
DESTYLACJA wymagania - chemia.biol-chem.uwb.edu.pl · DESTYLACJA FRAKCYJNA Do kolby okragłodennej...
27
DESTYLACJA – wymagania 1. Destylacja frakcyjna 2. Destylacja próżniowa 3. Destylacja azeotropowa 4. Destylacja z parą wodną I. Wymagania teoretyczne 1. Krzywa zależności temperatury od ilości destylatu w zależności od składu mieszaniny 2. Zasada działania kolumny rektyfikacyjnej 3. Zasada i zalety destylacji frakcyjnej, pod zmniejszonym ciśnieniem, azeotropowej, z parą wodną 4. Wpływ ciśnienia na temperaturę wrzenia 5. Substancje rozdzielane przez frakcyjną, próżniową, azeotropową, z parą wodną 6. Rodzaje destylacji pod zmniejszonym ciśnieniem II. Aparatura 1. Zestawy do destylacji 2. Rodzaje chłodnic, kolumn, pomp próżniowych i ich zastosowanie III. Pomiar temperatury wrzenia IV. BHP 1. Zapobieganie przegrzewaniu się cieczy 2. Zasady postępowania z palnymi rozpuszczalnikami 3. Praca z aparaturą pod zmniejszonym ciśnieniem
-
Upload
nguyenmien -
Category
Documents
-
view
220 -
download
1
Transcript of DESTYLACJA wymagania - chemia.biol-chem.uwb.edu.pl · DESTYLACJA FRAKCYJNA Do kolby okragłodennej...

DESTYLACJA – wymagania
1. Destylacja frakcyjna
2. Destylacja próżniowa
3. Destylacja azeotropowa
4. Destylacja z parą wodną
I. Wymagania teoretyczne
1. Krzywa zależności temperatury od ilości destylatu w zależności od składu mieszaniny
2. Zasada działania kolumny rektyfikacyjnej
3. Zasada i zalety destylacji frakcyjnej, pod zmniejszonym ciśnieniem, azeotropowej,
z parą wodną
4. Wpływ ciśnienia na temperaturę wrzenia
5. Substancje rozdzielane przez frakcyjną, próżniową, azeotropową, z parą wodną
6. Rodzaje destylacji pod zmniejszonym ciśnieniem
II. Aparatura
1. Zestawy do destylacji
2. Rodzaje chłodnic, kolumn, pomp próżniowych i ich zastosowanie
III. Pomiar temperatury wrzenia
IV. BHP
1. Zapobieganie przegrzewaniu się cieczy
2. Zasady postępowania z palnymi rozpuszczalnikami
3. Praca z aparaturą pod zmniejszonym ciśnieniem

EKSTRAKCJA – wymagania
1. Teoria ekstrakcji
a) prawo podziału Nernsta
b) efekt wysolenia
2. Rodzaje ekstrakcji
a) zwykła jednokrotna
b) zwykła wielokrotna
3. Ekstrakcja cieczy
a) prosta
b) ciągła
c) rozpuszczalnikami optycznie czynnymi
4. Ekstrakcja osadów
a) prosta
b) ciągła ciał stałych
5. Czynności i technika ekstrakcji
b) wybór rozpuszczalnika
c) stosowane środki suszące
d) metody zatężania
6. BHP (sposoby postępowania ze związkami palnymi i trującymi)

KRYSTALIZACJA I SUBLIMACJA – wymagania
1. Teoria i metody wykonywania krystalizacji i sublimacji
a) krystalizacja ze stopu
b) substancje organiczne i nieorganiczne ulegające sublimacji
2. Technika wykonywania krystalizacji
a) dobór rozpuszczalnika
b) mieszaniny chłodzące
c) sposoby przyspieszania krystalizacji
d) metody sączenia
e) pomiar temperatury topnienia
f) wpływ temperatury i czasu na przebieg krystalizacji
3. Aparatura
a) krystalizacja z rozpuszczalnika palnego
b) sublimacja pod normalnym i zmniejszonym ciśnieniem
4. BHP (sposoby postępowania z palnymi toksycznymi rozpuszczalnikami)

CHROMATOGRAFIA – wymagania
2. Chromatografia cienkowarstwowa (TLC)
a) absorbenty stosowane w chromatografii cienkowarstwowej
b) wykonanie chromatogramu:
- wybór rozpuszczalnika do rozwijania próbek (szereg eluotropowy rozpuszczalników)
- rozwijanie i wywołanie
- wartość Rf
c) zastosowanie chromatografii cienkowarstwowej
3. Chromatografia bibułowa i jej zastosowanie
4. Chromatografia kolumnowa
b) absorbenty stosowane w chromatografii kolumnowej
c) rodzaje kolumn chromatograficznych
d) wybór rozpuszczalnika do chromatografii absorbcyjnej
e) wypełnienie i nanoszenie substancji na kolumnę
f) rozwijanie chromatogramu
g) zastosowanie chromatografii kolumnowej
5. Chromatografia gazowa
6. BHP (praca z toksycznymi substancjami organicznymi)

DESTYLACJA PROSTA
Do kolby okrągłodennej o pojemności 250 mL wlać 130 mL mieszaniny i zmontować
zestaw do destylacji prostej. Po wrzuceniu kamyczków wrzennych kolbę ogrzewać i zbierać
destylat z szybkością 2-3 kropel na sekundę do cylindra miarowego.
Zanotować początkową temperaturę oraz temperatury po odebraniu każdych 5 mL
destylatu.
W opisie ćwiczenia umieścić tabelę z odczytami temperatury oraz wykres zależności
temperatury od objętości destylatu. Porównać wyniki dla destylacji prostej i frakcyjnej.
Podczas destylacji cieczy może nastąpić jej przegrzanie, tzn. ogrzanie powyżej
temperatury wrzenia. Wówczas, w wyniku wibracji lub obniżenia ciśnienia, zaczyna się
spontaniczne wrzenie, zwane potocznie „rzucaniem”. Dlatego też, podczas destylacji cieczy
należy intensywnie mieszać, np. mieszadłem magnetycznym, bądź też dodać „kamyczki
wrzenne”, np. wyprażony kaolin. Kaolin należy dodawać do zimnej jeszcze cieczy/ Po
jednorazowym użyciu „kamyczek wrzenny” traci swoje właściwości.

DESTYLACJA FRAKCYJNA
Do kolby okragłodennej o pojemności 250 mL wlać około 130 mL mieszaniny
i zmontować zestaw do destylacji frakcyjnej. Po wrzuceniu kamyczków wrzennych kolbę
ogrzewać i zbierać destylat do cylindra miarowego. Zanotować początkową temperaturę oraz
temperaturę po zebraniu każdych 5 mL destylatu.
W opisie ćwiczenia umieścić tabelkę z odczytami temperatury oraz wykres zależności
temperatury od objętości destylatu (zaznaczyć przedgon, frakcje główne, frakcje pośrednie,
pogon oraz temperatury wrzenia składników mieszaniny).
Rys. 6. Zestaw do destylacji frakcyjnej; a – kolumna typu Vigreux

DESTYLACJA AZEOTROPOWA
Do kolby o pojemności 250 mL zaopatrzoną w nasadkę azeotropową i chłodnicę wlać
mieszaninę rozpuszczalników: toluen:woda:etanol (1:1:1). Wrzucić kamyki wrzenne
i prowadzić destylację.
Obserwować zbierające się warstwy w nasadce azeotropowej.
Rys. 7. Zestaw do destylacji z nasadką azeotropową.

DESTYLACJA PRÓŻNIOWA
Do kolby destylacyjnej o pojemności 100 mL wlać 50 mL DMF. Przeprowadzić
destylacje próżniową zbierając poszczególne frakcje: przedgon, frakcja główna, pogon.
Zanotować temperaturę wrzenia i porównać z literaturową pod normalnym ciśnieniem.
Rys. 8. Zestaw do destylacji pod zmniejszonym ciśnieniem
Praca pod zmniejszonym ciśnieniem („pod próżnią”). Zmniejszone ciśnienie używane
jest podczas takich operacji jak: destylacja, sączenie, sublimacja, liofilizacja i suszenie
w eksykatorach próżniowych lub suszarkach próżniowych. Wszelkie prace pod
zmniejszonym ciśnieniem należy prowadzić pod wyciągiem lub za specjalnym ekranem,
zabezpieczającym przed odłamkami szkła w przypadku implozji. Podczas prowadzenia prac
pod zmniejszonym ciśnieniem należy bezwzględnie nosić okulary ochronne. Ze względu na
niebezpieczeństwo implozji do wszelkich prac prowadzonych: „pod próżnią” nie można
używać naczyń z płaskim dnem, takich jak np. kolby stożkowe. Wyjątek stanowią –
specjalnie wykonane z grubego szkła – kolby ssawkowe – stosowane do sączenia pod
zmniejszonym ciśnieniem. Większe aparaty lub eksykatory próżniowe bywają pokryte
warstwą przezroczystego tworzywa, które w przypadku awarii, podobnie jak w szybach
samochodowych, zabezpiecza przed powstaniem ostrych odłamków szkła.

DESTYLACJA Z PARĄ WODNĄ
W kolbie okrągłodennej umieścić mieszaninę: 2 g o-nitrofenolu i 2 g p-nitrofenolu,
rozpuszczoną w 30 mL wody. Dodać kamyki wrzenne i zmontować zestaw do destylacji
z parą wodną. Wytworzyć parę wodną w kociołku napełnionym do 2/3 objętości. W celu
niedopuszczenia do kondensowania się zbyt ilości pary wodnej w kolbie, powoli ogrzewamy
jej zawartość. Gdy woda w kociołku zacznie wrzeć, obserwować wydzielanie się
pęcherzyków pary wodnej w kolbie i destylowanie cieczy. Destylację prowadzić do momentu,
gdy wpływający do odbieralnika destylat jest klarowny.
Rys. 9. Zestaw do destylacji z para wodną

WYODRĘBNIANIE LIMONENU Z OWOCÓW
CYTRUSOWYCH ZA POMOCĄ DESTYLACJI
Z PARĄ WODNĄ
Limonen (węglowodór z grupy terpenów) jest głównym składnikiem olejku
eterycznego występującego w skórkach owoców cytrusowych. W przyrodzie występuje
izomer R limonenu.
CH3
C
H
H2C
CH3
Odczynniki: Sprzęt laboratoryjny:
skórka z dwu pomarańczy lub grapefruita kolby okragłodenne: 500 mL i 50 mL
chlorek metylenu 20 mL zestaw do destylacji z parą wodną
siarczan magnezu bezw. termometr
rozdzielacz stożkowy 100 mL
kolba stożkowa 50 mL – 2 szt.
moździerz
cylinder miarowy 100 mL
lejek szklany
Skórkę z 2 pomarańczy lub 1 grapefruita pokroić na drobne kawałki i rozetrzeć
w moździerzu wraz z 200 mL wody na miazgę. Miazgę przenieść do kolby destylacyjnej
na 500 mL, zmontować zestaw do destylacji prostej i destylować z para wodną do czasu
zebrania 50-60 mL destylatu. Destylat schłodzić do temperatury pokojowej, przenieść
do rozdzielacza i ekstrahować za pomocą 10 mL chlorku metylenu, którym uprzednio
przepłukano chłodnicę. Fazę organiczną zlać do suchej kolby stożkowej o pojemności 50 mL,
a pozostała w rozdzielaczu faze wodną ponownie ekstrahować 10 mL chlorku metylenu.
Połączone ekstrakty organiczne suszyć przez 15 min siarczanem magnezu, następnie
przesączyć przez sączek karbowany do suchej kolb i odparować chlorek metylenu.
Po odparowaniu pozostaje około 0,5 mL oleju o charakterystycznym zapachu.

EKSTRAKCJA JODU Z ROZTWORU JODKU POTASU
CHLORKIEM METYLENU
Do rozdzielacza o pojemności 100 mL wlać 20 mL roztworu jodku w jodku potasu
i ekstrahować 15 mL chlorku metylenu. Po dokładnym wytrząśnięciu pozostawić zawartość
do rozdzielenia się warstw. Warstwę organiczną przenieść do erlenmajerki.
Ekstrakcję powtórzyć 3-krotnie, obserwując zachodzące zmiany. Porównaj, czy
ekstrakcja 1-krotna (45 mL chlorku metylenu), czy 3-krotna tą samą ilością rozpuszczalnika
(3 x 15 mL) jest bardziej wydajna.
Rys. 3. Oddzielanie roztworów w rozdzielaczu: a – położenie rozdzielacza podczas
oddzielania dolnej warstwy, b – położenie rozdzielacza podczas wyrównywania ciśnienia
Proces ekstrakcji stosowany jest do wydzielania, np. z roztworu wodnego, substancji
lepiej rozpuszczającej się w cieczy, nie mieszającej się z wodą. Ekstrakcję prowadzi się
najczęściej w rozdzielaczach. Podczas mieszania się dwóch ciekłych faz w rozdzielaczu
bardzo często wytwarza się nadciśnienie, w związku z czym proces należy prowadzić
nadzwyczaj ostrożnie, usuwając nadciśnienie z wnętrza naczynia. W tym celu wylot
rozdzielacza należy skierować ku górze (rys. 3b), najlepiej pod wyciągiem, a następni
ostrożnie wyrównać ciśnienie, otwierając powoli kurek. Pod żadnym pozorem wylotu
rozdzielacza nie można kierować w kierunku laboratorium lub ku sąsiadom. Szczególnie
niebezpieczne są ekstrakcje fazy wodnej, zawierającej węglany, rozpuszczalnikami takimi jak
np. chloroform, zawierającymi niewielkie ilości chlorowodoru. Tworzy się wówczas
dwutlenek węgla, a powstałe nadciśnienie może wyrzucić zawartość naczynia na zewnątrz.
Podczas ekstrakcji należy zakładać okulary i rękawice ochronne.

EKSTRAKCJA SUBSTANCJI STAŁYCH
W APARACIE SOXHLETA
Aparat Soxhleta służący do ekstrakcji ciągłej substancji stałych gorącym
rozpuszczalnikiem pokazano na rys. 4a. Substancję przeznaczoną do ekstrakcji umieszcza się
w gilzie A wykonanej z twardej bibuły filtracyjnej. Gilzę wsuwa się do wewnętrznej rury
aparatu B, pod którym montuje się kolbę C wypełnioną rozpuszczalnikiem do ekstrakcji.
U góry aparatu montuje się chłodnicę zwrotną D. Kolbę z rozpuszczalnikiem ogrzewa się
do osiągnięcia stanu łagodnego wrzenia zawartości. Pary rozpuszczalnika przepływają
do chłodnicy, tam skraplają się i zostają zawrócone do gilzy. Po zebraniu takiej porcji
rozpuszczalnika w gilzie, że jego górny poziom osiąga wysokość bocznej rurki F, ekstrakt
zostaje przelany syfonem do kolby. Proces ten powtarza się automatycznie aż do zakończenia
ekstrakcji.
Po zakończeniu ekstrakcji rozpuszczalnik należy odparować na wyparce próżniowej
(rys. 4b) w całości. Wnioski z ćwiczeń umieścić w sprawozdaniu.
a) b)
Rys. 4. a - aparat Soxhleta; b – laboratoryjna wyparka obrotowa typu Unipan 350

EKSTRAKCJA Z WYKORZYSTANIEM
KWASOWO-ZASADOWYCH WŁAŚCIWOŚCI
WYODRĘBNIONEGO ZWIĄZKU
Często zdarza się, że w wyniku reakcji otrzymujemy mieszaninę, w której skład
wchodzą związki o właściwościach, kwasowych, zasadowych i obojętnych (w różnych
kombinacjach). Można je wówczas rozdzielić wykorzystując te właściwości, tzn. stosując
do ekstrakcji wodne roztwory kwasów lub zasad.
Związki o charakterze kwaśnym (fenole, kwasy karboksylowe) rozpuszczają się
w wodnych roztworach zasad, tworząc sole, i w ten sposób można je oddzielić od związków
obojętnych, które pozostają w fazie organicznej. Po rozdzieleniu faz kwasy możemy odzyskać
(o ile nam na nich zależy) przez zakwaszenie roztworu wodnego, a następnie odsączenie (jeśli
są to związki stałe) lub kolejną ekstrakcję, tym razem już rozpuszczalnikiem organicznym.
RCOOH + NaOH RCOO-Na+ + H2O
rozpuszczalne w wodzie
RCOO-Na+ + HCl RCOOH + NaCl
Związki o właściwościach zasadowych (aminy) rozpuszczają się w wodnych
roztworach kwasów i analogicznie można je oddzielić od związków obojętnych przez
ekstrakcję rozcieńczonym kwasem (związki obojętne pozostaną w fazie organicznej).
Po rozdzieleniu faz wolne aminy odzyskuje się przez potraktowanie roztworu ich soli zasadą
(np. NaOH), a następnie ekstrakcję rozpuszczalnikiem organicznym lub przez sączenie,
w zależności od właściwości fizycznych aminy.
RNH2 + NaOH RNH3+ + Cl- (chlorek amoniowy,
rozpuszczalny w wodzie)
RNH3+Cl- + NaOH RNH2 + NaCl + H2O
Jeżeli obecne w ekstrahowanej mieszaninie związki kwaśne lub zasadowe stanowią
jedynie zanieczyszczenie i nie są dla nas interesujące, to ich wodne ekstrakty odrzuca się bez
dalszego ich wyodrębniania. Mówimy wtedy, że kwas czy aminę odmywa się, np. wodnym
roztworem NaHCO3 (czy HCl).

Schemat stopniowego rozdzielania mieszaniny trzech związków O, Z i K
o właściwościach odpowiedni: obojętnych (O), zasadowych (Z) i kwaśnych (K) pomiędzy
fazą wodną i organiczną (np. chloroform) przedstawiono na rys. 5.
Rys. 5. Schemat rozdzielania mieszaniny związków o właściwościach kwasowych (K),
zasadowych (Z) i obojętnych (O)

KRYSTALIZACJA ACETANILIDU Z WODY
Odważyć 2 g acetanilidu i umieścić w zlewce o pojemności 250 mL, dodać 60 mL
wody i ogrzewać do wrzenia. Acetanilid staje się ciekły i tworzy olej w wodzie. Następnie
dolać porcjami gorącą wodę stale mieszając i ogrzać roztwór do łagodnego wrzenia,
aż acetanilid rozpuści się. Jeżeli roztwór nie jest bezbarwny, to należy nieznacznie go
ochłodzić, dodać węgla aktywnego i ogrzewać do wrzenia przez kilka minut, aby usunąć
barwne zanieczyszczenia. Prawie gorący roztwór przesączyć przez karbowany sączek,
umieszczony na lejku na płaszczu grzejnym. Przesącz zebrać do zlewki, nakryć szkiełkiem
zegarkowym i szybko schłodzić, energicznie mieszając. Następnie odstawić roztwór na
30 min, aby całkowicie wydzielił się osad. Kryształy odsączyć na lejku Büchnera, przemyć
dwukrotnie 5 mL wody (aby usunąć przylegający do kryształów macierzysty ług) i wycisnąć
na lejku za pomocą dużego korka szklanego. Lejek przewrócić na bibułę filtracyjną
o podwójnej grubości lub szkiełko zegarkowe i pozostawić kryształy do wysuszenia na
powietrzu do następnych zajęć. Przy suszeniu kryształów na powietrzu wskazane jest
przykrycie związku krążkiem z bibuły, podziurkowanej tak, aby umożliwić ulatnianie
rozpuszczalnika.
Wysuszoną substancję należy zważyć, obliczyć wydajność krystalizacji i oznaczyć
temperaturę topnienia.
a) b) c)
Rys. 10. Zestaw do sączenia: a – osadu; b – na gorąco; c - pod zmniejszonym ciśnieniem

KRYSTALIZACJA Z ROZPUSZCZALNIKA LOTNEGO
Odważyć 2,5 g p-acetanilidu (lub innego związku zgodnie z zaleceniem prowadzącego
ćwiczenia) i umieścić w kolbie okrągłodennej o pojemności 100 mL pod chłodnicą zwrotną
(rys.11a), dodać niewielką ilość rozpuszczalnika (etanolu lub innego w zależności
od krystalizowanej substancji) i ogrzewać do wrzenia. W przypadku gdyby substancja nie
uległa rozpuszczeniu, dodać następne porcje rozpuszczalnika, aż do momentu całkowitego
rozpuszczenia. Gorący roztwór przesączyć przez karbowany sączek. Przesącz ochłodzić,
wytrącony osad przesączyć na lejku Büchnera, a uzyskany produkt pozostawić do wysuszenia
na powietrzu do następnych zajęć.
Wysuszoną substancję należy zważyć, obliczyć wydajność krystalizacji i oznaczyć
temperaturę topnienia.
a) b)
Rys. 11. a - Zestaw do ogrzewania pod chłodnicą zwrotną; b – Aparat do mierzenia
temperatury topnienia typu Boetius: 1 - blok grzewczy, 2 - termometr w osłonie, 3 - lampa
oświetlająca próbkę od dołu, 4 - lampa oświetlająca termometr i układ optyczny, 5 - regulacja
ostrości, 6,7 - układ optyczny, 8 - regulacja jasności, 9 - przewód opornicy regulujący
ogrzewanie bloku, 10 - szklane pokrywy bloku i próbki, 11 - transformator oświetlenia,
12 - próbka

STRĄCANIE ROZPUSZCZALNIKIEM
1. Odważyć w małej zlewce 0,5 g glicyny. Następnie rozpuścić w niewielkiej ilości
wody. Wkraplać ostrożnie etanol aż do całkowitego wytrącenia osadu. Powstały osad
odsączyć na lejku Büchnera, wysuszyć i zważyć. Obliczyć wydajność strąconej
glicyny.
Podobnie postępować z:
1. kwas benzoesowy rozpuścić w acetonie – strącić wodą
2. kwas salicylowy rozpuścić w etanolu – strącić wodą
3. naftalen rozpuścić w acetonie – strącić wodą

KRYSTALIZACJA
Krystalizacja z H2O (na gorąco) – próbka 1-2 g
1. acetanilid
2. fenyloseryna
Krystalizacja z rozpuszczalników lotnych (na gorąco) – próbka 2 g
1. p-bromonitrobenzen z etanolu
2. benzanilid z etanolu
3. dicykloheksylidenoglukofuranoza z eteru naftowego
4. pentaacetylo-β-D-glukoza z metanolu
Strącanie z mieszaniny rozpuszczalnikiem
4. glicyna w wodzie – strącanie etanolem
5. kwas benzoesowy w acetonie – strącanie wodą
6. kwas salicylowy w etanolu – strącanie wodą
7. naftalen w acetonie – strącanie wodą

SUBLIMACJA
Parownicę porcelanową zawierającą 2,5 g naftalenu przykryć krążkiem bibuły
z małymi otworami i odwróconym lejkiem szklanym. Nóżkę lejka zatkać korkiem z waty.
Parownicę postawić w płaszczu grzejnym (rys. 12a). Po łagodnym ogrzaniu parownicy pary
czystej substancji przechodzą przez otwory w bibule i kondensują na wewnętrznych
ściankach lejka. Zebrać i zważyć sublimat.
Obliczyć wydajność sublimacji.
Rys. 12. Zestaw do sublimacji: a – pod normalnym ciśnieniem; b – pod zmniejszonym
ciśnieniem
Podczas sublimacji należy unikać przegrzania, ponieważ powoduje to stopienie
związku, czasami także jego rozkład, a tym samym straty. Do zbierania przesublimowanej
substancji można przystąpić dopiero po ochłodzeniu aparatury.

CHROMATOGRAFIA CIENKOWARSTWOWA (TLC)
I. Rozdzielanie aminokwasów
1) Przygotowanie komory chromatograficznej
Do komory chromatograficznej wyłożonej bibułą nalewa się mieszaninę
rozpuszczalników: propanol-1 – amoniak (7:3) (układ rozwijający), aby grubość warstwy
wyniosła 0,5 cm. Komorę zamknąć i odstawić na kilkanaście minut celem nasycenia jej
parami rozpuszczalników.
2) Przygotowanie chromatogramu
Na płytce chromatograficznej zaznaczyć delikatnie ołówkiem linie startu w odległości
ok. 1 cm wzdłuż krótszego boku płytki.
3) Nanoszenie substancji
Za pomocą cienkiej kapilary na linii startu w równych odległościach nanieść kolejne
roztwory wzorcowe aminokwasów i ich mieszaninę. Plamki można suszyć ostrożnie
zimnym strumieniem powietrza.
4) Rozwijanie chromatogramu
Płytkę włożyć do uprzednio przygotowanej komory chromatograficznej tak, aby dolna
krawędź była w momencie zanurzenia możliwie równoległa do powierzchni cieczy
w komorze. Należy zwrócić uwagę, aby krawędzie boczne nie dotykały ścian komory,
gdyż powoduje to nierównomierne wznoszenie się rozpuszczalnika na wysokość około
0,5 cm od góry. Płytkę należy następnie wyjąć, zaznaczyć czoło rozpuszczalnika
i wysuszyć dokładnie suszarką.
5) Wywołanie chromatogramu
Wysuszoną płytkę ustawić w pozycji pionowej i spryskać roztworem ninhydryny.
Następnie wstawić płytkę do suszarki o temp. 105˚C na około 10 min. Zaznaczyć plamki
odpowiednich aminokwasów.
ZESTAW AMINOKWASÓW:
I DL-alanina III L-lizyna
II L-leucyna IV mieszanina aminokwasów
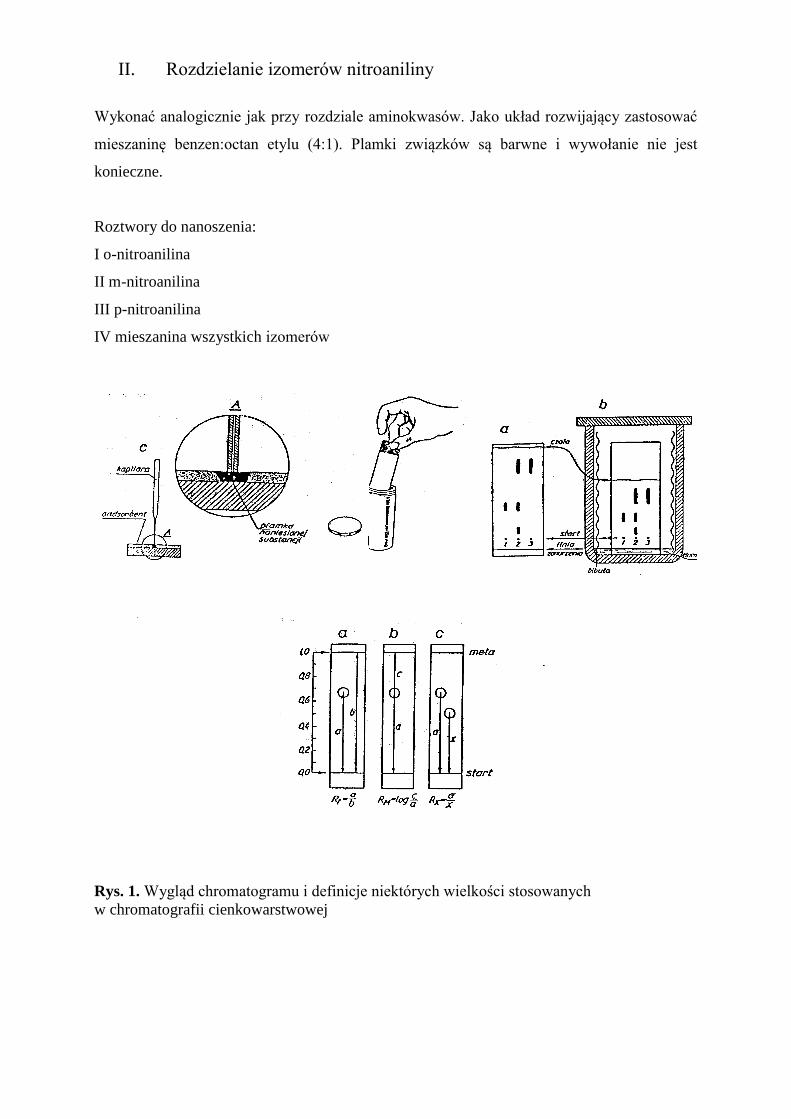
II. Rozdzielanie izomerów nitroaniliny
Wykonać analogicznie jak przy rozdziale aminokwasów. Jako układ rozwijający zastosować
mieszaninę benzen:octan etylu (4:1). Plamki związków są barwne i wywołanie nie jest
konieczne.
Roztwory do nanoszenia:
I o-nitroanilina
II m-nitroanilina
III p-nitroanilina
IV mieszanina wszystkich izomerów
Rys. 1. Wygląd chromatogramu i definicje niektórych wielkości stosowanych
w chromatografii cienkowarstwowej

CHROMATOGRAFIA KOLUMNOWA
Na dnie kolumny chromatograficznej umieszczamy niewielką ilość waty i napełniamy
silikażelem do ¾ wysokości. Na tak przygotowaną kolumnę wprowadzamy 1-1,5 mL
mieszaniny barwników. Chromatografię prowadzimy dodając kolejno następujące eluenty:
octan etylu, mieszanina etanolu i acetonu w stosunku 1:4. Rozdzielone barwniki zbieramy do
osobnych kolbek.
W opisie ćwiczenia podać barwę mieszaniny substancji, przebieg chromatografii oraz
barwy substancji wchodzących w skład mieszaniny.
Rys. 2. Zestaw aparatury do chromatografii kolumnowej

BADANIE SKŁADU BARWNIKÓW ROŚLIN
ZIELONYCH
W skład barwnika roślin zielonych wchodzą chlorofile: a i b oraz karotenoidy: karoten
i ksantofil.
Wykonanie: Zielone liście uciera się w moździerzu z odrobiną piasku (w celu
łatwiejszego zniszczenia tkanek komórkowych) i kilkoma kroplami acetonu, a następnie
za pomocą kapilary nanosi się na płytkę i rozwija w układzie aceton:toluen (1:2). Próbkę
należy nanosić kilkakrotnie w tym samym miejscu, zachowując jednocześnie małą średnicę
plamki. Po rozwinięciu należy szybko analizować, gdyż barwniki łatwo blakną. Plamki obu
chlorofili są blisko siebie, wyraźnie natomiast oddziela się od nich karoten i ksantofil.
Odczynniki wrażliwe na zanieczyszczenie powietrza w laboratorium: Produkty
stanowiące bardzo dobre adsorbenty takie jak na przykład silikażel czy też płytki do
chromatografii cienkowarstwowej (TLC) powinny być chronione przed atmosferą panującą
w laboratorium. Tego typu materiały należy przechowywać w specjalnych pojemnikach, gdyż
na przykład płytki do TLC znajdujące się w kontakcie z laboratoryjnym powietrzem
stopniowo tracą aktywność, co oczywiście wpływa na ich zdolności separacyjne.

PREPARAT do wyboru:
KWAS ACETYLOSALICYLOWY (ASPIRYNA)
COOH
OH
(CH3CO)2OH+
CH3COOH
COOH
OCOCH3
+ +
Odczynniki: Sprzęt laboratoryjny:
kwas salicylowy 2 g termometr
bezwodnik octowy 3 mL cylinder miarowy
kwas siarkowy stęż. 0.3 mL zlewka 250 mL
etanol kolba ssawkowa
lejek Büchnera
kolba stożkowa 100 mL
W kolbie stożkowej o pojemności 100 ml miesza się 2 g kwasu salicylowego,
3 mL bezwodnika octowego i 0.3 mL stęż. kwasu siarkowego. Kolbę stożkową ogrzewa się
na łaźni wodnej do temp. 60˚C w ciągu 20 min. Kontynuuje się temperaturę termometrem
umieszczonym w mieszaninie reakcyjnej i równocześnie miesza zawartość kolby. Mieszaninę
reakcyjną pozostawia się do ochłodzenia a następnie równocześnie mieszając, dodaje się
40 mL wody. Wytrącony surowy osad odsącza się pod zmniejszonym ciśnieniem
i krystalizuje z wodnego roztworu etanolu (1 objętość alkoholu na 3 objętości wody).
Wydajność ok. 2 g. Temperatura topnienia z rozkładem 128 – 135˚C.

NIKOTYNA Z TYTONIU
tytoñ
N
N
CH3
Odczynniki: Sprzęt laboratoryjny:
tytoń (np. z papierosów) 4 g zestaw do destylacji z parą wodną
3 N wodorotlenek sodu 67 mL cylinder miarowy 50 mL
chlorek sodu 25 g kolba stożkowa 100 mL – 3 szt.
eter dietylowy 60 mL rozdzielacz 100 mL
siarczan (VI) magnezu bezw. kolba okrągłodenna 100 mL
W kolbie okrągłodennej o pojemności 500 mL poddaje się destylacji z parą wodną
4 g tytoniu w 67 mL 3 N roztworu wodorotlenku sodu do chwili, aż destylat będzie
pozbawiony zapachu nikotyny (ok. 100 – 150 mL). Po oziębieniu destylatu dodaje się do
niego 14-25 g stałego chlorku sodu i ekstrahuje się produkt trzema porcjami eteru
dietylowego po 20 mL. Ekstrakty łączy się i przemywa wodą. Po osuszeniu ekstraktu bezw.
siarczanem(VI) magnezu oddestylowuje się rozpuszczalnik na wyparce obrotowej. Otrzymuje
się oleistą, surową nikotynę.

KOFEINA Z HERBATY
herbataCa(CO3)2/H2O
N
N N
N
O
O
CH3
CH3
H3C
kofeina
Odczynniki: Sprzęt laboratoryjny:
herbata 60 g kolba stożkowa 1000 mL
chloroform 180 mL cylinder miarowy 500 mL
węglan wapnia 60 g kolba stożkowa 100 mL
etanol rozdzielacz 250 mL
siarczan (VI) magnezu bezw. kolba okrągłodenna 100 mL
zlewka 250 mL – 2 szt.
W kolbie stożkowej z szeroką szyją o poj. 1000 mL umieszcza się 60 g herbaty
w torebkach (lub sypką w woreczku z gazy) i ogrzewa do wrzenia przez 20 minut z 600 mL
wody zawierającej 60 g sproszkowanego węglanu wapnia. Po przesączeniu na gorąco,
przemyciu herbaty gorącą wodą i oziębieniu dodaje się do przesączu 180 mL chloroformu.
Obie warstwy miesza się delikatnie (w celu uniknięcia powstania emulsji) przez 15 min,
stosując mieszadło magnetyczne. Ekstrakt chloroformowy oddziela się w rozdzielaczu, suszy
bezwodnym siarczanem magnezu i odparowuje na wyparce obrotowej. Surowy produkt
krystalizuje się benzenu lub etanolu, otrzymując 240 mg kofeiny o t,t, 225-228 ˚C.
Chromatografia TLC na żelu krzemionkowym:
1. Rf = 0.5-0.6, chloroform - etanol 99:1
2. Rf = 0.4, chlorek metylenu - octan etylu 1:1
Wykrywanie: płytkę spryskać roztworem jodu w etanolowym roztworze jodku potasu (1)
i po upływie 2 minut ponownie spryskać mieszaniną 25% kwasu solnego i etanolu w stosunku
1:1. W wyniku reakcji plama kofeiny barwi się na kolor ciemnobrunatny.
(1) 1 g jodu i 2 g jodku potasu rozpuścić w 100 mL etanolu.

EPISMILAGENINA
(redukcja epismilagenonu za pomocą NaBH4)
O
O
HO
O
O
H
NaBH4, THF
temp. pokojowa
HO
Odczynniki: Sprzęt laboratoryjny:
epismilagenon (M=414,62 g/mol) 50 mg kolba okrągłodenna x 2 szt.
NaBH4 (M=37,83 g/mol) 2 eq cylinder miarowy
chlorek metylenu (DCM) element mieszający
octan etylu (AcOEt) mieszadło magnetyczne
siarczan magnezu bezw. rozdzielacz
tetrahydrofuran (THF) 20 mL erlenmajerka 4 szt.
Heksan (Hex) lejek
komora chromatograficzna
Wykonanie:
Do roztworu epismilagenonu (100 mg) w THF-ie (50 mL), umieszczonego w kolbie
okrągłodennej na 100 mL, dodano 2 eq. NaBH4. Mieszaninę reakcyjną mieszano na mieszadle
magnetycznym w temperaturze pokojowej. Przebieg reakcji kontrolowano za pomocą
chromatografii cienkowarstwowej (TLC) w układzie rozpuszczalników AcOEt/Hex (3:7).
Po stwierdzeniu zakończenia reakcji redukcji, mieszaninę przeniesiono do
rozdzielacza, dodano wodę (20 mL), ekstrahowano trzema porcjami chlorku metylenu
(po 20 mL). Warstwę organiczną przemyto wodą, nasyconym roztworem NaCl
i na zakończenie wodą, osuszono nad bezw. siarczanem magnezu.
Oczyszczanie:
Przygotowano kilka układów rozpuszczalników do komór chromatograficznych
AcOEt/Hex w różnym stosunku. Należy wybrać najbardziej odpowiedni do efektywnego
oczyszczenia produktu na kolumnie chromatograficznej.
Kolumnę wypełniono silikażelem i naniesiono na nią osuszoną we wcześniejszym
etapie epismilageninę. Rozpoczęto eluowanie odpowiednio dobranym rozpuszczalnikiem.
Wymywanie związków monitorowano za pomocą TLC. Frakcje zawierające główny produkt
zebrano do kolby na 250 mL i odparowano na wyparce obrotowej. Osuszony związek
zważono i obliczono wydajność reakcji.
W sprawozdaniu należy umieścić:
schemat reakcji
tabelę zawierającą ilości stosowanych substancji, ich masy molowe, ilości moli,
ekwiwalentów
opis wykonania ćwiczenia
chromatogramy TLC
zasadę doboru rozpuszczalników do kolumny chromatograficznej
wydajność reakcji


















