
1. Terminologia, definicje, klasyfikacje - Zakład …. Metody oczyszczania koloid ów Dializa i...
50
Transcript of 1. Terminologia, definicje, klasyfikacje - Zakład …. Metody oczyszczania koloid ów Dializa i...


1.1. Terminologia, definicje, klasyfikacjeTerminologia, definicje, klasyfikacje
Układami koloidalnymi (w skrócie koloidami) nazywamy układy dyspersyjne,
najczęściej dwuskładnikowe, o wyglądzie układów fizycznie jednorodnych chociaŜw rzeczywistości oba składniki nie są ze sobą zmieszane cząsteczkowo.
Fazę ciągłą (np. rozpuszczalnik) nazywamy ośrodkiem dyspersyjnym (lub
rozpraszającym), a fazę rozproszoną lub zdyspergowaną tworzy drugi składnik
o wymiarach 1nm – 100, 500 lub 1000 nm (1µm).
Cząstki koloidalne zawarte są więc pomiędzy układami o rozdrobnieniu
cząsteczkowym (np. roztwory), a z drugiej – o rozdrobnieniu mechanicznym
(zawiesiny, suspensje).
Najpowszechniejszym podziałem koloidów jest zaproponowany przez Ostawlada
podział oparty na stanie skupienia ośrodka dyspersyjnego oraz fazy
rozproszonej (tabela 1).
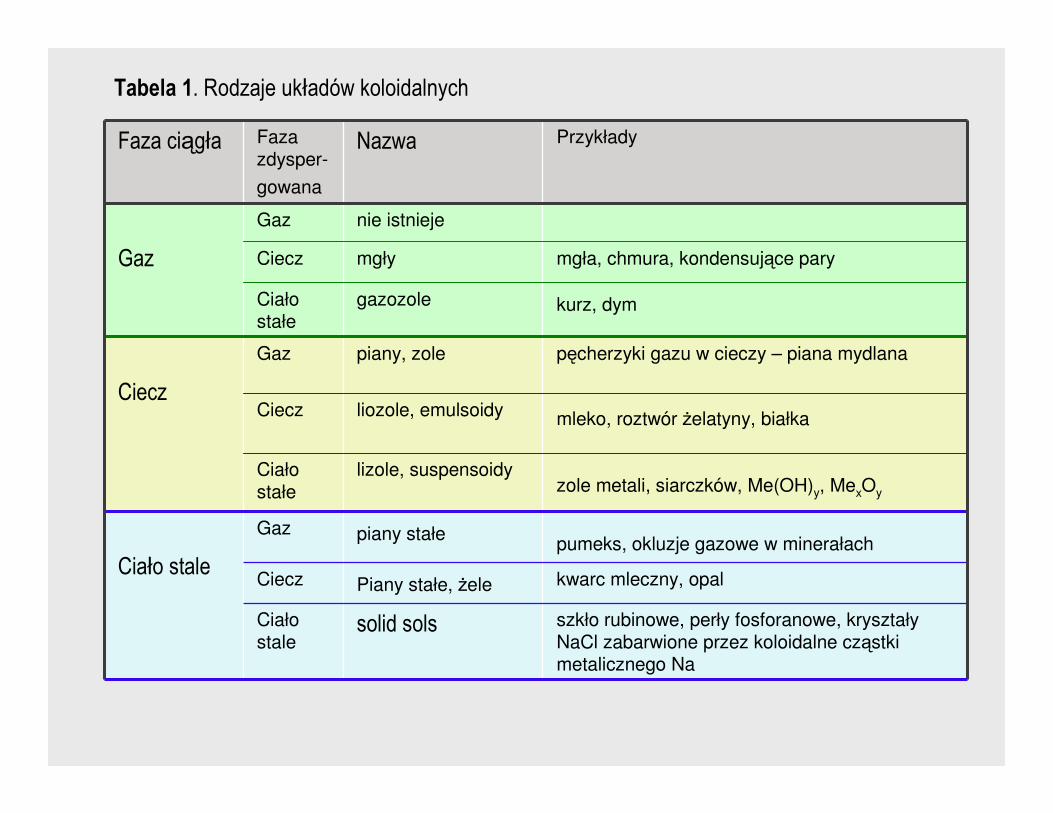
Faza ciągła Faza zdysper-
gowana
Nazwa Przykłady
Gaz
Gaz nie istnieje
Ciecz mgły mgła, chmura, kondensujące pary
Ciało stałe
gazozole kurz, dym
Ciecz
Gaz piany, zole pęcherzyki gazu w cieczy – piana mydlana
Ciecz liozole, emulsoidy mleko, roztwór Ŝelatyny, białka
Ciało stałe
lizole, suspensoidyzole metali, siarczków, Me(OH)y, MexOy
Ciało stale
Gaz piany stałepumeks, okluzje gazowe w minerałach
Ciecz Piany stałe, Ŝele kwarc mleczny, opal
Ciało stale
solid sols szkło rubinowe, perły fosforanowe, kryształy NaCl zabarwione przez koloidalne cząstki metalicznego Na
Tabela 1. Rodzaje układów koloidalnych

2. Przyk2. Przykłłady ukady ukłładadóów w koloidalnychkoloidalnych
W przyrodzie oŜywionej: białka, pektyny, węglowodany.
W przyrodzie nieoŜywionej: glina, mgła, smog, popioły wulkaniczne.
Syntetyczne substancje koloidalne: mydła, barwniki, koloidalna siarka,
uwodnione tlenki metali.
Związki chemiczne o cząsteczkach mających wymiary koloidalne – związki
wielkocząsteczkowe – koloidy cząsteczkowe: skrobia, celuloza, kauczuk,
keratyna, kolagen, glikogen, cząsteczki polimerowe (polistyren, polichlorek
winylu, poliamidy, poliwęglany, poliuretany.
).
Najbardziej rozpowszechnione są układy z fazą ciekłą dyspergującą – roztwory
koloidalne lub liozole czyli zole:
⇒ hydrozole (woda ośrodkiem dyspergującym),
⇒ organozole (ciecz organiczna ośrodkiem dyspergującym).

3. Podzia3. Podziałł ukukłładadóów w koloidalnychkoloidalnych
WyróŜnia się układy monodyspersyjne - koloidy w których cząstki fazy
rozproszonej mają jednakową wielkość oraz - polidyspersyjne, w których cząstki
mają róŜne wymiary.
cząstka fazy rozproszonej cząstka fazy rozproszonej

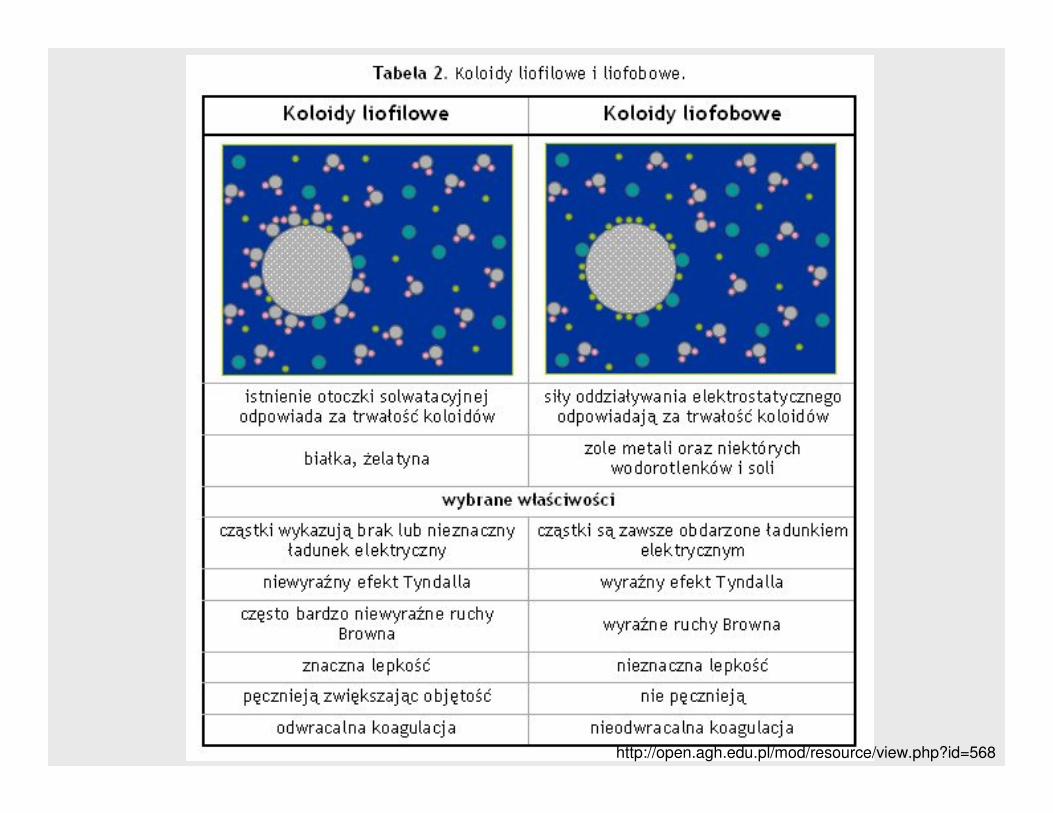
W zaleŜności od powinowactwa do rozpuszczalnika, koloidy dzieli się na:
⇒ liofilowe – silnie solwatują (hydratują) przez co układy takie są stabilne,
mniej wraŜliwe na czynniki koagulujące
⇒⇒⇒⇒ liofobowe – cząstki nie ulegają solwatacji (lub w bardzo małym stopniu)
cząsteczkami fazy rozpraszającej – jeśli woda to nie ulegają hydratacji,
jedynie na ich powierzchniach adsorbują się jony z roztworu
http://open.agh.edu.pl/mod/resource/view.php?id=568

Inny podziaInny podziałł ukukłładadóów koloidalnychw koloidalnych:
⇒ koloidy fazowe – cząstka koloidalna nie jest cząsteczką chemiczną
substancji rozproszonej, zwykle posiada ładunek elektryczny na powierzchni, np.
zole złota, srebra, tlenków metali, emulsje.
⇒ koloidy cząsteczkowe – w odpowiednich rozpuszczalnikach rozpuszczają
się cząsteczkowo ale wymiar cząsteczki jest tak duŜy (makrocząsteczka), Ŝe ma
właściwości układów koloidalnych, nie muszą posiadać ładunku elektrycznego,
np. białka, Ŝelatyna, polimery.
⇒ koloidy asocjacyjne (micelarne) – składają się z cząsteczek zasocjowanych,
które tworzą większą cząstkę, tzw. micelę, np. dodecylosiarczan sodowy (SDS).
Przy większych stęŜeniach łańcuchy oddziaływają na siebie siłami van der
Waalsa.
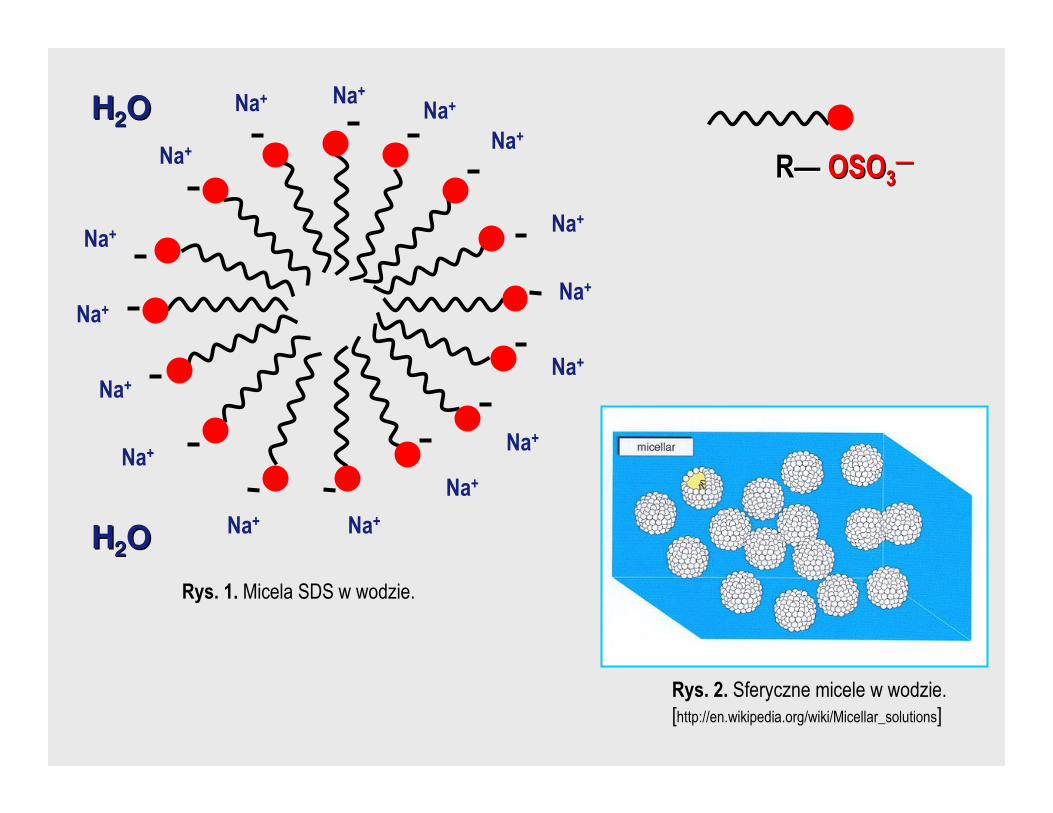
Rys. 1. Micela SDS w wodzie.
RR—— OSOOSO33——
Na+HH22OO
HH22OO
Na+
Na+
Na+
Na+
Na+
Na+
Na+
Na+ Na+Na+
Na+
Na+
Na+
Na+Na+
Rys. 2. Sferyczne micele w wodzie.
[http://en.wikipedia.org/wiki/Micellar_solutions]

44. . Metody otrzymywania koloidMetody otrzymywania koloidóów w
Dwie grupy metod: dyspersyjnedyspersyjne i kondensacyjnekondensacyjne..
Metody dyspersyjneMetody dyspersyjne:
♦♦♦♦ rozdrobnienie mechanicznerozdrobnienie mechaniczne – mielenie w młynach koloidalnych (mała wydajność).
♦♦♦♦ rozpylenie w rozpylenie w łłuku elektrycznymuku elektrycznym (zol metalu) lub za pomocą ultradźwięków,
naświetlanie promieniami.
♦♦♦♦ peptyzacjapeptyzacja – przeprowadzenie świeŜo wytrąconego osadu roztworem
odpowiedniego elektrolitu w zol.

Metody kondensacyjneMetody kondensacyjne (najczęściej metody chemiczne):
a)a) hydroliza solihydroliza soli – w wyŜszych temperaturach moŜna otrzymać hydrozole
wodorotlenków i tlenków a takŜe kwasu krzemowego, np.:
FeClFeCl33 + 3 H+ 3 H22O O ↔↔ Fe(OH)Fe(OH)33 + 3 HCl+ 3 HCl
NaNa22SiOSiO33 + 2 H+ 2 H22O O ↔↔ HH22SiOSiO3 3 + 2 NaOH+ 2 NaOH
b)b) reakcje wymianyreakcje wymiany – w odpowiednich warunkach w wyniku wymiany moŜna
wydzielić substancję koloidalną, np.:
2 HCl + Na2 HCl + Na22SiOSiO33 ↔↔ HH22SiOSiO33 + 2 NaCl+ 2 NaCl
c) reakcje redoxreakcje redox:
2 AuCl2 AuCl3 3 + 6 NaOH + HCOH + 6 NaOH + HCOH ↔↔ 2 Au + 6 NaCl + HCOOH + 3 H2 Au + 6 NaCl + HCOOH + 3 H22OO
HH22S + OS + O22 ↔↔ 2 H2 H22O + 2SO + 2S

d) d) reakcje polimeryzacjireakcje polimeryzacji, np. polimeryzacja dwuwinylu do kauczuku:
CHCH33––CHCH══CHCH──CHCH33 →→ ──CHCH22──CHCH══CHCH──CHCH22──CHCH22──CHCH══CHCH──CHCH22── →→((──CHCH22──CHCH══CHCH──CHCH22──CHCH22──CHCH══CHCH──CHCH22──)n)n
lub polimeryzacja aldehydu mrówkowego do polioksymetylenu::
H2C═O →─CH2─O─CH2─O─→ (─CH2O─)n
e) e) zmniejszenie rozpuszczalnozmniejszenie rozpuszczalnośścici – dodając ośrodka dyspergującego
(cieczy), w którym gorzej jest rozpuszczalny składnik koloidalny moŜna
otrzymać zol, np. wkraplanie roztworu etanolowego siarki do wody.
5. W5. Włłaaśściwociwośści ukci ukłładadóów koloidalnychw koloidalnych
WWłłaaśściwociwośści optyczneci optyczne
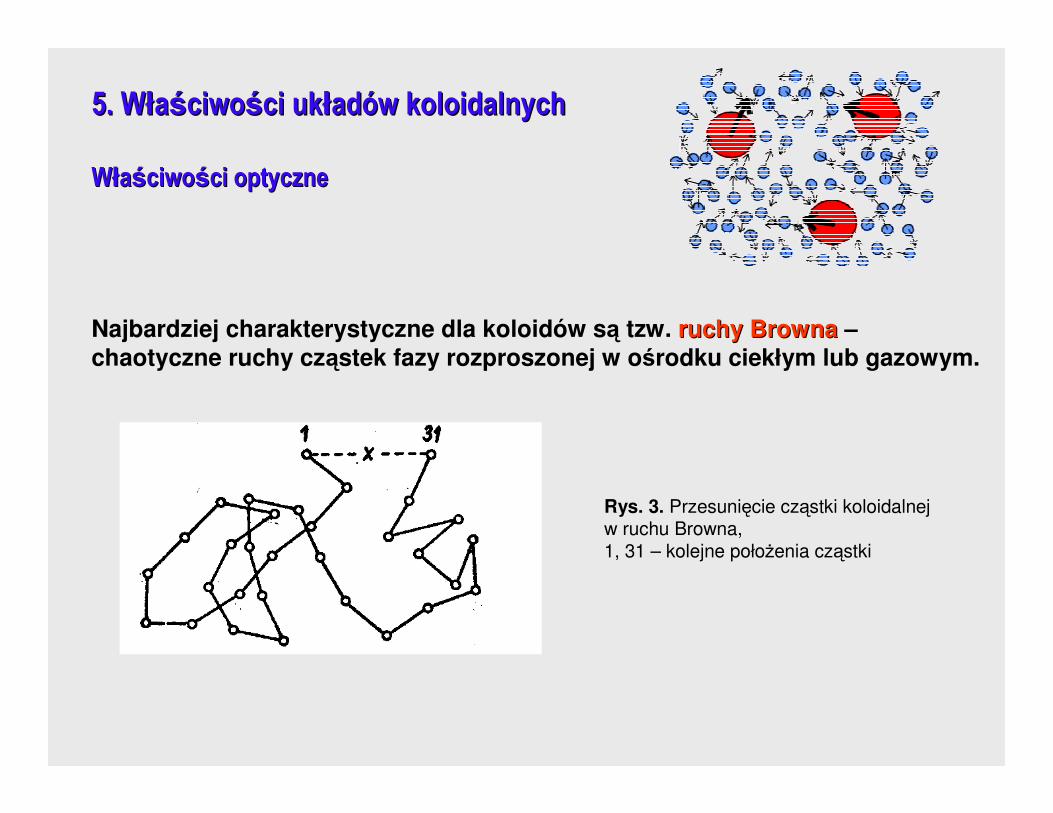
Najbardziej charakterystyczne dla koloidów są tzw. ruchy Brownaruchy Browna –chaotyczne ruchy cząstek fazy rozproszonej w ośrodku ciekłym lub gazowym.
Rys. 3. Przesunięcie cząstki koloidalnej w ruchu Browna, 1, 31 – kolejne połoŜenia cząstki

Teoretyczne uzasadnienie ruchów Browna, na bazie teorii kinetycznej materii,
podali EinsteinEinstein i SmoluchowskiSmoluchowski.
Ruchy Browna opisano kwadratem średniego przesunięcia cząstki w jednakowych
odstępach czasu:
(1)( )r3
t
N
RTx
r3
t
N
RTx
2
πη=∆⇒
πη=∆
gdzie: – średni rzut drogi cząstki na oś x, η – lepkość ośrodka,
r – promień cząstki dyfundującej, t – czas, N – liczba Avogadro;
np. T = 293 K, r = 1nm, η = 0,010 cP (g m-1 s-1), = 6,5·10-6 cm.
x∆
x∆
x∆

Efekt TyndallaEfekt Tyndalla – przepuszczane przez układ koloidalny promienie światła
ulegają rozproszeniu na cząstkach fazy rozproszonej, jeŜeli długość fali
padającego światła jest trochę większa od wymiarów cząstki., gdy średnice
cząstek wynoszą 40-900 nm, czyli nieco poniŜej długości fali światła
widzialnego (400 – 750 nm). Światło rozproszone łatwo zaobserwować w
świetle bocznym, prostopadłym do promienia padającego.
Rys. 4. Efekt Tyndalla

Efekt Efekt TyndallTyndallaa
Rys. 5. Efekt Tyndalla widoczny przy zastosowaniu światła laserowego
[http://silver-lightning.com/tyndall/]; [www.silverwell.com.au/Making-CS.html]
Woda zawiera 5 ppm Woda zawiera 5 ppm
koloidalnego srebrakoloidalnego srebraWoda z kranu z Woda z kranu z
ppęęcherzykami powietrzacherzykami powietrza Zol srebra

7. 7. MetMetody oczyszczania koloidody oczyszczania koloidóóww
DialDializa i elektrodializaiza i elektrodializa
DializaDializa polega na przepuszczeniu przez błonę półprzepuszczalną substancji o
charakterze cząsteczkowym.
Jony znajdujące się w roztworze koloidalnym w wyniku dyfuzji przechodzą przez
błonę dla nich przepuszczalną do przestrzeni z czystą wodą odnawianą w
przepływie ciągłym.
Podgrzewanie i mieszanie przyspiesza proces wymiany.
Proces oczyszczania z nadmiaru elektrolitów moŜna przyspieszyć w polu
elektrycznym w wyniku elektrodializy. Do tego celu wykorzystuje się urządzenia
zwane elektrodializatoremelektrodializatorem.
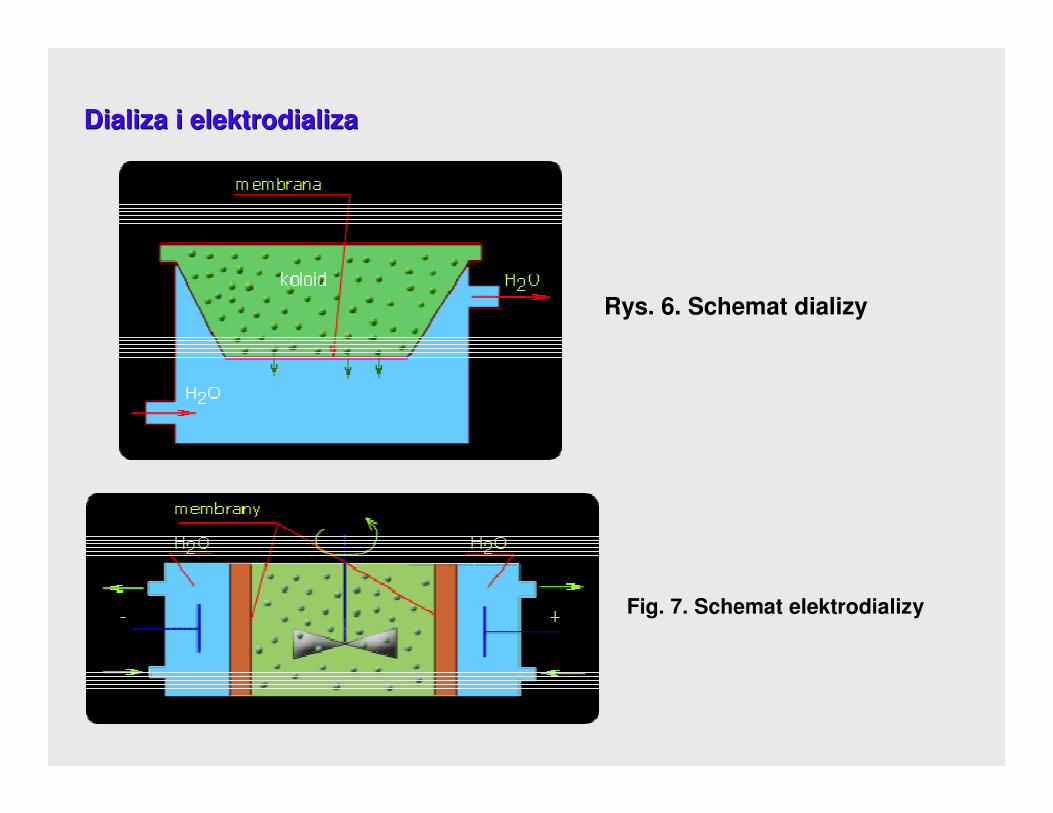
DialDializa i elektrodializaiza i elektrodializa
Rys. 6. Schemat dializy
Fig. 7. Schemat elektrodializy
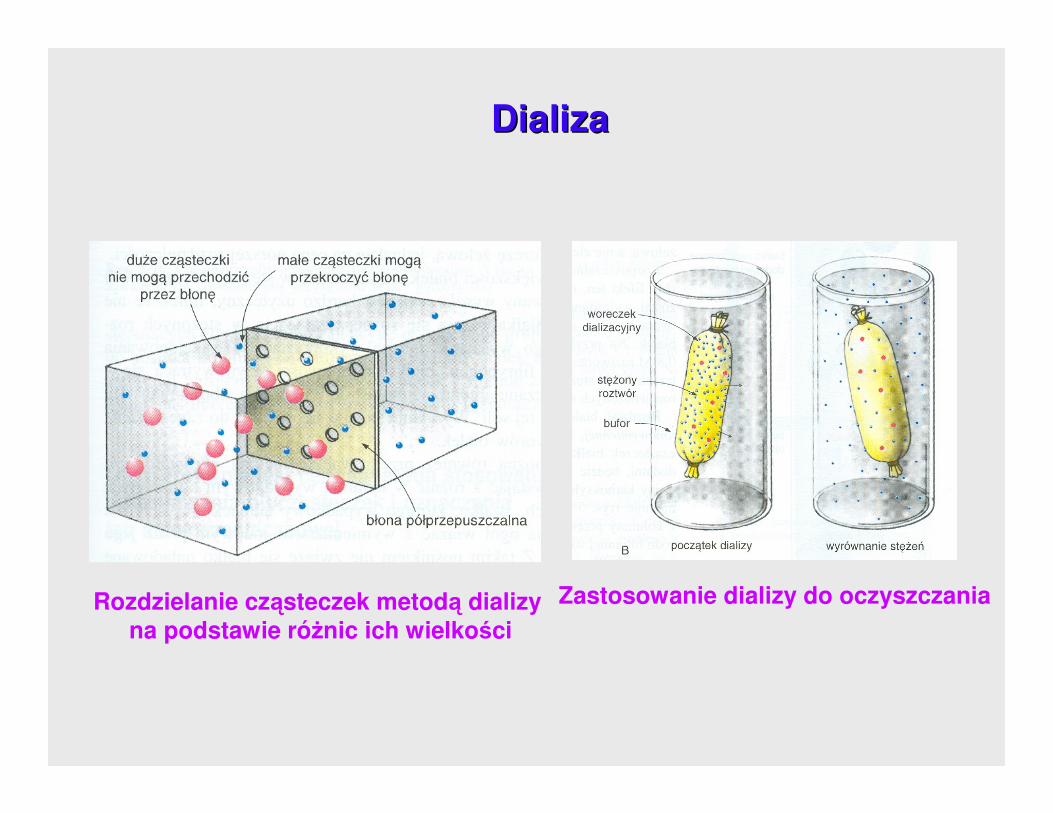
DializaDializa
Rozdzielanie cząsteczek metodą dializy na podstawie róŜnic ich wielkości
Zastosowanie dializy do oczyszczania
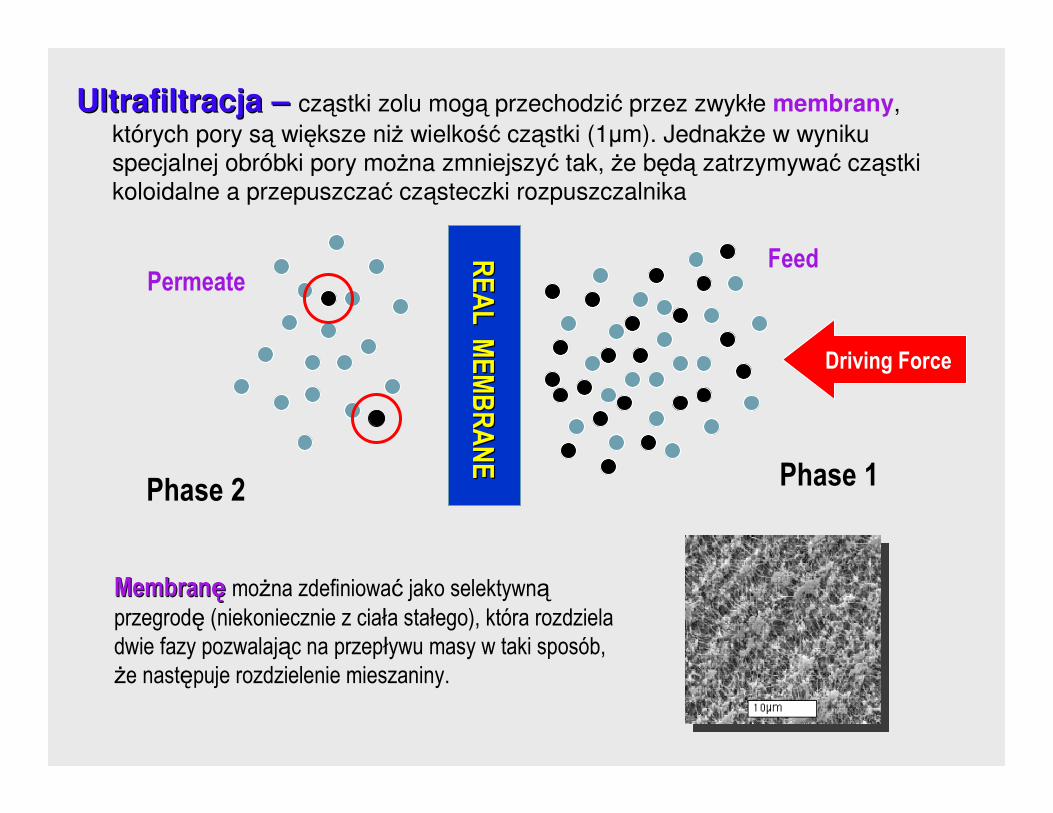
Ultrafiltracja Ultrafiltracja –– cząstki zolu mogą przechodzić przez zwykłe membrany,
których pory są większe niŜ wielkość cząstki (1µm). JednakŜe w wyniku
specjalnej obróbki pory moŜna zmniejszyć tak, Ŝe będą zatrzymywać cząstki
koloidalne a przepuszczać cząsteczki rozpuszczalnika
MembranMembranęę moŜna zdefiniować jako selektywnąprzegrodę (niekoniecznie z ciała stałego), która rozdziela
dwie fazy pozwalając na przepływu masy w taki sposób, Ŝe następuje rozdzielenie mieszaniny.
IDE
AL
ME
MB
RA
NE
PermeateFeed
Driving Force
REAL M
EMBRANE
REAL M
EMBRANE
Phase 1Phase 2

UltracentrUltracentryyfugfugowanie (Ultrawirowanie (Ultrawiróówkowanie)wkowanie)
Cząstki zolu nie sedymentują pod wpływem siły grawitacji ze względu
na energię kinetyczną (termiczną) ośrodka. JednakŜe stosując
wysokoobrotowe wirówki o obrotach 15 000 lub więcej na minutę,
cząstki koloidalne mogą ulec sedymentacji. Takie aparaty nazywa się
ultracentryfugami lub ultrawirówkami.

6. 6. . ŁŁadunek elektryczny czadunek elektryczny cząąstek koloidalnychstek koloidalnych
Ładunek elektryczny cząstki koloidalnej obok solwatacji – czynnik powodujący stabilność układu koloidalnego. Przyczyny powstawania ładunku na powierzchni cząstki koloidalnej:
� adsorpcja jonów,
� jonizacja grup powierzchniowych,
� przechodzenie jonów z powierzchni do roztworu.
Adsorpcja jonAdsorpcja jonóów w – róŜnych znaków zwykle nie jest jednakowa, cząstka
uzyskuje ładunek nadmiarowy – przewaŜnie ujemny (kationy są silniej
zhydratyzowane). Mogą to być jony tworzące cząstkę koloidalną i
bardzo często jony H+ i OH–.
JJonizaonizacja grup powierzchniowych cja grup powierzchniowych – grupy takie jak np. ―COO–, ―NH3+
powstają na powierzchni polimerów lub białek, stopień jonizacji silnie
zaleŜy od pH. Wartość pH, przy którym nie ma na powierzchni ładunku
nadmiarowego nazywa się punktem ładunku zerowego (point of zero
charge, pzc).

Przechodzenie jonów do fazy ciekłej (rozpuszczanie jonów) – w
przypadku koloidów mogących dysocjować na jony np. AgI – zachodzi
niejednakowe rozpuszczanie się jonów o róŜnych znakach, tu Ag+ i I–.
ZaleŜeć to będzie od stęŜenia tych jonów w roztworze i związane jest z
iloczynem rozpuszczalności, IR = aAg+·aJ- = 10-16. JeŜeli pAg = 5,5
(pI = 10,5) to powierzchnia AgI nie jest naładowana, występuje
wówczas punkt ładunku zerowego.
[Ag+]r = 10-5,5 [I-]r = 10-10,5 => pAgpzc = 5,5
Jony Ag+ i I– nazywa się jonami potencjałotwórczymi. W przypadku
tlenków i wodorotlenków rolę takich jonów spełniają H+ i OH–.
Wynikiem obecności ładunku nadmiarowego na powierzchni cząstki
koloidalnej jest powstanie wokół niej podwójnej warstwy elektrycznej.
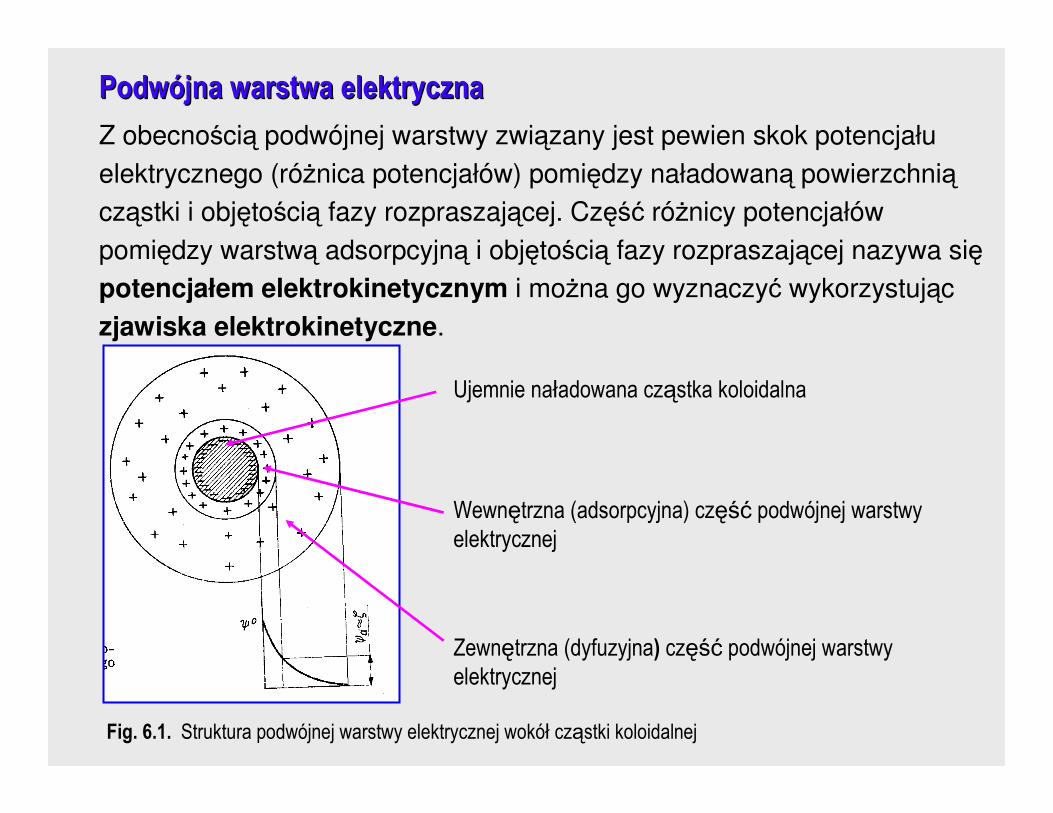
PodwPodwóójna warstwa elektrycznajna warstwa elektryczna
Z obecnością podwójnej warstwy związany jest pewien skok potencjału
elektrycznego (róŜnica potencjałów) pomiędzy naładowaną powierzchnią
cząstki i objętością fazy rozpraszającej. Część róŜnicy potencjałów
pomiędzy warstwą adsorpcyjną i objętością fazy rozpraszającej nazywa się
potencjałem elektrokinetycznym i moŜna go wyznaczyć wykorzystując
zjawiska elektrokinetyczne.
Fig. 6.1. Struktura podwójnej warstwy elektrycznej wokół cząstki koloidalnej
Ujemnie naładowana cząstka koloidalna
Wewnętrzna (adsorpcyjna) część podwójnej warstwy elektrycznej
Zewnętrzna (dyfuzyjna) część podwójnej warstwy elektrycznej

7. 7. StabilnoStabilnośćść ukukłładadóów koloidalnych w koloidalnych –– koagulacjakoagulacja
Jedną z najwaŜniejszych fizycznych właściwości układów koloidalnych
jest tendencja do agregacji cząstek. Główną przyczyną agregacji są
przyciągające siły van der Waalsa działające pomiędzy cząstkami.
Natomiast głównymi siłami odpychającymi są siły elektrostatycznego
oddziaływania cząstek naładowanych o tym samym znaku oraz
oddziaływania cząstka – rozpuszczalnik (ciecz).
TakŜe hydratacja (solwatacja) cząstek koloidalnych powoduje ich odpychanie w
roztworze, czyli stabilizuje system.
Liofobowe (hydrofobowe) zole są głównie stabilizowane siłami
podwójnej warstwy elektrycznej, dlatego są one bardzo wraŜliwe na
dodatek elektrolitu, który często juŜ przy małym stęŜeniu powoduje
wydzielanie się cząstek koloidalnych z ośrodka dyspersyjnego, co
określamy koagulacją czyli przejściem zolu w Ŝel.

Koagulacja jest procesem przejKoagulacja jest procesem przejśścia zolu w cia zolu w ŜŜel, natomiast odwrotnym procesem jest el, natomiast odwrotnym procesem jest
peptyzacjapeptyzacja
zolzol
coagulacja
ŜŜelelpeptizacja
Wynika to z „rozładowania” cząstki koloidalnej w wyniku adsorpcji
jonów. Najszybsza koagulacja zachodzi w pzc (punkt ładunku
zerowego) lub iep (punkt izoelektryczny, ζ = 0).
Te dwa parametry mogą wystąpić przy tej samej wartości pH.

Teoria Teoria DLVODLVO
Derjaguin i Landau oraz Vervey i Overbeek opracowali niezaleŜnie teorię
stabilności koloidów liofobowych, która pozwala obliczyć zmianę energii
podczas zbliŜania się cząstek do siebie. Teoria ta znana jest jako DLVO i
rozpatruje siły przyciągające (aktrakcyjne) van der Waalsa–Londona
i odpychające (repulsyjne) zachodzących na siebie podwójnych warstw
elektrycznych , czyli całkowita energia układu jest sumą energii
Lifshitza-van der Waalsa i energii elektrostatycznej:
LW121G∆
EL121G∆
EL
121
LW
121
TOT
121GGG ∆+∆=∆ (1)

JeŜeli maksimum przewyŜsza energię termiczną (kT) w danej temperaturze to
układ powinien być stabilny. Przy odpowiednio duŜych odległościach cząstek
występuje tzw. drugie minimum na krzywej energii potencjalnej i jeŜeli jest ono
wystarczająco duŜe w porównaniu z kT, to cząstki mogą tworzyć „luźne”
odwracalne agregaty.
Rys. 7.1 ZaleŜność energii
potencjalnej oddziaływania między
cząstkami koloidalnymi.
V(1) – silne odpychanie – słabe przyciąganie
(maksimum odpychania na średnich
odległościach), stan dyspersji trwały;
V(2) – silne przyciąganie – słabe odpychanie
(koagulacja) przy wszystkich odległościach
dominują siły przyciągające.

W przypadku koloidów liofilowych sytuacja komplikuje się, poniewaŜ
dochodzą jeszcze inne czynniki, jak energia desorpcji jonów, efekty
entropowe i mostkujące, które są trudne do ujęcia w matematycznym
opisie. W tych układach mały dodatek elektrolitu praktycznie nie wpływa
na ich stabilność. Przy duŜych stęŜeniach występuje efekt „wysalania” i
są one wytrącane.
Jony powodują desolwatację (dehyratację) cząstek ulegając same
hydratacji czyli zaleŜy to duŜo od energii hydratacji jonów. Kationy i
aniony tworzą szeregi liotropowe, np.:
Mg2+ > Ca2+ > Sr2+ > Be2+ > Li+ > Na+ > K+ > NH4+ > Rb+ > Cs+
oraz
cytrynian3– > SO42– > Cl– > NO3
– > J– > CNS–

W układach zdyspergowanych całkowita energia oddziaływania między
dwiema róŜnymi cząstkami determinowana jest energią odpychania
elektrostatycznego oraz oddziaływania siłami Londona-van der Waalsa
zgodnie z teorią DLVO. W przypadku oddziaływań pomiędzy powierzchniami
hydrofilowymi nie są one opisane w sposób kompletny przez tę teorię, dlatego
zaproponowano uwzględnienie solwatacyjnych lub hydratacyjno-
hydrofobowych oddziaływań strukturalnych, których pochodzenie moŜna
wiązać z obecnością specyficznych oddziaływań polarnych lub niepolarnych,
występujących w tych układach. Jedno z podejść stosowanych do opisu tych
sił zostało zaproponowane przez van Oss’a, Good’a i współ.
Całkowita energia oddziaływań pomiędzy dwiema róŜnymi cząstkami (1) i (2)
zdyspergowanymi w środowisku wodnym (3) moŜe być przedstawiona jako
suma trzech udziałów:
Rozszerzona teoria Rozszerzona teoria DLVODLVO

Dwa pierwsze oddziaływania są zaliczane są do klasycznego modelu DLVO,
natomiast składowa AB została wprowadzona w celu wyjaśnienia oddziaływań
pomiędzy cząstkami hydrofilowymi i hydrofobowymi w ośrodkach polarnych.
AB
132
LW
132
EL
132
TOT
132GGGG ∆+∆+∆=∆ (2)
W ośrodkach polarnych, szczególnie w wodzie swobodna energia
oddziaływań polarnych (AB) pomiędzy cząstkami rozproszonymi, jest zwykle
nawet 100 razy większa niŜ energia wynikająca z oddziaływań LW oraz ok. 10
razy przekraczają one oddziaływania EL.
� oddziaływań elektrostatycznych (EL), związanych z istnieniem na powierzchni
cząstek podwójnej warstwy elektrycznej,
� oddziaływań Lifshitza-van der Waalsa (LW)
� sił solwatacyjnych, które zgodnie z podejściem van Ossa i współ. mogą być
wynikiem oddziaływań kwasowo-zasadowych (AB) lub w ujęciu Lewisa elektrono-
akceptorowych i elektrono-donorowych, pomiędzy fazami będącymi w kontakcie.

Czasami dodatek substancji liofilowej powoduje zwiększenie stabilności
koloidu w wyniku adsorpcji na jego powierzchni – takie substancje nazywa
się koloidami ochronnymi lub substancjami ochronnymi. Działanie ich
polegać moŜe na: zwiększeniu ładunku i potencjału elektrycznego
koloidu (grupy jonizujące), obniŜenie oddziaływań van der Waalsa. JeŜeli
zastosuje się makromolekuły w postaci łańcuchów polimerowych to mogą
one rozciągać się w głąb fazy dyspergującej. W czasie zbliŜania się cząstek
występujące oddziaływania prowadzą do wzrostu uporządkowania czyli
spadku entropii.
JeŜeli zmiana entalpii ∆H jest niewielka i moŜna ją zaniedbać to zachodzą
dodatnie zmiany, ∆G = ∆H – T∆S, czyli zapobiega to procesowi
samorzutnej koagulacji. Efekt ten nazywamy steryczną lub entropową
stabilizacją.
∆∆G = G = ∆∆H H –– TT∆∆S > 0S > 0
WpWpłływ dodatku substancji na stabilnoyw dodatku substancji na stabilnośćść ukukłładu koloidalnegoadu koloidalnego

W niektórych układach koloidalnych mały dodatek zolu ochronnego do
koloidu liofobowego moŜe spowodować „uczulenie” na działanie
elektrolitu. Jest to sensybilizacja.
Trzeba jeszcze wspomnieć o dwu zjawiskach:
Tiksotropia – przejście zastygłego Ŝelu w stan ciekły, liozol pod wpływem
bodźców mechanicznych (mieszania, wytrząsania) i po pewnym czasie
ponownie w Ŝel, np. Fe(OH)3, glina.
Podobnym zjawiskiem jest synereza – wydzielanie się ośrodka
dyspersyjnego z Ŝelu, np. serwatka z kwaśnego mleka.


1. Typy emulsji1. Typy emulsji
Emulsja jest mieszaniną dwu lub więcej nierozpuszczalnych wzajemnie cieczy.
W emulsji jedna ciecz jest rozproszona (faza zdfspergowana) w drugiej cieczy (faza
ciągła).
MoŜna wyróŜnić dwa typy emulsji:
Olej– w – wodzie (O/WO/W) i woda – w –oleju (W/OW/O).
W emulsji O/W olej jest zdyspergowany w wodzie , która jest fazą ciągłą. A w emulsji
W/O odwrotnie.
Są takŜe emulsje wielokrotne typu woda /olej/woda i typu olej/woda/olej.
Przykłady pokazuje Rys. 1.1.
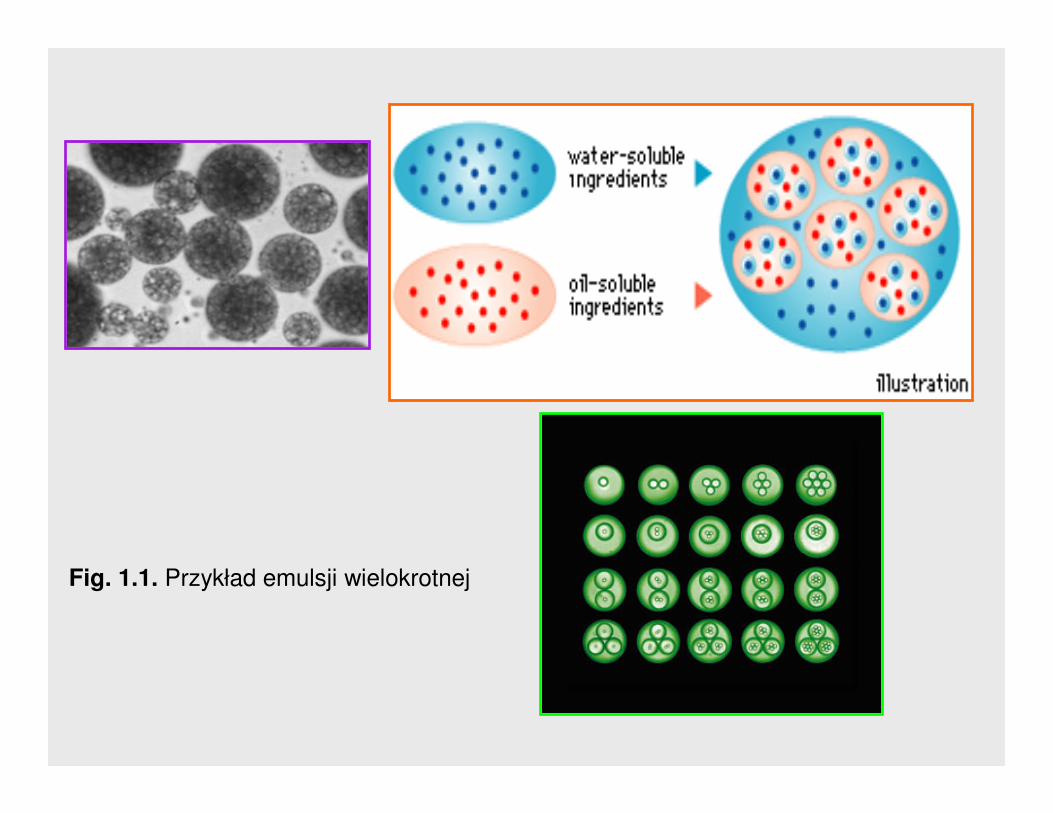
Fig. 1.1. Przykład emulsji wielokrotnej

Typ emulsji moTyp emulsji moŜŜna rozpoznana rozpoznaćć nastnastęępujpująącoco::
– Dodając pewną ilość jednej z cieczy tworzących emulsje obserwując jej rozpuszczalność.Np. Jeśli dodana woda rozpuszcza się w emulsji to jest ona typu olej-w-wodzie, O/W
– Stosując odpowiedni barwnik, który rozpuszcza się tylko w jednej cieczy ( w oleju lub
wodzie).
– Mierząc przewodnictwo elektryczne, które będzie znacznie większe w przypadku emulsji olej-w-wodzie, O/W.

2. Stabil2. Stabilnonośćść emulsemulsjiji
Emulsje są układami niestabilnymi, które po ich utworzeniu z uŜyciem siły mechanicznej (mieszanie), ciecze tworzące emulsjię szybko rozwarstwiająsię. Dlatego aby emulsja była trwała (lub przynajmniej trwalsza, stabilna) konieczny jest dodatek substancji stabilizującej – emulgatora (zwanego czasami stabilizatorem, stabilizator raczej wspomaga emulgatora).W przypadku emulsji olej-w-wodzie moŜna uŜyć surfaktantu mającego polarną głowę i łańcuch węglowodorowy. Część polarna będzie skierowana na zewnątrz kropelki oleju do fazy wodnej a łańcuch do jej wnętrza, co zapobiegnie szybkiej koalescencji.
Rys.1.2. Tworzenie emulsji olej-w-wodzie: A. Dwie niemieszające się ciecze przed emulsyfikacją.,.
B. Emulsja fazy II zdyspergowanej w Fazie I.
C. Progresywna separacja niestabilnej emulsji.
D. Cząsteczki surfaktantu wokół kropelek oleju stabilizujące emulsję.
(http://en.wikipedia.org/wiki/emulsion)

WyróŜnia się trzy typy niestabilności: flokulacja, śmietankowanie i
koalescencja.
Flokulacja – faza zdyspergowana wydziela się w postaci luźnych
agregatów (flakes)
Koalescencja – łączenie sę małych kropelek w większe.
Śmietankowanie – gromadzenie się ciekłej fazy zdyspergowanej na
wierzchu lub dnie naczynia, w zaleŜności od gęstości jednej fazy względem
drugiej.
Starzenie Ostwalda - spontaniczny proces wynikający z termodynamiki
poniewaŜ większe kropelki są energetycznie uprzywilejowane (mniejsza
powierzchnia) niŜ małe.
Nawet emulsje stabilizowane termodynamicznie są układami niestabilnymi
i powoli ulegają procesowi starzenia i powolnej koalescencji (łączenie się
małych kropelek w większe)
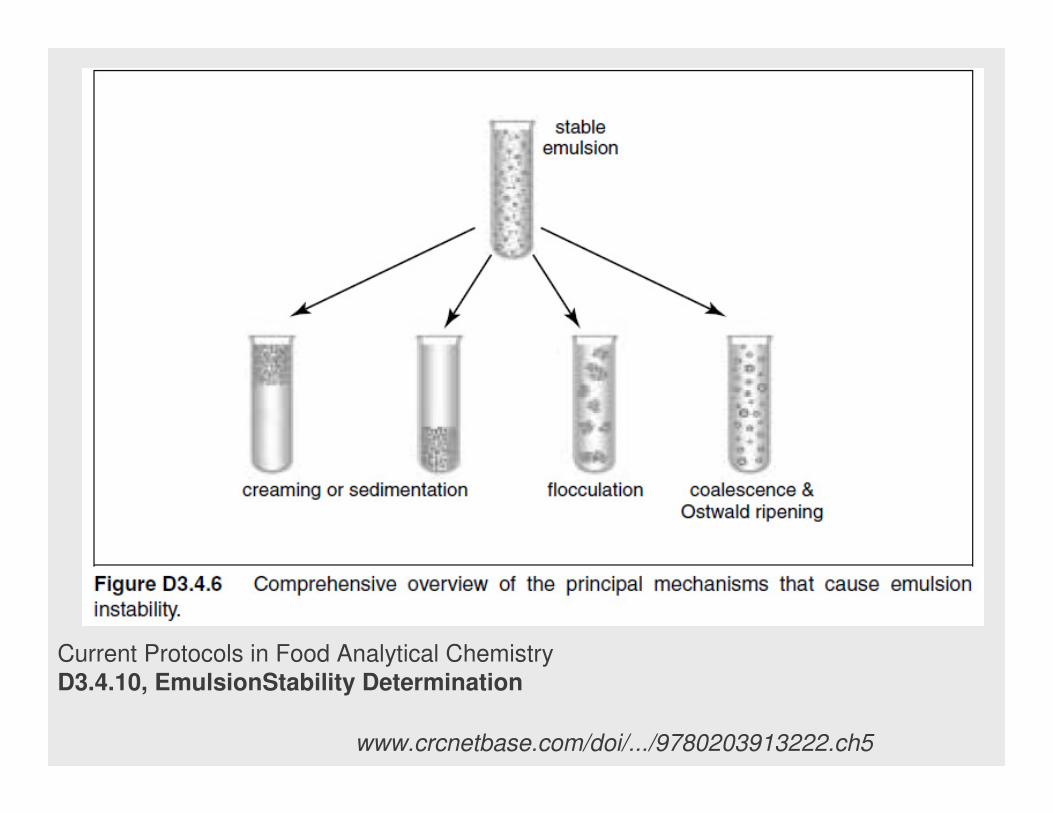
Current Protocols in Food Analytical Chemistry
D3.4.10, EmulsionStability Determination
www.crcnetbase.com/doi/.../9780203913222.ch5

Wynika on z faktu Ŝe cząsteczki na
powierzchni kropelki są energetycznie
mniej stabilne niŜ cząsteczki wewnątrz.
Surfaktanty jako molekuły amfifilowe posiadają grupę hydrofilową i
lipofilową. Bilans hydrofilowo-lipofilowy (HLB) jest to liczba stosowana jako
miara stosunku tych grup. Jej wartość wynosi pomiędzy 0-60 określając
powinowactwo surfaktantu do wody i oleju. Licba HLB obliczane dla
niejonowych surfaktantów zmienia się w zakresie 0-20.
HLB HLB -- hydrophilicity lipophilicity balancehydrophilicity lipophilicity balance
Bilans hydrofilowo-lipofilowy
Schemat procesu starzenia
Ostwalda
Klasyfikacja surfaktantów przy pomocy liczby HLB
Źródło: Wikipedia, the free encyclopedia)

Liczby HLB >10 oznaczają powinowatość do wody (hydrofilowe) a liczby <10
wskazują na powinowactwo do oleju (lipopofilowe). Surfaktanty jonowe
posiadają liczby HLB które mogą sięgać do 60.
Termin "HLB" po raz pierwszy został uŜyty przez pracowników Atlas Powder
Co. in America.
Oznacza on bilans pomiędzy częścią cząsteczki surfaktantu rozpuszczalnej w
oleju i części rozpuszczalnej w wodzie, co określa się jako Bilans
Hydrofilowo-Lipofilowy (Hydrophile-Liphophile Balance).
Emulgatory lepiej rozpuszczalne w oleju mają niŜszą liczbę HLB a lepiej
rozpuszczalne w wodzie mają wyŜszą liczbę HLB. Liczba HLB jest bardzo
uŜyteczna w dobieraniu emulgatorów, ale posiadają one kilka ograniczeń w
zastosowaniu do wszystkich surfaktantów.

Obliczanie liczby HLB dla mieszaniny surfaktantów
Liczbę HLB mieszaniny o składzie x% surfaktantu o liczbie HLB A i y%
surfaktantu o liczbie HLB B moŜna wyznaczyć z formuły:
HLB ( A + B ) = ( Ax + By ) / ( x + y )
Dyspersyjność w wodzie wg HLB
Typ dyspersji Przedział HLB
Brak dyspersji 1-4
Słaba dyspersja 3-6
Mleczna dyspersja po silnym mieszaniu 6-8
Stabilna mleczna dyspersja 8-10
Suspensja półprzeźroczysta do przeźroczystej 10-12
Klarowny roztwór 13+

Griffin w 1949 i 1954 opisał sposób obliczania Bilansu Hydrofilowo-
Lipofilowego cząsteczki surfaktantu poprzez obliczenie wartości tej liczby dla
części hydrofilowej i lipofilowej (czyli hydrofobowej) .
Inną metodę obliczania zaproponował w 1957 Davies.
Metoda Griffina dla niejonowych surfaktantów :
HLB = 20 ·Mh / M
Gdzie Mh jest masą cząsteczkową części hydrofilowej, a M masą
cząsteczkową całej cząsteczki, przyjmując umowną skalę od 0 do 20. HLB
równe 0 odpowiada cząsteczce całkowicie hydrofobowej a wartość 20
odpowiadałaby cząsteczce całkowicie hydrofilowej.

Metoda Davies’a polega na obliczeniu HLB grup chemicznych
występujących w cząsteczce. Zaletą tej metody jest to, Ŝe bierze się pod
uwagę wpływ silnych i słabych grup hydrofilowych zgodnie z równaniem:
HLB = 7 + m × Hh − n × Hl
gdzie:
m – ilość grup hydrofilowych w cząsteczce
Hh – liczba HLB grup hydrofilowych
n – ilość grup hydrofobowych w cząsteczce
Hl – HLB grup lipofilowych

Przykłady liczb HLB niezbędnych dla poszczególnych zastosowań.
HLB
zmieszanie dwu róŜnych olejów 1 do 3
utworzenie emulsji woda-w-oleju 4 to 6
zwilŜenie proszku olejem 7 to 9
tworzenie samo-emulgujących się olejów 7 do10
utworzenie emulsji oleju-w-wodzie 8 do16
utworzenie roztworu detergentu 13 do15
solubilizacja oleju (mikro-emulgowanie) w wodzie 13 do 18
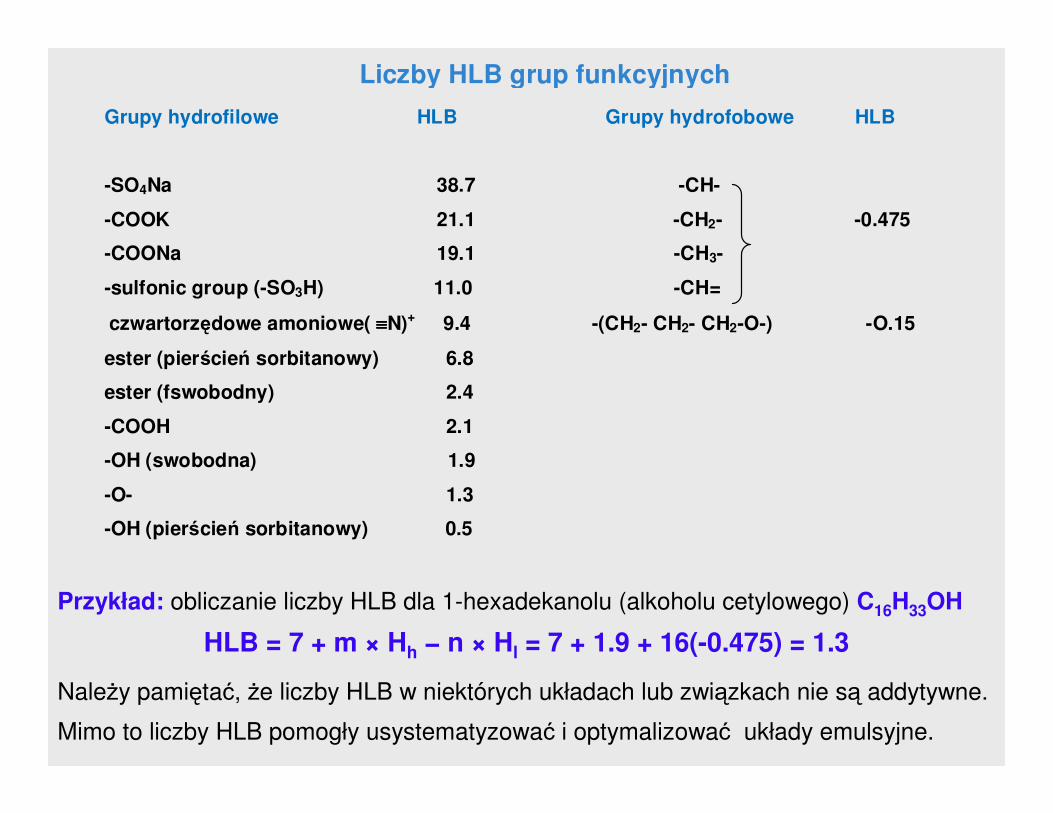
Liczby HLB grup funkcyjnych
Grupy hydrofilowe HLB Grupy hydrofobowe HLB
-SO4Na 38.7 -CH-
-COOK 21.1 -CH2- -0.475
-COONa 19.1 -CH3-
-sulfonic group (-SO3H) 11.0 -CH=
czwartorzędowe amoniowe( ≡≡≡≡N)+ 9.4 -(CH2- CH2- CH2-O-) -O.15
ester (pierścień sorbitanowy) 6.8
ester (fswobodny) 2.4
-COOH 2.1
-OH (swobodna) 1.9
-O- 1.3
-OH (pierścień sorbitanowy) 0.5
Przykład: obliczanie liczby HLB dla 1-hexadekanolu (alkoholu cetylowego) C16H33OH
HLB = 7 + m × Hh − n × Hl = 7 + 1.9 + 16(-0.475) = 1.3
NaleŜy pamiętać, Ŝe liczby HLB w niektórych układach lub związkach nie są addytywne.
Mimo to liczby HLB pomogły usystematyzować i optymalizować układy emulsyjne.

Mikroemulsjue są to przeźroczyste, stabilne, izotropowe mieszaniny cieczy
– oleju, wody i surfaktantu (często równieŜ kosurfaktantu). Faza wodna
moŜe zawierać sól (sole) i inne składniki a ‘olej’ moŜe w rzeczywistości być
mieszaniną róŜnych węglowodorów i olefin. W przeciwieństwie do zwykłych
emulsji, mikroemulsje tworzą się przez proste zmieszanie składników i nie
wymagają intensywnego mieszania. Dwa podstawowe typy mikroemulsji to
takŜe olej zdyspergowany w wodzie o/w i odwrotnie woda zdyspergowana w
oleju. Wielkość cząstek rozproszonych wynosi 5 - 200 nm i napięcie
międzyfazowe olej/woda jest bardzo niskie, 0,01 – 0,001 mN/m, co
‘kompensuje’ wzrost ∆G. Praca wykonana na obniŜenie napięcia
międzyfazowego związana jest ze wzrostem entropii ∆S układu związanej z
tworzeniem duŜej liczby małych kropelek. Warunki takie są spełnione gdy
surfaktant(y) tworzą ściśle upakowany film (warstewkę) na granicy faz
olej/woda. ∆G = ∆H – T∆S < 0
MIKROEMULSJE

Termin ‘mikroemulsje’ pierwszy raz uŜył w 1959 r. Jack H. Shulman, profesor
z Columbia University (Nowy Jork). Emulsje tego typu określa się takŜe jako:
przeźroczyste emulsje, powiększone micele, roztwory micelarne,
solubilizowane oleje (z ang. transparent emulsion, swollen micelle, micellar
solution, and solubilized oil). Termin ‘mikroemulsja’ jest mylący poniewaŜ
dotyczy jednej izotropowej fazy, która jest mieszaniną oleju, wody i
surfaktantu, lub jednej fazy (oleju bądź wody) będącej w nadmiarze i
równowadze z pozostałymi składnikami tej izotropowej fazy.
W takich trójskładnikowych układach jak mikroemulsje, gdzie dwie
niemieszające się fazy są obecne (woda i olej) z surfaktantem, cząsteczki
surfaktantu mogą tworzyć monowarstwę na granicy faz olej/woda, których
hydrofobowe łąńcuchy są „rozpuszczone” w fazie oleju a hydrofilowe
głowy w fazie wody.

Miniemulsja jest specjalnym typem emulsji.. Otrzymuje się je przez zdyspergowanie
dwu nierozpuszczalnych wzajemnie cieczy, surfaktantu i ko-surfaktantu. Typowym
przykładem jest tu heksadekan lub alkohol cetylowy.
W celu zdyspergowania stosuje się ultradźwięki lub wysokociśnieniowe
homogenizatory. W idealnej miniemulsji koalescencja i starzenie Ostwalda są
zminimalizowane poprzez obecność surfaktantu i ko-surfaktantu.
W tym procesie otrzymuje się stabilne kropelki o wymiarach pomiędzy 50 i 500 nm.
Taki proces otrzymywania miniemulsji szczególnie wykorzystuje się do wytwarzania
róŜnych nanomateriałów. Istnieje fundamentalna róŜnica pomiędzy tradycyjną
polimeryzacją emulsyjną (w emulsji) i polimeryzacją miniemulsyjną (w miniemulsji).
W przypadku emulsji cząstki (cząsteczki polimeru) tworzą się w wyniku micelarnej i
homogenicznej nukleacji, zaś w miniemulsji, cząstki tworzą się głównie poprzez
nukleację kropelek.
Źródło: http://en.wikipedia.org/wiki/Miniemulsion
MINIEMULSJE


















