
Ć ą (2012/2013) Cz ęść A - System ZEUS UMCS -...
7
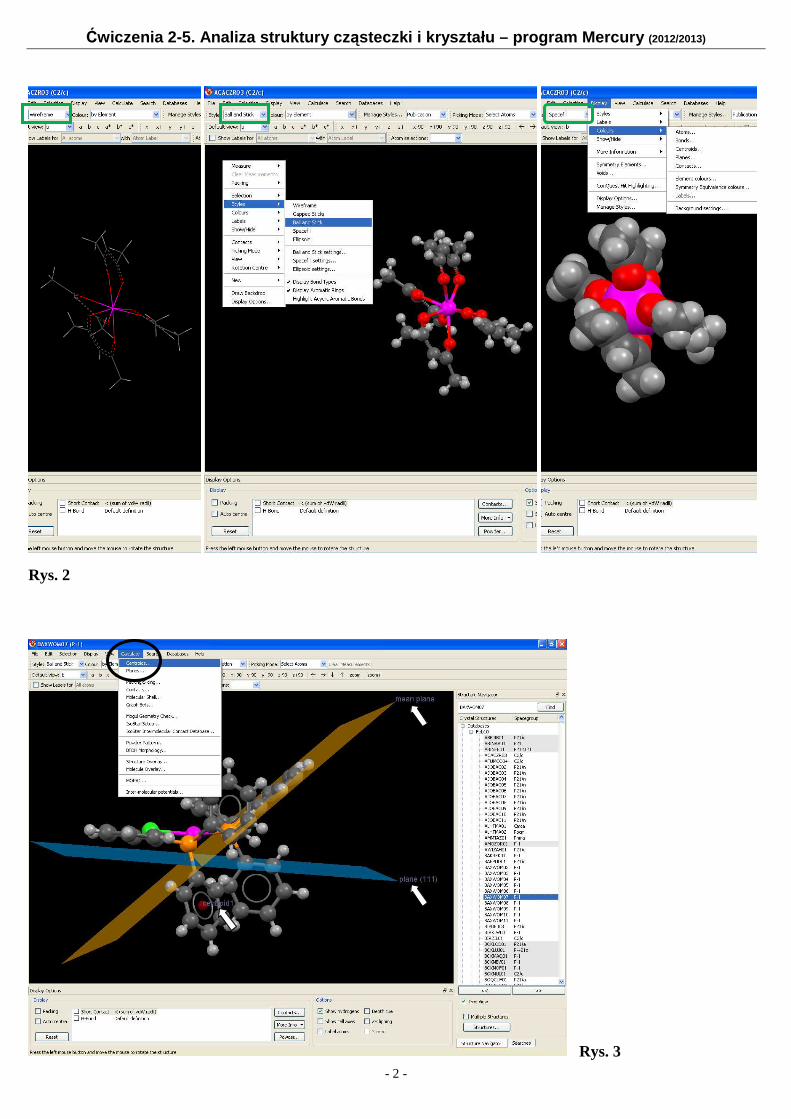
Ćwiczenia 2-5. Analiza struktury cząsteczki i krysztalu – program Mercury (2012/2013) - 1 - Część A – wprowadzenie do programu Mercury Mercury – program graficzny do wizualizacji i analizy geometrii cząsteczek i krysztalów. Program ten jest stale uzupelniany i modyfikowany, jednak jego podstawowe funkcje i zastosowanie pozostają. Zbiory wejściowe do tego programu o rozszerzeniu res, cif (mol lub pdb) zawierają wszystkie niezbędne informację o polożeniu atomów oraz geometrii sieci. Po wejściu do programu i odczytaniu danych strukturalnych z w/w zbiorów (File → → → Open) lub wybraniu struktury z krystalograficznej bazy danych (patrz Rys. 5 i strona 4) - CSD 1 - na ekranie otrzymamy rysunek cząsteczki, na którym atomy poszczególnych typów są zaznaczone kolorami im przypisanymi (Rys. 1). Rys. 1 Inne widoczne na Rys. 1 opcje pozwalają m. in. na: uzyskanie większej ilości informacji o strukturze (More Info), np. dane krystalograficzne (Structure Information), wzór strukturalny (Chemical Diagram), dlugości wiązań (Bond List), sprawdzenie nazw atomów – etykiet (Label atoms), sprawdzenie wartości dlugości wiązań (Bond List lub Picking Mode → → → Measure Distances ), kątów walencyjnych (Picking Mode → → → Measure Angles) i torsyjnych (Picking Mode → → → Measure Torsions), zaznaczanie atomów (Picking Mode → → → Select Atoms) ‘rozbudowywanie’ struktury (Picking Mode → → → Expand Contacts), itd... Na Rys. 2 przedstawiono wykorzystanie opcje menu do zmiany sposobu prezentacji graficznej cząsteczek: kreski (pokazane), cylindry, kulki-patyczki (pokazane), sfery van der Waalsa (pokazane) i elipsoidy drgań termicznych, (Display → → → Styles → → → Wireframe / Stick → → → Ball and stick / Spacefill / Ellipsoid) oraz zmiany kolorów atomów, tla, itp. (Display → → → Colours → → → Atoms/Bonds...). Innymi funkcjami programu są (Calculate → → → Centroids) i (Calculate → → → Planes), Rys. 3; pierwsza z nich umożliwia zdefiniowanie środka geometrycznego dla podzbioru wybranych atomów, druga natomiast pozwala na wyznaczenie plaszczyzny przechodzącej przez dane atomy (Mean plane), bądź plaszczyzny sieciowej o zadanych wskaźnikach Millera (hkl). Wygenerowaną plaszczyznę sieciową można edytować, np. zmieniać kolor, nazwę, ‘ukryć’, usunąć. 1 Cambridge Structural Database – krystalograficzna baza danych, w której każdej strukturze przypisany jest tzw. ‘refkod’
Transcript of Ć ą (2012/2013) Cz ęść A - System ZEUS UMCS -...

Ćwiczenia 2-5. Analiza struktury cz ąsteczki i kryształu – program Mercury (2012/2013)
- 1 -
Część A – wprowadzenie do programu Mercury Mercury – program graficzny do wizualizacji i analizy geometrii cząsteczek i kryształów. Program ten jest stale uzupełniany i modyfikowany, jednak jego podstawowe funkcje i zastosowanie pozostają. Zbiory wejściowe do tego programu o rozszerzeniu res, cif (mol lub pdb) zawierają wszystkie niezbędne informację o położeniu atomów oraz geometrii sieci. Po wejściu do programu i odczytaniu danych strukturalnych z w/w zbiorów (File →→→→ Open) lub wybraniu struktury z krystalograficznej bazy danych (patrz Rys. 5 i strona 4) - CSD1 - na ekranie otrzymamy rysunek cząsteczki, na którym atomy poszczególnych typów są zaznaczone kolorami im przypisanymi (Rys. 1). Rys. 1
Inne widoczne na Rys. 1 opcje pozwalają m. in. na: � uzyskanie większej ilości informacji o strukturze (More Info), np. dane krystalograficzne (Structure
Information), wzór strukturalny (Chemical Diagram), długości wiązań (Bond List), � sprawdzenie nazw atomów – etykiet (Label atoms), � sprawdzenie wartości długości wiązań (Bond List lub Picking Mode →→→→ Measure Distances ), kątów
walencyjnych (Picking Mode →→→→ Measure Angles) i torsyjnych (Picking Mode →→→→ Measure Torsions), zaznaczanie atomów (Picking Mode →→→→ Select Atoms) ‘rozbudowywanie’ struktury (Picking Mode →→→→ Expand Contacts), itd... Na Rys. 2 przedstawiono wykorzystanie opcje menu do zmiany sposobu prezentacji graficznej cząsteczek:
kreski (pokazane), cylindry, kulki-patyczki (pokazane), sfery van der Waalsa (pokazane) i elipsoidy drgań termicznych, (Display →→→→ Styles →→→→ Wireframe / Stick →→→→ Ball and stick / Spacefill / Ellipsoid) oraz zmiany kolorów atomów, tła, itp. (Display →→→→ Colours →→→→ Atoms/Bonds...). Innymi funkcjami programu są (Calculate →→→→ Centroids) i (Calculate →→→→ Planes), Rys. 3; pierwsza z nich umożliwia zdefiniowanie środka geometrycznego dla podzbioru wybranych atomów, druga natomiast pozwala na wyznaczenie płaszczyzny przechodzącej przez dane atomy (Mean plane), bądź płaszczyzny sieciowej o zadanych wskaźnikach Millera (hkl). Wygenerowaną płaszczyznę sieciową można edytować, np. zmieniać kolor, nazwę, ‘ukryć’, usunąć. 1 Cambridge Structural Database – krystalograficzna baza danych, w której każdej strukturze przypisany jest tzw. ‘refkod’
Ćwiczenia 2-5. Analiza struktury cz ąsteczki i kryształu – program Mercury (2012/2013)
- 2 -
Rys. 2
Rys. 3
Ćwiczenia 2-5. Analiza struktury cz ąsteczki i kryształu – program Mercury (2012/2013)
- 3 -
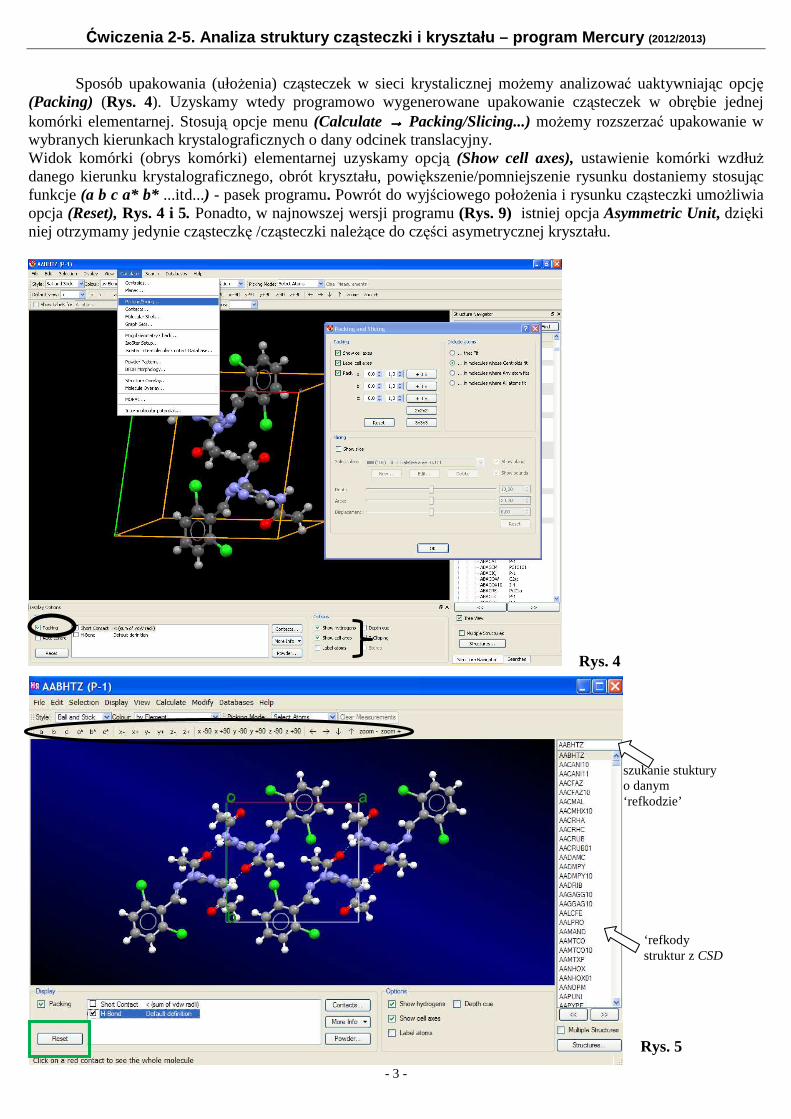
Sposób upakowania (ułożenia) cząsteczek w sieci krystalicznej możemy analizować uaktywniając opcję
(Packing) (Rys. 4). Uzyskamy wtedy programowo wygenerowane upakowanie cząsteczek w obrębie jednej komórki elementarnej. Stosują opcje menu (Calculate →→→→ Packing/Slicing...) możemy rozszerzać upakowanie w wybranych kierunkach krystalograficznych o dany odcinek translacyjny. Widok komórki (obrys komórki) elementarnej uzyskamy opcją (Show cell axes), ustawienie komórki wzdłuż danego kierunku krystalograficznego, obrót kryształu, powiększenie/pomniejszenie rysunku dostaniemy stosując funkcje (a b c a* b* ...itd...) - pasek programu. Powrót do wyjściowego położenia i rysunku cząsteczki umożliwia opcja (Reset), Rys. 4 i 5. Ponadto, w najnowszej wersji programu (Rys. 9) istniej opcja Asymmetric Unit, dzięki niej otrzymamy jedynie cząsteczkę /cząsteczki należące do części asymetrycznej kryształu.
Rys. 4
Rys. 5
‘refkody struktur z CSD
szukanie stuktury o danym ‘refkodzie’
Ćwiczenia 2-5. Analiza struktury cz ąsteczki i kryształu – program Mercury (2012/2013)
- 4 -
Aby przeanalizować np. stereochemię cząsteczki/strukturę kryształu z CSD (Databases →→→→ ...) o znanym
‘refkodzie’ (kod, pod którym cząsteczka zapisana jest w bazie), należy ją odnaleźć wśród wszystkich struktur poprzez wpisanie jej identyfikatora do odpowiedniego pola (Rys. 5); uzyskujemy wtedy wszystkie informacje o strukturze, które są zawarte w bazie.
Możemy także ‘wczytać’ do programu dane dowolnej struktury (nie zawartej w bazie) jeśli są one zapisane w postacie jednego ze zbiorów: res, cif (mol lub pdb), korzystamy wtedy z opcji menu (File →→→→ Open...).
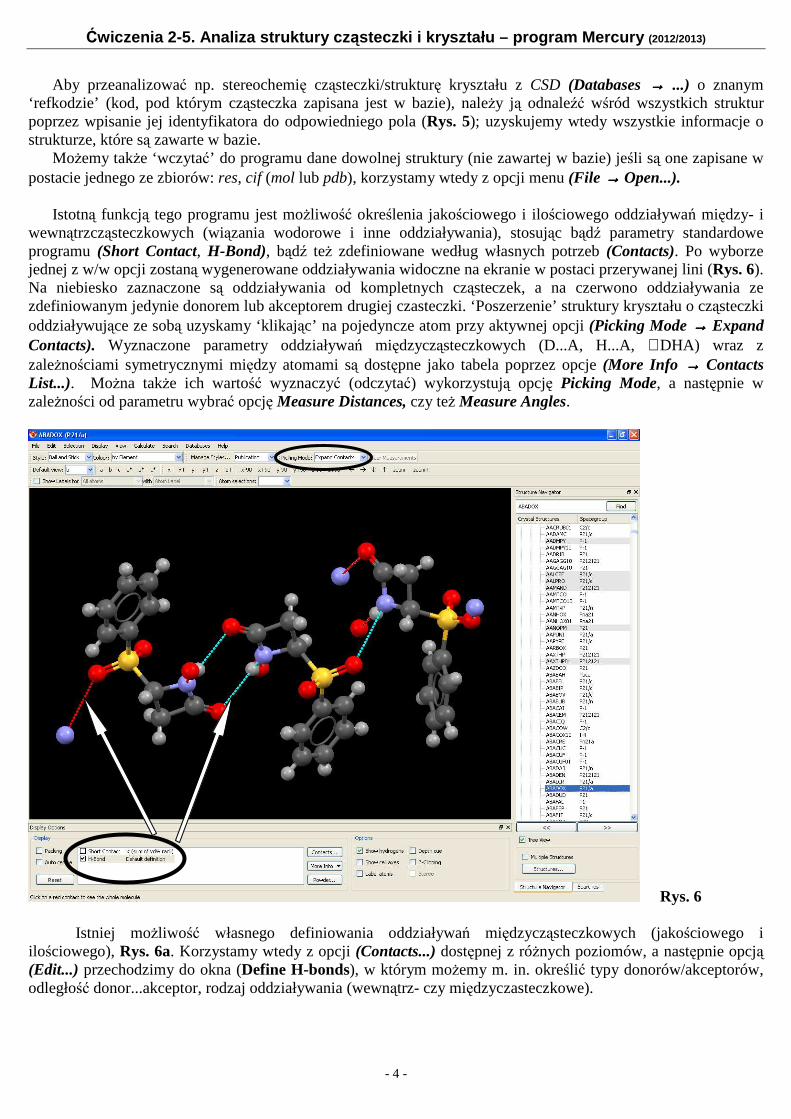
Istotną funkcją tego programu jest możliwość określenia jakościowego i ilościowego oddziaływań między- i
wewnątrzcząsteczkowych (wiązania wodorowe i inne oddziaływania), stosując bądź parametry standardowe programu (Short Contact, H-Bond), bądź też zdefiniowane według własnych potrzeb (Contacts). Po wyborze jednej z w/w opcji zostaną wygenerowane oddziaływania widoczne na ekranie w postaci przerywanej lini (Rys. 6). Na niebiesko zaznaczone są oddziaływania od kompletnych cząsteczek, a na czerwono oddziaływania ze zdefiniowanym jedynie donorem lub akceptorem drugiej czasteczki. ‘Poszerzenie’ struktury kryształu o cząsteczki oddziaływujące ze sobą uzyskamy ‘klikając’ na pojedyncze atom przy aktywnej opcji (Picking Mode →→→→ Expand Contacts). Wyznaczone parametry oddziaływań międzycząsteczkowych (D...A, H...A, ∠DHA) wraz z zależnościami symetrycznymi między atomami są dostępne jako tabela poprzez opcje (More Info →→→→ Contacts List...). Można także ich wartość wyznaczyć (odczytać) wykorzystują opcję Picking Mode, a następnie w zależności od parametru wybrać opcję Measure Distances, czy też Measure Angles.
Rys. 6
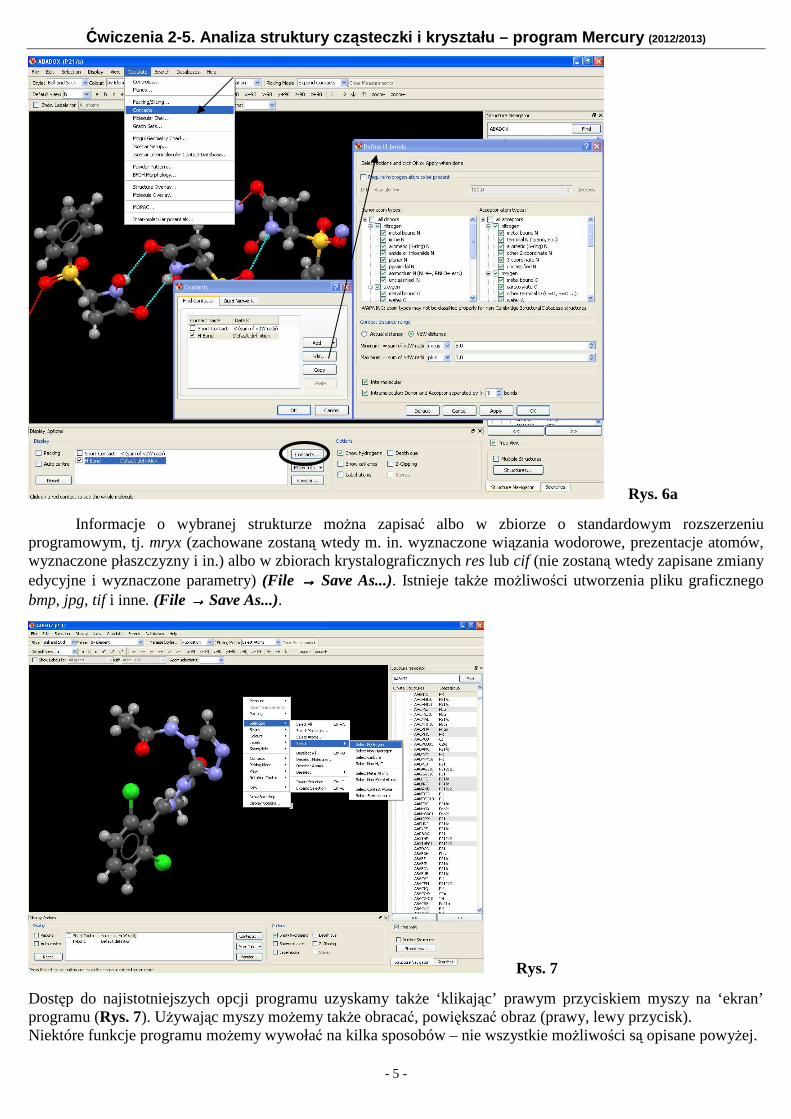
Istniej możliwość własnego definiowania oddziaływań międzycząsteczkowych (jakościowego i ilościowego), Rys. 6a. Korzystamy wtedy z opcji (Contacts...) dostępnej z różnych poziomów, a następnie opcją (Edit...) przechodzimy do okna (Define H-bonds), w którym możemy m. in. określić typy donorów/akceptorów, odległość donor...akceptor, rodzaj oddziaływania (wewnątrz- czy międzyczasteczkowe).
Ćwiczenia 2-5. Analiza struktury cz ąsteczki i kryształu – program Mercury (2012/2013)
- 5 -
Rys. 6a
Informacje o wybranej strukturze można zapisać albo w zbiorze o standardowym rozszerzeniu programowym, tj. mryx (zachowane zostaną wtedy m. in. wyznaczone wiązania wodorowe, prezentacje atomów, wyznaczone płaszczyzny i in.) albo w zbiorach krystalograficznych res lub cif (nie zostaną wtedy zapisane zmiany edycyjne i wyznaczone parametry) (File →→→→ Save As...). Istnieje także możliwości utworzenia pliku graficznego bmp, jpg, tif i inne. (File →→→→ Save As...).
Rys. 7
Dostęp do najistotniejszych opcji programu uzyskamy także ‘klikając’ prawym przyciskiem myszy na ‘ekran’ programu (Rys. 7). Używając myszy możemy także obracać, powiększać obraz (prawy, lewy przycisk). Niektóre funkcje programu możemy wywołać na kilka sposobów – nie wszystkie możliwości są opisane powyżej.
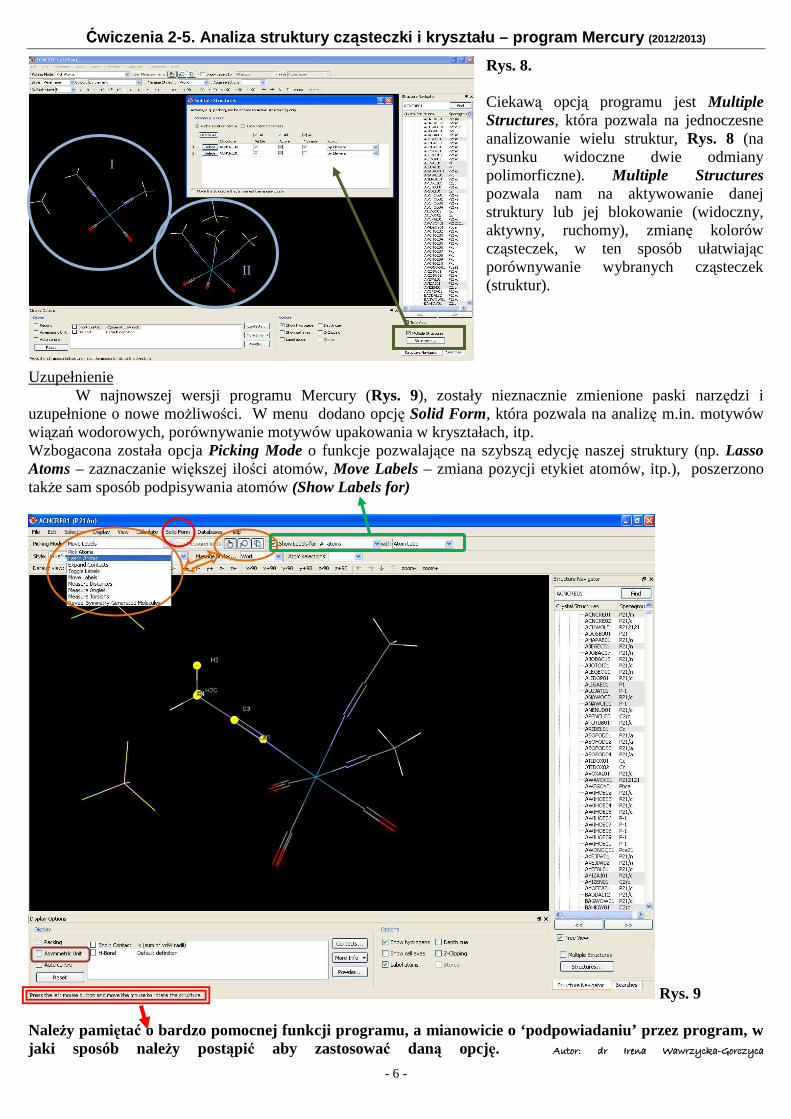
Ćwiczenia 2-5. Analiza struktury cz ąsteczki i kryształu – program Mercury (2012/2013)
- 6 -
Rys. 8. Ciekawą opcją programu jest Multiple Structures, która pozwala na jednoczesne analizowanie wielu struktur, Rys. 8 (na rysunku widoczne dwie odmiany polimorficzne). Multiple Structures pozwala nam na aktywowanie danej struktury lub jej blokowanie (widoczny, aktywny, ruchomy), zmianę kolorów cząsteczek, w ten sposób ułatwiając porównywanie wybranych cząsteczek (struktur).
Uzupełnienie W najnowszej wersji programu Mercury (Rys. 9), zostały nieznacznie zmienione paski narzędzi i
uzupełnione o nowe możliwości. W menu dodano opcję Solid Form, która pozwala na analizę m.in. motywów wiązań wodorowych, porównywanie motywów upakowania w kryształach, itp. Wzbogacona została opcja Picking Mode o funkcje pozwalające na szybszą edycję naszej struktury (np. Lasso Atoms – zaznaczanie większej ilości atomów, Move Labels – zmiana pozycji etykiet atomów, itp.), poszerzono także sam sposób podpisywania atomów (Show Labels for)
Rys. 9 Należy pamiętać o bardzo pomocnej funkcji programu, a mianowicie o ‘podpowiadaniu’ przez program, w jaki sposób należy postąpić aby zastosować daną opcję. AutorAutorAutorAutor: dr Irena Wawrzycka: dr Irena Wawrzycka: dr Irena Wawrzycka: dr Irena Wawrzycka----GorczycGorczycGorczycGorczycaaaa
I
II

Ćwiczenia 2-5. Analiza struktury cz ąsteczki i kryształu – program Mercury (2012/2013)
- 7 -
Zagadnienia teoretyczne obowiązujące do ćwiczeń Ćwiczenie 2
• Parametry strukturalne: długość wiązania, kąt walencyjny, kąt torsyjny. • Liczby i wielościany koordynacyjne. • Konformacja (układy cykliczne i acykliczne)
Ćwiczenie 3
• Struktura zbioru *.res. • Płaszczyzny i proste sieciowe (sposób wyznaczania). • Konformacja (układy cykliczne i acykliczne).
Ćwiczenie 4
• Struktura zbioru *.cif. • Polimorfizm. • Promienie van der Waalsa. • Część symetrycznie niezależna: Z i Z’.
Ćwiczenie 5
• Wiązanie wodorowe. • Typy asocjacji cząsteczek poprzez wiązania wodorowe. • Hydraty, solwaty (układy gość...gospodarz). • Komplementarność wiązań wodorowych.


















