
Spektroskopia molekularna - chemia.biol-chem.uwb.edu.pl · W. Kołos, J. Sadlej –Atom i...
78
Spektroskopia molekularna wykład 30 godzinny zakończony egzaminem pisemnym Dr hab. Alina T. Dubis Zakład Chemii Produktów Naturalnych ul. Ciołkowskiego 1K, pokój 1079 Tel. 85-7388034 Konsultacje : wtorek 11 - 13
Transcript of Spektroskopia molekularna - chemia.biol-chem.uwb.edu.pl · W. Kołos, J. Sadlej –Atom i...

Spektroskopia molekularna
wykład 30 godzinny zakończony
egzaminem pisemnym
Dr hab. Alina T. Dubis
Zakład Chemii Produktów Naturalnych
ul. Ciołkowskiego 1K, pokój 1079
Tel. 85-7388034
Konsultacje : wtorek 11 - 13
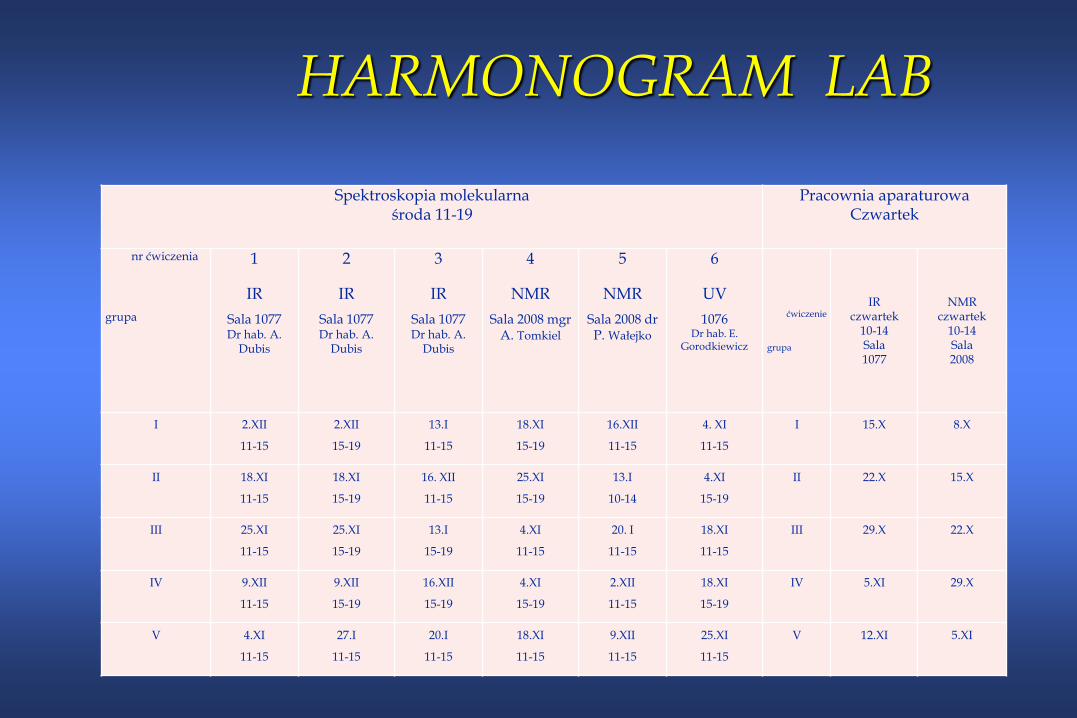
HARMONOGRAM LAB
Spektroskopia molekularna środa 11-19
Pracownia aparaturowaCzwartek
nr ćwiczenia
grupa
1
IR
Sala 1077Dr hab. A.
Dubis
2
IR
Sala 1077Dr hab. A.
Dubis
3
IR
Sala 1077Dr hab. A.
Dubis
4
NMR
Sala 2008 mgr A. Tomkiel
5
NMR
Sala 2008 dr P. Wałejko
6
UV
1076Dr hab. E.
Gorodkiewicz
ćwiczenie
grupa
IRczwartek
10-14Sala1077
NMRczwartek
10-14 Sala2008
I 2.XII
11-15
2.XII
15-19
13.I
11-15
18.XI
15-19
16.XII
11-15
4. XI
11-15
I 15.X 8.X
II 18.XI
11-15
18.XI
15-19
16. XII
11-15
25.XI
15-19
13.I
10-14
4.XI
15-19
II 22.X 15.X
III 25.XI
11-15
25.XI
15-19
13.I
15-19
4.XI
11-15
20. I
11-15
18.XI
11-15
III 29.X 22.X
IV 9.XII
11-15
9.XII
15-19
16.XII
15-19
4.XI
15-19
2.XII
11-15
18.XI
15-19
IV 5.XI 29.X
V 4.XI
11-15
27.I
11-15
20.I
11-15
18.XI
11-15
9.XII
11-15
25.XI
11-15
V 12.XI 5.XI

Tematyka
Wprowadzenie do spektroskopii (4 h)
Spektroskopia:
-Rotacyjna (4 h)
-Oscylacyjna (4 h)
-Ramana (4 h)
-Elektronowa (4 h + 4 h)
-NMR (4 h)
-EPR (2 h)

Literatura
Joanna Sadlej – Spektroskopia molekularna
Zbigniew Kęcki – Podstawy spektroskopii molekularnej
W. Kołos, J. Sadlej – Atom i Cząsteczka
David O. Hayward – Mechanika kwantowa dla chemików– seria Niezbędnik chemika
Peter W. Atkins – Chemia fizyczna


Spektroskopia nauka wykorzystująca światło do badania właściwości atomów,
cząsteczek i materiałów
Posługuje się kwantowo-mechanicznym modelem poziomów energetycznych do wydedukowania struktury cząsteczki czyli:
długości wiązań, ich mocy, stałych siłowych, symetrii orbitali molekularnych
Z pomiarów odczytujemy częstości zaabsorbowanego/emitowanego promieniowania oraz intensywności sygnałów
Pomiar częstości (energii) promieniowania zaabsorbowanego/emitowanego przez cząsteczkę dostarcza informacji o odległościach między skwantowanymi poziomami energetycznymi
Obserwowany rozkład intensywności informuje nas o:
- populacji cząsteczek
- „regułach wyboru” – czyli symetrii cząsteczek i funkcjach falowych
- właściwościach molekularnych takich jak moment dipolowy i polaryzowalność
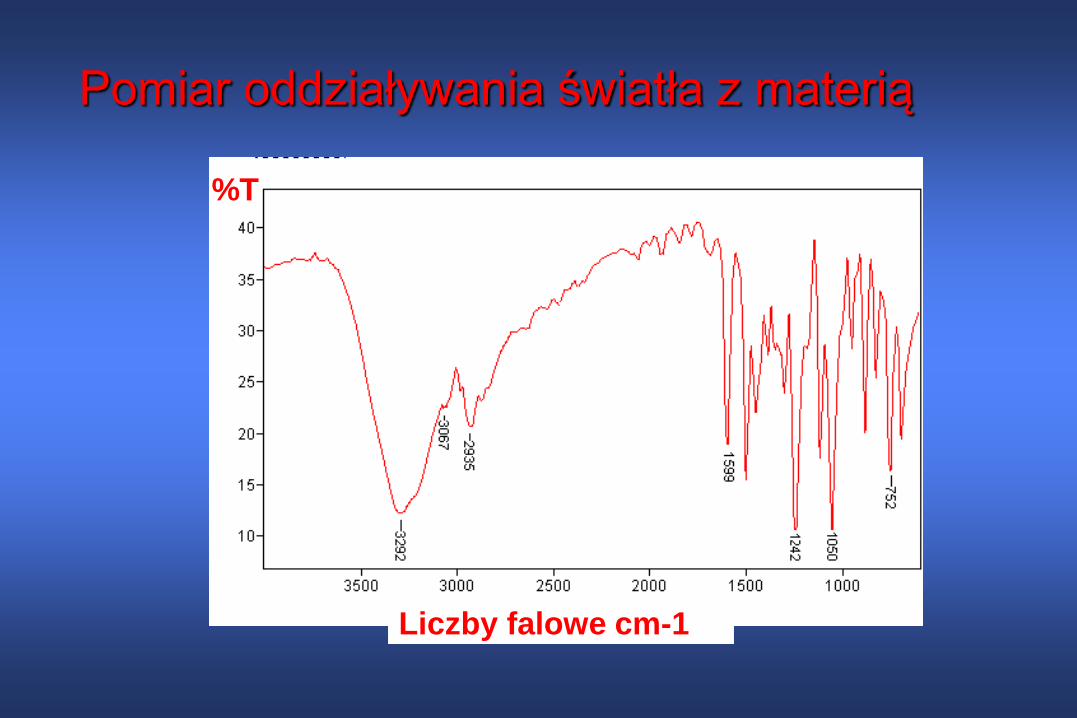
Pomiar oddziaływania światła z materią
Liczby falowe cm-1
%T

Jako wprowadzenie do dyskusji o spektroskopii dokonamy przeglądu dwóch
zagadnień:
Natury i właściwości światła
Podstawowych zagadnień kwantowo-mechanicznego
opisu cząsteczek
Połączymy je razem aby odpowiedzieć na pytanie:
Co powoduje przejścia spektroskopowe?

Fizyka na początku XX wieku
Teoria atomistyczna przyjęła, że materia zbudowana jest z cząstek zwanych
atomami których masy i rozmiary były w przybliżeniu znane
Elektrony są cząstkami o znanej masie i ładunku
Znana jest kinetyczna teoria gazów opisująca zachowanie się gazu
doskonałego pV=nRT
Układ okresowy został doświadczalnie odkryty… ale powód okresowości
(atomy w kolumnach wykazują podobne właściwości chemiczne) NIE był
zrozumiały (bo wymagało to znajomości mechaniki kwantowej)
Przyjęta była falowa natura światła w oparciu o takie zjawiska
odbicie światła
rozszczepienie w pryzmacie
interferencję
Falowa natura światła opisana została przez teorię Maxwella i jego równania
falowe
Ale… było kilka nierozwiązanych problemów…

Klasyczna falowa teoria światła
(James Clerk Maxwell, 1850)
Światło jest falą elektromagnetyczną poruszającą się ze skończoną
prędkością
Teoria Maxwella traktuje światło jako wielowymiarowy wektor, który
charakteryzuje się następującymi właściwościami:
oscylujące fale pola elektrycznego i magnetycznego przemierzają
przestrzeń z prędkością c=2.99792758 x108 m/s (w próżni)
linie sił pola elektrycznego są prostopadłe do pola magnetycznego
kierunek rozchodzenia się promieniowania jest prostopadły do wektorów pola elektrycznego i magnetycznego

Klasyczna falowa teoria światła
W zapisie matematycznym pole elektryczne można
przedstawić jako:
0 0( , ) cos( )E r t kE k r t
a odpowiadające mu pole magnetyczne jako:
0 0( , ) cos( )H r t k E k r t
jest wektorem o wielkości 2/ skierowanym w kierunku ruchukr określa położenie w przestrzeni
2 częstość oscylacji pola (radian/s)
/ 2 częstość oscylacji pola w cyklach/s
0E wektor określający polaryzację światła
0k E wektor prostopadły do wektorów k i E0 określający płaszczyznę
polaryzacji pola magnetycznego

Klasyczna falowa teoria światła
H
k
E
( , )E r t
( , )H r t
Ponieważ cząsteczki zbudowane są z dodatnio naładowanych jąder o dużej
gęstości oraz otaczających je ujemnych elektronów, ich elektryczne właściwości są
szczególnie ważne. W większości przypadków oscylujące pole elektryczne
wywiera większy efekt na jądra i elektrony niż towarzyszące mu pole magnetyczne.
Stosowane jednostki:
częstość oscylacji lub liczba oscylacji na sekundę s-1 lub Hz
długość fali – droga wzdłuż fali obejmująca jeden cykl, m, Å lub nm
Å = 10-10 m, nm = 10-9 m
liczba falowa – liczba długości fal jaka mieści się w jednostce długości
zazwyczaj cm-1 czyli liczba długości fal przypadających na 1 cm
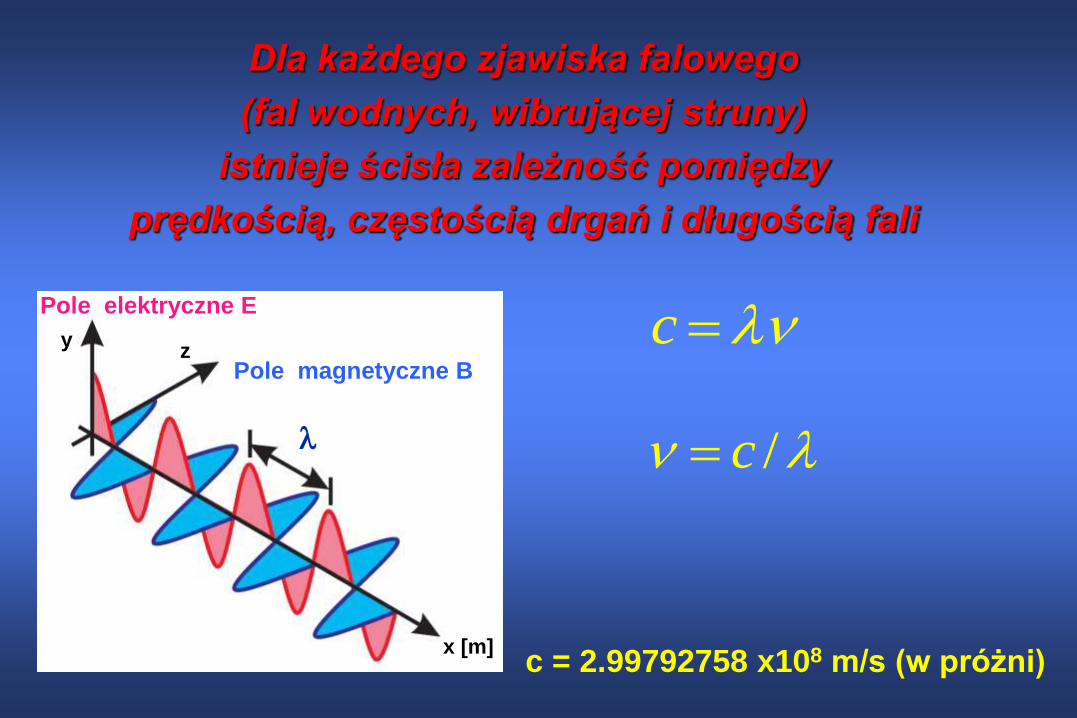
Dla każdego zjawiska falowego
(fal wodnych, wibrującej struny)
istnieje ścisła zależność pomiędzy
prędkością, częstością drgań i długością fali
c
/c
c = 2.99792758 x108 m/s (w próżni)
Pole elektryczne E
Pole magnetyczne B
x [m]
yz

Widmo
promieniowania elektromagnetycznego
X UV VIS IR Radiowe

Widmo promieniowania elektromagnetycznego
< 1 nm (104 – 106 eV)
X 1 – 50 nm (102 – 104 eV)
próżniowy UV 10 – 200 nm
bliski UV 200 – 350 nm
Widzialne 350 – 800 nm
bliska IR 0.8 m –
IR – 25 m
Mikrofale 400 m – 30 cm
Radiowe powyżej 100 cm

Falowa teoria światła
Np. Światło o długości fali =5000 Å = 500 nm
i częstości
=c/ = 2.99792458x108/500x10-9 = 5.9958x1014 s-1
= 5.9958x1014 Hz
Ma „energię” albo „częstość”
1120000cm

Dlaczego falowa teoria światła była
niewystarczająca?
Teoria ta nie potrafiła wyjaśnić natury dwóch bardzo istotnych zjawisk:
promieniowania ciała doskonale czarnego
efektu fotoelektrycznego
Rozwiązanie tych „zagadek” dało początek rewolucji, nazwanej
„mechaniką kwantową”,
która jest podstawą naszego nowoczesnego spojrzenia na świat fizyczny
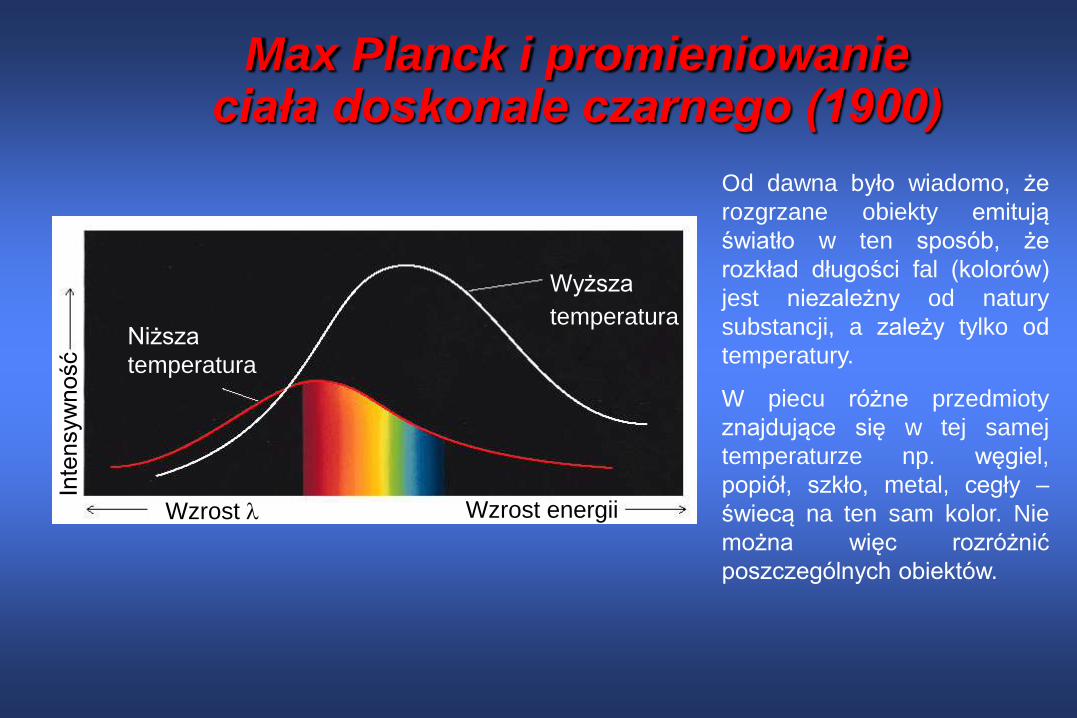
Max Planck i promieniowanie ciała doskonale czarnego (1900)
Wyższa
temperaturaNiższa
temperatura
Wzrost energiiWzrost
Inte
nsyw
ność
Od dawna było wiadomo, że
rozgrzane obiekty emitują
światło w ten sposób, że
rozkład długości fal (kolorów)
jest niezależny od natury
substancji, a zależy tylko od
temperatury.
W piecu różne przedmioty
znajdujące się w tej samej
temperaturze np. węgiel,
popiół, szkło, metal, cegły –
świecą na ten sam kolor. Nie
można więc rozróżnić
poszczególnych obiektów.
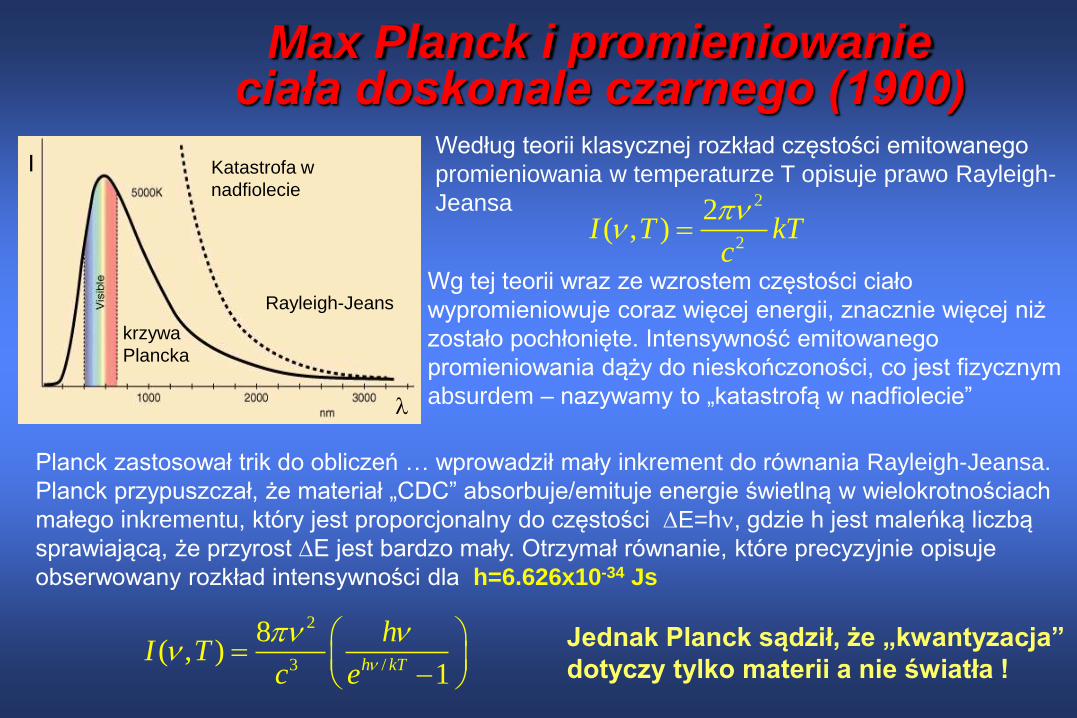
Max Planck i promieniowanie ciała doskonale czarnego (1900)
I
krzywa
Plancka
Rayleigh-Jeans
Katastrofa w
nadfiolecie2
2
2( , )I T kT
c
Według teorii klasycznej rozkład częstości emitowanego
promieniowania w temperaturze T opisuje prawo Rayleigh-
Jeansa
Wg tej teorii wraz ze wzrostem częstości ciało
wypromieniowuje coraz więcej energii, znacznie więcej niż
zostało pochłonięte. Intensywność emitowanego
promieniowania dąży do nieskończoności, co jest fizycznym
absurdem – nazywamy to „katastrofą w nadfiolecie”
Planck zastosował trik do obliczeń … wprowadził mały inkrement do równania Rayleigh-Jeansa.
Planck przypuszczał, że materiał „CDC” absorbuje/emituje energie świetlną w wielokrotnościach
małego inkrementu, który jest proporcjonalny do częstości E=h, gdzie h jest maleńką liczbą
sprawiającą, że przyrost E jest bardzo mały. Otrzymał równanie, które precyzyjnie opisuje
obserwowany rozkład intensywności dla h=6.626x10-34 Js
2
3 /
8( , )
1h kT
hI T
c e
Jednak Planck sądził, że „kwantyzacja”
dotyczy tylko materii a nie światła !

Zjawisko fotoelektryczne – następny eksperyment, którego fizyka klasyczna nie
potrafiła wyjaśnić
światło
elektrony = prąd
Hertz (1887) zauważył, że gdy światło padało na
płytkę metalową umieszczoną w próżni następowała
emisja elektronów a ponadto
liczba wyemitowanych elektronów rosła wraz z
natężeniem światła
energia kinetyczna emitowanych elektronów zależała
tylko od częstości padającego światła

Zjawisko fotoelektryczne
h=A+Ee
Brak
efektu
Potas potrzebuje 2eV energii do wybicia elektronu
Rezultaty powyższe nie były zgodne z
klasycznym ujęciem światła jako fali, która
może „wymywać” elektrony z powierzchni
jak fale wodne omywające skały na plaży.
Einstein (1905) stwierdził, że wszystkie
obserwacje da się wyjaśnić, jeżeli przyjmie
się, że światło o częstości jest
skwantowane w porcje nazwane fotonami
o wielkości Ef=h
W zderzeniu fotonu z elektronem energia fotonu zostaje całkowicie
odebrana przez elektron. Część tej energii zostaje zużyta na pracę
wyjścia (A), a reszta unosi elektron w postaci energii kinetycznej (Ek).

Zjawisko fotoelektryczne
Energia kinetyczna fotolelektronów
rośnie liniowo wraz ze wzrostem
częstości światła wywołującego
efekt. Dla danej częstości światła
określono maksymalną energię
kinetyczną emitowanych elektronów
(max)
[s-1]
Zmierzone nachylenie
prostych jest stałe i takie
same dla wszystkich
materiałów !
Częstość progowa jest różna
dla różnych materiałów
Eksperymentalnie wyznaczone
nachylenie prostych ma taką
samą wartość liczbową jak stała
w równaniu Plancka
(max) 0( )eE h

Planck i Einstein poprzez ukazanie korpuskularnej natury
światła dostarczyli dwóch argumentów przeciwko klasycznej
Maxwellowskiej teorii światła jako fali.
Trzeciego argumentu dostarczył …
Nagrody Nobla z fizyki
1918 r. Max Planck
1921 r. Albert Einstein

Efekt Comptona (1923)
Foton padający
Foton „odbity”
Elektron „odbity”
Gdy promieniowanie X (bardzo
małe długości fal) przechodzi
przez cienką folię metalową
cześć promieniowania „odbija
się” i zwiększa swoją długość.
Z punktu widzenia falowego
zjawisko to jest niezrozumiałe,
ale zostało wyjaśnione na
bazie kwantowego modelu
światła wprowadzonego przez
Einsteina

Efekt Comptona
Równanie Einsteina 2E mc h
hmc
c
hp
dla materii
dla światła
Po przekształceniu
Otrzymujemy pęd fotonów światła
Compton stwierdził, że fotony zderzają się elektronami podobnie jak
kule na stole bilardowym tracąc część pędu i energii, którą odbierają
elektrony.
Kwant światła o częstości posiada pęd p=h/ !

Właściwości światła - podsumowanie
Światło zachowuje się jak oscylująca fala elektromagnetyczna przemierzająca przestrzeń z prędkością c = 2.99792758 x 108 m/s . Fala posiada swoją częstość [s-1], długość [m] , liczbę falową i prędkość c=
Planck stwierdził, że światło wymienia energię z materią w porcjach zwanych kwantami o wielkości E=h, gdzie h=6.62606x10-34 Js jest mierzalną wielkością fizyczną
Einstein wyjaśnił zjawisko fotoelektryczne stwierdzając, że światło składa się z porcji zwanych fotonami o energii E=h. Na tej podstawie wyliczono, że zgodnie z doświadczeniem, maksymalna energia kinetyczna elektronu wybijanego z metalu przez światło wynosi:
Compton zauważył, że gdy fotony światła odbijają się od bardzo małych cząstek, zachowują się tak jakby miały pęd p=h/ . Na tej podstawie zasugerował korpuskularno-falową naturę światła
2
(max) 0
1( )
2e e eE m h
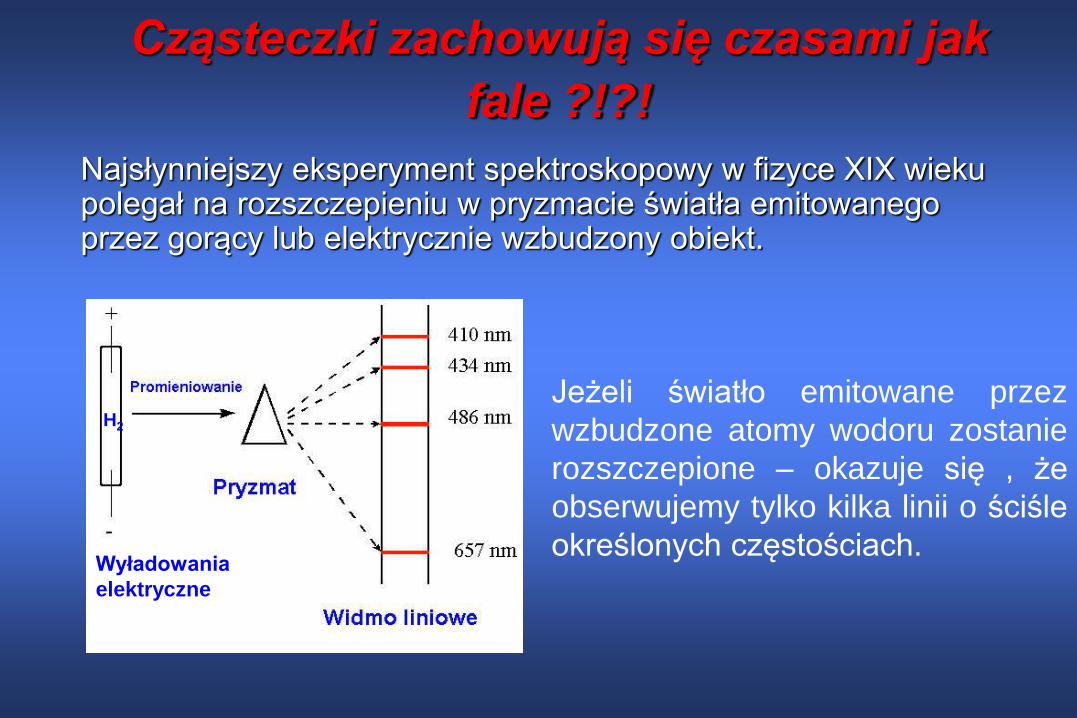
Cząsteczki zachowują się czasami jak
fale ?!?!
Najsłynniejszy eksperyment spektroskopowy w fizyce XIX wieku polegał na rozszczepieniu w pryzmacie światła emitowanego przez gorący lub elektrycznie wzbudzony obiekt.
Jeżeli światło emitowane przez
wzbudzone atomy wodoru zostanie
rozszczepione – okazuje się , że
obserwujemy tylko kilka linii o ściśle
określonych częstościach.
H2
Wyładowania
elektryczne

Widmo emisyjne
Szwedzki spektroskopista Johannes Rydberg zauważył (1890),
poprzez analizę wzorów widm liniowych, że częstość obserwowanych
linii można wyrazić wzorem:
RH nosi nazwę stałej Rydberga
n1= 1, 2, 3, 4 … n2= n1+1, n1+2, n1+3, …
Przez wiele lat to równanie było akceptowane jako piękny wzór
matematyczny – ale bez widocznego uzasadnienia
1 1
2 2
1 2
1 1[ ] 109737.31H HR cm gdzie R cm
n n
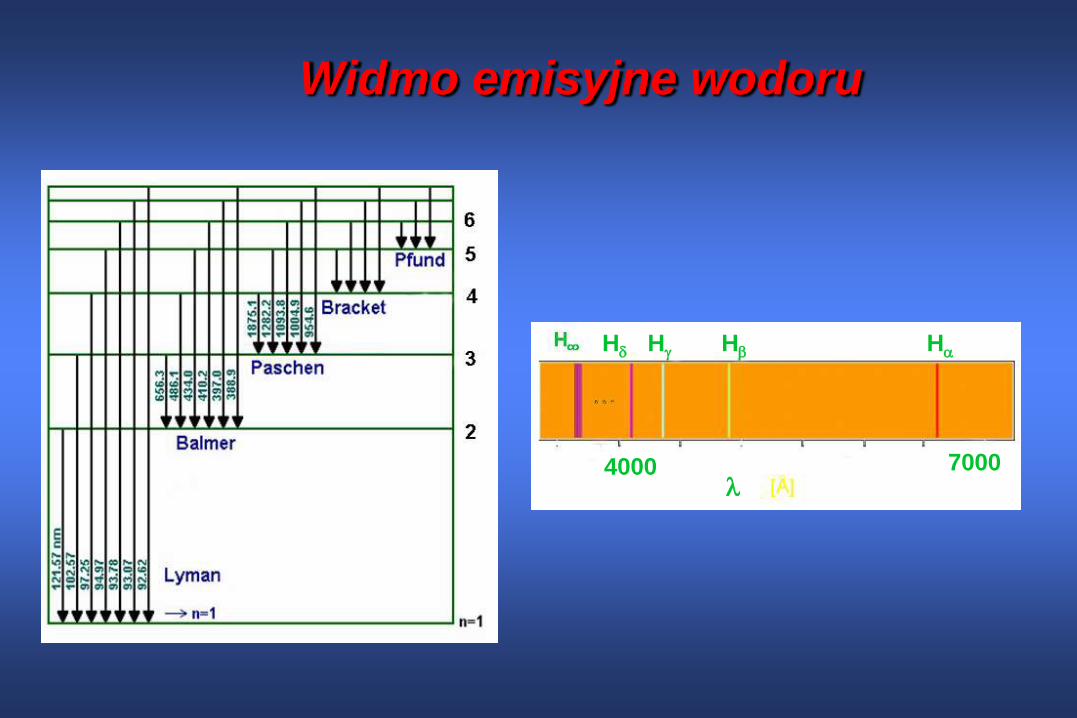
Widmo emisyjne wodoru
H H H H
70004000

Widmo emisyjne
1/n2 gwałtownie zmniejsza się ze wzrostem n
{1, 0.25, 0.111, 0.0625, 0.04, 0.0278, …}
Zbiór linii spektralnych dla jednakowych
wartości n1 jest zgrupowany w określonym
obszarze widmowym
n1 Nazwa grupy Obszar
widma
1 Lymana daleki UV
2 Balmera widzialny
3 Ritz-Passehna bliska IR
4 Bracketa średnia IR
5 Pfunda daleka IRZe wzoru wynika, że dla danej wartości n1
wraz ze wzrostem n2 linie zbliżają się do
siebie i nawarstwiają się przy wartości
granicznej
1 2
2
( ) HRn
n
Jak to wytłumaczyć?

Niels Bohr (1913-15) przedstawił „mechaniczny” model atomu
wodoru
Elektrony o ładunku ujemnym obiegają dodatnio naładowane jądro i siła
odśrodkowa ruchu obrotowego zrównoważona jest dokładnie siłąCoulombowskiego przyciągania ładunków dodatnich i ujemnych.
Planck wpadł na pomysł, że orbitalny moment pędu jest skwantowany
L n, , !!!
2
hgdzie n to liczba calkowita h stala Plancka
Z zasady zachowania energii wynika, że energia obserwowanego kwantu światła h
musi dokładnie pasować do odległości pomiędzy dozwolonymi poziomami
energetycznymi. Oznacza to, że poziomy energetyczne atomu są skwantowane

Stosując powyższe postulaty Bohr otrzymał:
2 2
2
2 2 2
2 2 2 2
22
2 2
2
0
4
4
L m r n
Zem
r r
Ze m n h
r r m r
hr n
mZe
nr a
Z
r – promień Bohra
dla n=1 r = a0= 0.529177 Å promień orbity elektronu w najniższym stanie
2 2
22
2 2 2
2
0
2
2 2
pot kinE E E
Ze mE k
r
Zem
r
Ze Z eE
r n a
E1 = -13.6 eV = -2.179 x 10-18 J
Zależności te precyzyjnie wyjaśniały widmo emisyjne wodoru, He+, Li2+ (systemów
jednoelektronowych), ale niczego poza tym. Żadne udoskonalenia matematyczne nie
przyniosły rezultatu (nawet wprowadzenie orbity eliptycznej).
Potrzeba było czegoś NOWEGO.
2 2 2 2
2 2
0 0
2 2
2 2
0
2 2
2 2
0
2 2
2 2
0
2 2
1 1
2
1 1
2
1 1
2
b a
b a
b a
a b
a b
a b
Z e Z eE E
n a n a
Z eE E
a n n
Z eh
a n n
Z e
ha n n

Fale de Broglie’a (1924)Na postawie rezultatów Einsteina, Plancka i Comptona, świadczących o
korpuskularno-falowej naturze promieniowania, de Broglie wysunął hipotezę, że
analogiczny dualizm występuje również w mechanice i że każda poruszająca się
cząstka np. elektron może wykazywać charakter falowy.
2
2
c cmc h
hcmc
h hmc stąd
mc
klasyczny pęd p m więc
h
p
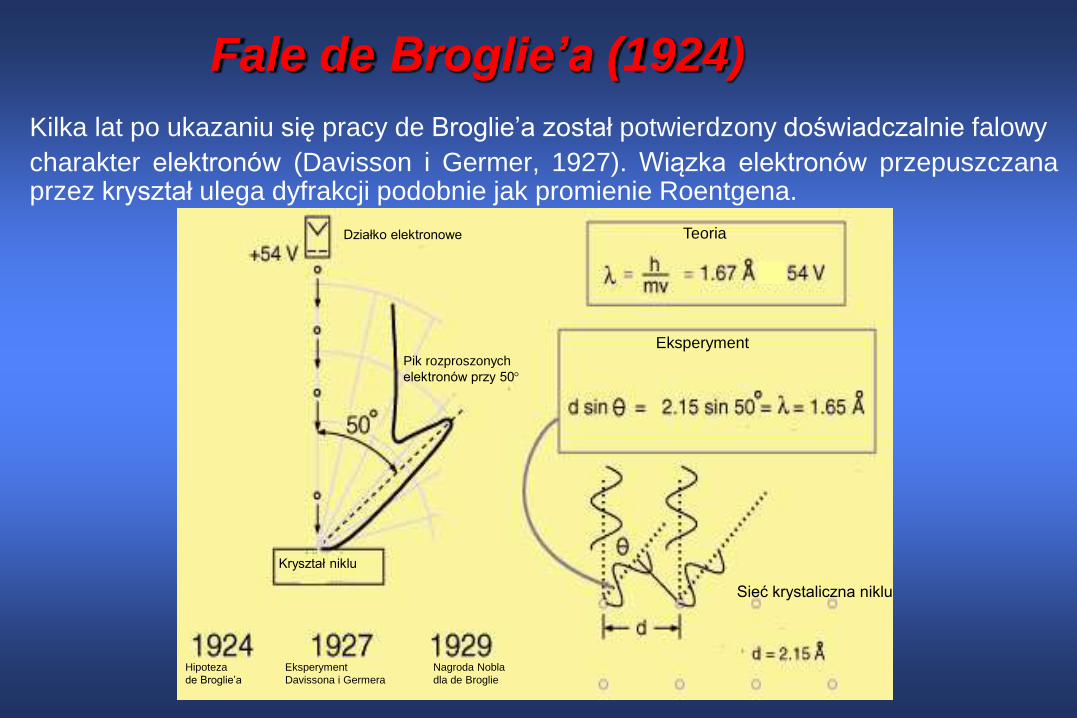
Fale de Broglie’a (1924)
Kilka lat po ukazaniu się pracy de Broglie’a został potwierdzony doświadczalnie falowy
charakter elektronów (Davisson i Germer, 1927). Wiązka elektronów przepuszczanaprzez kryształ ulega dyfrakcji podobnie jak promienie Roentgena.
Teoria
Eksperyment
Sieć krystaliczna niklu
Pik rozproszonych
elektronów przy 50
Kryształ niklu
Działko elektronowe
Hipoteza
de Broglie’a
Eksperyment
Davissona i Germera
Nagroda Nobla
dla de Broglie

Fale de Broglie’a (1924)
Teoria Bohra traktowała elektrony jako punkty materialne poruszające się po
określonych torach zgodnie z prawami mechaniki klasycznej, a warunek
kwantowania pozwalał jedynie wybrać niektóre spośród wszystkich możliwych
klasycznych torów. Wg hipotezy de Broglie’a dozwolonymi orbitami są tylko te na
których może zmieścić się całkowita wielokrotność długości fali elektronu.
2 1,2,3...n r dla n
Oczywistym się stało, że planetarny model był nieprawdziwy i musiał być zastąpiony
modelem uwzględniającym falowy charakter elektronów.
Orbita dozwolona Orbita niedozwolona

Przypomnienie wykładu 1
2
3 /
8( , )
1h kT
hI T
c e
Światło jako:
FALA
ugięcie
rozszczepienie
interferencja
KORPUSKUŁA
CDC (Planck 1890)
Efekt fotoelektryczny (Einstein 1905)
Efekt Comptona (1923) p=h/
Materia jako:
KORPUSKUŁA
Bohr kwantowanie orbitalnego
momentu pędu L=mvr=nh/2
FALA
Fale materii de Broglie’a (1924)
=h/p
2
(max) 0
1( )
2e e eE m h

Mechanika falowa i równanie Schrödingera
Erwin Schrödinger zaakceptował postulaty de Brogli’a i pracował nad równaniem falowym
opisującym cząstki atomowe. W najprostszym przypadku, dla cząstki o masie m poruszającej
się w jednym wymiarze, zamkniętej w pudle o wymiarze L, równanie niezależne od czasu ma
postać:
2 2 2'' 2
2 2 2
'' 2 2
2
( ) ( ) 2( ) ( ) lub
2
2
d y x d y x mEEy x to y x y k y
m dx dx
mEy k y gdzie k
Pytanie: Jaka funkcja ma drugą pochodną równą minus stała razy oryginalna funkcja?
Odpowiedź:
( ) sin lub ( ) cosy x kx y x kx
( ) sin cosy x A kx B kx
Skoro każda z nich może być rozwiązaniem dla tej samej wartości k to również może być
rozwiązaniem ich kombinacja liniowa wiec ogólne rozwiązanie ma postać:

Co to jest równanie różniczkowe?
Równanie , które zawiera jedną lub więcej pochodnych
Równanie pierwszego rzędu to równanie w którym pochodna jest 1 rzędu
' 3 lub 3dy
y x xdx
Równanie drugiego rzędu to równanie w którym pochodna jest 2 rzędu 2
''
23 lub 3
d yy x x
dx
Sprawdźmy – jako rozwiązanie równania Schrödingera weźmy jego pochodną
'
2'' 2 2
2
'' 2 2
( ) cos sin
( ) sin cos
sin cos
dyy x kA kx kB kx
dx
d y d dyy x k A kx k B kx
dx dx dx
y x k A kx B kx k y
Potwierdza to poprawność rozwiązania naszego równania dla dowolnych wartości A i B

Zasady mechaniki falowej
Dla każdego układu molekularnego istnieje „funkcja falowa”
- czyli y(x)
Funkcja falowa jest rozwiązaniem różniczkowego równania
Shchrödingera dla układu i zależy od wszystkich współrzędnych
przestrzennych (x, y, z) wszystkich cząstek w układzie (i od czasu ale
to pominiemy)
jest funkcją ciągłą wszystkich współrzędnych
2 jest proporcjonalny do prawdopodobieństwa, że układ posiada
daną konfigurację
Uwaga:
Jest matematyczną funkcją , która może mieć wartości ujemne lub
dodatnie, ale 2 ≥0
Dla cząstki poruszającej się w jednym wymiarze, rozwiązanie funkcji
sin lub cos oscyluje i ma wartości + i -

Jak powstało „kwantowanie”?
Zastosujmy przedstawione reguły i zobaczmy …
Dla cząstki o masie m w jednym wymiarze, zamkniętej w pudle o długości L rozwiązanie ma postać:
( ) ( ) sin cosy x x A kx B kx
En
erg
ia
•Jeżeli cząstka nie może uciec z pudła (ponieważ prawdopodobieństwo wynosi 0)
( ) 0 0x dla x i x L
•Skoro jest „ciągła”
czyli znika przy ściance ( 0) 0 sin 0 cos 0 0x A B B to B
•Stąd ogólne rozwiązanie ma postać: ( ) sinx A kx

Cząstka w pudle
Podobnie gdy jest ciągła w x=L wówczas:
( ) sin 0x L A kL
Równanie jest prawdziwe tylko gdy:
sin 0kL czyli 0,1,2,3,4...kL n dla n
Z definicji wynika, że:2
2mE nk
L
Poprawne rozwiązanie równania Schrödingera otrzymujemy gdy energia cząstki
wynosi:
22 2
2
2 22 8n
n hE E n
m L mL
I odpowiednia funkcja falowa dla każdego n ma postać:
( ) sinn x
x AL
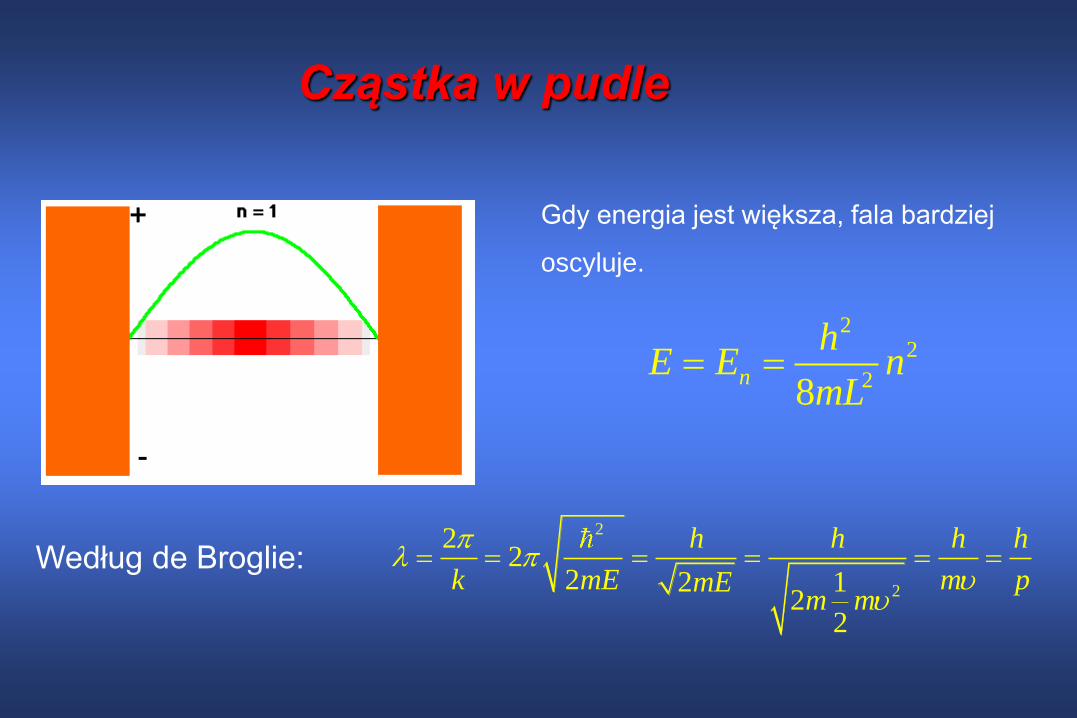
Cząstka w pudle
Gdy energia jest większa, fala bardziej
oscyluje.
2
2
22
2 2 12
2
h h h h
k mE m pmEm m
Według de Broglie:
22
28n
hE E n
mL

Cząstka w pudle
Wykres jest kombinacją dwóch typów zależności:
a) energia vs. odległość – pokazujący ściany pudła (gdzie
energia potencjalna dąży do ) wraz z dozwolonymi
poziomami energetycznymi
b) wartość funkcji n(x)=y(x) vs. x, pokazująca wartości zero
funkcji energii dla danego n
- punkty gdzie =0 są nazywane węzłami funkcji falowej
- im więcej węzłów tym wyższy poziom energetycznyx
E

Cząstka w pudle - wnioski
Kwantowanie energii cząstki wynika z tego, że rozwiązanie równania Schrödingera
dla cząstki w pudle dopuszcza ściśle określone wartości energii
22
28n
hE n
mL
dla cząstki w pudle (tj, elektronu w atomie uwięzionego przez siły columbowskiego
przyciągania lub oscylujących atomów utrzymywanych razem przez siły wiążące),
liczba węzłów funkcji (x) musi być liczbą całkowitą
im więcej węzłów tym wyższa energia
„kształt” ścian lub „miękkość” ścian pudła, określa odległości miedzy poziomami
energetycznymi
dla „twardych ścian” E wprost proporcjonalne do n2 – odległości zwiększają się
ze wzrostem n
dla atomu H gdzie elektron jest utrzymywany przez „ściany kulombowskie” E
jest ~ -1/n2 więc odległości pomiędzy poziomami zmniejszają się ze wzrostem n2 2
2
02
Z eE
n a

Czy elektron „orbituje” wokół jądra??
( 2 ) ( ) ( 4 ) ( 6 ) ...
( ) sin( )A l
m
Rozwiązaniem równania Schrödingera jest
oscylująca funkcja sinusoidalna. Jeśli zwiększymy
kąt do 2 (całkowity obrót wokół orbity) cząstka
powróci do punktu wyjściowego i stąd:
Jest to przyczyną kwantowania momentu pędu i
determinuje kształt orbitali atomowych.
Jeżeli funkcja falowa jest funkcją kąta
l to stała
( 2 ) sin 2 sin 2 sin cos 2 cos sin 2 sinA l A l l A l l l l A l
Równanie jest prawdziwe gdy: cos(2l)=1 i sin(2l)=0 oraz l jest liczbą całkowitą = 0,1,2,3,…

Funkcja falowa
0 2
0 2
2
()
(l=0)
(l=1)
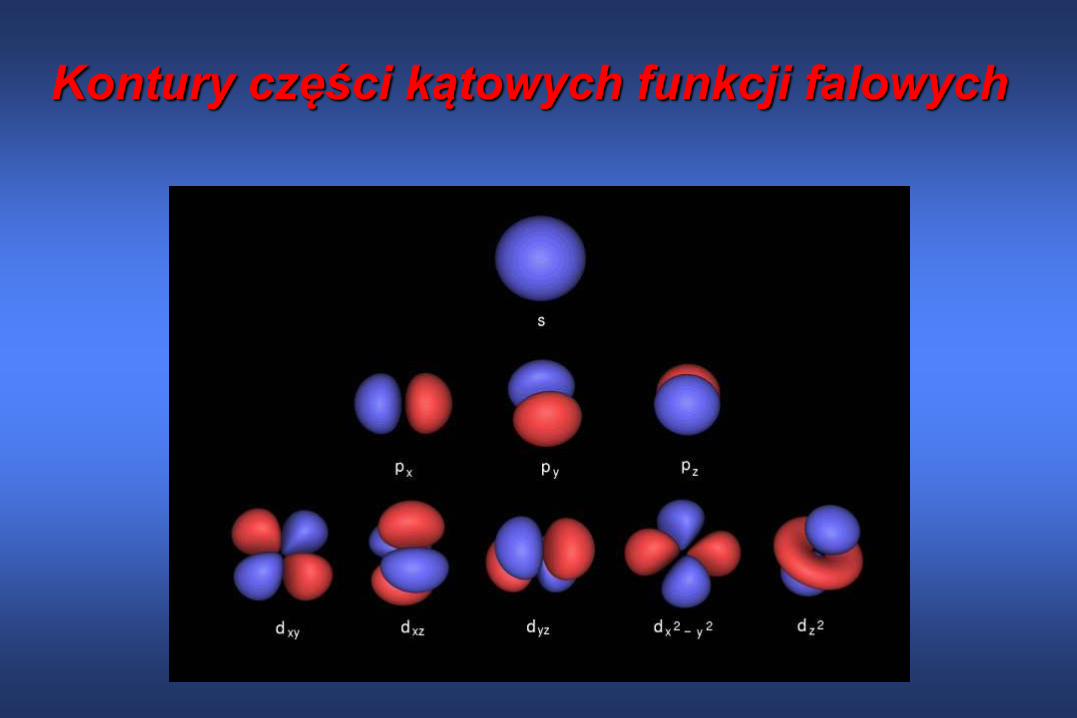
(l=2)Zależność funkcji falowej od kąta jest
przyczyną różnych kształtów orbitali
atomowych
l jest liczbą węzłów w funkcji falowej
elektronu w atomie wodoru
l=0 dla orbitalu s
l=1 dla orbitalu p
l=2 dla orbitalu d…

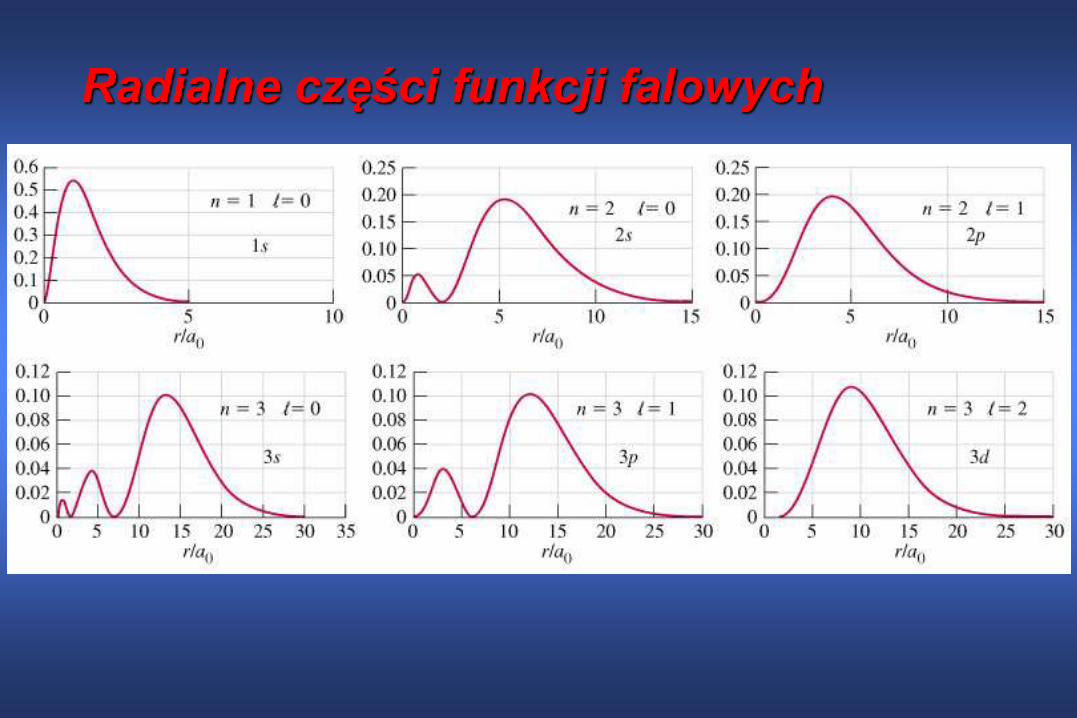
Funkcja falowa jako iloczyn funkcji radialnej i
kątowejCałkowita funkcja falowa zależy od
trzech współrzędnych elektronu:
kartezjańskich (x,y,z) lub
sferycznych (r,,) gdzie
x = r sincos
y = r sinsin
z = r cos
x
y
z
(x,y,z)(r,,)
Zazwyczaj jest przedstawiana w postaci
iloczynu funkcji radialnej R(r) zależnej tylko
od odległości elektronu od jadra oraz funkcji
kątowej Y(,) zależnej od współrzędnych
kątowych
, , ,ltotal n l l mR r Y
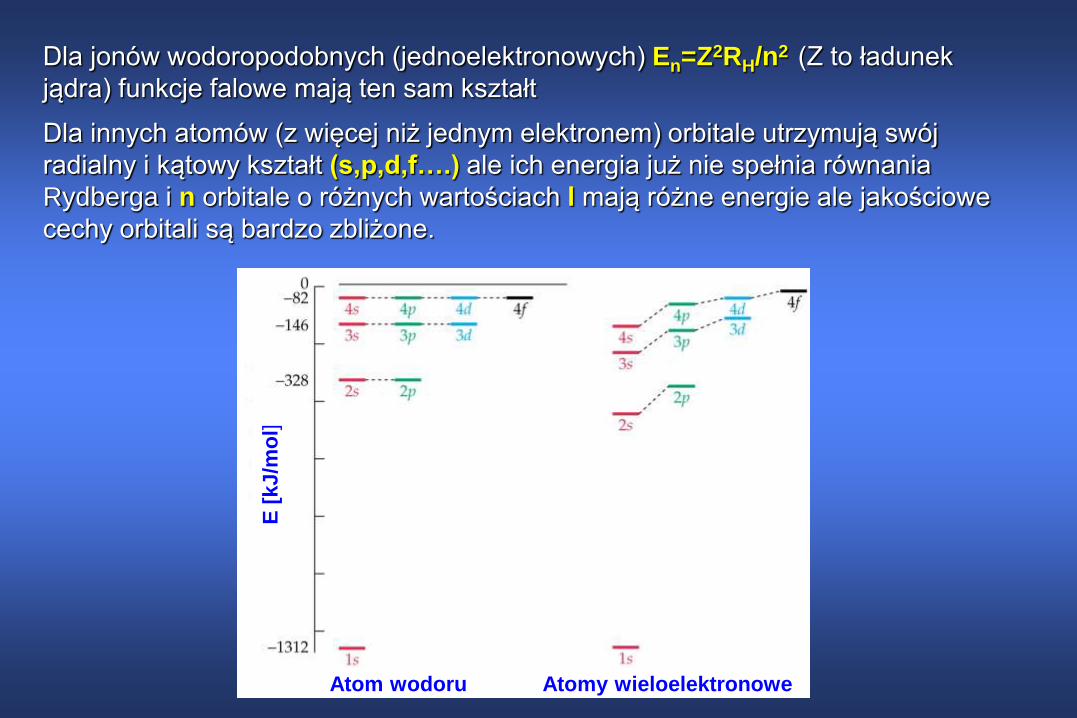
Dla jonów wodoropodobnych (jednoelektronowych) En=Z2RH/n2 (Z to ładunek
jądra) funkcje falowe mają ten sam kształt
Dla innych atomów (z więcej niż jednym elektronem) orbitale utrzymują swój
radialny i kątowy kształt (s,p,d,f….) ale ich energia już nie spełnia równania
Rydberga i n orbitale o różnych wartościach l mają różne energie ale jakościowe
cechy orbitali są bardzo zbliżone.
E [
kJ
/mo
l]
Atom wodoru Atomy wieloelektronowe
Radialne części funkcji falowych
Kontury części kątowych funkcji falowych

Poziomy energetyczne atomów od H do Na
Poziomy 2s i 2p rozsuwają się i rozsunięcie to wzrasta wraz ze
wzrostem rozmiaru atomu
1s
1s
1s
H He Li Be B C N O F Ne Na

Teoria Orbitali Molekularnych
Rozwiązując równanie Schrödingera dla ruchu nelektronów wokół jądra o ładunku n+ otrzymamy orbitalewodoropodobne.
Dla cząsteczek składających się z 2 lub więcej jąder,analogiczne rozwiązania równania Schrödingeranazwane są orbitalami molekularnymi (MO)
Istnieje szereg programów umożliwiających określenieorbitalnych funkcji falowych i energii wieloelektronowychcząsteczek. Jednakże bardzo użyteczny jest semi–ilościowy opis uzyskany przez przedstawienie orbitalimolekularnych jako Liniowej Kombinacji OrbitaliAtomowych (LCAO)

Wybrane właściwości Orbitali Molekularnych
Gdy atomy zbliżają się do siebie aby utworzyć cząsteczkę, orbitale atomowe o tej samej symetrii (o takim samym kształcie) i o takiej samej (lub zbliżonej) energii łączą się tworząc orbitale cząsteczkowe MO
Liczba MO w cząsteczce równa się sumie liczby orbitali atomowych obydwu atomów

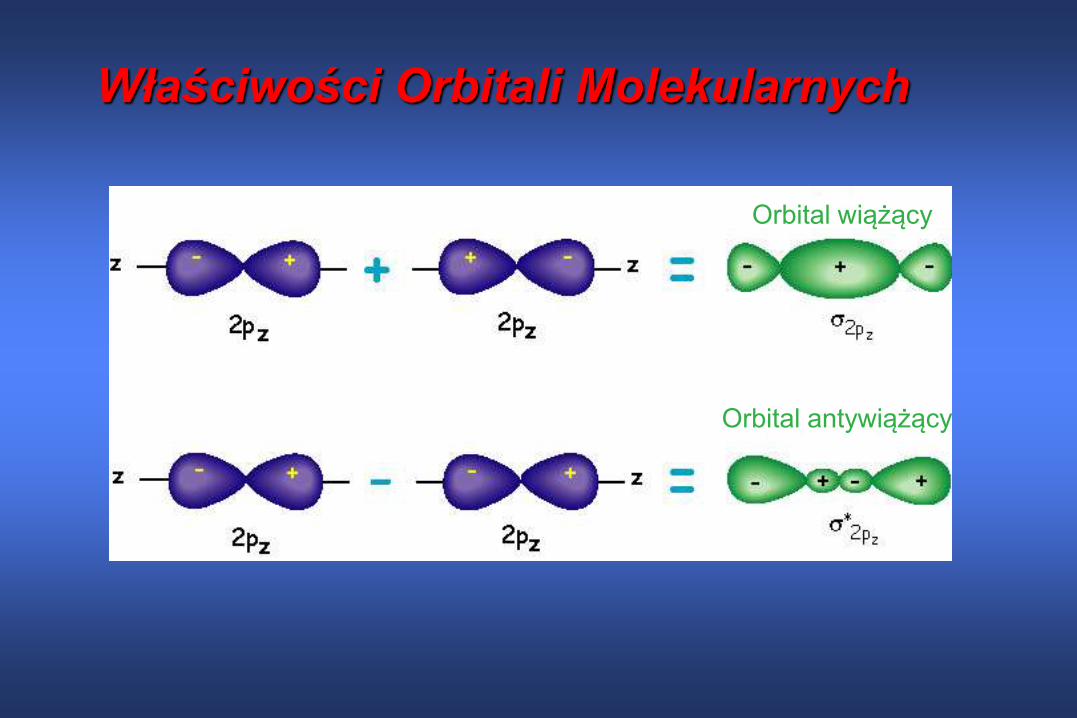
Wybrane właściwości Orbitali Molekularnych Orbitale MO są „antywiążące” i „wiążące” w zależności od tego czy (lub nie) MO
funkcja falowa:
- zmienia znak algebraiczny przy przejściu od jednego jądra do drugiego
- posiada płaszczyznę węzłową (gdzie MO=0) przecinającą oś wiązania
- jej gęstość elektronowa dąży do zera w niektórych miejscach pomiędzy jądrami
Dwie funkcje AO 1s dwóch atomów wodoru dają dwie funkcje MO:
+ + +-
+-
+ +
1 1 1s H A H Bs s *
1 1 1s H A H Bs s
WIĄŻĄCE ANTYWIĄŻĄCE
2

Wybrane właściwości Orbitali Molekularnych
Oznaczenie jest stosowane do orbitali mających symetrięcylindryczną wokół osi wiązania. Spoglądając wzdłuż osiwiązania, obydwa orbitale i * wyglądają tak samo
W porównaniu z energią oddzielnych atomów orbital wiążący ma niższą energię bo pomiędzy atomami znajduje sięobszar o zwiększonej gęstości elektronowej, podczas gdyorbital antywiażący * posiada większą energię ponieważfunkcje +/- znoszą się i w obszarze międzyjądrowymwystępuje zmniejszona gęstość elektronowa, więc jądra„widzą się” nawzajem i odpychają.

Wybrane właściwości Orbitali Molekularnych
Moc wiązania zależy od liczby elektronów wiążących i
antywiążących.
Rozważ OM cząsteczek: H2+, H2, He2
+, He2
Orbital antywiążący
Orbital wiążący
Orbitale atomoweE
E=431 kJ/mol

Wybrane właściwości Orbitali Molekularnych
Cząsteczka jest stabilna jeśli liczba elektronów wiążących jestwiększa od liczby elektronów antywiążących
Rząd wiązania = ½{(liczba elektronów wiążących)-(liczbaelektronów antywiążących)}
Im większy rząd wiązania tym silniejsze i krótsze jest wiązanie
Dla cząsteczek utworzonych z atomów wieloelektronowychobowiązują takie same reguły jak dla atomów jdnoelektronowych,ale sytuacja jest bardziej skomplikowana z powodu ekranowania iodpychania elektronów rozszczepiającego poziomy energetyczne
Orbitale atomowe 2s wiążą się tworząc orbitale 2s 2s*, tak samojak AO 1s.
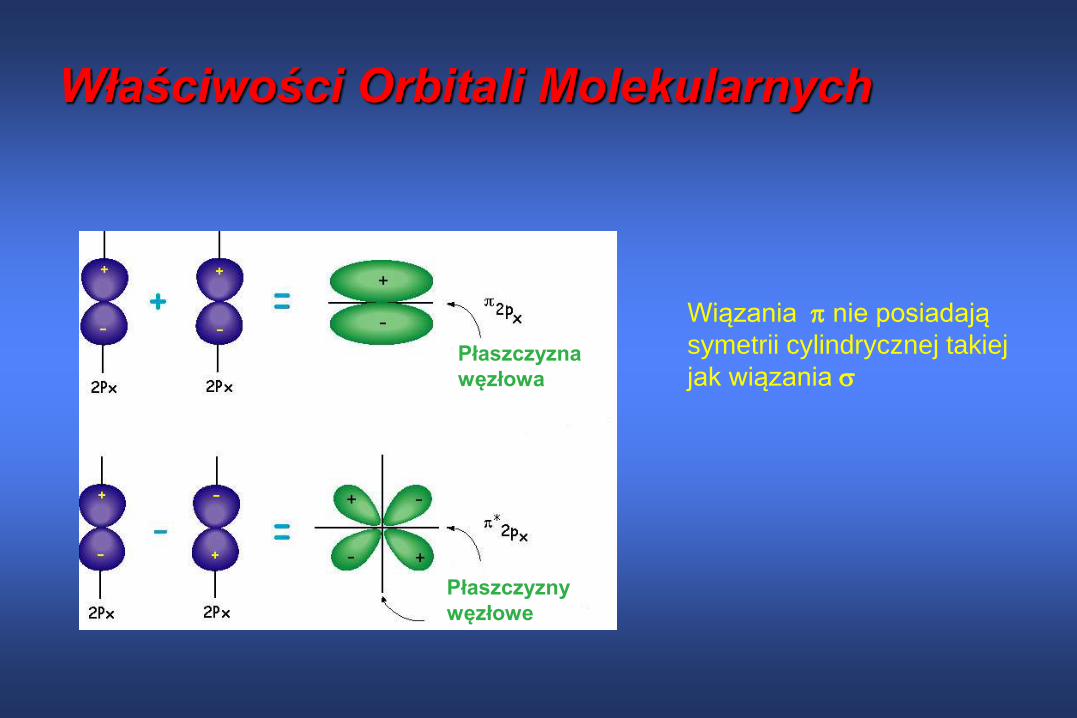
Orbitale 2p różnych atomów mogą się łączyć tylko wtedy gdy majątaka samą symetrię {2pz(A)± 2pz(B), 2px(A)± 2px(B), 2py(A)± 2py(B)}
Właściwości Orbitali Molekularnych
Orbital wiążący
Orbital antywiążący

Właściwości Orbitali Molekularnych
2 zp
2pz(A)± 2pz(B)
*
2 zp
Właściwości Orbitali Molekularnych
Płaszczyzna
węzłowa
Płaszczyzny
węzłowe
Wiązania nie posiadają
symetrii cylindrycznej takiej
jak wiązania

Właściwości Orbitali Molekularnych
W przypadku cząsteczek dwuatomowych homojądrowych
można oznaczać orbitale uwzględniając ich symetrię:
-Jeżeli funkcja falowa zmienia znak przez inwersje
przez środek wiązania wówczas posiada symetrię u
(ungerade)
-Jeżeli funkcja falowa nie zmienia znaku przez
inwersje przez środek wiązania wówczas posiada
symetrię g (gerade)
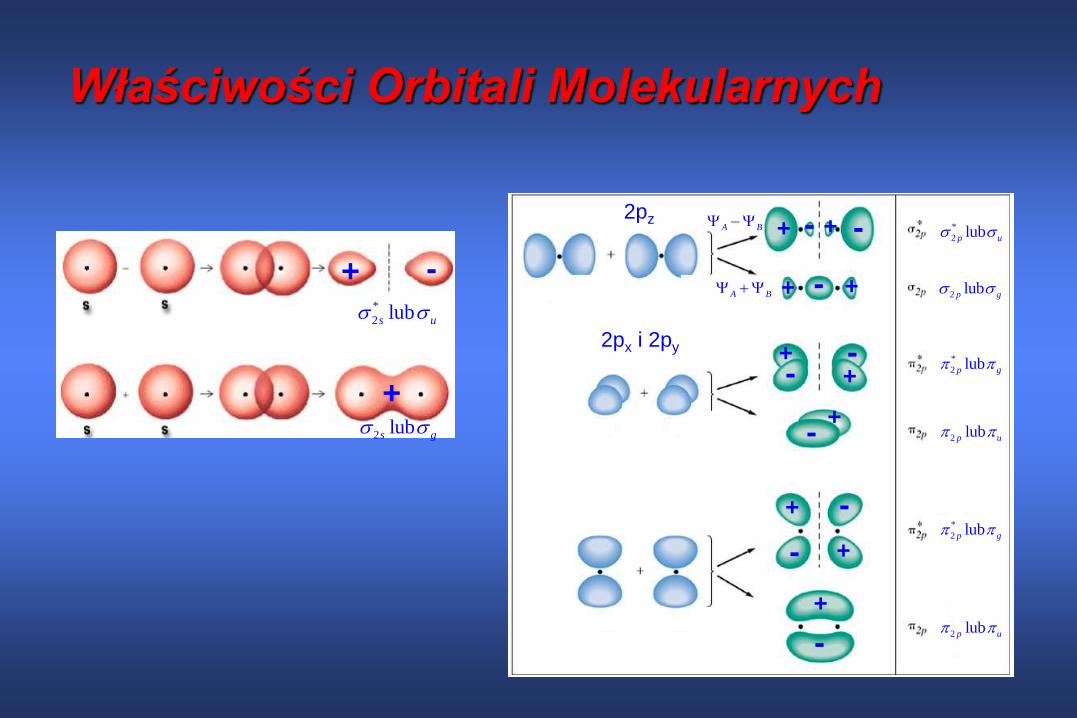
Właściwości Orbitali Molekularnych
*
2 lubs u
2 lubs g
+
+ -
2 lubp u
*
2 lubp g
2 lubp u
*
2 lubp g
2 lubp g
*
2 lubp u 2pz
2px i 2py
A B
A B -+ +
+
+
+
+
+
+
+
+
--
-
-
-
-
--

Właściwości Orbitali Molekularnych
Wszystkie wyjściowe orbitale p obu atomów mają taką samą
energię
Rozszczepienie orbitali każdego typu jest symetryczne (energia
orbitalu antywiążącego zwiększa się o taką samą wartość o jaką
obniża się energia orbitalu wiążącego)
Orbital 2p tworzy silniejsze wiązanie (tj. daje większe
rozszczepienie) niż orbitale 2p ponieważ gęstość elektronowa na
osi pomiędzy związanymi atomami jest większa i jądra atomowe
są silniej przyciągane ku sobie
Istnieją dwie równocenne pary orbitali orbitali u i g, pochodzące
od równocennych par orbitali atomowych px i py.

Właściwości Orbitali Molekularnych
Diagram energii orbitali molekularnych dwuatomowych cząsteczek
homojądrowych pierwszego i drugiego okresu.
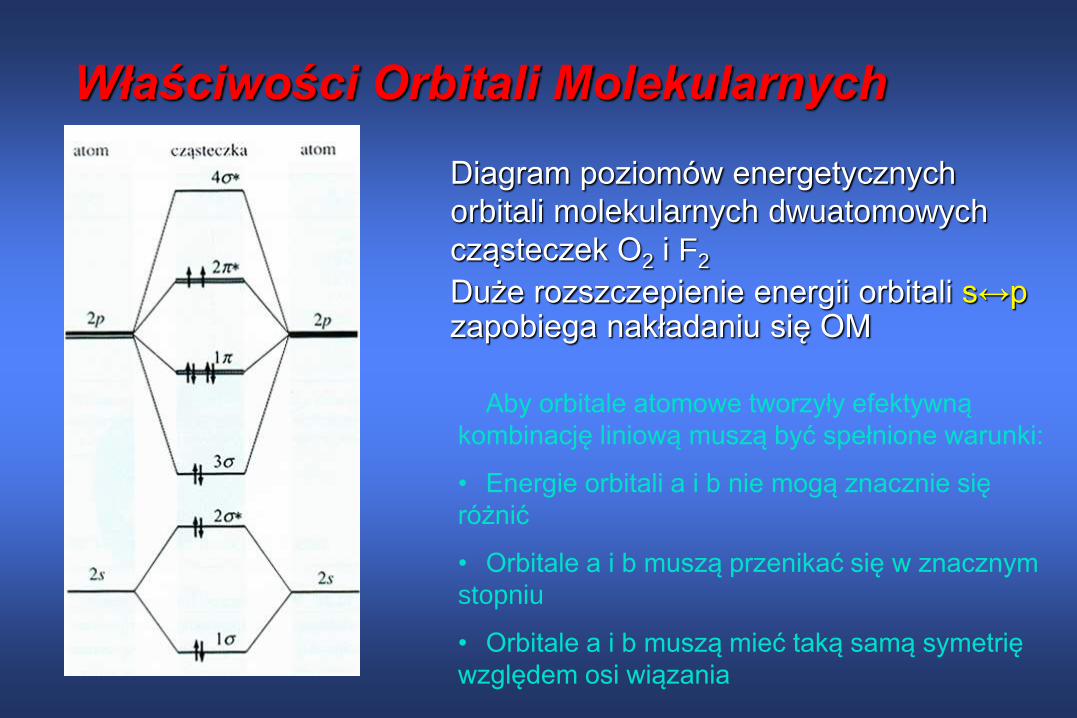
Właściwości Orbitali Molekularnych
Diagram poziomów energetycznych
orbitali molekularnych dwuatomowych
cząsteczek O2 i F2
Duże rozszczepienie energii orbitali s↔pzapobiega nakładaniu się OM
Aby orbitale atomowe tworzyły efektywną
kombinację liniową muszą być spełnione warunki:
• Energie orbitali a i b nie mogą znacznie się
różnić
• Orbitale a i b muszą przenikać się w znacznym
stopniu
• Orbitale a i b muszą mieć taką samą symetrię
względem osi wiązania
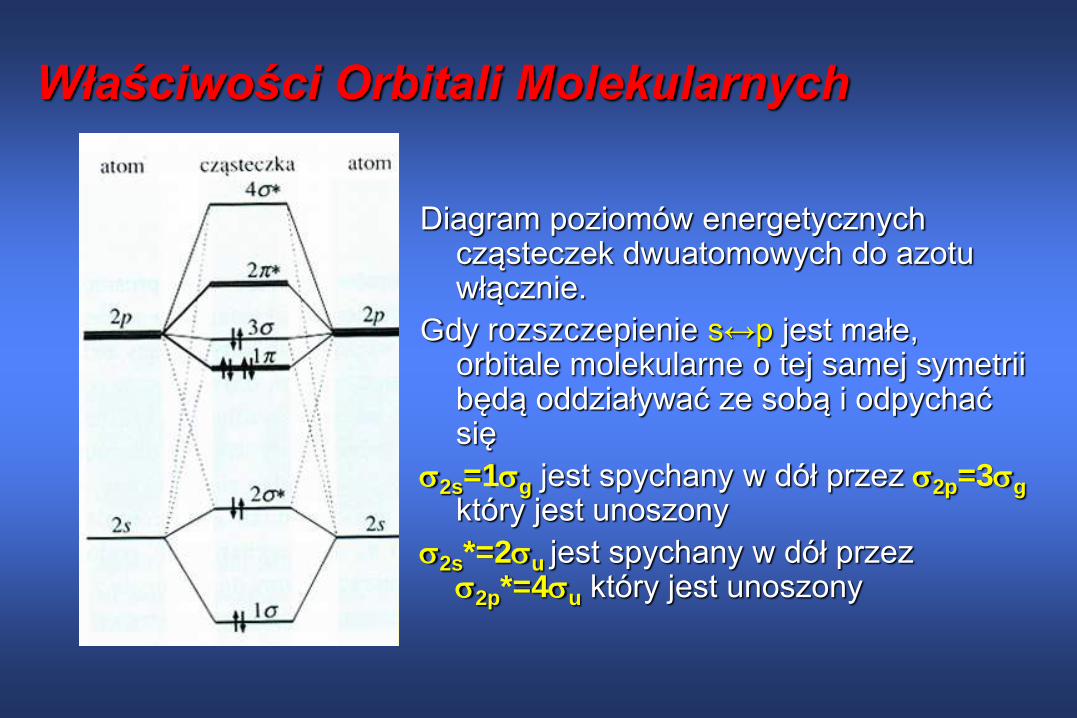
Właściwości Orbitali Molekularnych
Diagram poziomów energetycznych cząsteczek dwuatomowych do azotu włącznie.
Gdy rozszczepienie s↔p jest małe, orbitale molekularne o tej samej symetrii będą oddziaływać ze sobą i odpychać się
2s=1g jest spychany w dół przez 2p=3g
który jest unoszony
2s*=2u jest spychany w dół przez 2p*=4u który jest unoszony
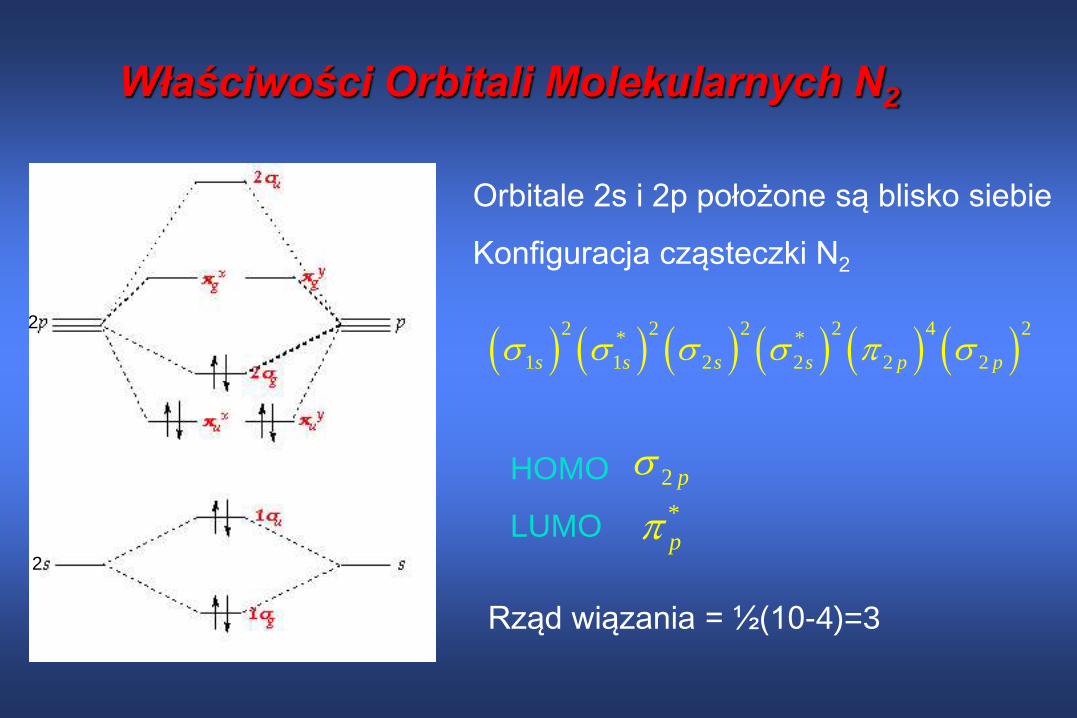
Właściwości Orbitali Molekularnych N2
2 2 2 2 4 2
* *
1 1 2 2 2 2s s s s p p
2 p
2
2
Orbitale 2s i 2p położone są blisko siebie
Konfiguracja cząsteczki N2
HOMO
LUMO *
p
Rząd wiązania = ½(10-4)=3
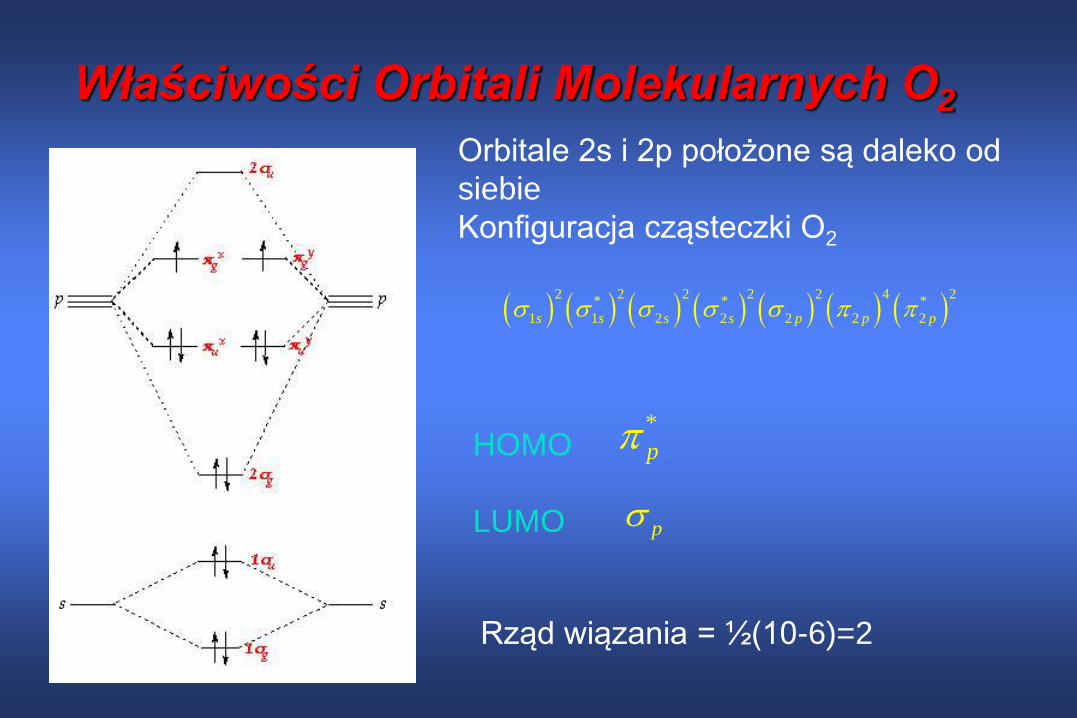
Właściwości Orbitali Molekularnych O2
2 2 2 2 2 4 2
* * *
1 1 2 2 2 2 2s s s s p p p
*
p
Orbitale 2s i 2p położone są daleko od
siebie
Konfiguracja cząsteczki O2
HOMO
LUMO p
Rząd wiązania = ½(10-6)=2
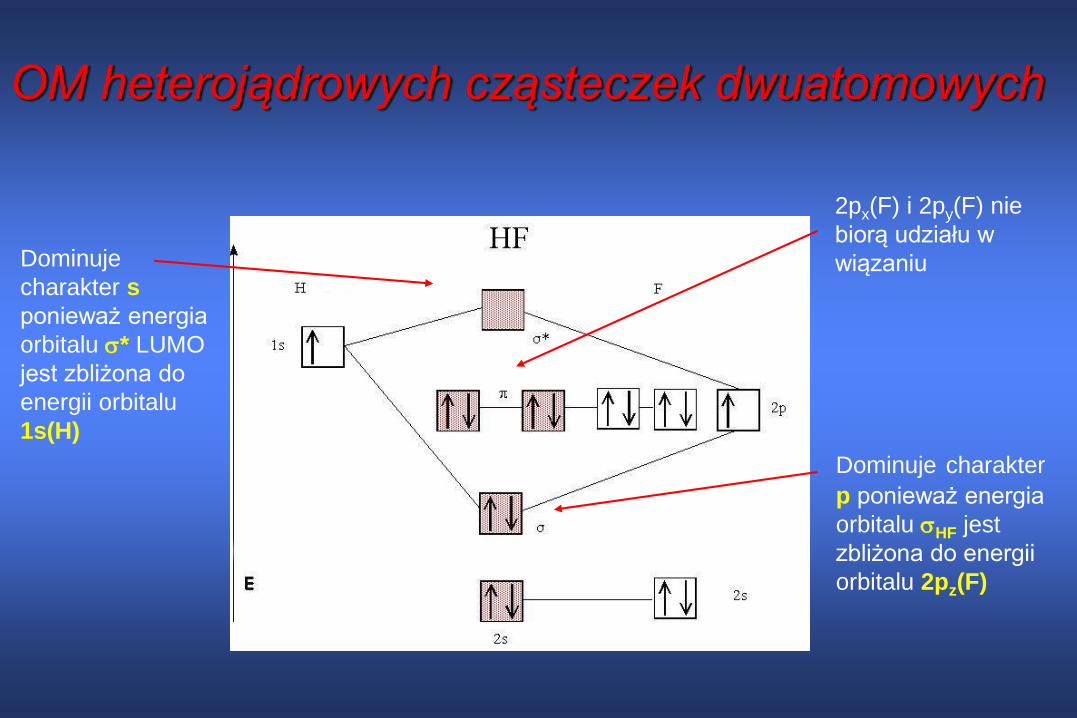
OM heterojądrowych cząsteczek dwuatomowych
Dominuje
charakter s
ponieważ energia
orbitalu * LUMO
jest zbliżona do
energii orbitalu
1s(H)
Dominuje charakter
p ponieważ energia
orbitalu HF jest
zbliżona do energii
orbitalu 2pz(F)
2px(F) i 2py(F) nie
biorą udziału w
wiązaniu

Przybliżenie Borna-OppenheimeraPomija wpływ ruchu jąder na ruch elektronów i rozdziela oscylacyjny ruch jąder od ruchu rotacyjnego
Tot Tot Tot TotH E
Całkowite równanie Schrödingera dla cząsteczki zależy od wszystkich współrzędnych
elektronów i jąder
W 1927 roku Born i Oppenheimer stwierdzili, że skoro elektrony są znacznie lżejsze
od jader (1837 razy) to poruszają się znacznie szybciej – tj. jądra nie nadążają za
ruchem elektronów. Operator Hamiltona ma postać:
Tot ele nucH H H
Dla jakiegokolwiek położenia jąder można oddzielnie rozwiązać równanie
elektronowe Schrödingera
Tot el nuc
el el el elH E
Atomy mają różne poziomy energii elektronowej i podobnie energia
cząsteczek zmienia się wraz z położeniem atomów !!!

Krzywa energii potencjalnej cząsteczki
Przybliżenie Borna-Oppenheimera
pozwala wybrać pewną odległość
międzyjądrową i rozwiązać równanie
Schrödingera dla elektronów przy
danej pozycji jąder. Prowadząc
obliczenia dla innych pozycji jąder
badamy jak zmienia się energia
cząsteczki wraz ze zmianą długości
wiązania (w złożonych cząsteczkach
również ze zmianą kątów).
Otrzymujemy krzywą energii
potencjalnej (energia kinetyczna
nieruchomych jąder równa się zero)
0
= Re
De

Krzywe energii potencjalnej dwóch stanów elektronowych jonu H2
+
Gdy jądra zbliżają się do siebie, następuje
wzrost energii rozszczepienia pomiędzy
orbitalem a *
Dla małych wartości R energia odpychania
jąder rośnie bardzo szybko
Odległość międzyatomowa odpowiadająca
minimum energii potencjalnej nazywa się
Re a energia wiążąca w tym minimum to De
(„e” = equilibrium = równowaga)
E
R
*
De
Krzywe energii elektronowej w zależności od odległości międzyatomowej są
funkcjami, które rządzą ruchem jąder – tak jak potencjał Coulumboowski V=-Ze2/r
zarządza ruchem obrotowym elektronu w atomie wodoru
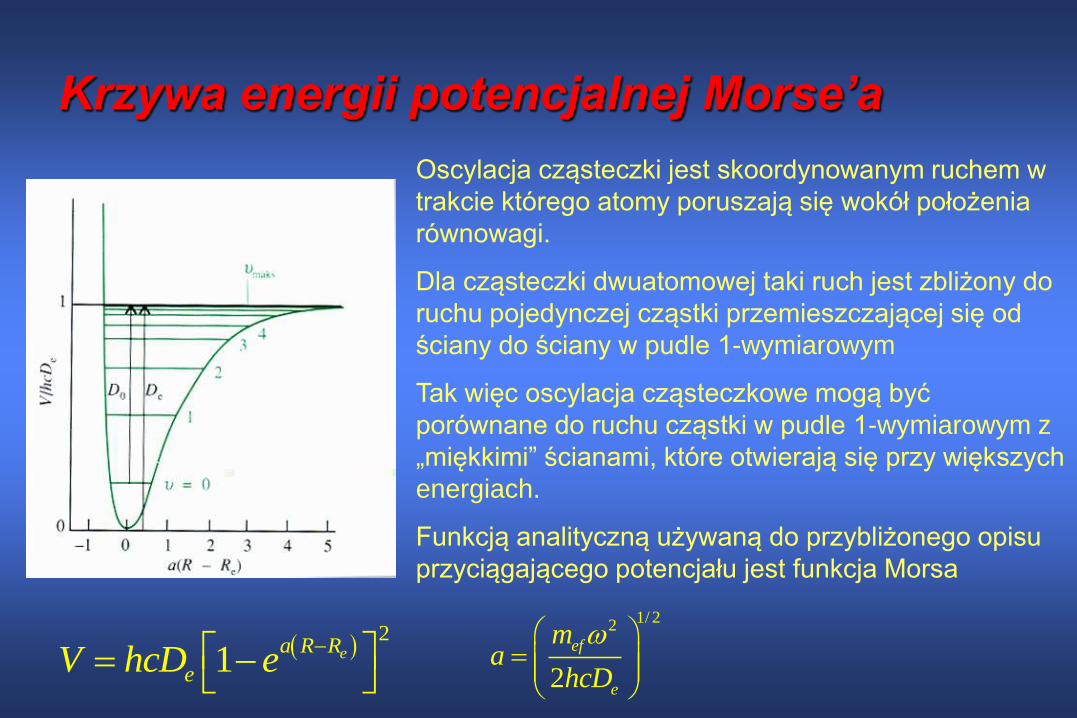
Krzywa energii potencjalnej Morse’a
2
1 ea R R
eV hcD e
Oscylacja cząsteczki jest skoordynowanym ruchem w
trakcie którego atomy poruszają się wokół położenia
równowagi.
Dla cząsteczki dwuatomowej taki ruch jest zbliżony do
ruchu pojedynczej cząstki przemieszczającej się od
ściany do ściany w pudle 1-wymiarowym
Tak więc oscylacja cząsteczkowe mogą być
porównane do ruchu cząstki w pudle 1-wymiarowym z
„miękkimi” ścianami, które otwierają się przy większych
energiach.
Funkcją analityczną używaną do przybliżonego opisu
przyciągającego potencjału jest funkcja Morsa
1/ 22
2
ef
e
ma
hcD
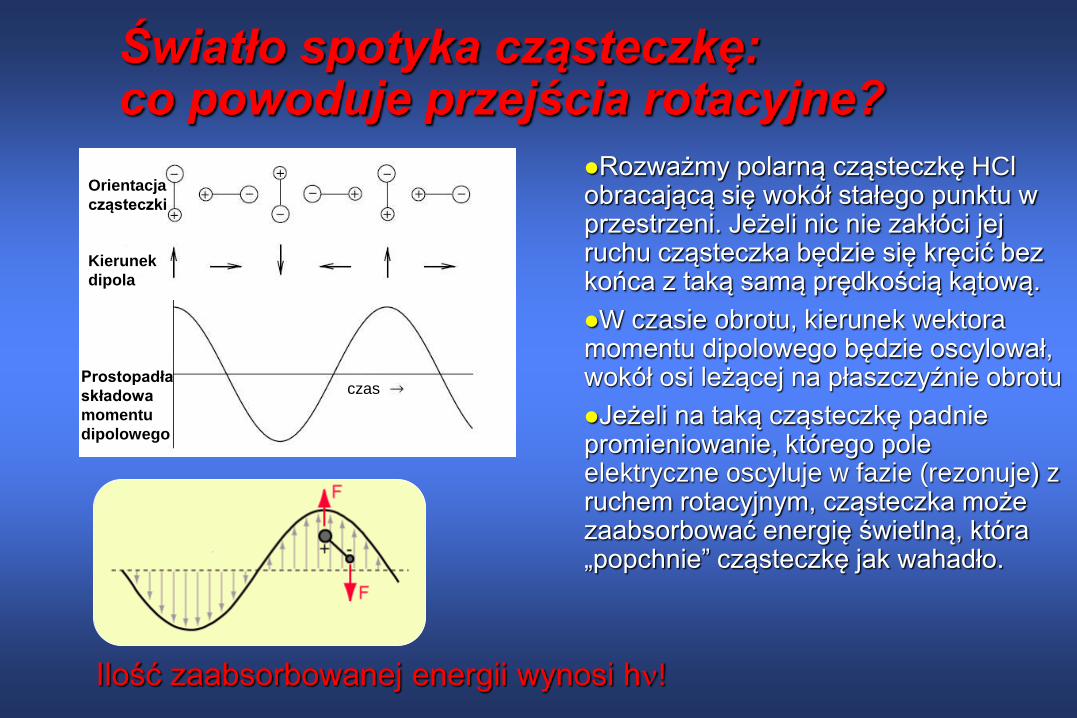
Światło spotyka cząsteczkę: co powoduje przejścia rotacyjne?
Rozważmy polarną cząsteczkę HClobracającą się wokół stałego punktu w przestrzeni. Jeżeli nic nie zakłóci jej ruchu cząsteczka będzie się kręcić bez końca z taką samą prędkością kątową.
W czasie obrotu, kierunek wektora momentu dipolowego będzie oscylował, wokół osi leżącej na płaszczyźnie obrotu
Jeżeli na taką cząsteczkę padnie promieniowanie, którego pole elektryczne oscyluje w fazie (rezonuje) z ruchem rotacyjnym, cząsteczka może zaabsorbować energię świetlną, która „popchnie” cząsteczkę jak wahadło.
Ilość zaabsorbowanej energii wynosi h!
czas
Kierunek
dipola
Orientacja
cząsteczki
Prostopadła
składowa
momentu
dipolowego

Światło spotyka cząsteczkę:
co powoduje przejścia rotacyjne?
Reguła wyboru: cząsteczki nie posiadające stałego momentu dipolowego (H2, N2, CH4, …) nie posiadają czystych widm „rotacyjnych” !!!
Cząsteczki posiadające stały moment dipolowy (wszystkie cząsteczki które nie są idealnie symetryczne) posiadają „czyste” widma rotacyjne
Uwaga
Przejścia rotacyjne są niskoenergetyczne 1-100 cm-1 i pojawiają się w obszarze spektralnym nazywanym „mikrofalowym” (MW)

Światło spotyka cząsteczkę: co powoduje przejścia oscylacyjne?
Cząsteczki posiadające stały
moment dipolowy, którego
wielkość zmienia się w
czasie drgania mogą
oddziaływać z polem
elektromagnetycznym, które
powoduje „popchnięcie”
cząsteczki
Jeżeli oscylujące pole
elektryczne światła jest w
fazie z oscylacją wiązania to
cząsteczka może
zaabsorbować światło!!
czas
Długość
wiązania
Wielkość
dipola
Prostopadła
składowa
dipola
Równowagowy
moment dipolowy

Światło spotyka cząsteczkę: co powoduje przejścia oscylacyjne?
Ruch oscylacyjny odbywa się zazwyczaj z większą częstością niż ruch rotacyjny, tak więc do wzbudzenia oscylacji potrzebne jest promieniowanie o większej energii leżące w obszarze zwanym „podczerwienią” (IR) 100 – 10 000 cm-1
Mówimy, że cząsteczki posiadające „czyste” widma w podczerwieni (np. HCl lub jakakolwiek cząsteczka dwuatomowa heterojądrowa), są „aktywne w podczerwieni”
Dwuatomowe cząsteczki homojądrowe (np. H2, Cl2, N2, O2…) nie posiadają widm w podczerwieni
Poliatomowe cząsteczki wykonują szereg różnych drgań oscylacyjnych –niektóre są aktywne w IR inne nieaktywne. Dotyczy to również cząsteczek bez stałego momentu dipolowego takich jak CO2
Drganie symetryczne rozciągające – nieaktywne w IR
Niesymetryczne rozciągające – aktywne w IR
Zginające (podwójnie zdegenerowane) aktywne w IR

Oddziaływanie promieniowania
elektromagnetycznego z cząsteczkamiZjawisko Obszar spektralny Długości fal
Elektrony wewnętrzne -
jonizacja
Promieniowanie X 0.1-1.0 nm
Elektrony walencyjne Ultrafiolet 0-400 nm
Oscylacje cząsteczek,
rozciąganie wiązań lub
rotacja
Podczerwień 0.8 m -25 m
Rotacja i orientacja spinu
elektronowego w polu
magnetycznym
Mikrofale 400 m – 30 cm
Orientacje spinów
jądrowych w polu
magnetycznym
Fale radiowe >100 cm





![Natan Glycynders Lachen - Autonomie und Chaos · titel TA NAJWAŻNIEJSZA CZĄSTECZKA [Das wichtigste Molekül]), deutsch unter dem titel N ATAN G LYCYNDERS L ACHEN in berlin/DDR 1986](https://static.fdocuments.pl/doc/165x107/5d4df1f588c99351698b92e1/natan-glycynders-lachen-autonomie-und-chaos-titel-ta-najwazniejsza-czasteczka.jpg)












