
Uniwersytet Przyrodniczy we Wrocławiu Wydział Medycyny ...
122
Uniwersytet Przyrodniczy we Wroclawiu Wydzial Medycyny Weterynaryjnej mgr inż. Patrycja Libako PRACA DOKTORSKA Ocena wplywu pozakomórkowego magnezu oraz interakcji magnez – wapń na odpowiedź immunologiczną typu komórkowego w warunkach in vitro Promotorzy: Prof. dr Andrzej Mazur, Directeur de Recherche Unité de Nutrition Humaine, Institut National de la Recherche Agronomique (INRA), Clermont-Ferrand/Theix et Université d'Auvergne, Clermont-Ferrand, Francja Prof. dr hab. Wojciech Nowacki, prof. zw. Katedra Immunologii, Patofizjologii i Prewencji Weterynaryjnej Wydzialu Medycyny Weterynaryjnej Uniwersytetu Przyrodniczego we Wroclawiu Wroclaw 2013
Transcript of Uniwersytet Przyrodniczy we Wrocławiu Wydział Medycyny ...

Uniwersytet Przyrodniczy we Wrocławiu
Wydział Medycyny Weterynaryjnej
mgr inż. Patrycja Libako
PRACA DOKTORSKA
Ocena wpływu pozakomórkowego magnezu oraz interakcji magnez – wapń na
odpowiedź immunologiczną typu komórkowego w warunkach in vitro
Promotorzy:
Prof. dr Andrzej Mazur, Directeur de Recherche
Unité de Nutrition Humaine, Institut National de la Recherche Agronomique (INRA), Clermont-Ferrand/Theix et Université d'Auvergne, Clermont-Ferrand, Francja
Prof. dr hab. Wojciech Nowacki, prof. zw.
Katedra Immunologii, Patofizjologii i Prewencji Weterynaryjnej Wydziału Medycyny Weterynaryjnej Uniwersytetu Przyrodniczego we Wrocławiu
Wrocław 2013

Dziękuję,
Promotorom:
prof. Andrzejowi Mazurowi
i prof. Wojciechowi Nowackiemu
za motywację opiekę naukową i pomoc w przygotowaniu pracy

Praca naukowa została wykonana w ramach projektu badawczego nr N N308 075234
finansowanego z funduszy MNiSW:
"Wpływ stężenia i stosunku Mg2+/Ca2+ na komórkową odpowiedź
immunologiczną in vitro".

4
SPIS TREŚCI
WYKAZ STOSOWANYCH SKRÓTÓW................................................................................. 8
STRESZCZENIE .................................................................................................................... .11
ABSTRACT ............................................................................................................................. 12
I WSTĘP................................................................................................................................... 13
1. Podstawowe informacje o magnezie w biologii komórki .................................................... 13
1.1 Metabolizm magnezu.............................................................................................. 13
1.2 Homeostaza magnezu w organizmie ...................................................................... 14
1.3 Homeostaza magnezu w komórce .......................................................................... 15
2. Niedobór magnezu................................................................................................................ 18
2.1 Niedobór magnezu jako czynnik chorób cywilizacyjnych ..................................... 19
2.1.1 Magnez i zespół metaboliczny................................................................. 19
2.1.2 Magnez i cukrzyca ................................................................................... 20
2.1.3 Magnez i choroby układu krążenia .......................................................... 21
3. Zależności pomiędzy wapniem i magnezem........................................................................ 21
4. Wapń jako sygnalizator komórkowy.................................................................................... 22
4.1 Mechanizm działania substancji blokujących kanały wapniowe ........................... 23
4.2 Blokery wapnia a układ odpornościowy................................................................. 24
5. Magnez i układ odpornościowy............................................................................................ 26
5.1 Niedobór magnezu jako czynnik stanu zapalnego u ludzi...................................... 26
5.2 Eksperymentalny niedobór magnezu...................................................................... 28
5.2.1 Zjawisko preaktywacji (priming) komórek układu odpornościowego .... 29
5.2.1.1 Niedobór magnezu jako pośredni czynnik preaktywujący ....... 30
5.2.1.2 Substancja P jako czynnik preaktywujący ................................ 31
II ZAŁOŻENIA I CEL PRACY............................................................................................... 32
III MATERIAŁY I METODY ................................................................................................. 33
PLAN DOŚWIADCZEŃ ......................................................................................................... 33
1. Wpływ blokerów wapnia na potencjał proliferacyjny ustalonych linii komórkowych:
J774.E oraz D10.G4.1 - wyznaczenie IC50 dla badanych związków........................................ 34
2. Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na aktywność
modelowych komórek fagocytarnych in vitro.......................................................................... 35
2.1 Ocena żywotności populacji granulocytów ............................................................ 37
2.2 Ocena czystości populacji granulocytów................................................................ 37

5
2.3 Ocena aktywności komórkowej i wzrostu komórek............................................... 37
2.4 Badanie endocytozy antygenu modelowego .......................................................... 38
2.5 Badanie produkcji reaktywnych form tlenu przez komórki fagocytarne ............... 39
2.6 Oznaczenie cytokin prozapalnych w nadsączach komórkowych testem ELISA ... 39
2.7 Oznaczenie ekspresji cytokin prozapalnych na poziomie transkryptu ................... 40
3. Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na wzrost
komórek limfocytarnych oraz ich aktywację przez komórki prezentujące antygen: komórki
dendrytyczne oraz makrofagi ................................................................................................... 42
3.1 Ocena aktywności komórek D10.G4.1 w monokulturze........................................ 42
3.2 Wyprowadzenie pierwotnych linii komórek dendrytycznych oraz makrofagów z
komórek szpiku kostnego myszy szczepu C3H/ HeN(H-2k) ....................................... 43
3.2.1 Określenie fenotypu makrofagów i komórek dendrytycznych
wyprowadzonych z komórek szpiku kostnego myszy C3H/HeN (H-2k)........ 44
3.2.2 Badanie endocytozy antygenu modelowego przez modelowe komórki
prezentujące antygen......................................................................................... 44
3.3 Wpływu pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na
prezentację antygenu – kokultury komórkowe............................................................. 45
3.3.1 Oznaczenie aktywności metabolicznej komórek w kokulturze ............... 46
3.3.2 Oznaczenie końcowej liczby komórek w kokulturze .............................. 46
3.3.3 Oznaczenie cytokin w nadsączach komórkowych testem ELISA........... 46
3.3.4 Wewnątrzkomórkowe znakowanie cytokin ............................................. 47
4. Zwierzęta doświadczalne...................................................................................................... 47
5. Analiza statystyczna wyników ............................................................................................. 48
6. Wykaz zastosowanych odczynników ................................................................................... 48
6.1 Wykaz przygotowanych roztworów i mediów ....................................................... 51
IV WYNIKI.............................................................................................................................. 54
1. Wpływ blokerów wapnia na potencjał proliferacyjny komórek ustalonych linii
komórkowych: J774.E oraz D10.G4.1 -wyznaczenie IC50 badanych związków .................... 54
2. Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na aktywność
modelowych komórek fagocytarnych in vitro.......................................................................... 54
2.1 Ocena wpływu magnezu i wapnia na potencjał proliferacyjny komórek linii J774.E
...................................................................................................................................... 54

6
2.2 Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na
produkcję reaktywnych form tlenu (RFT) przez modelowe komórki fagocytarne in
vitro............................................................................................................................... 56
2.3 Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na
endocytozę antygenu modelowego przez komórki makrofagowe J774.E.................... 57
2.4 Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na
wywołaną LPS produkcję cytokin prozapalnych (IL-1β, IL-6 oraz TNF-α) przez
komórki makrofagowe J774.E...................................................................................... 57
2.5 Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na
wywołaną LPS transkrypcję genów IL-1β, IL-6 oraz TNF-α przez komórki
makrofagowe J774.E .................................................................................................... 59
3. Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na wzrost
komórek limfocytarnych oraz ich aktywację przez komórki prezentujące antygen: komórki
dendrytyczne oraz makrofagi ................................................................................................... 60
3.1 Ocena wpływu magnezu i wapnia na wzrost komórek limfocytarnych D10.G4.1 w
monokulturze ................................................................................................................ 60
3.2 Określenie fenotypu komórek dendrytycznych i makrofagów wyprowadzonych z
komórek szpiku kostnego myszy C3H/HeN (H-2k)..................................................... 61
3.3 Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na
endocytozę antygenu modelowego przez komórki dendrytyczne i makrofagi
wyprowadzone z komórek szpiku kostnego myszy C3H/HeN (H-2k) ........................ 62
3.4 Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na
aktywność komórkową oraz liczbę komórek w kokulturze komórkowej .................... 63
3.5 Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na
produkcję cytokin IL-2, IL-4, IL-10 oraz interferonu-γ w modelowych kokulturach
komórkowych ............................................................................................................... 65
3.5.1 Wpływ zaburzonej równowagi magnezowo-wapniowej na produkcję i
sekrecję cytokin IL-2, IL-4, IL-10 oraz interferonu-γ w kokulturze
komórkowej: komórki D10.G4.1 : modelowe komórki dendrytyczne (BMDCs)
........................................................................................................................... 65
3.5.2 Wpływ zaburzonej równowagi magnezowo-wapniowej na produkcję i
sekrecję cytokin IL-2, IL-4, IL-10 oraz interferonu-γ w kokulturze
komórkowej: komórki D10.G4.1 : modelowe makrofagi (BMMFs) ............... 66

7
3.5.3 Wpływ zaburzonej równowagi magnezowo-wapniowej na produkcję
cytokin IL-4 oraz IL-10 w kokulturze komórkowej: komórki D10.G4.1:
modelowe komórki dendrytyczne (BMDCs).................................................... 67
V DYSKUSJA.......................................................................................................................... 68
VI WNIOSKI............................................................................................................................ 84
VII PIŚMIENNICTWO............................................................................................................ 85
8
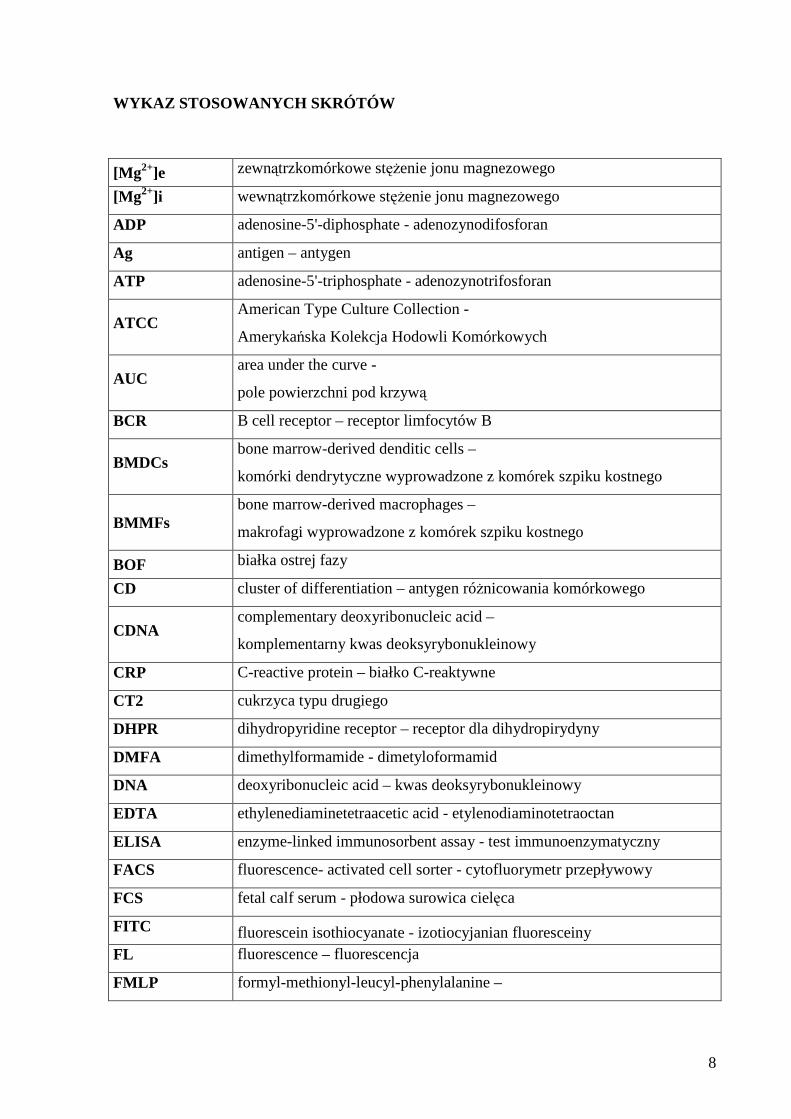
WYKAZ STOSOWANYCH SKRÓTÓW
[Mg2+]e zewnątrzkomórkowe stężenie jonu magnezowego
[Mg2+]i wewnątrzkomórkowe stężenie jonu magnezowego
ADP adenosine-5'-diphosphate - adenozynodifosforan
Ag antigen – antygen
ATP adenosine-5'-triphosphate - adenozynotrifosforan
ATCC American Type Culture Collection -
Amerykańska Kolekcja Hodowli Komórkowych
AUC area under the curve -
pole powierzchni pod krzywą
BCR B cell receptor – receptor limfocytów B
BMDCs bone marrow-derived denditic cells –
komórki dendrytyczne wyprowadzone z komórek szpiku kostnego
BMMFs bone marrow-derived macrophages –
makrofagi wyprowadzone z komórek szpiku kostnego
BOF białka ostrej fazy
CD cluster of differentiation – antygen różnicowania komórkowego
CDNA complementary deoxyribonucleic acid –
komplementarny kwas deoksyrybonukleinowy
CRP C-reactive protein – białko C-reaktywne
CT2 cukrzyca typu drugiego
DHPR dihydropyridine receptor – receptor dla dihydropirydyny
DMFA dimethylformamide - dimetyloformamid
DNA deoxyribonucleic acid – kwas deoksyrybonukleinowy
EDTA ethylenediaminetetraacetic acid - etylenodiaminotetraoctan
ELISA enzyme-linked immunosorbent assay - test immunoenzymatyczny
FACS fluorescence- activated cell sorter - cytofluorymetr przepływowy
FCS fetal calf serum - płodowa surowica cielęca
FITC fluorescein isothiocyanate - izotiocyjanian fluoresceiny FL fluorescence – fluorescencja
FMLP formyl-methionyl-leucyl-phenylalanine –
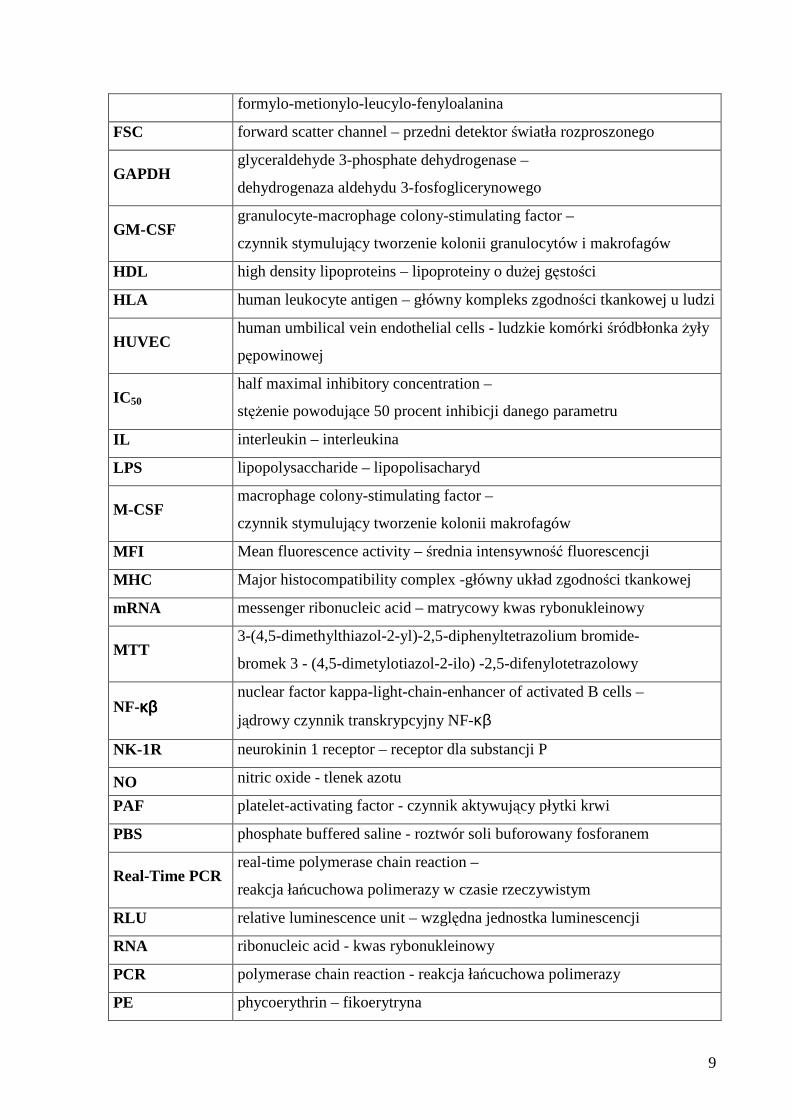
9
formylo-metionylo-leucylo-fenyloalanina
FSC forward scatter channel – przedni detektor światła rozproszonego
GAPDH glyceraldehyde 3-phosphate dehydrogenase –
dehydrogenaza aldehydu 3-fosfoglicerynowego
GM-CSF granulocyte-macrophage colony-stimulating factor –
czynnik stymulujący tworzenie kolonii granulocytów i makrofagów
HDL high density lipoproteins – lipoproteiny o dużej gęstości
HLA human leukocyte antigen – główny kompleks zgodności tkankowej u ludzi
HUVEC human umbilical vein endothelial cells - ludzkie komórki śródbłonka żyły
pępowinowej
IC50 half maximal inhibitory concentration –
stężenie powodujące 50 procent inhibicji danego parametru
IL interleukin – interleukina
LPS lipopolysaccharide – lipopolisacharyd
M-CSF macrophage colony-stimulating factor –
czynnik stymulujący tworzenie kolonii makrofagów
MFI Mean fluorescence activity – średnia intensywność fluorescencji
MHC Major histocompatibility complex -główny układ zgodności tkankowej
mRNA messenger ribonucleic acid – matrycowy kwas rybonukleinowy
MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide-
bromek 3 - (4,5-dimetylotiazol-2-ilo) -2,5-difenylotetrazolowy
NF-κκκκββββ nuclear factor kappa-light-chain-enhancer of activated B cells –
jądrowy czynnik transkrypcyjny NF-κβ
NK-1R neurokinin 1 receptor – receptor dla substancji P
NO nitric oxide - tlenek azotu
PAF platelet-activating factor - czynnik aktywujący płytki krwi
PBS phosphate buffered saline - roztwór soli buforowany fosforanem
Real-Time PCR real-time polymerase chain reaction –
reakcja łańcuchowa polimerazy w czasie rzeczywistym
RLU relative luminescence unit – względna jednostka luminescencji
RNA ribonucleic acid - kwas rybonukleinowy
PCR polymerase chain reaction - reakcja łańcuchowa polimerazy
PE phycoerythrin – fikoerytryna

10
PMA phorbol myristate acetate - octan mirystynianu forbolu
RDA recommended daily allowance –
rekomendowane dzienne zapotrzebowanie
RFT reaktywne formy tlenu
SDS sodium dodecyl sulfate - dodecylosiarczan sodu
SPF specific pathogen free – wolny od patogenów
SSC side scatter channel – boczny detektor światła rozproszonego
TCR T cell receptor – receptor limfocytów T
TG triglycerides - trójglicerydy
TLR toll-like receptor - receptor toll-like
TMB 3,3′,5,5′-Tetramethylbenzidine – 3,3’,5,5’- tetrametylobenzydyna
TMB-8 8-(Diethylamino)octyl 3,4,5-trimethoxybenzoate hydrochloride -
chlorowodorek 8-(dietyloamino)oktylo 3,4,5 trimetoksy benzoesanu
TNF Tumor necrosis factor - czynnik martwicy nowotworów
TRIS tris(hydroxymethyl)aminomethane - tri(hydroksymetylo)aminometan
WER Werapamil
ZM Zespół metaboliczny

11
STRESZCZENIE
Z uwagi na udział magnezu (Mg) w licznych procesach komórkowych, nawet
nieznaczne zaburzenia w homeostazie tego pierwiastka mogą stanowić przyczynę
wielokierunkowych zmian patofizjologicznych, m.in. zaburzonej aktywności układu
immunologicznego oraz - jako jej bezpośredniej konsekwencji - stanu zapalnego.
Niepodważalnych argumentów potwierdzających występowanie korelacji pomiędzy
niedoborem magnezu a wzmożoną reakcją zapalną dostarczyły rezultaty badań
doświadczalnych prowadzonych na zwierzętach, jak również liczne obserwacje kliniczne i
epidemiologiczne.
Dzisiejszy stan wiedzy nie pozwala jednoznacznie wyjaśnić, jaki jest bezpośredni wpływ
magnezu na powstawanie i rozwój stanu zapalnego jak również na samą aktywację komórek
układu odpornościowego. Wpływ magnezu polega w większości przypadków na
antagonizowaniu efektów wapnia (Ca), przy czym wapń pełni rolę wtórnego przekaźnika
sygnału łączącego szlaki aktywacyjne w leukocytach.
Celem pracy doktorskiej było zbadanie skojarzonego wpływu magnezu i wapnia oraz
ich wzajemnego stosunku na przebieg nieswoistej i swoistej odpowiedzi komórkowej in vitro.
Stwierdzono, że stężenie magnezu pozakomórkowego miało zróżnicowany wpływ na
aktywność modelowych komórek makrofagowych in vitro. Oceniono, że niskie stężenie
magnezu pozakomórkowego istotnie obniżyło potencjał proliferacyjny komórek J774.E. a
jednocześnie podwyższyło wywoływaną PMA produkcję reaktywnych form tlenu przez
komórki makrofagowe i neutrofile. Oceniono, że poziom magnezu pozakomórkowego nie ma
wpływu na proces endocytozy. Natomiast wysokie stężenie magnezu pozakomórkowego
zwiększało hamujący wpływ blokera wapnia na ekspresję IL-1β oraz IL-6 przez komórki
J774.E, przy czym nie miało istotnego wpływu na transkrypcję genów tych cytokin. Wysokie
stężenie magnezu pozakomórkowego, w obecności blokerów wapnia lub bez, istotnie
obniżyło produkcję IL-4 oraz IL-10 w kokulturach komórkowych (komórki prezentujące
antygen: limfocyty T) in vitro. Inhibitory kanałów wapniowych istotnie ograniczyły
antygenowo-zależną proliferację limfocytów T w kokulturze komórkowej, co dodatkowo
podkreśla kluczową rolę wapnia w prezentacji antygenu, przy czym efekt ten był niezależny
od stężenia magnezu pozakomórkowego

12
ABSTRACT
Magnesium (Mg) is involved in so many cellular pathways that even slight
disturbances in Mg homeostasis might lead to multiple pathophysiological consequences,
including an altered immune and inflammatory response. The strongest evidence of a close
connection between Mg and the inflammatory response derives from experimental animal
studies showing that Mg deficiency leads to the exacerbated inflammatory response. This
relationship between Mg status and inflammation is supported by clinical and epidemiological
observations.
The direct way the Mg affects the inflammation and immune cell’ activation remains
unclear. As Mg plays as the natural calcium (Ca) antagonist, the low extracellular Mg
concentration leads to the elevation of intracellular Ca. Meanwhile, Ca is an important second
messenger in signaling processes in leukocytes, as well as in maintaining normal functions of
these cells.
Thus, the purpose of this thesis work was to determine the consequence of Mg/Ca
balance or imbalance for the proper course of specific and non-specific immune system
defenses. This in the aim to contribute to better understating the role of Mg status in the
occurrence and in prevention of a broad spectrum of diseases with inflammatory component.
By using various Mg concentrations and Ca blockers in the present work we have
demonstrated that the extracellular Mg concentration had varied effects on the activity of
model phagocytes in vitro. Low extracellular Mg concentration significantly decreased the
proliferative potential of J774.E cells and at the same time increased the PMA-induced
reactive oxygen species by macrophage-like cells and neutrophils. The extracellular Mg level
did not affect the process of endocytosis. However, a high concentration of extracellular Mg
increased the inhibitory effect of Ca blocker on the expression of IL-1β and IL-6 by cells
J774.E, but had no significant effect on the transcription of these cytokines’ genes. High
extracellular concentration of Mg, in the presence or without Ca blockers, significantly
reduced production of IL-4 and IL-10 in co-cultures (antigen presenting cells and T cells) in
vitro. Calcium channel inhibitors significantly reduced the antigen-dependent T cell
proliferation in a co-cluture, which strongly underline the critical role of Ca in antigen
presentation, whereby the effect was independent of extracellular Mg concentration.

13
I WSTĘP
1. Podstawowe informacje o magnezie w biologii komórki
Magnez (Mg) jest pierwiastkiem niezbędnym dla wielu procesów biochemicznych
zachodzących w organizmie żywym. Magnez jest czwartym w kolejności kationem pod
względem całkowitej ilości w organizmie ssaków (Ca2+ > K+ > Na+ > Mg2+) a
wewnątrzkomórkowo drugim po potasie. Całkowite stężenie magnezu w większości komórek
ssaków waha się w zakresie 14-20 mM, przy czym stężenie jonu magnezowego (Mg2+) w
cytozolu wynosi około 0,5-0,7 mM i niezależnie od stanu fizjologicznego komórki pozostaje
na podobnym poziomie (Iseri and French, 1984; Saris et al., 2000). Pozakomórkowe (w
osoczu) stężenie magnezu u osób zdrowych oscyluje wokół 0,65-1,05 mM (Saris et al.,
2000).
W komórce magnez przede wszystkim pełni funkcję przeciwjonu dla związków
wysokoenergetycznych (głównie ATP) oraz kwasów nukleinowych (Ryc. 1). Magnez jest
kofaktorem ponad 300 reakcji enzymatycznych i wywiera wpływ na wydajność tych reakcji
poprzez: bezpośrednie wiązanie się z ATP (w reakcjach zależnych od ATP), wiązanie się z
miejscem aktywnym enzymu (np. enolazy, kinazy pirogronianowej), powodowanie zmian
konformacyjnych w trakcie katalizy (np. pompa sodowo-potasowa) oraz udział w tworzeniu
złożonych kompleksów enzymatycznych (np. dehydrogenza aldehydowa)(Altura and Altura,
1991; Quamme and Rabkin, 1990). Bardzo istotną właściwością magnezu jest konkurowanie
z wapniem o miejsce wiązania do białek lub antagonizowanie procesów wapniowych (Iseri
and French, 1984).
1.1 Metabolizm magnezu
U większości zwierząt zawartość magnezu szacuje się na ok. 0,4 g/kg masy ciała
(Maguire and Cowan, 2002), co oznacza, że w ciele zdrowego człowieka o masie ok. 70 kg
znajduje się ok. 28 g magnezu. Około 60% całkowitej puli magnezu znajduje się w kościach.
Około 40% zgromadzona jest w tkance mięśniowej oraz w innych tkankach miękkich. Mniej
niż 1% ogólnej puli magnezu to magnez pozakomórkowy – zlokalizowany w erytrocytach i
osoczu (stężenie u dorosłego człowieka: 0,65-1,05 mM (Tietz and Tietz 1990)). Magnez
pozakomórkowy występuje w trzech formach: wolnego jonu magnezowego, związanej z
białkami lub skompleksowanej z anionami (np. fosforanowym, węglanowym, siarczanowym).
Największą aktywnością biologiczną cechuje się magnez zjonizowany, którego stężenie u

14
dorosłego człowieka wynosi: 0,5-0,7 mM co stanowi ok. 70% puli magnezu znajdującego się
w osoczu (Altura and Altura, 1991).
Wewnątrzkomórkowe stężenie magnezu jest raczej stałe, przy czym dystrybucja magnezu w
komórce jest niejednorodna, a najniższe stężenie magnezu występuje w peryferyjnych
regionach komórki. Wolny magnez zjonizowany stanowi ok. 0,5-5% całkowitej puli magnezu
komórkowego; pozostała część związana jest z wysokoenergetycznymi ATP i ADP, białkami
oraz kwasami nukleinowymi lub jest magazynowana w mitochondriach. Dane literaturowe
podają, że stężenie magnezu wewnątrz komórki wynosi około 0,5 mM przy czym wartość ta
zależy od typu komórki i jest cechą gatunkową (Quamme and Rabkin, 1990). Podwyższone
stężenie magnezu wewnątrzkomórkowego odnotowuje się również w komórkach szybko
proliferujących, co może sugerować związek pomiędzy zwiększonym transportem magnezu a
wysoką aktywnością metaboliczną (Rude, 1993).
1.2 Homeostaza magnezu w organizmie
Wiele badań wskazuje, że wchłanianie magnezu w jelicie pozostaje w równowadze z
jego wydalaniem wraz z moczem (Kesteloot and Joossens, 1990; Spencer et al., 1980). W
okresie krótkotrwałego niedoboru magnezu (wynikającego np. z niskiej zawartości magnezu
w diecie) udostępniany jest magnez zgromadzony w kościach w celu utrzymania normalnego
stężenia w surowicy (Alfrey et al., 1974). Homeostaza magnezu w organizmie zależy zatem
glównie od trzech organów: jelita, gdzie następuje wchłanianie Mg2+; kości, które stanowią
główny rezerwuar magnezu oraz nerek, które są odpowiedzialne za jego wydalanie (Jeroen H.
F. de Baaij et al., 2012).
U zdrowych ludzi normalne stężenie magnezu w surowicy waha się w zakresie 0,65-1,05 mM
(Tietz and Tietz 1990). By utrzymać normalne stężenie Mg2+ w surowicy to według National
Research Council (USA) dorosła kobieta powinna spożywać dziennie ok. 280 mg a
mężczyzna ok. 350 mg magnezu. Normalnie ok. 30-50% magnezu przyjmowanego z
pożywieniem jest wchłaniana w jelitach, przy czym gdy zawartość magnezu w diecie jest
niska, jego absorpcja w jelicie rośnie - nawet do 80% (Graham et al., 1960). Wchłanianie
magnezu ma miejsce głównie w jelicie cienkim i okrężnicy (Kayne and Lee, 1993).
Zidentyfikowano dwie drogi wchłaniania magnezu w jelicie cienkim ssaków (Quamme,
2008). Międzykomórkowy transport wymaga wchłaniana magnezu przez małe przestrzenie
pomiędzy komórkami nabłonka i jest to mechanizm bierny sterowany głównie gradientem
elektrochemicznym. Około 80-90% magnezu w jelicie jest wchłaniana biernie. Mechanizm

15
aktywny zakłada transport śródkomórkowy magnezu do krwi przez komórki nabłonkowe;
mechanizm ten podlega ścisłej regulacji (Jeroen H. F. de Baaij et al., 2012; Quamme, 2008).
1.3 Homeostaza magnezu w komórce
W większości tkanek i komórek ssaków całkowite stężenie magnezu wynosi od około
14 do 20 mM (Grubbs and Maguire, 1987; Romani and Scarpa, 1992). Zmierzono całkowitą
zawartość magnezu w poszczególnych organellach komórek HL-60 (ludzka ustalona linia
komórek ostrej białaczki promielocytowej) i wynosiła ona od około 100 do 120 mmol/kg
suchej masy komórkowej w jądrze komórkowym, mitochondriach oraz siateczce
śródplazmatycznej (Di Francesco et al., 1998). Podobny poziom i analogiczną koncentrację
(skumulowanie) magnezu w komórce oznaczono m.in. w miocytach (Shuman and Somlyo,
1987). Magnez w komórce (ok. 90%) występuje głównie w formie związanej z chromatyną i
DNA (jądro komórkowe), rybosomami i fosfolipidami (retikulum endo- i sarkoplazmatyczne)
oraz ATP (macierz mitochondrialna), stąd ta charakterystyczna dystrybucja magnezu
pomiędzy organella komórkowe. Wolny jon magnezowy [Mg2+] i stanowi niewielką cześć
całkowitej puli magnezu wewnątrzkomórkowego, a jago stężenie np. w macierzy
mitochondrialnej waha się od 0,8 do 1,2 mM (Jung and Brierley, 1986; Rutter et al., 1990).
Zachowanie homeostazy magnezu w komórce polega na utrzymaniu relatywne stałego
stężenia [Mg2+] i przy wysokich wahaniach całkowitego poziomu magnezu
wewnątrzkomórkowego, które mogą być rezultatem np. wzmożonej aktywności
metabolicznej komórki lub stymulacji hormonalnej (Gunther, 1993). Z danych literaturowych
wynika, że w warunkach fizjologicznych, nawet przy intensywnym przepływie magnezu
przez błony komórkowe zmiany w stężeniu [Mg2+] i są niewielkie, a rolę elementów
buforujących pełni głównie ATP (Cittadini and Scarpa, 1983) oraz białka cytoplamatyczne
(np. kalmodulina (Ohki et al., 1997)).
Napływ magnezu do komórki odbywa się głównie na drodze dyfuzji, zgodnie z potencjałem
błonowym komórki (po stronie cytozolu błona komórkowa ma ujemny ładunek co promuje
przechodzenie kationów) (Flatman, 1991). Utrzymanie stałego wewnątrzkomórkowego
stężenia [Mg2+] i wymaga sprawnego systemu transporterów wyspecjalizowanych w
przenoszeniu tego pierwiastka przez błonę komórkową, jak i redystrybucję tego pierwiastka
pomiędzy organella komórkowe (Ryc. 2). Przepływ magnezu w komórce może zachodzić na
drodze zależnej i niezależnej od jonów sodu. Mechanizm zależny od jonów sodu to por
wymienny Na+/ Mg2+ (Gunther, 1993), który zidentyfikowany został m.in. w erytrocytach
wielu gatunków zwierząt (Gunther and Vormann, 1985; Gunther et al., 1990), w komórkach

16
mięśniowych (Tashiro and Konishi, 1997) oraz komórkach układu odpornościowego
(Gunther et al., 1992; Wolf et al., 1997). Mechanizm transportowy niezależny od jonów
sodowych zakłada wymianę wewnątrzkomórkowego Mg2+ na kationy pozakomórkowe np.
Mn2+ (zidentyfikowany w erytrocytach (Feray and Garay, 1987; Ludi and Schatzmann, 1987))
Ca2+ (zidentyfikowany w kardiomiocytach (Romani et al., 1993b) i hepatocytach (Fagan and
Romani, 2001; Romani et al., 1993a) lub aniony np. Cl- lub HCO3- (oba zidentyfikowane w
erytrocytach (Gunther and Vormann, 1990)). W tkance sercowej, mózgu, wątrobie, nerce i
jelicie zidentyfikowano transportery kationów dwuwartościowych należące do rodziny
SLC41 (ang. solute carrier family 41): m.in. SLC41A1 oraz SLC41A2 (Goytain and
Quamme, 2005b, 2005c; Wabakken et al., 2003). Przez SLC41A1 przenoszone są m.in. Fe2+,
Cu2+ oraz Zn2+; ten transporter jest nieprzepuszczalny dla Ca2+ (jony wapniowe nie blokują
transportu Mg2+). SLC41A1 należy do grupy transporterów bramkowanych napięciem
(Goytain and Quamme, 2005a), przy czym badania wykonane przez Koliska i wsp. wskazują,
że SLC41A1 ma większy udział w transporcie Mg2+ na zewnątrz niż do wewnątrz komórki
(Kolisek et al., 2008). Zwiększoną ekspresję tego transportera zaobserwowano przy diecie
ubogiej w magnez (Goytain and Quamme, 2005a). Strukturalnie SLC41A1 oraz SLC41A2 są
w 70% homologicznymi białkami (Sahni et al., 2007), ale oba transportery różnią się istotnie
w zdolności przenoszenia kationów dwuwartościowych (Goytain and Quamme, 2005c). Przez
SLC1A2 transportowane są m.in. Ni2+, Fe2+ oraz Mn2+ , przy czym transporter jest
nieprzepuszczalny m.in. dla jonów Ca2+ które, inaczej niż w przypadku SLC1A1, blokują
transport Mg2+. Potwierdzono obecność SLC1A2 w wielu tkankach ssaków, a jego ekspresja
wydaje się nie być zależna od ilości magnezu dostarczanego z dietą (Goytain and Quamme,
2005c). Kolejny transporter jonowy którego ekspresja w komórce rośnie gdy zawartość
magnezu w diecie jest niska to ACDP2 (ang. ancient conserved domain protein) (Goytain and
Quamme, 2005a). Transportery z rodziny ACDP są powszechnie obecne w tkankach
człowieka (Wang et al., 2003; Wang et al., 2004), przy czym są to białka wysoko
konserwatywne cechujące się 50% homologią strukturalną z białkiem CorC, transporterem
jonowym regulującym przepływ magnezu u procaryota (Kehres and Maguire, 2002). ACDP2
jest sterowany potencjałem i przepuszczalny dla wielu kationów dwuwartościowych (m.in.
Mg2+, Cu2+, Fe2+, Sr2+, Ba2+) przy czym jony Zn2+ hamują jego aktywność (Goytain and
Quamme, 2005a). W ostatnich latach zidentyfikowano rodzinę transporterów błonowych
TRPM (ang. transient receptor potential cation channel, subfamily M (melastatin)), gdzie
spośród wszystkich członków, szczególnie dwa kanały TRPM6 (Schlingmann et al., 2002)
oraz TRPM7 (Nadler et al., 2001) wydają się pełnić kluczową rolę w utrzymaniu homeostazy

17
magnezu w komórce. TRPM6 jest ekspresjonowany przede wszystkim w jelicie cienkim i
nerkach, co sugeruje jego znaczący udział jednocześnie w absorpcji magnezu dostarczanego z
pożywieniem oraz reabsorbcji magnezu (Ryc. 2). Poziom ekspresji TRPM6 jest regulowany
zmianami w stężeniu Mg2+ oraz kompleksu Mg-ATP w komórce (Voets et al., 2004). TRPM7
cechuje obecność we wszystkich tkankach ssaków, jest przepuszczalny dla jonów wapnia i
magnezu. Podobnie jak TRPM6, ekspresja TRPM7 jest regulowana poprzez
wewnątrzkomórkowe stężenie magnezu i kompleksu Mg-ATP (Demeuse et al., 2006).
TRPM7 warunkuje homeostazę magnezu na poziomie komórki, a jego brak lub dysfunkcja są
dla komórki letalne. Usunięcie genu TRPM7 skutkuje obniżeniem żywotności i
zahamowaniem wzrostu komórek in vitro, przy czym zmiany te mogą zostać cofnięte przez
podwyższenie stężenia magnezu pozakomórkowego do wartości ponadfizjologicznych
(10mM) (Schmitz et al., 2003), co sugeruje że w określonych warunkach efektywny transport
magnezu do komórki może odbywać się niezależnie od TRPM7 np. przez SLC4A1/2 oraz
MagT1. MagT1 (ang. magnesium transporter 1) należy do grupy transporterów błonowych
bramkowanych napięciem, odznaczający się wysokim powinowactwem do jonów
magnezowych przy czym jest właściwie nieprzepuszczalny dla innych kationów
dwuwartościowych. MagT1 jest obecny we wszystkich tkankach ssaków, a poziom jego
ekspresji jest regulowany przez zewnątrzkomórkowe stężenie magnezu (Goytain and
Quamme, 2005d). Za redystrybucję magnezu wewnątrz komórki odpowiadają m.in. Mrs2
(ang. mitochondrial RNA splicing 2 protein) pełniący kluczową rolę w transporcie magnezu
do mitochondriów (Kolisek et al., 2003), transportery MMgT1 oraz MMgT2 (ang. membrane
Mg2+ transporter 1/2) (Goytain and Quamme, 2008) których obecność została potwierdzona w
aparacie Golgiego (Goytain and Quamme, 2008). Mechanizmy które rządzą przepływem
magnezu w komórce są ciągłym przedmiotem badań. Wypływ magnezu z komórki reguluje
głównie zależny od ATP wymiennik Na+ /Mg2+ przy zachowaniu bilansu ładunku po obu
stronach błony komórkowej (Romani, 2011). Wydajność wymiany magnezu pomiędzy
osoczem a tkanką zależy od typu tkanki oraz ponownie jest cechą gatunkową (Maguire and
Cowan, 2002). Szybkość transportu magnezu przez błony komórkowe do osocza np. w sercu
(Rogers and Mahan, 1959), nerkach (Rogers and Mahan, 1959), wątrobie (Rogers and
Mahan, 1959) i adipocytach (Elliott and Rizack, 1974) ssaków jest wysoka tzn. według
danych literaturowych w sytuacjach stresowych w przeciągu 3-4 godzin całkowita pula
magnezu wewnątrz komórki może być wytransportowana do osocza. Stymulacja hormonalna
wydaje się być najbardziej dynamicznym mechanizmem indukującym wypływ magnezu z
komórki, która może skutkować gwałtownym zmniejszeniem wewnątrzkomórkowego

18
stężenia magnezu nawet o 10-15% (Romani, 2011). Podobny efekt wywołują substancje/stany
komórki które znacząco obniżają poziom i produkcję ATP np. cyjanki (Harman et al., 1990),
fruktoza (Gaussin et al., 1997), etanol (Tessman and Romani, 1998) oraz niedotlenienie
(Gasbarrini et al., 1992)). Obniżenie stężenia lub degradacja (do ADP lub AMP) głównego
czynnika wiążącego magnez powoduje dysocjację kompleksów ATP-Mg2+, istotny wzrost
[Mg2+] i w cytoplazmie i transport magnezu na zewnątrz w celu utrzymania stałego stężenia
magnezu w komórce (Romani, 2011).
2. Niedobór magnezu
Hypomagnezemia oznacza niską zawartość magnezu w surowicy krwi. Klinicznie,
według danych literaturowych, hypomagnezemię stwierdza się u pacjentów u których stężenie
magnezu w surowicy było ≤ 0,61mM (1,5mg/dL) (Guerrero-Romero et al., 2004; Hashizume
and Mori, 1990; Wong et al., 1983) lub ≤0,75mM (Chernow et al., 1989; Whang and Ryder,
1990). Pierwotny niedobór magnezu wynika z niedostatecznej podaży magnezu w diecie.
Wtórny niedobór magnezu wynika najczęściej z zaburzeń we wchłanianiu magnezu przez
układ pokarmowy i niewystarczającej reabsorbcji magnezu w nerkach, zaburzeń
hormonalnych lub jest skutkiem ubocznym terapii. Największy wpływ na ograniczenie
zwrotnego wchłaniania magnezu mają diuretyki pętlowe, ponieważ punkt ich działania
znajduje się w tym samym miejscu pętli Henlego. Szacuje się, że nawet 11%
hospitalizowanych pacjentów cierpi na hypomagnezemię (Wong et al., 1983), przy czym
wśród pacjentów przebywających na oddziałach intensywnej terapii medycznej wartość ta
wzrasta nawet do 60-65% (Ryzen et al., 1985). Zaobserwowano, że hypomagnezemia była
częstą reperkusją leczenia gdzie stosowano: aminoglikozydy, cytostatyki (np.cisplatynę),
digoksynę, diuretyki pętlowe (np. furosemid) oraz niektóre antybiotyki (np. amfoterycyna B)
lub cyklosporynę A, pomimo że mechanizm wyjaśniający te zależności często nie jest znany
(Chernow et al., 1989; Hashizume and Mori, 1990) (Jahnen-Dechent, 2012).
Niedobór magnezu jest często powiązany z innymi zaburzeniami gospodarki mineralnej,
przez co objawy hypomagnezemii są często niejednoznaczne. W 1969 Shils objawy
wywołanego niedoboru magnezu u grupy ochotników opisał jako ogólne osłabienie, brak
apetytu którym towarzyszyły hypokalemia i hypokalcemia oraz pozytywne objawy Chvostka
(gwałtowne skurcze mięśni mimicznych twarzy) i Trousseau (przykurcz palców dłoni) (Shils,
1969a, 1969b).
Szczególnym przykładem choroby związanym z hypomagnezemią jest tężyczka pastwiskowa
(Grass tetany). Tężyczka pastwiskowa zaliczana jest do chorób metabolicznych, występuje u

19
krów w okresie wysokiej laktacji głównie wiosną i jej przyczyną jest niedobór magnezu
(zawartość magnezu w szybko rosnącej trawie jest niska) i/lub może wynikać ze złej
proporcji składników w diecie (niska zawartość magnezu/wysoka zawartość potasu oraz azotu
w paszy) (Fontenot et al., 1989). Tężyczka pastwiskowa może występować w postaci ostrej
(drgawki, gorączka oraz trudnościami w oddychaniu które są wynikiem skurczy mięśni
oddechowych) lub w postaci podklinicznej, która objawia się obniżeniem mleczności,
zmniejszeniem apetytu, sztywnym i niezręcznym chodem, drgawkami i lękliwością. By
ograniczyć ryzyko zachorowań na tężyczkę pastwiskową zaleca się stopniowe wprowadzanie
bydła na pastwiska w okresie po zimowym oraz dodatek magnezu do paszy (Harris et al.,
1983; Odette, 2005).
2.1 Niedobór magnezu jako czynnik chorób cywilizacyjnych
Już w 1981 roku Karppanen (Karppanen, 1981) opublikował dane które wskazywały,
że spożycie magnezu w krajach wysoko uprzemysłowionych było niewystarczające, co
korelowało z wysoką zapadalnością na chorobę niedokrwienną serca, natomiast utrzymanie
właściwego statusu magnezowego mogło zmniejszać ryzyko zachorowań na schorzenia
układu sercowo-naczyniowego. W latach 1999-2000 National Health and Nutrition
Examination Survey przeprowadziło badania (King et al., 2005) na reprezentatywnej grupie
dorosłych Amerykanów gdzie wykazano, że średnie dzienne spożycie magnezu wynosi 328
mg ± 293 mg, przy czym 68 % osób spożywało mniej niż RDA a 45 % badanych
przyjmowało aż o 25 % mniej magnezu niż wynosiła RDA. Niewystarczające spożycie
magnezu zaobserwowano również w wysoko rozwiniętych krajach europejskich (Galan et al.,
1997).
2.1.1 Magnez i zespół metaboliczny
Zespół metaboliczny (zespół polimetaboliczny, zespół X, zespół insulinooporności)
jest wypadkową czynników istotnie zwiększających ryzyko rozwoju chorób metabolicznych i
schorzeń układu sercowo-naczyniowego. Składają się na niego: otyłość typu brzusznego,
nadciśnienie tętnicze, hiperglikemia oraz hiperlipidemia (głownie zwiększone stężenie
trójglicerydów (TG) w surowicy i zmniejszone stężenie frakcji cholesterolu o dużej gęstości
(ang. high density lipoprotein (HDL)) (Eckel et al., 2005).
Zespół metaboliczny (ZM) stanowi coraz większy problem kliniczny i epidemiologiczny.
Ford i współ. opublikowali dane (Ford et al., 2002) z których wynika, że w Stanach
Zjednoczonych 13% młodzieży, 24% osób w średnim wieku oraz ponad 40% osób które

20
ukończyły siedemdziesiąt lat cierpi na zespół metaboliczny. W Polsce według badań
populacyjnych z 2004 roku na ZM cierpi 22,6% kobiet oraz 18% mężczyzn, przy czym
szacuje się, ze zapadalność na ZM na świecie będzie rosła, przez co profilaktyka i szybkie
rozpoznanie stają się kluczowymi etapami w terapii ZM (Zdrojewski T. et al., 2004).
Guerrero-Romero i współ. w badaniach populacyjnych (Guerrero-Romero and Rodriguez-
Moran, 2002a) wykazali niezależny związek pomiędzy niskim stężeniem magnezu w
surowicy u ludzi (niskim statusem magnezowym) a zwiększonym występowaniem ZM. Na
podstawie danych zgromadzonych w latach 1988-1994 w Stanach Zjednoczonych (Ford et al.,
2007) stwierdzono, że istnieje odwrotna zależność pomiędzy przyjmowaniem magnezu a
ryzykiem wystąpienia ZM u ludzi, przy czym dieta bogata w magnez jest rekomendowana dla
utrzymania dobrego zdrowia metabolicznego oraz przy zapobieganiu schorzeniom układu
krążenia. Badania prospektywne, które prowadzono przez 15 lat na reprezentatywnej grupie
młodych Amerykanów (przedział wiekowy: 18-30 lat w momencie rozpoczęcia badań)
wykazały, że wyższe spożycie magnezu korelowało ze zmniejszonym ryzykiem rozwoju
poszczególnych składowych ZM (He et al., 2006).
2.1.2 Magnez i cukrzyca
Niedobór magnezu często towarzyszy cukrzycy (Pham et al., 2007; Simmons et al.)
przy czym niskie spożycie magnezu jest określane jako czynnik wysokiego ryzyka dla
rozwoju cukrzycy typu drugiego (CT2) niezależnie od wieku, indeksu masy ciała, spożycia
alkoholu czy częstości występowania insulinooporności w rodzinie (Larsson and Wolk,
2007). Szerokie badania prospektywne prowadzone przez Women’s Health Institute (Song et
al., 2004) oraz Nurses’ Health Study wspólnie z Health Professionals’ Follow-up Study
(Lopez-Ridaura et al., 2004) wskazały, że istnieje odwrotna zależność pomiędzy spożyciem
magnezu a ryzykiem rozwoju CT2. Nadler i wsp. dowiedli, że wraz z postępującym
niedoborem magnezu (wywołanym dietą ubogą w magnez) zmniejsza się wrażliwość tkanek
na insulinę u osób zdrowych (Nadler et al., 1993). U osób u których wykazano
insulinooporność (bez zdiagnozowania CT2) oraz deficyt magnezu (stężenie magnezu w
surowicy mniejsze lub równe 0,74 mM) chlorek magnezu przyjmowany doustnie (2,5
g/dzień) przez trzy miesiące normalizował poziom magnezu w surowicy, co korelowało z
wyższą wrażliwością tkanek na insulinę (Guerrero-Romero et al., 2004). Przyjmowanie
chlorku magnezu (2,5 g/dzień przez 16 tygodni) przez osoby ze zdiagnozowaną cukrzycą typu
2 i niskim statusem magnezowym skutkowało ponownie normalizacją poziomu magnezu we
krwi oraz zwiększeniem wrażliwości tkanek na insulinę (Rodriguez-Moran and Guerrero-

21
Romero, 2003). Szlaki jakimi niedobór magnezu (hypomagnezemia) wpływa na rozwój
cukrzycy nie są poznanie. Obecny stan wiedzy zakłada, że 1) deficyt magnezu zwiększając
wewnątrzkomórkowe stężenie Ca2+ w komórce może osłabiać działanie insuliny (Elamin and
Tuvemo, 1990; Tosiello, 1996); 2) magnez bezpośrednio reguluje metabolizm glukozy w
komórce jako kofaktor wielu enzymów oraz przekaźnik drugiego rzędu dla insuliny (Laughlin
and Thompson, 1996; Paolisso et al., 1992); 3) niskie wewnątrzkomórkowe stężenie magnezu
może upośledzać transport glukozy do komórki (Kandeel et al., 1996; Takaya et al., 2004).
2.1.3 Magnez i choroby układu krążenia
Rola deficytu magnezu jako czynnika rozwoju chorób układu sercowo-naczyniowego
jest dyskutowana od dawna. Badania kliniczne wykazały, że niedobór magnezu w diecie
może prowadzić do zaburzeń pracy serca (Klevay and Milne, 2002; Nielsen et al., 2007) jak
również niskie stężenie magnezu we krwi jest istotnym prognostykiem schorzeń układu
krążenia (Reffelmann et al., 2010; Reffelmann et al., 2011).
3. Zależności pomiędzy wapniem i magnezem
Jedną z funkcji biologicznych magnezu jest antagonizowanie procesów indukowanych
przez wapń (Iseri and French, 1984), co wynika w dużym stopniu z właściwości
fizykochemicznych obu pierwiastków i odmiennej koncentracji w organizmie.
Wewnątrzkomórkowe stężenie wolnego jonu magnezowego w cytoplazmie wynosi około 0,5-
0,7 mM i zmienia się jedynie w wąskim zakresie stężeń. Pozakomórkowe stężenie magnezu
oscyluje wokół 0,7 – 1,1 mM (Iseri and French, 1984; Saris et al., 2000). Niski gradient
stężeń po obu stronach błony komórkowej potwierdza założenie, że magnez odgrywa
uzupełniającą rolę jako długoterminowy element regulacyjny (Grubbs and Maguire, 1987).
Cytozolowe stężenie wapnia zjonizowanego (Ca2+) wynosi około 100 nM i jest o kilka
rzędów wielkości niższe od jego stężenia pozakomórkowego (Berridge et al., 2000). Ten
wysoki gradient chemiczny stwarza komórkom możliwość szybkiego podwyższenia
wewnątrzkomórkowego poziomu wapnia poprzez przejściowe otwarcie błonowych kanałów
wapniowych lub uwolnienie wewnątrzkomórkowych zasobów wapnia w celu przekazania
sygnału i aktywacji komórki (Berridge et al., 2000).
Różnice w budowie otoczki hydratacyjnej magnezu i wapnia (Ryc. 3) oraz zdolność
wiązania się tych jonów do różnych grup chemicznych sprawiają, że w przeciwieństwie do
wapnia, magnez wiąże się słabiej i nie dociera do głębiej usytuowanych miejsc wiążących w
białkach. Dodatkowo wąskie kanały transportowe w błonie komórkowej przez które jony

22
wapniowe są swobodnie transportowane nie są osiągalne dla jonów magnezowych (Barry and
Bridge, 1993; Carafoli, 1987; Williams, 1970).
Stosunek Mg/Ca w tkankach miękkich wynosi 3:2, przy czym zmiany w tej zależności mogą
prowadzić do nienormalnych zmian w funkcjonowaniu komórki (Schroeder et al., 1969).
Dane literaturowe wskazują, że magnez pozkomórkowy znacząco wpływa na wychwyt,
dystrybucję i stężenie wapnia w tkance mięśniowej (Altura and Altura, 1974; Turlapaty and
Altura, 1978). Zaobserwowano również odwrotną zależność liniową pomiędzy zawartością
magnezu w diecie a stężeniem wapnia w surowicy zwierząt doświadczalnych (Zimmermann
et al., 2000). U szczurów z niską zawartością magnezu w diecie (normalna zawartość wapnia)
przekrwienie uszu i ogona pojawiało się szybciej i było silniejsze niż u zwierząt z niską
zawartością magnezu i wapnia w diecie (Bussiere et al., 2002b). U myszy z niedoborem
magnezu zaobserwowano korzystny wpływ antagonisty wapnia, nifedipiny, na przeżywalność
zwierząt (Sakaguchi et al., 1992). Mechanizmy definiujące zależności pomiędzy wapniem i
magnezem nie zostały jeszcze wyjaśnione, przy czym zakłada się że 1) wapń i magnez mogą
konkurować o to samo miejsce w systemie transportowym 2) wapń może wpływać na
przepuszczalność błon komórkowych dla magnezu 3) wapń pośrednio moduluje absorpcję
magnezu poprzez bezpośredni wpływ na gospodarkę hormonalną (Hardwick et al., 1991).
Przedstawione dane literaturowe sugerują, by rozpatrywać skojarzony wpływ obu
pierwiastków na funkcjonowanie komórki i całego organizmu.
4. Wapń jako sygnalizator komórkowy
Wapń (Ca2+) jest uniwersalnym sygnalizatorem który inicjuje/kontroluje wiele
procesów komórkowych. Całkowite stężenie wapnia w komórce wynosi około 10 µM, przy
czym wapń w formie związanej skoncentrowany jest głównie w organellach komórkowych.
Stężenie wolnego jonu wapniowego w komórce w stanie spoczynku wynosi około 100 nM.
Dane literaturowe podają, że dla zainicjowania procesów biochemicznych w komórce
niezbędne jest podwyższenie stężenia Ca2+ w cytoplazmie do 500 – 1000 nM (Berridge et al.,
2000). Wysokie stężenie Ca2+ w cytoplazmie może być osiągnięte poprzez wnikanie Ca2+ z
przestrzeni pozakomórkowej i/lub uwalnianie wapnia z wewnątrzkomórkowych magazynów
błonowych tj. siateczki sarkoplazmatycznej (w komórkach mięśniowych) lub równoważnego
organellum, siateczki śródplazmatycznej (w pozostałych typach komórek) (Berridge et al.,
2000). Transport Ca2+ ze środowiska pozakomórkowego jest regulowany przez kilka typów
kanałów w tym: kanały sterowane napięciem (ang. voltage-operated channels), kanały które
otwierają się po aktywowaniu receptora przez np. acetylocholinę lub ATP (ang. receptor-

23
operated channels) oraz kanały które otwierają się gdy istotnie obniży się poziom wapnia w
magazynach błonowych (ang. store-operated channel). Uwolnienie Ca2+ ze źródeł
wewnątrzkomórkowych jest ściśle kontrolowane, przy czym główną rolę w tym procesie
pełnią rodziny kanałów: receptora dla 1,4,5-trifosfatydyloinozytolu oraz receptora
rianodynowego (Berridge, 1993; Clapham, 2007). Aktywatorem tych kanałów jest
bezpośrednio jon wapniowy, przy czym sam proces jest określany jako „indukowane Ca2+
uwalnianie Ca2+”. Napływ Ca2+ do komórki ze środowiska indukuje związanie agonisty z
receptorem (m.in. 1,4,5-trifosfatydyloinozytolu ze swoistym receptorem, cyklicznej ADP-
rybozy do receptora rianodynowego (Clapper et al., 1987)) i uwolnienie wapnia z magazynów
wewnątrzkomórkowych. Przy wyciszaniu sygnału wapniowego Ca2+ są szybko usuwane z
cytoplazmy przez system pomp i wymienników jonowych. Ca2+ są transportowane na
zewnątrz komórki przez ATPazę wapniową (Ca2+-ATPaza) oraz wymiennik Na+/Ca2+ gdy
np. w miocytach ATPaza w siateczce sarkoplazmatycznej steruje gromadzeniem wapnia
wewnątrz komórki (Blaustein and Lederer, 1999; Pozzan et al., 1994).
4.1 Mechanizm działania substancji blokujących kanały wapniowe
Miejscem działania stosowanych klinicznie blokerów wapnia są kanały wapniowe
zależne od potencjału (Janiec and Krupinska, 2005). Kanały wapniowe bramkowane
napięciem pośredniczą w napływie Ca2+ do komórki w odpowiedzi na depolaryzację błony i
pośrednio regulują procesy wewnątrzkomórkowe takie jak skurcz, neuroprzekaźnictwo i
ekspresja genów w różnych typach tkanek. Ich właściwe funkcjonowanie jest niezbędne do
sprzęgania sygnałów elektrycznych na powierzchni komórki i zdarzeń fizjologicznych w
komórkach (Catterall et al., 2005). Ze względu na budowę strukturalną, próg pobudliwości
oraz szybkość inaktywacji kanały wapniowe podzielono na podtypy: LP/Q, R, N
[aktywowane przy silnej depolaryzacji (około: +30 do +50 mV)] (Calin-Jageman and Lee,
2008; Catterall et al., 2005), oraz T [aktywowany przy słabej depolaryzacji (około: −55 do
−20 mV)](Iftinca and Zamponi, 2009; Talavera and Nilius, 2006). Kanały wapniowe zależne
od potencjału mogą występować w postaci spoczynkowej, otwartej lub nieaktywnej, co wiąże
się bezpośrednio z procesami depolaryzacji i repolaryzacji błony komórkowej.
Antagoniści wapnia blokują/hamują napływ jonów wapnia do komórki poprzez wiązanie się
do kanału jonowego, przy czym w stopniu marginalnym mogą wpływać na bezpośrednią
interakcję wolnych jonów wapniowych z białkami. Inhibitory kanałów wapniowych
stosowane w leczeniu, hamują przepływ wapnia głównie w kanałach typu L (Janiec and
Krupinska, 2005), przy czym jak donoszą ostatnie badania werapamil może hamować

24
przepływ Ca2+ również w kanałach typu T (Bergson et al., 2011) . Antagoniści wapnia mogą
zmniejszać częstotliwość z jaką jony wapnia są transportowane przez kanał lub ograniczają
częstotliwość otwierania się kanału wapniowego. Blokery wapnia hamujące wybiórczo
kanały wapniowe wiążą się do kanałów typu L tylko w ich określonym stanie
czynnościowym np. kiedy są otwarte (leki z grupy werapamilu) lub w stanie inaktywacji (leki
z grupy nifedipiny). Po ustąpieniu deoplaryzacji inhibitory wapnia odłączają się od kanałów
wapniowych (Janiec and Krupinska, 2005).
4.2 Blokery wapnia a układ odpornościowy
Substancje blokujące wybiórczo kanały wapniowe znalazły zastosowanie przede
wszystkim w terapii schorzeń układu naczyniowo-sercowego, w tym choroby niedokrwiennej
serca (choroba wieńcowa), nadciśnienia tętniczego i płucnego czy niewydolności serca w
przebiegu kardiomiopatii (Eisenberg et al., 2004). Efekty jakie wywierają inhibitory kanałów
wapniowych na komórki mięśniowe są powszechnie znane, przy czym w ostatnich latach
szczególnie wzrosło zainteresowanie immunosupresyjnymi właściwościami blokerów wapnia
również w kontekście schorzeń układu krwionośnego. W mięśniu sercowym w wyniku
uszkodzenia tkanek mogą występować procesy przypominające klasyczne reakcje zapalne
(Baumgarten et al., 2001; Kapadia et al., 1995; Satoh et al., 1996). Miocyty sercowe są zdolne
do produkcji cytokin prozapalnych, chemokin oraz mediatorów komórkowych (tlenek azotu)
co jest reakcją na bezpośrednie uszkodzenie tkanek lub rozpoznanie wzorców molekularnych
związanych z patogenami (LPS, enterowirusy) przez receptory rozpoznające wzorce (CD14,
TLR2, TLR3, TLR4, TLR6) obecne na powierzchni komórek mięśniowych (Cowan et al.,
2000; Dong et al.; Frantz et al., 2001). Przedłużająca się nadekspresja mediatorów zapalenia
(cytokin prozapalnych) w mięśniu sercowym może prowadzić do trwałego uszkodzenia
tkanek i skutkować dekompensacją serca (Mann et al., 2010). W literaturze przedstawiono
immunosupresyjne a jednocześnie kardioprotektywne właściwości blokerów kanałów
wapniowych (Topkara et al., 2011).
O ile znaczenie i przepływ wapnia w komórce immunologicznej został już dobrze poznany i
opisany (Ma and Beaven, 2011; Oh-hora and Rao, 2008; Tintinger et al., 2005), to proces
wyciszania sygnału wapniowego w poszczególnych komponentach układu
immunologicznego od lat stanowi przedmiot zainteresowania wielu grup badawczych.
Blokery kanałów wapniowych znacząco zmniejszają aktywność wielu elementów
komórkowych układu odpornościowego. Tanzaki i wsp. wykazali, że nifedipina znacząco
obniża wychwyt wapnia i sekrecję histaminy przez uczulone (tutaj albuminą jaja kurzego)

25
komórki tuczne szczurów (Tanizaki et al., 1983a; Tanizaki et al., 1983b) co potwierdzają
wyniki Ennis’a i wsp. gdzie nifedipina i werapamil hamują indukowaną sekrecję histaminy z
otrzewnowych komórek tucznych szczurów, przy czym efekt ten nie został zaburzony przez
zwiększenie stężenia wapnia pozakomórkowego (Ennis et al., 1983). Preinkubacja
makrofagów z werapamilem lub nifedipiną skutkowała zmniejszeniem indukowanej PMA
produkcji reaktywnych form tlenu i tlenku azotu przez makrofagi, przy czym efekt był
zależny od dawki zastosowanych blokerów kanałów wapniowych (Shen et al., 1995).
Inhibitory kanałów wapniowych mogą hamować indukowaną przez cytokiny
wielokierunkową aktywację (m.in. migrację i produkcję reaktywnych form tlenu) ludzkich
granulocytów obojętnochłonnych (Shima et al., 2008). Birx i wsp. dowiedli, że zastosowanie
inhibitorów kanałów wapniowych (nifedipina, werapamil, diltiazem) hamuje aktywację
limfocytów T oraz indukowaną przez IL-2 proliferację komórek (Birx et al., 1984).
Dodatkowo wykazano, że te same inhibitory wolnych kanałów wapniowych hamują
indukowaną cytokinami migrację limfocytów T w stopniu zależnym od dawki substancji
(Bacon et al., 1989). Jednocześnie werapamil selektywnie tłumi funkcje efektorowe
cytotoksycznych limfocytów T, limfocytów T pomocniczych oraz limfocytów B (Singh et al.,
1990). W zakresie wpływu jaki wywierają inhibitory wolnych kanałów wapniowych na
aktywację i funkcje poszczególnych typów komórek immunologicznych szczególnie
interesujące wydają się być dane na temat ich wpływu na limfocyty T pomocnicze (które ze
względu na odmienne funkcje i różny panel produkowanych cytokin zostały podzielone na
dwie subpopulacje komórkowe: Th1 oraz Th2 (ang. T helper cells 1/2 ))(Romagnani, 1999).
Dane literaturowe wskazują, że przepływ i szlaki sygnałowe Ca2+ są różne w komórkach Th1
i Th2 (Sloan-Lancaster et al., 1997). W stanie spoczynku wewnątrzkomórkowe stężenie
wolnego jonu wapniowego jest wyższe w komórkach Th2 niż w Th1, a po aktywacji komórek
T za pośrednictwem receptora dla komórek T (TCR, ang. T-cell receptor) poziom Ca2+ jest
niższy w komórkach Th2 niż Th1 (Fanger et al., 2000). Dodatkowo stwierdzono, że receptor
dla dihydropirydyny (podjednostka α1 kanału wapniowego typu L (DHPR, ang.
dihydropyridne receptor)) jest ekspresjonowany na powierzchni limfocytów Th2, przy czym
nie stwierdzono jego obecności na komórkach Th1, co sugeruje że komórki Th2 są
wrażliwsze na działanie antagonistów DHPR takich jak nifedipina lub nikardipina. Receptory
DHPR mogą być selektywnymi markerami subpopulacji komórek Th2 w tylko w obrębie
komórek T, dlatego że ich obecność potwierdzono m.in. w komórkach dendrytycznych (Poggi
et al., 1998) i limfocytach B (Sadighi Akha et al., 1996).

26
5. Magnez i układ odpornościowy
Dane doświadczalnie jednoznacznie wskazują, że indukowanemu niedoborowi
magnezu towarzyszy wzmożona aktywność poszczególnych komponentów komórkowych
układu immunologicznego i rozwój reakcji zapalnej, przez co markery zapalenia (m.in. wolne
rodniki, cytokiny, białka ostrej fazy) są często podstawowymi parametrami określanymi w
badaniach nad niedoborem magnezu.
5.1 Niedobór magnezu jako czynnik stanu zapalnego u ludzi
CRP (ang. C-reactive protein) jest białkiem należącym do grupy białek ostrej fazy.
Produkowane jest głównie w wątrobie i wydzielane do krwi. Wysokie stężenie CRP jest
wynikiem doznanego urazu, infekcji lub zapalenia, przy czym stężenie CRP szybko obniża
się po ustąpieniu przyczyny wzmożonej ekspresji tego białka (Du Clos, 2000). W diagnostyce
klinicznej utrzymujące się podwyższone stężenie CRP jest wiązane z przewlekłym stanem
zapalnym (Ridker, 2007). W ostatnich latach wzrosło zainteresowanie bezpośrednim
związkiem pomiędzy niskim statusem magnezowym/niskim spożyciem magnezu a
podwyższonym poziomem CRP u ludzi. Ta pozytywna zależność jest szczególnie istotna,
ponieważ wysoki poziom CRP bezpośrednio koreluje z podwyższonym ryzykiem wystąpienia
schorzeń układu sercowo-naczyniowego (Pearson et al., 2003) oraz cukrzycą i otyłością
(Ford, 1999; Pradhan et al., 2001).
King i wsp. (King et al., 2005) wykazali, że u osób spożywających mniej magnezu niż RDA
zaobserwowano podwyższony poziom CRP, przy czym dane te zostały znormalizowane
względem parametrów mających istotny wpływ na wynik analizy (m.in. wiek, rasa, płeć,
indeks masy ciała, palenie papierosów, spożywanie alkoholu, ogólna kondycja zdrowotna i
całkowita ilość spożywanych kalorii). W wybranej grupie dorosłych (ukończone 40lat,
indeks masy ciała powyżej 25) u osób które przyjmowały mniej niż połowa zalecanej dla
magnezu RDA podwyższone stężenie CRP we krwi występowało ponad 2 razy częściej niż u
osób które spożywały zalecaną ilość tego pierwiastka (King et al., 2005). Korelacja pomiędzy
niskim poziom magnezu a wysokim stężeniem CRP w osób dorosłych została wykazana
również przez Guerrero-Romero i wsp. (Guerrero-Romero and Rodriguez-Moran, 2002b).
Dodatkowo doustne przyjmowanie magnezu (cytrynian magnezu, 300 mg/dzień przez 5
tygodni) istotnie obniżyło poziom CRP u osób ze zdiagnozowaną niewydolnością serca w
porównaniu do grupy chorych która otrzymywała placebo (Almoznino-Sarafian et al., 2007).
W latach 1999-2002 National Health and Nutrition Examination Survey przeprowadziło
badania obejmujące grupę 5007 dzieci i młodzieży (przedział wieku: 6-17 lat) z których

27
wynikało, że u osób które spożywały mniej niż 75% zalecanej dla magnezu RDA, stężenie
CRP było prawie dwukrotnie wyższe niż u tych badanych, którzy przyjmowali więcej
magnezu niż RDA (King et al., 2007). Badania przeprowadzone na grupie 488 dzieci
(przedział wiekowy: 10-13lat) wykazały, że 109 (22,3%) z nich miało podwyższone stężenie
CRP we krwi; u 101 (20,7%) odnotowano obniżone stężenie magnezu w surowicy, przy czym
łącznie w obrębie tych dwóch grup u 183 (87,1%) dzieci odnotowano jednocześnie:
podwyższone stężeniu białka i obniżone stężenie magnezu we krwi (Rodriguez-Moran and
Guerrero-Romero, 2008). Women's Health Initiative Observational Study (USA) analizując
dane od 3713 kobiet (przedział wiekowy: 50-79 lat; u osób biorących udział w badaniu w
momencie rozpoczęcia nie zdiagnozowano żadnej z chorób układu krążenia, cukrzycy ani
nowotworu) wykazało, że istnieje odwrotna zależność pomiędzy odpowiednim spożyciem
magnezu a podwyższonym stężeniem trzech markerów zapalenia: CRP, TNF-α oraz IL-6 w
surowicy (Chacko et al., 2010). Song i wsp. wykazali, że niskie spożycie magnezu w diecie
(badania przeprowadzone na grupie 657 zdrowych kobiet w wieku 43-69 lat) jest powiązane z
podwyższonym stężeniem niektórych markerów zapalenia i systematycznej dysfunkcji
śródbłonka u kobiet zdrowych. Dane znormalizowane o takie parametry jak: palenie tytoniu,
spożywanie alkoholu, aktywność fizyczna, indeks masy ciała czy stosowanie zastępczej
terapii hormonalnej, wykazały związek pomiędzy niskim statusem magnezowym a
podwyższonym poziomem CRP i E-selektyn u badanych kobiet (Song et al., 2007).
Wykazano również, że niedobór magnezu może wpływać na stan zapalny i stres oksydacyjny
wywołane przez inne czynniki, w tym zakłócenia lub brak snu (Nielsen et al., 2010). Badania
przeprowadzone na grupie 100 osób u których stwierdzono niską jakość snu (wskaźnik
jakości snu według Pittsburg Sleep Quality Index wynosił więcej niż 5) wykazały, że
przyjmowanie magnezu (cytrynian magnezu, 320 mg/dzień przez 7 tygodni) istotnie obniżyło
poziom CRP i znormalizowało poziom magnezu w surowicy tylko u osób u których
pierwotnie rozpoznano niedobór magnezu (niski status magnezowy) w porównaniu do osób z
niedoborem magnezu które przyjmowały placebo (cytrynian sodu). U wszystkich biorących
udział w badaniu odnotowano poprawę jakości snu, co sugeruje: 1) zależność pomiędzy
statusem magnezowym a jakością snu; 2) niski status magnezowy może być przyczyną lub
skutkiem złej jakości snu (Nielsen et al., 2010).

28
5.2 Eksperymentalny niedobór magnezu
Dieta uboga w magnez skutkuje szybkim obniżeniem poziomu tego pierwiastka we
krwi zwierząt doświadczalnych. W ósmym dniu diety deficytowej, stężenie magnezu w
surowicy szczurów młodych wynosiło około 0,14 mM podczas gdy w grupie kontrolnej
wynosiło ono ok. 0,81 mM (Malpuech-Brugere et al., 2000). Indukowanemu niedoborowi
magnezu towarzyszyły klasyczne objawy zapalenia tj. m.in. rozszerzenie peryferyjnych
naczyń krwionośnych oraz leukocytoza (Malpuech-Brugere et al., 2000). Do zamian
patologicznych będących reperkusją wysokiego deficytu magnezu u szczurów zaliczana jest
splenomegalia (wywołana głównie naciekiem tego narządu przez granulocyty
obojętnochłonne i makrofagi, zaobserwowano również zmianę w proporcjach subpopulacji
limfocytów śledzionowych w odniesieniu do grupy kontrolnej (Malpuech-Brugere et al.,
1998a) oraz wzmożoną syntezę DNA, co jest wiązane ze zwiększoną proliferacją limfocytów
w śledzionie (Malpuech-Brugere et al., 1998a; Zieve et al., 1977)) i inwolucja grasicy (już w
trakcie drugiego dnia diety niedoborowej zaobserwowano zwiększoną liczbę neutrofili i
makrofagów w rdzeniu narządu oraz wzmożoną apoptozę komórek w porównaniu do grupy
kontrolnej (Malpuech-Brugere et al., 1999a)). Wykazano, że w trakcie niedoboru magnezu
neutrofile wykazują cechy komórek aktywowanych (Bussiere et al., 2002d). Eozynofilia,
wysoki poziom histaminy oraz IgE w surowicy zwierząt niedoborowych sprawia, że wysoki
deficyt magnezu jest wiązany z występowaniem nadwrażliwości typu I (Claverie-Benureau et
al., 1980). Wykazano, że dieta niedoborowa w magnez skutkuje wzrostem stężenia cytokin
prozapalnych IL-1, IL-6 (Malpuech-Brugere et al., 2000), TNF-α (Weglicki and Phillips,
1992; Weglicki et al., 1992) oraz mediatorów zapalenia (m.in. substancji P) (Weglicki and
Phillips, 1992) w surowicy zwierząt doświadczalnych. Wykazano, że wzrost IL-6 we krwi
zwierząt niedoborowych występuje już w czwartym dniu eksperymentu (Malpuech-Brugere et
al., 2000), co prowadzi do indukcji syntezy białek ostrej fazy w wątrobie (Ceciliani et al.,
2002). Po tygodniu diety niedoborowej w magnez, odnotowano wzrost stężenia dodatnich
białek ostrej fazy: α2- makroglobuliny, α1-kwaśnej glikoproteiny oraz fibrynogenu we krwi
szczurów doświadczalnych, przy czym dla dwóch pierwszych białek odnotowano również
zwiększoną transkrypcję genu w tkankach wątroby (Malpuech-Brugere et al., 2000). IL-1
oraz TNF-α indukują syntezę składników dopełniacza (Frank and Fries, 1991). W pierwszym
tygodniu postępującego niedoboru magnezu u zwierząt doświadczalnych również
odnotowano podwyższony poziom C3 składnika dopełniacza (stwierdzono jednocześnie
wzrost poziomu mRNA w tkankach wątroby oraz białka w osoczu) (Bussiere et al., 2003) .
29
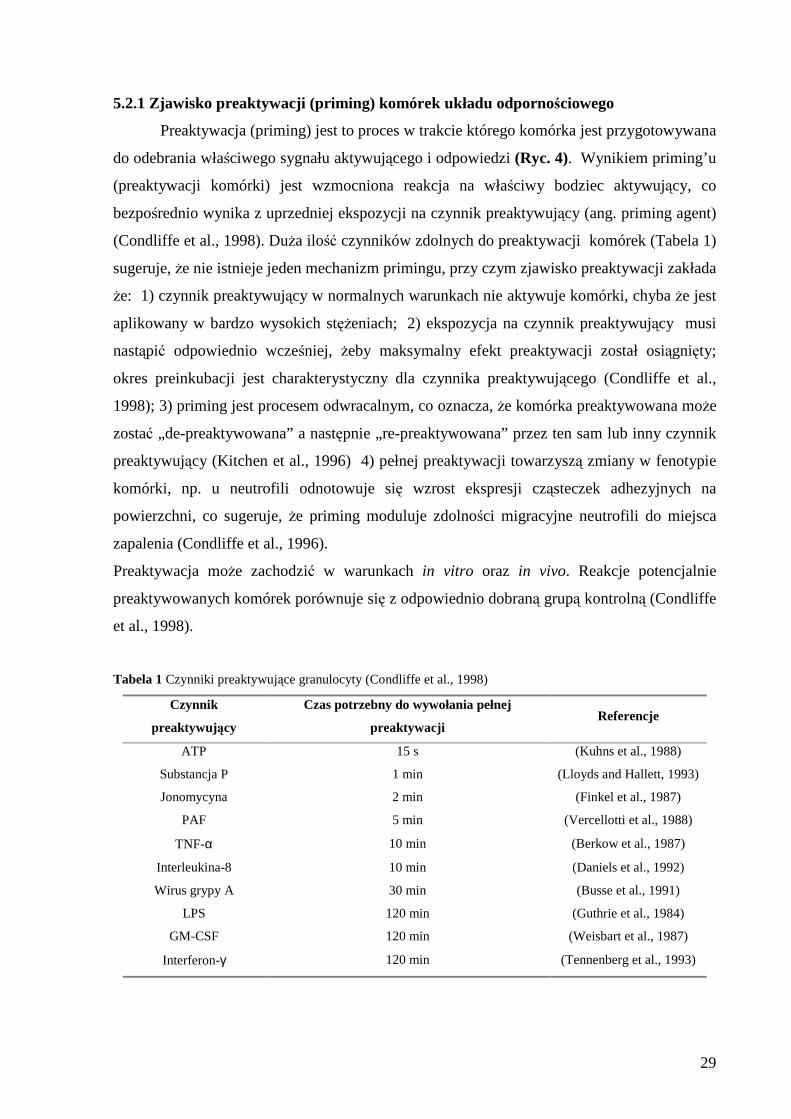
5.2.1 Zjawisko preaktywacji (priming) komórek układu odpornościowego
Preaktywacja (priming) jest to proces w trakcie którego komórka jest przygotowywana
do odebrania właściwego sygnału aktywującego i odpowiedzi (Ryc. 4). Wynikiem priming’u
(preaktywacji komórki) jest wzmocniona reakcja na właściwy bodziec aktywujący, co
bezpośrednio wynika z uprzedniej ekspozycji na czynnik preaktywujący (ang. priming agent)
(Condliffe et al., 1998). Duża ilość czynników zdolnych do preaktywacji komórek (Tabela 1)
sugeruje, że nie istnieje jeden mechanizm primingu, przy czym zjawisko preaktywacji zakłada
że: 1) czynnik preaktywujący w normalnych warunkach nie aktywuje komórki, chyba że jest
aplikowany w bardzo wysokich stężeniach; 2) ekspozycja na czynnik preaktywujący musi
nastąpić odpowiednio wcześniej, żeby maksymalny efekt preaktywacji został osiągnięty;
okres preinkubacji jest charakterystyczny dla czynnika preaktywującego (Condliffe et al.,
1998); 3) priming jest procesem odwracalnym, co oznacza, że komórka preaktywowana może
zostać „de-preaktywowana” a następnie „re-preaktywowana” przez ten sam lub inny czynnik
preaktywujący (Kitchen et al., 1996) 4) pełnej preaktywacji towarzyszą zmiany w fenotypie
komórki, np. u neutrofili odnotowuje się wzrost ekspresji cząsteczek adhezyjnych na
powierzchni, co sugeruje, że priming moduluje zdolności migracyjne neutrofili do miejsca
zapalenia (Condliffe et al., 1996).
Preaktywacja może zachodzić w warunkach in vitro oraz in vivo. Reakcje potencjalnie
preaktywowanych komórek porównuje się z odpowiednio dobraną grupą kontrolną (Condliffe
et al., 1998).
Tabela 1 Czynniki preaktywujące granulocyty (Condliffe et al., 1998)
Czynnik
preaktywuj ący
Czas potrzebny do wywołania pełnej
preaktywacji Referencje
ATP 15 s (Kuhns et al., 1988)
Substancja P 1 min (Lloyds and Hallett, 1993)
Jonomycyna 2 min (Finkel et al., 1987)
PAF 5 min (Vercellotti et al., 1988)
TNF-α 10 min (Berkow et al., 1987)
Interleukina-8 10 min (Daniels et al., 1992)
Wirus grypy A 30 min (Busse et al., 1991)
LPS 120 min (Guthrie et al., 1984)
GM-CSF 120 min (Weisbart et al., 1987)
Interferon-γ 120 min (Tennenberg et al., 1993)

30
5.2.1.1 Niedobór magnezu jako pośredni czynnik preaktywuj ący
Bussiere i wsp. potwierdzili, że wysoki niedobór magnezu skutkuje rozwojem
klasycznej reakcji zapalnej u szczurów (Bussiere et al., 2002a). W ósmym dniu diety
deficytowej odnotowano istotny spadek stężenia magnezu oraz stężania wolnego jonu
magnezowego w surowicy zwierząt doświadczalnych. Zmianom tym towarzyszył wzrost
poziomu wapnia, istotnie wyższe stężenie IL-6 oraz tlenku azotu w surowicy zwierząt
niedoborowych. Zaobserwowano też wysoką leukocytozę, głównie we frakcji neutrofili
(Bussiere et al., 2002a). Granulocyty obojętnochłonne pozyskane od zwierząt niedoborowych
wykazywały cechy „częściowej aktywacji” - bez dodatkowej stymulacji produkowały istotnie
więcej reaktywnych form tlenu w odniesieniu do neutrofili pozyskanych od zwierząt z grupy
kontrolnej. Po stymulacji PMA produkcja reaktywnych form tlenu przez neutrofile zwierząt
deficytowych była wielokrotnie wyższa niż u komórek pozyskanych od zwierząt z
odpowiednią zawartością magnezu w diecie (Bussiere et al., 2002a). Granulocyty od zwierząt
niedoborowych cechowała również wyższa aktywność fagocytarna oraz większa zdolność
przylegania do szkła w porównaniu do leukocytów zwierząt z grupy kontrolnej (Bussiere et
al., 2002a). Rolę magnezu jako czynnika modulującego odpowiedź komórek układu
immunologicznego potwierdzają dane eksperymentalne, które wskazują że wysokie stężenie
magnezu w medium hodowlanym obniża aktywność fagocytów. Wysokie pozakomórkowe
stężenie magnezu in vitro (8,0 mM vs 0,8 mM) istotnie zmniejszyło indukowaną PMA lub
fMLP produkcję reaktywnych form tlenu przez ludzkie granulocyty obojetnochłonne
(odpowiednio o 15% i 31%). Jednocześnie wysoki poziom magnezu pozakomórkowego (8,0
mM vs 0,8 mM) istotnie zmniejszył indukowaną PMA produkcję reaktywnych form tlenu
przez eozynofile pozyskane od pacjentów u których występował proces zapalny (Bussiere et
al., 2002c).
Weglicki i wsp. przedstawili hipotezę, że w rozwoju procesu zapalnego który towarzyszy
postępującemu niedoborowi magnezu u zwierząt doświadczalnych pośredniczą neuropeptydy
(Weglicki and Phillips, 1992). Zaobserwowano, że w trakcie pierwszego tygodnia diety
niedoborowej w magnez znacząco wzrósł poziom substancji P (inna nazwa: neurokinina 1;
neuropeptyd zaliczany do tachykinin, stymuluje rozwój m.in. lokalnej reakcji zapalnej i
ekspresję cytokin (O'Connor et al., 2004)) a w trzecim tygodniu niedoboru wykazano
maksymalny poziom cytokin prozapalnych (IL-1, IL-6, TNF-α) u zwierząt niedoborowych
(Weglicki and Phillips, 1992).

31
5.2.1.2 Substancja P jako czynnik preaktywujący
Substancja P jest dobrze opisanym w literaturze bezpośrednim czynnikiem
preaktywującym (Lloyds and Hallett, 1993; Perianin et al., 1989). Substancja P może
wpływać na limfocyty B i T determinując ich różnicowanie i aktywację (Bost and Pascual,
1992; Stanisz et al., 1987). Weglicki i współ. wykazali, że limfocyty T pozyskane od myszy
z niedoborem magnezu produkowały więcej cytokin (IL)-2, 4, 5, 10, 12, 13 oraz Interferon-γ
po godzinnej inkubacji w medium zawierającym substancję P w porównaniu do komórek
pozyskanych od zwierząt z grupy kontrolnej (Weglicki et al., 1996). Wiadomo, że IFN-γ
preaktywuje makrofagi m.in. poprzez zwiększenie ich wrażliwości na LPS, stymuluje
ekspresję receptorów Fc oraz cząsteczek MHC II (Fernandez and Caperos, 1977).
Preinkubacja eozynofili z IL-5 wzmacnia indukowany wybuch tlenowy, przy czym eozynofile
pozbawione dostępności wapnia pozostają nadal preaktywowane po inkubacji z IL-10
(Koenderman et al., 1989) (Ryc. 5).
Związek pomiędzy podwyższonym poziomem substancji P przy doświadczalnym niedoborze
magnezu jest ciągle tematem badań. Dane literaturowe wskazują, że podwyższony poziom
substancji P w surowicy wynika z obniżonej aktywności endopeptydazy u zwierząt z
deficytem magnezu (Weglicki et al., 2009). Aktywacja receptora dla substancji P, NK-1R
(ang. neurokinin receptor 1) prowadzi do aktywacji czynnika transkrypcyjnego NF-κβ (ang.
nuclear factor κβ) co skutkuje wzmożoną produkcją cytokin przez makrofagi (Bardelli et al.,
2005). Ponadto w trakcie niedoboru magnezu u zwierząt doświadczalnych zaobserwowano
wzmożoną ekspresję NK-1R na powierzchni komórek układu odpornościowego (Weglicki et
al., 1996).

32
II ZAŁO ŻENIA I CEL PRACY
Z uwagi na udział magnezu (Mg) w wielu procesach komórkowych, zaburzenia w
homeostazie tego pierwiastka mogą stanowić przyczynę zaburzeń w funkcjonowaniu
organizmu. Dzisiejszy stan wiedzy, przedstawiony w skrócie we wstępie, nie pozwala
jednoznacznie wyjaśnić, jaki jest bezpośredni wpływ zaburzonej homeostazy magnezu na
powstawanie i rozwój stanu zapalnego jak również na samą aktywację komórek układu
immunologicznego. Według najnowszych danych epidemiologicznych i klinicznych,
schorzeniom układu krążenia oraz zaburzeniom metabolicznym powiązanym z przewlekłym
stanem zapalnym często towarzyszy hypomagnezemia. Dlatego wyjaśnienie bezpośredniej
zależności pomiędzy stężeniem magnezu pozakomórkowego a właściwą aktywacją komórek
układu odpornościowego może mieć duże znaczenie dla zdrowia człowieka.
Biorąc pod uwagę wpływ magnezu na nieswoiste i swoiste mechanizmy
odpornościowe należy uwzględnić działanie wapnia (Ca). Magnez jest naturalnym
antagonistą wapnia, przy czym wapń pełni rolę wtórnego przekaźnika sygnału łączącego
szlaki aktywacyjne w leukocytach. Dodatkowo dane literaturowe wskazują, że wpływ
stężenia magnezu na układ odpornościowy może w dużym stopniu zależeć od dostępności
wapnia pozakomórkowego, regulacji jego przepływu i uwalniania zasobów
wewnątrzkomórkowych.
Celem pracy doktorskiej było zbadanie wpływu magnezu pozakomórkowego oraz
zaburzonej równowagi magnezowo-wapniowej na przebieg nieswoistej i swoistej odpowiedzi
komórkowej:
1). analiza skojarzonego wpływu magnezu pozakomórkowego i wapnia na wybrane
parametry aktywności modelowej komórki fagocytarnej in vitro
2). zbadanie wpływu pozakomórkowego magnezu i wapnia na prezentację antygenu,
w tym funkcjonowanie modelowych komórek prezentujących antygen oraz ich zdolność do
aktywacji limfocytu T in vitro
Zastosowanie inhibitorów transportu wapnia w błonie komórkowej i/lub siateczce
śródplazmatycznej pozwoliło określić istotność dostępności wapnia poza- i
wewnątrzkomórkowego dla właściwego funkcjonowania komórek układu odpornościowego.

33
III MATERIAŁY I METODY
PLAN DOŚWIADCZE Ń
Przeprowadzone doświadczenia miały na celu zbadanie wpływu pozakomórkowego
stężenia magnezu i zaburzonej równowagi magnezowo-wapniowej na nieswoistą i swoistą
odpowiedź komórkową in vitro.
W pierwszym etapie badań zbadano wpływ magnezu pozakomórkowego i wapnia,
oraz ich wzajemnych oddziaływań, na aktywność modelowych komórek fagocytarnych in
vitro (Ryc. 6). Doświadczenia przeprowadzono na dwóch mysich rodzajach komórek:
ustalonej mysiej linii komórek makrofagowych J774.E oraz granulocytach pozyskanych z
krwi obwodowej myszy szczepu Balb/C.
Drugi eksperyment pozwolił na oszacowanie skojarzonego wpływu magnezu i wapnia
na aktywację limfocytu T przez komórki mające zdolność do prezentowania antygenu:
komórki dendrytyczne oraz makrofagi (Ryc. 7). W celu określenia wpływu
pozakomórkowego stężenia magnezu i zaburzonej równowagi magnezowo-wapniowej na
prezentację antygenu prowadzono hodowlę mieszaną dwóch typów komórek (kokulturę
komórkową): modelowych limfocytów T (komórki linii ustalonej D10.G4.1, genetycznie
predysponowane do rozpoznania modelowego antygenu: konalbuminy (białko z grupy
albumin występujące w białku jaja kurzego, wykazuje właściwości antybakteryjne dzięki
wysokiemu powinowactwu do jonów metali m.in. żelaza, (Trziszka, 2000)) z komórkami
dendrytycznymi lub makrofagami prezentującymi modelowy antygen. Oceniono potencjał
proliferacyjny komórek D10.G4.1 i produkcję wybranych cytokin: IL-2, IL-4, IL-10 oraz
interferonu-γ jako miarę prezentacji i rozpoznania konalbuminy.
Stężenie magnezu pozakomórkowego w układzie eksperymentalnym było
warunkowane poprzez dodatek inaktywowanej FCS (10%) i siarczanu magnezu do medium
testowego (skład mediów hodowlanych i testowych właściwych dla określonego typu
komórek przedstawiono w wykazie zastosowanych odczynników).
W medium deficytowym stężenie magnezu pozakomórkowego wynosiło 0,1 mM - najniższe
stężenie magnezu jakie zmierzono w surowicy szczurów po ośmiu dniach diety niedoborowej

34
w magnez (Malpuech-Brugere et al., 1999b). W medium kontrolnym stężenie magnezu
pozakomórkowego wynosiło 1,0 mM i było zbliżone do normalego poziomu magnezu w
surowicy (Tietz and Tietz 1990). W medium o wysokiej zawartości magnezu stężenie
wynosiło 5,0 mM - najwyższe stężenie magnezu jakie oznaczono w surowicy po podaniu
parenteralnym soli magnezu (Maier et al., 2004).
Stężenie wapnia pozakomórkowego było stałe (około 0,25 mM) i wynikało z dodatku
inaktywowanej FCS (10%) do medium. Przepływ wapnia w komórce był regulowany poprzez
zastosowanie blokerów wapnia: werapamilu i/lub TMB-8. Werapamil jest inhibitorem
kanałów wapniowych w błonie komórkowej (Striessin, 2004). TMB-8 jest antagonistą wapnia
wewnątrzkomórkowego (Chiou and Malagodi, 1975; Kumar and Chakrabarti, 2000).
1. Wpływ blokerów wapnia na potencjał proliferacyjny ustalonych linii komórkowych:
J774.E oraz D10.G4.1 - wyznaczenie IC50 dla badanych związków
Komórki linii J774.E naniesiono na 96-dołkową płaskodenną płytkę testową w ilości
4x104 komórek/mL medium; komórki linii D10.G4.1 naniesiono w ilości 6x104 komórek/mL
odpowiedniego medium. Komórki były hodowane przez 48 godzin w medium właściwym dla
każdego typu komórek przy różnym stężeniu testowym werapamilu lub TMB-8 w zakresie 0-
200 µM. Po zakończeniu inkubacji oceniono aktywność komórek wykonując test MTT.
Po zakończeniu inkubacji dodano roztwór wodny MTT (5 µg/mL) w ilości 20 µL/dołek płytki
testowej. Całość inkubowano przez 2 godziny w standardowych warunkach hodowlanych.
Następnie do każdej próby dodano bufor lizujący w ilości 80 µL/dołek płytki testowej i
umieszczono w inkubatorze (5% CO2, 37°C). Po upływie 4 godzin zmierzono gęstość
optyczną próbek przy długości fali 570 nm.
Założono, że miarą aktywności metabolicznej/potencjału proliferacyjnego komórek jest
zdolność redukcji MTT do formazanu.
Żywe komórki redukują MTT (barwa żółta) do formazanu (barwa fioletowa). Gęstość
optyczna badanych próbek przy długości fali 570 nm jest proporcjonalna do liczby
aktywnych metabolicznie komórek w próbie. Próbę negatywną stanowi roztwór pochodzący z
dołków zawierających wyjściowo tylko medium hodowlane.

35
Otrzymane wartości zmierzonej dla poszczególnych prób testowych absorbancji (Ap)
przeliczono na wartości określające procent zahamowania aktywności komórkowej, według
wzoru:
% zahamowania aktywności komórkowej = 100100 −
⋅
−−
mk
mp
AA
AA
gdzie:
Am - wartość absorbancji otrzymanej dla próby negatywnej
Ak - wartość absorbancji otrzymanej dla próby kontrolnej gdzie komórki zawieszone były w
medium o stałej zawartości magnezu (1,0 mM) bez inhibitorów kanałów wapniowych
Ap – wartość absorbancji otrzymanej dla prób testowych gdzie komórki J774.E zawieszone
były w odpowiednim medium testowym (1,0 mM Mg) w obecności werapamilu lub TMB-8
(stężenia testowe: 10-200 µM)
Z uzyskanych wartości zahamowania aktywności komórkowej /ograniczenia potencjału
proliferacyjnego komórek wykreślone zostały krzywe zależności efektu od stężenia
zastosowanego inhibitora kanałów wapniowych (przykład krzywej na rysunku poniżej).
Na osi X umieszczono stężenia badanego związku w skali logarytmicznej (logarytmy
dziesiętne), na osi Y – stopień inhibicji badanego parametru. Wykreślona została prosta na
poziomie 50 % zahamowania badanego parametru. Z punktu przecięcia krzywej wyznaczonej
doświadczalnie i „linii 50 %” odczytane zostało stężenie związku, przy którym zahamowanie
aktywności metaboliczne/potencjału proliferacyjnego wynosiło 50 % (Ryc. 8).
2. Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na aktywność
modelowych komórek fagocytarnych in vitro
Badania przeprowadzono na dwóch typach komórek: 1) ustalonej linii komórek
makrofagowych J774.E (Diment et al., 1987; Porcaro et al., 2003) (pochodzacej z Kolekcji
Komórkowych Instytutu Immunologii i Terapii Doświadczalnej im Ludwika Hirszfelda PAN
we Wrocławiu, udostępnionej do badań przez Pana Profesora dr hab. Janusza Boratyńskiego)
oraz 2) populacji granulocytów uzyskanych z krwi obwodowej myszy Balb/C.
Hodowla komórek linii J774.E prowadzona była w naczyniach wykonanych z
polistyrenu przeznaczonych dla hodowli komórkowych w standardowych warunkach
hodowlanych (5 % CO2, 37 °C) w medium hodowlanym. Komórki linii J774.E w trakcie

36
wzrostu silnie adherują do dna naczyń hodowlanych. Przy pasażowaniu komórek linii J774.E
stosowano sterylny roztwór trypsyny wieprzowej (0,25 %) z dodatkiem EDTA (0,2 g/L).
Gęstość wyjściowa hodowli wynosiła 2x104 komórek/mL medium hodowlanego J1 /cm2
powierzchni wzrostu. Komórki linii J774.E były pasażowane w odstępach 48- lub 72-
godzinnych przy czym hodowla komórkowa nie była prowadzona dłużej niż 4 tygodnie.
Granulocyty zostały uzyskane z krwi obwodowej myszy szczepu Balb/C według
zmodyfikowanego protokołu Englisha i Andersona (English and Andersen, 1974).
Jednostopniowa metoda izolacji komórek wielojądrzastych na gradiencie gęstości jest oparta
na założeniu, że gradient gęstości jest utworzony przez objętościowo równe dwie warstwy
nietoksycznych mediów do izolacji: Histopaque-1077 i Histopaque-1119. Krew pobrana na
antykoagulant była starannie nawarstwiana na Histopaque-1077. Podczas wirowania
erytrocyty tworzyły agregaty i sedymentowały. Granulocyty znajdowały się na granicy faz
Histopaque-1077 / Histopaque-1119, natomiast limfocyty i monocyty znajdowały się na
granicy faz Histopaque-1077 / osocze.
Szczegółowy protokół izolacji:
1. Do sterylnej probówki typu falcon przeznaczonej do wirowania o objętości 15 mL dodano
3 mL Histopaque-1119 (20 °C).
2. Na powierzchnię Histopaque-1119 naniesiono 3 mL Histopaque-1077 (20 °C).
3. 6 mL pełnej krwi pobranej na chlorek heparyny (40 U/mL krwi) naniesiono na
powierzchnię gradientu gęstości.
4. Całość wirowano przy 700 g przez 30 minut w temperaturze pokojowej.
5. Usunięto trzy najwyższe warstwy wyodrębnione po rozdziale: osocze, komórki
mononuklearne oraz medium izolacyjne: Histopaque-1077. Warstwę granulocytów oraz
medium izolacyjne: Histopaque-1119 w objętości do 2 mL przeniesiono do sterylnej
stożkowej probówki przeznaczonej do wirowania o objętości 15 mL.
6. Probówkę uzupełniono do objętości 15 mL izotonicznym roztworem PBS pozbawionym
jonów wapnia i magnezu. Całość wirowano przy 200 g przez 10 minut w temperaturze
pokojowej.
7. Supernatant odrzucono. Osad komórek zawieszono w 15 mL izotonicznego roztworu PBS
bez jonów wapnia i magnezu. Całość wirowano przy 200 g przez 10 minut w temperaturze
pokojowej. Czynność powtórzono dwukrotnie.
8. Osad komórek zawieszono w 10 mL medium hodowlanego J1. Całość przenoszono do
naczynia przeznaczonego dla hodowli komórkowych i umieszczono w inkubatorze (5% CO2,

37
37°C). Przed rozpoczęciem kolejnego etapu eksperymentu oceniano żywotność uzyskanych
granulocytów.
2.1 Ocena żywotności populacji granulocytów
Do zawiesiny komórek dodano 0,4 % roztwór błękitu trypanu w buforze PBS w
stosunku 1:1. Po upływie 3 minut 10 µL mieszaniny umieszczono w komorze Thoma. Pod
mikroskopem świetlnym policzono całkowitą ilość komórek i komórki martwe (zabarwione
na niebiesko-fioletowo) a następnie wyliczono procent żywych komórek w preparacie.
Żywotność granulocytów po każdej izolacji wynosiła nie mniej niż 90 %.
2.2 Ocena czystości populacji granulocytów
Mysie komórki wielojądrzaste (1x106 komórek/mL) zawieszono w 100 µL buforu do
znakowania przeciwciałami. Cząsteczkę powierzchniową: CD11c znakowano przeciwciałem
monoklonalnym anty-CD11c z fikoerytryną zgodnie z wytycznymi producenta stosując
wskazaną kontrolę izotypową dla przeciwciała. Całość inkubowano w ciemności przez 30
minut w temperaturze 4 °C. Analizy cytometrycznej dokonano przy użyciu oprogramowania
Win MDI 2.9 (http://www.cyto.purdue.edu/flowcyt/software/Winmdi.html). Komórki o
fenotypie CD11c+ stanowiły zawsze nie mniej niż 85 % komórek pozyskanych na gradiencie
gęstości.
2.3 Ocena aktywności komórkowej i wzrostu komórek
Komórki mysiej linii makrofagowej J774.E naniesiono na 96-dołkową płaskodenną
płytkę przeznaczoną do hodowli komórkowych w wyjściowym stężeniu 4x104 komórek /mL
(200 µL zawiesiny komórek/dołek płytki testowej) w medium testowym J1T.
Komórki J774.E hodowano przez 24, 48 lub 72 godziny w mediach z różną zawartością
magnezu w zakresie 0,1 mM – 5,0 mM i odpowiednich blokerów kanałów wapniowych:
werapamilu (inhibitor kanałów wapniowych w błonie komórkowej) oraz TMB-8
(wewnątrzkomórkowy inhibitor kanalów wapniowych) o testowym stężeniu 25 µM. Do
oceny aktywności komórek w badanych warunkach eksperymentalnych zastosowano test
MTT (według protokołu zamieszczonego w paragrafie: 1).

38
Otrzymane wartości zmierzonej dla poszczególnych prób testowych absorbancji (Ap)
przeliczono na wartości określające procent zahamowania aktywności komórkowej/potencjału
proliferacyjnego, według wzoru:
% zahamowania aktywności komórkowej = 100100 −
⋅
−−
mk
mp
AA
AA
gdzie:
Am - wartość absorbancji otrzymanej dla próby negatywnej
Ak - wartość absorbancji otrzymanej dla próby kontrolnej gdzie komórki zawieszone były w
medium o stałej zawartości magnezu (1,0 mM) bez blokerów wapnia
Ap – wartość absorbancji otrzymanej dla prób testowych gdzie komórki J774.E zawieszone
były w medium o różnej zawartości magnezu w zakresie od 0,1-5,0 mM w obecności
werapamilu i/lub TMB-8.
2.4 Badanie endocytozy antygenu modelowego
Komórki linii J774.E naniesiono na 24-dołkową płytkę hodowlaną (5x104
komórek/mL/dołek). Komórki inkubowano (5% CO2, 37 °C) przez 6 godzin w obecności
koniugatu albuminy jaja kurzego z fluoresceiną (owoalbumina-FITC) o stężeniu testowym 1
mg/ml w medium testowym J1T o różnej zawartości magnezu w zakresie 0,1 mM – 5,0 mM i
odpowiednich blokerów wapnia: werapamilu oraz TMB-8 o testowym stężeniu 25 µM. Po
zakończeniu inkubacji komórki przemyto trzykrotnie zimnym (4 °C) buforem PBS z
dodatkiem inaktywowanej FCS (10 %). W celu oznaczenia/wykluczenia oddziaływań
niespecyficznych eksperyment kontrolny prowadzono równolegle w temperaturze 4 °C. Przy
użyciu półilościowej analizy fluorocytometrycznej określono poziom pobrania owoalbuminy-
FITC przez komórki makrofagowej linii J774.E. Analizy dokonano przy użyciu
oprogramowania Win MDI 2.9. Dla każdej badanej próby obliczono wartość średniej
intensywności fluorescencji MFI (ang. mean fluorescence activity). Wartość MFI obliczono
ze wzoru:
MFI=MFI ova-fitc- MFI k
gdzie MFI oznacza średnią intensywność fluorescencji dla komórek które pobrały
ovoalbuminę-FITC z medium w 37°C, zaś MFI k oznacza średnią intensywność fluorescencji
dla komórek z analogicznej do eksperymentalnej próby kontrolnej (4°C) (Ryc. 9).

39
2.5 Badanie produkcji reaktywnych form tlenu przez komórki fagocytarne
Modelowe komórki fagocytarne: komórki makrofagowej linii ustalonej J774.E oraz
komórki wielojądrzaste uzyskane z krwi naniesiono na 96-dołkową płytkę (1x106
komórek/mL) przeznaczoną do pomiaru luminescencji w czasie rzeczywistym (biała, sterylna,
płaskodenna płytka do hodowli komórkowych). Komórki inkubowano (5% CO2, 37 °C)
przez 90 minut w medium bez czerwieni fenolowej o różnej zawartości magnezu w zakresie
0,1 mM – 5,0 mM i odpowiednich blokerów wapnia : werapamilu oraz TMB-8 o testowym
stężeniu 25 µM. Po zakończeniu inkubacji komórki były stymulowane PMA (stężenie
testowe: 100 mM; rozcieńczony w DMSO) oraz dodawano soli sodowej luminolu (stężenie
testowe: 500 mM, rozpuszczona w wodzie). Stężenie testowe DMSO nie przekraczało 2 %.
Następnie przez co najmniej 90 minut w temperaturze 37 °C w odstępach jednominutowych
mierzono luminescencję w poszczególnych próbach doświadczalnych. Ocena produkcji
reaktywnych form tlenu w opisanym modelu eksperymentalnym opiera się na pomiarze
luminescencji w czasie jako wynik utleniania luminolu przez reaktywne formy tlenu
produkowane przez komórki fagocytarne. Podstawową jednostką pomiaru była relatywna
jednostka luminescencji na minutę (RLU/min, ang. relative luminescence unit). Jednostką
końcowo opisującą proces produkcji reaktywnych form tlenu było pole powierzchni pod
krzywą luminescencji (AUC, ang. area under the curve).
2.6 Oznaczenie cytokin prozapalnych w nadsączach komórkowych testem ELISA
Komórki linii J774.E naniesiono na 24-dołkową płytkę hodowlaną (5x105
komórek/mL/dołek) w medium testowym J1T o różnej zawartości magnezu w zakresie 0,1
mM – 5,0 mM i odpowiednich inhibitorów kanałów wapniowych: werapamilu oraz TMB-8 o
testowym stężeniu 50 µM lub 100 µM. Komórki stymulowano przez 6 godzin LPS (E.coli
O111:B4) o stężeniu testowym 100 ng/mL. Całość umieszczono w inkubatorze. Po
zakończeniu stymulacji zebrano nadsącze komórkowe i przechowywano je w temperaturze -
80°C do wykonania oznaczeń. Stężenie cytokin IL-1β, IL-6 oraz TNF-α oceniono testem
immunoenzymatycznym ELISA. Do oznaczeń użyto zestawów komercyjnych firmy Becton
Dickinson: Mouse IL1- β ELISA Set nr kat. 559603, Mouse IL-6 ELISA Set nr kat. 555240,
oraz Mouse TNF-α ELISA Set nr kat. 555268.
Płytki 96-dołkowe typu MaxiSorp opłaszczano przeciwciałem wychwytującym dla każdej z
badanych cytokin, rozcieńczonym zgodnie z zaleceniami producenta. Płytki inkubowano w
temperaturze 40C przez co najmniej 12 godzin. Po odpłukaniu niezwiązanego przeciwciała,

40
przy użyciu buforu PBS z dodatkiem 0,05 % Tween 20, płytki blokowano buforem do
blokowania (bufor PBS z dodatkiem inaktywowanej FCS (10 %)) przez 60 minut w
temperaturze pokojowej. Następnie rozcieńczono nadsącze komórkowe (zastosowano
rozcieńczenia: 1:100 dla IL-6, 1:2 dla IL-1β oraz TNF-α) oraz przygotowano roztwory
przeciwciał do krzywych standardowych zgodnie z certyfikatem analizy producenta i
nakładano je na wcześniej przygotowane płytki testowe w objętości 100 µL/dołek. Płytki
inkubowano przez 2 godziny w temperaturze pokojowej. Po usunięciu supernatantu płytki
trzykrotnie odlpukano. Do wykrycia badanych antygenów naniesiono rozcieńczone według
zaleceń producenta przeciwciała drugiej warstwy w objętości 100 µL/dołek i całość
inkubowano przez 60 minut w temperaturze pokojowej. Po zakończeniu inkubacji płytki
odpłukiwano pięciokrotnie. Wszystkie przeciwciała drugiej warstwy skoniugowane były z
enzymem peroksydazą chrzanową. Reakcję enzymatyczną wywołano dodając substrat TMB
w objętości 100 µL na dołek, po czym płytki testowe inkubowano przez 10 minut w
ciemności w temperaturze pokojowej. Reakcję zatrzymywano przez dodanie 1M kwasu
siarkowego VI w objętości 50 µL/dołek. Gęstość optyczną próbek odczytano przy długości
fali 450 nm za pomocą spektrofotometru Spektra II (SLT Labinstruments, Grödig, Austria).
Dla krzywych standardowych dopasowano krzywą regresji, wyznaczono matematyczne
równanie krzywej według którego obliczano stężenia danej cytokiny w badanych próbach
(Ryc. 10).
2.7 Oznaczenie ekspresji cytokin prozapalnych na poziomie transkryptu
Komórki linii J774.E naniesiono na 24-dołkową płytkę hodowlaną (5x106
komórek/mL/dołek) w medium testowym J1T o różnej zawartości magnezu w zakresie 0,1
mM – 5,0 mM i odpowiednich blokerów wapnia: werapamilu oraz TMB-8 o testowym
stężeniu 50 µM lub 100 µM. Komórki stymulowano przez 6 godzin LPS (E.coli O111:B4) o
stężeniu testowym 100 ng/mL. Całość umieszczono w inkubatorze. Po zakończeniu
stymulacji usunięto nadsącze komórkowe. Płytkę odpłukano trzykrotnie roztworem PBS bez
jonów wapania i magnezu (40C ) w objętości 2 mL/dołek. Następnie na płytkę dodawano
odczynnika TRIzol w objętości 800 µL/dołek. Lizaty komórkowe przechowywano w
temperaturze -80°C do wykonania oznaczeń.
Izolacji całkowitego RNA dokonano zgodnie z zaleceniami producenta zestawu Total RNA
Mini kit. W celu określenia wydajności izolacji RNA oraz sprawdzenia jego czystości
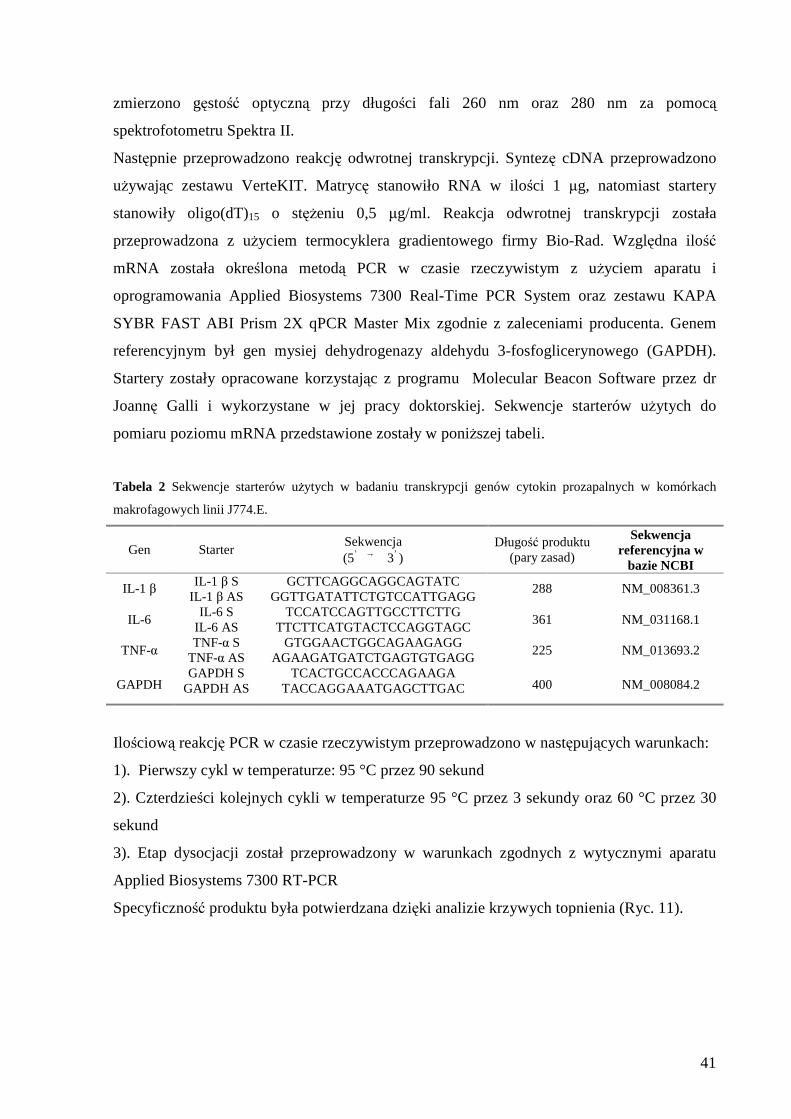
41
zmierzono gęstość optyczną przy długości fali 260 nm oraz 280 nm za pomocą
spektrofotometru Spektra II.
Następnie przeprowadzono reakcję odwrotnej transkrypcji. Syntezę cDNA przeprowadzono
używając zestawu VerteKIT. Matrycę stanowiło RNA w ilości 1 µg, natomiast startery
stanowiły oligo(dT)15 o stężeniu 0,5 µg/ml. Reakcja odwrotnej transkrypcji została
przeprowadzona z użyciem termocyklera gradientowego firmy Bio-Rad. Względna ilość
mRNA została określona metodą PCR w czasie rzeczywistym z użyciem aparatu i
oprogramowania Applied Biosystems 7300 Real-Time PCR System oraz zestawu KAPA
SYBR FAST ABI Prism 2X qPCR Master Mix zgodnie z zaleceniami producenta. Genem
referencyjnym był gen mysiej dehydrogenazy aldehydu 3-fosfoglicerynowego (GAPDH).
Startery zostały opracowane korzystając z programu Molecular Beacon Software przez dr
Joannę Galli i wykorzystane w jej pracy doktorskiej. Sekwencje starterów użytych do
pomiaru poziomu mRNA przedstawione zostały w poniższej tabeli.
Tabela 2 Sekwencje starterów użytych w badaniu transkrypcji genów cytokin prozapalnych w komórkach
makrofagowych linii J774.E.
Gen Starter Sekwencja (5’ → 3’ )
Długość produktu (pary zasad)
Sekwencja referencyjna w
bazie NCBI IL-1 β S GCTTCAGGCAGGCAGTATC IL-1 β
IL-1 β AS GGTTGATATTCTGTCCATTGAGG 288 NM_008361.3
IL-6 S TCCATCCAGTTGCCTTCTTG IL-6
IL-6 AS TTCTTCATGTACTCCAGGTAGC 361 NM_031168.1
TNF-α S GTGGAACTGGCAGAAGAGG TNF-α
TNF-α AS AGAAGATGATCTGAGTGTGAGG 225 NM_013693.2
GAPDH S TCACTGCCACCCAGAAGA GAPDH GAPDH AS TACCAGGAAATGAGCTTGAC 400 NM_008084.2
Ilościową reakcję PCR w czasie rzeczywistym przeprowadzono w następujących warunkach:
1). Pierwszy cykl w temperaturze: 95 °C przez 90 sekund
2). Czterdzieści kolejnych cykli w temperaturze 95 °C przez 3 sekundy oraz 60 °C przez 30
sekund
3). Etap dysocjacji został przeprowadzony w warunkach zgodnych z wytycznymi aparatu
Applied Biosystems 7300 RT-PCR
Specyficzność produktu była potwierdzana dzięki analizie krzywych topnienia (Ryc. 11).

42
Na podstawie krzywych fluorescencji oszacowano wydajność reakcji i dla poszczególnych
genów obliczono względny poziom ekspresji według metody Livaka i Schmittgena (Livak
and Schmittgen, 2001) gdzie:
Obliczenia wykonane zostały dla prób badawczych i kalibracyjnych:
∆Ct (próby badawcza) = Ct genu badanego – Ct genu referencyjnego
∆Ct (próba kalibracyjna) = Ct genu badanego – Ct genu referencyjnego
Wyznaczana zostła wartość ∆∆Ct dla każdej próby:
∆∆Ct = ∆Ct (próba badawcza) - ∆Ct (próba kalibracyjna)
Znormalizowaną wartość względnego poziomu ekspresji badanego genu w próbie badanej
względem kalibratora (R) wyznaczono według wzoru:
R=2-∆∆Ct
3. Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na wzrost
komórek limfocytarnych oraz ich aktywację przez komórki prezentujące antygen:
komórki dendrytyczne oraz makrofagi
Limfocyty T były reprezentowane przez ustaloną linię komórek D10.G4.1 Linia
komórek D10.G4.1 została zakupiona w Amerykańskiej Kolekcji Kultur Komórkowych
(ATTC nr katalogowy: TIB-224). Linia ustalona D10.G4.1(H-2k) została scharakteryzowana
jako linia komórek limfocytarnych Th2, wyprowadzona z węzła chłonnego myszy szczepu
AKR/J. Komórki D10.G4.1 swoiście rozpoznają konalbuminę (albumina występująca w
białku jaja kurzego).
Hodowla komórek limfocytarnych D10.G4.1 prowadzona była w naczyniach hodowlanych
wykonanych z polistyrenu przeznaczonych dla hodowli komórkowych w standardowych
warunkach hodowlanych (5% CO2, 37 °C) w medium D1. Komórki linii D10.G4.1 cechuje
nieadherentny (zawiesinowy) charakter wzrostu. Gęstość wyjściowa hodowli wynosiła 2x105
komórek/mL medium hodowlanego. Komórki linii modelowej były pasażowane w odstępach
48- lub 72-godzinnych przy czym hodowla komórkowa nie była prowadzona dłużej niż 4
tygodnie.
3.1 Ocena aktywności komórek D10.G4.1 w monokulturze
Komórki mysiej linii limfocytarnej D10.G4.1 naniesiono na 96-dołką płaskodenną
płytkę przeznaczoną do hodowli komórkowych o wyjściowym stężeniu 6x104 komórek /mL

43
(200 µL zawiesiny komórek/dołek płytki testowej) w medium testowym D1T. Komórki
D10.G4.1 hodowano przez 24, 48 lub 72 godziny w mediach z różną zawartością magnezu w
zakresie 0,1 mM – 5,0 mM iblokerów wapnia: werapamilu oraz TMB-8 o testowym stężeniu
25 µM.
Do oceny aktywności komórek w badanych warunkach eksperymentalnych zastosowano test
MTT (wg protokołu zamieszczonego w paragrafie: 1).
Otrzymane wartości zmierzonej dla poszczególnych prób testowych absorbancji (Ap)
przeliczono na wartości określające procent zahamowania aktywności metabolicznej (wg
protokołu dla komórek linii makrofagowej J774.E, paragraf: 2.3).
3.2 Wyprowadzenie pierwotnych linii komórek dendrytycznych oraz makrofagów z
komórek szpiku kostnego myszy szczepu C3H/ HeN (H-2k)
Myszy szczepu C3H/HeN zostały poddane eutanazji i pobrano od nich kości udowe
oraz piszczelowe. Po całkowitym usunięci tkanek miękkich i ścięgien, kości umieszczono w
alkoholu etylowym (70%) na około 3 minuty. Następnie kości zostały przeniesione do
sterylnej szalki hodowlanej z medium I1. Głowy kości zostały usunięte przy użyciu skalpela.
Mysie komórki prekursorowe zostały uzyskane poprzez aseptyczne wypłukanie szpiku
kostnego z kości długich przy użyciu igły o grubości 23 G oraz strzykawki o objętości 5 mL.
Kości były przepłukiwane zimnym medium do izolacji. Po zakończeniu izolacji zawiesina
komórek była wirowana przy 125 g przez 10 minut w 4 °C (Ryc. 12).
Komórki dendrytyczne (BMDCs) wyprowadzono z komórek szpiku kostnego według
protokołu opracowanego przez Lutza i współpracowników (Lutz et al., 1999). W dniu „0”
komórki szpiku kostnego zostały zawieszone w 10mL kompletnego medium hodowlanego R1
w stężeniu 2x105 komórek/mL. Do hodowli zastosowano bakteriologiczne szalki Petriego o
średnicy 100 mm. W trzecim dniu eksperymentu do hodowli komórkowej dodano 10 mL
medium R1. W szóstym oraz ósmym dniu hodowli pobrano 10mL zawiesiny komórek, całość
odwirowano (125 g, 10 minut, 4°C), osad komórek zawieszono w 10 mL medium R1 i
przeniesiono z powrotem na szalkę hodowlaną. Od 10-go do 12-go dnia hodowli
nieadherentne komórki zebrano, określono ich fenotyp i przekazano do kolejnego etapu
eksperymentu.
Makrofagi (BMMFs) wyprowadzono z komórek szpiku kostnego w oparciu o protokoły
Stanley’a (Stanley, 1997) oraz Weischenfeldt’a i Porse’go (Weischenfeldt and Porse, 2008).
W dniu „0” komórki prekursorowe szpiku kostnego zostały zawieszone w 10 mL

44
kompletnego medium hodowlanego R2 w liczbie 2x106 komórek/mL. Do hodowli
zastosowano szalki dla hodowli komórkowej o zwiększonej przyczepności. Po 24 godzinach
nieadherentne komórki usunięto, do hodowli dodano 20 mL medium R2. W dniach trzecim,
szóstym i ósmym z hodowli usuwano 10 ml medium i dodawano 10 mL medium R2. Od 10-
go do 12-go dnia hodowli adherentne komórki zdejmowano z szalki przy użyciu
nieenzymatycznego odczynnika (ang. cell dissociation solution non-enzymatic) oraz
określono ich fenotyp (Ryc. 13).
3.2.1 Określenie fenotypu makrofagów i komórek dendrytycznych wyprowadzonych z
komórek szpiku kostnego myszy C3H/HeN (H-2k)
Fenotyp komórek prezentujących antygen (APCs) poprzez ocenę poziomu ekspresji
cząsteczek kostymulujących na powierzchni komórek dendrytycznych i makrofagów
określono przy pomocy analizy cytometrycznej (Ryc. 13). Komórki dendrytyczne i
makrofagi znakowano przeciwciałami anty-CD40, anty-CD86 oraz anty-MHC II gdzie
zastosowano PE jako fluorochrom, oraz przeciwciałami anty-CD11c, anty-CD80 oraz anty-
F4/80 gdzie jako fluorochrom zastosowano FITC. Dla każdego przeciwciała zastosowano
odpowiednią kontrolę izotypową. Fenotypowane komórki (5x106 komórek/mL) zawieszono
w objętości 100 µL buforu do znakowania przeciwciałami. Całość inkubowano w ciemności
przez 30 minut w temperaturze 4°C. Wszystkie przeciwciała były dodawane zgodnie z
wytycznymi producenta. Po zakończeniu inkubacji komórki opłukano trzykrotnie w buforze
do znakowania przeciwciałami i zawieszono w objętości 300 µl roztworu formaldehydu (1
%). Odczytu dokonywano przy użyciu cytometru przepływowego FACSCalibur (Becton
Dickinson, Franklin Lakes, NJ, USA) przy stałych ustawieniach czułości detektorów oraz
stałych parametrach kompensacji dla wszystkich badanych prób. Fenotyp dwóch populacji
modelowych komórek prezentujących antygen został przedstawiony w następnym rozdziale.
3.2.2 Badanie endocytozy antygenu modelowego przez modelowe komórki prezentujące
antygen
Pierwotne mysie komórki dendrytyczne i makrofagi naniesiono na 24-dołkową płytkę
hodowlaną (5x104 komórek/mL/dołek). Komórki inkubowano (5% CO2, 37 °C) przez 6
godzin w obecności koniugatu albuminy jaja kurzego z FITC (owoalbumina-FITC) o stężeniu
testowym 1 mg/mL w medium (odpowiednio R1T lub R2T) o różnej zawartości magnezu w
zakresie 0,1 mM – 5,0 mM i odpowiednich blokerów wapnia: werapamilu oraz TMB-8 o

45
testowym stężeniu 25 µM. Po zakończeniu inkubacji komórki przemyto trzykrotnie zimnym
(4 °C) buforem PBS z dodatkiem inaktywowanej FCS (2 %). W celu oznaczenia/wykluczenia
oddziaływań niespecyficznych eksperyment kontrolny prowadzono równolegle w
temperaturze 4 °C. Po dokonaniu półilościowej analizy fluorocytometrycznej przy użyciu
cytometru przepływowego FACSCalibur określono poziom pobrania owoalbuminy-FITC
przez pierwotne komórki prezentujące antygen. Sposób analizy uzyskanych wyników został
opisany w paragrafie 2.4.
3.3 Wpływu pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na
prezentację antygenu – kokultury komórkowe
Komórki prezentujące antygen: BMDCs lub BMMFs naniesiono na szalkę hodowlaną
w stężeniu 1x105 komórek/mL w objętości 10 mL medium hodowlanego (odpowiednio R1
lub R2) i traktowano inhibitorem replikacji DNA - mitomycyną C (S. caespitosus) (50
µL/mL). Całość inkubowano w ciemności przez 30 minut, 37 °C. Po zakończeniu inkubacji
komórki pięciokrotnie opłukiwano zimnym buforem PBS bez jonów wapnia i magnezu z
dodatkiem inaktywowanej FCS (10 %). Następnie modelowe komórki dendrytyczne lub
makrofagi umieszczano na 24- lub 96-o dołkowej płaskodennej płytce testowej w stężeniu
5x104 komórek/mL w medium hodowlanym odpowiednio R1 lub R2 z dodatkiem
konalbuminy (300 µg/mL). Po zakończeniu 6-o godzinnej inkubacji usunięto medium
zawierające konalbuminę, która nie została pochłonięta przez modelowe APCs. Komórki
odpłukano pięciokrotnie sterylnym, zimnym buforem PBS bez jonów wapnia i magnezu z
10% dodatkiem inaktywowanej FCS (wirowanie przy 125 g przez 10 min w temperaturze
4°C). Na płytkę testową na której znajdowały się modelowe komórki dendrytyczne lub
makrofagi naniesiono komórki linii limfocytarnej D10.G4.1, tak by ostatecznie stosunek
modelowych komórek prezentujących antygen do komórek linii D10.G4. 1 wynosił 1:5.
Kokulturę komórkową prowadzono w standardowych warunkach hodowlanych przez 24, 48,
72 godziny w celu określenia wzrostu komórek w kokulturze komórkowej lub 12 lub 24
godziny w celu oznaczenia cytokin w nadsączach komórkowych. Modelowe kokultury
prowadzono odpowiednio w mediach C1 i C2. Medium w którym prowadzono kokulturę
cechowała różna zawartość magnezu w zakresie 0,1 mM – 5,0 mM i obecność blokerów
wapnia: werapamilu oraz TMB-8 o testowym stężeniu 50 µM.

46
3.3.1 Oznaczenie aktywności metabolicznej komórek w kokulturze
Po zakończeniu inkubacji oceniono aktywność metaboliczną komórek w kokulturze
wykonując test MTT (według protokołu przedstawionego w paragrafie: 1).
Otrzymane wartości zmierzone dla poszczególnych prób testowych absorbancji (Ap)
przeliczono na wartości określające procent zahamowania aktywności metablocznej (według
protokołu dla komórek linii makrofagowej J774.E, paragraf: 2.3).
3.3.2 Oznaczenie końcowej liczby komórek w kokulturze
Po zakończeniu inkubacji komórki były przenoszone z płytki testowej do probówki
testowej i zawieszane w 500 µL buforu PBS bez jonów wapnia i magnezu. Do zawiesiny
komórek (50 µL) dodano 0,4 % roztwór błękitu trypanu w buforze PBS w stosunku 1:1. Po
upływie 3 minut 10 µL zawiesiny komórek umieszczono w komorze Thoma. Pod
mikroskopem świetlnym obliczono liczbę komórek żywych (nie zabarwionych).
* Przed rozpoczęciem kokultury modelowe komórki prezentujące antygen traktowano
inhibitorem replikacji DNA - mitomycyną C (S. caespitosus), przez co wzrost całkowitej
liczby komórek w kokulturze jest wynikiem proliferacji komórek linii limfocytarnej
D10.G4.1.
3.3.3 Oznaczenie cytokin w nadsączach komórkowych testem ELISA
Stężenie cytokin IL-2, IL-4, IL-10 oraz interferonu-γ w nadsączach komórkowych
oceniono testem immunoenzymatycznym ELISA (Ryc. 14). Do oznaczeń użyto zestawów
komercyjnych firmy Becton Dickinson: Mouse IL-2 ELISA Set nr kat. 555148, Mouse IL-4
ELISA Set nr kat. 555232, Mouse IL-10 ELISA Set nr kat. 555252, Mouse Interferon-γ
ELISA Set nr kat. 555138. Test ELISA został wykonana wg zaleceń producenta przy czym
szczegółowa procedura wykonania testu i analizy wyników została wykonana analogicznie do
tej przedstawionej w paragrafie 2.6.
W celu oznaczenia interferonu-gamma nie rozcieńczano nadsączy komórkowych, dla
oznaczenia pozostałych cytokin nadsącze rozcieńczono 200-krotnie w PBS bez jonów wapnia
i magnezu z dodatkiem FCS (10 %).

47
3.3.4 Wewnątrzkomórkowe znakowanie cytokin
Kokulturę komórek dendrytycznych i komórek limfocytarnych linii D10.G4.1
prowadzono w sposób opisany w paragrafie 3.3. Na sześć godzin przed zakończeniem
inkubacji do hodowli dodano brefedrynę A - związek hamujący aktywność aparatów
Golgiego (w stężeniu 10 µg/mL). Po zakończeniu inkubacji usunięto medium hodowlane,
komórki umieszczono w buforze do permabilizacji błony komórkowej i odwirowano
trzykrotnie (125 g, 10 min, 4 °C). Komórki w stężeniu 5x106 komórek/mL zawieszono w 100
µL buforu do znakowania przeciwciałami. Cząsteczki powierzchniowe: CD3 oraz CD4
znakowano przeciwciałami monoklonalnymi anty-CD3-FITC oraz anty-CD4-Alexa_647
zgodnie z wytycznymi producenta stosując wskazane kontrole izotypowe dla każdego z
przeciwciał (Ryc. 15). Po zakończeniu inkubacji w ciemności (30 minut, 4 °C) próbki
zawieszano każdorazowo w 5 mL buforu permeabilizującego i trzykrotnie odwirowano (125
g, 10 min, 4 °C). Ostatecznie osad komórek zawieszono w 100 µL buforu do permeabilizacji i
dodano przeciwciał monoklonalnych anty-IL4-PE lub anty-IL10-PE lub zgodnie z
wytycznymi producenta; zastosowano wskazane kontrole izotypowe dla każdego z
przeciwciał. Przeciwciała rozcieńczano w buforze do permeabilizacji komórek. Całość
inkubowano w ciemności przez 30 minut w temperaturze 4°C. Po końcowym odpłukaniu
komórki utrwalano zawieszając je w roztworze formaldehydu (1 %) w objętości 300 µL.
Odczytu dokonywano przy użyciu cytometru przepływowego FACSCalibur (Becton
Dickinson) przy stałych ustawieniach czułości detektorów oraz identycznej kompensacji dla
wszystkich badanych prób (Ryc. 15).
4. Zwierzęta doświadczalne
W eksperymentach przedstawionych w pracy doktorskiej wykorzystano:
1). Komórki granulocytarne wyizolowane z krwi obwodowej myszy szczepu Balb/C.
Zwierzęta (samice w wieku 4-6 tygodni) zakupiono w Centrum Medycyny Doświadczalnej
Uniwersytetu Medycznego w Białymstoku. Zwierzęta były hodowane w warunkach SPF
(ang. specyfic pathogen free) w Instytucie Immunologii i Terapii Doświadczalnej PAN we
Wrocławiu.
2). Komórki szpiku kostnego zostały pobrane od myszy szczapu C3H/HeN (H-2k). Zwierzęta
(samice w wieku 4-6 tygodni) zostały zakupione w Charles Rivers Laboratory (Monachium,
Niemcy).

48
Zwierzęta były hodowane w warunkach SPF w Instytucie Immunologii i Terapii
Doświadczalnej PAN we Wrocławiu.
Na przeprowadzenie doświadczenia uzyskano zgodę (51/03) I Lokalnej Komisji Etycznej ds.
Doświadczeń na Zwierzętach we Wrocławiu.
5. Analiza statystyczna wyników
Analizę statystyczną przeprowadzono metodami parametrycznej lub
nieparametrycznej analizy wariancji (ANOVA) w zależności od charakteru rozkładu danych
doświadczalnych. W przypadku uzyskania rozkładów normalnych i spełnienia warunków
homogenności wariancji przeprowadzono jednoczynnikową ANOVA (dla grup niezależnych)
lub ANOVA z powtarzanymi pomiarami (dla grup zależnych). Jako testu post-hoc użyto testu
t-Studenta. Dla porównań grup zależnych w analizie nieparametrycznej zastosowano test
Friedmana i test Wilcoxona z poprawką Bonferroniego, natomiast w przypadku analizy grup
niezależnych wykorzystano test Kruskala-Wallisa oraz test Mana-Whitneya. Za poziom
istotności we wszystkich analizach przyjęto p=0,05. Obliczeń dokonano przy pomocy
oprogramowania STATISTICA 6.0.
6. Wykaz zastosowanych odczynników
Odczynnik Pochodzenie
2-betamerkaptoetanol Sigma Aldrich, St. Louis, MO, USA,
6xDNA loading Dye Fermentas, St. Leon-Rot, Niemcy
Agaroza Prona, Madryt, Hiszpania
Azydek sodu Sigma Aldrich, St. Louis, MO, USA
Brefedryna A eBioscience, San Diego, CA, USA
Bromek etydyny Sigma Aldrich, St. Louis, MO, USA
Bufor do permeabilizacji (permeabilization
buffer (10X)) eBioscience, San Diego, CA,USA
Cell dissociation solution non-enzymatic Sigma Aldrich, St. Louis, MO, USA
Chlorek heparyny Sigma Aldrich, St. Louis, MO, USA
DMFA Sigma Aldrich, St. Louis, MO, USA
DMSO Sigma Aldrich, St. Louis, MO, USA
Etanol 96% POCh, Gliwice, Polska
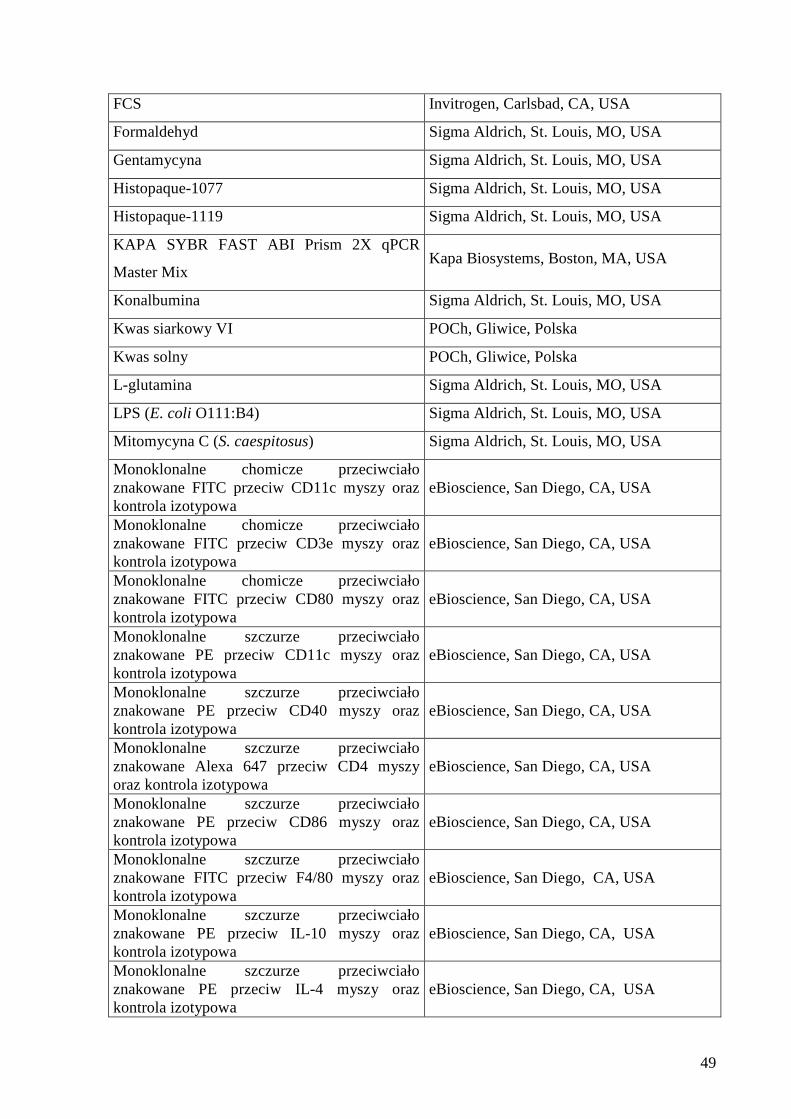
49
FCS Invitrogen, Carlsbad, CA, USA
Formaldehyd Sigma Aldrich, St. Louis, MO, USA
Gentamycyna Sigma Aldrich, St. Louis, MO, USA
Histopaque-1077 Sigma Aldrich, St. Louis, MO, USA
Histopaque-1119 Sigma Aldrich, St. Louis, MO, USA
KAPA SYBR FAST ABI Prism 2X qPCR
Master Mix Kapa Biosystems, Boston, MA, USA
Konalbumina Sigma Aldrich, St. Louis, MO, USA
Kwas siarkowy VI POCh, Gliwice, Polska
Kwas solny POCh, Gliwice, Polska
L-glutamina Sigma Aldrich, St. Louis, MO, USA
LPS (E. coli O111:B4) Sigma Aldrich, St. Louis, MO, USA
Mitomycyna C (S. caespitosus) Sigma Aldrich, St. Louis, MO, USA
Monoklonalne chomicze przeciwciało znakowane FITC przeciw CD11c myszy oraz kontrola izotypowa
eBioscience, San Diego, CA, USA
Monoklonalne chomicze przeciwciało znakowane FITC przeciw CD3e myszy oraz kontrola izotypowa
eBioscience, San Diego, CA, USA
Monoklonalne chomicze przeciwciało znakowane FITC przeciw CD80 myszy oraz kontrola izotypowa
eBioscience, San Diego, CA, USA
Monoklonalne szczurze przeciwciało znakowane PE przeciw CD11c myszy oraz kontrola izotypowa
eBioscience, San Diego, CA, USA
Monoklonalne szczurze przeciwciało znakowane PE przeciw CD40 myszy oraz kontrola izotypowa
eBioscience, San Diego, CA, USA
Monoklonalne szczurze przeciwciało znakowane Alexa 647 przeciw CD4 myszy oraz kontrola izotypowa
eBioscience, San Diego, CA, USA
Monoklonalne szczurze przeciwciało znakowane PE przeciw CD86 myszy oraz kontrola izotypowa
eBioscience, San Diego, CA, USA
Monoklonalne szczurze przeciwciało znakowane FITC przeciw F4/80 myszy oraz kontrola izotypowa
eBioscience, San Diego, CA, USA
Monoklonalne szczurze przeciwciało znakowane PE przeciw IL-10 myszy oraz kontrola izotypowa
eBioscience, San Diego, CA, USA
Monoklonalne szczurze przeciwciało znakowane PE przeciw IL-4 myszy oraz kontrola izotypowa
eBioscience, San Diego, CA, USA

50
Monoklonalne szczurze przeciwciało znakowane PE przeciw MHC II myszy oraz kontrola izotypowa
eBioscience, San Diego, CA, USA
Mouse IL-1β ELISA Set Becton Dickinson, Franklin Lakes, NJ, USA
Mouse IL-10 ELISA Set Becton Dickinson, Franklin Lakes, NJ, USA
Mouse IL-2 ELISA Set Becton Dickinson, Franklin Lakes, NJ, USA
Mouse IL-4 ELISA Set Becton Dickinson, Franklin Lakes, NJ, USA
Mouse IL-6 ELISA Set Becton Dickinson, Franklin Lakes, NJ, USA
Mouse Interferon - γ ELISA Set Becton Dickinson, Franklin Lakes, NJ, USA
Mouse TNF-α ELISA Set Becton Dickinson, Franklin Lakes, NJ, USA
MTT Sigma Aldrich, St. Louis,MS, USA
Ovoalbumina-FITC Invitrogen, Carlsbad, CA, USA
PBS bez Mg2+ i Ca2+ IITD PAN we Wrocławiu
Penicylina G Sigma Aldrich, St. Louis, MO, USA
PMA Sigma Aldrich, St. Louis, MO, USA
Rekombinowana mysia IL-1α Invitrogen, Carlsbad, CA, USA
Rekombinowana mysia IL-2 Invitrogen, Carlsbad, CA, USA
Rekombinowany mysi GM-CSF Invitrogen, Carlsbad, CA, USA
Rekombinowany mysi M-CSF Invitrogen, Carlsbad, CA, USA
RPMI 1640 (1) Sigma Aldrich, St. Louis, MO, USA
RPMI 1640 bez Mg2+ i Ca2+ oraz bez czerwieni
fenolowej (2) IITD PAN we Wrocławiu
SDS Sigma Aldrich, St. Louis, MO, USA
Siarczan magnezu Sigma Aldrich, St. Louis, MO, USA
Sól sodowa luminolu Sigma Aldrich, St. Louis, MO, USA
Streptomycyna Sigma Aldrich, St. Louis, MO, USA
TMB Sigma Aldrich, St. Louis, MO, USA
TMB-8 Sigma Aldrich, St. Louis, MO, USA
Total RNA Mini kit A&A Biotechnology, Gdynia, Polska
TRIzol Reagent Invitrogen, Carlsbad, CA, USA
Błękit trypanu (roztwór 0,4%) Sigma Aldrich, St. Louis, MO, USA
Trypsyny wieprzowa z EDTA (roztwór 0,4%) Sigma Aldrich, St. Louis, MO, USA
Tween 20 Sigma Aldrich, St. Louis, MO, USA

51
VerteKIT Novazym, Poznań, Polska
Werapamil Sigma Aldrich, St. Louis, MO, USA
6.1 Wykaz przygotowanych roztworów i mediów
Bufor lizujący (skład ilościowy)
225 ml DMFA, 275 ml wody demineralizowanej, 67,5 g SDS
Bufor do znakowania przeciwciałami
PBS bez Mg2+ i Ca2+ z dodatkiem inaktywowanej FCS (10%) i azydku sodu (1%).
Medium do izolacji (medium I1)
Podstawę medium do izolacji stanowiło RPMI 1640 (1) wzbogacone o inaktywowaną* FCS
(10%), L-glutaminę (2,0 mM) oraz antybiotyki: penicylinę G (40U/mL) oraz streptomycynę
(100 µg/mL)
Media hodowlane
1). Medium hodowlane dla komórek ustalonej linii makrofagowej J774.E (medium J1)
Podstawę medium hodowlanego stanowiło medium RPMI 1640 (1) wzbogacone o
wzbogacone o inaktywowaną* FCS (10%), L-glutaminę (2,0 mM) oraz gentamycynę (50
mg/L).
2). Medium hodowlane dla komórek ustalonej linii limfocytarnej D10.G4.1 (medium D1)
Podstawę medium hodowlanego stanowiło medium RPMI 1640 (1) wzbogacone o
inaktywowaną* FCS (10%), L-glutaminę (2,0 mM), 2-betamerkaptoetanol (0,05 mM) oraz
antybiotyki: penicylinę G (40 U/mL) oraz streptomycynę (100 µg/mL). Kompletne medium
hodowlane zawierało również rekombinowaną mysią IL-2 (2 ng/mL) oraz rekombinowaną
mysią IL-1α (10 pg/mL).
3). Medium hodowlane dla komórek dendrytycznych które wyprowadzono z komórek szpiku
kostnego myszy – BMDCs (medium R1)

52
Podstawę medium hodowlanego stanowiło: medium RPMI 1640 (1) wzbogacone o
inaktywowaną* FCS (10%), L-glutaminę (2.0 mM), 2-betamerkaptoetanol (0,05 mM),
antybiotyki: penicylinę G (40U/mL) i streptomycynę (100 µg/mL) oraz rekombinowaną
mysią cytokinę GM-CSF (20 ng/mL).
4). Medium hodowlane dla makrofagów które wyprowadzono z komórek szpiku kostnego
myszy – BMMFs (medium R2)
Podstawę kompletnego medium hodowlanego stanowiło: medium RPMI 1640 (1)
wzbogacone o inaktywowaną* FCS (10%), L-glutaminę (2,0 mM), 2-betamerkaptoetanol
(0,05 mM), antybiotyki: penicylinę G (40 U/mL) i streptomycynę (100 µg/mL) oraz
rekombinowaną mysią cytokinę M-CSF (40 ng/mL).
Media testowe
1). Medium testowe dla komórek ustalonej linii makrofagowej J774.E (medium J1T)
Podstawę medium testowego stanowiło medium RPMI 1640 (2) wzbogacone o wzbogacone o
inaktywowaną* FCS (10%), L-glutaminę (2,0 mM) oraz gentamycynę (50,0 mg/L).
2). Medium testowe dla komórek ustalonej linii limfocytarnej D10.G4.1 (medium D1T)
Podstawę medium testowego stanowiło medium RPMI 1640 (2) wzbogacone o
inaktywowaną* FCS (10%), L-glutaminę (2,0 mM), 2-betamerkaptoetanol (0,05 mM) oraz
antybiotyki: penicylinę G (40U/mL) oraz streptomycynę (100 µg/mL). Kompletne medium
hodowlane zawierało również rekombinowaną mysią IL-2 (2 ng/mL) oraz rekombinowaną
mysią IL-1α (10 pg/mL).
3). Medium testowe dla komórek dendrytycznych które wyprowadzono z komórek szpiku
kostnego myszy – BMDCs (medium R1T)
Podstawę medium testowego stanowiło: medium RPMI 1640 (2) wzbogacone o
inaktywowaną* FCS (10%), L-glutaminę (2,0 mM), 2-betamerkaptoetanol (0,05 mM),
antybiotyki: penicylinę G (40U/mL) i streptomycynę (100 µg/mL) oraz rekombinowaną
mysią cytokinę GM-CSF (20 ng/mL).

53
4). Medium testowe dla makrofagów które wyprowadzono z komórek szpiku kostnego myszy
– BMMFs (medium R2T)
Podstawę kompletnego medium testowego stanowiło: medium RPMI 1640 (2) wzbogacone o
inaktywowaną* FCS (10%), L-glutaminę (2,0 mM), 2-betamerkaptoetanol (0,05 mM),
antybiotyki: penicylinę G (40 U/mL) i streptomycynę (100 µg/mL) oraz rekombinowaną
mysią cytokinę M-CSF (40 ng/mL).
5). Medium testowe dla kokultury komórkowej: komórki D10.G4.1: BMDCs (medium C1)
Podstawę medium testowego stanowiło: medium RPMI 1640 (2) wzbogacone o
inaktywowaną* FCS (10%), L-glutaminę (2,0 mM), 2-betamerkaptoetanol (0,05 mM),
antybiotyki: penicylinę G (40U/mL) i streptomycynę (100 µg/mL), rekombinowaną mysią
cytokinę GM-CSF (20 ng/mL), rekombinowaną mysią IL-2 (2 ng/mL) oraz rekombinowaną
mysią IL-1α (10 pg/mL).
6). Medium testowe dla kokultury komórkowej: komórki D10.G4.1: BMMFs (medium C2)
Podstawę kompletnego medium testowego stanowiło: medium RPMI (2) wzbogacone o
inaktywowaną* FCS (10%), L-glutaminę (2,0 mM), 2-betamerkaptoetanol (0,05 mM),
antybiotyki: penicylinę G (40 U/mL) i streptomycynę (100 µg/mL), rekombinowaną mysią
cytokinę M-CSF (40 ng/mL), rekombinowaną mysią IL-2 (2 ng/mL) oraz rekombinowaną
mysią IL-1α (10 pg/mL).
Stężenie Mg2+ w medium testowym:
- 0,1 mM (medium deficytowe) – zawartość magnezu wynikała z dodatku FCS (10%)
- 1,0 mM (medium kontrolne) - zawartość magnezu wynikała z dodatku FCS (10%) i
siarczanu magnezu
- 5,0 mM (medium o wysokiej zawartości magnezu) - zawartość magnezu wynikała z
dodatku FCS (10%) i siarczanu magnezu
-
Stężenie Ca2+ w medium było stałe (około 0,25 mM) i wynikało z dodatku FCS (10%).
* FCS inaktywowano w temperaturze 56°C przez 30 minut.

54
IV WYNIKI
1. Wpływ blokerów wapnia na potencjał proliferacyjny komórek ustalonych linii
komórkowych: J774.E oraz D10.G4.1 -wyznaczenie IC50 badanych związków
IC50 werapamilu dla komórek D10.G4.1 wyniosła 58,2 ± 7,5 µM, natomiast dla
komórek J774.E wartość ta wynosiła około 48,3 ± 7,6 µM. IC50 dla TMB-8 to około 37,3 ±
8,1 µM podczas gdy analogiczna wartość dla modelowych komórek makrofagowych
kształtowała się na poziomie 32,6 ± 5,4 µM. IC50 werapamilu było zdecydowanie wyższe niż
IC50 dla TMB-8 w obu modelach komórkowych, co sugeruje że dopływ wapnia z zasobów
wewnątrzkomórkowych jest ważniejszy dla właściwego funkcjonowania i wzrostu komórek
niż wapń pozakomórkowy. Oceniono, że czas podwojenia populacji komórek makrofagowych
J774.E w standardowych warunkach hodowlanych in vitro to około 18-22 godziny;
analogiczna wartość dla komórek limfocytarnych D10.G4.1 to około 22-28 godzin. Czas
podwojenia populacji komórek wpłynął na określenie punktu czasowego w którym
wyznaczono IC50 dla każdego z blokerów wapnia (48 godzin hodowli w obecności
werapamilu i/lub TMB-8). Uzyskane wyniki sugerują, że im wyższy jest „początkowy”
potencjał proliferacyjny komórki tym IC50 dla badanych blokerów wapnia jest niższe a
niedobór wapnia wywiera większy efekt na właściwe funkcjonowanie i wzrost komórek.
Przedstawione dane są średnią wartością z co najmniej czterech niezależnych eksperymentów
± odchylenie standardowe.
2. Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na aktywność
modelowych komórek fagocytarnych in vitro
2.1 Ocena wpływu magnezu i wapnia na potencjał proliferacyjny komórek linii J774.E
Długotrwały niedobór magnezu w medium hodowlanym skutkował zmniejszeniem
potencjału proliferacyjnego komórek J774.E w ponad 40%. Wysokie pozakomórkowe
stężenie magnezu (5,0 mM) nie miało znaczącego wpływu na potencjał proliferacyjny
komórek J774.E. Stwierdzono, że obecność blokerów wapnia– werapamilu i TMB-8 istotnie
wpływała na wzrost komórek J774.E; efekt ten był szczególnie widoczny w trzecim dniu
hodowli w zmiennych warunkach dostępności magnezu i wapnia (tabela 3, Ryc. 16).
Inhibitor wolnych kanałów wapniowych w błonie komórkowej – werapamil w istotny sposób
zmniejszył (o około 25%) potencjał proliferacyjny komórek, przy czym efekt ten był
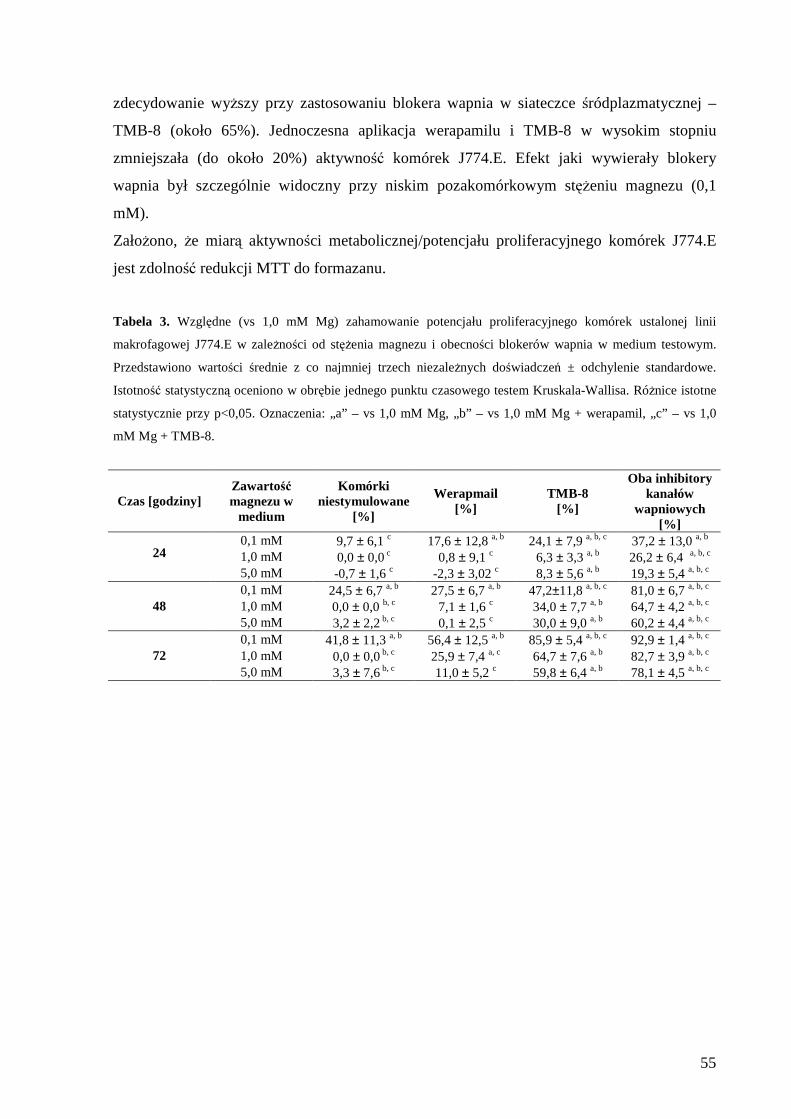
55
zdecydowanie wyższy przy zastosowaniu blokera wapnia w siateczce śródplazmatycznej –
TMB-8 (około 65%). Jednoczesna aplikacja werapamilu i TMB-8 w wysokim stopniu
zmniejszała (do około 20%) aktywność komórek J774.E. Efekt jaki wywierały blokery
wapnia był szczególnie widoczny przy niskim pozakomórkowym stężeniu magnezu (0,1
mM).
Założono, że miarą aktywności metabolicznej/potencjału proliferacyjnego komórek J774.E
jest zdolność redukcji MTT do formazanu.
Tabela 3. Względne (vs 1,0 mM Mg) zahamowanie potencjału proliferacyjnego komórek ustalonej linii
makrofagowej J774.E w zależności od stężenia magnezu i obecności blokerów wapnia w medium testowym.
Przedstawiono wartości średnie z co najmniej trzech niezależnych doświadczeń ± odchylenie standardowe.
Istotność statystyczną oceniono w obrębie jednego punktu czasowego testem Kruskala-Wallisa. Różnice istotne
statystycznie przy p<0,05. Oznaczenia: „a” – vs 1,0 mM Mg, „b” – vs 1,0 mM Mg + werapamil, „c” – vs 1,0
mM Mg + TMB-8.
Czas [godziny] Zawartość magnezu w
medium
Komórki niestymulowane
[%]
Werapmail [%]
TMB- 8 [%]
Oba inhibitory kanałów
wapniowych [%]
0,1 mM 9,7 ± 6,1 c 17,6 ± 12,8 a, b 24,1 ± 7,9 a, b, c 37,2 ± 13,0 a, b 1,0 mM 0,0 ± 0,0 c 0,8 ± 9,1 c 6,3 ± 3,3 a, b 26,2 ± 6,4 a, b, c 24
5,0 mM -0,7 ± 1,6 c -2,3 ± 3,02 c 8,3 ± 5,6 a, b 19,3 ± 5,4 a, b, c 0,1 mM 24,5 ± 6,7 a, b 27,5 ± 6,7 a, b 47,2±11,8 a, b, c 81,0 ± 6,7 a, b, c 1,0 mM 0,0 ± 0,0 b, c 7,1 ± 1,6 c 34,0 ± 7,7 a, b 64,7 ± 4,2 a, b, c 48 5,0 mM 3,2 ± 2,2 b, c 0,1 ± 2,5 c 30,0 ± 9,0 a, b 60,2 ± 4,4 a, b, c 0,1 mM 41,8 ± 11,3 a, b 56,4 ± 12,5 a, b 85,9 ± 5,4 a, b, c 92,9 ± 1,4 a, b, c 1,0 mM 0,0 ± 0,0 b, c 25,9 ± 7,4 a, c 64,7 ± 7,6 a, b 82,7 ± 3,9 a, b, c 72 5,0 mM 3,3 ± 7,6 b, c 11,0 ± 5,2 c 59,8 ± 6,4 a, b 78,1 ± 4,5 a, b, c

56
2.2 Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na
produkcj ę reaktywnych form tlenu (RFT) przez modelowe komórki fagocytarne in vitro
Analizując kinetykę produkcji reaktywnych form tlenu w czasie przez dwa typy
komórek fagocytarnych – granulocyty (Ryc. 17A) oraz komórki makrofagowe J774.E (Ryc.
17B) stwierdzono, że niskie pozakomórkowe stężenie magnezu (0,1 mM) w istotny sposób
wzmacniało produkcję reaktywnych form tlenu w porównaniu do warunku podstawowego
(1,0 mM Mg) w obu modelach komórkowych. Granulocyty jak również komórki J774.E
hodowane w medium o normalnym stężeniu magnezu (1,0 mM) produkowały prawie o 30%
mniej pochodnych tlenowych niż te hodowane w medium niedoborowym (0,1 mM Mg).
Wysokie pozakomórkowe stężenie magnezu (5,0 mM) nie miało istotnego wpływu na
produkcję RFT w odniesieniu do warunku podstawowego (1,0 mM Mg). Werapamil hamował
produkcję RFT w istotnym stopniu przy czym dopływ wapnia z siateczki śródplazmatycznej
wydawał się istotniejszy dla wydajności produkcji RFT. Kiedy werapamil i TMB-8 były
dodawane osobno lub w kombinacji, wydajność wywołanej PMA produkcji pochodnych
tlenowych była znacząco niższa w odniesieniu do warunku podstawowego (1,0 mM Mg,
normalna dostępność wapnia) w obu modelach komórkowych. Można wnioskować, że niskie
pozakomórkowe stężenie magnezu (0,1 mM) wzmacniało efekt jaki wywierały blokery
wapnia na indukowaną produkcję RFT przez komórki fagocytarne in vitro. Przy niskim
stężeniu magnezu pozakomórkowego (0,1 mM Mg) produkcja RFT przez granulocyty była
ograniczona w około 15% przez werapamil, 20% przez TMB-8 i 40% przez blokery wapnia
aplikowane razem w stosunku do warunku kontrolnego (0,1 mM Mg); analogiczne wartości
hamowania odpowiedzi dla komórek J774.E wynoszą około: 20%, 30% oraz 40%.
Jest istotna różnica w indukowanej produkcji reaktywnych form tlenu pomiędzy dwoma
typami komórek fagocytarnych. W podstawowym warunku eksperymentalnym (1,0 mM Mg,
bez dodatku blokerów wapnia) komórki makrofagowej linii ustalonej J774.E produkowały
RFT na poziomie około 2300 AUC podczas gdy granulocyty produkowały RFT na poziomie
około 3500 AUC.

57
2.3 Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na
endocytozę antygenu modelowego przez komórki makrofagowe J774.E
Zbadano wpływ pozakomórkowego stężenia magnezu oraz ograniczonej dostępności
wapnia na pobranie antygenu modelowego – albuminy jaja kurzego (koniugat białka z
fluorochromem - FITC) przez modelowe makrofagi. Wykazano, że pozakomórkowe stężenie
magnezu nie ma istotnego wpływu na aktywność endocytarną komórek J774.E (Ryc. 18B).
Blokery wapnia, werapamil i TMB-8, w podobnym stopniu ograniczyły endocytozę
przeprowadzaną przez komórki J774.E o około 40-45 %, przy czym ten efekt był niezależny
od pozakomórkowego stężenia magnezu. Jednoczesna obecność werapamilu i TMB-8
obniżała endocytozę nawet o 50% w odniesieniu do warunku podstawowego (1,0 mM Mg).
2.4 Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na
wywołaną LPS produkcję cytokin prozapalnych (IL-1ββββ, IL-6 oraz TNF-αααα) przez
komórki makrofagowe J774.E
Pozakomórkowe stężenie magnezu nie miało istotnego wpływu na spontaniczną
produkcję cytokin IL-1β, IL-6 lub TNF-α (Ryc. 4). Komórki J774.E hodowane w medium o
obniżonym stężeniu magnezu (0,1 mM) produkowały o około 15% więcej IL-1β niż komórki
hodowane w medium o normalnej (1,0 mM) zawartości magnezu (Ryc. 19A).
Stymulacja LPS powodowała znaczący wzrost stężenia każdej z badanych cytokin w
nadsączach komórkowych, przy czym dodatek blokerów wapnia w istotny sposób ograniczał
indukowaną LPS produkcję cytokin prozapalnych przez komórki J774.E. Efekt wywierany
przez inhibitor kanałów wapniowych w błonie komórkowej na indukowaną LPS produkcję
cytokin prozapalnych był w części wzmacniany przez wysokie pozakomórkowe stężenie
magnezu (5,0 mM). Efekt blokera wapnia w siateczce śródplazmatycznej na wywoływaną
LPS produkcję cytokin IL-1β, IL-6 oraz TNF-α był niezależny od zawartości magnezu w
medium testowym. Werapamil, zależnie od dawki, w istotny sposób zmniejszał produkcję IL-
1β. Dodatek werapamilu (100 µM) obniżył stężenie IL-1β w nadsączu komórkowym z około
300pg/ml do 230 pg/ml w odniesieniu do warunku kontrolnego (1,0 mM Mg, LPS). Efekt
werapamilu był wzmacniany przez wysokie pozakomórkowe stężenie magnezu (5,0 mM).
Werapamil w stężeniu testowym 50 µM przy wysokim stężeniu magnezu w medium (5,0
mM) obniżał produkcję IL-1β o około 25 % w odniesieniu do warunku kontrolnego
(werapamil 50 µM, LPS, 1,0mM Mg) podczas gdy werapamil w stężeniu 100 µM (LPS,
5,0mM Mg) obniżał stężenie tej cytokiny w nadsączu komórkowy prawie o połowę w

58
porównaniu do warunku kontrolnego (LPS, werapamil 100 µM, 1,0 mM Mg). TMB-8 (50
µM, 100 µM) zmniejszał kilkukrotnie indukowaną LPS produkcję IL-1β, przy czym efekt ten
był niezależny od pozakomórkowego stężenia magnezu. Aplikacja werapamilu i TMB-8
obniżała wywołaną LPS produkcję IL-1β nawet w około 85% w odniesieniu do warunku
podstawowego (LPS, 1,0 mM Mg); efekt ten był niezależny od stężenia magnezu w medium
testowym.
Stężenie magnezu w medium testowym nie miało istotnego wpływu na indukowaną LPS
produkcję IL-6 przez komórki linii J774.E (Ryc. 19B). Werapamil w stężeniu testowym 100
µM zmniejszył indukowaną LPS produkcję IL-6 o około 25 % w stosunku do warunku
kontrolnego (LPS, 1,0 mM Mg). Efekt jaki wywierał werapamil na produkcję IL-6 był
podwyższany przez wysokie pozakomórkowe stężenie magnezu. TMB-8 (50 µM, 100 µM)
zmniejszał w około 80-85% wywołaną LPS produkcję IL-6, przy czym efekt ten był, jak w
przypadku indukowanej LPS produkcji IL-1β, niezależny od pozakomórkowego stężenia
magnezu. Jednoczesny zastosowanie obu blokerów wapnia obniżało stężenie IL-6 w nadsączu
komórkowym nawet o 90 % i efekt ten był niezależny od pozakomórkowego stężenia
magnezu.
Pozakomórkowe stężenie magnezu nie miało istotnego wpływu na poziom TNF-α w
nadsączach komórkowych (Ryc. 19C). Werapamil obniżał indukowaną LPS produkcję TNF-
α przez komórki linii makrofagowej nawet o 30 % w odniesieniu do warunku kontrolnego
(LPS, 1,0 mM Mg) przy czym efekt ten był niezależny od pozakomórkowego stężania
magnezu. TMB-8 obniżał poziom TNF-α (TMB-8 100 µM, LPS, 1,0 mM Mg) nawet o 60 %
w odniesieniu do warunku kontrolnego (LPS, 1,0 mM Mg) i efekt ten jak w przypadku IL-1β
oraz IL-6 był niezależny pod pozakomórkowego poziomu magnezu. Jednoczesna aplikacja
werapamilu i TMB-8 powodowała obniżenie indukowanej LPS produkcji TNF-α nawet w 90
% i ponownie efekt ten był niezależny od zawartości magnezu w medium testowym

59
2.5 Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na
wywołaną LPS transkrypcję genów IL-1ββββ, IL-6 oraz TNF-αααα przez komórki
makrofagowe J774.E
Stymulacja LPS zwiększyła transkrypcję genów dla IL-1β, IL-6 oraz TNF-α w
porównaniu do prób niestymulowanych. Pozakomórkowe stężenie magnezu nie miało
istotnego wpływu na spontaniczną lub wywołaną LPS transkrypcję badanych genów.
Inhibitor kanałów wapniowych w błonie komórkowej w istotnym stopniu hamował
indukowaną LPS ekspresję (mRNA) wskazanych genów, przy czym dopływ wapnia ze źródeł
wewnątrzkomórkowych wydawał się istotniejszy w kontekście transkrypcji genów cytokin
prozapalnych. Efekt jaki wywoływały werapamil i TMB-8 był niezależny od
pozakomórkowego stężenia magnezu.
Werapamil w stężeniu testowym 100 µM obniżył indukowany LPS poziom transkryptu dla
IL-1α o około 45 % w odniesieniu do warunku podstawowego (1,0 mM Mg, LPS)(Ryc. 20A).
Działanie werapamilu było całkowicie niezależne od stężenia magnezu w medium testowym.
TMB-8 (50 µM, 100 µM) obniżał wywoływany LPS poziom mRNA badanego genu o 60-80
% w odniesieniu do warunku kontrolnego (LPS, 1,0 mM). Jednoczesna aplikacja dwóch
blokerów wapnia obniżyła indukowaną transkrypcję IL-1α nawet w 95 % w odniesieniu do
warunku kontrolnego (LPS, 1,0 mM). Efekt ten był 1) zależny od stężenia testowego
inhibitorów kanałów wapniowych 2) niezależny od pozakomórkowego stężenia magnezu.
Werapamil (50 µM, 100 µM) obniżył wywołany LPS poziom mRNA IL-6 prawie
dwukrotnie w porównaniu do warunku kontrolnego (LPS, 1,0 mM) (Ryc. 20B). TMB-8
obniżył poziom transkryptu badanago genu w 80% w stosunku do warunku kontrolnego
(LPS, 1,0 mM). Jednoczesna aplikacja werapamilu i TMB-8 ograniczała wywołaną LPS
transkrypcję genu IL-6 w około 90% w stosunku warunku kontrolnego (LPS, 1,0 mM).
Werapamil (100 µM) i TMB-8 (100 µM) obniżyły indukowaną LPS transkrypcję TNF-α w
około 50% i 65% w odniesieniu do warunku kontrolnego (LPS, 1,0 mM) (Ryc. 20C).
Podobnie jak w przypadku cytokin IL-1β, IL-6, jednoczesna aplikacja werapamilu i TMB-8 w
nawet 90% obniżała wywoływaną LPS transkrypcję genu TNF-α w stosunku do warunku
kontrolnego (LPS, 1,0 mM). Efekt ten, podobnie jak poprzednio, był całkowicie niezależny
od pozakomórkowego stężenia magnezu.

60
3. Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na wzrost
komórek limfocytarnych oraz ich aktywację przez komórki prezentujące antygen:
komórki dendrytyczne oraz makrofagi
3.1 Ocena wpływu magnezu i wapnia na wzrost komórek limfocytarnych D10.G4.1 w
monokulturze
Niskie pozakomórkowe stężenie magnezu (0,1 mM) ograniczyło aktywność
metaboliczną komórek limfocytarnych o około 10% w porównaniu do komórek hodowanych
w medium o normalnej zawartości magnezu (1,0 mM) dopiero w trzecim dniu eksperymentu.
Wysoki poziom magnezu (5,0 mM) w medium testowym nie miał istotnego wpływu na
aktywność metaboliczną komórek limfocytarnych. W ciągu pierwszych 24 godzin
eksperymentu nie odnotowano istotnego wpływu blokerów wapnia na aktywność
metaboliczną komórek D10.G4.1 (tabela 4). Wykazano, że podobnie jak w przypadku
komórek makrofagowych J774.E, obecność blokerów wapnia, werapamilu jak również TMB-
8, istotnie zmniejsza potencjał proliferacyjny komórek limfocytarnych D10.G4.1 w trzecim
dniu eksperymentu. Werapamil ograniczył potencjał proliferacyjny komórek nawet o 25-30 %
w odniesieniu do warunku podstawowego (1,0 mM Mg). TMB-8 ograniczał potencjał
proliferacyjny komórek D10.G4.1 niemal dwukrotnie w stosunku do komórek hodowanych w
medium o normalnej zawartości magnezu (1,0 mM Mg). Jednoczesny dodatek werapamilu i
TMB-8 ograniczał aktywność metaboliczną komórek w około 70 % w porównaniu do
warunku podstawowego (1,0 mM Mg). Efekt jaki wywierały blokery wapnia na aktywność
metaboliczną komórek D10.G4.1 w monokulturze był całkowicie niezależny od
pozakomórkowego stężenia magnezu.
Założono, że miarą aktywności metabolicznej/potencjału proliferacyjnego komórek D10.G4.1
jest zdolność redukcji MTT do formazanu.
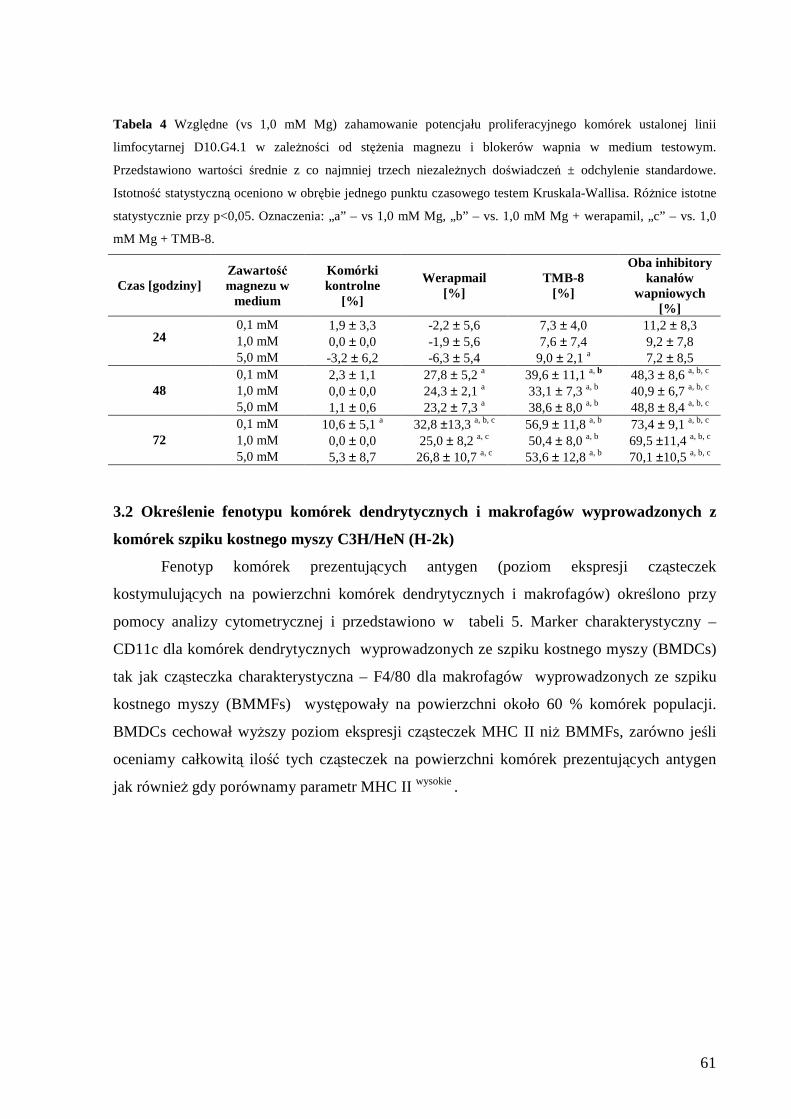
61
Tabela 4 Względne (vs 1,0 mM Mg) zahamowanie potencjału proliferacyjnego komórek ustalonej linii
limfocytarnej D10.G4.1 w zależności od stężenia magnezu i blokerów wapnia w medium testowym.
Przedstawiono wartości średnie z co najmniej trzech niezależnych doświadczeń ± odchylenie standardowe.
Istotność statystyczną oceniono w obrębie jednego punktu czasowego testem Kruskala-Wallisa. Różnice istotne
statystycznie przy p<0,05. Oznaczenia: „a” – vs 1,0 mM Mg, „b” – vs. 1,0 mM Mg + werapamil, „c” – vs. 1,0
mM Mg + TMB-8.
Czas [godziny] Zawartość magnezu w
medium
Komórki kontrolne
[%]
Werapmail [%]
TMB- 8 [%]
Oba inhibitory kanałów
wapniowych [%]
0,1 mM 1,9 ± 3,3 -2,2 ± 5,6 7,3 ± 4,0 11,2 ± 8,3 1,0 mM 0,0 ± 0,0 -1,9 ± 5,6 7,6 ± 7,4 9,2 ± 7,8 24
5,0 mM -3,2 ± 6,2 -6,3 ± 5,4 9,0 ± 2,1 a 7,2 ± 8,5 0,1 mM 2,3 ± 1,1 27,8 ± 5,2 a 39,6 ± 11,1 a, b 48,3 ± 8,6 a, b, c 1,0 mM 0,0 ± 0,0 24,3 ± 2,1 a 33,1 ± 7,3 a, b 40,9 ± 6,7 a, b, c 48 5,0 mM 1,1 ± 0,6 23,2 ± 7,3 a 38,6 ± 8,0 a, b 48,8 ± 8,4 a, b, c 0,1 mM 10,6 ± 5,1 a 32,8 ±13,3 a, b, c 56,9 ± 11,8 a, b 73,4 ± 9,1 a, b, c 1,0 mM 0,0 ± 0,0 25,0 ± 8,2 a, c 50,4 ± 8,0 a, b 69,5 ±11,4 a, b, c 72 5,0 mM 5,3 ± 8,7 26,8 ± 10,7 a, c 53,6 ± 12,8 a, b 70,1 ±10,5 a, b, c
3.2 Określenie fenotypu komórek dendrytycznych i makrofagów wyprowadzonych z
komórek szpiku kostnego myszy C3H/HeN (H-2k)
Fenotyp komórek prezentujących antygen (poziom ekspresji cząsteczek
kostymulujących na powierzchni komórek dendrytycznych i makrofagów) określono przy
pomocy analizy cytometrycznej i przedstawiono w tabeli 5. Marker charakterystyczny –
CD11c dla komórek dendrytycznych wyprowadzonych ze szpiku kostnego myszy (BMDCs)
tak jak cząsteczka charakterystyczna – F4/80 dla makrofagów wyprowadzonych ze szpiku
kostnego myszy (BMMFs) występowały na powierzchni około 60 % komórek populacji.
BMDCs cechował wyższy poziom ekspresji cząsteczek MHC II niż BMMFs, zarówno jeśli
oceniamy całkowitą ilość tych cząsteczek na powierzchni komórek prezentujących antygen
jak również gdy porównamy parametr MHC II wysokie .
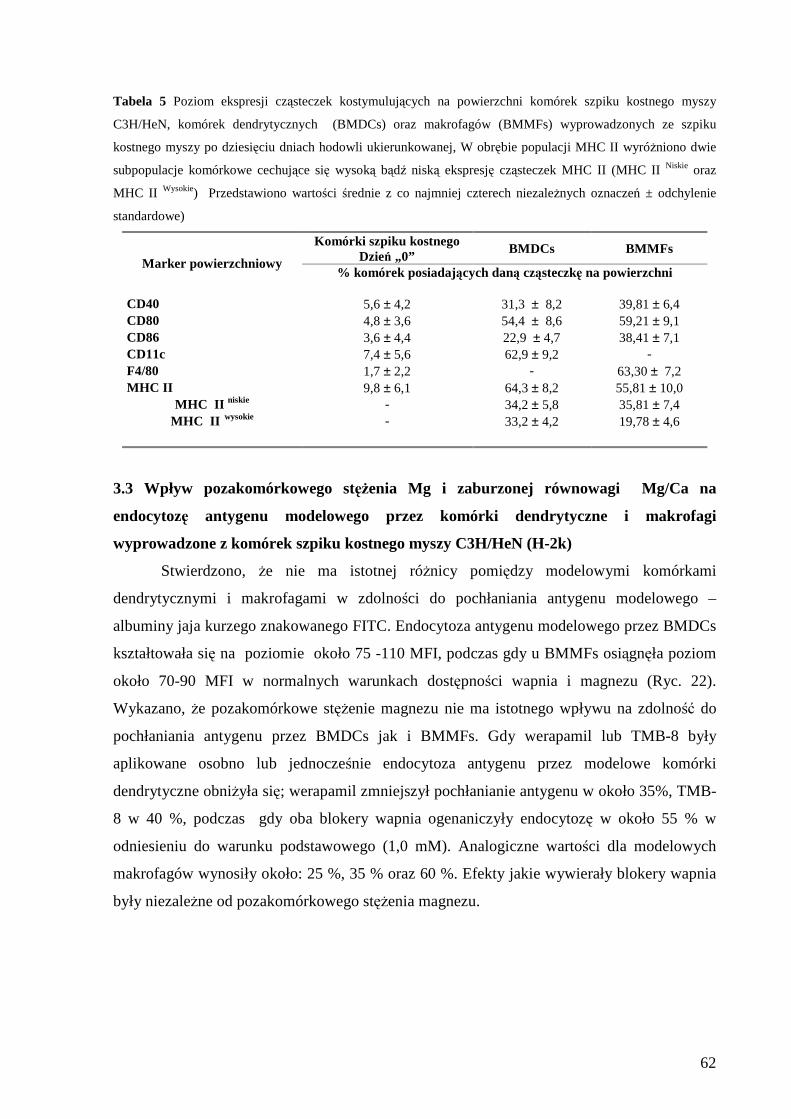
62
Tabela 5 Poziom ekspresji cząsteczek kostymulujących na powierzchni komórek szpiku kostnego myszy
C3H/HeN, komórek dendrytycznych (BMDCs) oraz makrofagów (BMMFs) wyprowadzonych ze szpiku
kostnego myszy po dziesięciu dniach hodowli ukierunkowanej, W obrębie populacji MHC II wyróżniono dwie
subpopulacje komórkowe cechujące się wysoką bądź niską ekspresję cząsteczek MHC II (MHC II Niskie oraz
MHC II Wysokie) Przedstawiono wartości średnie z co najmniej czterech niezależnych oznaczeń ± odchylenie
standardowe)
Komórki szpiku kostnego Dzień „0”
BMDCs BMMFs Marker powierzchniowy
% komórek posiadających daną cząsteczkę na powierzchni
CD40 5,6 ± 4,2 31,3 ± 8,2 39,81 ± 6,4 CD80 4,8 ± 3,6 54,4 ± 8,6 59,21 ± 9,1 CD86 3,6 ± 4,4 22,9 ± 4,7 38,41 ± 7,1 CD11c 7,4 ± 5,6 62,9 ± 9,2 - F4/80 1,7 ± 2,2 - 63,30 ± 7,2 MHC II 9,8 ± 6,1 64,3 ± 8,2 55,81 ± 10,0
MHC II niskie - 34,2 ± 5,8 35,81 ± 7,4 MHC II wysokie - 33,2 ± 4,2 19,78 ± 4,6
3.3 Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na
endocytozę antygenu modelowego przez komórki dendrytyczne i makrofagi
wyprowadzone z komórek szpiku kostnego myszy C3H/HeN (H-2k)
Stwierdzono, że nie ma istotnej różnicy pomiędzy modelowymi komórkami
dendrytycznymi i makrofagami w zdolności do pochłaniania antygenu modelowego –
albuminy jaja kurzego znakowanego FITC. Endocytoza antygenu modelowego przez BMDCs
kształtowała się na poziomie około 75 -110 MFI, podczas gdy u BMMFs osiągnęła poziom
około 70-90 MFI w normalnych warunkach dostępności wapnia i magnezu (Ryc. 22).
Wykazano, że pozakomórkowe stężenie magnezu nie ma istotnego wpływu na zdolność do
pochłaniania antygenu przez BMDCs jak i BMMFs. Gdy werapamil lub TMB-8 były
aplikowane osobno lub jednocześnie endocytoza antygenu przez modelowe komórki
dendrytyczne obniżyła się; werapamil zmniejszył pochłanianie antygenu w około 35%, TMB-
8 w 40 %, podczas gdy oba blokery wapnia ogenaniczyły endocytozę w około 55 % w
odniesieniu do warunku podstawowego (1,0 mM). Analogiczne wartości dla modelowych
makrofagów wynosiły około: 25 %, 35 % oraz 60 %. Efekty jakie wywierały blokery wapnia
były niezależne od pozakomórkowego stężenia magnezu.

63
3.4 Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na
aktywność komórkową oraz liczbę komórek w kokulturze komórkowej
Proliferację komórek w każdym modelu doświadczalnym oceniono porównując liczbę
komórek po 24, 48 lub 72 godzinach względem początkowej ich liczby w dniu rozpoczęcia
eksperymentu. Przed rozpoczęciem kokultury, komórki prezentujące antygen traktowano
inhibitorem replikacji DNA - mitomycyną C (S. caespitosus), przez co wzrost całkowitej
liczby komórek w kokulturze był wynikiem proliferacji komórek linii limfocytarnej
D10.G4.1. Kokulturę komórkową tworzyły komórki prezentujące antygen (komórki
dendrytyczne (BMDCs) lub makrofagi (BMMFs) wyprowadzone ze szpiku kostnego myszy)
oraz komórki linii limfocytarnej D10.G4.1 hodowane w stosunku wyjściowym 1:5.
Całkowita liczba komórek w kokulturze w dniu rozpoczęcia eksperymentu wynosiła 15x104
/ml właściwego medium testowego.
Efekty wywierane przez werapamil i TMB-8 na aktywność metaboliczną (założono, że miarą
aktywności metabolicznej jest zdolność redukcji MTT do formazanu) i całkowitą liczbę
komórek w kokulturze były analogiczne. W pierwszych 24 godzinach eksperymentu obecność
blokerów wapnia nie miała istotnego wpływu na aktywność metaboliczną (Ryc. 23B, 23D)
ani całkowitą liczbę komórek (Ryc. 23A, 23C) w żadnym z dwóch modeli kokultur
komórkowych. Znaczący niekorzystny wpływ werapamilu na aktywność metaboliczną
kokultur komórkowych odnotowano w trzecim dniu eksperymentu (Ryc. 23B, 23D) przy
czym dopływ wapnia z wewnątrzkomórkowego źródła wydaje się istotniejszy, co
zaobserwowano przy użyciu TMB-8 i znacznym obniżeniu jednocześnie aktywności
metabolicznej jak i całkowitej liczny komórek w odniesieniu do warunku podstawowego (1,0
mM Mg) w każdym modelu kokultury komórkowej. Wpływ blokerów wapnia na aktywność
komórkowąmodelowych kokultur był niezależny od pozakomórkowego stężenia magnezu
(Ryc. 23B, 23D). Niskie pozakomórkowe stężenie magnezu (0,1 mM) w istotny sposób
zmniejsza całkowitą liczbę komórek już w drugim dniu eksperymentu (Ryc. 23A, 23C) w
porównaniu do warunku kontrolnego (1,0 mM); efekt ten jest również obserwowany w
trzecim dniu eksperymentu. Negatywny wpływ inhibitorów kanałów wapniowych na
całkowitą liczbę komórek jest najlepiej widoczny w trzecim dniu eksperymentu (jednoczesny
dodatek werapamilu i TMB-8 obniża całkowitą liczbę komórek o około 30% w modelowych
kokulturach w porównaniu do warunku podstawowego (1,0 mM Mg)). Negatywny efekt jaki
wywierają inhibitory wapnia na proliferację komórek D10.G4.1 jest wzmacniany przez niskie
stężenie magnezu w medium testowym (Ryc. 23A, 23C). W trzecim dniu eksperymentu
kokultura komórkowa w której komórki prezentujące antygen są reprezentowane przez

64
modelowe komórki dendrytyczne (Ryc. 23B) cechuje się wyższą aktywnością metaboliczną
(gęstość optyczna próby wynosi ok. 0,7 przy 1,0 mM Mg) w odniesieniu do modelu gdzie
komórkami prezentującymi antygen są modelowe makrofagi (gęstość optyczna próby wynosi
ok. 0,55 przy 1,0 mM Mg) (Ryc. 23D). Również całkowita liczba komórek w kokulturze z
komórkami dendrytycznymi w trzecim dniu doświadczenia (Ryc. 23A) jest wyższa (około
6,5x105 /ml przy 1,0 mM Mg) niż w drugim modelu (Ryc. 23C) gdzie antygen jest
prezentowany przez modelowe makrofagi (około 5x105/ml przy 1,0 mM Mg).

65
3.5 Wpływ pozakomórkowego stężenia Mg i zaburzonej równowagi Mg/Ca na
produkcj ę cytokin IL-2, IL-4, IL-10 oraz interferonu- γγγγ w modelowych kokulturach
komórkowych
Stężenie cytokin w nadsączach z kokultur oceniono po 12 i 24 godzinach hodowli,
dlatego że w tych punktach czasowych blokery wapnia nie miały istotnego wpływu na
całkowitą liczbę komórek ani na aktywność metaboliczną w żadnym z dwóch modeli
kokultury komórkowej. Nie udało się ocenić poziomu interferonu-γ w medium testowym
testem ELISA o czułości 10 pg/ml. Nie wskazano różnic w stężeniu IL-2 ze względu na
wysoki poziom wyjściowy IL-2 w medium testowym (komórki linii limfocytarnej D10.G4.1
wymagają dla właściwego wzrostu i aktywności obecności IL-2 w stężeniu 2 ng/ml).
3.5.1 Wpływ zaburzonej równowagi magnezowo-wapniowej na produkcj ę i sekrecję
cytokin IL-2, IL-4, IL-10 oraz interferonu- γγγγ w kokulturze komórkowej: komórki
D10.G4.1 : modelowe komórki dendrytyczne (BMDCs)
Niskie pozakomórkowe stężenie magnezu (0,1 mM) nie miało istotnego wpływu na
produkcję IL-4 ani IL-10 (Ryc. 24A, 24B). Stwierdzono, że w ciągu 12 i 24 godzin
eksperymentu wysokie pozakomórkowe stężenie magnezu (5,0 mM) w istotnym stopniu
obniżyło poziom IL-4 w medium testowym, odpowiednio o około 10% i 40% w odniesieniu
do warunku podstawowego (1,0 mM) (Ryc. 24A). W ciągu pierwszych 12 godzin werapamil
nie wpływał istotnie na produkcję IL-4, podczas gdy TMB-8 obniżył poziom tej cytokiny w
medium testowym o około 45 %, a blokery wapnia dodane jednocześnie obniżyły produkcję
IL-4 o około 65 % w odniesieniu do warunku podstawowego (1,0 mM Mg), Wysoki
pozakomórkowy poziom magnezu (5,0 mM) wzmacniał negatywny efekt jaki wywierały
blokery wapnia na produkcję IL-4. Poziom IL-4 został obniżony o około 40%-60% przez
wysokie stężenie magnezu (5,0 mM) w odniesieniu do normalnego poziomu magnezu w
medium (1,0 mM) w obecności werapamilu i/lub TMB-8. W ciągu 24 godzin inhibitory
kanałów wapniowych ograniczyły produkcję IL-4 o około 25 % (werapamil), 35 % (TMB-8) i
55 % (bloker wapnia dodane jednocześnie). Efekt jaki wywierały blokery wapnia na
produkcję IL-4 był istotnie wzmacniany przez wysokie pozakomórkowe stężenie magnezu
(5,0 mM Mg).
Wysokie pozakomórkowe stężenie magnezu (5,0 mM) obniżyło produkcję IL-10 o około 20
% w porównaniu do warunku podstawowego (1,0 mM). W trakcie pierwszych 12 godzin
werapamil i TMB-8 obniżyły poziom IL-10 odpowiednio o około 30 % i 60 % (Ryc. 24B),

66
natomiast jednoczesna aplikacja obu blokerów wapnia obniżyła poziom IL-10 w medium
testowym o około 70 % w odniesieniu do warunku podstawowego (1,0 mM Mg). W ciągu 24
godzin werapamil zmniejszył poziom IL-10 w nadsączu komórkowym o około 45 %, TMB-8
o około 60 % a oba blokery wapnia obniżyły poziom tego białka o około 75 % w odniesieniu
do warunku kontrolnego (1,0 mM Mg). Negatywny efekt jaki wywierały blokery wapnia na
produkcję IL-10, tak jak w przypadku IL-4, był zwiększony przez wysokie pozakomórkowe
stężenie magnezu (5,0 mM).
3.5.2 Wpływ zaburzonej równowagi magnezowo-wapniowej na produkcj ę i sekrecję
cytokin IL-2, IL-4, IL-10 oraz interferonu- γγγγ w kokulturze komórkowej: komórki
D10.G4.1 : modelowe makrofagi (BMMFs)
Niskie pozakomórkowe stężenie magnezu (0,1 mM) nie miało istotnego wpływu na
produkcję badanych cytokin w tym modelu doświadczalnym. Wykazano, że wysokie
pozakomórkowe stężenie magnezu (5,0 mM) istotnie obniżało poziom zarówno IL-4 jak i
(Ryc. 25A) jak i IL-10 (Ryc. 25B) w medium testowym w odniesieniu do normalnego
stężenia magnezu (1,0 mM) dopiero po 24 godzinach trwania eksperymentu. Werapamil w
istotnym stopniu obniżł poziom IL-4 (o około 25 %) w stosunku do warunku bazowego (0,1
mM Mg) dopiero po 24 godzinach prowadzenia kokultury. TMB-8 obniżył poziom badanego
białka o około 30% po 12 godzinach hodowli i około 50 % po 24 godzinach hodowli w
odniesieniu do warunku podstawowego (1,0 mM Mg). Jednoczesna aplikacja dwóch
blokerów wapnia przez 24 godziny obniżyła poziom IL-4 w medium testowym o prawie 70
% w porównaniu do warunku podstawowego (0,1 mM Mg). Efekty jakie wywarły blokery
wapnia na produkcję IL-4 były statystycznie niezależne od pozakomórkowego stężenia
magnezu.
Podobnie jak przy IL-4, wysokie stężenie magnezu w medium testowym (5,0 mM) obniżyło
poziom IL-10 o około 15% po 24 godzinach prowadzenia kokultury. TMB-8 już po 12
godzinach eksperymentu obniżył poziom IL-10 o około 55%-60% w odniesieniu do warunku
podstawowego (0,1 mM Mg) przy czym podobny efekt odnotowano również w 24-ej
godzinie eksperymentu. Jednoczesny dodatek werapamilu i TMB-8 do medium testowego
ograniczał produkcję IL-10 nawet w 75%. Negatywne efekty jakie wywarły werapamil i/lub
TMB-8 na produkcję IL-10 były niezależne od pozakomórkowego stężenia magnezu.
Po 24 godzinach hodowli stężenia cytokin IL-4 oraz IL-10 są o około 10 %-15 % wyższe w
medium gdzie komórki prezentujące antygen są reprezentowane przez komórki dendrytyczne

67
(przy 1,0 mM Mg) w odniesieniu do modelu gdzie komórkami prezentującymi antygen są
modelowe makrofagi (1,0 mM Mg).
3.5.3 Wpływ zaburzonej równowagi magnezowo-wapniowej na produkcj ę cytokin IL-4
oraz IL-10 w kokulturze komórkowej: komórki D10.G4.1 : modelowe komórki
dendrytyczne (BMDCs)
Pozakomórkowe stężenie magnezu nie miało istotnego wpływu na produkcję
badanych cytokin przez komórki limfocytarne D10.G4.1. Obecność blokerów wapnia w
istotnym stopniu obniża produkcję cytokin IL-4 (Ryc. 26A) oraz IL-10 (Ryc. 26B), przy czym
efekt ten był niezależny od pozakomórkowego stężenia magnezu. Werapamil w ciągu 12
godzin obniżył produkcję IL-4 o około 40 %, podczas gdy TMB-8, podobnie jak jednoczesny
dodatek obublokerów wapnia, obniżył poziom badanego białka o około 55 % w odniesieniu
do poziomu IL-4 w komórkach nie traktowanych inhibitorami kanałów wapniowych.
Werapamil i/lub TMB-8 obniżał poziom IL-4 po 24 godzinach eksperymentu o około 65 %
w porównaniu do poziomu IL-4 w komórkach nie traktowanych inhibitorami kanałów
wapniowych. W podstawowym warunku doświadczalnym (1,0 mM Mg) produkcja IL-4 przez
komórki D10,G4 po 24 godzinach eksperymentu była nieznaczne wyższa niż po 12 godzinach
(160 MFI vs, 140 MFI),
Werapamil obniżył poziom IL-10 o około 50% i 60% odpowiednio po 12 i 24 godzinach
hodowli w odniesieniu do warunku kontrolnego (1,0 mM Mg). Analogiczne wartości dla
TMB-8 oraz dla obu blokerów wapnia dodawanych jednocześnie, kształtowały się na
poziomie około 60 % i 70 %. Nie odnotowano istotnych różnic w poziomie IL-10 w
komórkach D10.G4.1 po 12 i 24 godzinach hodowli w warunkach kokultury z modelowymi
komórkami dendrytycznymi.

68
V DYSKUSJA
Magnez pozakomórkowy oraz zaburzona równowaga magnezowo-wapniowa miały
zróżnicowany wpływ na funkcje komórek fagocytarnych in vitro. Podstawowym modelem
doświadczalnym była ustalona mysia linia komórek makrofagowych J774.E. Niskie stężenie
magnezu pozakomórkowego ograniczyło proliferację komórek J774.E., przy czym
podwyższyło wywoływaną PMA produkcję reaktywnych form tlenu przez komórki
fagocytarne. Stężenie magnezu pozakomórkowego nie miało istotnego wpływu na
endocytozę antygenu modelowego ani wywołaną LPS produkcję niektórych cytokin
prozapalnych przez komórki J774.E. Szczególnie interesująca wydaje się być interakcja
pomiędzy wysokim pozakomórkowym stężeniem magnezu oraz werapamilem - wysokie
stężenie magnezu pozakomórkowego zwiększało hamujący wpływ blokera wapnia na
ekspresję IL-1β oraz IL-6 przez komórki J774.E, przy czym nie miało znaczącego wpływu na
transkrypcję genów tych cytokin.
Badania rozpoczęto od określenia wpływu magnezu i wapnia na potencjał
proliferacyjny komórek linii J774.E. Niska dostępność magnezu pozakomórkowego (0,1 mM)
nie miała istotnego wpływu na potencjał proliferacyjny komórek linii J774.E w pierwszych
24 godzinach eksperymentu. Potencjał proliferacyjny komórek linii J774.E był obniżony o
prawie 25% i 45% odpowiednio po 48 i 72 godzinach hodowli w medium o niskim stężeniu
magnezu (0,1 mM). Jednocześnie oceniono, że czas podwojenia populacji komórek
makrofagowych J774.E w standardowych warunkach hodowlanych (normalna dostępność
magnezu i wapnia ze środowiska pozakomórkowego i zasobów wewnątrzkomórkowych) in
vitro to około 18-22 godziny.
Z przeglądu piśmiennictwa wynika, że magnez pozakomórkowy jest niezbędny dla
właściwego wzrostu komórek in vitro. Obniżenie stężenia magnezu w medium hodowlanym
(z 0,8 mM do 0,1 mM) zahamowało proliferację fibroblastów zarodków kurzych, przy czym
normalizacja poziomu magnezu w medium hodowlanym skutkowała przywróceniem
normalnej szybkości proliferacji (Rubin and Chu, 1978). Tymczasem wysokie stężenie
magnezu w medium stymulowało proliferację mysich komórek naskórka (Tennenbaum et al.,
1990), przy czym podniesienie pozakomórkowego stężenia magnezu do 10 mM skutkowało
dwukrotnie większą liczbą komórek HUVEC po 48 godzinach hodowli niż w medium
kontrolnym z zawartością magnezu 1,0 mM (Maier et al., 2004). Szybkość proliferacji
komórek HUVEC rosła liniowo wraz ze stężeniem magnezu w medium hodowlanym (Maier
et al., 2004). W badaniach własnych nie zaobserwowano wpływu wysokiego

69
pozakomórkowego stężenia magnezu (5,0 mM) na proliferację komórek makrofagowych
J774.E.
Wykazano, że niektóre typy komórek mogą adaptować się trwale lub czasowo do
niskiego stężenia magnezu pozakomórkowego in vitro. Komórki linii ustalonej HL-60
zachowały normalne tempo wzrostu gdy obniżono poziom magnezu w medium hodowlanym
z 0,5 mM do 0,03 mM (Covacci et al., 1998). Komórki HL-60 hodowane w medium gdzie
zawartość magnezu wynosiła 0,01 mM obumierały po 6-7 dniach (Covacci et al., 1998). Nie
odnotowano zamiany szybkości wzrostu komórek ustalonej linii nowotworowej MCF-7
(komórki nabłonkowe gruczołu sutkowego myszy) in vitro przy obniżeniu poziomu magnezu
w medium hodowlanym z 0,5 mM do 0,05 mM, przy czym w tych warunkach szybkość
proliferacji oraz synteza DNA komórek ustalonej linii normalnej HC11 (komórki nabłonkowe
gruczołu sutkowego myszy) zmniejszyły się o połowę (Sgambato et al., 1999). Zależność
pomiędzy szybkością proliferacji komórek in vitro a stężeniem magnezu pozakomórkowego
wydaje się być cechą charakterystyczną dla typu komórek. Całkowita zawartość magnezu w
komórce proliferującej jest wyższa niż w komórce nie proliferującej co sugeruje stymulujący
wpływ magnezu na syntezę DNA i białek komórkowych oraz wysokie zapotrzebowanie
komórek o dużym tempie wzrostu na magnez (Cameron and Smith, 1989; Rubin et al., 1979).
Ze względu na różnorodność modeli komórkowych trudno będzie jednoznacznie określić
wpływ magnezu na cykl komórkowy i proliferację komórek (Wolf and Cittadini, 1999).
Większość z transporterów przenosi szereg kationów dwuwartościowych, przy czym według
naszej wiedzy u ssaków jedynie dwa systemy transportowe: MagT1 (ang. magnesium
transporter 1) oraz NIPA2 (ang. nonimprinted in Prader-Willi/Angelman syndrome subtype 2)
są selektywne dla Mg2+ (Quamme, 2010), co oznacza że konsekwencją zmian w homeostazie
magnezu mogą być zaburzenia w przepływie innych jonów i ich wpływ na proliferację i
funkcjonowanie komórek.
Z przeprowadzonych badań wynika, że ograniczenie dostępności wapnia również znacząco
obniża potencjał proliferacyjny komórek makrofagowych J774.E. Werapamil (Chen et al.,
2007; Hoffman et al., 1998; McMillen et al., 1985) podobnie jak TMB-8 (Kumar and
Chakrabarti, 2000; Lijnen et al., 1999; Tiemann et al., 1999) znacząco obniżają normalną oraz
indukowaną proliferację wielu typów komórek in vitro, co pośrednio potwierdza uniwersalną
i kluczową rolę wapnia w procesie wzrostu i proliferacji komórek (Berridge, 1995). W
przeprowadzonych badaniach wykazano, że w ciągu 72 godzin werapamil (25 µM)
zmniejszył proliferację komórek J774.E o około 25% podczas gdy TMB-8 (25 µM) o około
65%, przy czym zawartość magnezu w medium hodowlanym była normalna (1,0 mM). Dane

70
te pośrednio wskazują, że dostępność wapnia z wewnątrzkomórkowych źródeł
(zablokowanych przez TMB-8) wydaje się ważniejsza niż dostępność wapnia ze środowiska
pozakomórkowego (zablokowane przez werapamil) dla zachowania normalnego tempa
wzrostu komórek makrofagowych J774.E. Deficyt magnezu w medium hodowlanym (0,1
mM) oraz jednocześnie zastosowane blokery wapnia znacząco ograniczyły proliferację
komórek J774.E przy czym efekt ten był już widoczny po 24 godzinach. Wynik ten wydaje
się być zrozumiały tj. niedostępność dwóch kluczowych dla wzrostu komórki pierwiastków
znacząco ogranicza proliferację i aktywność komórkową. Szczególnie interesująca wydaje się
być interakcja pomiędzy werapamilem oraz wysokim pozakomórkowym stężeniem magnezu
(5,0 mM) oraz jej wpływ na potencjał proliferacyjny modelowych komórek makrofagowych.
Werapamil zastosowany przy normalnym stężeniu magnezu w medium hodowlanym (1,0
mM) obniżył potencjał proliferacyjny komórek o około 25%, przy czym werapamil przy
wysokim poziomie magnezu w medium (5,0mM) zmniejszył proliferację komórek J774.E o
nieco ponad 10% po 72 godzinach hodowli. Z przedstawionych danych wynika, że wysoki
poziom pozakomórkowego magnezu ma dodatni wpływ na proliferację modelowych komórek
fagocytarnych, gdy zablokowany zostanie dopływ wapnia z medium hodowlanego
(zastosowanie werapamilu). Ta zależność nie jest jasna. Według naszej wiedzy taka interakcja
pomiędzy magnezem a werapamilem oraz jej wpływ na proliferację komórek fagocytarnych
in vitro nie została wcześniej zaobserwowana. Interakcje pomiędzy werapamilem a wysokim
pozakomórkowym stężeniem magnezu będą szerzej dyskutowane w dalszej części rozdziału.
Jedną z najlepiej widocznych konsekwencji postępującego niedoboru magnezu u
zwierząt doświadczalnych jest stres oksydacyjny (Hans et al., 2002), który przejawia się m.in.
obniżeniem poziomu antyoksydantów, takich jak wit. E, w surowicy i zwiększoną
peroksydacją lipidów m.in. w mięśniu sercowym (Bussiere et al., 2002a). Dodatkowo
wykazano, że jedną z najszybciej występujących konsekwencji wywołanego dietą niedoboru
magnezu u zwierząt doświadczalnych jest leukocytoza oraz infiltracja tkanek i narządów
przez fagocyty, z dominującą neutrofilią, przy czym te komórki wykazywały cechy komórek
preaktywowanych. Założono, że niedobór magnezu może peraktywować neutrofile 1) na
drodze primingu poprzez indukcję/stymulację produkcji czynników praktywujących komórkę
fagocytarną do odpowiedzi na właściwy bodziec oraz 2) prowadząc do wzrostu stężenia
wapnia w komórce, który jest ważnym sygnalizatorem drugiej linii w komórce układu
odpornościowego (Libako et al., 2009). Wielokrotnie zaobserwowano, że postępujący
niedobór magnezu u zwierząt doświadczalnych skutkuje wzrostem poziomu ważnych
czynników preaktywujących komórkę m.in. substancji P i cytokin prozapalnych do produkcji

71
reaktywnych form tlenu (Weglicki and Phillips, 1992; Weglicki et al., 1992). Wykazano , że
komórki pozyskane z jamy otrzewnowej (oznaczono, ze nie mniej niż 60% zawiesiny
komórek to były makrofagi) zwierząt z deficytem magnezu po stymulacji PMA produkowały
o około 400% więcej reaktywnych form tlenu niż komórki zwierząt z grupy kontrolnej
(Malpuech-Brugere et al., 1998b). Komórki otrzewnowe (pozyskane od szczurów o
normalnym statusie magnezowym) które były preinkubowane z surowicą szczurów z
deficytem magnezu produkowały po stymulacji PMA o około 25% więcej RFT niż komórki
preinkubowane z surowicą szczurów z grupy kontrolnej (Malpuech-Brugere et al., 1998b).
Wyniki obu cytowanych eksperymentów zasugerowały, że w warunkach in vivo istnieje
dodatkowy czynnik/mechanizm preaktywujący komórki fagocytarne. Wielokrotnie
wykazano, że u zwierząt z wywołanym deficytem magnezu, przy niskim stężeniu tego
pierwiastka w surowicy, obserwowany jest wzrost poziomu wapnia we krwi. Eksperymenty
in vitro wykazały, że po stymulacji PAF stężenie wolnego jonu wapniowego w komórkach
otrzewnowych pozyskanych od zwierząt z deficytem magnezu było prawie trzykrotnie
wyższe w porównaniu do grupy kontrolnej (Malpuech-Brugere et al., 1998b), co
zasugerowało że deficyt magnezu in vivo wpływa na homeostazę wapnia w komórce,
aktywność oksydazy NADPH i produkcję reaktywnych form tlenu. Dowiedziono, że wysokie
pozakomórkowe stężenie magnezu (8,0 mM) istotnie obniżyło indukowaną PMA lub fMLP
produkcję reaktywnych form tlenu przez ludzkie neutrofile w warunkach in vitro w
porównaniu do warunku kontrolnego (normalne stężenie magnezu w medium hodowlanym;
0,8 mM). Dodatkowo wykazano, że niskie pozakomórkowe stężenie magnezu (0,2 mM)
wzmacniało indukowaną produkcję reaktywnych form tlenu przez granulocyty
obojętnochłonne w odniesieniu do warunku kontrolnego (Bussiere et al., 2002c). Jak
wiadomo, aktywacja leukocytów bezpośrednio koresponduje ze wzrostem stężenia wapnia w
komórce. Magnez, jako naturalny antagonista wapnia (Iseri and French, 1984), może
konkurować z nim o miejsce w systemie transportowym błon komórkowych. Wysokie
stężenie magnezu w środowisku pozakomórkowym może więc hamować napływ wapnia do
komórki i w ten sposób hamować/opóźniać aktywację komórki immunologicznej. Wyniki
własne wykazały, że wysokie pozakomórkowe stężenie magnezu (5,0 mM) nie miało wpływu
na indukowaną PMA produkcję reaktywnych form tlenu przez mysie granulocyty
obojętnochłonne oraz komórki makrofagowe J774.E. Być może stężenie magnezu 5,0 mM w
środowisku pozakomórkowym jest zbyt niskie by widocznie zaburzyć/zmniejszyć produkcję
reaktywnych form tlenu przez komórki modelowe. Jednocześnie wyniki własne wskazują, że
niski poziom magnezu w medium hodowlanym stymuluje indukowaną PMA produkcję

72
reaktywnych form tlenu przez komórki modelowe. Ograniczenie dopływu wapnia ze
środowiska pozakomórkowego (zastosowanie werapamilu) oraz ze źródeł
wewnątrzkomórkowych (dodatek TMB-8) w istotny sposób zmniejszyło indukowaną PMA
produkcję reaktywnych form tlenu. Nie zaobserwowano istotnego skojarzonego wpływu
pomiędzy inhibitorami kanałów wapniowych a pozakomórkowym stężeniem magnezu.
Można zaobserwować tendencję wskazującą, że niski poziom magnezu w medium
hodowlanym (0,1 mM) w odniesieniu do normalnego stężenia magnezu w medium (1,0 mM)
wzmacnia hamujący wpływ blokerów kanałów wapniowych na indukowaną PMA produkcję
RFT, przy czym efekt ten jest lepiej widoczny u neutrofili. Jednoczesne zastosowanie
werapamilu i TMB-8 istotnie obniżyło proporcjonalną do ilości RFT luminescencję w modelu
neutrofili mysich w porównaniu do warunków gdzie został zastosowany tylko jeden z
blokerów wapnia (normalne stężenie magnezu w medium hodowlanym (1,0 mM)), co
pośrednio sugeruje, że w trakcie aktywacji komórki i produkcji reaktywnych form tlenu,
podwyższone stężenie wapnia w komórce jest wynikiem uwalniania tego pierwiastka z
zasobów wewnątrzkomórkowych oraz jego napływu ze środowiska pozakomórkowego.
Wyniki własne oraz dane literaturowe pokazują, że zablokowanie wolnych kanałów
wapniowych w błonie komórkowej wyraźnie zmniejszyło aktywność tlenową neutrofili (Lee
et al., 2005). Potencjał makrofagów do produkcji RFT zależy od aktywności kanałów
wapniowych typu L (Azenabor and Chaudhry, 2003) przy czym magnez pozakomórkowy
może bezpośrednio modulować aktywność tych kanałów (Sonna et al., 1996). Dodatkowo
zaproponowano, że magnez pozakomórkowy może zaburzać transport jonów do komórki
poprzez blokowanie pompy Na+/Ca2+. Simchowitz i wsp. wykazali, że indukowana fMLP
produkcja reaktywnych form tlenu przez ludzkie neutrofile in vitro została istotnie obniżona
przez obecność w środowisku pozakomórkowym kationów dwu- i trzywartościowych: La3+,
Zn2+, Sr2+, Cd2+, Ba2+, Co2+, Ni2+ oraz Mg2+, przy czym zdolność określonych kationów do
inhibicji produkcji RFT korespondowała z obniżaniem wydajności transportu wapnia przez
pompę Na+/Ca2+ w obecności ww. kationów przy stałym poziomie wapnia w środowisku
pozakomórkowym (1,0mM) (Simchowitz et al., 1990).
Magnez pozakomórkowy może również modulować aktywność endocytarną
makrofagów i neutrofili. Wykazano, że w trakcie indukowanego deficytu magnezu u
zwierząt doświadczalnych zaobserwowano wzmożoną aktywność fagocytów (Bussiere et al.,
2002a). Granulocyty obojętnochłonne uzyskane od zwierząt w ósmym dniu diety
niedoborowej w magnez (stężenie magnezu w surowicy szczurów wynosiło 0,17 mM)
pochłaniały na drodze fagocytozy ośmiokrotnie więcej komórek E. coli niż komórki

73
pozyskane od zwierząt z grupy kontrolnej (stężenie magnezu w surowicy szczurów wynosiło
około 0,8 mM) (Bussiere et al., 2002a). Czynnikiem wzmacniającym reaktywność komórki in
vivo mogła być wysoka dostępność wapnia pozakomórkowego (stężenie wapnia w surowicy
zwierząt z indukowanym dietą deficytem magnezu była istotnie wyższa niż w grupie
kontrolnej (Bussiere et al., 2002a)). W trakcie postępującego niedoboru magnezu w surowicy
zwierząt doświadczalnych pojawiają się mediatory zapalenia oraz czynniki preaktywujące
komórkę m.in. cytokiny prozapalne (w tym IL-6, której wyższy poziom zaobserwowano już
w czwartym dniu diety niedoborowej (Malpuech-Brugere et al., 2000)) co sugeruje
preaktywację neutrofili w kierunku fagocytozy (Bussiere et al., 2002a). Ishiguro i wsp.
wykazali, że niskie stężenie magnezu pozakomórkowego in vitro hamuje przeprowadzaną
przez makrofagi fagocytozę, przy czym ponad fizjologiczny poziom magnezu w medium
hodowlanym nie miał istotnego wpływu na aktywność fagocytarną modelowych komórek.
Inhibitor Mg2+-ATPazy (enzym pełniący istotną rolę w utrzymaniu asymetrii błony
komórkowej) zahamował aktywność żerną komórek niestymulowanych i stymulowanych
LPS (Ishiguro et al., 2000). Niskie stężenie magnezu w medium hodowlanym, podobnie jak
zastosowanie inhibitora Mg2+-ATPazy, podniosło poziom wolnego jonu wapniowego w
komórce (Ishiguro et al., 2000). Niskie stężenie magnezu pozakomórkowego znacząco
zmniejszyło aktywność fagocytarną ludzkich granulocytów obojętnochłonnych in vitro, przy
czym efekt ten nie był kompensowany przez wapń pozakomórkowy (Roschger et al., 1990).
Dosogne i wsp. wykazali, że niskie pozakomórkowe stężenie wapnia oraz magnezu skutkuje
zahamowaniem przeprowadzanej przez neutrofile bydlęce fagocytozy oraz fagocytozy
immunologicznej komórek E. coli (Dosogne et al., 1998). Zablokowanie pozakomórkowego
źródła wapnia (poprzez dodatek EDTA do środowiska pozakomórkowego) ograniczyło
aktywność żerną komórek w 95 %. Dodatkowo wskazano, że fagocytoza immunologiczna jest
bardziej zależna od dostępności wapnia (Dosogne et al., 1998). Nishio i wsp. wykazali, że
makrofagi uzyskane od szczurów po 30 dniach diety ubogiej w magnez (stężenie magnezu w
surowicy wynosiło około 0,16 mM) cechowały się istotnie niższą aktywnością żerną in vitro
niż komórki zwierząt z grupy kontrolnej (stężenie magnezu w surowicy wynosiło około 0,76
mM) (Nishio et al., 2001). Obniżenie aktywności fagocytarnej makrofagów pozyskanych od
zwierząt niedoborowych w trzydziestym dniu eksperymentu było potęgowane przez niskie
pozakomórkowe stężenie magnezu (0,098 mM) w warunkach in vitro. Autorzy zasugerowali,
że obniżona aktywność fagocytarna makrofagów jest podyktowana długotrwałym niedoborem
magnezu, czego bezpośrednim skutkiem jest zaburzenie równowagi wapniowo-magnezowej
(Nishio, et al., 2001). Podsumowując, z analizy danych literaturowych wynika, że

74
eksperymentalny niedobór magnezu może istotnie wpływać na aktywność endocytarną
fagocytów. In vivo rozwijającemu się niedoborowi magnezu u zwierząt doświadczalnych
towarzyszy pojawienie się mediatorów zapalenia (Malpuech-Brugere et al., 2000) co może
preaktywować komórki do wzmożonej aktywności żernej (Bussiere et al., 2002a), przy czym
długotrwały niedobór magnezu może obniżać aktywność endocytarną komórek (Nishio et al.,
2001). In vitro niskie pozakomórkowe stężenie magnezu (i wapnia) obniża aktywność
fagocytarną makrofagów i granulocytów (Roschger et al., 1990, Dosogne et al., 1998). Niskie
pozakomórkowe stężenie magnezu i inhibitor Mg2+-ATPazy podwyższyły poziomu Ca2+ w
komórce fagocytarnej, co wskazuje na możliwe interakcje pomiędzy tymi jonami (Ishiguro et
al., 2000). Wyniki własne sugerują, że endocytoza antygenu modelowego przez komórki
makrofagowe J774.E (albumina jaja kurzego, pochłaniana głównie na drodze endocytozy
zależnej od receptorów (Autenrieth and Autenrieth, 2009)) nie zależy od pozakomórkowego
stężenia magnezu, przy czym zdolność do pochłaniania albuminy jaja kurzego przez
modelowe komórki fagocytarne została znacząco obniżona przez blokery wapnia w błonie
komórkowej (werapamil) i siateczce śródplazmatycznej (TMB-8), co wskazuje
jednoznacznie, że ten proces jest zależny od dostępności wapnia. Blank i wsp. wykazali, że
werapamil hamuje adhezję, fagocytozę oraz migrację ludzkich granulocytów
obojętnochłonnych (Blank et al., 1987) przy czym TMB-8 istotnie obniżył aktywność
bakteriobójczą ludzkich komórek fagocytarnych wyizolowanych z siary (Morceli et al.,
2013). Na podstawie danych literaturowych i wyników własnych można wywnioskować, że
dostępność wapnia ma kluczowe znaczenie dla endocytozy, przy czym konsekwencją zmian
w homeostazie magnezu są zaburzenia w przepływie wapnia (Iseri and French, 1984), co
sugeruje by rozpatrywać skojarzony wpływ obu pierwiastków na funkcjonowanie komórki
fagocytarnej. Wyjaśnienie bezpośredniego wpływu magnezu pozakomórkowego na proces
endocytozy wymaga dalszych badań.
Wysoki poziom cytokin prozapalnych towarzyszy postępującemu niedoborowi
magnezu u zwierząt doświadczalnych. Podwyższone stężenie IL-6 oznaczono już w czwartym
dniu diety niedoborowej w magnez (Malpuech-Brugere et al., 2000), przy czym maksymalne
stężenie cytokin: IL-1, IL-6 oraz TNF-α u szczurów odnotowano w trzecim tygodniu
eksperymentu (Weglicki and Phillips, 1992). Zaproponowano, że wzmożona produkcja
cytokin prozapalnych w trakcie wywołanego dietą niedoboru magnezu może być wynikiem
aktywacji np. przez substancję P (Weglicki and Phillips, 1992) lub reperkusją zaburzonej
równowagi magnezowo-wapniowej w organizmie (Malpuech-Brugere et al., 1998b).
Nieliczne dane wskazują, że stężenie pozakomórkowe magnezu in vitro może istotnie

75
wpływać na produkcję cytokin prozapalnych przez komórki fagocytarne. Oono i wsp.
wykazali, że niskie pozakomórkowe stężenie magnezu stymuluje indukowaną LPS
transkrypcję genów dla IL-1β oraz TNF-α przez makrofagi pierwotne pozyskane od szczurów
z normalnym statusem magnezowym (Oono et al., 2002). Badania nad wpływem magnezu
pozakomórkowego na produkcję cytokin prozapalnych były kontynuowane przez tę samą
grupę badawczą, przy czym nowym celem prowadzonych doświadczeń było wykazanie, czy
magnez pozakomórkowy w analogiczny sposób wpływa ma poziom mRNA dla IL-1β oraz
TNF-α co na produkcję tych cytokin (Shogi et al., 2003). Shogi i wsp. wykazali, że niskie
pozakomórkowe stężenie magnezu istotnie wpływa na wywołaną LPS produkcję cytokin
prozapalnych przez szczurze makrofagi pierwotne (IL-1β oraz TNF-α) – stężenie badanych
białek w nadsączach komórkowych było zdecydowanie wyższe niż w grupie kontrolnej
(normalne stężenie magnezu pozakomórkowego) (Shogi et al., 2003). Wyniki własne
wskazują, że niskie pozakomórkowe stężenie magnezu nie ma istotnego wpływu na
spontaniczną oraz indukowaną LPS transkrypcję genów (poziom mRNA) oraz syntezę IL-1β,
IL-6, TNF-α (poziom białka w supernatancie komórkowym) przez komórki makrofagowe
linii ustalonej J774.E. Różnice pomiędzy wynikami własnymi a wynikami zaprezentowanymi
przez Oono i wsp. (Oono et al., 2002) oraz Shogi i wsp. (Shogi et al., 2003) mogą wynikać z
różnic w warunkach eksperymentalnych. Shogi i wsp. jako niskie pozakomórkowe stężenie
magnezu przyjęli wartość: 0,021 mM, przy czym za normalne pozakomórkowe stężenie
magnezu uznano wartość: 0,39 mM (Shogi et al., 2003), co oznacza że stężenie magnezu w
medium deficytowym było ponad 18-krotnie niższe niż w medium o założonym normalnym
poziomie magnezu. W badaniach własnych stężenie magnezu w medium deficytowym było
10-krotnie niższe niż w medium o założonym normalnym stężeniu tego pierwiastka (0,1 mM
vs 1,0 mM). Stężenie magnezu w medium deficytowym zastosowanym przez Shogi i wsp.
(0,021 mM) (Shogi et al., 2003) było prawie 5-krotnie niższe niż w medium zastosowanym w
badaniach własnych (0,1 mM). Kolejną różnicą w warunkach doświadczalnych był czas
ekspozycji modelowych komórek na „pozakomórkowy deficyt magnezu”. Shogi i wsp.
hodowali makrofagi przez 22 godziny w medium o obniżonej zawartości magnezu (0,021
mM) a następnie stymulowali LPS przez 2 godziny. W badaniach własnych komórki
makrofagowe nie były pre-eksponowane na „deficyt magnezu w medium testowym” a
stymulacja LPS trwała 6 godzin. W badaniach Shogi i wsp. (Shogi et al., 2003; Shogi et al.,
2002) oraz Oono i wsp. (Oono et al., 2002) modelem komórkowym były pierwotne makrofagi
pęcherzykowe szczurów (szczep Wistar), podczas gdy w badaniach własnych modelem

76
komórkowym były mysie komórki makrofagowe ustalonej linii J774.E (szczep Balb/C).
Shogi i wsp. (Shogi et al., 2003) w swoich badaniach poddali modelowe makrofagi działaniu
ekstremalnych warunków niedoboru magnezu pozakomórkowego (według naszej wiedzy
najniższy poziom magnezu jaki oznaczono w surowicy szczurów szczepu Wistar z
wywołanym dietą niedoborem tego pierwiastka to ok. 0,07 mM (Malpuech-Brugere et al.,
1999b)) co mogło być niezbędne do wykazania bezpośredniego wpływu niskiego
pozakomórkowego stężenia magnezu na spontaniczną oraz stymulowaną ekspresję genów
cytokin prozapalnych. W badaniach własnych nie eksponowano komórek J774.E na
„pozakomórkowy niedobór magnezu” dlatego że jak wykazano, 24-godzinna hodowla tych
komórek w medium o obniżonej zawartości magnezu (0,1 mM) skutkowała obniżeniem ich
aktywności komórkowej i proliferacji o około 10%. Z przeglądu piśmiennictwa wynika, że
niskie pozakomórkowe stężenie magnezu zwiększa spontaniczną oraz indukowaną LPS
ekspresję genów cytokin prozapalnych, przy czym efekt ten jest istotnie obniżany przez
nifedipinę (inhibitor wolnych kanałów wapniowych typu L) (Shogi et al., 2002). Shogi i wsp.
wykazali, że 1) werapamil istotnie obniżył indukowaną LPS produkcję i sekrecję IL-1β, przy
czym nie miał wpływu na wywołaną niskim pozakomórkowym stężeniem magnezu (0,021
mM) produkcję IL-1β ani TNF-α przez pierwotne makrofagi pęcherzykowe szczurów w
warunkach in vitro; 2) TMB-8 istotnie obniżył indukowaną LPS produkcję TNF-α, przy
czym nie miał istotnego wpływu na poziom IL-1β w nadsączu komórkowym (Shogi et al.,
2003). Wyniki własne wskazały że: 1) werapamil oraz TMB-8 istotnie obniżyły indukowaną
LPS ekspresję genów (poziom mRNA oraz białka) cytokin prozapalnych: IL-1β, IL-6, TNF-α
w komórkach linii J774.E, przy czym efekt był zależny od stężenia testowego inhibitora
kanałów wapniowych; 2) stymulowana LPS ekspresja cytokin prozapalnych była w większym
stopniu ograniczona przez zablokowanie wewnątrzkomórkowych źródeł wapnia
(zastosowanie TMB-8) niż pozakomórkowych źródeł wapnia (dodatek werapamilu). Różnice
w danych prezentowanych przez Shogi i wsp. i wynikach własnych mogą wynikać ponownie:
z różnic w warunkach doświadczalnych. Shogi i wsp. eksponowali pierwotne makrofagi
pęcherzykowe szczurów na inhibitory kanałów wapniowych: werapamilu lub TMB-8 przez
22 godziny, następnie stymulowali komórki LPS w obecności ww. związków (Shogi et al.,
2003). W doświadczeniach własnych nie pre-inkubowano komórek makrofagowych J774.E z
werapamilem ani TMB-8, ze względu na to, ze w ciągu 24-godzinnej ekspozycji na ww.
związki, przy normalnej dostępności magnezu pozakomórkowego, aktywność
komórkowa/potencjał proliferacyjny komórek J774.E zostały obniżone o ponad 15% przez

77
werapamil i około 25% przez TMB-8) oraz dobranych modeli komórkowych. Watanabe i
wsp. jednoznacznie wykazali, że indukowana LPS produkcja TNF-α przez komórki
makrofagowe linii ustalonej J774.1 została wyraźnie ograniczona przez TMB-8 (ograniczenie
dopływu wapnia ze źródeł wewnątrzkomórkowych) oraz EDTA (ograniczenie dostępności
wapnia w środowisku pozakomórkowym) (Watanabe et al., 1996) co koresponduje z
wynikami własnymi. Wykazano, że wysokie pozakomórkowe stężenie magnezu (5,0 mM)
istotnie ograniczało indukowaną LPS produkcję IL-1β przez komórki linii J774.E.
Zaobserwowano również, że wysokie pozakomórkowe stężenie magnezu (5,0 mM)
zwiększało hamujący wpływ werapamilu na produkcję IL-1β oraz IL-6 (poziom białek w
nadsączu komórkowym). Lin i wsp. wykazali, że wysokie pozakomórkowe stężenie magnezu
in vitro (5,0 mM oraz 20,0 mM) istotnie hamowało indukowaną LPS produkcję IL-1β, IL-6,
TNF-α oraz tlenku azotu przez komórki ustalonej mysiej linii makrofagowej RAW264.7,
przy czym efekt inhibicji zależał liniowo od stężenia magnezu w medium testowym (Lin et
al., 2010). Lin i wsp. założyli, że efekt jaki wywierał magnez na indukowaną LPS odpowiedź
komórek makrofagowych opierał się na antagonizowaniu efektów wapnia i/lub bezpośrednim
hamowaniu wolnych kanałów wapniowych typu L (miejsce działania m.in. werapamilu),
ponieważ hamujący wpływ magnezu na produkcję cytokin prozapalnych, tlenku azotu oraz
aktywację czynnika transkrypcyjnego NF-κβ był częściowo odwracany przez zwiększenie
stężenia wapnia w środowisku pozakomórkowym (dodatek chlorku wapnia, 20 mM) lub
zastosowanie aktywatora wolnych kanałów wapniowych typu L: BAY-K8644 (Lin et al.,
2010). Dodatkowo dane literaturowe potwierdzają, ze magnez pozakomórkowy może istotnie
wpływać na aktywność wolnych kanałów wapniowych typu L (Sonna et al., 1996), miejsce
działania wielu klinicznie stosowanych blokerów wapnia, co sugeruje by uwzględnić wpływ
pozakomórkowego stężenia magnezu przy stosowaniu związków farmakologicznych
modulujących aktywność kanałów wapniowych typu L w warunkach in vitro.
Celem tego projektu była ocena wpływu zaburzonej równowagi magnezowo-
wapniowej na przebieg nieswoistej i swoistej odpowiedzi komórkowej, dlatego że te dwa
rodzaje odpowiedzi są ze sobą ściśle powiązane.
Deficyt magnezu może istotnie wpłynąć na aktywność limfocytów T. U zwierząt
doświadczalnych z wywołanym dietą niedoborem magnezu zaobserwowano inwolucję
grasicy i zwiększoną apoptozę komórek T (Malpuech-Brugere et al., 1999a) oraz
splenomegalię, czemu towarzyszyła zmiana w proporcjach subpopulacji limfocytów
śledzionowych w odniesieniu do grupy kontrolnej (Malpuech-Brugere et al., 1998a). Opisano

78
też istotny wpływ magnezu oraz niedoboru magnezu na przebieg nadwrażliwości typu I oraz
nadwrażliwości kontaktowej typu IV (Blach et al., 2007). Nieliczne dane wskazują, że w
warunkach in vitro limfocyty T pozyskane od myszy z eksperymentalnym niedoborem
magnezu produkowały więcej cytokin (IL)-2, 4, 5, 10, 12, 13 oraz interferonu-γ po godzinnej
ekspozycji na substancję P (mediator zapalenia, którego podwyższony poziom oznaczono w
surowicy zwierząt eksperymentalnych z niedoborem magnezu) w porównaniu do komórek
zwierząt z grupy kontrolnej (Weglicki et al., 1996). Celem drugiego etapu prezentowanych
badań było oszacowanie zaburzonej równowagi magnezowo - wapniowej na proliferację oraz
aktywację modelowych komórek T. Z piśmiennictwa wynika, że deficyt magnezu w medium
testowym istotnie obniżył proliferację pierwotnych limfocytów T pozyskanych ze śledzony
myszy szczepu C57Bl/6 po 72 godzinach hodowli (Flynn, 1984). Uzyskane wyniki wskazują,
że niskie pozakomórkowe stężenie magnezu (0,1 mM) nie miało istotnego wpływu na
potencjał proliferacyjny komórek mysiej linii ustalonej D10.G4.1 (modelowe komórki Th2)
przez pierwsze 48 godzin hodowli. 72-godzinna ekspozycja modelowych komórek
limfocytarnych na pozakomórkowy niedobór magnezu skutkowała obniżeniem potencjału
proliferacyjnego komórek o około 10% w odniesieniu do komórek modelowych hodowanych
w medium o normalnym stężeniu magnezu (1,0 mM). Dane literaturowe (Abboud et al.,
1985; Modiano et al., 1988) i wyniki własne korespondują ze sobą i wskazują, że długotrwała
ekspozycja na niskie pozakomórkowe stężenie magnezu obniża proliferację limfocytów T w
różnych modelach doświadczalnych. Wykazano, że komórki makrofagowe linii ustalonej
J774.E były bardziej wrażliwe na niskie pozakomórkowe stężenie magnezu (0,1 mM) niż
komórki linii D10.G4.1. Po 72 godzinach hodowli w medium deficytowym w magnez
aktywność komórkowa/potencjał proliferacyjny komórek J774.E zostały obniżone o około
40%, podczas gdy analogiczna wartość dla modelowych komórek limfocytarnych wynosiła
około 10%, co potwierdza, że „wrażliwość” na „niedobór magnezu pozakomórkowego” in
vitro jest cechą charakterystyczną dla określonego typu komórek. Komórki makrofagowe
J774.E cechowały się wyższą szybkością proliferacji (doświadczalnie wyznaczony czas
podwojenia populacji tych komórek w standardowych warunkach hodowli in vitro to około
18-22 godziny) niż komórki limfocytarne D10.G4.1 (doświadczalnie wyznaczony czas
podwojenia populacji modelowych komórek T w standardowych warunkach hodowli in vitro
to około 22-28 godzin) co może predysponować komórki J774.E do wyższej wrażliwości na
deficyt magnezu w medium hodowlanym.
Ograniczenie dostępności wapnia ze środowiska pozakomórkowego (poprzez zastosowanie
werapamilu) i/lub wapnia wewnatrzkomórkowego (zastosowanie TMB-8) istotnie obniżyło

79
potencjał proliferacyjny komórek limfocytarnych D10.G4.1. Podobnie jak przy
niedostępności magnezu pozakomórkowego, ograniczenie dostępności wapnia miało większy
wpływ na zahamowanie wzrostu komórek J774.E niż komórek D10.G4.1.
Wykazano, że podobnie jak w przypadku modelowych komórek makrofagowych J774.E,
pozakomórkowe stężenie magnezu nie miało wpływu na pochłanianie antygenu modelowego
(albumina jaja kurzego jest pochłaniana na drodze endocytozy zależnej od receptorów
(Autenrieth and Autenrieth, 2009)) przez modelowe pierwotne komórki prezentujące antygen:
komórki dendrytyczne (BMDCs) oraz makrofagi (BMMFs) wyprowadzone w trakcie hodowli
ukierunkowanej in vitro z komórek szpiku kostnego myszy C3H/HeN. Z przeglądu
piśmiennictwa wynika, że komórki prezentujące antygen tego samego typu (tutaj komórki
dendrytyczne) ale pozyskane od myszy różnych szczepów, mogą znacząco różnić się w
zakresie zdolności do pochłaniania tego samego antygenu (Autenrieth and Autenrieth, 2009).
Z badań własnych wynika, że w identycznych warunkach eksperymentalnych, modelowe
BMDCs oraz BMMFs cechuje podobna zdolność do endocytozy antygenu modelowego (w
standardowych warunkach hodowli in vitro wynosi ona około 80-90 MFI; wartość MFI rośnie
wraz z ilością pochłanianego antygenu), przy czym komórki makrofagowe linii ustalonej
J774.E (uzyskane od myszy szczepu Balb/C) cechuje istotnie niższa zdolność do endocytozy
albuminy jaja kurzego (na poziomie około 40 MFI) niż pozostałe dwa modele komórkowe, co
koresponduje z danymi literaturowymi, że wydajność pochłaniania tego samego antygenu jest
inna w różnych modelach komórkowych. Podobnie jak w przypadku komórek
makrofagowych J774.E, ograniczenie dostępności wapnia wewnątrzkomórkowego
(zastosowanie TMB-8) i/lub wapnia pozakomórkowego (zastosowanie werapamilu) istotnie
obniżyło endocytozę albuminy jaja kurzego przez BMDCs oraz BMMFs in vitro.
Z analizy piśmiennictwa wynika, że wpływ magnezu pozakomórkowego na prezentację
antygenu był badany sporadycznie. Według naszej wiedzy badania dotyczące skojarzonego
wpływu magnezu pozakomórkowego i wapnia na prezentację antygenu w kokulturze in vitro
nie zostały dotychczas opublikowane.
W doświadczaniu w którym badano wpływ środowiska pozakomórkowego na efektywność
prezentacji antygenu, BMDCs lub BMMFs pochłaniały konalbuminę w normalnych
warunkach dostępności magnezu i wapnia in vitro. Modelowe komórki Th2 (komórki
D10.G4.1) są genetycznie predysponowane do rozpoznania konalbuminy. W modelu
kokultury komórkowej in vitro (modelowe komórki prezentujące antygen BMDCs lub
BMMFs były hodowane z komórkami D10.G4.1 w stosunku 1:5) oceniono wpływ zaburzonej
równowagi magnezowo-wapniowej na prezentację antygenu modelowego i aktywację

80
limfocytu T. Założono, że miarą efektywnej prezentacji antygenu i aktywacji modelowych
limfocytów T (komórki D10.G4.1) będzie: 1) antygenowo-zależna proliferacja komórek
D10.G4.1 w kokulturze z modelowymi komórkami prezentującymi antygen (BMDCs lub
BMMFs) oraz 2) produkcja cytokin IL-4 oraz IL-10 w kokulturze komórkowej.
Wykazano, że niskie pozakomórkowe stężenie magnezu (0,1 mM) miało istotny
wpływ na antygenowo-zależną proliferację modelowych komórek Th2 w kokulturze
komórkowej zarówno z BMDCs jak i BMMFs dopiero po 72 godzinach (wzrost liczby
komórek w kokulturze komórkowej jest wiązany bezpośrednio z proliferacją modelowych
komórek T, ponieważ przed rozpoczęciem kokultury komórki prezentujące antygen
traktowano inhibitorem replikacji DNA - mitomycyną C (S. caespitosus)). Odnotowano, że
ograniczenie dostępności wapnia (poprzez zastosowanie werapamilu i/lub TMB-8) istotnie
ograniczyło wzrost komórek D10.G4.1 w obu modelach kokultury komórkowej dopiero po 48
i 72 godzinach. W kokulturze gdzie BMDCs prezentują antygen modelowym komórkom T
tempo proliferacji komórek D10.G4.1 było wyższe niż w kokulturze gdzie modelowymi
komórkami prezentującymi antygen były BMMFs, co sugeruje, że modelowe komórki
dendrytyczne są lepszymi aktywatorami komórek D10.G4.1 niż modelowe makrofagi
(według badań własnych modelowe komórki dendrytyczne ekspresjonują m.in. więcej
cząsteczek MHC II na swojej powierzchni niż modelowe makrofagi; różnice są widoczne
zwłaszcza w obrębie parametru: MHC IIwysokie). Jeśli założymy, że poziom cytokin IL-4 oraz
IL-10 w supernatancie komórkowym koresponduje liniowo ze zdolnością do aktywacji
komórek D10.G4.1 to ponownie modelowe komórki dendrytyczne są bardziej „efektywne”
niż modelowe makrofagi. Ograniczenie dostępności wapnia (poprzez zastosowanie
werapamilu i/lub TMB-8) istotnie obniżyło produkcję IL-4 oraz IL-10 w obu modelach
kokultur komórkowych w każdym z badanych punktów czasowych; hamujący wpływ
werapamilu i TMB-8 na produkcję ww. cytokin był istotnie podwyższany przez wysokie
pozakomórkowe stężenie magnezu (5,0 mM), przy czym efekt ten był szczególnie dobrze
widoczny w modelu kokultury komórkowej gdzie komórkami prezentującymi antygen były
modelowe komórki dendrytyczne. Oznaczanie stężeń ww. cytokin dokonano po 12 i 24
godzinach hodowli w warunkach kokultury komórkowej in vitro, dlatego że według
wyników własnych, w tych punktach czasowych blokery wapnia nie miały istotnego wpływu
na całkowitą liczbę komórek ani na aktywność metaboliczną kokultury. Dodatkowo z
przeprowadzonych badań własnych wynika, że poziomy cytokin IL-4 oraz IL-10 są istotnie
obniżane przez wysokie pozakomórkowe stężenie magnezu (5,0 mM), przy czym efekt ten

81
ponownie był szczególnie widoczny w modelu kokultury komórkowej gdzie komórkami
prezentującymi antygen były modelowe komórki dendrytyczne.
Według naszej wiedzy, w literaturze nie został dotychczas opisany skojarzony wpływ
wapnia i magnezu i/lub zaburzonej równowagi tych pierwiastków na prezentację antygenu i
aktywację limfocytów T. Z nielicznych danych wynika, że magnez pozakomórkowy obniża
zdolność ludzkich komórek Langerhansa (komórki dendrytyczne w skórze) do prezentacji
antygenu i aktywacji limfocytów w warunkach in vivo oraz in vitro. Dodatek chlorku
magnezu w medium testowym (1%) korespondował z obniżoną ekspresją MHC II oraz
cząsteczek kostymulotorowych CD80/CD86 (B7-1/B7-2) na powierzchni komórek
Langerhansa, odnotowano również obniżoną produkcją ekspresjonowanego konstytutywnie
przez te komórki TNF-α w warunkach in vitro (Schempp et al., 2000). Inhibitory wolnych
kanałów wapniowych typu L (nifedipina i werapamil) obniżają zdolność komórek
Langerhansa do prezentacji antygenu modelowego w warunkach in vitro, przy czym efekt ten
był zależny od dawki inhibitora kanałów wapniowych. Zarówno nifedipina jak i werapamil
istotnie obniżały ekspresję cząsteczek MHC II oraz CD80 na powierzchni komórek w
warunkach in vitro (Katoh et al., 1997). Wykazano, że receptor dla dihydropirydyny
(podjednostka α1 kanału wapniowego typu L) jest ekspresjonowany na powierzchni
limfocytów Th2, co bezpośrednio wskazuje, że inhibitory wolnych kanałów wapniowych typu
L mogą istotnie wpływać na aktywność tych komórek (Gomes et al., 2007; Savignac et al.,
2004). Aktywność wolnych kanałów wapniowych typu L jest również modulowana przez
magnez pozakomórkowy (Sonna et al., 1996). Aktywacja limfocytów T wydaje się być
szczególnie zależna od Mg2+. Li i wsp. zaproponowali, że magnez pozakomórkowy
synergistycznie z wapniem reguluje aktywację limfocytów T, przy czym nie ma istotnego
wpływu na aktywację limfocytów B (Li et al., 2011). Aktywacji komórki z udziałem TCR
(lecz nie BCR) wymaga przejściowego (krótkotrwałego) napływu Mg2+ przez selektywny dla
magnezu transporter MagT1 (ang. magnesium transporter 1), czemu towarzyszy aktywacja
fosfolipazy Cγ1 i następujący po niej napływ Ca2+ do komórki. Dodatkowo wykazano, że
indukowany mitogenem (m.in. ConA, PHA, przeciwciało aktywujące αCD3) napływ Mg2+ do
komórki T przez MagT1 nie ma wpływu na homeostazę magnezu (Li et al., 2011). Rolę tego
transportera w aktywacji komórek T wykazano pośrednio przez usunięcie genu MagT1 w
normalnych komórkach T, przy czym sprzężoną z chromosomem X mutację genu MagT1
potwierdzono u osób cierpiących na wrodzony niedobór odporności cechujący się m.in.

82
limfocytopenią w obrębie komórek Th i niepełnowartościową aktywacją tych komórek (Li et
al., 2011).
Z badań własnych i piśmiennictwa wynika, że wysokie stężenie magnezu
pozakomórkowego istotnie wpływa na proces prezentacji antygenu. Dodatkowo wysokie
pozakomórkowe stężenie może wzmacniać efekt jaki wywierają antagoniści wolnych
kanałów wapniowych typu L na proces prezentacji antygenu i aktywacji komórek T w
warunkach in vitro.
Z zaprezentowanych danych wynika, że magnez pozakomórkowy może w istotny
sposób wpływać na komponenty komórkowe układu immunologicznego w warunkach in
vitro. Zakłada się, że niskie pozakomórkowe stężenie magnezu skutkuje wzrostem stężenia
wapnia w komórce, przy czym wysokie pozakomórkowe stężenie magnezu wpływa na
napływ wapnia do komórki. Wapń jest ważnym sygnalizatorem drugiej linii, a wzrost stężenia
wapnia w cytoplazmie koresponduje z aktywacją komórki immunologicznej.
Wewnątrzkomórkowy poziom magnezu zmienia się w wąskim zakresie stężeń, przez co
prawdopodobnie nie ma istotnego wpływu na uwalnianie (hamowanie uwalniania) wapnia z
zasobów wewnątrzkomórkowych. Wydaje się, że wysokie stężenie magnezu
pozakomórkowego może hamować/opóźniać aktywację komórki immunologicznej poprzez
ograniczenie napływu wapnia ze środowiska pozakomórkowego in vitro. Zaobserwowano
interesującą zależność, gdzie pozakomórkowe stężenie magnezu często wzmacnia efekt jaki
na komórkę układu odpornościowego wywierał werapamil (hamuje napływ wapnia ze
środowiska pozakomórkowego), przy czym jak wynika z danych literaturowych, magnez
może w istotny sposób modulować aktywność wolnych kanałów wapniowych typu L, miejsca
działania werapamilu.
Zakldajac hipotezę, że wysokie stężenie magnezu pozakomórkowego jest efektywnym
antagonistą wapnia (m.in. przez hamowanie aktywności kanałów wapniowych typu L).
Wówczas: 1) Szczególnie interesującym wydaje się być zagadnienie, czy komórka układu
odpornościowego (której aktywacja zależy bezpośrednio od dostępności wapnia), będzie
cechowała się wzmożoną ekspresją alternatywnych systemów transportowych dla wapnia w
błonie komórkowej w warunkach wysokiego stężenia magnezu pozakomórkowego, co
jednocześnie sprawi, że będzie bardziej wrażliwa na działanie innych blokerów wapnia. 2)
Wyjaśnienia wymagałby również mechanizm, w jaki sposób wysokie pozakomórkowe
stężenie magnezu może potęgować działanie inhibitorów kanałów wapniowych typu L (np.
czy magnez istotnie wpływa na częstotliwość otwierania/zamykania się kanałów wapniowych

83
lub czy magnez może bezpośrednio wpływać na zdolność wiązania się antagonistów wapnia
do systemów transportowych).

84
VI WNIOSKI
Równowaga magnezowo-wapniowa ma istotny wpływ na kluczowe etapy odpowiedzi
swoistej i nieswoistej w warunkach in vitro.
1). Niskie stężenie magnezu pozakomórkowego zaburza proliferację badanych komórek
układu odpornościowego in vitro.
2). Niskie pozakomórkowe stężenie magnezu podwyższa wywoływaną PMA produkcję
reaktywnych form tlenu przez modelowe komórki fagocytarne in vitro. Obniżenie produkcji
reaktywnych form tlenu po zastosowaniu blokerów wapnia podkreśla ważną rolę wapnia w
tym procesie.
3). Pozakomórkowe stężenie magnezu nie ma istotnego wpływu na endocytozę
przeprowadzaną przez modelowe komórki prezentujące antygen in vitro, przy czym efekt ten
jest niezależny od dostępności wapnia.
4). Dostępność wapnia jest kluczowa dla stymulowanej LPS ekspresji cytokin prozapalnych
in vitro. Wysokie pozakomórkowe stężenie magnezu potencjalizuje wpływ werapamilu na
produkcję niektórych cytokin prozapalnych in vitro.
5). Wysokie stężenie magnezu pozakomórkowego istotnie obniża produkcję cytokin oraz
potencjalizuje wpływ blokerów wapnia na produkcję cytokin w modelowych kokulturach
komórkowych (komórki dendrytyczne : limfocyty T oraz makrofagi : limfocyty T) in vitro, co
oznacza, że zaburzona równowaga magnezowo-wapniowa w środowisku pozakomórkowym
istotnie wpływa na prezentację antygenu.

85
VII PI ŚMIENNICTWO
Abboud CN, Scully SP, Lichtman AH, Brennan JK, Segel GB. 1985. The requirements for ionized calcium and magnesium in lymphocyte proliferation. J Cell Physiol 122:64-72.
Alfrey AC, Miller NL, Trow R. 1974. Effect of age and magnesium depletion on bone magnesium pools in rats. J Clin Invest 54:1074-1081.
Almoznino-Sarafian, Berman S, Mor A, Shteinshnaider M, Gorelik O, Tzur I, Alon I, Modai D, N. C. 2007. Magnesium and C-reactive protein in heart failure: an anti-inflammatory effect of magnesium administration? Eur J Nutr. 46:230-237.
Altura BM, Altura BT. 1974. Magnesium and contraction of arterial smooth muscle. Microvasc Res 7:145-155.
Altura BT, Altura BM. 1991. Measurement of ionized magnesium in whole blood, plasma and serum with a new ion-selective electrode in healthy and diseased human subjects. Magnes Trace Elem 10:90-98.
Autenrieth SE, Autenrieth IB. 2009. Variable antigen uptake due to different expression of the macrophage mannose receptor by dendritic cells in various inbred mouse strains. Immunology 127:523-529.
Azenabor AA, Chaudhry AU. 2003. Effective macrophage redox defense against Chlamydia pneumoniae depends on L-type Ca2+ channel activation. Med Microbiol Immunol 192:99-106.
Bacon KB, Westwick J, Camp RD. 1989. Potent and specific inhibition of IL-8-, IL-1 alpha- and IL-1 beta-induced in vitro human lymphocyte migration by calcium channel antagonists. Biochem Biophys Res Commun 165:349-354.
Bardelli C, Gunella G, Varsaldi F, Balbo P, Del Boca E, Bernardone IS, Amoruso A, Brunelleschi S. 2005. Expression of functional NK1 receptors in human alveolar macrophages: superoxide anion production, cytokine release and involvement of NF-kappaB pathway. Br J Pharmacol 145:385-396.
Barry WH, Bridge JH. 1993. Intracellular calcium homeostasis in cardiac myocytes. Circulation 87:1806-1815.
Baumgarten G, Knuefermann P, Nozaki N, Sivasubramanian N, Mann DL, Vallejo JG. 2001. In vivo expression of proinflammatory mediators in the adult heart after endotoxin administration: the role of toll-like receptor-4. J Infect Dis 183:1617-1624.
Bergson P, Lipkind G, Lee SP, Duban ME, Hanck DA. 2011. Verapamil block of T-type calcium channels. Mol Pharmacol 79:411-419.
Berkow RL, Wang D, Larrick JW, Dodson RW, Howard TH. 1987. Enhancement of neutrophil superoxide production by preincubation with recombinant human tumor necrosis factor. J Immunol 139:3783-3791.
Berridge MJ. 1993. Inositol trisphosphate and calcium signalling. Nature 361:315-325. Berridge MJ. 1995. Calcium signalling and cell proliferation. Bioessays 17:491-500. Berridge MJ, Lipp P, Bootman MD. 2000. The versatility and universality of calcium
signalling. Nat Rev Mol Cell Biol 1:11-21. Birx DL, Berger M, Fleisher TA. 1984. The interference of T cell activation by calcium
channel blocking agents. J Immunol 133:2904-2909. Blach J, Nowacki W, Mazur A. 2007. [Magnesium in skin allergy]. Postepy Hig Med Dosw
(Online) 61:548-554. Blank I, Baudisch W, Burmeister J, Franz U. 1987. [Effect of verapamil and nifedipine on the
adhesion, migration and phagocytosis of human neutrophilic granulocytes]. Folia Haematol Int Mag Klin Morphol Blutforsch 114:117-125.

86
Blaustein MP, Lederer WJ. 1999. Sodium/calcium exchange: its physiological implications. Physiol Rev 79:763-854.
Bost KL, Pascual DW. 1992. Substance P: a late-acting B lymphocyte differentiation cofactor. Am J Physiol 262:C537-545.
Busse WW, Vrtis RF, Steiner R, Dick EC. 1991. In vitro incubation with infuenza virus primes human polymorphonuclear leukocyte generation of superoxide. Am J Respir Cell Mol Biol 4:347-354.
Bussiere FI, Gueux E, Rock E, Girardeau JP, Tridon A, Mazur A, Rayssiguier Y. 2002a. Increased phagocytosis and production of reactive oxygen species by neutrophils during magnesium deficiency in rats and inhibition by high magnesium concentration. Br J Nutr 87:107-113.
Bussiere FI, Gueux E, Rock E, Mazur A, Rayssiguier Y. 2002b. Protective effect of calcium deficiency on the inflammatory response in magnesium-deficient rats. Eur J Nutr 41:197-202.
Bussiere FI, Mazur A, Fauquert JL, Labbe A, Rayssiguier Y, Tridon A. 2002c. High magnesium concentration in vitro decreases human leukocyte activation. Magnes Res 15:43-48.
Bussiere FI, Tridon A, Zimowska W, Mazur A, Rayssiguier Y. 2003. Increase in complement component C3 is an early response to experimental magnesium deficiency in rats. Life Sci 73:499-507.
Bussiere FI, Zimowska W, Gueux E, Rayssiguier Y, Mazur A. 2002d. Stress protein expression cDNA array study supports activation of neutrophils during acute magnesium deficiency in rats. Magnes Res 15:37-42.
Calin-Jageman I, Lee A. 2008. Ca(v)1 L-type Ca2+ channel signaling complexes in neurons. J Neurochem 105:573-583.
Cameron IL, Smith NK. 1989. Cellular concentration of magnesium and other ions in relation to protein synthesis, cell proliferation and cancer. Magnesium 8:31-44.
Carafoli E. 1987. Intracellular Calcium Homeostasis. Annual Review of Biochemistry 56:395-433.
Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. 2005. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev 57:411-425.
Ceciliani F, Giordano A, Spagnolo V. 2002. The systemic reaction during inflammation: the acute-phase proteins. Protein Pept Lett 9:211-223.
Chacko SA, Song Y, Nathan L, Tinker L, de Boer IH, Tylavsky F, Wallace R, Liu S. 2010. Relations of dietary magnesium intake to biomarkers of inflammation and endothelial dysfunction in an ethnically diverse cohort of postmenopausal women. Diabetes Care 33:304-310.
Chen J, Zhang H, Wang H. 2007. Experimental study on the inhibitory effects of verapamil on the proliferation of meningiomas cells. J Huazhong Univ Sci Technolog Med Sci 27:88-90.
Chernow B, Bamberger S, Stoiko M, Vadnais M, Mills S, Hoellerich V, Warshaw AL. 1989. Hypomagnesemia in patients in postoperative intensive care. Chest 95:391-397.
Chiou CY, Malagodi MH. 1975. Studies on the mechanism of action of a new Ca-2+ antagonist, 8-(N,N-diethylamino)octyl 3,4,5-trimethoxybenzoate hydrochloride in smooth and skeletal muscles. Br J Pharmacol 53:279-285.
Cittadini A, Scarpa A. 1983. Intracellular Mg2+ homeostasis of Ehrlich ascites tumor cells. Arch Biochem Biophys 227:202-209.
Clapham DE. 2007. Calcium signaling. Cell 131:1047-1058.

87
Clapper DL, Walseth TF, Dargie PJ, Lee HC. 1987. Pyridine nucleotide metabolites stimulate calcium release from sea urchin egg microsomes desensitized to inositol trisphosphate. J Biol Chem 262:9561-9568.
Claverie-Benureau S, Lebel B, Gaudin-Harding F. 1980. Magnesium deficiency allergy-like crisis in hairless rats: a suggested model for inflammation studies. J Physiol (Paris) 76:173-175.
Condliffe AM, Chilvers ER, Haslett C, Dransfield I. 1996. Priming differentially regulates neutrophil adhesion molecule expression/function. Immunology 89:105-111.
Condliffe AM, Kitchen E, Chilvers ER. 1998. Neutrophil priming: pathophysiological consequences and underlying mechanisms. Clin Sci (Lond) 94:461-471.
Covacci V, Bruzzese N, Sgambato A, Di Francesco A, Russo MA, Wolf FI, Cittadini A. 1998. Magnesium restriction induces granulocytic differentiation and expression of p27Kip1 in human leukemic HL-60 cells. J Cell Biochem 70:313-322.
Cowan DB, Poutias DN, Del Nido PJ, McGowan FX, Jr. 2000. CD14-independent activation of cardiomyocyte signal transduction by bacterial endotoxin. Am J Physiol Heart Circ Physiol 279:H619-629.
Daniels RH, Finnen MJ, Hill ME, Lackie JM. 1992. Recombinant human monocyte IL-8 primes NADPH-oxidase and phospholipase A2 activation in human neutrophils. Immunology 75:157-163.
Demeuse P, Penner R, Fleig A. 2006. TRPM7 channel is regulated by magnesium nucleotides via its kinase domain. J Gen Physiol 127:421-434.
Di Francesco A, Desnoyer RW, Covacci V, Wolf FI, Romani A, Cittadini A, Bond M. 1998. Changes in magnesium content and subcellular distribution during retinoic acid-induced differentiation of HL60 cells. Arch Biochem Biophys 360:149-157.
Diment S, Leech MS, Stahl PD. 1987. Generation of macrophage variants with 5-azacytidine: selection for mannose receptor expression. J Leukoc Biol 42:485-490.
Dong JW, Vallejo JG, Tzeng HP, Thomas JA, Mann DL. Innate immunity mediates myocardial preconditioning through Toll-like receptor 2 and TIRAP-dependent signaling pathways. Am J Physiol Heart Circ Physiol 298:H1079-1087.
Dosogne H, Burvenich C, Paape MJ. 1998. Effect of extracellular ionic calcium and magnesium on opsonic and non-opsonic phagocytosis of escherichia coli bovine blood polymorphonuclear leucocytes. Comparative Haematology International 8:82-86.
Du Clos TW. 2000. Function of C-reactive protein. Ann Med 32:274-278. Eckel RH, Grundy SM, Zimmet PZ. 2005. The metabolic syndrome. Lancet 365:1415-1428. Eisenberg MJ, Brox A, Bestawros AN. 2004. Calcium channel blockers: an update. Am J
Med 116:35-43. Elamin A, Tuvemo T. 1990. Magnesium and insulin-dependent diabetes mellitus. Diabetes
Res Clin Pract 10:203-209. Elliott DA, Rizack MA. 1974. Epinephrine and adrenocorticotropic hormone-stimulated
magnesium accumulation in adipocytes and their plasma membranes. J Biol Chem 249:3985-3990.
English D, Andersen BR. 1974. Single-step separation of red blood cells. Granulocytes and mononuclear leukocytes on discontinuous density gradients of Ficoll-Hypaque. J Immunol Methods 5:249-252.
Ennis M, Ind PW, Pearce FL, Dollery CT. 1983. Calcium antagonists and histamine secretion from rat peritoneal mast cells. Agents Actions 13:144-148.
Fagan TE, Romani A. 2001. alpha(1)-Adrenoceptor-induced Mg2+ extrusion from rat hepatocytes occurs via Na(+)-dependent transport mechanism. Am J Physiol Gastrointest Liver Physiol 280:G1145-1156.

88
Fanger CM, Neben AL, Cahalan MD. 2000. Differential Ca2+ influx, KCa channel activity, and Ca2+ clearance distinguish Th1 and Th2 lymphocytes. J Immunol 164:1153-1160.
Feray JC, Garay R. 1987. A one-to-one Mg2+:Mn2+ exchange in rat erythrocytes. J Biol Chem 262:5763-5768.
Fernandez JG, Caperos JR. 1977. [Styrene exposure. An experimental study of pulmonary absorption and excretion (author's transl)]. Int Arch Occup Environ Health 40:1-12.
Finkel TH, Pabst MJ, Suzuki H, Guthrie LA, Forehand JR, Phillips WA, Johnston RB, Jr. 1987. Priming of neutrophils and macrophages for enhanced release of superoxide anion by the calcium ionophore ionomycin. Implications for regulation of the respiratory burst. J Biol Chem 262:12589-12596.
Flatman PW. 1991. Mechanisms of magnesium transport. Annu Rev Physiol 53:259-271. Flynn A. 1984. Control of in vitro lymphocyte proliferation by copper, magnesium and zinc
deficiency. J Nutr 114:2034-2042. Fontenot JP, Allen VG, Bunce GE, Goff JP. 1989. Factors influencing magnesium absorption
and metabolism in ruminants. J Anim Sci 67:3445-3455. Ford ES. 1999. Body mass index, diabetes, and C-reactive protein among U.S. adults.
Diabetes Care 22:1971-1977. Ford ES, Giles WH, Dietz WH. 2002. Prevalence of the metabolic syndrome among US
adults: findings from the third National Health and Nutrition Examination Survey. JAMA 287:356-359.
Ford ES, Li C, McGuire LC, Mokdad AH, Liu S. 2007. Intake of dietary magnesium and the prevalence of the metabolic syndrome among U.S. adults. Obesity (Silver Spring) 15:1139-1146.
Frank MM, Fries LF. 1991. The role of complement in inflammation and phagocytosis. Immunol Today 12:322-326.
Frantz S, Kelly RA, Bourcier T. 2001. Role of TLR-2 in the activation of nuclear factor kappaB by oxidative stress in cardiac myocytes. J Biol Chem 276:5197-5203.
Galan P, Preziosi P, Durlach V, Valeix P, Ribas L, Bouzid D, Favier A, Hercberg S. 1997. Dietary magnesium intake in a French adult population. Magnes Res 10:321-328.
Gasbarrini A, Borle AB, Farghali H, Bender C, Francavilla A, Van Thiel D. 1992. Effect of anoxia on intracellular ATP, Na+i, Ca2+i, Mg2+i, and cytotoxicity in rat hepatocytes. J Biol Chem 267:6654-6663.
Gaussin V, Gailly P, Gillis JM, Hue L. 1997. Fructose-induced increase in intracellular free Mg2+ ion concentration in rat hepatocytes: relation with the enzymes of glycogen metabolism. Biochem J 326 ( Pt 3):823-827.
Gomes B, Cabral MD, Gallard A, Savignac M, Paulet P, Druet P, Mariame B, Moreau M, Leclerc C, Guery JC, Pelletier L. 2007. Calcium channel blocker prevents T helper type 2 cell-mediated airway inflammation. Am J Respir Crit Care Med 175:1117-1124.
Goytain A, Quamme GA. 2005a. Functional characterization of ACDP2 (ancient conserved domain protein), a divalent metal transporter. Physiol Genomics 22:382-389.
Goytain A, Quamme GA. 2005b. Functional characterization of human SLC41A1, a Mg2+ transporter with similarity to prokaryotic MgtE Mg2+ transporters. Physiol Genomics 21:337-342.
Goytain A, Quamme GA. 2005c. Functional characterization of the mouse [corrected] solute carrier, SLC41A2. Biochem Biophys Res Commun 330:701-705.
Goytain A, Quamme GA. 2005d. Identification and characterization of a novel mammalian Mg2+ transporter with channel-like properties. BMC Genomics 6:48.

89
Goytain A, Quamme GA. 2008. Identification and characterization of a novel family of membrane magnesium transporters, MMgT1 and MMgT2. Am J Physiol Cell Physiol 294:C495-502.
Graham LA, CAESAR J, BUEGEN ASV. 1960. Gastrointestinal absorption and excretion of Mg28 in man. Metabolism 9:646-659.
Grubbs RD, Maguire ME. 1987. Magnesium as a regulatory cation: criteria and evaluation. Magnesium 6:113-127.
Guerrero-Romero F, Rodriguez-Moran M. 2002a. Low serum magnesium levels and metabolic syndrome. Acta Diabetol 39:209-213.
Guerrero-Romero F, Rodriguez-Moran M. 2002b. Relationship between serum magnesium levels and C-reactive protein concentration, in non-diabetic, non-hypertensive obese subjects. Int J Obes Relat Metab Disord 26:469-474.
Guerrero-Romero F, Tamez-Perez HE, Gonzalez-Gonzalez G, Salinas-Martinez AM, Montes-Villarreal J, Trevino-Ortiz JH, Rodriguez-Moran M. 2004. Oral magnesium supplementation improves insulin sensitivity in non-diabetic subjects with insulin resistance. A double-blind placebo-controlled randomized trial. Diabetes Metab 30:253-258.
Gunther T. 1993. Mechanisms and regulation of Mg2+ efflux and Mg2+ influx. Miner Electrolyte Metab 19:259-265.
Gunther T, Vormann J. 1985. Mg2+ efflux is accomplished by an amiloride-sensitive Na+/Mg2+ antiport. Biochem Biophys Res Commun 130:540-545.
Gunther T, Vormann J. 1990. Characterization of Na(+)-independent Mg2+ efflux from erythrocytes. FEBS Lett 271:149-151.
Gunther T, Vormann J, Hollriegl V. 1990. Characterization of Na(+)-dependent Mg2+ efflux from Mg2(+)-loaded rat erythrocytes. Biochim Biophys Acta 1023:455-461.
Gunther T, Vormann J, Hollriegl V. 1992. Isoproterenol-induced Mg2+ uptake in liver. FEBS Lett 307:333-336.
Guthrie LA, McPhail LC, Henson PM, Johnston Jr RB. 1984. Priming of neutrophils for enhanced release of oxygen metabolites: evidence for increased activity of the superoxide-producing enzyme. J Exp Med 160:1656-1671.
Hans CP, Chaudhary DP, Bansal DD. 2002. Magnesium deficiency increases oxidative stress in rats. Indian J Exp Biol 40:1275-1279.
Hardwick LL, Jones MR, Brautbar N, Lee DB. 1991. Magnesium absorption: mechanisms and the influence of vitamin D, calcium and phosphate. J Nutr 121:13-23.
Harman AW, Nieminen AL, Lemasters JJ, Herman B. 1990. Cytosolic free magnesium, ATP and blebbing during chemical hypoxia in cultured rat hepatocytes. Biochem Biophys Res Commun 170:477-483.
Harris DJ, Lambell RG, Oliver CJ. 1983. Factors predisposing dairy and beef cows to grass tetany. Aust Vet J 60:230-234.
Hashizume N, Mori M. 1990. An analysis of hypermagnesemia and hypomagnesemia. Jpn J Med 29:368-372.
He K, Liu K, Daviglus ML, Morris SJ, Loria CM, Van Horn L, Jacobs DR, Jr., Savage PJ. 2006. Magnesium intake and incidence of metabolic syndrome among young adults. Circulation 113:1675-1682.
Hoffman S, Gopalakrishna R, Gundimeda U, Murata T, Spee C, Ryan SJ, Hinton DR. 1998. Verapamil inhibits proliferation, migration and protein kinase C activity in human retinal pigment epithelial cells. Exp Eye Res 67:45-52.
Iftinca MC, Zamponi GW. 2009. Regulation of neuronal T-type calcium channels. Trends Pharmacol Sci 30:32-40.

90
Iseri LT, French JH. 1984. Magnesium: nature's physiologic calcium blocker. Am Heart J 108:188-193.
Ishiguro S, Miyamoto A, Tokushima T, Ueda A, Nishio A. 2000. Low extracellular Mg2+ concentrations suppress phagocytosis in vitro by alveolar macrophages from rats. Magnes Res 13:11-18.
Jahnen-Dechent WaK, M. 2012. Magnesium basics. Clinical Kidney Journal 5:i3-i14. Janiec W, Krupinska J. 2005. Farmakodynamika. Podręcznik dla studentów farmacji.
Wydawnictwo Lekarskie PZWL. Jeroen H. F. de Baaij, J. JG, Bindels HaRJM. 2012. Regulation of magnesium balance:
lessons learned from human genetic disease. Clinical Kidney Journal 5:i15-i24. Jung DW, Brierley GP. 1986. Matrix magnesium and the permeability of heart mitochondria
to potassium ion. J Biol Chem 261:6408-6415. Kandeel FR, Balon E, Scott S, Nadler JL. 1996. Magnesium deficiency and glucose
metabolism in rat adipocytes. Metabolism 45:838-843. Kapadia S, Lee J, Torre-Amione G, Birdsall HH, Ma TS, Mann DL. 1995. Tumor necrosis
factor-alpha gene and protein expression in adult feline myocardium after endotoxin administration. J Clin Invest 96:1042-1052.
Karppanen H. 1981. Epidemiological studies on the relationship between magnesium intake and cardiovascular diseases. Artery 9:190-199.
Katoh N, Hirano S, Kishimoto S, Yasuno H. 1997. Calcium channel blockers suppress the contact hypersensitivity reaction (CHR) by inhibiting antigen transport and presentation by epidermal Langerhans cells in mice. Clin Exp Immunol 108:302-308.
Kayne LH, Lee DB. 1993. Intestinal magnesium absorption. Miner Electrolyte Metab 19:210-217.
Kehres DG, Maguire ME. 2002. Structure, properties and regulation of magnesium transport proteins. Biometals 15:261-270.
Kesteloot H, Joossens JV. 1990. The relationship between dietary intake and urinary excretion of sodium, potassium, calcium and magnesium: Belgian Interuniversity Research on Nutrition and Health. J Hum Hypertens 4:527-533.
King DE, Mainous AG, 3rd, Geesey ME, Ellis T. 2007. Magnesium intake and serum C-reactive protein levels in children. Magnes Res 20:32-36.
King DE, Mainous AG, 3rd, Geesey ME, Woolson RF. 2005. Dietary magnesium and C-reactive protein levels. J Am Coll Nutr 24:166-171.
Kitchen E, Rossi AG, Condliffe AM, Haslett C, Chilvers ER. 1996. Demonstration of reversible priming of human neutrophils using platelet-activating factor. Blood 88:4330-4337.
Klevay LM, Milne DB. 2002. Low dietary magnesium increases supraventricular ectopy. Am J Clin Nutr 75:550-554.
Koenderman L, Yazdanbakhsh M, Roos D, Verhoeven AJ. 1989. Dual mechanisms in priming of the chemoattractant-induced respiratory burst in human granulocytes. A Ca2+-dependent and a Ca2+-independent route. J Immunol 142:623-628.
Kolisek M, Launay P, Beck A, Sponder G, Serafini N, Brenkus M, Froschauer EM, Martens H, Fleig A, Schweigel M. 2008. SLC41A1 is a novel mammalian Mg2+ carrier. J Biol Chem 283:16235-16247.
Kolisek M, Zsurka G, Samaj J, Weghuber J, Schweyen RJ, Schweigel M. 2003. Mrs2p is an essential component of the major electrophoretic Mg2+ influx system in mitochondria. EMBO J 22:1235-1244.
Kuhns DB, Wright DG, Nath J, Kaplan SS, Basford RE. 1988. ATP induces transient elevations of [Ca2+]i in human neutrophils and primes these cells for enhanced O2- generation. Lab Invest 58:448-453.

91
Kumar S, Chakrabarti R. 2000. [8-(Diethylamino)octyl-3,4,5-trimethoxybenzoate, HCl], the inhibitor of intracellular calcium mobilization, blocked mitogen-induced T cell proliferation by interfering with the sustained phase of protein kinase C activation. J Cell Biochem 76:539-547.
Larsson SC, Wolk A. 2007. Magnesium intake and risk of type 2 diabetes: a meta-analysis. J Intern Med 262:208-214.
Laughlin MR, Thompson D. 1996. The regulatory role for magnesium in glycolytic flux of the human erythrocyte. J Biol Chem 271:28977-28983.
Lee C, Xu DZ, Feketeova E, Kannan KB, Fekete Z, Deitch EA, Livingston DH, Hauser CJ. 2005. Store-operated calcium channel inhibition attenuates neutrophil function and postshock acute lung injury. J Trauma 59:56-63; discussion 63.
Li FY, Chaigne-Delalande B, Kanellopoulou C, Davis JC, Matthews HF, Douek DC, Cohen JI, Uzel G, Su HC, Lenardo MJ. 2011. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature 475:471-476.
Libako P, Nowacki W, Rock E, Rayssiguier Y, Mazur A. 2009. Phagocyte priming by low magnesium status: input to the enhanced inflammatory and oxidative stress responses. Magnes Res 23:1-4.
Lijnen P, Fagard R, Petrov V. 1999. Mibefradil-induced inhibition of proliferation of human peripheral blood mononuclear cells. J Cardiovasc Pharmacol 33:595-604.
Lin CY, Tsai PS, Hung YC, Huang CJ. 2010. L-type calcium channels are involved in mediating the anti-inflammatory effects of magnesium sulphate. Br J Anaesth 104:44-51.
Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402-408.
Lloyds D, Hallett MB. 1993. Activation and priming of the human neutrophil oxidase response by substance P: distinct signal transduction pathways. Biochim Biophys Acta 1175:207-213.
Lopez-Ridaura R, Willett WC, Rimm EB, Liu S, Stampfer MJ, Manson JE, Hu FB. 2004. Magnesium intake and risk of type 2 diabetes in men and women. Diabetes Care 27:134-140.
Ludi H, Schatzmann HJ. 1987. Some properties of a system for sodium-dependent outward movement of magnesium from metabolizing human red blood cells. J Physiol 390:367-382.
Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, Schuler G. 1999. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods 223:77-92.
Ma HT, Beaven MA. Regulators of Ca(2+) signaling in mast cells: potential targets for treatment of mast cell-related diseases? Adv Exp Med Biol 716:62-90.
Ma HT, Beaven MA. 2011. Regulators of Ca(2+) signaling in mast cells: potential targets for treatment of mast cell-related diseases? Adv Exp Med Biol 716:62-90.
Maguire ME, Cowan JA. 2002. Magnesium chemistry and biochemistry. Biometals 15:203-210.
Maier JA, Bernardini D, Rayssiguier Y, Mazur A. 2004. High concentrations of magnesium modulate vascular endothelial cell behaviour in vitro. Biochim Biophys Acta 1689:6-12.
Malpuech-Brugere C, Kuryszko J, Nowacki W, Rock E, Rayssiguier Y, Mazur A. 1998a. Early morphological and immunological alterations in the spleen during magnesium deficiency in the rat. Magnes Res 11:161-169.

92
Malpuech-Brugere C, Nowacki W, Daveau M, Gueux E, Linard C, Rock E, Lebreton J, Mazur A, Rayssiguier Y. 2000. Inflammatory response following acute magnesium deficiency in the rat. Biochim Biophys Acta 1501:91-98.
Malpuech-Brugere C, Nowacki W, Gueux E, Kuryszko J, Rock E, Rayssiguier Y, Mazur A. 1999a. Accelerated thymus involution in magnesium-deficient rats is related to enhanced apoptosis and sensitivity to oxidative stress. Br J Nutr 81:405-411.
Malpuech-Brugere C, Nowacki W, Rock E, Gueux E, Mazur A, Rayssiguier Y. 1999b. Enhanced tumor necrosis factor-alpha production following endotoxin challenge in rats is an early event during magnesium deficiency. Biochim Biophys Acta 1453:35-40.
Malpuech-Brugere C, Rock E, Astier C, Nowacki W, Mazur A, Rayssiguier Y. 1998b. Exacerbated immune stress response during experimental magnesium deficiency results from abnormal cell calcium homeostasis. Life Sci 63:1815-1822.
Mann DL, Topkara VK, Evans S, Barger PM. 2010. Innate immunity in the adult mammalian heart: for whom the cell tolls. Trans Am Clin Climatol Assoc 121:34-50; discussion 50-31.
McMillen MA, Lewis T, Jaffe BM, Wait RB. 1985. Verapamil inhibition of lymphocyte proliferation and function in vitro. J Surg Res 39:76-80.
Modiano JF, Kelepouris E, Kern JA, Nowell PC. 1988. Requirement for extracellular calcium or magnesium in mitogen-induced activation of human peripheral blood lymphocytes. J Cell Physiol 135:451-458.
Morceli G, Honorio-Franca AC, Fagundes DL, Calderon IM, Franca EL. 2013. Antioxidant effect of melatonin on the functional activity of colostral phagocytes in diabetic women. PLoS One 8:e56915.
Nadler JL, Buchanan T, Natarajan R, Antonipillai I, Bergman R, Rude R. 1993. Magnesium deficiency produces insulin resistance and increased thromboxane synthesis. Hypertension 21:1024-1029.
Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ, Kurosaki T, Kinet JP, Penner R, Scharenberg AM, Fleig A. 2001. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature 411:590-595.
Nielsen, LK J, H. Z. 2010. Magnesium supplementation improves indicators of low magnesium status and inflammatory stress in adults older than 51 years with poor quality sleep. Magnes Res. 23:158-168.
Nielsen FH, Milne DB, Klevay LM, Gallagher S, Johnson L. 2007. Dietary magnesium deficiency induces heart rhythm changes, impairs glucose tolerance, and decreases serum cholesterol in post menopausal women. J Am Coll Nutr 26:121-132.
Nishio A, Kikuchi K, Ueda A, Miyamoto A, Ishiguro S. 2001. Phagocytosis by alveolar macrophages during dietary magnesium deficiency in adult rats. Advances in magnesium research: nutrition and health 297-300.
O'Connor TM, O'Connell J, O'Brien DI, Goode T, Bredin CP, Shanahan F. 2004. The role of substance P in inflammatory disease. J Cell Physiol 201:167-180.
Odette O. 2005. Grass tetany in a herd of beef cows. Can Vet J 46:732-734. Oh-hora M, Rao A. 2008. Calcium signaling in lymphocytes. Curr Opin Immunol 20:250-
258. Ohki S, Ikura M, Zhang M. 1997. Identification of Mg2+-binding sites and the role of Mg2+
on target recognition by calmodulin. Biochemistry 36:4309-4316. Oono H, Nakagawa M, Miyamoto A, Ishiguro S, Nishio A. 2002. Mechanisms underlying the
enhanced elevation of IL-1beta and TNF-alpha mRNA levels following endotoxin challenge in rat alveolar macrophages cultured with low-Mg2+ medium. Magnes Res 15:153-160.

93
Paolisso G, Sgambato S, Gambardella A, Pizza G, Tesauro P, Varricchio M, D'Onofrio F. 1992. Daily magnesium supplements improve glucose handling in elderly subjects. Am J Clin Nutr 55:1161-1167.
Pearson TA, Mensah GA, Alexander RW, Anderson JL, Cannon RO, 3rd, Criqui M, Fadl YY, Fortmann SP, Hong Y, Myers GL, Rifai N, Smith SC, Jr., Taubert K, Tracy RP, Vinicor F. 2003. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: A statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation 107:499-511.
Perianin A, Snyderman R, Malfroy B. 1989. Substance P primes human neutrophil activation: a mechanism for neurological regulation of inflammation. Biochem Biophys Res Commun 161:520-524.
Pham PC, Pham PM, Pham SV, Miller JM, Pham PT. 2007. Hypomagnesemia in patients with type 2 diabetes. Clin J Am Soc Nephrol 2:366-373.
Poggi A, Rubartelli A, Zocchi MR. 1998. Involvement of dihydropyridine-sensitive calcium channels in human dendritic cell function. Competition by HIV-1 Tat. J Biol Chem 273:7205-7209.
Porcaro I, Vidal M, Jouvert S, Stahl PD, Giaimis J. 2003. Mannose receptor contribution to Candida albicans phagocytosis by murine E-clone J774 macrophages. J Leukoc Biol 74:206-215.
Pozzan T, Rizzuto R, Volpe P, Meldolesi J. 1994. Molecular and cellular physiology of intracellular calcium stores. Physiol Rev 74:595-636.
Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. 2001. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 286:327-334.
Quamme GA. 2008. Recent developments in intestinal magnesium absorption. Curr Opin Gastroenterol 24:230-235.
Quamme GA. 2010. Molecular identification of ancient and modern mammalian magnesium transporters. Am J Physiol Cell Physiol 298:C407-429.
Quamme GA, Rabkin SW. 1990. Cytosolic free magnesium in cardiac myocytes: identification of a Mg2+ influx pathway. Biochem Biophys Res Commun 167:1406-1412.
Reffelmann T, Dorr M, Ittermann T, Schwahn C, Volzke H, Ruppert J, Robinson D, Felix SB. 2010. Low serum magnesium concentrations predict increase in left ventricular mass over 5 years independently of common cardiovascular risk factors. Atherosclerosis 213:563-569.
Reffelmann T, Ittermann T, Dorr M, Volzke H, Reinthaler M, Petersmann A, Felix SB. 2011. Low serum magnesium concentrations predict cardiovascular and all-cause mortality. Atherosclerosis 219:280-284.
Ridker PM. 2007. Inflammatory biomarkers and risks of myocardial infarction, stroke, diabetes, and total mortality: implications for longevity. Nutr Rev 65:S253-259.
Rodriguez-Moran M, Guerrero-Romero F. 2003. Oral magnesium supplementation improves insulin sensitivity and metabolic control in type 2 diabetic subjects: a randomized double-blind controlled trial. Diabetes Care 26:1147-1152.
Rodriguez-Moran M, Guerrero-Romero F. 2008. Serum magnesium and C-reactive protein levels. Arch Dis Child 93:676-680.
Rogers TA, Mahan PE. 1959. Exchange of radioactive magnesium in the rat. Proc Soc Exp Biol Med 100:235-239.
Romagnani S. 1999. Th1/Th2 cells. Inflamm Bowel Dis 5:285-294.

94
Romani A, Marfella C, Scarpa A. 1993a. Hormonal stimulation of Mg2+ uptake in hepatocytes. Regulation by plasma membrane and intracellular organelles. J Biol Chem 268:15489-15495.
Romani A, Marfella C, Scarpa A. 1993b. Regulation of magnesium uptake and release in the heart and in isolated ventricular myocytes. Circ Res 72:1139-1148.
Romani A, Scarpa A. 1992. Regulation of cell magnesium. Arch Biochem Biophys 298:1-12. Romani AM. 2011. Cellular magnesium homeostasis. Arch Biochem Biophys 512:1-23. Roschger P, Graninger W, Klima H. 1990. Magnesium-dependent induction of phagocytosis-
associated chemiluminescence of adherent human polymorphonuclear leukocytes by non-opsonized zymosan. J Biolumin Chemilumin 5:31-36.
Rubin AH, Chu B. 1978. Reversible regulation by magnesium of chick embryo fibroblast proliferation. J Cell Physiol 94:13-19.
Rubin AH, Terasaki M, Sanui H. 1979. Major intracellular cations and growth control: correspondence among magnesium content, protein synthesis, and the onset of DNA synthesis in BALB/c3T3 cells. Proc Natl Acad Sci U S A 76:3917-3921.
Rude RK. 1993. Magnesium metabolism and deficiency. Endocrinol Metab Clin North Am 22:377-395.
Rutter GA, Osbaldeston NJ, McCormack JG, Denton RM. 1990. Measurement of matrix free Mg2+ concentration in rat heart mitochondria by using entrapped fluorescent probes. Biochem J 271:627-634.
Ryzen E, Wagers PW, Singer FR, Rude RK. 1985. Magnesium deficiency in a medical ICU population. Crit Care Med 13:19-21.
Sadighi Akha AA, Willmott NJ, Brickley K, Dolphin AC, Galione A, Hunt SV. 1996. Anti-Ig-induced calcium influx in rat B lymphocytes mediated by cGMP through a dihydropyridine-sensitive channel. J Biol Chem 271:7297-7300.
Sahni J, Nelson B, Scharenberg AM. 2007. SLC41A2 encodes a plasma-membrane Mg2+ transporter. Biochem J 401:505-513.
Sakaguchi H, Sakaguchi R, Ishiguro S, Nishio A. 1992. Beneficial effects of a calcium antagonist, nifedipine, on death and cardiovascular calcinosis induced by dietary magnesium deficiency in adult mice. Magnes Res 5:121-125.
Saris NE, Mervaala E, Karppanen H, Khawaja JA, Lewenstam A. 2000. Magnesium. An update on physiological, clinical and analytical aspects. Clin Chim Acta 294:1-26.
Satoh M, Tamura G, Segawa I, Tashiro A, Hiramori K, Satodate R. 1996. Expression of cytokine genes and presence of enteroviral genomic RNA in endomyocardial biopsy tissues of myocarditis and dilated cardiomyopathy. Virchows Arch 427:503-509.
Savignac M, Gomes B, Gallard A, Narbonnet S, Moreau M, Leclerc C, Paulet P, Mariame B, Druet P, Saoudi A, Fournie GJ, Guery JC, Pelletier L. 2004. Dihydropyridine receptors are selective markers of Th2 cells and can be targeted to prevent Th2-dependent immunopathological disorders. J Immunol 172:5206-5212.
Schempp CM, Dittmar HC, Hummler D, Simon-Haarhaus B, Schulte-Monting J, Schopf E, Simon JC. 2000. Magnesium ions inhibit the antigen-presenting function of human epidermal Langerhans cells in vivo and in vitro. Involvement of ATPase, HLA-DR, B7 molecules, and cytokines. J Invest Dermatol 115:680-686.
Schlingmann KP, Weber S, Peters M, Niemann Nejsum L, Vitzthum H, Klingel K, Kratz M, Haddad E, Ristoff E, Dinour D, Syrrou M, Nielsen S, Sassen M, Waldegger S, Seyberth HW, Konrad M. 2002. Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat Genet 31:166-170.

95
Schmitz C, Perraud AL, Johnson CO, Inabe K, Smith MK, Penner R, Kurosaki T, Fleig A, Scharenberg AM. 2003. Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell 114:191-200.
Schroeder HA, Nason AP, Tipton IH. 1969. Essential metals in man. Magnesium. J Chronic Dis 21:815-841.
Sgambato A, Wolf FI, Faraglia B, Cittadini A. 1999. Magnesium depletion causes growth inhibition, reduced expression of cyclin D1, and increased expression of P27Kip1 in normal but not in transformed mammary epithelial cells. J Cell Physiol 180:245-254.
Shen H, Wiederhold MD, Ou DW. 1995. The suppression of macrophage secretion by calcium blockers and adenosine. Immunopharmacol Immunotoxicol 17:301-309.
Shils ME. 1969a. Experimental human magnesium depletion. Medicine (Baltimore) 48:61-85. Shils ME. 1969b. Experimental production of magnesium deficiency in man. Ann N Y Acad
Sci 162:847-855. Shima E, Katsube M, Kato T, Kitagawa M, Hato F, Hino M, Takahashi T, Fujita H, Kitagawa
S. 2008. Calcium channel blockers suppress cytokine-induced activation of human neutrophils. Am J Hypertens 21:78-84.
Shogi T, Miyamoto A, Ishiguro S, Nishio A. 2003. Enhanced release of IL-1beta and TNF-alpha following endotoxin challenge from rat alveolar macrophages cultured in low-mg(2+) medium. Magnes Res 16:111-119.
Shogi T, Oono H, Nakagawa M, Miyamoto A, Ishiguro S, Nishio A. 2002. Effects of a low extracellular magnesium concentration and endotoxin on IL-1beta and TNF-alpha release from, and mRNA levels in, isolated rat alveolar macrophages. Magnes Res 15:147-152.
Shuman H, Somlyo AP. 1987. Electron energy loss analysis of near-trace-element concentrations of calcium. Ultramicroscopy 21:23-32.
Simchowitz L, Foy MA, Cragoe EJ, Jr. 1990. A role for Na+/Ca2+ exchange in the generation of superoxide radicals by human neutrophils. J Biol Chem 265:13449-13456.
Simmons D, Joshi S, Shaw J. Hypomagnesaemia is associated with diabetes: Not pre-diabetes, obesity or the metabolic syndrome. Diabetes Res Clin Pract 87:261-266.
Singh AB, Hiehle K, Casale P, Gerber S, Nayar S, Mann RA. 1990. Modulation of murine in vitro immune response by verapamil (V). Immunopharmacology 20:165-174.
Sloan-Lancaster J, Steinberg TH, Allen PM. 1997. Selective loss of the calcium ion signaling pathway in T cells maturing toward a T helper 2 phenotype. J Immunol 159:1160-1168.
Song, Li TY, van Dam RM, Manson JE, FB H. 2007. Magnesium intake and plasma concentrations of markers of systemic inflammation and endothelial dysfunction in women. Am J Clin Nutr. 85:1068-1074.
Song Y, Manson JE, Buring JE, Liu S. 2004. Dietary magnesium intake in relation to plasma insulin levels and risk of type 2 diabetes in women. Diabetes Care 27:59-65.
Sonna LA, Hirshman CA, Croxton TL. 1996. Role of calcium channel blockade in relaxation of tracheal smooth muscle by extracellular Mg2+. Am J Physiol 271:L251-257.
Spencer H, Lesniak M, Gatza CA, Osis D, Lender M. 1980. Magnesium absorption and metabolism in patients with chronic renal failure and in patients with normal renal function. Gastroenterology 79:26-34.
Stanisz AM, Scicchitano R, Dazin P, Bienenstock J, Payan DG. 1987. Distribution of substance P receptors on murine spleen and Peyer's patch T and B cells. J Immunol 139:749-754.
Stanley ER. 1997. Murine bone marrow-derived macrophages. Methods Mol Biol 75:301-304.

96
Takaya J, Higashino H, Kobayashi Y. 2004. Intracellular magnesium and insulin resistance. Magnes Res 17:126-136.
Talavera K, Nilius B. 2006. Biophysics and structure-function relationship of T-type Ca2+ channels. Cell Calcium 40:97-114.
Tanizaki Y, Akagi K, Lee KN, Townley RG. 1983a. Inhibitory effect of nifedipine and cromolyn sodium on skin reactions and 45Ca uptake and histamine release in rat mast cells induced by various stimulating agents. Int Arch Allergy Appl Immunol 72:102-109.
Tanizaki Y, Komagoe H, Sudo M, Ohtani J, Kimura I, Akagi K, Townley RG. 1983b. Inhibitory effect of the CA2+ antagonist nifedipine on histamine release from rat peritoneal mast cells. Acta Med Okayama 37:207-211.
Tashiro M, Konishi M. 1997. Na+ gradient-dependent Mg2+ transport in smooth muscle cells of guinea pig tenia cecum. Biophys J 73:3371-3384.
Tennenbaum T, Yuspa SH, Kapitulnik J. 1990. Magnesium and phosphate enrichment of culture medium stimulates the proliferation of epidermal cells from newborn and adult mice. J Cell Physiol 143:431-438.
Tennenberg SD, Fey DE, Lieser MJ. 1993. Oxidative priming of neutrophils by interferon-c. J Leukocyte Biol 53:301-308.
Tessman PA, Romani A. 1998. Acute effect of EtOH on Mg2+ homeostasis in liver cells: evidence for the activation of an Na+/Mg2+ exchanger. Am J Physiol 275:G1106-1116.
Tiemann U, Neels P, Pohland R, Walzel H, Lohrke B. 1999. Influence of inhibitors on increase in intracellular free calcium and proliferation induced by platelet-activating factor in bovine oviductal cells. J Reprod Fertil 116:63-72.
Tintinger G, Steel HC, Anderson R. 2005. Taming the neutrophil: calcium clearance and influx mechanisms as novel targets for pharmacological control. Clin Exp Immunol 141:191-200.
Topkara VK, Evans S, Zhang W, Epelman S, Staloch L, Barger PM, Mann DL. 2011. Therapeutic targeting of innate immunity in the failing heart. J Mol Cell Cardiol 51:594-599.
Tosiello L. 1996. Hypomagnesemia and diabetes mellitus. A review of clinical implications. Arch Intern Med 156:1143-1148.
Turlapaty PD, Altura BM. 1978. Extracellular magnesium ions control calcium exchange and content of vascular smooth muscle. Eur J Pharmacol 52:421-423.
Vercellotti GM, Yin HQ, Gustafson KS, Nelson RD, Jacob HS. 1988. Platelet-activating factor primes neutrophil responses to agonists: role in promoting neutrophil-mediated endothelial damage. Blood 71:1100-1107.
Voets T, Nilius B, Hoefs S, van der Kemp AW, Droogmans G, Bindels RJ, Hoenderop JG. 2004. TRPM6 forms the Mg2+ influx channel involved in intestinal and renal Mg2+ absorption. J Biol Chem 279:19-25.
Wabakken T, Rian E, Kveine M, Aasheim HC. 2003. The human solute carrier SLC41A1 belongs to a novel eukaryotic subfamily with homology to prokaryotic MgtE Mg2+ transporters. Biochem Biophys Res Commun 306:718-724.
Wang CY, Shi JD, Yang P, Kumar PG, Li QZ, Run QG, Su YC, Scott HS, Kao KJ, She JX. 2003. Molecular cloning and characterization of a novel gene family of four ancient conserved domain proteins (ACDP). Gene 306:37-44.
Wang CY, Yang P, Shi JD, Purohit S, Guo D, An H, Gu JG, Ling J, Dong Z, She JX. 2004. Molecular cloning and characterization of the mouse Acdp gene family. BMC Genomics 5:7.

97
Watanabe N, Suzuki J, Kobayashi Y. 1996. Role of calcium in tumor necrosis factor-alpha production by activated macrophages. J Biochem 120:1190-1195.
Weglicki WB, Chmielinska JJ, Tejero-Taldo I, Kramer JH, Spurney CF, Viswalingham K, Lu B, Mak IT. 2009. Neutral endopeptidase inhibition enhances substance P mediated inflammation due to hypomagnesemia. Magnes Res 22:167S-173S.
Weglicki WB, Dickens BF, Wagner TL, Chmielinska JJ, Phillips TM. 1996. Immunoregulation by neuropeptides in magnesium deficiency: ex vivo effect of enhanced substance P production on circulating T lymphocytes from magnesium-deficient mice. Magnes Res 9:3-11.
Weglicki WB, Phillips TM. 1992. Pathobiology of magnesium deficiency: a cytokine/neurogenic inflammation hypothesis. Am J Physiol 263:R734-737.
Weglicki WB, Phillips TM, Freedman AM, Cassidy MM, Dickens BF. 1992. Magnesium-deficiency elevates circulating levels of inflammatory cytokines and endothelin. Mol Cell Biochem 110:169-173.
Weisbart RH, Kwan L, Golde DW, Gasson JC. 1987. Human GM-CSF primes neutrophils for enhanced oxidative metabolism in response to the major physiological chemoattractants. Blood 69:18-21.
Weischenfeldt J, Porse B. 2008. Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. CSH Protoc 2008:pdb prot5080.
Whang R, Ryder KW. 1990. Frequency of hypomagnesemia and hypermagnesemia. Requested vs routine. JAMA 263:3063-3064.
Williams RPJ. 1970. The biochemistry of sodium, potassium, magnesium, and calcium. Q. Rev. Chem. Soc. 24:331-365.
Wolf FI, Cittadini A. 1999. Magnesium in cell proliferation and differentiation. Front Biosci 4:D607-617.
Wolf FI, Di Francesco A, Covacci V, Cittadini A. 1997. Regulation of magnesium efflux from rat spleen lymphocytes. Arch Biochem Biophys 344:397-403.
Wong ET, Rude RK, Singer FR, Shaw ST, Jr. 1983. A high prevalence of hypomagnesemia and hypermagnesemia in hospitalized patients. Am J Clin Pathol 79:348-352.
Zdrojewski T., Bandosz P., P. S. 2004. Rozpowszechnienie głównych czynników ryzyka chorób układu sercowo-naczyniowego w Polsce. Wyniki badania NATPOL PLUS. Kardiol Pol 61:1-26.
Zieve FJ, Freude KA, Zieve L. 1977. Effects of magnesium deficiency on protein and nucleic acid synthesis in vivo. J Nutr 107:2178-2188.
Zimmermann P, Weiss U, Classen HG, Wendt B, Epple A, Zollner H, Temmel W, Weger M, Porta S. 2000. The impact of diets with different magnesium contents on magnesium and calcium in serum and tissues of the rat. Life Sci 67:949-958.

Ryc. 1 Wewnątrzkomórkowa dystrybucja Mg2+. Opracowanie własne. Schemat został wykonany z
wykorzystaniem materiałów Servier Medical Art
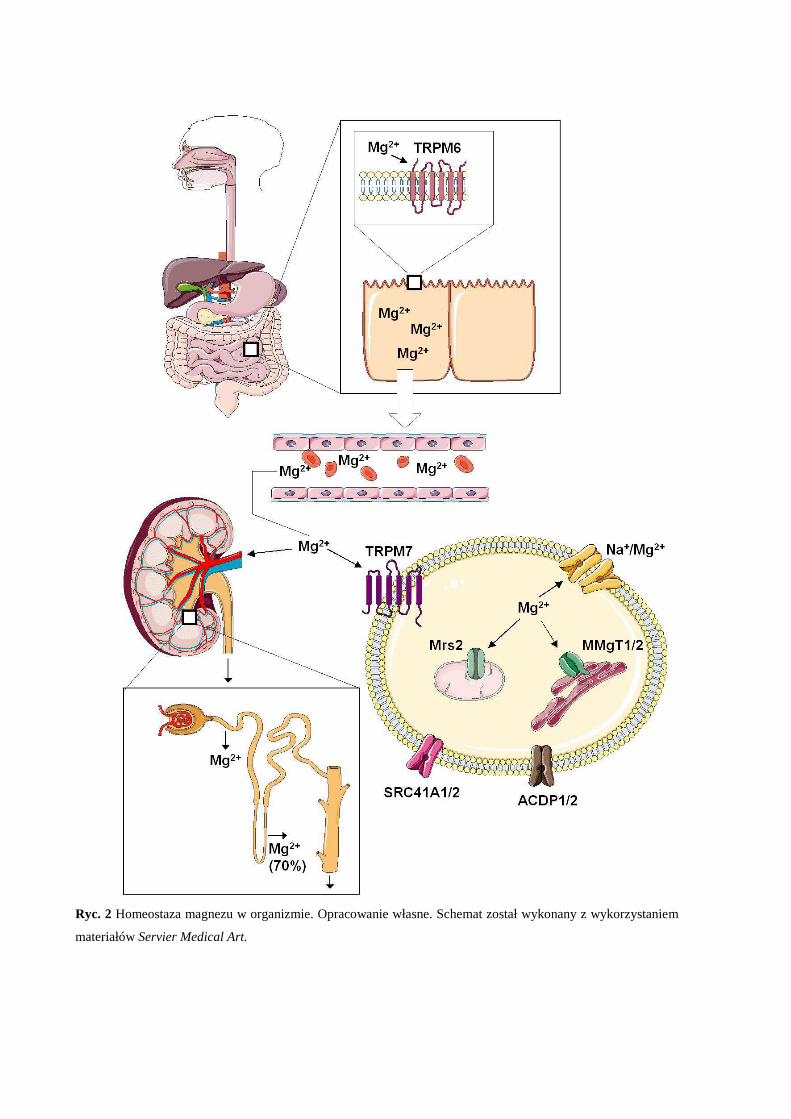
Ryc. 2 Homeostaza magnezu w organizmie. Opracowanie własne. Schemat został wykonany z wykorzystaniem
materiałów Servier Medical Art.
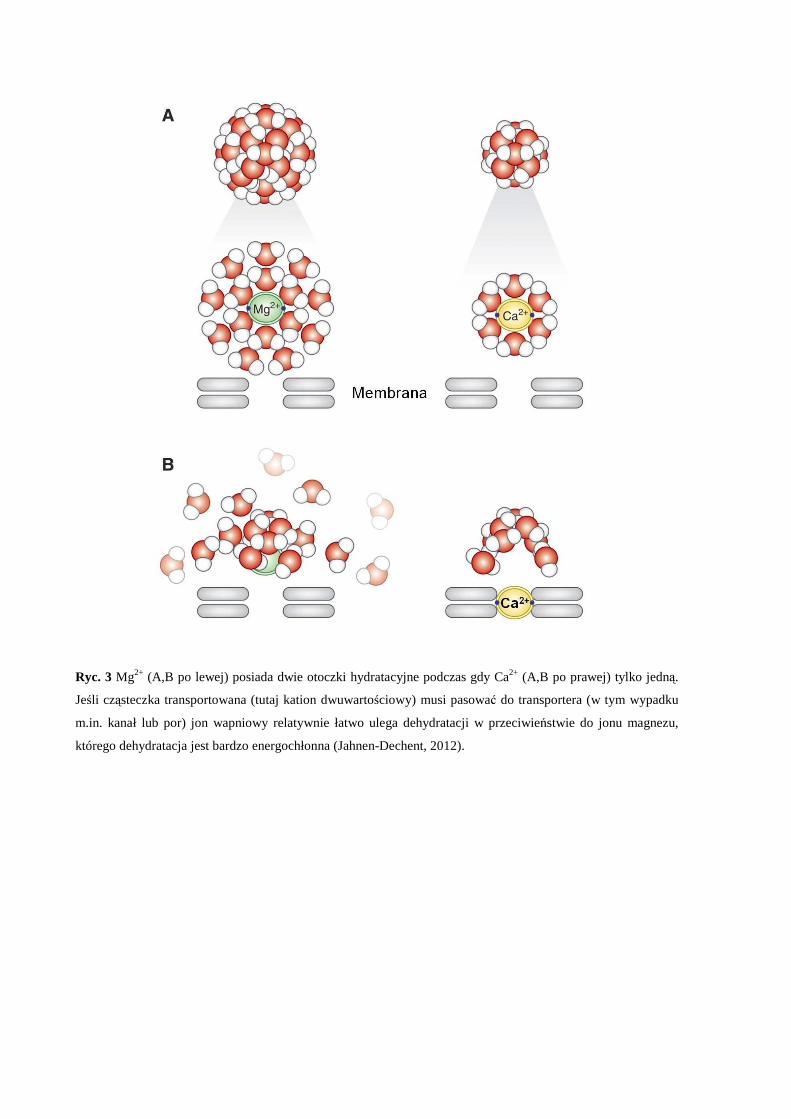
Ryc. 3 Mg2+ (A,B po lewej) posiada dwie otoczki hydratacyjne podczas gdy Ca2+ (A,B po prawej) tylko jedną.
Jeśli cząsteczka transportowana (tutaj kation dwuwartościowy) musi pasować do transportera (w tym wypadku
m.in. kanał lub por) jon wapniowy relatywnie łatwo ulega dehydratacji w przeciwieństwie do jonu magnezu,
którego dehydratacja jest bardzo energochłonna (Jahnen-Dechent, 2012).

Ryc. 4 Preaktywacja (priming) i aktywacja komórki fagocytarnej. Opracowanie własne. Schemat został
wykonany z wykorzystaniem materiałów Servier Medical Art.
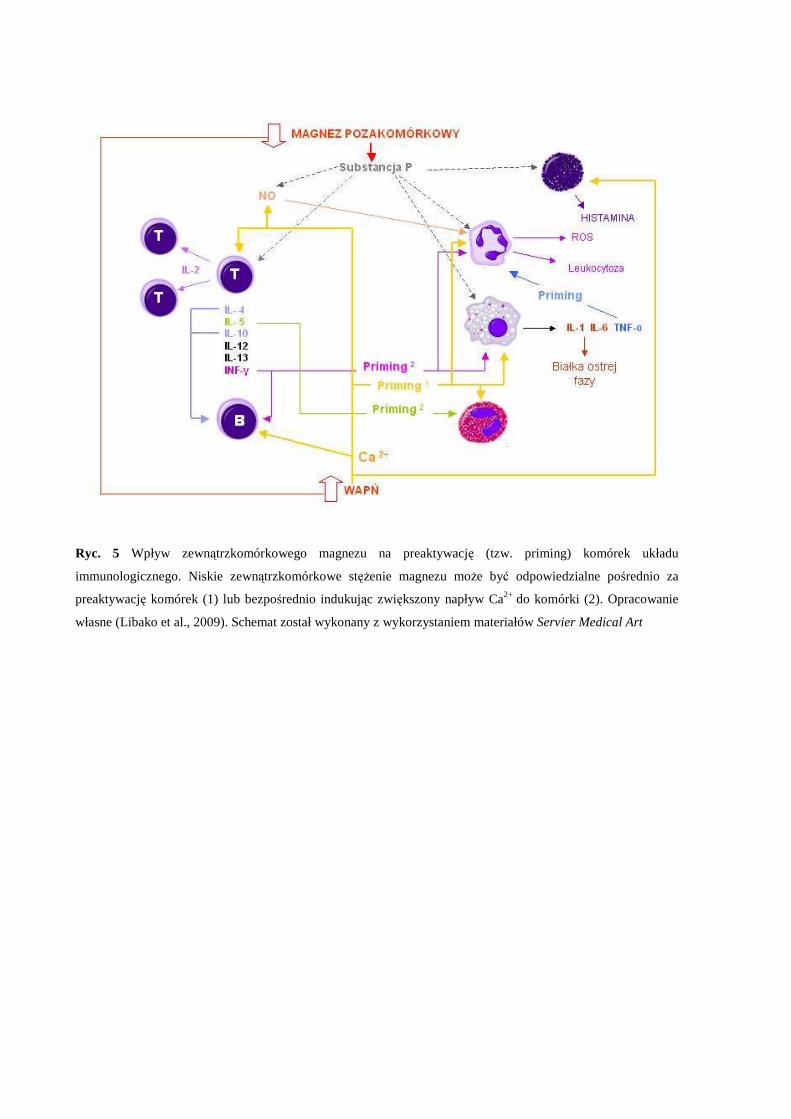
Ryc. 5 Wpływ zewnątrzkomórkowego magnezu na preaktywację (tzw. priming) komórek układu
immunologicznego. Niskie zewnątrzkomórkowe stężenie magnezu może być odpowiedzialne pośrednio za
preaktywację komórek (1) lub bezpośrednio indukując zwiększony napływ Ca2+ do komórki (2). Opracowanie
własne (Libako et al., 2009). Schemat został wykonany z wykorzystaniem materiałów Servier Medical Art
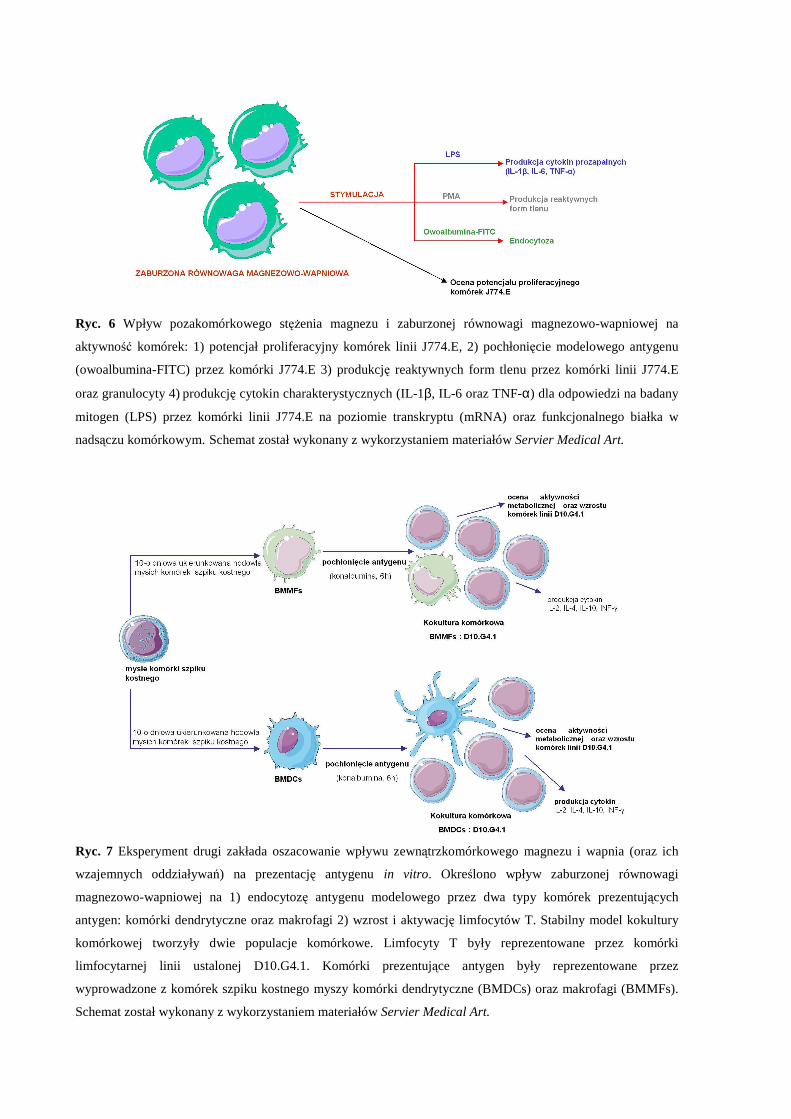
Ryc. 6 Wpływ pozakomórkowego stężenia magnezu i zaburzonej równowagi magnezowo-wapniowej na
aktywność komórek: 1) potencjał proliferacyjny komórek linii J774.E, 2) pochłonięcie modelowego antygenu
(owoalbumina-FITC) przez komórki J774.E 3) produkcję reaktywnych form tlenu przez komórki linii J774.E
oraz granulocyty 4) produkcję cytokin charakterystycznych (IL-1β, IL-6 oraz TNF-α) dla odpowiedzi na badany
mitogen (LPS) przez komórki linii J774.E na poziomie transkryptu (mRNA) oraz funkcjonalnego białka w
nadsączu komórkowym. Schemat został wykonany z wykorzystaniem materiałów Servier Medical Art.
Ryc. 7 Eksperyment drugi zakłada oszacowanie wpływu zewnątrzkomórkowego magnezu i wapnia (oraz ich
wzajemnych oddziaływań) na prezentację antygenu in vitro. Określono wpływ zaburzonej równowagi
magnezowo-wapniowej na 1) endocytozę antygenu modelowego przez dwa typy komórek prezentujących
antygen: komórki dendrytyczne oraz makrofagi 2) wzrost i aktywację limfocytów T. Stabilny model kokultury
komórkowej tworzyły dwie populacje komórkowe. Limfocyty T były reprezentowane przez komórki
limfocytarnej linii ustalonej D10.G4.1. Komórki prezentujące antygen były reprezentowane przez
wyprowadzone z komórek szpiku kostnego myszy komórki dendrytyczne (BMDCs) oraz makrofagi (BMMFs).
Schemat został wykonany z wykorzystaniem materiałów Servier Medical Art.
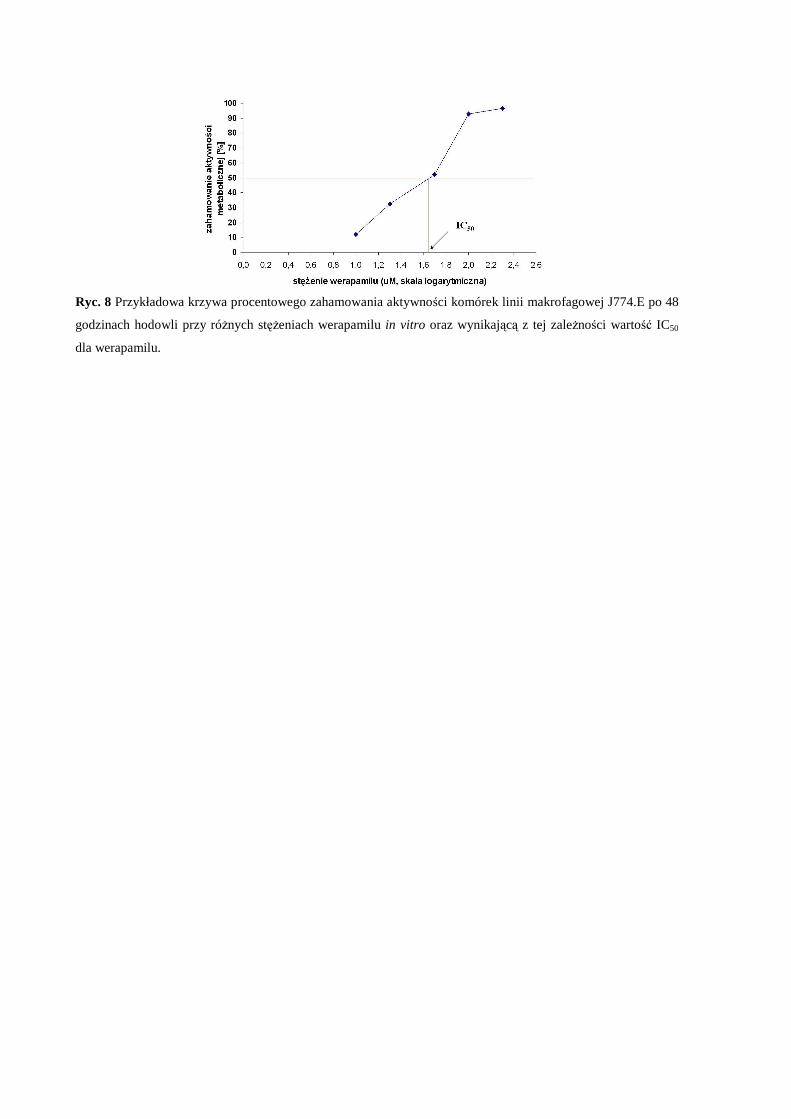
Ryc. 8 Przykładowa krzywa procentowego zahamowania aktywności komórek linii makrofagowej J774.E po 48
godzinach hodowli przy różnych stężeniach werapamilu in vitro oraz wynikającą z tej zależności wartość IC50
dla werapamilu.
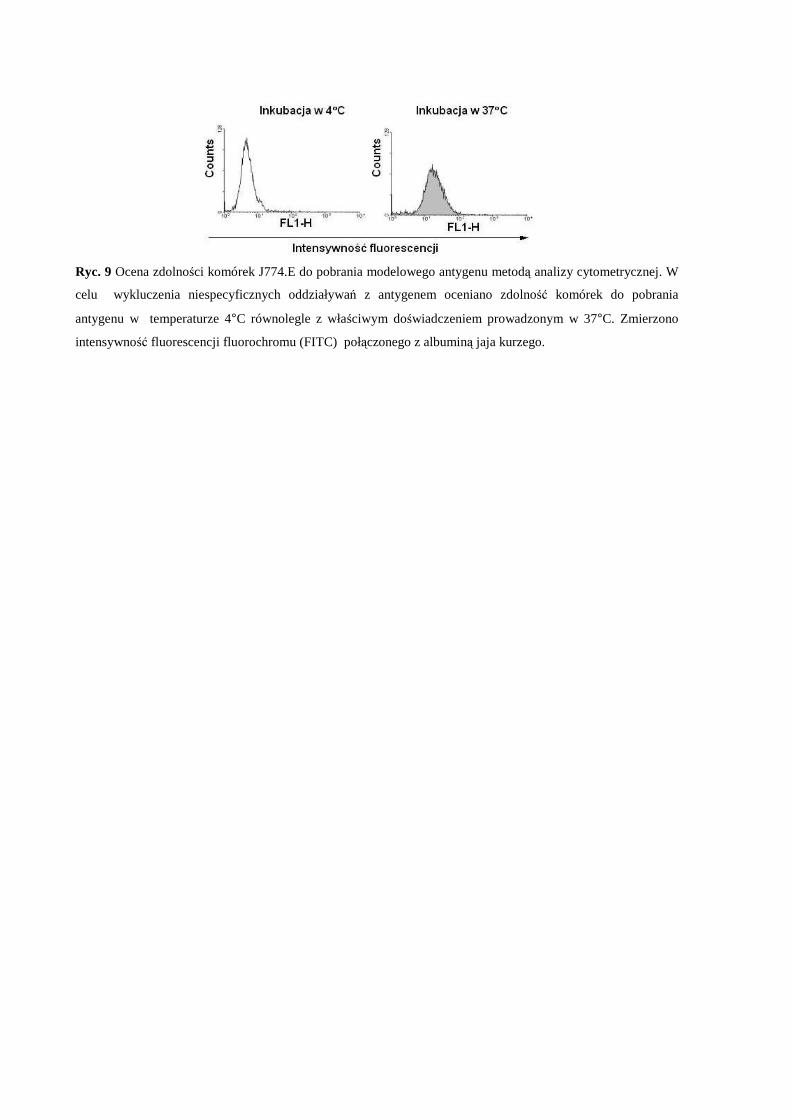
Ryc. 9 Ocena zdolności komórek J774.E do pobrania modelowego antygenu metodą analizy cytometrycznej. W
celu wykluczenia niespecyficznych oddziaływań z antygenem oceniano zdolność komórek do pobrania
antygenu w temperaturze 4°C równolegle z właściwym doświadczeniem prowadzonym w 37°C. Zmierzono
intensywność fluorescencji fluorochromu (FITC) połączonego z albuminą jaja kurzego.
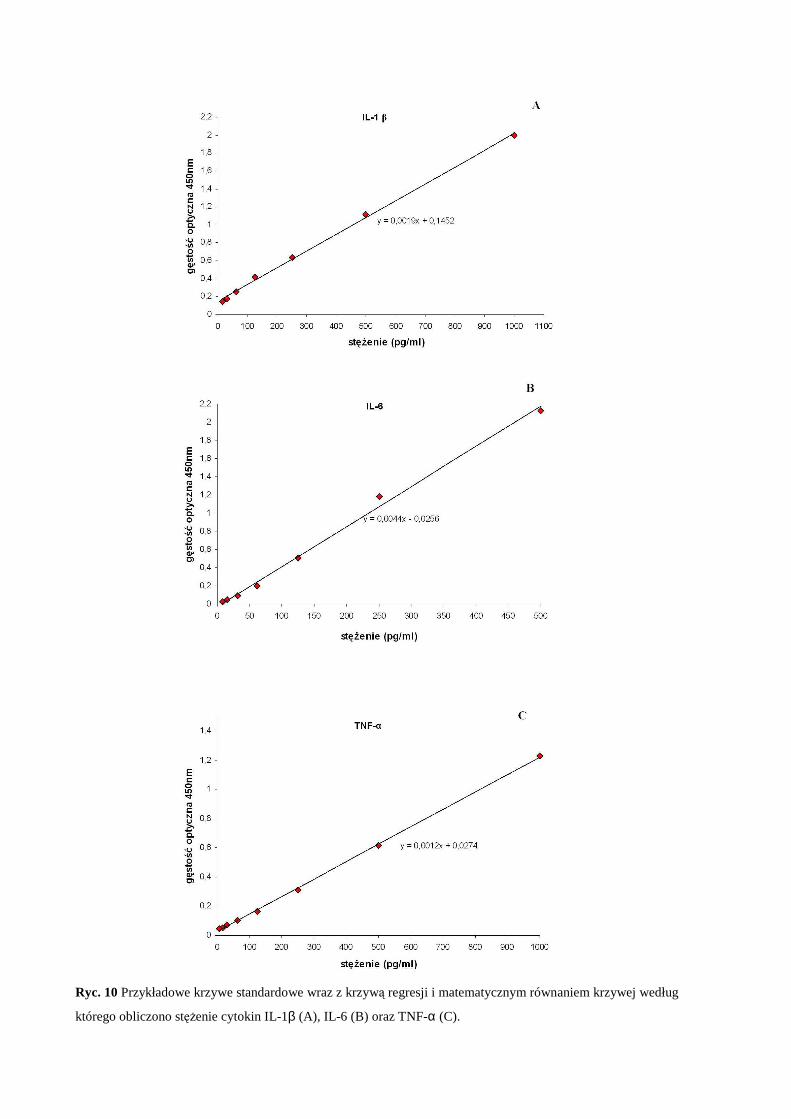
Ryc. 10 Przykładowe krzywe standardowe wraz z krzywą regresji i matematycznym równaniem krzywej według
którego obliczono stężenie cytokin IL-1β (A), IL-6 (B) oraz TNF-α (C).
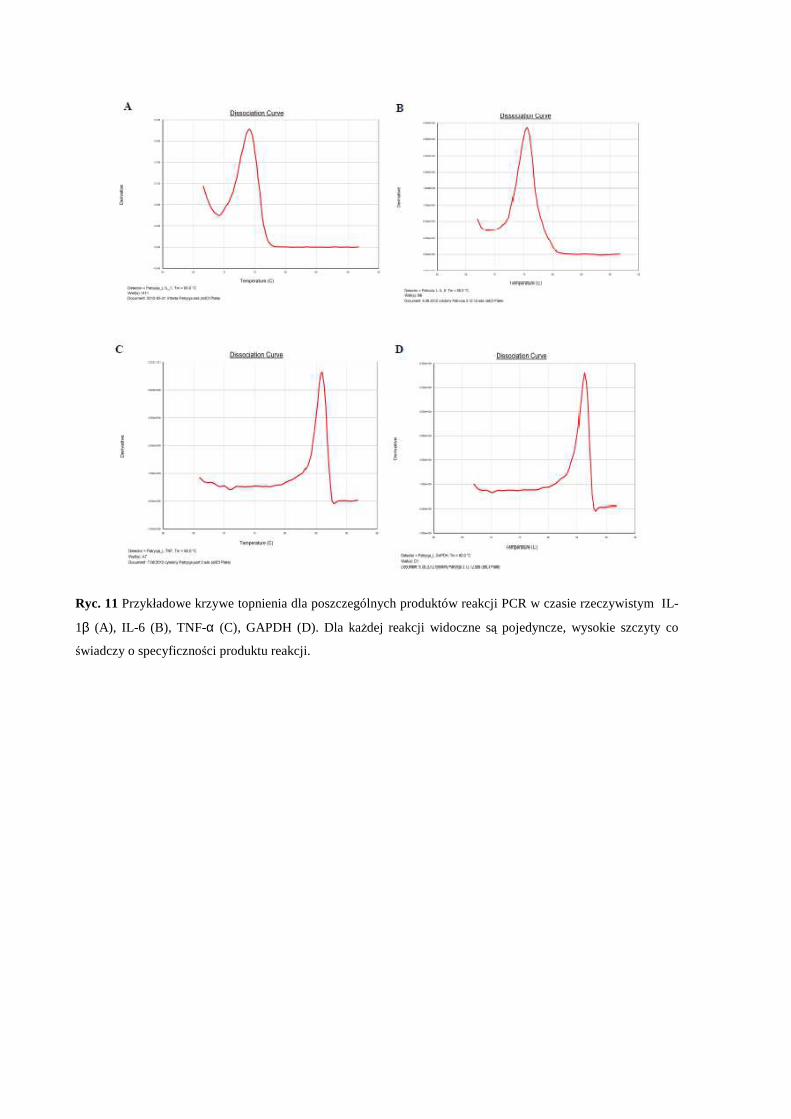
Ryc. 11 Przykładowe krzywe topnienia dla poszczególnych produktów reakcji PCR w czasie rzeczywistym IL-
1β (A), IL-6 (B), TNF-α (C), GAPDH (D). Dla każdej reakcji widoczne są pojedyncze, wysokie szczyty co
świadczy o specyficzności produktu reakcji.

Ryc. 12 Hodowla ukierunkowana komórek szpiku kostnego myszy (szczep C3H/HeN(H-2k)) – pozyskanie
modelowych komórek dendrytycznych (BMDCs) oraz makrofagów (BMMFs). Opracowanie własne. Schemat
został wykonany z wykorzystaniem materiałów Servier Medical Art.
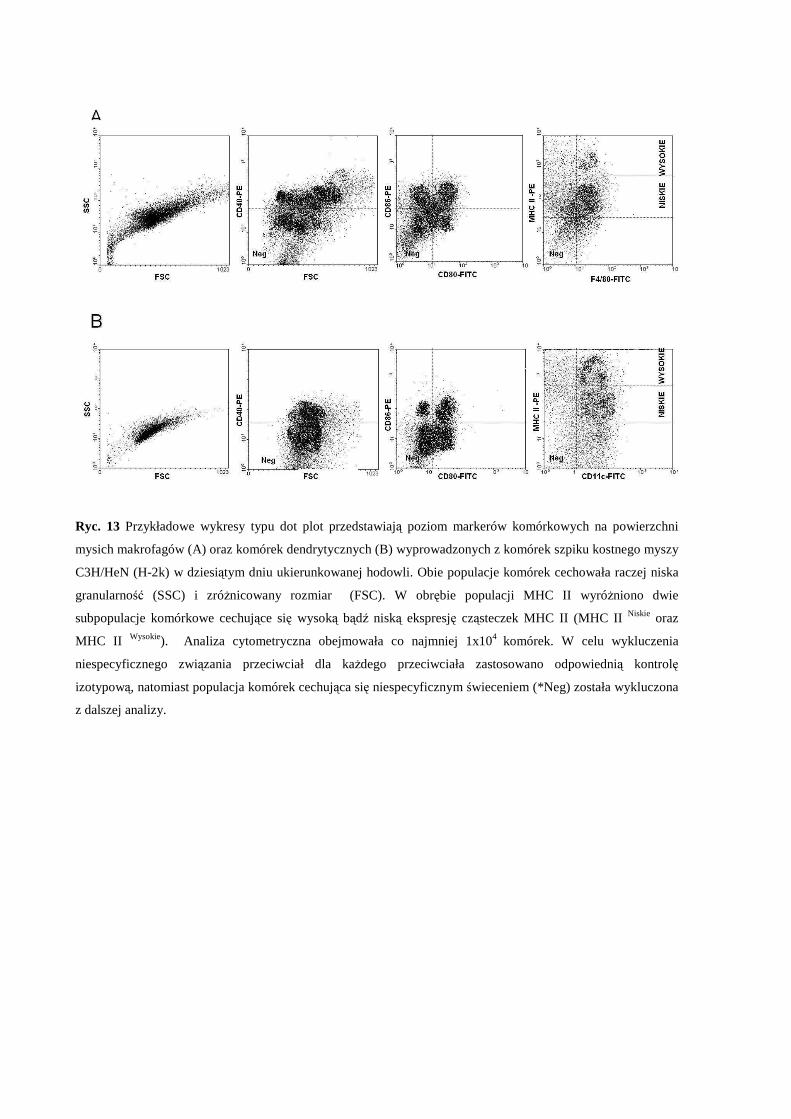
Ryc. 13 Przykładowe wykresy typu dot plot przedstawiają poziom markerów komórkowych na powierzchni
mysich makrofagów (A) oraz komórek dendrytycznych (B) wyprowadzonych z komórek szpiku kostnego myszy
C3H/HeN (H-2k) w dziesiątym dniu ukierunkowanej hodowli. Obie populacje komórek cechowała raczej niska
granularność (SSC) i zróżnicowany rozmiar (FSC). W obrębie populacji MHC II wyróżniono dwie
subpopulacje komórkowe cechujące się wysoką bądź niską ekspresję cząsteczek MHC II (MHC II Niskie oraz
MHC II Wysokie). Analiza cytometryczna obejmowała co najmniej 1x104 komórek. W celu wykluczenia
niespecyficznego związania przeciwciał dla każdego przeciwciała zastosowano odpowiednią kontrolę
izotypową, natomiast populacja komórek cechująca się niespecyficznym świeceniem (*Neg) została wykluczona
z dalszej analizy.
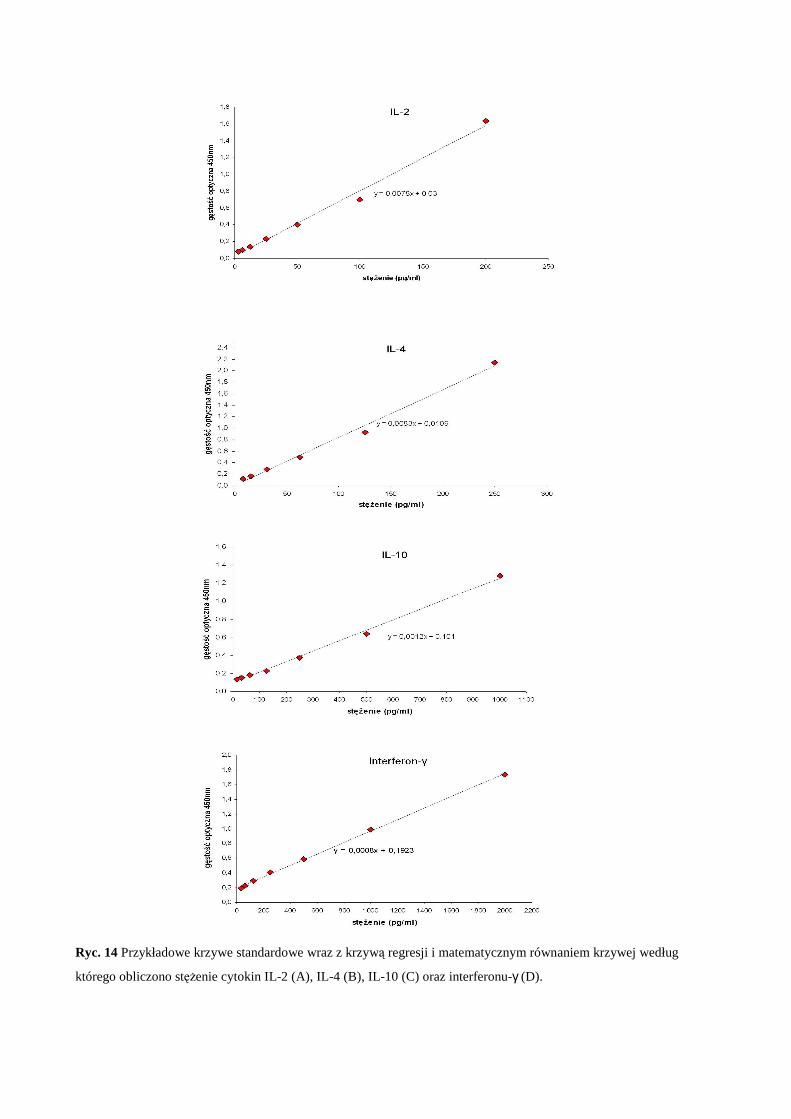
Ryc. 14 Przykładowe krzywe standardowe wraz z krzywą regresji i matematycznym równaniem krzywej według
którego obliczono stężenie cytokin IL-2 (A), IL-4 (B), IL-10 (C) oraz interferonu-γ (D).
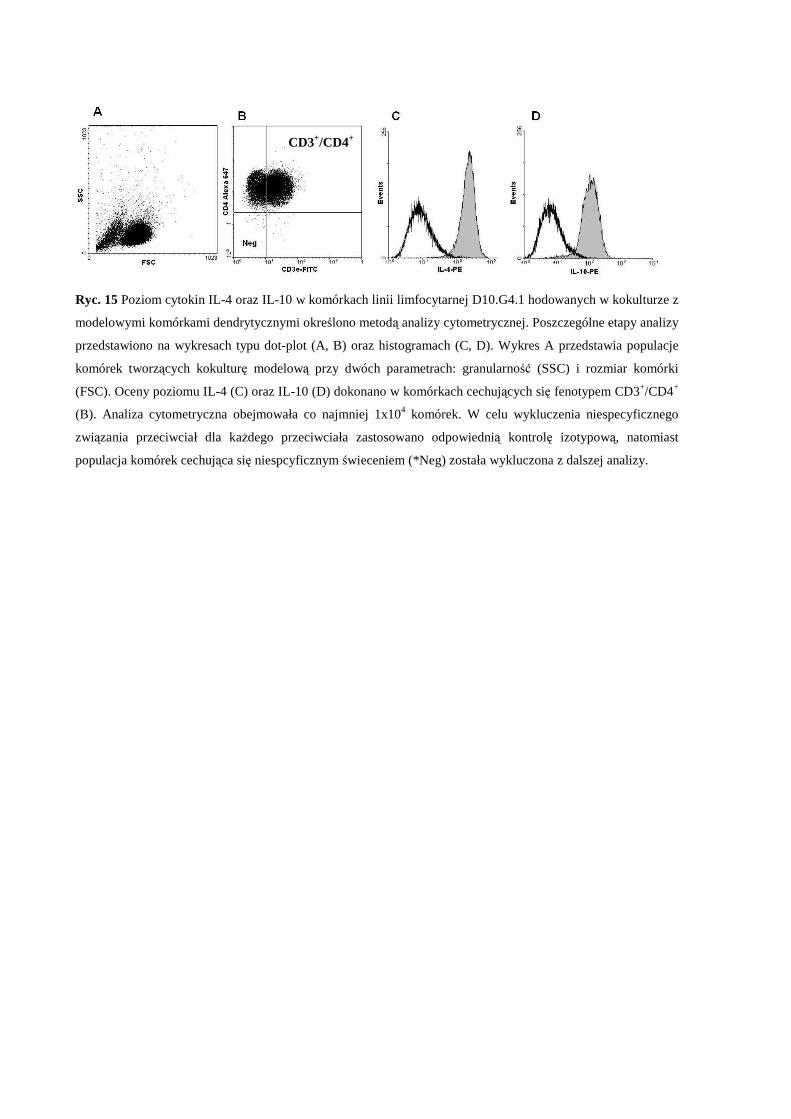
Ryc. 15 Poziom cytokin IL-4 oraz IL-10 w komórkach linii limfocytarnej D10.G4.1 hodowanych w kokulturze z
modelowymi komórkami dendrytycznymi określono metodą analizy cytometrycznej. Poszczególne etapy analizy
przedstawiono na wykresach typu dot-plot (A, B) oraz histogramach (C, D). Wykres A przedstawia populacje
komórek tworzących kokulturę modelową przy dwóch parametrach: granularność (SSC) i rozmiar komórki
(FSC). Oceny poziomu IL-4 (C) oraz IL-10 (D) dokonano w komórkach cechujących się fenotypem CD3+/CD4+
(B). Analiza cytometryczna obejmowała co najmniej 1x104 komórek. W celu wykluczenia niespecyficznego
związania przeciwciał dla każdego przeciwciała zastosowano odpowiednią kontrolę izotypową, natomiast
populacja komórek cechująca się niespcyficznym świeceniem (*Neg) została wykluczona z dalszej analizy.
CD3+/CD4+

Ryc. 16 Obraz przedstawia populację komórek J774.E hodowanych przez 24, 48 i 72 godziny w zmiennych
warunkach dostępności wapnia przy stałym stężeniu magnezu w medium (1,0 mM). Zdjęcia zostały wykonane
pod mikroskopem odwróconym (powiększenie x100) bezpośrednio przed oceną aktywności metabolicznej
komórek testem MTT. Obecność blokerów wapnia - werapamilu i TMB-8 w znaczący sposób ograniczyła
wzrost komórek J774.E w trzecim dniu eksperymentu w odniesieniu do warunku podstawowego (1,0 mM Mg).
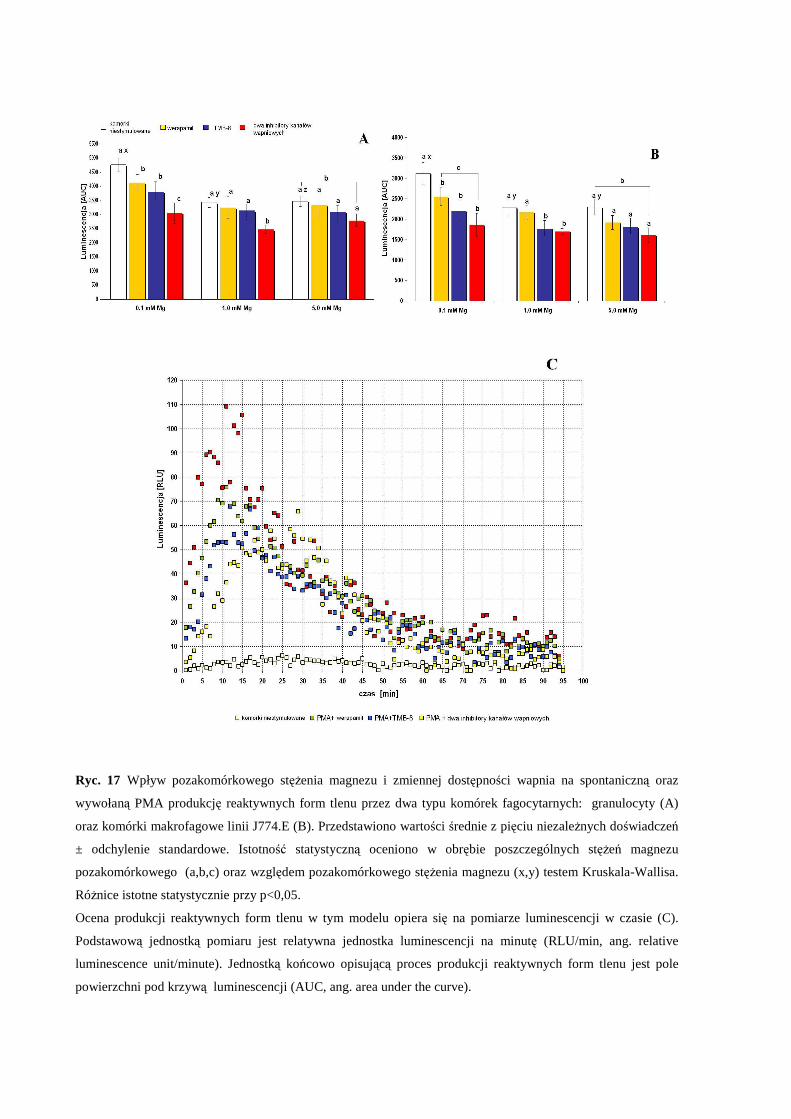
Ryc. 17 Wpływ pozakomórkowego stężenia magnezu i zmiennej dostępności wapnia na spontaniczną oraz
wywołaną PMA produkcję reaktywnych form tlenu przez dwa typu komórek fagocytarnych: granulocyty (A)
oraz komórki makrofagowe linii J774.E (B). Przedstawiono wartości średnie z pięciu niezależnych doświadczeń
± odchylenie standardowe. Istotność statystyczną oceniono w obrębie poszczególnych stężeń magnezu
pozakomórkowego (a,b,c) oraz względem pozakomórkowego stężenia magnezu (x,y) testem Kruskala-Wallisa.
Różnice istotne statystycznie przy p<0,05.
Ocena produkcji reaktywnych form tlenu w tym modelu opiera się na pomiarze luminescencji w czasie (C).
Podstawową jednostką pomiaru jest relatywna jednostka luminescencji na minutę (RLU/min, ang. relative
luminescence unit/minute). Jednostką końcowo opisującą proces produkcji reaktywnych form tlenu jest pole
powierzchni pod krzywą luminescencji (AUC, ang. area under the curve).
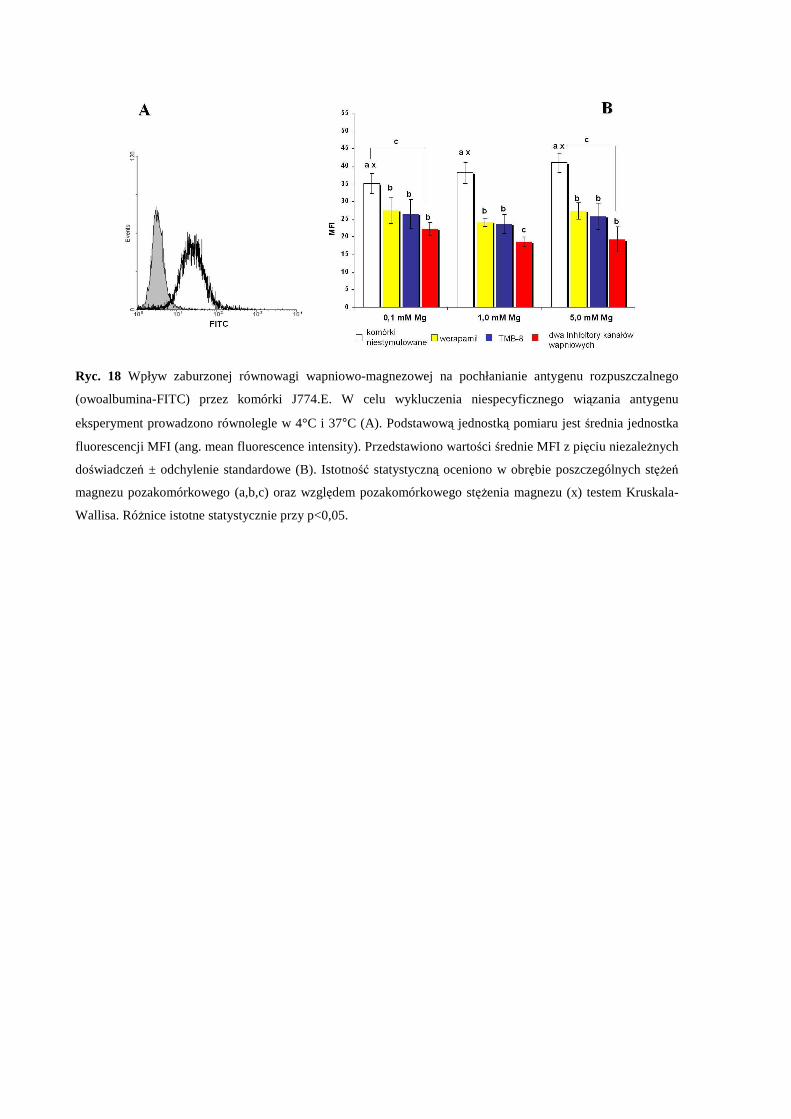
Ryc. 18 Wpływ zaburzonej równowagi wapniowo-magnezowej na pochłanianie antygenu rozpuszczalnego
(owoalbumina-FITC) przez komórki J774.E. W celu wykluczenia niespecyficznego wiązania antygenu
eksperyment prowadzono równolegle w 4°C i 37°C (A). Podstawową jednostką pomiaru jest średnia jednostka
fluorescencji MFI (ang. mean fluorescence intensity). Przedstawiono wartości średnie MFI z pięciu niezależnych
doświadczeń ± odchylenie standardowe (B). Istotność statystyczną oceniono w obrębie poszczególnych stężeń
magnezu pozakomórkowego (a,b,c) oraz względem pozakomórkowego stężenia magnezu (x) testem Kruskala-
Wallisa. Różnice istotne statystycznie przy p<0,05.
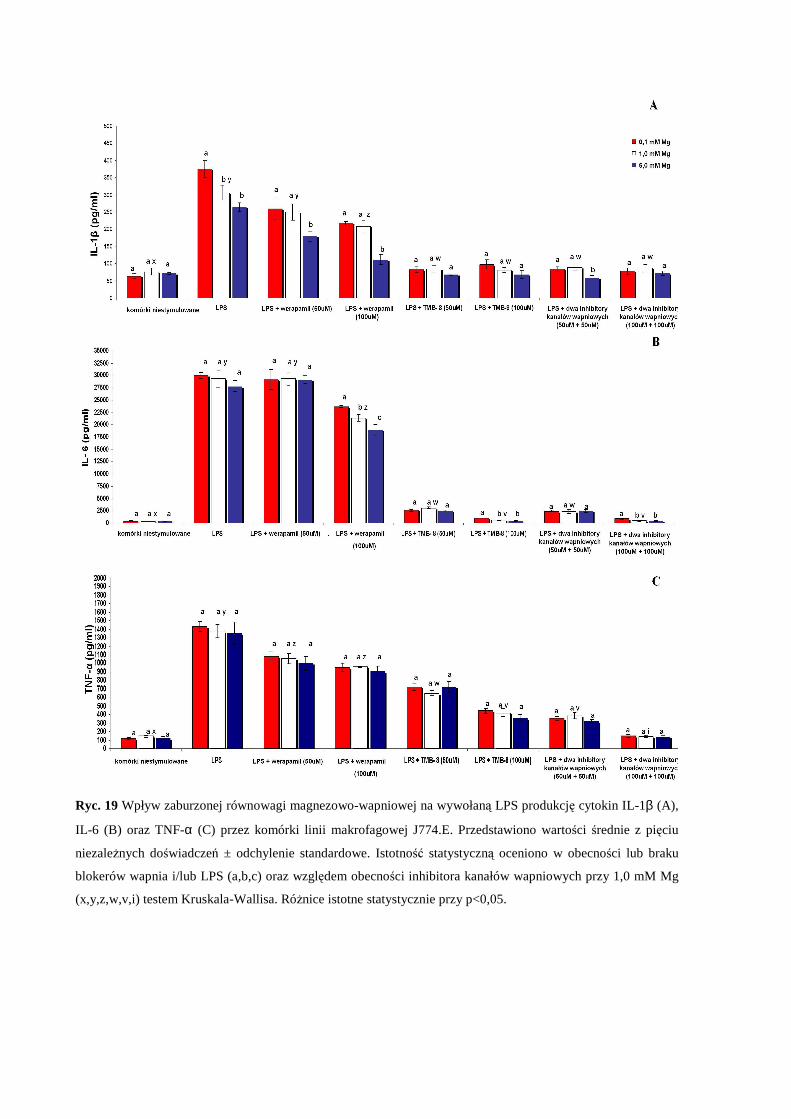
Ryc. 19 Wpływ zaburzonej równowagi magnezowo-wapniowej na wywołaną LPS produkcję cytokin IL-1β (A),
IL-6 (B) oraz TNF-α (C) przez komórki linii makrofagowej J774.E. Przedstawiono wartości średnie z pięciu
niezależnych doświadczeń ± odchylenie standardowe. Istotność statystyczną oceniono w obecności lub braku
blokerów wapnia i/lub LPS (a,b,c) oraz względem obecności inhibitora kanałów wapniowych przy 1,0 mM Mg
(x,y,z,w,v,i) testem Kruskala-Wallisa. Różnice istotne statystycznie przy p<0,05.
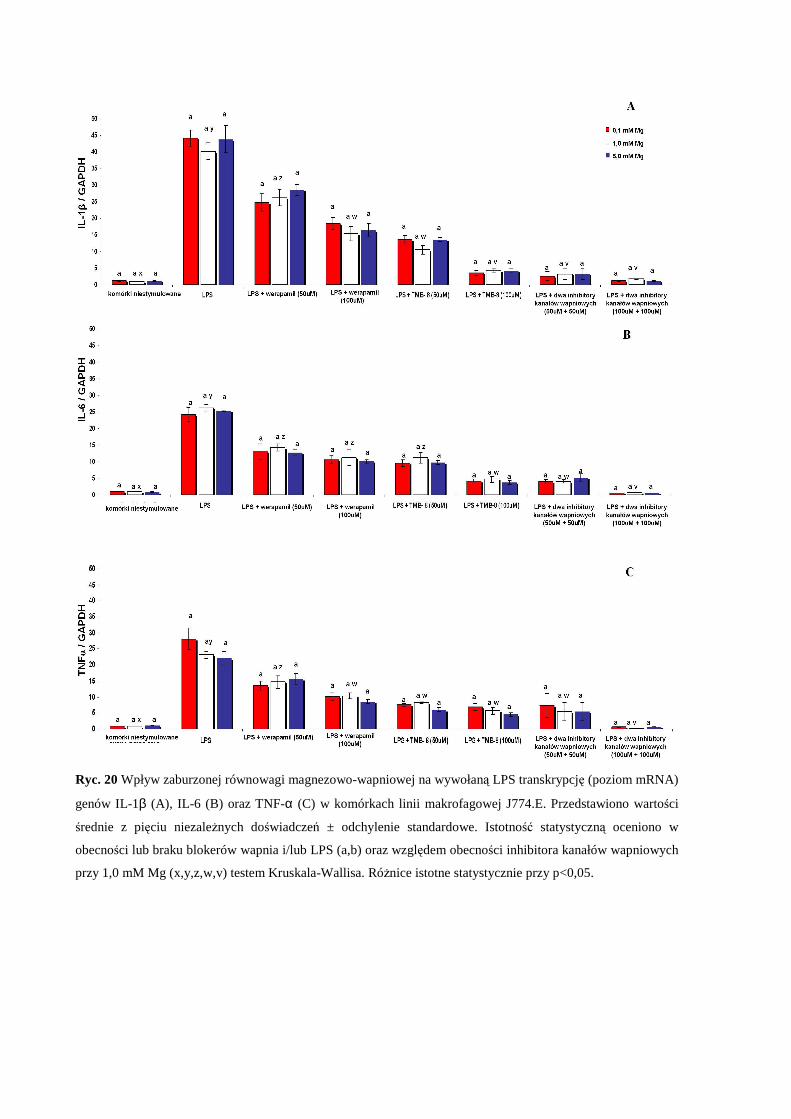
Ryc. 20 Wpływ zaburzonej równowagi magnezowo-wapniowej na wywołaną LPS transkrypcję (poziom mRNA)
genów IL-1β (A), IL-6 (B) oraz TNF-α (C) w komórkach linii makrofagowej J774.E. Przedstawiono wartości
średnie z pięciu niezależnych doświadczeń ± odchylenie standardowe. Istotność statystyczną oceniono w
obecności lub braku blokerów wapnia i/lub LPS (a,b) oraz względem obecności inhibitora kanałów wapniowych
przy 1,0 mM Mg (x,y,z,w,v) testem Kruskala-Wallisa. Różnice istotne statystycznie przy p<0,05.
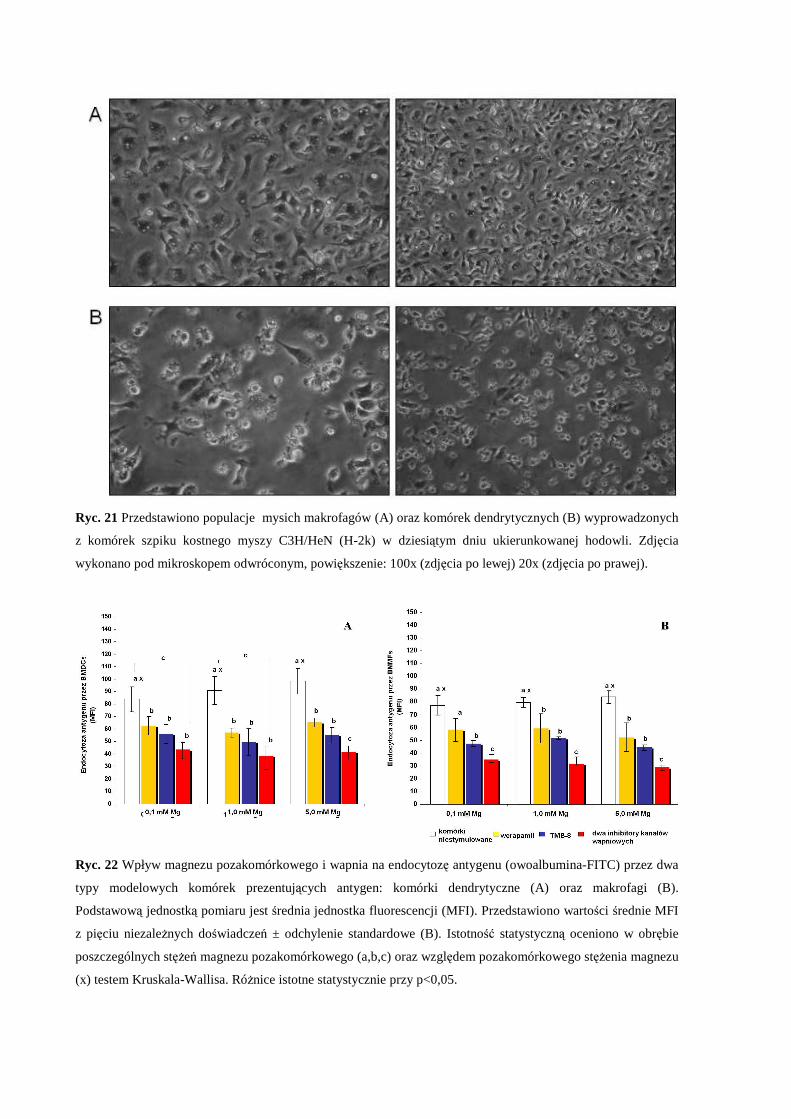
Ryc. 21 Przedstawiono populacje mysich makrofagów (A) oraz komórek dendrytycznych (B) wyprowadzonych
z komórek szpiku kostnego myszy C3H/HeN (H-2k) w dziesiątym dniu ukierunkowanej hodowli. Zdjęcia
wykonano pod mikroskopem odwróconym, powiększenie: 100x (zdjęcia po lewej) 20x (zdjęcia po prawej).
Ryc. 22 Wpływ magnezu pozakomórkowego i wapnia na endocytozę antygenu (owoalbumina-FITC) przez dwa
typy modelowych komórek prezentujących antygen: komórki dendrytyczne (A) oraz makrofagi (B).
Podstawową jednostką pomiaru jest średnia jednostka fluorescencji (MFI). Przedstawiono wartości średnie MFI
z pięciu niezależnych doświadczeń ± odchylenie standardowe (B). Istotność statystyczną oceniono w obrębie
poszczególnych stężeń magnezu pozakomórkowego (a,b,c) oraz względem pozakomórkowego stężenia magnezu
(x) testem Kruskala-Wallisa. Różnice istotne statystycznie przy p<0,05.
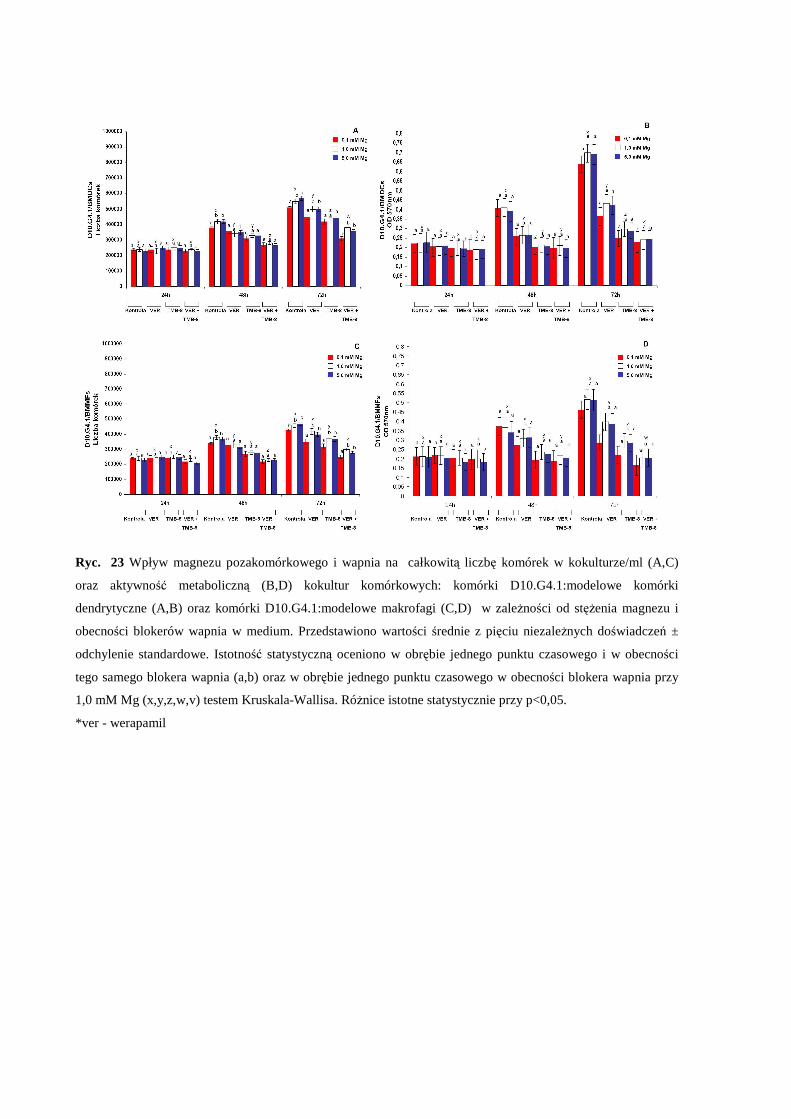
Ryc. 23 Wpływ magnezu pozakomórkowego i wapnia na całkowitą liczbę komórek w kokulturze/ml (A,C)
oraz aktywność metaboliczną (B,D) kokultur komórkowych: komórki D10.G4.1:modelowe komórki
dendrytyczne (A,B) oraz komórki D10.G4.1:modelowe makrofagi (C,D) w zależności od stężenia magnezu i
obecności blokerów wapnia w medium. Przedstawiono wartości średnie z pięciu niezależnych doświadczeń ±
odchylenie standardowe. Istotność statystyczną oceniono w obrębie jednego punktu czasowego i w obecności
tego samego blokera wapnia (a,b) oraz w obrębie jednego punktu czasowego w obecności blokera wapnia przy
1,0 mM Mg (x,y,z,w,v) testem Kruskala-Wallisa. Różnice istotne statystycznie przy p<0,05.
*ver - werapamil
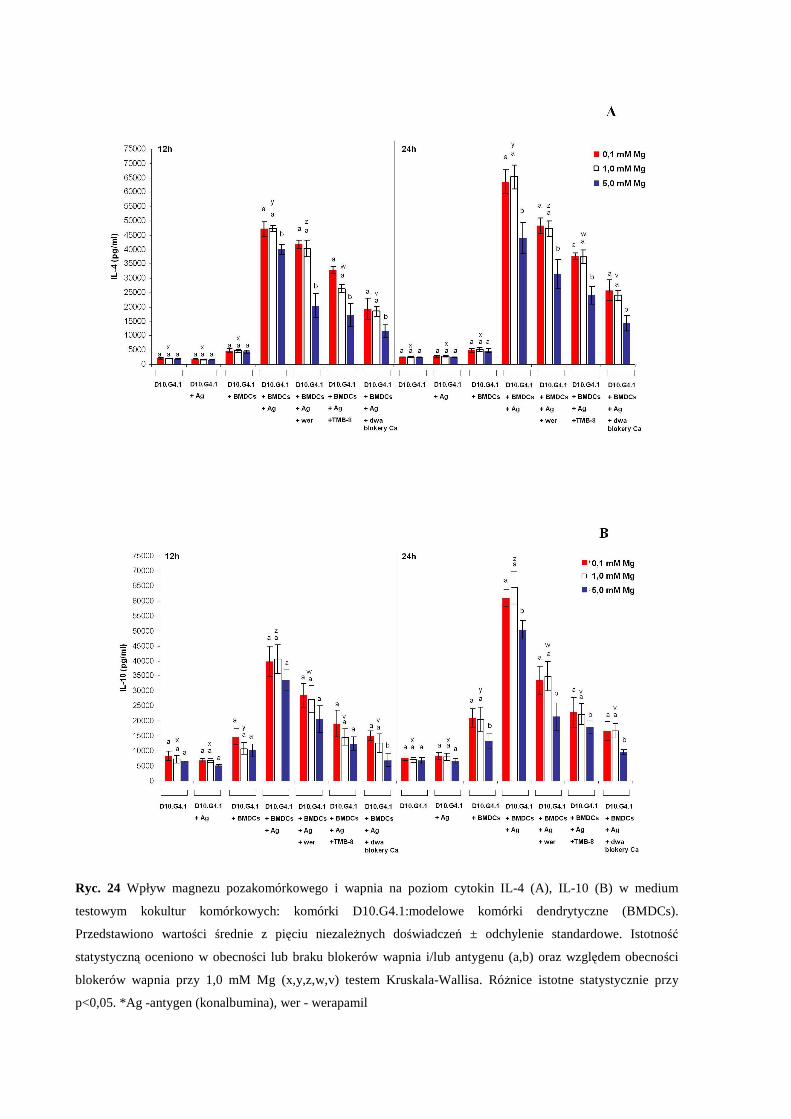
Ryc. 24 Wpływ magnezu pozakomórkowego i wapnia na poziom cytokin IL-4 (A), IL-10 (B) w medium
testowym kokultur komórkowych: komórki D10.G4.1:modelowe komórki dendrytyczne (BMDCs).
Przedstawiono wartości średnie z pięciu niezależnych doświadczeń ± odchylenie standardowe. Istotność
statystyczną oceniono w obecności lub braku blokerów wapnia i/lub antygenu (a,b) oraz względem obecności
blokerów wapnia przy 1,0 mM Mg (x,y,z,w,v) testem Kruskala-Wallisa. Różnice istotne statystycznie przy
p<0,05. *Ag -antygen (konalbumina), wer - werapamil
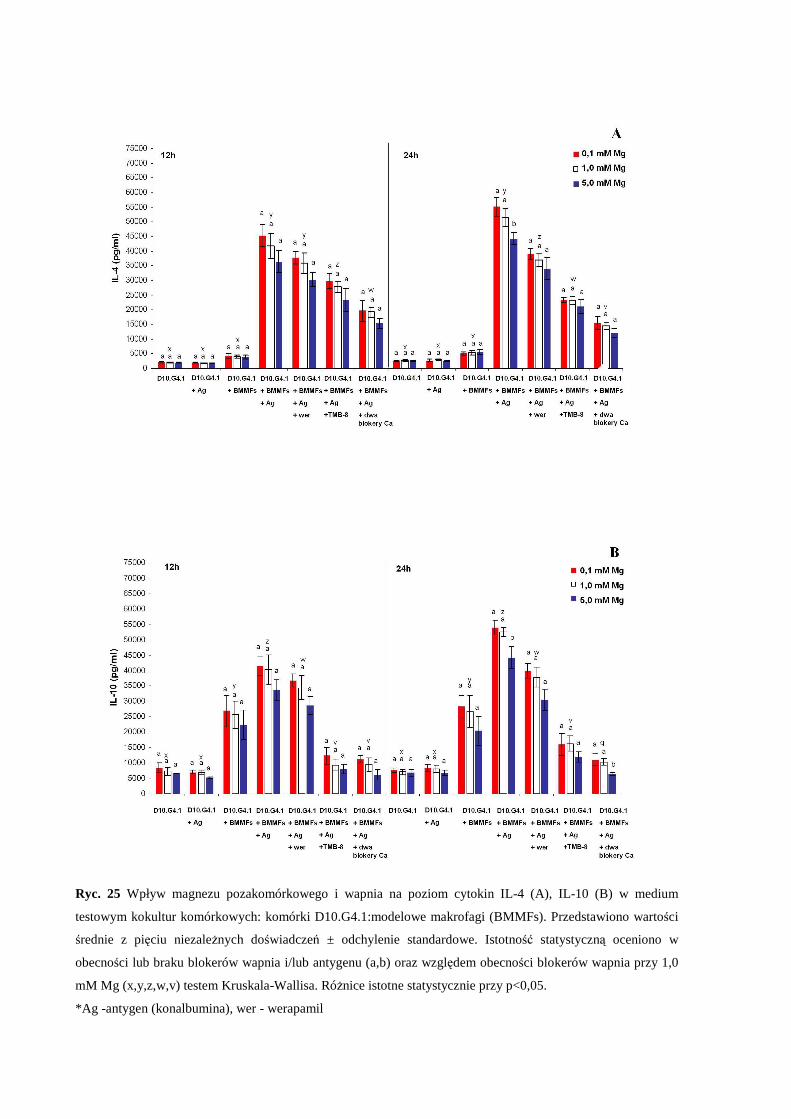
Ryc. 25 Wpływ magnezu pozakomórkowego i wapnia na poziom cytokin IL-4 (A), IL-10 (B) w medium
testowym kokultur komórkowych: komórki D10.G4.1:modelowe makrofagi (BMMFs). Przedstawiono wartości
średnie z pięciu niezależnych doświadczeń ± odchylenie standardowe. Istotność statystyczną oceniono w
obecności lub braku blokerów wapnia i/lub antygenu (a,b) oraz względem obecności blokerów wapnia przy 1,0
mM Mg (x,y,z,w,v) testem Kruskala-Wallisa. Różnice istotne statystycznie przy p<0,05.
*Ag -antygen (konalbumina), wer - werapamil
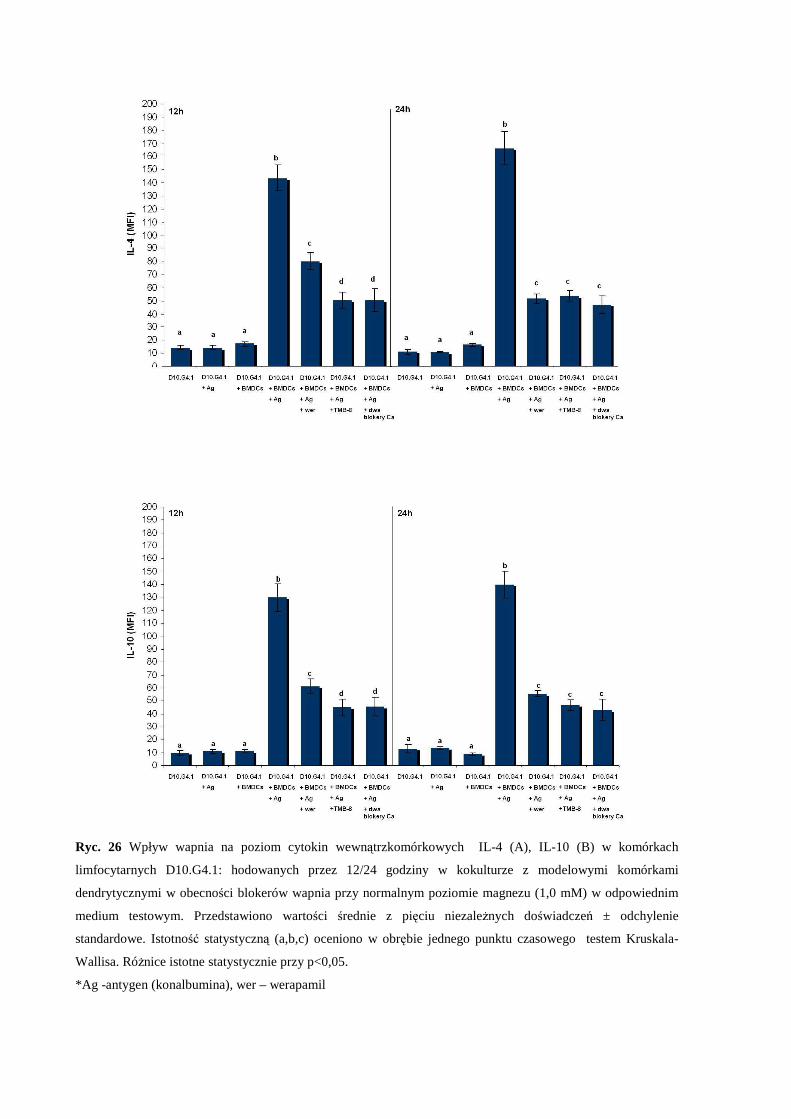
Ryc. 26 Wpływ wapnia na poziom cytokin wewnątrzkomórkowych IL-4 (A), IL-10 (B) w komórkach
limfocytarnych D10.G4.1: hodowanych przez 12/24 godziny w kokulturze z modelowymi komórkami
dendrytycznymi w obecności blokerów wapnia przy normalnym poziomie magnezu (1,0 mM) w odpowiednim
medium testowym. Przedstawiono wartości średnie z pięciu niezależnych doświadczeń ± odchylenie
standardowe. Istotność statystyczną (a,b,c) oceniono w obrębie jednego punktu czasowego testem Kruskala-
Wallisa. Różnice istotne statystycznie przy p<0,05.
*Ag -antygen (konalbumina), wer – werapamil

Ryc. 27 Cząsteczki biorące udział w interakcji pomiędzy komórkami T a komórkami prezentującymi antygen
(APC) – model uproszczony. Chlorek magnezu obniżył ekspresję cząsteczek CD80/CD86 oraz MHC II na
powierzchni APC (Schempp et al., 2000). Inhibitory wolnych kanałów wapniowych typu L (nifedipina i
werapamil) obniżały ekspresję cząsteczek MHC II oraz CD80 na powierzchni APC (Katoh et al., 1997).
Opracowanie własne. Schemat został wykonany z wykorzystaniem materiałów Servier Medical Art.
*Ag - antygen


















