
Niedobór żelaza: opathology.wum.edu.pl/system/files/konspekt_f_pf_1_1.pdf · Enzym cyklu...
46
Niedobór żelaza: o Okres przedutajony - ujemny bilans żelaza: dzienna utrata tego pierwiastka przekracza możliwości absorpcji utrata 10-20 ml krwinek czerwonych dziennie. o Okres utajony - Wyczerpanie zapasów żelaza: poziom ferrytyny <15ug/l oznacza wyczerpanie zapasów żelaza. Dokładne badanie rozmazu krwi obwodowej ujawni obecność mikrocytow. o Okres jawny - Umiarkowana niedokrwistość: Hb 10-13 g/dl. Niewielka odpowiedź szpiku. o Objawy: męczliwość, bladość, cheilitis – zapalenie kącików ust- pęknięcia. Koilonychia – wklęśnięcie paznokci. o Różnicowanie: Talasemie (mikrocytoza, ale Fe prawidłowe), ACD- choroby przewlekłe-zwykle normo- chromiczna i –cytarna. Zespoły mielodysplastyczne – Fe prawidłowe mimo mikrocytozy i hipochromii. ACD o Skutek przewlekłych stanów zapalnych, zakażeń, ran, wydzielania cytokin pozapalnych (IL1, 6, TNFalfa). o Objawy: patrz wyżej. Zwykle przebieg jest łagodny. Niedokrwistość w chorobach nerek: o Przebieg zależny od ciężkości uszkodzenia nerek (torbielowatość!). EPO, ale także skrócenia czasu przeżycia erytrocytów (zespół hemolityczno – mocznicowy). o Stany hipometaboliczne: o Głodzenie: spada zapotrzebowanie na O 2 , na co reagują nerki obniżając produkcję o Niedobór androgenów, hormonów tarczycy, przysadki. Choroba Addisona. o Choroby wątroby: skrócenie czasu przeżycia erytrocytów. Hemoglobinopatie: o Hb oprócz transportu O 2 wpływa także na kształt, elastyczność i lepkość krwi. o Hemoglobinopatie to zaburzenia struktury Hb, których skutkiem mogą być: niedokrwistość, erytrocytoza, sinica lub powstawanie zatorów w naczyniach. o Gen a globiny - chromosom 16 o Gen b i d globiny - chromosom 11 o Transport tlenu zależy od tetramerycznej struktury hemoglobiny, modyfikacje powinowactwa Hb do tlenu: o efekt Bohra (mniejsze powinowactwo w niższym pH); o 2,3DPG o Hb F nie wiąże się z 2,3DPG Grupy hemoglobinopatii: o Strukturalne – HbS(a 2 b 2 6Glu Va1 ) Objawy: anemia hemolityczna, mikrokatory, Ostry zespół piersiowy:ból w klp.; tachypnoe, gorączka, kaszel, obniżenie saturacji o Talasemie – niedobór poszczególnych łańcuchów globiny – skrócenie życia erytrocyta. Beta: hypochromia, mikrocytoza, mała ilość prawidłowych tetrametrów. Stymulacja wydzielania EPO prowadzi do nadmiernego rozrostu szpiku i w związku z tym upośledzenia rozwoju kości o Alfa: może przypominać talasemię beta, o mało nasilonych objawach. Łańcuchy beta formują tetrametry (HbH o Nieprawidłowe warianty strukturalne o Przetrwała hemoglobina płodowa o Hemoglobinopatie nabyte – wpływ czynnikow toksycznych Pod względem funkcji: o Hb o zwiększonym powinowactwie do O2 o Hb o zmniejszonym powinowactwie do O2 o Hb niestabilne Niedokrwistości megaloblastyczne: Spowodowane zaburzeniami syntezy DNA. Kwas foliowy: Witamina B12:
Transcript of Niedobór żelaza: opathology.wum.edu.pl/system/files/konspekt_f_pf_1_1.pdf · Enzym cyklu...

Niedobór żelaza: o Okres przedutajony - ujemny bilans żelaza: dzienna utrata tego pierwiastka przekracza możliwości
absorpcji utrata 10-20 ml krwinek czerwonych dziennie.
o Okres utajony - Wyczerpanie zapasów żelaza: poziom ferrytyny <15ug/l oznacza wyczerpanie
zapasów żelaza. Dokładne badanie rozmazu krwi obwodowej ujawni obecność mikrocytow.
o Okres jawny - Umiarkowana niedokrwistość: Hb 10-13 g/dl. Niewielka odpowiedź szpiku.
o Objawy: męczliwość, bladość, cheilitis – zapalenie kącików ust- pęknięcia. Koilonychia – wklęśnięcie
paznokci.
o Różnicowanie: Talasemie (mikrocytoza, ale Fe prawidłowe), ACD- choroby przewlekłe-zwykle
normo- chromiczna i –cytarna. Zespoły mielodysplastyczne – Fe prawidłowe mimo mikrocytozy i
hipochromii.
ACD
o Skutek przewlekłych stanów zapalnych, zakażeń, ran, wydzielania cytokin pozapalnych (IL1, 6,
TNFalfa).
o Objawy: patrz wyżej. Zwykle przebieg jest łagodny.
Niedokrwistość w chorobach nerek:
o Przebieg zależny od ciężkości uszkodzenia nerek (torbielowatość!). EPO, ale także skrócenia czasu
przeżycia erytrocytów (zespół hemolityczno – mocznicowy).
o Stany hipometaboliczne:
o Głodzenie: spada zapotrzebowanie na O2 , na co reagują nerki obniżając produkcję
o Niedobór androgenów, hormonów tarczycy, przysadki. Choroba Addisona.
o Choroby wątroby: skrócenie czasu przeżycia erytrocytów.
Hemoglobinopatie: o Hb oprócz transportu O2 wpływa także na kształt, elastyczność i lepkość krwi.
o Hemoglobinopatie to zaburzenia struktury Hb, których skutkiem mogą być: niedokrwistość,
erytrocytoza, sinica lub powstawanie zatorów w naczyniach.
o Gen a globiny - chromosom 16
o Gen b i d globiny - chromosom 11
o Transport tlenu zależy od tetramerycznej struktury hemoglobiny, modyfikacje powinowactwa Hb do
tlenu:
o efekt Bohra (mniejsze powinowactwo w niższym pH);
o 2,3DPG
o Hb F nie wiąże się z 2,3DPG
Grupy hemoglobinopatii:
o Strukturalne – HbS(a2b26Glu Va1
)
Objawy: anemia hemolityczna, mikrokatory, Ostry zespół piersiowy:ból w klp.; tachypnoe,
gorączka, kaszel, obniżenie saturacji
o Talasemie – niedobór poszczególnych łańcuchów globiny – skrócenie życia erytrocyta.
Beta: hypochromia, mikrocytoza, mała ilość prawidłowych tetrametrów. Stymulacja
wydzielania EPO prowadzi do nadmiernego rozrostu szpiku i w związku z tym upośledzenia
rozwoju kości
o Alfa: może przypominać talasemię beta, o mało nasilonych objawach. Łańcuchy beta formują
tetrametry (HbH
o Nieprawidłowe warianty strukturalne
o Przetrwała hemoglobina płodowa
o Hemoglobinopatie nabyte – wpływ czynnikow toksycznych
Pod względem funkcji:
o Hb o zwiększonym powinowactwie do O2
o Hb o zmniejszonym powinowactwie do O2
o Hb niestabilne
Niedokrwistości megaloblastyczne:
Spowodowane zaburzeniami syntezy DNA.
Kwas foliowy:
Witamina B12:

Krew IKrew I
Michał Michał PyzlakPyzlak
www.pathophysiology.amwaw.edu.plwww.pathophysiology.amwaw.edu.pl

Czym jest krew?Czym jest krew?
�Elementy morfotyczne
ErytrocytyLeukocytyPłytki
�Osocze
WodaElektrolityBiałkaAlbuminy, Globuliny, Czynniki krzepni ęcia
ErytropoezaErytropoeza
�Komórka macierzysta = erytroblast�Dojrzały Erytrocyt ma 8 mikrometrów średnicy, 120 dni Ŝycia i przebywa
drog ę o długo ści 300 mil w naczyniach.�Eliminacja
w układzie siateczkowo – śródbłonkowym…
Proerytroblast(Pronormoblast)
ErytroblastZasadochłonny
ErytroblastPolichromatofilny
ErytroblastOrtochromatyczny
Retikulocyt
Erytrocyt
Wczesne Pośrednie Dojrzałe

Istotne dane laboratoryjneIstotne dane laboratoryjne�Mean Corpuscular Volume – Średnia obj ętość krwinki
MCV (norma 82-92fL )MCV < 80 = MikrocytMCV >92 = Makrocyt
�Mean Cell Haemoglobin – Średnia zawarto ść hemoglobiny w krwinceMCH (norma 27-31pg)MCH < 27 = HipochromiaMCH > 31 = Hiperchromia
�Mean Cell Hemoglobin Concentration – Średnie st ęŜenie hemoglobinyHgb/Hct
�TIBC(µmol/l) = 0,025 x transferyna (mg/l) [45 -73 µmol/l]
�Transferryna: 2 – 4 g/l
�Ferrytyna: 20 – 200 µg/l
Niedokrwisto śćNiedokrwisto ść�StęŜenie hemoglobiny (Hgb) mniejsze ni Ŝ�13.0g/dl u m ęŜczyzn (13.0-18.0) lub �11.5g/dl u kobiet (11.5-16.0)
�Zwykle powi ązana ze zmniejszeniem liczby erytrocytów (M 4.2-5.6 x 10^12/l
K 3.5-5.6 x 10^12/l) oraz hematokrytu (Hct): (M 40-54% K 37-48%)

Mechanizm niedokrwisto ściMechanizm niedokrwisto ściZaburzenia hemopoezy :
Uszkodzenie komórek szpiku-Leki, Promieniowanie, Infekcje, Substancje toksycz ne
Niedobór:-śelaza (Synteza hemu)-Witaminy B 12 (Synteza DNA)-Kwasu foliowego (Synteza DNA)
Skrócenie prze Ŝycia krwinek :Niszczenie krwinek
-Niedokrwisto ści hemolityczne
Utrata krwi:Krwawienia i krwotoki…
ObjawyObjawyOgólne osłabienieTachycardia, tachypnoeBól zamostkowy w czasie wysiłkuZawroty głowyDepresja, pogorszenie zdolno ści intelektualnychBrak łaknieniaBlado ść
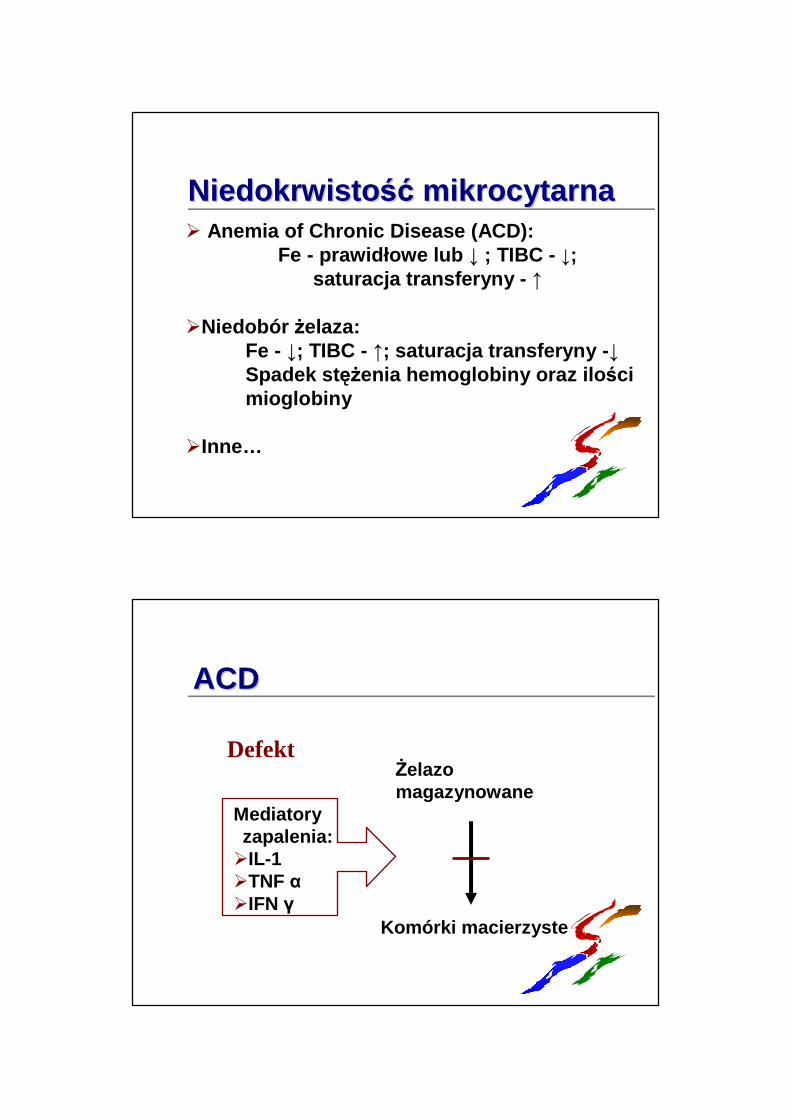
Niedokrwisto ść Niedokrwisto ść mikrocytarnamikrocytarna� Anemia of Chronic Disease (ACD):
Fe - prawidłowe lub ↓ ; TIBC - ↓; saturacja transferyny - ↑
�Niedobór Ŝelaza:Fe - ↓; TIBC - ↑; saturacja transferyny - ↓Spadek st ęŜenia hemoglobiny oraz ilo ści mioglobiny
�Inne…
ACDACD
Defekt
Mediatoryzapalenia:�IL-1�TNF α�IFN γ
Komórki macierzyste
śelazomagazynowane

Niedokrwisto ść z niedoboru Niedokrwisto ść z niedoboru ŜelazaŜelaza
�Niedobarwliwa, mikrocytarna
�Przyczyny- Przewlekłe krwawienie (500ml krwi = 250mg Fe)- DuŜe zapotrzebowanie (u młodych osób), ci ąŜa,
laktacja, krwawienia miesi ączkowe.
Metabolizm Ŝelaza Metabolizm Ŝelaza -- dystrybucjadystrybucja
Szpik
Transferryna
Ukł. Siateczkowo-śródbłonkowy 0.5-
1.5g(makrofagi)
Hemoglobina „krąŜąca”1.7-2.4g
Absorbcja UtrataTkanki
20 mg 20 mg
20 mg 20 mg
1 mg 1 mg

Objawy niedoboru ŜelazaObjawy niedoboru Ŝelaza
�Skóra i błony śluzowe: pr ąŜkowanie paznokci, zaklęśnięcie płytki paznokciowej, łamliwo ść włosów. „Zajady” w k ącikach ust - cheilitis
�Zespół Plummera-Vinsona (atrofia gardła j ęzyka, przełyku)
�Zaburzenia neurologiczne
�Objawy wspólne dla niedokrwisto ści, niezale Ŝnie od typu
Przyczyny
�(Niedobór witaminy B12)�(Niedobór kwasu foliowego)
�Nieprawidłowy metabolizm ww.�drug induced – polekowa (antymetabolity kwasu
foliowego)�idiopatyczny lub nabyty po gastrektomii lub
w chorobach jelita cienkiego
�Niedobory dietetyczne w diecie np. wega ńskiej
�Nieprawidłowa synteza DNA wrodzonaalkohol, Hydroksymocznik, Cytarabina
Niedokrwisto ść Niedokrwisto ść makrocytarnamakrocytarna

Niedobory Niedobory witwit . B12 i kwasu foliowego. B12 i kwasu foliowego
�Objawy niedoboru wit. B 12:Triada: 1 hematologiczne,
2 neurologiczne, 3 gastroenterologiczne
�Objawy niedoboru kwasu foliowego:Podobne jw.Brak objawów neurologicznych
Rola Rola witwit . B. B1212 oraz oraz folianówfolianów
�Witamina B12 jest te Ŝ koenzymem wymaganym podczas metylacji homocysteiny do metioninyoraz (jako deoksyadenozylokobalamina) podczas powstawania bursztynylo-koenzymu A
Syntetazatymidylanowa
DNA
FH4 (Tetrahydrofolian)
dTMP
N5 - Metylo FH 4
Metylokobalamina
Kobalamina(Witamina B 12)
dUMP

Niedokrwisto ść Niedokrwisto ść megaloblastycznamegaloblastyczna (I)(I)�Makrocytarna, normo- lub hipochromiczna�Dotyczy wszystkich komórek
�Obni Ŝony poziom B 12 we krwi - powody róŜnicujemy testem Schillinga
�Witamina B 12
- W diecie 7-30 µg- Zapasy 2-3 mg (na 2-4 lat)- Wchłaniana w jelicie kr ętym- Wchłanianie wymaga obecno ści czynnika wewn ętrznego
(Castle IF)
Niedokrwisto ść Niedokrwisto ść megaloblastycznamegaloblastyczna (II)(II)Kwas foliowy
� Niezbędny do:-przenoszenia reszt metylowych-syntezy nukleotydów purynowych-syntezy nukleotydów pyrimidynowych
�W diecie 600-1000 µg/24h
�Zapas 5 mg (na 3 miesi ące)
�Wchłaniana w dwunastnicy i jelicie czczym

Niedokrwisto ść złośliwaNiedokrwisto ść złośliwaChoroba Addisona – Biermera, Pernicious anaemia
�90% obecno ść przeciwciał przeciw komórkom okładzinowym
�35% obecno ść przeciwciał przeciwtarczycowych�Częściej kobiety (cho ć nie ma co do tego pewno ści),
bielactwo, choroby tarczycy oraz rak Ŝołądka
Objawy: �zapalenie zanikowe błony śluzowej Ŝołądka�achlorhydria
�zdradziecki pocz ątek, Ŝółtaczka, wzrost [LDH],zapalenie j ęzyka (Hunter), zapalenie k ącików ust.
Niedokrwisto ść Niedokrwisto ść aplastycznaaplastyczna�Związana z niewydolno ścią szpiku –- jego aplazj ą lub hipoplazj ą�Rzadka: 0,2/100000 mieszka ńców�Stwierdzono zwi ązek z HLA DR2 (tylko niektóre populacje)
Etiologia:�Wrodzona – np. zespół Fanconi, zespół Diamond�Nabyte:
Polekowe (chloramfenikol, fenylbutazon, inne NLPZ, cytostatyki prowadzą do niedokrwistości jatrogennej)
Substancje chemiczne (np. benzen)
Promieniowanie jonizuj ąceZakaŜenia (WZW, Herpeswirusy, Parwowirus B19…)

Niedokrwisto ść Niedokrwisto ść aplastycznaaplastyczna ––patomechanizmpatomechanizm
�Komórki hemopoezy gin ą, szpik jest zast ępowany przez tkank ę tłuszczow ą uwidacznian ą w biopsji oraz w rezonansie magnetycznym.
�W wypadku niedokrwisto ści Fanconiego – autosomalna, recesywna mutacja (gen FANCA ).
� Łamliwo ść chromosomów, pancytopenia, niski wzrost, plamy cafe au lait ...
�Niedokrwisto ść aplastyczna nie jest skutkiem uszkodzenia komórek zr ębu szpiku, lub niedoboru cytokin.
�Uszkodzenie komórek hemopoezy obserwujemy tak Ŝe w: mielodysplazji, izolowanej aplazji (granulocytarnej, trombocytarneji erytrocytarnej (PRCA,zesp. Diamond-Blackfan))
Objawy niedokrwisto ści Objawy niedokrwisto ści aplastycznejaplastycznej
�Objawy: �Pancytopenia – główny objaw ci ęŜkiegouszkodzenia szpiku
Objawy skazy krwotocznej:
petechiae, podbiegnięcia,
krwawienia
ZakaŜenia – często oportunistyczne, utrzymująca się
gorączka
Objawy typowe dla niedokrwistości:
…..
TrombocytopeniaGranulocytopeniaErytrocytopenia
�Często wła śnie skaza krwotoczna jest pierwszym objawem choroby!

Przypadek 1Przypadek 1�85 letnia emerytka�ChNS, AO�SkarŜy si ę na wysiłowe bóle zamostkowe, męczliwo ść�Hb - 9.7g/dl.�Inne badania dodatkowe:
Ht 30%MCV 78fLMCH 25pg
�Jaki typ niedokrwisto ści ?
Przypadek 2Przypadek 2�54 letni wykładowca uniwersytecki�Niedawno zwolniony z pracy�SkarŜy si ę na: bolesno ść języka, m ęczliwo ść�Hb - 7.6g/dl�Inne badania dodatkowe:
Ht 38%MCV 103fLMCH 30pgPłytki krwi 130 x 10 9/l Leukocyty 4 x 10 9/l
�Wątroba i śledziona powi ększone�Jaki typ niedokrwisto ści ?

Niedokrwisto ści hemolityczneNiedokrwisto ści hemolityczne�Klasyfikacja niedokrwisto ści hemolitycznych
I. czynniki wewn ątrzkrwinkoweA. Dziedziczne
1. Defekty błony komórkowej2. Defekty metaboliczne 3. Hemoglobinopatie
B. Nabyte1. Napadowa nocna hemoglobinuria
II. Czynniki pozakrwinkoweA. immunologiczne
1. Autoimmunologiczne2. Transfuzje niezgodnej grupowo krwi
B. INNE1. Chemikalia 2. ZakaŜenia (bakterie, paso Ŝyty)3. Hemoliza wskutek uszkodzenia fizycznego 4. Hipersplenizm
Klasyfikacja niedokrwisto ści Klasyfikacja niedokrwisto ści hemolitycznychhemolitycznych
1. Wewnątrznaczyniowe - krwinki s ą uszkadzane w świetle naczynia
• Przykłady:
hemoliza poprzetoczeniowa oparzenianocna napadowa hemoglobinuriahemoliza jako skutek mikroangiopatii urazy fizyczne infekcje
• wska źniki laboratoryjne :
hiperbilirubinemiapobudzenie szpikumethemoalbuminemiahemoglobinuriazmniejszenie st ęŜenia wolnej haptoglobinyhemosyderynuria

Klasyfikacja niedokrwisto ści Klasyfikacja niedokrwisto ści hemolitycznychhemolitycznych
2. Zewnątrznaczyniowy - uszkodzenie (eliminacja ) krwinek następuje w układzie siateczkowo- śródbłonkowym
• Przykłady:
hemoliza z przyczyn immunologicznychhemoglobinopatiedziedziczna sferocytozahipersplenizmhemoliza w chorobach w ątroby
• wska źniki laboratoryjne:
hiperbilirubinemiapobudzenie szpikuhemosyderoza
Niedokrwisto ści hemolityczne Niedokrwisto ści hemolityczne --objawyobjawy
•• W badaniu przedmiotowymW badaniu przedmiotowym-- blado ść blado ść -- ŜółtaczkaŜółtaczka-- splenomegaliasplenomegalia
• Badania dodatkowe:1. Niedokrwisto ść hemolityczna
- normocytarna/makrocytarna, hiperchromiczna- retikulocytoza- zwiększenie st ęŜenia Fe w surowicy-wykrycie obecno ści przeciwciał przeciwko erytrocytom
2. Rozmaz krwi- poikilocytoza, sferocytoza- obecno ść erytroblastów- schistiocyty
3. Rozmaz szpiku- aktywacja hemopoezy

Defekty błony komórkowejDefekty błony komórkowej --Dziedziczna Dziedziczna sferocytozasferocytoza (I.A.1) (I.A.1)
1. Patofizjologiazaburzenie powstawania spektryny
2. Inne przypadki w rodzinie
3. Klinicznie- splenomegalia
4. W laboratorium - anemia hemolityczna- sferocyty- obni Ŝona oporno ść na zmiany
ciśnienia osmotycznego
siła ścinaj ąca
Uszkodzenia błony
sferocytsplenomegalia
Niedokrwisto ść hemolityczna
spadek wytrzymało ści błony
komórkowej
Niedobory enzymatyczne Niedobory enzymatyczne (I.A.2)(I.A.2)
� Dehydrogenaza Glukozo-6-fosforanowa :� Enzym cyklu pentozofosforanowego - brak enzymu
to:mniej NADPH → zaburzenia redukcji glutationu
� Niedobór (>400 wariantów)SprzęŜony z X, MęŜczyźni > Kobiety
� Bezobjawowa do czasu wywołania stresu oksydacyjnego :
Leki (prymachina, sulfonamidy), nasiona bobu [NO]↑
� Zdenaturowana hemoglobina precypituje (ciałka Heinza), narastaj ą zaburzenia cyklu pentozowegopowoduj ąc skrócenie czasu Ŝycia krwinek i ich eliminacj ę przez śledzion ę
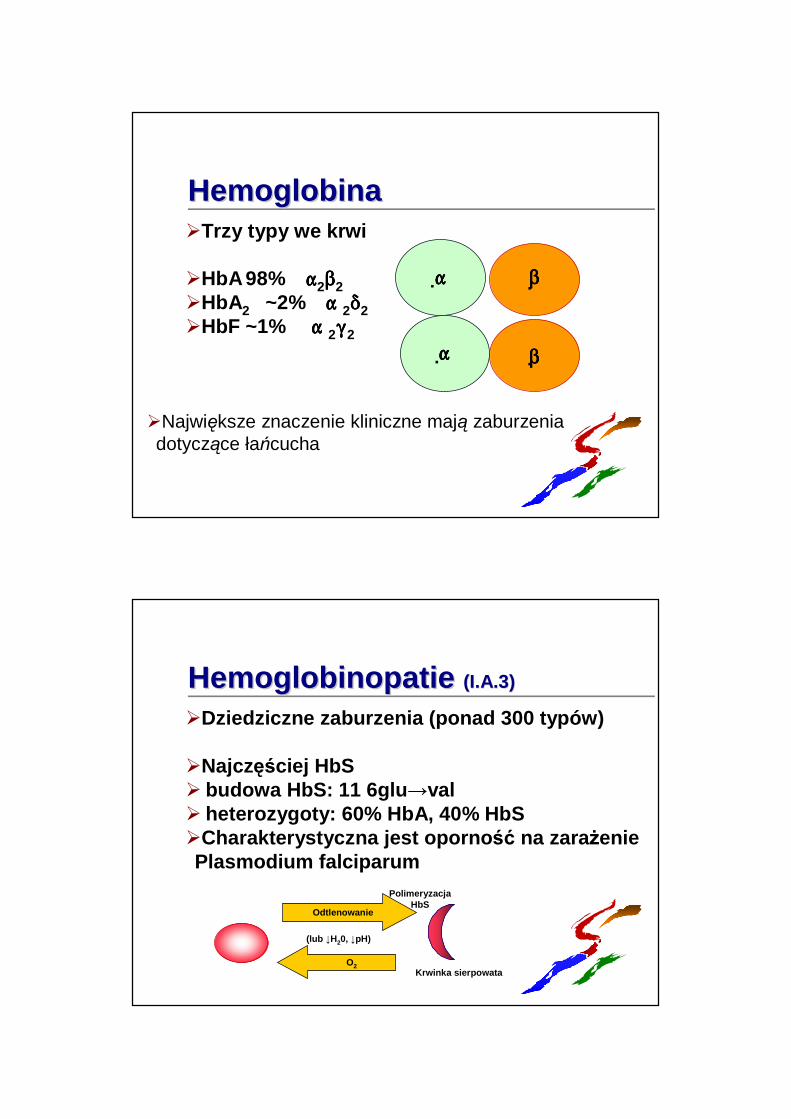
HemoglobinaHemoglobina�Trzy typy we krwi
�HbA98% """"2$$$$2�HbA2 ~2% """" 2****2�HbF ~1% """" 2((((2
�Największe znaczenie kliniczne mają zaburzenia dotyczące łańcucha
""""
""""
$$$$
$$$$
HemoglobinopatieHemoglobinopatie (I.A.3)(I.A.3)
�Dziedziczne zaburzenia (ponad 300 typów)
�Najczęściej HbS� budowa HbS: 11 6glu →val� heterozygoty: 60% HbA, 40% HbS�Charakterystyczna jest oporno ść na zaraŜeniePlasmodium falciparum
Odtlenowanie
PolimeryzacjaHbS
Krwinka sierpowataO2
(lub ↓H20, ↓pH)

Niedokrwisto ść Niedokrwisto ść sierpowatokrwinkowasierpowatokrwinkowa�Krwinki sierpowate s ą niszczone w śledzionie (splenomegalia)
�Mogą takŜe tworzy ć mikrozatory:- Niedokrwienie tkanek, mikrozawały, ból- Autosplenektomia (u dorosłych) – wskutek niedokrwienia
�Podatno ść na infekcje - Salmonella
�Przełom aplastyczny mo Ŝe wyst ąpić � po zaka Ŝeniu Parvovirusem B19
Nocna napadowa Nocna napadowa hemoglobinuriahemoglobinuria (I.B.1)(I.B.1)
1. Patogeneza – mutacja dotycz ąca genu Pig-A ( gen „kotwicy”) w jednej z komórek macierzystych prowadzi do niedoboru dwóch białek:DAF (CD55) – inhibitor szlaku aktywacji dopełniacza (zarówno klasycznej jak i alternatywnej) oraz MIRL (CD59) – inhibitor przekształcania podjednostek C9 w kanał atakuj ący błon ę komórki.
• Wspomniany niedobór czyni erytrocyty bardziej wra Ŝliwymi na działanie dopełniacza.
• Płytki krwi równie Ŝ nie posiadaj ą inhibitorów – aktywacja dopełniacza pobudza je do agregacji, co wydaje si ę być główn ą przyczyn ą zakrzepicy (która wyst ępuje u ok 40% pacjentów)
2. Objawy: chory co rano oddaje ciemno zabarwiony m ocz.3. Laboratorium: - pancytopenia
- obni Ŝenie Fe w surowicy- hemoglobinuria-- hemosyderynuriahemosyderynuria

AutoimmunologicznaAutoimmunologicznaanemia hemolityczna anemia hemolityczna (II.A.1)(II.A.1)
A.A. wywołana przeciwciałami ciepłymi klasy wywołana przeciwciałami ciepłymi klasy IgGIgG::
I. Pierwotna II. Wtórna:
1. Ostra: infekcje wirusowe, leki ( αααα-Metyldopa, Penicylina, Chinina, Chinidyna)
2. Przewlekła : Tocze ń, Chłoniak nieziarniczy
B. wywołana aglutyninami zimnymi klasy wywołana aglutyninami zimnymi klasy IgMIgM: :
I. I. Samoistna II. Wtórna: infekcje (mykoplazmy, wirusy), choroba
Waldenstroema
Przypadek 3Przypadek 3• WBC x 109/L 7.4 [4-11] • Hb g/L 104 [140-180] • MCV fl 69 [80-100]• Płytki x 10 9 /L 512 [150-450]• Neutrofile x 109 /L 5.3 [2-7.5] • Limfocyty x 10 9 /L 1.7 [1.5-4] • Monocyty x 10 9 /L 0.4 [0.2-0.8] • Eozynofile x 10 9 /L 0 [0-0.7] • Bazofile x 109/L 0 [0-0.1] • Blasty w szpiku x 109/L 0 [0] • Promielocyty x 109/L 0 [0] • Mielocyty x109/L 0 [0]• Metamielocyty x 109/L 0 [0] •• Komentarz hipochromia+ mikrocytoza++• Fe w surowicy prawidłowe 13-35 µmol/l Wynik u pacjenta 8 µmol/l• TIBC: prawidłowe 38-81 µmol/l Wynik u pacjenta 90 µ mol/l• Ferrytyna: prawidłowe 10-300 µg/l Wynik u pacjenta 6 µ g/l

Przypadek 4Przypadek 4• WBC 9.3 x 109/L [4-11]• Hb g/L 102 [120-160]• MCV fl 68 [79-98]• Płytki x 109/L 270 [150-450] • Neutrofile x 109/L 6.0 [2-7.5] • Limfocyty x 109/L 2.8 [1.5-4] • Monocyty x 109/L 0.5 [0.2-0.8] • Eozynofile x 109/L 0 [0-0.7] • Bazofile x 109/L 0 [0-0.1] • Blasty w szpiku x 109/L 0 [0] • Promielocyty x 109/L 0 [0] • Mielocyty x109/L 0 [0] • Metamielocyty x 109/L 0 [0] •• Komentarz mikrocytoza+ hipochromia+• Fe w surowicy prawidłowe 13-35 µmol/l Wynik u pacjenta 28 µmol/l • TIBC: prawidłowe 38-81 µmol/l Wynik u pacjenta 70 µmol/l• Ferrytyna: prawidłowe 10-300 µg/l Wynik u pacjenta 42 µg/l
Przypadek 5Przypadek 5• WBC x 109/L 3.1 [4-11]• Hb g/L 64 [120-160]• MCV fl 136 [79-98]• Płytki x 109/L 110 [150-450]• Neutrofile x 109/L 2.2 [2-7.5]• Limfocyty x 109/L 0.7 [1.5-4] • Monocyty x 109/L 0.2 [0.2-0.8] • Eozynofile x 109/L 0 [0-0.7] • Bazofile x 109/L 0 [0-0.1] • Blasty w szpiku x 109/L 0 [0] • Promielocyty x 109/L 0 [0] • Mielocyty x109/L 0 [0] • Metamielocyty x 109/L 0 [0] •• Komentarz owalne makrocyty++ hypersegmentacja neutrofili• Fe w surowicy prawidłowe 13-35 µmol/l Wynik u pacjenta 30 µmol/l • TIBC: prawidłowe 38-81 µmol/l Wynik u pacjenta 48 µmol/l• Ferrytyna: prawidłowe 10-300 µg/l Wynik u pacjenta 70 µg/l

HematopatologiaHematopatologia
Leukocytoza/Leukocytopenia
Ostre białaczki
Zespoły mieloproliferacyjne
Krwinki białeKrwinki białeWBC-white blood cells – ilo ść krwinek białychNorma: 4-10 x 10 9/lWBC < 4x10 9/l = LeukopeniaWBC > 10x 10 9/l = Leukocytoza
% l.bezwzgl ędna (/µl)Neutrofile 65 4550Limfocyty 30 2100Monocyty 3 300Eozynofile 1 200Bazofile 1 20
Odczyn białaczkowy - WBC > 50000/mm3 z „odczynowym przesuni ęciem w lewo”
Białaczka – WBC > 100000/mm3 z „patologicznym przesuni ęciem w lewo”

LeukocytozyLeukocytozyNEUTROFILIA: Ostre stany zapalne (zakaŜenia bakteryjne, martwica tkanek,
nowotwory); Czynniki fizyczne (temperatura); Leki (adrenalina, glikokortykosteroidy); Emocje; Białaczki szpikowe
EOZYNOFILIA: Choroby alergiczne; Przewlekłe choroby skóry (pęcherzyca, łuszczyca); Choroby paso Ŝytnicze; Zaka Ŝenia (bruceloza); Białaczka eozynofilowa
BAZOFILIA: Białaczka bazofilowa; Przewlekłe stany zapalne prze wodu pokarmowego; Niedoczynno ść tarczycy
LIMFOCYTOZA: ZakaŜenia wirusowe; Przewlekłe stany zapalne; W zakaŜeniu EBV (mononukleoza zakaźna - liczne „duŜe” [aktywowane] limfocyty)
MONOCYTOZA: ZakaŜenia pierwotniakami; Gru źlica i sarkoidoza; Zapalenie wsierdzia; Ziarnica zło śliwa; Białaczka monocytowa
LeukopenieLeukopenieZaburzenia produkcji (aplazja szpiku, zwłóknienie
szpiku, białaczki, przerzuty nowotworowe, leki cytotoksyczne…)
Niszczenie komórek we krwi (choroby autoimmunologiczne, leki…)
Choroby zaka źne (ze zuŜycia)
Stres (uraz, wysiłek)
Wpływ hormonów - steroidy

Ostre białaczkiOstre białaczkiZłośliwy rozrost nowotworowy, o charakterze klonalnym, wywodz ący si ę z róŜnych linii komórkowych układu krwiotwórczego.
Zwykle powy Ŝej 15-20% (30% wg FAB) blastów w rozmazie szpiku kostnego
Ostra białaczka limfoblastyczna(AcuteLymphoblasticLeukaemia) – dzieci
Szczyt zachorowa ń ok. 4 r. Ŝ.
Ostra białaczka szpikowa (AcuteMyelogenousLeukaemia) – doro śli
Szczyt zachorowa ń ok. 50 r. Ŝ.
Ostre białaczki Ostre białaczki -- patofizjologiapatofizjologiaKomórki macierzyste
Transformacja
Proliferacja, nacieki
Krew obwodowaTrzewa
Szpik:obecno ść blastówcytopenia

Nagły pocz ątekUtrata prawidłowej funkcji szpiku
Niedokrwisto ść, zmęczenie, gor ączka, zaka Ŝenia, krwawienia
Nacieki w narz ądach wewn ętrznychHepatomegalia, splenomegalia
Zaburzenia funkcjonowania OUN:Bóle głowyNudno ści, wymiotyPoraŜenia nerwów
Ostre białaczki Ostre białaczki -- przebiegprzebieg
Zespoły Zespoły mieloproliferacyjnemieloproliferacyjneNowotworowe proliferacje komórek uładu
krwiotwórczego, klasyfikowane na podstawie linii komórkowej, z której si ę wywodz ą
Najczęściej u osób mi ędzy 20 a 50 r. Ŝ.Przebieg przewlekły, charakterystyczne jest
przechodzenie jednej postaci w drug ą.
Zespoły mieloproliferacyjne:czerwienica prawdziwa – erytrocytyprzewlekła białaczka szpikowa (CML) - granulocytynadpłytkowo ść samoistna - trombocytysamoistne zwłóknienie szpiku – O steoMieloFibroza –
dotyczy wszystkich komórek szpiku

SplenomegaliaFaza komórkowa:
Szpik bogatokomórkowyWzrost ilo ści krwinek danego szeregu we
krwi obwodowej
Zwłóknienie szpiku lub ostra białaczka :post ępuj ącezwłóknienie szpiku (mielofibroza)AML lub ALL (w 20%)
Zespoły Zespoły mieloproliferacyjnemieloproliferacyjne
Chromosom Chromosom PhiladelphiaPhiladelphiaTranslokacja wzajemna
t(9;22)Skutkuje fuzj ą genów
bcr/abl
c-abl chromosom 9
bcr (break point cluster region) chromosom 22
Białko o aktywno ści kinazytyrozynowej – odgrywa wiod ącą rol ę w patogenezie

Czerwienica prawdziwaCzerwienica prawdziwaZwiększona ilo ść
erytrocytów:Wzrost:
HematokrytuObjętości krwiGęsto ści (lepko ści) krwi
Zaburzenia czynno ści płytek– zakrzepica lub krwawienia
Przebieg powolnyPost ępujące zwłóknienie
szpikuPrzejście w ostr ą białaczk ę
w 2-15%
MielofibrozaMielofibrozaZwłóknienie jam szpikowych
– proliferacja fibroblastów jest stymulowana czynnikami wzrostu, produkowanymi przez komórki szeregu megakariocytowego (tych jest najwi ęcej)
Hemopoeza pozaszpikowa(wątroba i śledziona)
Hepato- i splenomegaliaObecno ść blastów we krwi
obwodowejPoczątkowo nadkrwisto ść,
później (post ęp włóknienia) – niedokrwisto ść i „Puste biopsje”
Przejście w AML w 10%

Dziękuj ęDziękuj ę

2008-11-26
1
KREW IIZABURZENIA HEMOSTAZY
HEMOSTAZA
DEFINICJA
�Całość procesów związanych z
utrzymaniem krwi w stanie płynnym w
obrębie łożyska naczyniowego

2008-11-26
2
HEMOSTAZA
ZAŁOŻENIA
�Mechanizmy hemostazy są aktywowane
� Jedynie w miejscu w którym są niezbędne
� Jedynie w czasie kiedy są potrzebne
�Po zakończeniu naprawy mechanizmy są
„unieczynniane” , a przepływ krwi
przywrócony
MECHANIZMY HEMOSTAZY
�Hemostaza pierwotna:
� Skurcz naczynia
� Tworzenie czopu płytkowego
�Hemostaza wtórna (mechanizmy osoczowe)
�Fibrynoliza

2008-11-26
3
MECHANIZMY HEMOSTAZY
CZOP PŁYTKOWY
• Uszkodzenie naczynia
• Ekspozycja kolagenu ściany naczynia
• Połączenie płytkowego receptora IB czynnikiem von
Willebranda (vWF)
• Skurcz naczyń oraz aktywacja płytek zależne od tromboksanu
MECHANIZMY HEMOSTAZY
OSOCZOWY MECHANIZM KRZEPNIĘCIA
� Stabilizuje powstały czop płytkowy
� Skutkuje zamianą fibrynogenu w fibrynę
� Ma trzy główne etapy:
1. Powstanie aktywatora protrombiny
2. Przekształcenie protrombiny w trombinę
3. Przekształcenie fibrynogenu w fibrynę (włóknik)

2008-11-26
4
CZYNNIKI KRZEPNIĘCIA – CECHY SZCZEGÓLNE
�Fosfolipidy ściany naczynia aktywują czynniki
krzepnięcia
�Część czynników krzepnięcia jest zależna od
witaminy K (II, VII, IX, X)
�Labilne czynniki V i VIII ułatwiają kontrolę
�Czynnik VIII & czynnik von Willebranda
CZYNNIK VIII
� Występuje w kompleksie:
VIII : C� część aktywna
Czynnik von Willebranda (vWF)� Uczestniczy w adhezji płytek krwi
� Stabilizuje czynnik VIII
� Jego niedobór prowadzi do zaburzen płytkowych i osoczowych
Choroba von Willebranda a Hemofilia A:
hemofilia A VIII:C
vWD type 1 VIII:C vwF
vWD type 2 vwF

2008-11-26
5
ANTYTROMBINA III (AT III)
�Antykoagulant osoczowy
�Wiąże się z czynnikami: IXa, Xa, XIa, XIIa
�Jej działanie przyspiesza heparyna
�Heparyna nie ma „własnej” aktywności
przeciwzakrzepowej i jej skuteczność zależy
od obecności ATIII
CZAS
Czynnik t ½
Fibrynogen (I) 72-120
Protrombina (II) 60-70
V 12-16
VII 3-6
VIII 8-12
IX 18-24
X 30-40
Czynnik t ½
XI 52
XII 60
Białko C 6
Białko S 42
Czynnik tkankowy
--
Trombomodulina --
Antytrombina 72

2008-11-26
6
FIBRYNOLIZA
� Plazminogen → plazmina
� Uwolnienie tkankowego aktywatora plazminogenu
(tPA) przez komórki śródbłonka
� Proteoliza skrzepu→ FDP / FSP
� Udrożnienie naczynia
BADANIA LABORATORYJNE
CZAS PROTROMBINOWY (PT)
� Zewnątrzpochodny i wspólny szlak mechanizmu
krzepnięcia
� „Zależny” od witaminy K - (czynniki II, VII, IX, X)
� Zasada pomiaru:
tromboplastyna + Ca+2 + osocze
� Wartość referencyjna: 12-14 s
� International Normalized Ratio (INR)
� Standaryzacja pomiaru PT
� Wartość referencyjna: 0.8 -1.2

2008-11-26
7
BADANIA LABORATORYJNE
CZAS PROTROMBINOWY (PT)
�Ocena terapii doustnymi antykoagulantami
�Ocena funkcji czynnika VII
�Ocena działania przeciwzakrzepowego:
Czynniki II i X
BADANIA LABORATORYJNE
CZAS KAOLINOWO-KEFALINOWY (APTT)
� Wewnątrzpochodny i wspólny szlak mechanizmu
krzepnięcia
� Nie zależy od czynników VII i XIII
� Wartość referencyjna: 25 – 35 s.
� Zasada pomiaru:
Osocze (cytrynian) + fosfolipidy

2008-11-26
8
BADANIA LABORATORYJNE
CZAS KAOLINOWO – KEFALINOWY (APTT)
� Ocena działania przeciwzakrzepowego
heparyn oraz w diagnostyce hemofilli
� Wydłużony pod wpływem: heparyn,
inhibitorów trombiny, FDP
� Wydłużenie, gdy aktywność czynników
krzepnięcia spada poniżej 30 % normy
CZAS KRWAWIENIA
�Ocena hemostazy pierwotnej
�Niespecyficzny test funkcji płytek krwi
�Niska czułość dla łagodnych zaburzeń
dotyczących płytek
�Brak standaryzacji

2008-11-26
9
Badanie Znaczenie Wartości
Czas krwawienia Hemostaza pierwotna 3 - 10 min
PT I, II, V, VII, IX, X 12 - 14 s
PTT I, II, V, VIII, IX, X, XI, XII 24 - 35 s
Czas trombinowy I, II 12 - 20 s
DIAGNOSTYKA SKAZ KRWOTOCZNYCH
� WYWIAD� BADANIE PRZEDMIOTOWE� LABORATORYJNE BADANIA PODSTAWOWE� LABORATORYJNE BADANIA SPECJALISTYCZNE

2008-11-26
10
SKAZY KRWOTOCZNE
•••• PIERWOTNE = WRODZONE•••• WTÓRNE = NABYTE
SKAZY KRWOTOCZNE
•••• PŁYTKOWE •••• OSOCZOWE•••• NACZYNIOWE

2008-11-26
11
OBJAWY KLINICZNE SKAZ NACZYNIOWYCH
� Plamica
� Wybroczyny
� Podbiegnięcia
� Wyjątkowo dochodzi do rozległych krwawień – jako skutek uszkodzenia dużych naczyń krwionośnych
SKAZY KRWOTOCZNE NACZYNIOWE
WRODZONE
� Choroba Rendu – Oslera – Webera
� Zespół Marfana
� Zespół Ehlers – Danlos

2008-11-26
12
SKAZY KRWOTOCZNE NACZYNIOWE
NABYTE
� Polekowe
� Poinfekcyjne
� Szkorbut
� Zespół Schönleina - Henocha
DIAGNOSTYKA SKAZ KRWOTOCZNYCH
NACZYNIOWYCH
� Badania koagulologiczne w normie
� Morfologia krwi obwodowej w normie
� Test opaskowy – dodatni
� Inne możliwości diagnostyczne - biopsja skóry

2008-11-26
13
OBJAWY KLINICZNE SKAZ PŁYTKOWYCH
� Mnogie wybroczyny
� Krwawienia i wybroczyny w miejscach dorbnych urazów
� Nadmierne krwawienia
� Krwawienia z błon śluzowych
� BRAK krwawień o dużym nasileniu do tkanek miękkich i jam ciała oraz jam stawowych
SKAZY KRWOTOCZNE PŁYTKOWE
� Ilościowe� Zależne od produkcji � Zależne od eliminacji (niezależne od produkcji)
� Immunologiczne� Nieimmunologiczne
� Jakościowe� Pierwone (wrodzone)� Wtórne (nabyte)

2008-11-26
14
SKAZY KRWOTOCZNE PŁYTKOWE ILOŚCIOWE
ZALEŻNE OD PRODUKCJI
� Cytostatyki, radioterapia
� Narażenie na środki chemiczne
� Niedobory żywieniowe – np. wit. B12
� Alkoholizm
� Zespoły mieloproliferacyjne
� Ostre białaczki
� Zakażenia wirusowe
SKAZY PŁYTKOWE ILOŚCIOWE NIEZALEŻNE OD
PRODUKCJI IMMUNOLOGICZNE
� Immunologiczna plamica małopłytkowa
� Polekowa plamica małopłytkowa
� Potransfuzyjna plamica małopłytkowa
� Poinfekcyjna plamica małopłytkowa

2008-11-26
15
SKAZY PŁYTKOWE ILOŚCIOWE NIEZALEŻNE OD
PRODUKCJI NIEIMMUNOLOGICZNE
� Sztuczne zastawki
� Krążenie pozaustrojowe
� Małopłytkowość poalkoholowa
� Hipersplenizm
� Zespół hemolityczno – mocznicowy (HUS)
� Zespół rozsianego wykrzepiania wewnątrznaczyniowego (DIC)
SKAZY KRWOTOCZNE PŁYTKOWE
CZYNNOŚCIOWE PIERWOTNE
� Choroba Barnarda – Souliera
� Trombastenia Glanzmana

2008-11-26
16
SKAZY KRWOTOCZNE PŁYTKOWE
CZYNNOŚCIOWE NABYTE
� Zespoły mieloproliferacyjne
� Szpiczak
� Makroglobulinemia Waldenstroema
� Mocznica
� Marskość wątroby
� Polekowe zaburzenia czynności płytek
� Krążenie pozaustrojowe
DIAGNOSTYKA SKAZ KRWOTOCZNYCH
PŁYTKOWYCH
� Wydłużenie czasu krwawienia
� Morfologia krwi obwodowej
� Badania koagulologiczne w normie

2008-11-26
17
OBJAWY KLINICZNE SKAZ OSOCZOWYCH
� Krwawienia wtórne / odroczone – często jako powikłanie zabiegów chirurgicznych
� Samoistne krwawienia z nosa
� Samoistne krwawienia� Podskórne� Do mięśni i przestrzeni międzypowięziowych� Do stawów
� Przedłużone i nawracające krwawienia z pępowiny u noworodków
SKAZY KRWOTOCZNE OSOCZOWE WRODZONE
� Hemofilia A
� Hemofila B
� Niedobór innych czynników krzepnięcia
� Choroba von Willebrandta

2008-11-26
18
SKAZY KRWOTOCZNE OSOCZOWE NABYTE
� Niedobór witaminy K
� Zaburzenia czynnosci watroby
� DIC
� Zaburzenia polekowe – antagoniści witaminy K
� Zatrucia
DIAGNOSTYKA SKAZ KRWOTOCZNYCH
OSOCZOWYCH
� Duża gama objawów np.:� Przedłużenie czasu krzepnięcia� Prawidłowy czas krwawienia

2008-11-26
19
DICDISSEMINATED INTRAVASCULAR COAGULATION
ROZSIANE WYKRZEPIANIE WEWNĄTRZNACZYNIOWE
CZYNNIKI ETIOLOGICZNE DIC
� Uwolnienie / „wtargnięcie” czynnika tkankowego lub substancji o podobnej aktywności
� Uszkodzenie śródbłonka naczyń krwionośnych
� Bezpośrednia aktywacja czynnika X
� Bezpośrednia aktywacja protrombiny


















