MIKROSATELITY - jkpiechota.pljkpiechota.pl/PDFmarkery/wyklad2-pdf.pdf · - mikrosatelitów jest...
43
MIKROSATELITY Stanowią 3% ludzkiego genomu – to dużo, czy mało? Występują stosunkowo rzadko w regionach kodujących białka. Tylko trój- i sześcionukleotydy występują częściej w egzonach - wynika to z negatywnej selekcji skierowane przeciwko przesunięciom ramki odczytu. - u ludzi powtórzenia trójnukleotydów powodują czasami choroby (np. choroba Huntingtona) najprawdopodobniej związane z obecnością długich ciągów tej samej reszty aminokwasowej w białku U wielu gatunków większość mikrosatelitów to dwunukleotydy (48-67%) - u pierwotniaków występują głównie ciągi mononukleotydowe - u ludzi najwięcej jest ciągów mononukleotydowych , ąle łącznie najliczniejsze są dwu-, cztero- i sześcionukleotydy - ciągi dwunukleotydowe występują w 5’ lub 3’ częściach genów, które nie ulegają transkrypcji 14% wszystkich białek zawiera sekwencje powtórzone, jest ich trzy razy więcej u organizmów eukariotycznych w porównaniu do prokariotycznych Wzory dystrybucji różnych typów mikrosatelitów (posiadających różną długość i różne motywy) w sekwencjach kodujących i niekodujących są filogenetycznie uwarunkowane. - mikrosatelitów jest więcej i są dłuższe u kręgowców niż u bezkręgowców.
Transcript of MIKROSATELITY - jkpiechota.pljkpiechota.pl/PDFmarkery/wyklad2-pdf.pdf · - mikrosatelitów jest...

MIKROSATELITY Stanowią 3% ludzkiego genomu – to dużo, czy mało?
Występują stosunkowo rzadko w regionach kodujących białka.
Tylko trój- i sześcionukleotydy występują częściej w egzonach - wynika to z negatywnej selekcji skierowane przeciwko przesunięciom ramki odczytu.- u ludzi powtórzenia trójnukleotydów powodują czasami choroby (np. choroba
Huntingtona) najprawdopodobniej związane z obecnością długich ciągów tej samej reszty aminokwasowej w białku
U wielu gatunków większość mikrosatelitów to dwunukleotydy (48-67%)- u pierwotniaków występują głównie ciągi mononukleotydowe- u ludzi najwięcej jest ciągów mononukleotydowych , ąle łącznie najliczniejsze są
dwu-, cztero- i sześcionukleotydy- ciągi dwunukleotydowe występują w 5’ lub 3’ częściach genów,które nie ulegają transkrypcji
14% wszystkich białek zawiera sekwencje powtórzone, jest ich trzy razy więcej u organizmów eukariotycznych w porównaniu do prokariotycznych
Wzory dystrybucji różnych typów mikrosatelitów (posiadających różną długość i różne motywy) w sekwencjach kodujących i niekodujących są filogenetycznie uwarunkowane.- mikrosatelitów jest więcej i są dłuższe u kręgowców niż u bezkręgowców.

Lokalizacja sekwencji mikrosatelitarnych na chromosomach genomu człowieka

Egzony
Introny
Regiony międzygenowe
Odmienność chromosomów
19 i Y

Najczęściej występujące mikrosatelity:
A, AT, AC, AAT, AAC, AAG, AGC, AAAC, AAAT, AAAG, AAGG, AGAT
Najrzadziej występujące mikrosatelity:
C, CG, ACT, ACG, AACC, AACG, AACT, AAGC, AAGT, ACCC, ACCG, ACCT, CCCG, CCGG

Pętle ułatwiające rozplatanieLub rozpoznawane przez białka
„hot spots”, dinukleotydyrozpoznawane przez enzymy
Zatrzymanie replikacji,Gen CHK1i
Mikrosatelity występują w genach enzymów tego systemu i są podatne na mutacje

Struktury tworzone przez mikrosatelitarne trimery ułatwiające ich ekspansję w chorobach neurologicznych
Zespół łamliwegochromosomu X
Dystrofia miotoniczna
Choroba Huntingtona

Nić o sekwencji (GCC)n łatwo tworzy strukturę spinki do włosówSpinka może wydłużać się (SLIP)lub przesuwać się (SLIDE)
Konsekwencje:
1. Ekspansja lub skracanie

2. Metylacja prowadzącado obniżenia lubzahamowania ekspresjigenu FMR1
A
GT
A
G
A
A
TG
GA
A
T
G
G A
A
T
G
G
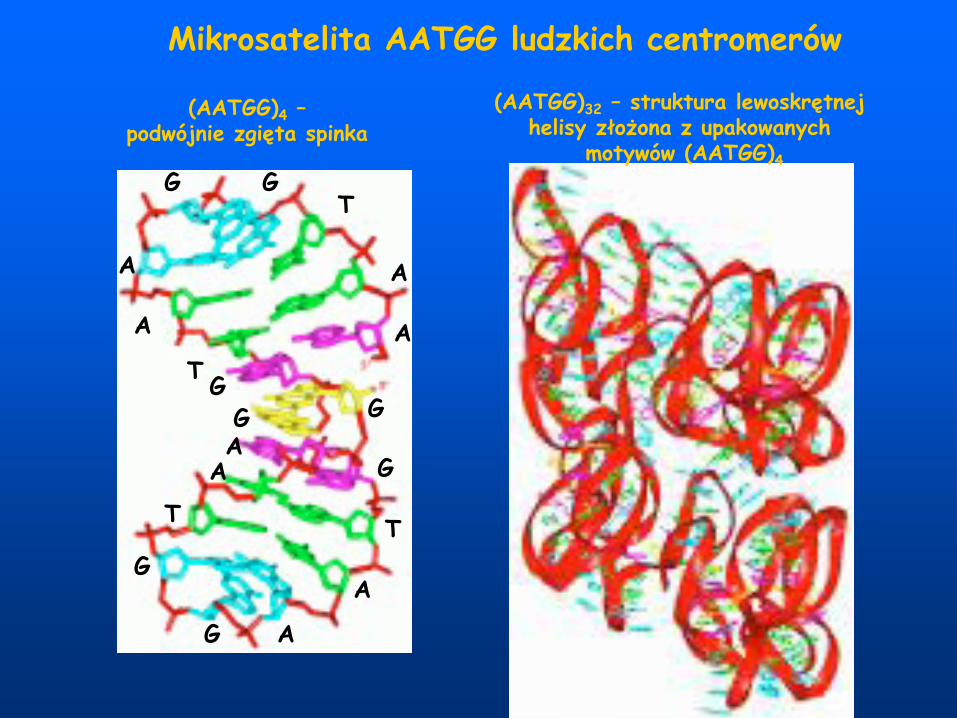
Mikrosatelita AATGG ludzkich centromerów
(AATGG)4 –podwójnie zgięta spinka
(AATGG)32 – struktura lewoskrętnej helisy złożona z upakowanych
motywów (AATGG)4

Na 3’ końcach telomerów znajdują się jednoniciowe fragmenty, w których 30 do 100 razy jest powtórzony motyw TTAGGG tworzący kwartety G Rozpoznawane przez białka stabilizujące

Rozróżnienie alleli zawierających różną liczbę powtórzeń sekwencji mikrosatelitarnych za pomocą reakcji PCR

Główne obszary zastosowań polimorfizmów STR:• Ustalanie pokrewieństwa;• Identyfikacja osób;• Ustalanie pochodzenia śladów biologicznych (wykrywanie sprawców
przestępstw);
Zalety stosowania polimorfizmów STR:• Mniej pracochłonna i mniej kosztowna technika;• Krótszy czas oczekiwania na wynik;• Mniejsza ilość DNA potrzebna do analizy;• DNA może być gorszej jakości (możliwość użycia próbek DNA
otrzymanych ze starego i zniszczonego materiału biologicznego a nawet z niezbyt starych próbek archeologicznych.

Ewolucja analiz STR typu multiplex na przykładzie firmy zestawu PowerPlex™ firmy Genscript Promega®
GenePrint®

Ewolucja analiz STR typu multiplex na przykładzie firmy zestawu PowerPlex™ firmy Genscript Promega®
PowerPlex® 1.2 System

Ewolucja analiz STR typu multiplex na przykładzie firmy zestawu PowerPlex™ firmy Genscript Promega®
PowerPlex® 2.1 System

Ewolucja analiz STR typu multiplex na przykładzie firmy zestawu PowerPlex™ firmy Genscript Promega®
PowerPlex® 16


Identyfikacja sprawcy gwałtu
KP M P Podejrzani
CSF1P0
TPOX
TH01
F13A01
FESFPS
KP M P Podejrzani Podejrzani P M
vWA

CODIS – działa w ramach FBI, umożliwia federalnym, stanowym i lokalnym laboratoriom medycyny sądowej elektroniczną wymianę i porównywanie profili DNA.

Several Indexes Categorize the Profiles Entered into CODIS
• Convicted Offender
contains profiles of individuals convicted of a crime
• Forensic
contains DNA profiles developed from crime scene evidence such as
semen stain or blood.
• Arrestees
contains profiles of arrested persons (if state law permits the collection
of arrestee samples).
• Missing Persons
contains DNA reference profiles from missing persons.
• Unidentified Human Remains
contains DNA profiles developed from unidentified human remains.
• Biological Relatives of Missing Persons
contains DNA profiles voluntarily contributed from relatives of missing
persons.

Collected samples of:
• Nuclear DNA
mostly STR - 13 locus STR : CSF1PO, FGA, TH01, TPOX, vWA, D3S1358,
D5S818, D7S820, D8S1179, D13S317, D16S539, D18S51, D21S11
• Mitochondrial DNA
Mostly SNPs.

Measuring Success
The National DNA Index (NDIS) contains over 8,080,941 offender profiles
and 311,560 forensic profiles as of March 2010. Ultimately, the success of the
CODIS program will be measured by the crimes it helps to solve. CODIS's
primary metric, the "Investigation Aided," tracks the number of criminal
investigations where CODIS has added value to the investigative process. As
of March 2010, CODIS has produced over 114,300 hits assisting in more
than 112,300 investigations.

Identyfikacja szczątków rodziny Romanowów

Nowotwory oraz choroby genetyczne związane z mikrosatelitami
15% sporadycznych przypadków raka jelita grubego koreluje z niestabilnością powtórzeń mikrosatelitarnych
Niestabilność mikrosatelitów przyczynia się również do wystąpienia nowotworów przewodu pokarmowego
14 typów zaburzeń neurologicznych wiąże się z ekspansją mikrosatelitów w sekwencjach kodujących lub niekodujących
Ekspansja trójnukleotydu CTG powoduje dystrofię mięśniową, chorzy mają powyżej 50 kopii tego motywu
Ekspansja trójnukleotydu CAG powoduje powstawanie białek z ciągami poliglutaminowymi – choroba Huntingtona oraz Machado Josepha
Długość ciągu powtórzeń (CA)n w 5’ regionie niekodującym reduktazy aldozowej jest związana z retinopatią cukrzycową
Długość (CA)n w pierwszym intronie genu γ- interferonu jest związana z występowaniem zwłóknienia płuc

BPES, blepharophimosis, ptosis and epicanthus inversusCCD, cleidocranial dysplasiaCCHS, congenital central hypoventilation syndromeDM, myotonic dystrophyDRPLA, dentatorubral–pallidoluysian atrophyEPM1, progressive myoclonic epilepsy 1FRAXA, fragile X syndromeFRAXE, fragile X mental retardation associated with FRAXE siteFRDA, Friedreich's ataxiaFXTAS, fragile X tremor and ataxia syndromeHD, Huntington's diseaseHDL2, Huntington's-disease-like 2HFG, hand–foot–genital syndromeHPE5, holoprosencephaly 5ISSX, X-linked infantile spasm syndromeMRGH, mental retardation with isolated growth hormone deficiencyOPMD, oculopharyngeal muscular dystrophySBMA, spinal and bulbar muscular atrophySCA, spinocerebellar ataxiaSPD, synpolydactyly.

Dystrofia miotoniczna - CTG
• Obejmuje wiele układów, głównie wpływa na mięśnie
• Miotonia – mięśnie mogą się kurczyć, ale trudno je rozluźnić (trudności z otworzeniem zaciśniętej dłoni)
- osłabienie mięśni (twarz, szyja,kończyny)
• Dotyka 1 na 20 000 osób
• Zaburzenie autosomalne dominujące
• Penetracja - wysoka
• Ekspresyjność – różnorodna nawet w obrębie rodziny
• Antycypacja – skłonność do przybierania coraz poważniejszego przebiegu w kolejnych pokoleniach

Podłoże molekularne
• Gen warunkujący (1992) – na 19 chromosomie
- kinaza DM – ekspresja w mózgu, mięśniach, sercu i jądrach (narządy i tkanki uszkodzone)
- domeny specyficzne dla kinaz białkowych, które fosforylują reszty serynowe i treoninowe
- mutacja w pobliżu 3’ końca genu – ulega transkrypcji, ale nie translacji
- prawdopodobna przyczyna fenotypu –obniżenie ekspresji enzymu
Region kodujący CTG…CTG
Transkrypcja
Translacja

CTG…CTG – najczęściej 5 – 27 powtórzeń- osoby chore 50 lub więcej
Wzrost liczby powtórzeń niestabilność regionu dalszy wzrost
antycypacja – cięższy przebieg choroby z wcześniejszymi objawami w kolejnych pokoleniach
Wystąpienie antycypacji zależy od tego, od którego z rodziców dziedziczony jest wadliwy allel.Antycypacja DM następuje w dziedziczeniu mutacji od matki.
Poważna dziecięca postać dystrofii miotonicznej – objawy rozpoczynają się w momencie narodzin – u 20% potomstwa chorej matki, które odziedziczyło mutację, prawdopodobieństwo rośnie wraz z nasileniem objawów u matki
W dziedziczeniu od ojca – kontrakcja

Hybrydyzacja typu SouthernPCR
DIAGNOSTYKA

Choroba Huntingtona
• Opisana w 1872 przez Huntingtona – pląsawica – niekontrolowane ruchy, gr. chorea
• Występuje u 1 na 100 tys. osób• Pojawia się między 35 a 50 rokiem życia• Choroba ośrodkowego układu nerwowego, zwyrodnienie neuronów• Niszczy obszary mózgu odpowiedzialne za koordynacje ruchów,
życie emocjonalne, funkcje intelektualne (myślenie, percepcja, pamięć)
• Objawy – niewytłumaczalne zmiany nastroju: rozdrażnienie –apatia, złość – depresja- trudności w uczeniu się, przypominaniuniekontrolowane ruchy stóp, palców, twarzy- trudności z utrzymaniem równowagi i chodzeniem
Z czasem objawy pogłębiają się, choroba trwa 10 – 30 lat
Antycypacja w dziedziczeniu od ojca – niestabilność (CAG)n w spermatogenezie.

Podłoże molekularne• 1993 – gen na chromosomie 4 - HD lub IT-15
• Mutacja – ekspansja powtórzenia CAG w eksonie 1:
- Normalne allele – ≤ 26 CAG - nie powodują HD i nie wykazują ekspansji CAG u potomstwa
- Normalne allele z tendencją do mutacji – 27 – 35 CAG (1.9% populacji)Nie powodują choroby u nosiciela, ale wykazują ekspansję CAG u potomstwa. Ryzyko ekspansji jest większe jeśli nosicielem jest mężczyzna.
- Allele powodujące chorobę - ≥ 36 CAG – powodują HD, zwłaszcza ≥ 40.≥ 55 – postać młodzieńcza

• Jedna z 9 chorób neurodegeneracyjnych spowodowanych przez powtórzenia CAG – każda z tych chorób związana z innym genem
• Ciągi CAG w genie – bloki poliglutaminowe w białku (ang. huntingtin)
• Formy białka z długimi blokami polyQ nie ulegają poprawnemu zwinięciu i tworzą wysokocząsteczkowe agregaty
• Choroba Huntingtona jest chorobą konformacyjną (ch. Alzheimera i Parkinsona, ch. prionowe)

CAG x N
1 2 3 4 5
26 kopii
35 kopii40 kopii

Polimorfizmy punktowe SNP (ang. single nucleotide polymorphism)

1.2%, które czyni człowieka i 0.1%, które czyni różnicę
1.2%0.1%

• 3 miliony SNP odróżnia dwie osoby, SNP co 1200 pz• przypuszcza się, że w genomie człowieka jest 10 000 000 SNP, w 2005 roku w
publicznych bazach danych było ich 9 000 000.• 75% - obszary międzygenowe, 24% - introny, 1% - eksony• Źródła SNP – delecje, insercje, duplikacje, substytucje• SNP ma mniej możliwości wariantów niż markery mini- i mikrosatelitarne• Substytucja – teoretycznie 4 warianty w jednym locus, praktycznie – 2• Różna częstość wariantów – niska heterozygotyczność• W celach identyfikacyjnych należy oznaczyć nawet do 100 loci • Zalety SNP – olbrzymia liczba, metody detekcji nie wymagające elektroforezy
w żelu i łatwe w automatyzacji
• Istnieją bazy danych SNP: HGBASE, dbSNP, SNP Consortium
• SNP Consortium – powstało w 1999 roku aby stworzyć i udostępnić katalog 300 000 SNP, w 2001 dysponowało już 1 000 000 SNP
• Celem działania baz danych SNP jest stworzenie mapy SNP w ludzkim genomie (high-density SNP map) w celu identyfikacji mutacji powodujących choroby, związanych z długością życia


Mitochondrialne SNP i starzenie

Farmakogenetyka (ang. Pharmacogenetics) –
bada genetyczne podstawy indywidualnych różnic w odpowiedzi na leki,
jej celem jest opracowanie leków dopasowanych do genotypu pacjenta
1959 – Vogel proponuje termin „farmakogenetyka”
1962 – pierwsza książka „Pharmacogenetics”Heredity and the Response to Drugs”

Famakogenetyka a Farmakogenomika
• Dziedziczność różnic w odpowiedzi pacjentów na lek i struktura populacji
• Jeden lek – wiele genomów (pacjentów)
• Badanie SNP i poziomu ekspresji
• Studiuje genom -różnice pomiędzy komórkami różnych tkanek
• Jeden genom – wiele leków
• Profile ekspresji genów

Farmakokinetyka• Metabolizm leków – ich degradacja, aktywacja za
pomocą enzymów
• Wyróżniamy 2 fazy metabolizmu leków:
I faza – reakcje funkcjonalizacji, np: hydroksylacja przez CYP450
II faza – reakcje sprzęgania, np: acetylacja przez NAT

Wolni acetylatorzy w populacjach kaukaskiej i orientalnej
Populacja kaukaska• Włosi• Szwedzi• Ludność biała USA• Brytyjczycy• Norwegowie• Niemcy• Francuzi• Kanadyjczycy• Czesi i Słowacy• Finowie• Polacy
Populacja orientalna• Eskimosi• Koreańczycy• Japończycy• Ajnowie• Chińczycy• Ludność Riukiu• Tajowie• Chińczycy z Singapuru• Chińczycy z Tajwanu• Filipińczycy• Chińczycy z Tajlandii
%49
51-5852-5753-62
565759
59-7060
61-6448-62
%5
117-12
1313151822222834
Bönicke i Reif – polimorfizm acetylacji izoniazydu
(leku tuberkulostatycznego)
izoniazyd objawy neuropatii wśród chorych leczonych lekiem tuberkulostatycznym
- polimorfizm acetylacji przez N-acetylotransferazę NAT2
- w populacji kaukaskiej 40-60% ma fenotyp „wolno metabolizujący”, w populacji Japońskiej 10%

Kalow – polimorfizm hydrolizy prokainy (anastetyk) i sukcynylocholiny (zwiotczanie mięśni)
sukcynylocholina przedłużenie z kilku minut do 3 godzin blokady nerwowo-mięśniowej Przyczyna: polimorfizm hydrolizyWinna: pseudoesteraza cholinowa