25 - matlosz1995

of 5
-
Upload
linda-yampolski -
Category
Documents
-
view
213 -
download
0
Transcript of 25 - matlosz1995
-
8/19/2019 25 - matlosz1995
1/9
jPergamon
Elrcr rochmca Acm Vol 40 No 4 393 401p 1995
Copyright < 1995 Elsewermnce Ltd.
Prmted reatmu.
llnghrs
reserved
0013 686;95$9.50000
h
ch
c,
C*,
D
F
i
10
m
RO
RI
4
4,
Timary
R”“if”nn
n
t
U
V
Z
MODELING OF IMPEDANCE MECHANISMS IN
ELECTROPOLISHING
MICHAEL MATLOSZ
Laboratoire des sciences du genie chimique CNRS, Ecole nationale suparieure des industries chimiques,
1, rue Grandville B. P. 451, F-54001 Nancy, France
(Received 21 April 1994)
Abstract-Recent theoretical results concerning mechanisms for mass-transport-limited electrochemical
polishing are summarized and discussed. The underlying physical bases for both salt-film and acceptor
models are presented with particular emphasis on the differences
in their alternating-current impedance
behavior. Direct comparison of the dependence on operating conditions of characteristic features of the
impedance diagrams in the high-frequency range provides a clear experimental basis to distinguish
among the various mechanisms that can lead to the polishing phenomenon in specific cases.
Key words: mathematical modeling, alternating current impedance, electropolishing, anodic dissolution,
kinetic mechanism.
NOMENCL TURE
Greek symbols
Tafel constant
bulk acceptor concentration/mol cmS3
characteristic
capacity of the high-
frequency loop/F cm- ’
double-layer
capacity of the metal/
electrolyte interface/F cm- ’
dif n coefficient of the acceptor species/
Faraday constant/96 487 C molt ’
current density/A cm- *
pre-exponential
factor (solid-state
conduction), or exchange-current density
(Tafel kinetics)/A cm _ 2
steady-state current density/A cm - ’
rate constant for cation solvation by the
acceptor/s
’
number of acceptor molecules needed to
solvate a single cationic species
high-frequency impedance limit/ohm-cm2
diameter of the high-frequency impedance
loop/ohm-cm*
resistance of the porous film (per unit
a
Y
6
parameter in the solid-state conduction
expression/cm V- ’
dimensionless parameter in the model
developed in reference[6]
diffusion-layer thickness/cm
porous-film porosity
electrical permittivity of the compact layer/
Fcm-’
surface coverage of adsorbed cations
thickness of the compact film/cm
thickness of the porous film/cm
potential at the inner limit of the porous
layer or potential at the metal/electrolyte
interface/V
potential at the outer limit of the porous
layer/V
potential at the reference
electrode
position/V
angular frequency/rad s-
’
INTRODUCTION
length)/ohm-cm
Electropolishing is a surface finishing process
ohmic resistance of the electrolyte/ohm-
cm2
based on anodic dissolution of a metal or alloy in an
appropriately chosen electrolyte. Applications of the
ohmic resistance for a primary current
distribution/ohm-cm2
technique are numerous and range from the pol-
ohmic resistance for a uniform current
ishing of stainless steel cutlery to the preparation of
distribution/ohm-cm2
samples for transmission electron microscopy. The
polishing phenomenon is characterized by the elimi-
time/s
nation of micro-roughness (leveling) and the absence
c/c,, dimensionless surface concentration
of crystallographic and grain-boundary attack
of the acceptor
(brightening) and results in the production of
electrode potential/V
smooth, bright surfaces. A complete summary and
metal cation valence
discussion of the scientific literature concerning elec-
393
-
8/19/2019 25 - matlosz1995
2/9
394
M.
MATLOSZ
tropolishing from its patented publication by
Jacquet in 1930[1-33 up to and including research
results from the mid-1980s can be found in the
review by Landolt[4].
Mass-transport limitations for anodic dissolution
are generally believed to be responsible for electro-
polishing, and this view is supported by the observ-
ation in numerous experimental systems of polishing
for anodic dissolution along a limiting-current
plateau. Leveling behavior for anodic dissolution at
the limiting current can be interpreted as the prefer-
ential dissolution of protrusions on the order of the
diffusion layer due to their greater accessibility for
diffusive transport. Brightening can also be inter-
preted as a result of mass-transport control, but on a
smaller scale where diffusion is essentially isotropic
and independent of the crystallographic orientation
and grain structure of the metallic surface.
Whether or not anodic dissolution is mass-
transport controlled depends on the experimental
system. Unlike cathodic limiting-current plateaux in
electrodeposition, which are the inevitable result of
the depletion of metal cations in the diffusion layer
near the electrode surface, anodic limiting-current
plateaus do not necessarily appear with increasing
overpotential in all cases since the surface concentra-
tion of dissolving metal ions will generally rise with
increasing anodic current. Mass-transport-limited
anodic dissolution requires therefore the presence of
an additional mechanistic step, such as the precipi-
tation of a salt film (which limits the surface concen-
tration to the saturation value of the metal cations)
or a diffusion limitation for transport of an acceptor
molecule necessary for solvation.
It is of consideral scientific and technological
interest to be able to determine clearly which of the
possible mechanisms is at work in a given polishing
system in order to understand the chemistry
involved and the role of the various operating
parameters. For this purpose, several studies of salt-
film and acceptor systems have been undertaken in
the past decade, including theoretical work on the
shapes and sizes of the characteristic loops of the
impedance diagrams measured along the hmiting-
current plateau. The results of two of these studies,
summarized here, yield considerable insight into the
transport mechanisms involved and provide a solid
basis for determining the likely mechanism in a given
experimental system.
The two models chosen for discussion have been
studied theoretically in some detail over the past
several years and represent special limiting cases of
the salt-film and acceptor approaches. The first, the
duplex salt-film model proposed by Grimm et aI.[S],
attempts to characterize the role of compact and
porous layers in the frequency response of complex
precipitate films. The second, the adsorbate-acceptor
mechanism proposed by Matlosz et a/.[6], examines
the role of adsorbed intermediates and acceptor-
molecule transport in the behavior of polishing
systems in the absence of films. Both types of mecha-
nism have been observed and studied in experimen-
tal polishing systems.
The objective of this article is to present the
underlying physical bases of the models and the
principal results of the theoretical analyses in order
to obtain a clear pictue of the differences and simi-
larities in the approaches. For this reason, quaht-
ative arguments will be favored and mathematical
developments limited to the greatest extent possible.
In the interest of clarity, some notation has been
modified slightly with respect to the original refer-
ences, and model behavior has been simplified some-
what in certain cases. In particular, the intermediate
surface reaction step discussed in the acceptor model
in [6], and which is not strictly necessary for pol-
ishing, has been eliminated in the present discussion
(an approach equivalent to the case of y = 0 in[6]).
More detailed mathematical treatments and addi-
tional theoretical support for the conclusions report-
ed here can be found in[5] and[6].
THE DUPLEX SALT-FILM MODEL
A physical picture of the duplex salt-film model is
represented schematically in Fig. 1. The origin of the
anodic limiting current is the salt-film precipitate
DUPLEX SALT FILM MODEL
oxidation
Fe + Fe+* +
2 e
precipitation
dissolution
‘r
r’
w
(diffusion) Cl -
sat
L
Reference
k:
- Cbulk
compact
dielectric
film
diffusion
layer
Fig. 1. Schematic illustration of the duplex salt-film model.
-
8/19/2019 25 - matlosz1995
3/9
Modeling of impedance mechanisms
395
which fixes the metal cation concentration at its
saturation value at the salt-film/electrolye interface.
The rate of transport of the cations across the diffu-
sion layer into the bulk electrolyte limits the anodic
dissolution rate. The precipitate itself is composed of
two regions. In the porous-film region, the pores of
the precipitate are filled with electrolyte solution (at
the saturation concentration) and the mobile charge
carriers (anions and cations) transport the current by
migration in the electric field in the pores. In order
to explain the high electric resistance of the precipi-
tate films, the porous layers are generally attributed
a rather low porosity and taken to be several
microns thick. In fact, however, it is not possible to
distinguish clearly whether an increase in film resist-
ance is due to an increase or decrease in porous film
resistance is due soley to a change in film thickness.
In the compact-film region, the precipitate forms a
solid dielectric barrier through which the cations are
transported by solid-state ionic conduction in the
presence of a much higher electric field. Due to the
lower mobility of the ions for solid-state transport,
compact films are generally considered to have
thicknesses on the order of 10 nm, much thinner than
corresponding porous layers. Limiting cases of inter-
est for the duplex model arise if one or the other of
the two regions can be neglected. In such cases, one
may speak of a porous-film model or a compact-film
model, and most of the results presented here will be
restricted to one or the other of these limiting cases.
Although the duplex model is viewed here as two
distinct layers, it is possible to consider an alterna-
tive (and perhaps more realistic) interpretation as a
single film of variable properties with a gradual
change from a region of high porosity to a region of
low porosity with simultaneous solid-state and
liquid-state transport. Both interpretations result in
essentially the same behavior, however, with the
presentation as two separate regions offering the
advantage of clearly separating solid-state and
liquid-state effects. For this reason, the description
below will be restricted to the two-film approach.
Discussion of the physical basis of the model is
somewhat simplified if one considers the dissolution
of a single metal into a binary electrolyte. Following
Grimm et 0/.[5], the presentation here will focus on
the dissolution of Fe into an electrolyte of FeCI,.
Transport processes are taking place in three distinct
layers: the electrolyte diffusion layer, the porous-film
layer, and the compact-film layer. In the electrolyte
diffusion layer, metal cations Fe’* are transported
outward toward the bulk electrolyte by both diffu-
sion in the concentration field and migration in the
electric held, whereas the counterions Cl --, whose net
steady-state fux must be zero, are drawn simulta-
neously outward by diffusion and inward by migra-
tion. The migration of both ions contributes to the
electrolyte conductivity and to net current flow. The
resulting steady-state concentration profile is consis-
tent with electroneutrality and shows a rise in FeCI,
concentration from its bulk value up to a saturation
concentration at the salt-flim/electrolyte interface.
Transport processes in the binary electrolyte in the
pores of the outer salt film are somewhat different
from those in the liquid diffusion layer. Contrary to
the diffusion layer, the concentration in the pores of
the outer film cannot vary with position, since rapid
equtlibratton with the solid prectpttate and the
requirement of the electroneutrality force the con-
centration of the electrolyte in the pores to remain at
the same saturation value throughout. As a result,
diffusional transport for both ions is eliminated, only
migration of the ions in the electric field remains,
and the porous layer acts as a simple ohmic resistor.
Since Fe”
cations and Cll anions have similar
mobilities, it is reasonable to assume that migration
will cause cations to be transported outward
through the film by migration, whereas anions will
tend to be drawn inward. There is clearly no net
transport of chloride ions into the dissolving iron
electrode, and the chloride transported into the
porous film must therefore precipitate out at the
bottom of the pores. Since (for constant film
porosity) the film thickness must remain constant at
steady state, one sees clearly that the precipitation of
freshly formed FeCl, beneath the film must be com-
pensated by equivalent dissolution of the precipitate
at the outer-film/electrolyte interface. Physically, the
porous-film model represents a dynamic process
with a salt film of constant thickness continually
being renewed by precipitation in the inner region
beneath the pores and dissolution at the outer edge
along the film/electrolyte interface. The fact that the
layer thickness is conserved by precipitation of
FeCI, solid beneath the film is an interesting result,
and one might speculate as to whether the mechani-
cal pressure built up by the precipitating layer
beneath the film may not be responsible in some way
for cracks and fissures leading to film porosity.
Under the transient conditions resulting from
potential modulation, the dynamic steady state of
film renewal is perturbed, and the resulting imped-
ance response depends on the nonzero rates of chlo-
ride transport and the corresponding variations in
film thickness with time. During a growth phase, for
example, a net flux of Cl- is drawn toward the elec-
trode surface by an increase in migration that is not
totally compensated by diffusion since the concentra-
tion profiles are fixed by the saturation and bulk
values. During a thinning phase, the net flux of Cl
is reversed and the thickness of the film decreases.
For the inner compact layer, it is possible to
imagine that only the Fe+2 cations are capable of
significant mobility in the solid phase (transference
number of Cll zero in the solid). In that case, the
migration flux of the iron cations corresponds to the
net anodic current and the film is not renewed as in
the case of the porous outer region. If chloride ions
do have significant mobility in the soild, their trans-
port through the compact film will result in renewal
of the film at constant steady-state thickness similar
to that described for the porous layer. In the most
general case of a complete duplex film with Cl
mobility in both layers, the renewal rates of the
layers will depend on the solid-state and liquid-state
transference numbers for chloride ions and material
balances at the interface between the two regions.
Figure 2 shows the relative contributions of the
porous layer, compact layer and electrolyte solution
to the overall potential drop observed in an experi-
mental polishing system. In the example shown, the
overpotential for Fe dissolution at the inner metal/
-
8/19/2019 25 - matlosz1995
4/9
396
M. MATLOSZ
anode
$0
POTENTIAL DROP : SALT FILM MODEL
Reference
Fig. 2. Sources of potential drop in salt-film systems.
compact-film interface has been neglected, and the
role of the potential drop across the compact layer
has been emphasized. For typical high-field conduc-
tion of metallic cations, the steady-state current
density transported across the compact layer can be
represented as follows,
i,, = i,
exp(“(‘, ‘O))
where
(V - +o)/
denotes the electric field strength
across the compact layer of thickness A,, x a con-
stant characterstic of the jump spacing for solid-state
ionic transport in the precipitate crystal, and i, a
kinetic constant related to the number of charge car-
riers present in the compact film.
For operation along the limiting-current plateau,
increasing the applied potential does not change the
current density (which is limited by the transport of
Fe+’ across the electrolyte diffusion layer), and con-
sequently the electric field strength in equation (1)
must be conserved. This is accomplished by an
increase in the thickness 1, which compensates for
any increase in
V - qSo
in the absence of a porous
layer.
For transient operation, the charging of the dielec-
tric layer must be taken into account along with the
high-field conduction. The thicker the dielectric film,
the smaller the charging current necessary to estab-
lish the steady-state field. With this interpretation,
the current density under transient or modulated
conditions can be expressed by addition of a second
term to equation (I) as follows:
where Em_,,,
denotes the electrical permittivity of the
compact film.
When a porous layer is present, the current
density through the porous region can be expressed
by the equivalent of Ohm’s law:
where R, denotes the porous-film resistance (per unit
length), and 1, the porous-film thickness. The resist-
ance R, is a function of the electrolyte conductivity
and film porosity and tortuosity. The total potential
drop
V - q5p
across a duplex film is divided between
the compact and porous layers, with an increase in
applied potential generally resulting in an increase in
the thickness of both regions.
In the special case of a porous film only, the over-
potential for charge transfer at the metal electrolyte
solution within the pores is generally not negligible.
In this case, one can reinterpret the potential 4. as
that of an appropriate reference electrode located in
the electrolyte solution in the pores just outside the
electric double layer at the metal/electrolyte inter-
face. For Tafel kinetics, the following steady-stae
expression can be used
t =
i, exp(b( V - ))
(4)
where b denotes a Tafel constant,
i,
an appropriate
exchange-current density, and t: the film porosity
(included to represent the fact that charge transfer
occurs only on the fraction of the metal surface
exposed to electrolyte solution in the pores). Use of
the same symbols
i,
and b. is intended to emphasize
the considerable similarity between the expressions
for high-field conduction (compact film) and Tafel
kinetics (porous film). Despite these similarities, there
are important differences between the two limiting
cases. In the case of a porous layer alone with Tafel
kinetics, for example, it is not possible to alter the
value of the Tafel costant h by changing the film
thickness, whereas in a compact film model the
equivalent constant r/i, in the solid-state conduc-
tion expression is inversely proportional to thick-
ness. This difference is essential in the determination
of the size of the high-frequency loop in the imped-
ance diagrams because of its influence on the effec-
tive charge-transfer resistance.
In the absence of a compact film, the value of
V - 4. cannot vary along the limiting-current
plateau, and all of the necessary potential drop must
be obtained by thickening of the porous layer alone.
-
8/19/2019 25 - matlosz1995
5/9
Modeling of impedance mechanisms
397
In addition, the double-layer capacity in the absence
of a compact film no longer represents the charging
of a solid dielectric material, but rather the charging
of a metal-electrolyte interface on the exposed area
beneath the pores. This charging contribution can be
added to the Tafel expression in the porous film
model to yield
:
i
- = i0 ew(W - I) +
c,,
d(V - 44
E
dr
(9
where C,, denotes the capacity of the electric double
layer at the metal/electrolyte interface.
A final source of potential drop is the electrolyte
solution between the electrode surface and the posi-
tion of the reference electrode used to measure the
applied potential. The current density can be
expressed in terms of the electrolyte resistance as
follows:
(6)
where R, represents the effective ohmic resistance of
the electrolyte from the dissolving metal electrode to
the position of the reference electrode in the bulk
electrolyte. This ohmic resistance depends on the
geometry of the experimental system and on the con-
ductivity of the electrolyte, and can be affected by
current-distribution effects which differ considerably
between the porous-film and compact-film models.
In particular, in high-frequency impedance measure-
ments,
R,
represents essentially the ohmic resistance
for the passage of the double-layer charging current
through the electrolyte. In the case of a totally
compact dielectric film, it is the salt film itself which
is charged to create the electric field necessary for
solid-state ionic conduction. As a result, the surface to
be charged is in direct contact with the ohmic resist-
ance of the bulk electrolyte solution. The resistance
R, should correspond in that case to the equivalent
resistance measured in the absence of a film and
should not be affected by the thickness of the
compact layer.
The case of a porous or duplex film is different. If
the porous layer is thick enough to present a signifi-
cant ohmic resistance, that resistance will be placed in
series with the bulk electrolyte since double-layer
charging takes place beneath the porous layer. This
additional resistance has two effects. The first effect
is a linear increase in the high-frequency ohmic
resistance with increasing film thickness, an increase
not observed for compact films. The second effect is
more subtle and is related to the influence of the film
resistance on the distribution of the current lines in
the bulk electrolyte. In the case of a disk electrode,
for example, the presence of a significant surface
resistance in series with double-layer charging will
tend to create a more uniform distribution of current
lines by drawing more current toward the center of
the disk. In the limit where the porous film resistance
is significant enough to create a completely uniform
distribution along the outer film surface, the effective
ohmic resistance for the electrolyte alone (for a refer-
ence placed far from the surface) will increase by
approximately 27 per cent compared to the primary
resistance in the absence of a film. This effect has
been examined experimentally by Grimm et aI.[5] by
extrapolation of the measured ohmic resistance in
the presence of the film to the limit of zero film
thickness and comparison of the result with the mea-
sured electrolyte resistance in the absence of a film.
THE ADSORBATE ACCEPTOR MODEL
Figures 3 and 4 present schematically the
adsorbate-acceptor model in a similar manner to the
duplex film model in Figs 1 and 2. The system pre-
sented shows uniform anodic dissolution of a metal-
lic alloy M (for example Fe-15Cr) in an electrolyte
containing a small quantity of acceptor species A.
One can imagine (as in[6]) that the electrolyte is a
concentrated solution of phosphoric and sulfuric
ADSORBATE ACCEPTOR MODEL
solvation :
Reference
I-
' dl
d
adsorbate
double
diffusion
layer
layer
‘MA bulk
M = Fe-i 50
A = H,O
Fig. 3. Schematic illustration of the adsorbate-acceptor model
-
8/19/2019 25 - matlosz1995
6/9
398
M. MATLOSZ
POTENTIAL DROP : ACCEPTOR MODEL
Fig. 4. Sources of potential drop in acceptor systems.
acid, and that the acceptor is water or a water-related
species necessary for solvation of the dissolving
cations. Contrary to the salt-film models, where the
surface concentration is fixed by a saturation value,
in this case the situation corresponds much more
closely to the traditional limiting current observed in
cathodic metal deposition. The limiting current is
reached when the concentration of the acceptor
species A drops to near zero at the electrode/solution
interface. Due to the absence of the salt films, the
transport processes are limited to diffusion of the
acceptor species through the electrolyte diffusion
layer.
At the electrode surface, the dissolution mecha-
nism consists of oxidation of the metal to adsorbed
cations followed by solvation of the adsorbed ions
by the acceptor species. Whereas in the salt-film
model it is the thickening of the salt layer which
leads to an increase in potential drop along the
limiting-current plateau, in the adsorbate-acceptor
model it is the accumulation of adsorbed ions on the
surface which leads to blocking of the surface and a
subsequent increase in overpotential for metal disso-
lution.
For transient measurements, a clear consequence
of the absence of solid films on the electrode surface
can be observed in the double-layer charging of the
interface. The situation is similar to the porous-film
model in the absence of a compact layer in that the
charging phenomenon is related to the double-layer
capacity of the metal/electrolyte interface. In the
adsorbate acceptor model, however, the entire elec-
trode surface is available and the double-layer con-
tribution can be expressed simply as follows:
4, = C,,
d(I’ - 40)
dt
(7)
where C,, denotes the double-layer capacity of the
metal/electrolyte interface in the presence of the
adsorbed cations. The presence of the adsorbed
cations is not expected to alter dramatically the
value of C,, in comparison with a typical metal/
electrolvte interface. however. and the exnerimental
measurements in[7] tend to support this view for the
Fe-15Cr system. In salt-film systems, on the other
hand, the measured capacities depend on film
properties (thickness, porosity, dielectric constant)
and can be much lower. In the experimental studies
reported in[5], for example, the values of effective
double-layer capacity (and the corresponding RC
time constants) were so low that complete measure-
ment of the high-frequency loops in the impedance
diagrams could not be achieved in the frequency
range of the experimental apparatus.
The adsorbate-acceptor model exhibits specific
characteristic behavior for the charge-transfer resist-
ance as well. For metal dissolution with Tafel
kinetics, for example, but with the additional
requirement that free surface sites must be available
for the adsorbed oxidized species, the steady-state
rate of anodic oxidation will drop with increasing
coverage as follows:
i,, = i, exp(b(V - &J) . (1 - 0)
(8)
where 0 denotes the surface coverage by the
adsorbed intermediates. The current density at a
given potential depends not only on the kinetic
expression, but also on the mass balance for adsorb-
ate and acceptor species. At steady state, the rate of
production of adsorbed species by equation (5) must
equal their rate of consumption by solvation. In par-
ticular, with kinetics for solvation first order in both
acceptor molecules and adsorbed cations:
s= c uO
where z denotes the cation valence, F the Faraday
constant,
k
a rate constant for solvation (assumed
results from the equivalence of acceptor transport
independent of potential), cb the bulk concentration
of acceptor molecules, and u the ratio of surface to
bulk acceptor concentration. Equation (6) illustrates
the coupling that occurs in the acceptor mechanism
between the rate of mass transport of acceptor
species and the rate of cation solvation. Determi-
nation of the surface concentration of acceptor (u)
-
8/19/2019 25 - matlosz1995
7/9
Modeling of impedance mechanisms
399
and the steady-state current:
43s
DC ~ - u)
-=
i F
rnd
where D denotes the acceptor diffusion coefficient, 6
a diffusion-layer thickness and M the number of
acceptor molecules needed to solvate a single
cationic species.
Along the limiting-current plateau (u < l), the
current density is essentially constant and limited by
the rate of arrival of acceptor molecules at the elec-
trode surface. The surface coverage rises quickly, and
the group 1 - U (the fraction of free surface sites)
drops exponentially with increasing potential:
1 - 0 = 5 exp(-b(V - &)).
As a result, and contrary to what one would expect
for normal Tafel kinetics in the absence of surface
blocking, the effective charge-transfer resistance does
not drop with increasing potential, but instead
remains perfectly constant along the limiting-current
plateau. This feature is characteristic of a blocking
mechanism and can be observed in porous-salt-film
models as well. In the porous-film models, however,
the effect is not due to a rise in surface overpotential.
Rather, a change in applied external potential simply
does not alter the true surface overpotential beneath
the film, since all of the potential difference is elimi-
nated by additional ohmic drop due to growth of the
porous layer.
The situation for a compact salt film is qualit-
atively different. Increasing applied potential in a
compact film system results in an increase in thick-
ness 1,) and a corresponding increase in the charge-
transfer resistance through the group &/u. As a
result, a change in applied potential results in a
strong variation in charge-transfer resistance which
is observed neither in porous-film models nor in
acceptor models.
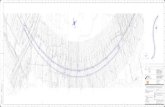
NYQUIST DIAGRAM
SALT FILM MODEL
DISTINGUISHING FEATURES OF THE
IMPEDANCE DIAGRAMS
The features pointed out in the discussion above
concerning ohmic resistances, charge-transfer resist-
ances, and double-layer capacities can be examined
directly with impedance measurements, and can be
used to establish experimental criteria to distinguish
between the different models. Figures 5 and 6 present
the general shapes of the impedance diagrams
obtained with the salt-film and adsorbate-acceptor
systems, respectively. The most important overall
feature of the diagrams concerns the difference in
low-frequency behavior between the adsorbate-
acceptor model and the salt-film models. Despite the
complexity of the transport processes involved, the
impedance behavior of the salt-film models is rela-
tively simple. At high frequencies, charge-transfer
processes (Tafel kinetics for the porous film, solid-
state ionic conduction for the compact film) domi-
nate the response since the film does not have time
to grow or shrink and remains essentially unchanged
during modulation. As the lower frequencies are
approached, film thickening and thinning processes
begin to play an ever greater role, and at the very
lowest frequencies the system behaves essentially as a
pure capactor with the film growing and shrinking in
response to the oscillating signal.
The adsorbate-acceptor system is quite different.
At the highest frequencies charge-transfer and
double-layer processes dominate, but as the fre-
quency is lowered additional features related to time
constants for the rate of change of surface coverage
begin to appear. At the very lowest frequencies, a
large third loop appears containing contributions
from an underlying Warburg-Nernst impedance.
The Warburg-Nernst loop is due to variations in
surface acceptor concentration as a result of the
oscillations in surface flux. It is interesting to note
that such effects are impossible in the salt-film
models, since the surface cation concentration in
those models is fixed at its saturation value and
Fig. 5. General shape of the Nyquist diagrams in salt-film systems.
EA 40 4 O
-
8/19/2019 25 - matlosz1995
8/9
400
M. MATLOSZ
NYQUIST DIAGRAM : ACCEPTOR MODEL
Fig. 6. General shape of the Nyquist diagrams in acceptor systems.
cannot be affected by the oscillating flux. This differ-
ence between the salt-film and acceptor models is
very striking, but it can only be observed with low-
frequency data that may be difficult to obtain in a
polishing system undergoing high-rate dissolution. It
is therefore of interest to compare more easily
obtainable and reproducible data that can be mea-
sured at high frequencies.
At high frequencies, all of the models yield
Nyquist diagrams of essentially the same shape, a
semicircle displaced to the right of the origin. The
position of the high-frequency limit of the semicircle
R,,
the diameter of the circle R, and the effective
capacity C,, determined from the angular frequency
at the top of the semicircle w = l/R,C,, provide
three characteristic values that can be measured
easily and compared in light of the theory for the
different models. Table 1 summarizes the results
from the theoretical analyses in [S] and [6].
Since all three models yield similar diagrams at
high frequency, it is not possible to distinguish
between them based on the shape of a single imped-
ance spectrum. The results in Table 1 can be used,
however, to determine expected trends for targeted
experiments aimed at comparing impedance spectra
obtained at different steady-state operating condi-
tions. Consider, for example, a series of impedance
diagrams obtained at different total applied poten-
Table 1. Characteristic features of the high-frequency loops
Salt-Film Models
Porous Film Compact Film
Adsorbate-
Acceptor
Model
tials along the limiting-current plateau. Under condi-
tions of constant
convective diffusion, the
limiting-current value is essentially unchanged. For
compact or porous-film models, increasing total
applied potential at constant limiting current density
must result in an increase in the corresponding film
thicknesses. Similarly, for the adsorbate-acceptor
model, an increase in applied potential should result
in an increase in the adsorbate surface coverage. The
results in Table 1 for an increase in 1, and 1, indi-
cate an increase in
R,
but no change in
R,
and C,
for a porous-film model, whereas for a compact film
the same experiment should yield an increase in
R,,
a decrease in C, but no change in
R,.
For an
adsorbate-acceptor model, none of the parameters
should vary.
An alternative series of impedance diagrams can
be obtained at a fixed applied potential (corrected
for the ohmic drop i,,
R,),
but at varying convective
diffusion conditions. For a rotating disk electrode,
for example, one can increase the disk rotation
speed, which will result in an increase in the steady-
state current density i,, (proportional to the square
root of disk rotation speed). For constant applied
potential, the incease in steady-state current will lead
to a decrease in salt-film thickness and both factors
will play a role in the behavior of the diagrams. For
a porous-film system, Table 1 reveals that
R,
and
R,
should decrease while C, should remain unchanged.
For a compact film,
R,
should remain unchanged,
C, increase and
R,
decrease. Finally, for the
adsorbate-acceptor model,
R,
and C, should remain
unchanged, while
R,
should decrease (propor-
tionately to the square root of the disk rotation
speed). Each of these effects has a clear explanation
in the underlying physical models as described above
and can be related directly to the parameters of the
experimental system under study. Experimental
investigations of this type have been undertaken on
both salt-film and acceptor systems and have pro-
vided considerable support for the physical pictures
of the polishing mechanisms described above.
-
8/19/2019 25 - matlosz1995
9/9
Modeling of impedance mechanisms 401
CONCLUSIONS
tion will be of general use in the study of a wide
range of electropolishing systems of practical and
Theoretical analyses of the underlying physical scientific importance.
bases of simple salt-film and acceptor models have
resulted in identification of important qualitative dif-
ferences between the models that are independent of
the specific property parameters for a given electro-
polishing system. Targeted experiments aimed at
studying the variation in alternating current imped-
ance spectra as a function of steady-state operating
conditions such as applied potential and hydrody-
namic conditions can be used to elucidate the
mechanism. The results of such experimental studies
can provide fundamental information concerning the
mechanisms of electropolishing on a qualitative basis
without recourse to extensive fitting of the model
parameters. It is hoped that the physical picture of
the polishing process presented here and the corre-
sponding methodology for experimental investiga-
REFERENCES
1. H. Figour and P. A. Jacquet, French Patent No. 707526
(1930).
2. P. A. Jacquet, Nature 135, 1076 (1935).
3. P. A. Jacquet, Trans. Electrochemical Society 69, 639
(1936).
4. D. Landolt, Electrochim. Acta 32, 1 (1987).
5. R.-D. Grimm, A. West and D. Landolt, J. electrochem.
Sot., 139, 1622 (1992).
6. M. Matlosz, S. Magaino and D. Landolt, J. electrochem.
sot., 141,410 (1994).
7. S. Magaino, M. Matlosz and D. Landolt, J. electrochem.
Sot. 140, 1365 (1993).


















