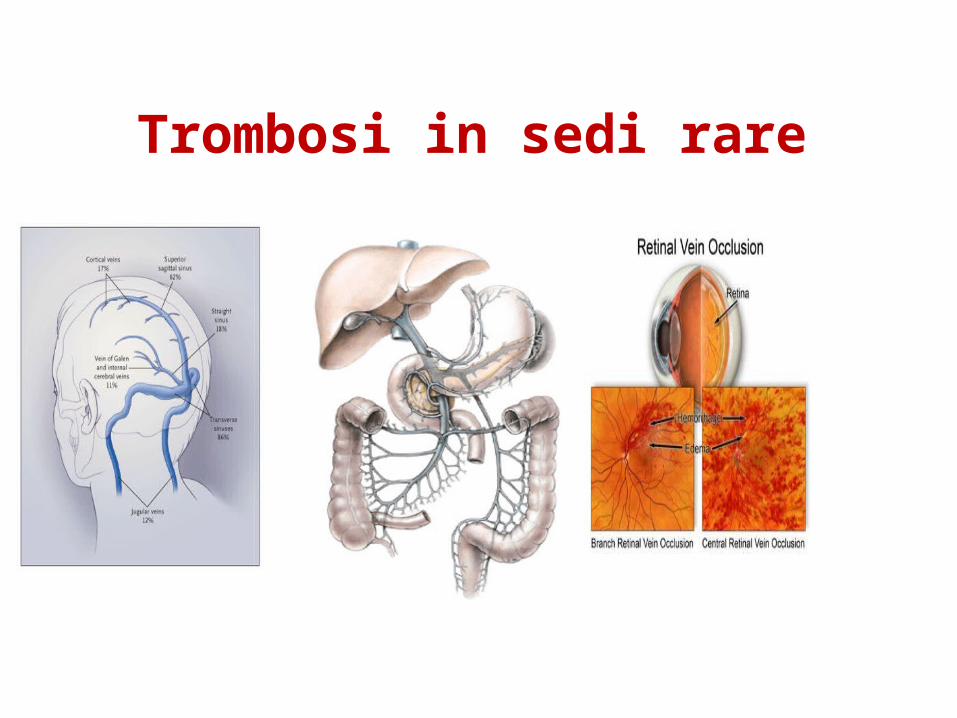
Trombosi in sedi rare

46

Trombosi nella circolazione placentare
• Formazione di microtrombi che portano a insufficiente apporto di sangue con ipoperfusione. Rischi di aborto, morte intrauterina, preeclapsia o ritadi di crescita del bambino

48
Recurrent Thrombosis in PV or ETRecurrent Thrombosis in PV or ET
0 24 48 72 96 120 144 1680
102030405060708090
100
overall recurrences
arterial recurrences
venous recurrences
A
months from the index thrombosis
cum
ulat
ive
prob
abili
ty o
fre
curr
ent t
hrom
bosi
s %
Time after first thrombosis (months)
5.6%pt-yr 3.4%pt-yr 2.2%pt-yrC
umul
ativ
e pr
obab
ility
(%
)
49.9%
De Stefano V et al, Haematologica 2007; 93:372-380

AA 2010-11 Lezioni di Ematologia prof A.M. Vannucchi
49
Multifactorial pathogenesis of Multifactorial pathogenesis of thrombosis in MPNthrombosis in MPN

La trombosi nella malattie mieloproliferative si previene anche con una buon controllo dei fattori di rischio cardiovascolari

51
Risk category Age >60
orhistory of thrombosis
Cardiovascular risk
factors (*)
Low NO NO
Intermediate NO YES
High YES----
Current Risk Stratification in Current Risk Stratification in PV and ETPV and ET
(*) Cardiovascular risk factors include diabetes, hypertension, smoking habitus

Possibili complicanze (II)• evoluzione naturale, nel lungo termine, a mielofibrosi (circa 5% a 15 anni dalla diagnosi di PV)
per confermare la diagnosi necessaria la biopsia osteomidollare
Evoluzione a leucemia acuta: stimata 5-8% a lungo termine (ma non sempre!)
- ridotta necessità di salassi o riduzione del dosaggio dei farmaci utilizzati- possibile anemizzazione- incremento del volume della milza (più raramente del fegato)- comparsa di sintomi costituzionali prima assenti- comparsa di globuli rossi dalla forma alterata, con alcuni elementi immaturi- possibile aumento/riduzione del numero di globuli bianchi con alcune forme
immature- possibile aumento/riduzione del numero di piastrine

Cosa sono ?
0
0.2
0.4
0.6
0.8
1
01 2 3 4 5 6 7 8 9 10 11 12 13 14 15
Sopravvivenza (anni)
Prob
abili
tà
ItaliaPV (studio GISP)
Durata di sopravvivenza dei pazienti con Policitemia Vera in confronto alla popolazione normale italiana di pari sesso ed età
Durata di sopravvivenza dei pazienti con Policitemia Vera in confronto alla popolazione normale italiana di pari sesso ed
età
PV (studio GISP)Popolazione generale
PV (studio GISP)
SOPRAVVIVENZA (ANNI)
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
Probabilita’
0.2
0.4
0.6
0.8
1

54
CAUSE DI MORTE PIÙ FREQUENTI NELLA PV e ET
• Trombotiche (arteriose-IMA; venose-cerebrali,emboliche, splancniche)
• Emorragiche (gastrointestinali, cerebrali)• Evoluzione in mielofibrosi• Evoluzione in leucosi mieloide acuta • Neoplasie secondarie (mammella, colon,
polmone)

Barbui T, JCO 2011; 29:761
Terapia di PV e ET
Scopo della terapiaEvitare il primo episodio o la ricorrenza di trombosis e di emorragiaMinimizzare il rischio di evoluzione in Leucemia Acuta o mielofibrosi
Controllare I sintomi sistemici
Trattare le complicazioni ( Trombosi e emorragie)
Ottimizzare la terapia di situazioni a rischio ( esempio: gravidanza, chirurgia)

56
La terapia è correlata alla classe di rischio
Risk category PV ET
Low • Phlebotomies to target Hct
• ASA
• nil
• ASA (no consensus)Intermediate
High
• Myelosuppression (HuOH; Interferon)• Phlebotomies to target Hct • ASA
• Myelosuppression (HuOH; Interferon)
• ASA
Target Hct: <42%, <45%; ASA means low-dose ASA