
Struktura chromatyny rejonu - Strona Wydziału Biologii UW · Struktura chromatyny rejonu...
85
Praca magisterska Struktura chromatyny rejonu promotorowego genu CTA1 drożdży Saccharomyces cerevisiae Takao Ishikawa Uniwersytet Warszawski 2003
Transcript of Struktura chromatyny rejonu - Strona Wydziału Biologii UW · Struktura chromatyny rejonu...

Praca magisterska
Struktura chromatyny rejonu promotorowego genu CTA1 drożdży
Saccharomyces cerevisiae
Takao Ishikawa
Uniwersytet Warszawski
2003

Struktura chromatyny rejonu promotorowego genu CTA1 drożdży
Saccharomyces cerevisiae
Takao Ishikawa
Praca magisterska wykonana
pod kierunkiem dr. hab. Jana Fronka
Zakład Biologii Molekularnej Instytut Biochemii, Wydział Biologii
Uniwersytet Warszawski
2003

Serdecznie dziękuję
dr. hab. Janowi Fronkowi za opiekę, życzliwość, cierpliwość i zawsze konstruktywne rady
mgr Urszuli Bulkowskiej
dr Joannie Trzcińskiej-Danielewicz za życzliwość i praktyczne rady
Agacie Jacewicz
za nieocenioną pomoc w pracy laboratoryjnej
wszystkim pracownikom i studentom Zakładu Biologii Molekularnej
za stworzenie wspaniałej atmosfery pracy

Spis treści
1. Streszczenie...................................................................................................................1 2. Wstęp.........................................................................................................................2 3. Założenia i cel pracy................................................................................................13 4. Materiały i metody...................................................................................................14 Informacje ogólne....................................................................................................14 Szczepy Saccharomyces cerevisiae wykorzystywane do doświadczeń.....14 Pojemność stosowanych kolb Erlenmeyera i objętość hodowli........................15 Badanie aktywności promotora genu CTA1 za pomocą genu reporterowego.................16 Konstrukcja genu reporterowego oraz wektory.................................................16 Otrzymywanie DNA plazmidowego z bakterii.................................................17 Transformacja S.cerevisiae i selekcja transformantów......................................18 Pożywki użyte do regulacji aktywności genu CTA1.........................................19 Hodowla S.cerevisiae.........................................................................................19 Schemat doświadczenia.....................................................................................21 Sporządzenie ekstraktów...................................................................................22 Oznaczenie poziomu aktywności promotora genu CTA1..................................23 Oznaczanie stężenia białka w otrzymanym ekstrakcie........................23 Oznaczanie aktywności β-galaktozydazy............................................24 Obliczanie aktywności właściwej β-galaktozydazy............................25 Analiza statystyczna............................................................................26 Badanie struktury chromatyny rejonu promotora genu CTA1.......................................27 Pożywki użyte w części doświadczenia dotyczącej ustalania struktury chromatyny w rejonie promotora genu CTA1....................................................28 Hodowla S.cerevisiae.........................................................................................28 Izolacja jąder komórkowych S.cerevisiae i trawienie chromatyny nukleazą z micrococcus i DNazą I.......................................................................................30 Usuwanie komponentów białkowych z preparatu chromatyny i uzyskanie oczyszczonego DNA.........................................................................................33 Elektroforeza DNA w żelu agarozowym mająca na celu sprawdzenie jakości preparatu............................................................................................................34 Analiza produktów trawienia DNazy I i nukleazy z Micrococcus....................35 Trawienie endonukleazą restrykcyjną MboI......................................................35

Elektroforeza DNA w żelu agarozowym preparatów gotowych do przeniesienia na membranę......................................................................................................36 Przenoszenie DNA z żelu agarozowego na membranę.....................................36 Znakowanie sondy do hybrydyzacji..................................................................37 Hybrydyzacja typu Southern.............................................................................38 Autoradiografia..................................................................................................39 5. Wyniki.........................................................................................................................40 Aktywność transkrypcyjna promotora genu CTA1 i kinetyka indukcji............................40 Komórki z plazmidem episomalnym Hodowla w logarytmicznej fazie wzrostu.........................................................45 Komórki z plazmidem episomalnym Hodowla w fazie stacjonarnej............................................................................46 Komórki z plazmidem integracyjnym Hodowla w logarytmicznej fazie wzrostu.........................................................47 Komórki z plazmidem integracyjnym Hodowla w fazie stacjonarnej............................................................................48 Reorganizacja struktury chromatyny w rejonie promotora genu CTA1..........................51 Ustalenie miejsc wrażliwych na nukleazę z Micrococcus w szczepie kontrolnym (MHY501α)...................................................................................53 Analiza miejsc wrażliwych na nukleazę z Micrococcus w szczepie pip2.........56 Analiza miejsc wrażliwych na nukleazę z Micrococcus w szczepach adr1 i oaf1 w warunkach represji i derepresji genu CTA1...................................................58 Próba porównania położenia miejsc wrażliwych na nukleazę z Micrococcus w szczepach MHY501α, adr1, oaf1 i pip2 w warunkach derepresji genu CTA1..................................................................................................................60 Analiza miejsc nadwrażliwych na DNazę I w szczepie kontrolnym (MHY501α) i szczepie oaf1 w warunkach indukcji genu CTA1............................................61 6. Dyskusja.....................................................................................................................63 Aktywność transkrypcyjna promotora genu CTA1 i kinetyka indukcji..............63 Reorganizacja struktury chromatyny w rejonie promotora genu CTA1............69 7. Podsumowanie...........................................................................................................72 8. Literatura...................................................................................................................73

1
1. Streszczenie
Gen CTA1 kodujący katalazę A występującą w peroksysomach drożdży
Saccharomyces cerevisiae podlega trójpoziomowej regulacji ekspresji zależnej od
dostępnego źródła węgla. Ważną rolę w regulacji genu CTA1, a także innych genów
peroksysomalnych, odgrywają czynniki transkrypcyjne Oaf1p i Pip2p. Ich rola w
indukcji genów peroksysomalnych nie została jednak jednoznacznie wyjaśniona. W
derepresji, a prawdopodobnie także w indukcji genu CTA1 bierze również udział
czynnik transkrypcyjny Adr1p, regulujący transkrypcję wielu genów podlegających,
podobnie jak gen CTA1, represji glukozowej.
Badano wpływ wymienionych czynników transkrypcyjnych na ekspresję genu
CTA1 śledząc poziom indukcji genu CTA1 przy użyciu wprowadzonej do komórek
S.cerevisiae konstrukcji będącej fuzją obszaru promotora genu CTA1 z genem
reporterowym, a także analizując strukturę nukleosomową rejonu promotora badanego
genu w warunkach represji, derepresji i indukcji.
Przeprowadzone badania sugerują, iż istotną rolę w procesie indukcji genu
CTA1 pełni czynnik transkrypcyjny Oaf1p, natomiast czynniki transkrypcyjne Pip2p i
Adr1p wydają się nie pełnić istotnej roli w indukcji genu CTA1.
Uzyskane wyniki wskazują na subtelną reorganizację struktury chromatyny
zachodzącą podczas indukcji w rejonie promotora genu CTA1. Nie zaobserwowano
natomiast istotnych zmian struktury chromatyny w procesie derepresji genu CTA1. Jak
dotąd nie udało się jednoznacznie określić wpływu poszczególnych czynników
transkrypcyjnych na strukturę chromatyny rejonu promotora badanego genu.

2
2. Wstęp
W każdym organizmie żywym informacja genetyczna zawarta jest w kwasie
deoksyrybonukleinowym (DNA). We wszystkich organizmach jest odpowiedzialna za
rozmaite procesy życiowe, ponadto w organizmach wielokomórkowych odpowiada ona
także za proces rozwojowy, prowadzący do ukształtowania dojrzałego organizmu z
zygoty powstałej w wyniku zapłodnienia.
Organizmy żyjące dzieli się na prokarionty i eukarionty. Jedną z
najważniejszych cech różniących te dwie grupy jest rozmieszczenie DNA w komórce. U
prokariontów DNA znajduje się w cytoplazmie, w przeciwieństwie do eukariontów,
których materiał genetyczny znajduje się w jądrze komórkowym. Warto tutaj
wspomnieć, że komórka eukariotyczna jest tworem wysoce uporządkowanym.
Występuje w niej wiele wyspecjalizowanych struktur zbudowanych z błon lipidowych,
których funkcje są ściśle zdefiniowane. Można do nich zaliczyć m.in. jądro komórkowe
przechowujące materiał genetyczny, siateczkę śródplazmatyczną będącą m.in. miejscem
syntezy białek, aparat Golgiego odpowiedzialny za transport i sekrecję odpowiednich
białek i peroksysomy, w których przede wszystkim zachodzą procesy związane z
detoksykacją komórki.
Funkcje jądra komórkowego nie ograniczają się do fizycznej izolacji materiału
genetycznego. Jest to miejsce, w którym przebiega transkrypcja oraz różne procesy
prowadzące do powstania ostatecznej cząsteczki mRNA transportowanej następnie do
cytoplazmy, gdzie zachodzi translacja. Fakt, że istnieje bariera oddzielająca materiał
genetyczny wprowadza dodatkowy etap umożliwiający regulację wyrażania informacji
genetycznej.

3
DNA w jądrze komórki eukariotycznej występuje w skondensowanej formie
związany z silnie konserwowanymi ewolucyjnie białkami histonowymi, tworząc
chromatynę. Podstawową jednostką strukturalną chromatyny jest nukleosom,
zbudowany z oktameru histonowego składającego się z histonów H2A, H2B, H3 i H4
oraz fragmentu DNA o długości 146pz [1, 2]. Taki kompleks DNA i białek histonowych
kondensując dalej tworzy tzw. włókno 30nm będące kolejnym stopniem upakowania
materiału genetycznego. Włókna 30nm uważa się za najczęściej występującą formę
chromatyny w jądrze niedzielącej się komórki. Warto przy tym wspomnieć, że w
procesie kondensowania chromatyny bierze udział również histon H1, który wiąże się z
DNA łączącym poszczególne nukleosomy [3]. W dzielących się komórkach dalsza
kondensacja chromatyny ostatecznie prowadzi do powstania chromosomów
metafazowych będących najsilniej skondensowaną formą materiału genetycznego.
Histony H2A, H2B, H3 i H4 wchodzące w skład rdzenia nukleosomu mają
identyczny schemat budowy charakteryzujący się silnie zasadową częścią rdzeniową
tych białek oraz ogonami nie przyjmującymi konkretnych struktur przestrzennych.
Domena rdzeniowa histonów, ze względu na swoje właściwości fizyczne, bardzo
efektywnie oddziałuje z DNA umożliwiając wspomnianą wcześniej jego kondensację
[4]. Część ogonowa histonów ulega różnym modyfikacjom posttranslacyjnym:
acetylacji [5], fosforylacji [6], metylacji [7] i ubikwitynylacji [8, 9]. Uważa się, że
enzymy modyfikujące reszty aminokwasowe tego rejonu mają ułatwiony dostęp dzięki
brakowi zwartych i zdefiniowanych struktur przestrzennych. Modyfikacje
posttranslacyjne ogonów histonowych pełnią istotną rolę w regulacji stopnia
kondensacji chromatyny. Acetylacja powoduje obniżenie ładunku pozytywnego
histonów i osłabia oddziaływanie DNA z tymi białkami, a w konsekwencji powoduje

4
rozluźnienie struktury chromatyny [10]. Jednak modyfikacje te nie służą jedynie
regulacji struktury chromatyny. Hipoteza kodu histonowego zakłada, że pewne
kombinacje modyfikacji posttranslacyjnych mogą powodować selektywną indukcję lub
represję niektórych obszarów genoforu [11].
Niewątpliwie formowanie chromatyny służy kondensacji DNA, ale jej funkcje,
jak wcześniej wspomniano, nie ograniczają się jedynie do fizycznego zmniejszenia
długości nici DNA. Dzięki badaniom prowadzonym w ciągu ostatnich kilkunastu lat
okazało się, że chromatyna pełni także funkcje regulujące transkrypcję genów, ponadto
bierze ona udział w wielu procesach komórkowych związanych z DNA, takich jak
naprawa czy rekombinacja [12].
Często spotykany w ostatnich latach termin „epigenetyka” definiuje się jako
„dziedziczne zmiany ekspresji genów niezależne od sekwencji nukleotydowej DNA”
[13]. Mechanizmy epigenetycznej regulacji transkrypcji genów uważa się zatem za
niezależne w sposób bezpośredni od sekwencji nukleotydowej DNA. Głównymi
mechanizmami takiej regulacji transkrypcji genów jest metylacja DNA i
posttranslacyjne modyfikacje ogonów histonów powodujące reorganizację struktury
chromatyny [14]. Obszary DNA, które uległy metylacji są zwykle nieaktywne
transkrypcyjnie [13], znajduje to potwierdzenie również w przypadku wyciszenia
transkrypcji genów w jednej z kopii chromosomów X u samic ssaków [15]. Metylacja
DNA i modyfikacje ogonów histonów są ze sobą powiązane w słabo jak dotąd poznany
sposób. Istnieje jednak kilka przykładów wskazujących na związek tych zjawisk.
Jednym z nich jest Neurospora crassa. W organizmie tym stwierdzono, iż do metylacji
DNA konieczna jest metylacja określonych reszt aminokwasowych w obrębie ogonów
histonowych [16]. Istnieje również głęboki związek między metylacją DNA i

5
deacetylacją histonów. Badania wykazały, że metylacja DNA jest znacznikiem
rozpoznawanym przez deacetylazy histonów, które przeprowadzają deacetylację
ogonów histonów znajdujących się w nukleosomach w danym rejonie [17]. Powoduje
to kondensację DNA i ogólną represję transkrypcji genów. Natomiast regulacja
transkrypcji genów w drodze reorganizacji struktury chromatyny opiera się, najogólniej
rzecz biorąc, na przesuwaniu nukleosomów i modulowaniu dostępności określonych
sekwencji promotorowych dla czynników transkrypcyjnych i maszynerii
transkrypcyjnej. W tym przypadku, podobnie jak w przypadku metylacji, stwierdzono
głęboki związek z modyfikacjami posttranslacyjnymi ogonów histonowych. Okazało się
bowiem, że rekrutacja kompleksów reorganizujących strukturę chromatyny, takich jak
Swi/Snf, uzależniona jest od acetylacji ogonów histonowych przeprowadzonej przez
acetylazy histonowe [18, 19]. Wszystkie przedstawione przykłady wskazują na to, że
mechanizmy epigenetycznej regulacji transkrypcji genów mają silne powiązania.
Znanych jest wiele genów, których aktywność transkrypcji regulowana jest
przez zmianę rozmieszczenia nukleosomów w rejonie promotorowym. Pod tym
względem dokładnie poznano m.in. geny z grupy GAL (białka umożliwiające
wykorzystanie galaktozy) [20, 21] i PHO (fosfataza kwaśna oraz białka regulatorowe)
[22], gen ADH2 (dehydrogenaza alkoholowa II) [23, 24, 25] i gen TOP1
(topoizomeraza I) [26]. Wszystkie te badania zostały przeprowadzone na drożdżach
piekarniczych Saccharomyces cerevisiae. Jest to najprostszy dobrze poznany organizm
eukariotyczny, a łatwość manipulacji genetycznych i znajomość pełnej sekwencji
nukleotydowej jego genomu czyni go bardzo atrakcyjnym organizmem modelowym do
różnych badań. Ponadto posiada on wiele wspólnych cech z wyższymi organizmami
eukariotycznymi. Można wśród nich wymienić m.in. ogólne zasady organizacji

6
struktury chromatyny, modyfikacje posttranslacyjne histonów i obecność kompleksów
zmieniających strukturę chromatyny w sposób zależny od ATP [27]. Warto jednak
pamiętać o szczególnych cechach S.cerevisiae. Genom S.cerevisiae jest niezwykle mały
i prawie w całości aktywny transkrypcyjnie, podczas gdy w genomach wyższych
organizmów eukariotycznych jedynie niewielka jego część ulega transkrypcyji [28].
Niektóre obszary metabolizmu komórkowego również są nieco odmienne w
S.cerevisiae. Przykładem tego może być β-oksydacja, która zachodzi wyłącznie w
peroksysomach [29]. To organellum pełni więc istotną rolę w degradacji kwasów
tłuszczowych w komórkach drożdżowych, a dzięki temu S.cerevisiae staje się
atrakcyjnym organizmem do prowadzenia badań nad tymi procesami.
Podobnie jak w przypadku genów z grupy GAL i genu ADH2, geny
peroksysomalne są regulowane przez warunki środowiska zewnętrznego. Zarówno dla
genów z grupy GAL i genu ADH2, jak i dla genów peroksysomalnch jest nim źródło
węgla. Geny z grupy GAL ulegają transkrypcji, gdy w środowisku znajduje się
galaktoza [20], natomiast gen ADH2 jest aktywowany przez etanol lub brak glukozy
[23]. W obu przypadkach regulacja jest dwupoziomowa (represja i derepresja). Sytuacja
staje się bardziej skomplikowana w przypadku genów peroksysomalnych [30]. Kiedy w
środowisku jest dostatecznie wysokie stężenie glukozy, która jest dla S.cerevisiae
najdogodniejszym źródłem węgla, geny peroksysomalne ulegają represji [31]. Wynika
to z faktu, iż w warunkach optymalnych komórka nie przeprowadza biogenezy
peroksysomów. Derepresja tych genów jest powodowana obecnością związków
niefermentowalnych, takich jak etanol. W stanie derepresji geny te ulegają transkrypcji,
ale na bardzo niskim poziomie. Dopiero w środowisku bogatym w kwasy tłuszczowe
geny peroksysomalne ulegają pełnej indukcji [32]. Można zatem stwierdzić, że jest to

7
regulacja trójpoziomowa.
Geny peroksysomalne można podzielić na dwie grupy: geny kodujące białka
odpowiedzialne za proliferację peroksysomów i geny kodujące enzymy katalizujące
reakcje chemiczne zachodzące w peroksysomach. W przypadku S.cerevisiae do
pierwszej grupy zalicza się około 20 genów PEX [33]. Fakt, iż mutacje genów
ortologicznych u człowieka powodują choroby genetyczne, takie jak syndrom
Zellwegera [34], wyraźnie wskazuje na to, że są to geny kodujące białka odgrywające
istotne role w podstawowym metabolizmie komórki. Do drugiej grupy zalicza się takie
geny jak CTA1 (katalaza A) [35], POX1 (oksydaza acyloCoA), ECI1 (izomeraza
enoiloCoA), FOX2 (kompleks β-oksydacji) i FOX3 (tiolaza 3-oksoacyloCoA) [36].
Obok enzymów degradujących kwasy tłuszczowe, w peroksysomach występują także
enzymy przeprowadzające detoksykację komórki. Wśród nich szczególnie istotną rolę
wydaje się pełnić katalaza A kodowana przez gen CTA1, która jest odpowiedzialna za
utylizację cząsteczki H2O2 powstającej m.in. w procesie rozkładu kwasów tłuszczowych
w peroksysomach.
Transkrypcja genu CTA1 jest regulowana nie tylko przez źródło węgla, ale
także przez tlen oraz hem [37]. Zarówno hodowla S.cerevisiae w warunkach
beztlenowych, jak i brak hemu powodują znaczne obniżenie poziomu mRNA genu
katalazy A. Podobny sposób regulacji dotyczy także innych genów, ale w przypadku
genu CTA1 obecność hemu nie znosi zahamowania transkrypcji spowodowanego
hodowlą w warunkach beztlenowych. Jest to cecha wyróżniająca gen CTA1 spośród
innych genów regulowanych przez tlen i hem. Poziom ekspresji genu CTA1 nie zmienia
się także pomimo zmiany poziomu Hap1p, czynnika transkrypcyjnego zależnego od
hemu [38]. Jest to zaskakujące, ponieważ w rejonie promotorowym genu CTA1

8
stwierdzono obecność sekwencji wykazującej duży stopień podobieństwa do sekwencji
wiążącej Hap1p i można było się spodziewać wpływu tego białka na poziom ekspresji
genu CTA1 [35]. Wydaje się więc prawdopodobne, że istnieje niepoznany jeszcze
mechanizm regulujący transkrypcję genu CTA1 przy udziale tlenu i hemu.
Jak wcześniej wspomniano, regulacja transkrypcji genu CTA1 zależna od
dostępnego źródła węgla jest trójpoziomowa, a jej mechanizm jest dużo lepiej poznany
w porównaniu z mechanizmem regulacji przy udziale tlenu i hemu. Zidentyfikowane
zostały sekwencje nukleotydowe w rejonie promotora genu CTA1, z którymi wiążą się
odpowiednie białka regulujące transkrypcję tego genu, m.in. Oaf1p, Pip2p i Adr1p.
Funkcje niektórych z tych białek nie zostały jednak jednoznacznie określone.
Sekwencja nukelotydowa ORE Fragment sekwencji
promotora genu CTA1 CGGCTTTAACAAATATAAACTCCG
Konsensus CGGNNNTNA(N.....9 - 12)CCG
Tab. 2-1 Porównanie sekwencji ORE występującej w rejonie od -209 do -185 promotora genu CTA1 z
konsensusem tej sekwencji. Zaadaptowano z [39].
Charakterystyczną sekwencją nukleotydową występującą w rejonie
promotorów genów regulowanych przez kwasy tłuszczowe jest ORE (oleate response
element) [39]. Sekwencja ta odpowiedzialna jest za wiązanie białek aktywujących
transkrypcję wielu genów peroksysomalych, w tym także genu CTA1. W przypadku
tego genu, sekwencja ORE występuje w rejonie od -209 do -185 (numerem 1
oznaczono początek sekwencji nukleotydowej kodującej Cta1p) [35]. Tuż obok
znajduje się inna istotna sekwencja nukleotydowa biorąca udział w regulacji
transkrypcji genu CTA1. Jest to sekwencja UAS1 (upstream activating sequence type 1)

9
występująca w rejonie od -184 do -156 [40]. Jest ona miejscem wiązania białka
odpowiedzialnego za derepresję genu CTA1 [32].
Stosunkowo dobrze poznana została rola czynników transkrypcyjnych Oaf1p,
Pip2p i Adr1p w regulacji transkrypcji genu CTA1. Pip2p zostało opisane również jako
Oaf2p [41]. Porównanie Oaf1p i Pip2p wykazało około 40% homologię sekwencji
aminokwasowych, szczególnie wysoką w części N-końcowej tych białek, gdzie
znajdują się motywy „palców cynkowych” (Zn2Cys6) odpowiedzialne za wiązanie tych
białek z sekwencją ORE [42, 43, 44]. Białka te wydają się wiązać z sekwencją ORE
jako heterodimer Oaf1p-Pip2p [43, 45, 46], jednak istnieją badania sugerujące
możliwość wiązania się homodimeru (Oaf1p)2 z sekwencją ORE przy braku Pip2p [39,
45]. Niedobór Pip2p może być spowodowany różnicą w ekspresji genów kodujących te
białka. W przeciwieństwie do genu OAF1 wyrażanego konstytutywnie, transkrypcja
genu PIP2 jest aktywowana przez kwasy tłuszczowe, a w rejonie promotorowym genu
PIP2 występuje sekwencja ORE [45]. Niektóre prace sugerują jednak konieczność
występowania heterodimeru Oaf1p-Pip2p do indukcji genu CTA1 [43, 45, 46], tak więc
mechanizm aktywacji transkrypcji tego genu wymaga dalszych badań.
Adr1p jest czynnikiem transkrypcyjnym aktywującym transkrypcję genów
związanych z wykorzystywaniem niefermentowalnych źródeł węgla, takich jak etanol,
glicerol czy kwasy tłuszczowe, kiedy stężenie glukozy w pożywce jest zbyt niskie [31].
Pod jego kontrolą jest wiele genów kodujących enzymy związane z metabolizmem
niefermentowalnych źródeł węgla, w tym gen CTA1 [41]. Adr1p wiąże się z sekwencją
UAS1 występującą w rejonie od -184 do -156 genu CTA1 i pełni istotną rolę w
derepresji tego genu [32]. Niektóre prace sugerują także udział tego białka w indukcji
genu CTA1 [32, 47]. Istnieje jednak praca, która stwierdza brak oddziaływania między

10
Adr1p i sekwencją ORE [32]. Wydają się więc niezbędne dalsze badania mające na celu
wyjaśnienie mechanizmu ewentualnej indukcji genu CTA1 przez Adr1p§.
CTA1 ORE UAS1
Oaf1p P ip2p Adr1p
Oaf1p Oaf1p
lub
Indukcja Derepresja
?
-209 -185 -184 -156 +1
Ryc. 2-1 Schemat rejonu promotora genu CTA1. Zaznaczono istotne sekwencje nukleotydowe biorące udział w
regulacji tego genu oraz oddziałujące z nimi białka.
Regulacja genów związanych z wykorzystaniem niefermentowalnych źródeł
węgla może również być kontrolowana na wyższym poziomie. W procesie tym biorą
udział takie białka jak Snf1p i Glc7p [47, 48]. Snf1p jest kinazą białkową działającą
wydajnie w warunkach niskiego stężenia glukozy w komórce. Aktywna forma Snf1p
fosforyluje wcześniej wspomniany czynnik transkrypcyjny Adr1p i umożliwia jego
działanie jako aktywatora transkrypcji genów związanych z wykorzystywaniem
niefermentowalnych źródeł węgla. Glc7p zaś jest fosfatazą białkową aktywną w
warunkach wysokiego stężenia glukozy w komórce i działając na Adr1p uniemożliwia
§ W ostatnim czasie stwierdzono, że ekspresja genu PIP2 jest zależna od Adr1p [66]. W sugerowanej przez
niektórych autorów indukcji genu CTA1 przez Adr1p może więc pośredniczyć gen PIP2. Nie poznano jednak
ewentualnego mechanizmu indukcji genu CTA1 przez Adr1p.

11
jego działanie jako czynnika transkrypcyjnego [48]. Można stwierdzić, że Adr1p pełni
rolę swoistego „przełącznika” odpowiednich genów, w tym genu CTA1, włączającego
lub wyłączającego transkrypcję w zależności od stężenia glukozy w środowisku
zewnętrznym.
Istnieje inny, równie ważny mechanizm regulacji transkrypcji genów, który
najprawdopodobniej jest zaangażowany także w regulację transkrypcji genu CTA1. Jest
nim reorganizacja struktury chromatyny. Czasowe wyłączenie syntezy histonu H4 w
specjalnie skonstruowanym szczepie S.cerevisiae spowodowało aktywację transkrypcji
ok. 15% genów zawartych w genomie [49]. Jako przyczynę tego zjawiska podano
niemożność formowania poprawnej struktury chromatyny spowodowaną brakiem
histonu H4, a w konsekwencji ogólne rozluźnienie chromatyny. W ten sposób białka
inicjujące transkrypcję genów uzyskują łatwiejszy dostęp do istotnych w tym procesie
sekwencji nukleotydowych. Wśród genów, w których nastąpiła aktywacja transkrypcji
znajduje się również gen CTA1. Może to świadczyć o tym, że istnieje związek struktury
chromatyny i regulacji transkrypcji genu CTA1. Jednak rozluźnienie struktury
chromatyny może powodować również zmiany poziomu ekspresji innych genów,
których produkty wpływają na transkrypcję genu CTA1, tak więc może to być jedynie
efekt pośredni.
Dzięki stosunkowo dobrze poznanym wpływom czynników transkrypcyjnych
oraz prawdopodobnemu udziałowi struktury chromatyny w regulacji ekspresji genu
CTA1, gen ten wydaje się atrakcyjnym modelem do badań nad wpływem struktury
chromatyny na ekspresję genów. Pełne poznanie mechanizmów regulacji transkrypcji
genów w komórkach S.cerevisiae powinno ułatwić także zrozumienie podobnych
procesów w innych organizmach. Dzięki temu możliwe będzie dokładne poznanie

12
mechanizmów rozwoju organizmu, przyczyn wielu chorób, metod ich leczenia i innych
zagadnień związanych z wyrażaniem informacji genetycznej.

13
3. Założenia i cel pracy
Aktywność transkrypcyjna promotora genu CTA1 regulowana jest w zależności
od dostępnego źródła węgla. Mechanizm ten został dość dobrze poznany, jednak rola
niektórych czynników transkrypcyjnych biorących udział w regulacji transkrypcji genu
CTA1 nie została jednoznacznie wyjaśniona. Nie została także poznana kinetyka
indukcji genu CTA1.
Prawdopodobna natomiast jest regulacja aktywności transkrypcji genu CTA1
przez reorganizację struktury chromatyny. Jednak do tej pory nie zostały
przeprowadzone badania mające na celu stwierdzenie udziału tego mechanizmu w
regulacji transkrypcji genu CTA1.
Celem tej pracy było:
• Ustalenie wpływu nieobecności czynników transkrypcyjnych Adr1p, Oaf1p i Pip2p
na regulację aktywności transkrypcyjnej genu CTA1.
• Zbadanie kinetyki indukcji genu CTA1.
• Wykazanie udziału reorganizacji struktury chromatyny w regulacji aktywności
transkrypcyjnej genu CTA1 oraz wpływu czynników transkrypcyjnych Adr1p,
Oaf1p i Pip2p na reorganizację struktury chromatyny.

14
4. Materiały i metody
Informacje ogólne
Jeżeli nie zaznaczono inaczej, stosowano odczynniki cz.d.a. z firm: Sigma,
POCh i Merck.
Szczepy Saccharomyces cerevisiae wykorzystywane do doświadczeń
Symbol szczepu
Symbol usuniętej
ORF
Nazwa usuniętego
genu Genotyp
MHY501α MATα; his3-∆200; leu2-3; 112 ura3-52 trp1-1 BY4742 MATα; his3∆1; leu2∆0; lys2∆0; ura3∆0 Y10355
oaf1 YAL051w OAF1
BY4739; MATα; leu2∆0; lys2∆0; ura3∆0; YAL051w::kanMX4
Y11660 pip2
YOR363c PIP2 BY4742; MATα; his3∆1; leu2∆0; lys2∆0; ura3∆0; YOR363c::kanMX4
Y13575 adr1
YDR216w ADR1 BY4742; MATα; his3∆1; leu2∆0; lys2∆0; ura3∆0; YDR216w::kanMX4
Wszystkie wyżej wymienione szczepy pochodzą z EUROSCARF (European
Saccharomyces cerevisiae archive for functional analysis) z wyjątkiem szczepu
MHY501α, który został opisany w [50].

15
Pojemność stosowanych kolb Erlenmeyera i objętość hodowli
Pojemność kolby Objętość hodowli 100ml 25ml 250ml 50ml lub 75ml 2000ml 500ml

16
Badanie aktywności promotora genu CTA1 za pomocą genu
reporterowego
Badanie aktywności promotora genu CTA1 prowadzono przy użyciu
wprowadzonej do komórek S.cervisiae konstrukcji będącej fuzją natywnego obszaru
promotora genu CTA1 z genem kodującym β-galaktozydazę. Po sporządzeniu ekstraktu
z odpowiednich hodowli S.cerevisiae oznaczano zawartość białka oraz aktywność
β-galaktozydazy niżej opisanymi metodami. Obliczona w sposób niżej opisany
aktywność właściwa β-galaktozydazy jest pośrednią miarą aktywności transkrypcyjnej
promotora genu CTA1.
Konstrukcja genu reporterowego oraz wektory
Doświadczenie prowadzono przy użyciu genu reporterowego będącego fuzją
obszaru promotorowego genu CTA1 z S.cerevisiae (sekwencja nukleotydowa od -819 do
+3, numerem 1 oznaczono początek sekwencji nukleotydowej kodującej Cta1p) i
sekwencji genu lacZ z Escherichia coli kodującej β-galaktozydazę. Ten gen fuzyjny
został wprowadzony na wektor episomalny i integracyjny. Wektor episomalny,
pochodna plazmidu YEp357 [51], niesie ori 2µ i dzięki temu występuje w komórce w
wielu kopiach. Wektor integracyjny zaś występuje w komórce w jednej kopii i jest
pochodną plazmidu YIp357, który integruje z genomem w locus URA3 [51]. Oba
plazmidy niosą marker uracylowy przydatny w selekcji transformantów drożdżowych,
ponadto oba plazmidy są bifunkcyjne, funkcjonują zarówno w komórkach S.cerevisiae

17
jak i E.coli. Plazmidy te zostały zaprojektowane, wykonane i udostępnione przez dr. M.
Skonecznego z Instytutu Biochemii i Biofizyki PAN w Warszawie. Plazmid episomalny
i integracyjny nazwano odpowiednio p149 i p150.
Otrzymywanie DNA plazmidowego z bakterii
Plazmidy udostępnione przez dr. M. Skonecznego znajdowały się w
komórkach E.coli XL-1 Blue MRF’ (∆(mcrA)183 ∆(mcrCB-hsdSMR-mrr)173 endA1
supE44 thi-1 recA1 gyrA96 relA1 lac [F’ proAB lacIqZ∆M15 Tn10 (Tetr)]). Plazmidowy
DNA został wyizolowany metodą lizy alkalicznej [52]. Dodatkowo do buforu, w
którym zawieszano bakterie, dodano RNazęA (Sigma) do końcowego stężenia
0,5mg/ml.
W celu identyfikacji otrzymanych plazmidów część wyizolowanego preparatu
poddano trawieniu endonukleazami restrykcyjnymi EcoRI/BamHI i ApaI/BamHI
(Fermentas), a następnie przeprowadzono elektroforezę w 0,8% żelu agarozowym.
Uzyskano fragmenty DNA zgodne z oczekiwanymi na podstawie map restrykcyjnych
tych plazmidów (elektroforetogramów nie przedstawiono).
Plazmid integracyjny został poddany trawieniu endonukleazą restrykcyjną
ApaI (Fermentas), której jedyne miejsce rozpoznawane znajduje się w obrębie genu
URA3. W ten sposób plazmid integracyjny został przygotowany do transformacji i
integracji z genomem S.cerevisiae w locus genu URA3.

18
Transformacja S.cerevisiae i selekcja transformantów
W celu wprowadzenia plazmidów p149 i p150 do wszystkich wyżej
wymienionych szczepów S.cerevisiae, zastosowano metodę wysokowydajnej
transformacji drożdży [53]. S.cerevisiae szczepiono na pożywkę YPD2% (1% ekstrakt
drożdżowy, 1% pepton, 2% glukoza) i inkubowano w 300C z wytrząsaniem
(250obr./min.) przez noc. Po sprawdzeniu gęstości hodowli (liczenie komórek w
komorze Bürkera), przeszczepiano odpowiednią objętość hodowli w ten sposób, aby w
nowej pożywce YPD2% (obj. 50ml) gęstość hodowli wynosiła 5·106komórek/ml.
Prowadzono hodowlę w 300C z wytrząsaniem (250obr./min.) do momentu uzyskania
gęstości hodowli 2·107komórek/ml. Hodowlę wirowano następnie przy 3000g przez
5min., płukano wodą i zawieszano w 1,0ml 100mM octanu litu. Po wirowaniu w
mikrowirówce i usunięciu supernatantu zawieszano osad komórek w 100mM octanu
litu tak, aby objętość końcowa wynosiła 500µl. Do 50µl w ten sposób przygotowanych
komórek S.cerevisiae dodawano 350µl buforu do transformacji (240µl PEG3350 (50%
w/v), 36µl 1M octanu litu, 15µl ssDNA (10mg/ml), 59µl wody) oraz ok. 1µg DNA
plazmidowego. Po dokładnym wymieszaniu mieszaninę inkubowano w 300C przez
30min., a następnie poddawano szokowi cieplnemu w 420C przez 30min. Poprzez
wirowanie w mikrowirówce usuwano bufor do transformacji, a osad komórek
zawieszano w 300µl wody. Całość zawiesiny komórek wysiewano na szalkę z pożywką
ω02% (0,67% YNB (zestaw soli mineralnych i witamin, ang. Yeast Nitrogen Base
without Aminoacids, Difco), 2% glukoza) zawierającą odpowiednie uzupełnienia
aminokwasów (10-50µg/ml), ale bez uracylu, ponieważ selekcja transformantów
opierała się na zniesieniu wymagania uracylu do wzrostu.

19
Pożywki użyte do regulacji aktywności genu CTA1
Represja Derepresja Indukcja ω010% ω0EtOH ω0Kw.Ol. 0,67% YNB 10% glukoza
0,67% YNB 0,5% glukoza 2% etanol
0,67% YNB 0,5% kwas olejowy
Hodowla S.cerevisiae
Dzień 1
Odpowiedni szczep drożdży S.cerevisiae szczepiono na pożywkę ω02% (obj.
50ml) i hodowano w 300C z wytrząsaniem (250obr./min.) przez noc do osiągnięcia
wartości OD600=2,5-3,0 (fazy stacjonarnej).
Dzień 2
Z hodowli założonej w dniu poprzednim szczepiono do pożywek ω010% (obj.
50ml) i ω0EtOH (dwie hodowle o obj. 75ml), odpowiednio po 100-400µl i 1,0-6,0ml
hodowli. Zaszczepione pożywki umieszczano w takich samych warunkach. Hodowle
prowadzono przez noc.
Dzień 3
Hodowle z poprzedniego dnia prowadzono do momentu osiągnięcia wartości
OD600=1,5 (logarytmiczna faza wzrostu) i OD600=2,5 (faza stacjonarna). Po osiągnięciu
planowanych wartości OD600, z hodowli prowadzonych w pożywce ω010% sporządzano

20
ekstrakt (w sposób niżej opisany), natomiast dwie identyczne hodowle prowadzone w
pożywce ω0EtOH o objętości 75ml w pożywce mieszano ze sobą uzyskując w ten
sposób 150ml hodowli. Po rozdzieleniu hodowli na 6 porcji po 25ml, poddawano je
wirowaniu (5000g, 2min.), następnie po usunięciu pożywki w sterylnych warunkach
przenoszono komórki do pożywki ω0Kw.Ol w porcjach po 25ml. Z jednej z sześciu tak
przeszczepionych hodowli sporządzano ekstrakt natychmiast po przeszczepieniu, a
pozostałe pięć hodowli umieszczano w 300C i prowadzono hodowlę z wytrząsaniem
(250obr./min.) przez 3, 6, 9, 12 i 24 godziny. Po hodowli prowadzonej przez określony
czas sporządzano ekstrakt.
Hodowle mające na celu indukcję genu CTA1 przeszczepiano po osiągnięciu
planowanej wartości OD600 w pożywce ω0EtOH, ponieważ komórki S.cerevisiae nie
dzielą się w pożywce ω0Kw.Ol.
Opisana procedura oraz schemat hodowli S.cerevisiae (Ryc. 4-1) dotyczą
pełnego doświadczenia mającego na celu zbadanie zarówno procesu indukcji, jak i
procesu derepresji genu CTA1. W dalszej części pracy zostaną jednak przedstawione
wyłącznie wyniki dotyczące procesu indukcji badanego genu.

21
Ryc. 4-1 Schemat hodowli S.cerevisiae prowadzonych w celu sporządzania ekstraktów drożdżowych, w których
następnie oznaczano aktywność promotora genu CTA1 mierząc aktywność β-galaktozydazy. Linią przerywaną
zaznaczono część doświadczenia, której wyniki zostaną przedstawione w dalszej części pracy.
ω02%
50ml
ω010% 50ml
do OD600=1,5
ω010% 50ml
do fazy
stacjonarnej
ω0EtOH 2 x 75ml
do OD600=1,5
ω0EtOH 2 x 75ml
do fazy stac.
ω0Kw.Ol. 6 x 25ml
ω0Kw.Ol. 6 x 25ml
Ekstrakt
Ekstrakt po
hodowli
przez 3, 6,
9,12 i 24h
Zaraz po
przeszczep.
ekstrakt
Zaraz po
przeszczep.
ekstrakt
Ekstrakt po
hodowli
przez 3, 6,
9,12 i 24h
Ekstrakt

22
Sporządzenie ekstraktów
Hodowlę poddawano wirowaniu przy 5000g przez 2min., w 40C. Osad
komórek uzyskany w wyniku wirowania zawieszano w 25ml sterylnej wody w celu
usunięcia resztek pożywki. Komórki następnie poddawano wirowaniu jak wcześniej, a
uzyskany osad komórek zawieszano w 400µl (dla hodowli o obj. 25ml) lub 800µl (dla
hodowli o obj. 50ml) 50mM buforu potasowo-fosforanowego (50mM KPi pH7,0). Do
zawiesiny dodawano równą objętość szklanych kulek (∅0,45-0,50mm), a następnie
zamrażano w -200C. Na tym etapie przechowywano uzyskane komórki od kilku dni do
miesiąca. Po rozmrożeniu w temperaturze pokojowej komórki rozbijano przez
sześciominutowe wytrząsanie na mikrowstrząsarce typu vortex w temperaturze 40C. W
celu usunięcia nierozbitych komórek i reszt ścian komórkowych, uzyskany homogenat
poddawano wirowaniu przy 10000g przez 5min. w 40C. Otrzymaną w supernatancie
frakcję cytoplazmatyczną wykorzystywano bezpośrednio do oznaczeń stężenia białka
oraz aktywności β-galaktozydazy.
Stosowane podczas sporządzania ekstraktów szklane kulki płukano przed
użyciem w stężonym kwasie solnym. Po ok. 4 godzinach przemywano wodą
doprowadzając do pH7, a następnie suszono przez 3 godziny w 1800C.

23
Oznaczenie poziomu aktywności promotora genu CTA1
A. Oznaczanie stężenia białka w otrzymanym ekstrakcie
Oznaczanie przeprowadzano metodą Bradford [54], zgodnie z niniejszym
schematem. Do oznaczeń używano odczynnika Bradford z firmy Sigma.
Próba
kontrolna 1ml H2O 1ml odczynnika
Bradford Próba
oznaczana 990µl H2O + 10µl ekstraktu rozcieńczonego
2x lub 5x w 50mM KPi 1ml odczynnika
Bradford
Po dodaniu odczynnika Bradford próby pozostawiano na 10min., a następnie
oznaczano spektrofotometrycznie przy długości fali 595nm. Rozcieńczenia
ekstraktu dobierano tak, aby odczyt spektrofotometryczny mieścił się w przedziale
0,2-0,7. Każdy ekstrakt oznaczano w dwóch powtórzeniach, a stężenie białka w
ekstrakcie obliczano na podstawie uśrednionych wartości z dwóch pomiarów.
Podstawę obliczeń stężenia białka stanowiła krzywa wzorcowa wykonana przy
użyciu roztworu albuminy o stężeniu 1mg/ml. Roztwór ten rozcieńczano wodą tak,
aby uzyskać stężenia albuminy 1,67µg/ml, 3,33µg/ml, 5,0µg/ml, 10µg/ml, 15µg/ml i
20µg/ml. Wszystkie wymienione roztwory albuminy oznaczano w dwóch
powtórzeniach i odczyty uśredniano, podobnie jak w przypadku oznaczenia stężenia
białka w próbach. Dla każdej nowej butelki odczynnika Bradford sporządzano nową
krzywą wzorcową.

24
B. Oznaczanie aktywności β-galaktozydazy
Oznaczanie aktywności β-galaktozydazy prowadzono wg [55] przy użyciu
o-nitrofenylogalaktopiranozydu (ONPG). Związek ten rozkładany jest przez
β-galaktozydazę na galaktozę i barwny o-nitrofenol.
Do 800µl buforu Z (60mM NaH2PO4, 40mM Na2HPO4, 10mM KCl, 1mM
MgSO4, pH7,0) dodawano 50µl ekstraktu odpowiednio rozcieńczonego w 50mM
KPi. W próbie kontrolnej zamiast ekstraktu dodawano 50mM KPi. Przygotowaną
próbkę inkubowano w 300C przez 5min. Następnie dodawano 160µl 0,4% ONPG
(substrat do reakcji, rozpuszczony w 50mM Tris-HCl, pH8) i inkubowano w takich
samych warunkach, mierząc czas inkubacji, do uzyskania widocznego gołym okiem
jasno żółtego zabarwienia. Reakcję zatrzymywano przez dodanie 400µl 1M węglanu
sodu i umieszczano w lodzie.
Oznaczanie zawartości o-nitrofenolu prowadzono spektrofotometrycznie przy
długości fali 420nm.
Oznaczenie aktywności β-galaktozydazy dla każdego ekstraktu prowadzono w
dwóch powtórzeniach. Następnie wyniki pomiaru spektrofotometrycznego
uśredniano w celu prowadzenia dalszych obliczeń. Czas trwania reakcji wahał się od
3 do 46min., natomiast rozcieńczenia ekstraktów od 1 do 500 razy. Różnice
zarówno w czasie reakcji, jak i w rozcieńczeniu ekstraktu wynikają z poziomu
aktywności β-galaktozydazy w poszczególnych ekstraktach.
Reakcja ta jest liniowa w czasie i w stosunku do rozcieńczeń ekstraktu [56].

25
C. Obliczanie aktywności właściwej β-galaktozydazy
Aktywność właściwą β-galaktozydazy obliczano stosując następujący wzór
matematyczny:
tvpluAx
⋅⋅⋅⋅⋅
=ε
gdzie:
x aktywność właściwa β-galaktozydazy [µmol·mg-1·min-1]
A wartość absorbancji przy długości fali 420nm
u całkowita objętość, w której prowadzono reakcję. W stosowanym
układzie doświadczalnym wartość ta wynosiła 1,41 [ml]
ε współczynnik absorbancji o-nitrofenolu przy długości fali 420nm.
Wartość współczynnika wynosi 0,0045 [µM-1·cm-1]
l grubość kuwety. W doświadczeniu używano kuwet o grubości 1 [cm]
p stężenie białka w ekstrakcie [mg/ml]
v objętość nierozcieńczonego ekstraktu dodana do reakcji [ml]
t czas trwania reakcji [min.]

26
Na podstawie otrzymanych wyników dla każdego szczepu, w logarytmicznej
fazie wzrostu oraz w fazie stacjonarnej, obliczano względny poziom indukcji
promotora genu CTA1, wyrażany następującym wzorem:
0h) (Kw.Ol. EtOH
xhKw.Ol.
Akt.Akt.
gdzie:
Akt.Kw.Ol. xh aktywność β-galaktozydazy po x godzinach hodowli w
warunkach indukcji (gdzie x = 3, 6, 9, 12 lub 24 godziny)
Akt.EtOH (Kw.Ol. 0h) aktywność β-galaktozydazy w warunkach derepresji (wartości
uzyskane dla ekstraktów sporządzanych z komórek tuż po ich
przeniesieniu do pożywki ω0Kw.Ol.)
D. Analiza statystyczna
W celu sprawdzenia istotności statystycznej różnic poziomu indukcji genu
CTA1 między szczepem kontrolnym i szczepami badanymi przeprowadzono test
sprawdzający równość wariancji oraz dwustronny test t Studenta różnic między
średnimi przy różnych wariancjach. W obu testach za próg istotności statystycznej
przyjęto wartość p=0,055.

27
Badanie struktury chromatyny rejonu promotora genu CTA1
Przy badaniu struktury chromatyny rejonu promotora genu CTA1 stosowano
metodę pośredniego znakowania końców [57, 58, 59]. Wyizolowane jądra komórkowe
poddawano trawieniu nukleazą z Micrococcus i DNazą I o różnych stężeniach. Po
zakończeniu reakcji odbiałczano DNA i przecinano wyczerpująco odpowiednio dobraną
endonukleazą restrykcyjną. Wybrano endonukleazę restrykcyjną, która nie ma miejsca
cięcia w obszarze, którego strukturę chromatyny analizowano, ale tnącą DNA w pobliżu
tego obszaru. W przypadku genu CTA1 dogodną endonukleazą restrykcyjną jest MboI,
która ma miejsce cięcia zarówno po stronie 5', jak i 3' obszaru promotorowego CTA1.
Po trawieniu enzymem restrykcyjnym DNA poddaje się elektroforezie w żelu
agarozowym, przenosi na filtr i po denaturacji hybrydyzuje z krótką, wyznakowaną
radioaktywnie sondą o sekwencji nukleotydowej odpowiadającej sekwencji
zlokalizowanej blisko miejsca działania użytej endonukleazy restrykcyjnej. Z sondą
hybrydyzują odcinki DNA o różnej długości, jednak jeden ich koniec jest zawsze ten
sam, powstały w wyniku trawienia endonukleazy restrykcyjnej. Natomiast drugi koniec
powstaje w wyniku trawienia nukleazą z Micrococcus lub DNazą I. Miejscem działania
nukleazy z Micrococcus jest DNA znajdujący się między nukleosomami, nie chroniony
przez oktamery histonowe. Zatem długości hybrydyzujących odcinków określają
odległość miejsc wrażliwych na użytą nukleazę od miejsca cięcia MboI, a co za tym
idzie, rozmieszczenie nukleosomów w badanym rejonie chromatyny. W przypadku
działania DNazą I można zidentyfikować tzw. miejsca nadwrażliwe. Są one jednak
często w innym miejscu niż miejsca wrażliwe na nukleazę z Micrococcus. Substratem
dla DNazy I są miejsca na nici DNA o zmienionej strukturze przestrzennej. DNaza I

28
atakuje miejsca te nawet wtedy, gdy znajdują się one na nukleosomach. Interesujący jest
również fakt, iż miejsca te są charakterystyczne dla obszarów promotorowych i często
odpowiadają miejscom wiązania czynników transkrypcyjnych [60, 61].
Pożywki użyte w części doświadczenia dotyczącej ustalania struktury
chromatyny w rejonie promotora genu CTA1
Hodowlę mającą na celu odświeżenie i namnożenie komórek S.cerevisiae
prowadzono w pożywce pełnej YPD2%, następnie hodowle przeszczepiano na pożywki
zapewniające regulację aktywności promotora genu CTA1.
Represja Derepresja Indukcja YPD10% YPDE YPO 1% ekstrakt drożdżowy 1% pepton 10% glukoza
1% ekstrakt drożdżowy 1% pepton 0,5% glukoza 2% etanol
1% ekstrakt drożdżowy 1% pepton 0,5% kwas olejowy
Hodowla S.cerevisiae
Dzień 1
Odpowiedni szczep drożdży S.cerevisiae szczepiono na pożywkę YPD2% (obj.
25ml) i hodowano w 300C z wytrząsaniem (250obr./min.) przez noc do osiągnięcia
wartości OD600=9,0-10,0 (fazy stacjonarnej).

29
Dzień 2
Niewielką objętość hodowli (300µl-900µl) założonej poprzedniego dnia
szczepiono do trzech kolb z pożywką YPD10% (represja) i sześciu kolb z pożywką
YPDE (derepresja i indukcja) o objętości 500ml. Pożywki te uzupełniano
streptomycyną i ampicyliną (odpowiednio 40µg/ml i 30µg/ml). Zaszczepione pożywki
umieszczano w takich samych warunkach i prowadzono hodowle przez noc.
Dzień 3
Hodowlę z poprzedniego dnia prowadzono do momentu osiągnięcia OD600=2,5
(logarytmiczna faza wzrostu). Po osiągnięciu planowanej wartości OD600 hodowle z
trzech kolb z jednakową pożywką łączono, a następnie prowadzono izolację jąder
komórkowych (represja i derepresja) lub przenoszono do pożywki YPO (indukcja). W
tym przypadku poddawano hodowlę wirowaniu przy 3000g przez 5min. w temperaturze
pokojowej. Następnie osad komórek przenoszono sterylnie do trzech kolb z pożywką
YPO (obj. 500ml). Hodowlę prowadzono w 300C z wytrząsaniem (250obr./min.) przez
noc.
Dzień 4 (dotyczy tylko hodowli prowadzonej w warunkach indukcji genu CTA1)
Po trwającej 24 godziny hodowli w pożywce YPO rozpoczynano izolację jąder
komórkowych.
Hodowlę mającą na celu indukcję genu CTA1 przeszczepiano po osiągnięciu
planowanej wartości OD600 w pożywce YPDE, ponieważ komórki S.cerevisiae nie
dzielą się w pożywce YPO.

30
Wartości OD600 wskazujące na osiągnięcie przez hodowlę odpowiedniej fazy
wzrostu różnią się w zależności od stosowanej pożywki. Przy hodowli prowadzonej w
pożywce minimalnej ω0 (stosowana w części dotyczącej oznaczenia aktywności
właściwej β-galaktozydazy) wartość OD600=2,5 odpowiada najczęściej fazie
stacjonarnej, w pożywce pełnej YPD wartość ta jednak odpowiada logarytmicznej fazie
wzrostu.
Izolacja jąder komórkowych S.cerevisiae i trawienie chromatyny
nukleazą z Micrococcus i DNazą I
Izolację jąder przeprowadzano metodą wirowania w gradiencie gęstości fikolu
lizatu sferoplastów otrzymanych w wyniku traktowania komórek drożdży zymoliazą
[22]. Hodowlę o odpowiedniej wartości OD600 poddawano wirowaniu przy 3000g przez
5min. w temperaturze pokojowej. Osad komórek zawieszano w równej objętości wody*
i wirowano w takich samych warunkach. Następnie osad zawieszano w 60ml roztworu
1* (20mM EDTA pH7,4, 0,7M β-merkaptoetanol) i umieszczano w 300C. Po inkubacji
trwającej 30min. wirowano zawiesinę komórek przy 3000g przez 5min. w temperaturze
pokojowej. Po zawieszeniu osadu komórek w 50ml 1M sorbitolu*, wirowano jak
wcześniej. Uzyskany osad komórek zawieszano w 20ml roztworu 2* (1M sorbitol, 5mM
β-merkaptoetanol) i przenoszono do kolbki o pojemności 100ml, którą umieszczano w
* Symbolem tym oznaczono roztwory, do których dodawano glukozę do stężenia 5% (w/v) w przypadku
otrzymywania jąder komórkowych z hodowli prowadzonych w pożywce YPD10%. Dodatek ten służył utrzymaniu
stanu represji glukozowej.

31
inkubatorze z wytrząsaniem (300C, 200obr./min.). Następnie dodawano 10-20mg
zymoliazy 100T z Arthrobacter luteus (Seikagaku) zawieszonej w 1ml 50mM KPi
pH7,5 i prowadzono inkubację przez ok. 35min. Stopień strawienia ściany komórkowej
S.cerevisiae oceniano na podstawie obserwacji w mikroskopie świetlnym wykorzystując
właściwości sferoplastów (komórek pozbawionych ściany komórkowej), które
rozpadają się w wodzie na skutek ciśnienia osmotycznego. Po pobraniu niewielkiej
objętości z zawiesiny komórek umieszczano ją w wodzie oraz 1M sorbitolu i
porównywano obraz obu zawiesin. Po stwierdzeniu braku sferoplastów w wodzie
przystępowano do dalszego etapu procedury. W zależności od stopnia strawienia ściany
komórkowej, czas trawienia zymoliazą 100T przedłużano do półtorej godziny
prowadząc obserwację mikroskopową co 15min.
Procedurę otrzymania jąder komórkowych z S.cerevisiae po uzyskaniu
sferoplastów prowadzono w 40C. Sferoplasty wirowano przy 3600g przez 10min.,
uzyskany osad zawieszano w 15ml 1M sorbitolu*. Zawiesinę poddawano wirowaniu w
takich samych warunkach, uzyskując osad sferoplastów gotowych do lizy. Sferoplasty
zawieszano w 100ml roztworu do lizy* (18% ficoll 400 (Pharmacia Fine Chemicals),
20mM KPi pH6,8, 0,25mM EGTA, 0,25mM EDTA, 1mM MgCl2, 1mM PMSF) i
homogenizowano ręcznie w lodzie przy pomocy homogenizatora typu
Pottera-Elvehjema. Homogenat poddawano wirowaniu przy 2500g przez 5min.,
następnie supernatant przenoszono do nowych probówek i wirowano przy 30000g przez
25min (15749obr./min. w wirówce Beckman J-30I Avanti z rotorem JA30.50Ti).
Połowę uzyskanego w ten sposób osadu jąder S. cerevisiae zawieszano w 15ml buforu
do DNazyI* (15mM Tris pH7,4, 75mM NaCl, 3mM MgCl2, 0,05mM CaCl2, 1mM
β-merkaptoetanol), a drugą połowę w 15ml buforu do nuklezay z Micrococcus* (15mM

32
Tris pH8,0, 50mM NaCl, 1,4mM CaCl2, 0,2mM EGTA, 0,2mM EDTA, 5mM
β-merkaptoetanol). Zawiesinę jąder wirowano przy 2500g przez 5min. Osad jąder
zawieszano ponownie w 15ml odpowiedniego buforu i wirowano w takich samych
warunkach. Uzyskany w ten sposób osad jąder zawieszano w 2ml odpowiedniego
buforu. Następnie w celu ustalenia zawartości DNA w otrzymanych preparatach
wprowadzano po 5µl zawiesiny jąder do 2ml roztworu do pomiaru stężenia DNA (1%
SDS, 150mM NaCl, 1mM EDTA, 10mM Tris-HCl, pH7,5). Zawartość DNA w
preparatach ustalano spektrofotometrycznie przy długości fali 260nm. W przypadku
uzyskania odczytów większych niż A260=240 dla nierozcieńczonych preparatów
(A260=0,6 dla preparatu 400 razy rozcieńczonego przy pomiarze) rozcieńczano je
odpowiednim buforem do wartości A260=240, natomiast przy odczytach większych niż
A260=24 dla nierozcieńczonych preparatów (A260=0,06 dla preparatu 400 razy
rozcieńczonego przy pomiarze) rozcieńczano je odpowiednim buforem do wartości
A260=24 (procedura ustalona empirycznie przez dr Joannę Trzcińską-Danielewicz).
Rozcieńczone preparaty dzielono na 6 porcji i poddawano trawieniu DNazą I
(Calbiochem, stężenie końcowe 0,01-2,0U/ml z roztworu 2,2U/µl w wodzie) i nukleazą
z Micrococcus (Sigma, stężenie końcowe 0,00033-2,43U/ml z roztworu 0,2U/µl w
50mM Tris-HCl pH8,0, 0,05mM CaCl2, 20% glicerol) w 370C przez 20min. Reakcję
zatrzymywano przez dodanie buforu STOP 5x (12,5mM Tris pH7,5, 0,375M NaCl,
37,5mM EDTA, 0,75% SDS) do końcowego stężenia 1x. Do preparatów dodawano
proteinazę K (Sigma) do stężenia końcowego 5,0-10µg/ml i inkubowano przez noc w
370C.

33
W stosowanej metodzie bardzo trudno było przewidzieć zawartość DNA w
uzyskanym preparacie na podstawie pomiarów wartości A260 oraz wynikających z nich
obliczeń, ponieważ zaobserwowano brak zależności liniowej między wartością A260 a
zawartością DNA w preparacie. Na podstawie licznych doświadczeń stwierdzono, że
przyjęty sposób postępowania zapewnia poprawną ocenę zawartości DNA w preparacie
i dlatego stosowano go w celu uzyskania odpowiedniego stężenia DNA. Ponadto
dzielono preparaty na 6 porcji, które następnie poddawano działaniu DNazy I lub
nukleazy z Micrococcus o różnym stężeniu. Umożliwiało to późniejszy wybór
preparatów o odpowiednim stopniu strawienia, które poddawano dalszej analizie.
Usuwanie komponentów białkowych z preparatu chromatyny i
uzyskanie oczyszczonego DNA
Do preparatów po trawieniu proteinazą K dodawano równą objętości fenolu
nasyconego 1M Tris-HCl pH8,0. Po energicznym wstrząsaniu wirowano w
mikrowirówce w temperaturze pokojowej. Zbierano fazę wodną do nowej probówki.
Czynność powtarzano dwukrotnie. Następnie dodawano równą objętości chloroformu i
wstrząsano energicznie. Preparat poddawano wirowaniu w mikrowirówce w
temperaturze pokojowej, a do zebranej fazy wodnej dodawano RNazę A do końcowego
stężenia 50-100µg/ml i inkubowano w 370C przez 2 godziny. Po inkubacji
przeprowadzano ekstrakcję DNA dwukrotnie fenolem i chloroformem, w taki sam
sposób jak opisano wcześniej. Po ekstrakcji chloroformem dodawano równą objętość
izopropanolu i wytrącano DNA w -200C w ciągu 30min. Preparat wirowano przy

34
14500obr./min. w 40C w celu uzyskania osadu wytrąconego DNA. Osad płukano w
200µl 70% etanolu. Po wirowaniu i usunięciu 70% etanolu, osad DNA osuszano przez
15min. DNA rozpuszczano w 100µl wody.
Elektroforeza DNA w żelu agarozowym mająca na celu sprawdzenie
jakości preparatu
Przygotowywano żel agarozowy o stężeniu 2% w buforze TAE (40mM
Tris-octan, 1mM EDTA, pH7,2). Elektroforezie poddawano 5µl każdego preparatu
DNA wraz z barwnikiem OrangeG. Elektroforezę w żelu agarozowym o długości 10cm
prowadzono przy napięciu 80V przez ok. półtorej godziny do momentu dotarcia
barwnika naniesionego wraz z preparatem do końca żelu. Następnie żel przenoszono do
roztworu bromku etydyny (0,5µg/ml) na 15min. w celu zabarwienia DNA, który
obserwowano na transiluminatorze UV. Fotografię żelu agarozowego wykonywano
aparatem fotograficznym Polaroid DS34 z filmem Studio B/W o czułości ISO3000 tej
samej firmy (czas ekspozycji 1s, wartość przesłony 4,5). Na podstawie
elektroforetogramu wybierano preparaty do dalszej analizy. Odrzucano zarówno
preparaty o zbyt niskim, jak i zbyt wysokim stopniu strawienia.

35
Analiza produktów trawienia DNazy I i nukleazy z Micrococcus
Trawienie endonukleazą restrykcyjną MboI
Całość wybranego preparatu poddawano trawieniu endonukleazą restrykcyjną
MboI (Fermentas) w 370C przez noc. Dodawano 4µl endonukleazy restrykcyjnej MboI
(10U/µl) w celu przeprowadzenia wyczerpującego trawienia. Reakcję zatrzymywano,
zgodnie z zaleceniami producenta, przez umieszczenie preparatu w 650C na 20min.
Następnie wytrącano DNA 96% etanolem w -200C w ciągu 30min. W celu uzyskania
osadu DNA, preparat poddawano wirowaniu przy 14500obr./min. w 40C. Osad płukano
Numer ścieżki
elektroforetogramu Nazwa enzymu
Stężenie enzymu
w próbce (U/ml)
1 0,025
2 0,05
3 0,1
4 0,2
5 0,4
6
DNaza I
0,8
7 0,00033
8 0,001
9 0,003
10 0,009
11 0,027
12
Nukleaza z
Micrococcus
(MNaza)
0,081
Ryc. 4-2 Elektroforetogram preparatu DNA uzyskanego ze szczepu pip2 hodowanego w pożywce YPDE,
następnie poddanego trawieniu DNazą I i MNazą o różnych stężeniach. Do dalszej analizy wybrano preparaty
ze ścieżek 3, 4 i 5 (po trawieniu DNazą I) oraz 8, 9 i 10 (po trawieniu MNazą). Elektroforezie poddawano 1/20
obj. każdej próbki.
1 2 3 4 5 6 7 8 9 10 11 12
DNaza I MNaza

36
w 200µl 70% etanolu. Po wirowaniu i usunięciu 70% etanolu, osad DNA osuszano
przez 15min. Następnie rozpuszczano DNA w 100µl wody.
Elektroforeza DNA w żelu agarozowym preparatów gotowych do
przeniesienia na membranę
Przygotowywano żel agarozowy o stężeniu 2% w buforze TAE. Elektroforezie
poddawano 15-25µl preparatu DNA wraz z 3-5µl barwnika OrangeG. Do skrajnych
kieszonek nanoszono 2µl wskaźnika wielkości (DNA bakteriofaga φX174 poddany
trawieniu endonukleazą restrykcyną HaeIII, Fermentas) wraz z 20µl roztworu
barwników (60% glicerol, 0,09% błękit bromofenolowy, 0,09% cyjanol ksylenu, 60mM
EDTA, Fermentas). Barwniki te migrowały z ruchliwością odpowiadającą odcinkom
DNA o długości 300pz i 4000pz. Elektroforezę w żelu o długości ok. 16cm prowadzono
przy napięciu 20V przez 18-20 godzin do momentu dotarcia błękitu bromofenolowego
do końca żelu. Następnie żel przenoszono do roztworu bromku etydyny na 15min. w
celu zabarwienia DNA, który obserwowano na transiluminatorze UV. Fotografię żelu
agarozowego wykonywano w sposób opisany wcześniej.
Przenoszenie DNA z żelu agarozowego na membranę
DNA z żelu agarozowego przenoszono na membranę metodą elektrotransferu.
Z żelu odcinano skrajne ścieżki, 1,0-1,5cm części dolnej, a także fragment żelu

37
znajdujący się powyżej studzienek. Następnie wycinano membranę nylonową
(Zeta-probe, Amersham) odpowiadającą wymiarom przyciętego żelu agarozowego oraz
dwa kawałki bibuły Whatman 3 nieco większe niż żel agarozowy. Wyciętą membranę,
bibuły i gąbki z aparatu do elektrotransferu nawilżano w buforze 0,5x TBE (45mM
Tris-boran, 1mM EDTA, pH8,0) i usuwano wszelkie pęcherzyki powietrza. Następnie
układano poszczególne elementy w następującej kolejności: [katoda] - gąbka - bibuła -
żel agarozowy (wierzchnią stroną do bibuły) - membrana - bibuła - gąbka - [anoda].
Przy układaniu poszczególnych elementów obficie polewano je buforem 0,5x TBE i
rolowano bagietką usuwając w ten sposób pęcherzyki powietrza. Przygotowaną
„kanapkę” następnie umieszczano w aparacie do elektrotransferu wypełnionym buforem
0,5x TBE. Procedurę elektrotransferu przeprowadzano przy napięciu 10V przez 18
godzin w 40C. Następnie w celu utrwalenia membrany z DNA umieszczano ją stroną
DNA do góry na bibule Whatman 3 nasączonej 0,4M roztworem wodorotlenku sodu. Po
10min. płukano membranę dwukrotnie buforem SSC 2x (0,3M NaCl, 0,03M cytrynian
sodu, pH7,0). Wilgotną membranę zawijano w folię spożywczą i przechowywano w
-200C.
Znakowanie sondy do hybrydyzacji
Sondy do hybrydyzacji znakowano radioaktywnie metodą losowych starterów
stosując Megaprime DNA Labelling System (Amersham) i fragment Klenowa
polimerazy I DNA, zgodnie z instrukcją producenta. Ilość matrycowego DNA sondy,
którą poddawano znakowaniu oceniano przeprowadzając elektroforezę w żelu

38
agarozowym. Umieszczano ok. 25ng matrycowego DNA sondy (ilość odpowiadająca
dobrze widocznemu prążkowi na elektroforetogramie) wraz z 5µl starterów w probówce
typu Eppendorf i przeprowadzano denaturację matrycowego DNA w 1000C w ciągu
5min. Następnie przygotowywano mieszaninę składników pozwalających na
znakowanie sondy, dodając po 4µl nieznakowanych dCTP, dGTP i dTTP, 5µl buforu do
reakcji, 2U fragmentu Klenowa i 5µl [α-32P]dATP (3000Ci/mmol, Amersham)
doprowadzając objętość wodą do 50µl. Reakcję znakowania prowadzono w 370C przez
10min., następnie zatrzymywano przez dodanie 5µl roztworu 0,2M EDTA, 10mg/ml
tRNA z E.coli i wytrącano DNA dodając 100µl 96% EtOH, a następnie umieszczając w
-200C na 30 min. Preparat poddawano wirowaniu w mikrowirówce (14500obr./min.,
temp. pokojowa, 10min.). Przed użyciem sondę rozpuszczano w 500µl buforu do
hybrydyzacji (patrz niżej) i denaturowano przez podgrzanie do 1000C na 5 min.
Używano sondy o sekwencji nukleotydowej całkowicie zgodnej z rejonem od
+68 do +366 sekwencji nukleotydowej genu CTA1 (długość sondy wynosi 298pz,
numerem 1 oznaczono pierwszy nukleotyd, który ulega translacji). Matrycowy DNA
sondy został namnożony metodą PCR przez dr Joannę Trzcińską-Danielewicz.
Hybrydyzacja typu Southern
Do pojemnika do hybrydyzacji (zakręcane, grubościenne szklane fiolki o
pojemności 200ml) wkładano membranę zwiniętą stroną DNA do wewnątrz, a następnie
wlewano ok. 15ml buforu do hybrydyzacji o składzie 0,5M bufor sodowo-fosforanowy

39
(NaPi), 7% SDS, 1mM EDTA, pH7,5 i prowadzono prehybrydyzację w 650C przez 1-3
godziny. Następnie do pojemnika wprowadzano całość zdenaturowanej sondy i
prowadzono hybrydyzację przez 12-18 godzin w takich samych warunkach. Po
hybrydyzacji, dokładnie zlewano bufor do hybrydyzacji wraz z niezwiązaną sondą, a
następnie membranę płukano w 650C:
• dwa razy po 10min. w 0,2M NaPi, 1% SDS, pH7,5
• dwa razy po 10min. w 0,1M NaPi, 1% SDS, pH7,5
• dwa razy po 5min. w 0,04M NaPi, 0,1% SDS, pH7,5.
W zależności od wielkości i liczby filtrów za każdym razem używano od 50 do
150ml roztworu do płukania. Po przeprowadzonych płukaniach sprawdzano sygnał
membrany przy użyciu licznika Geigera-Müllera.
Autoradiografia
Membranę po hybrydyzacji umieszczano w kasecie wraz z filmem
rentgenowskim i prowadzono ekspozycję w -700C przez 1-7 dni. Stosowano filmy
XS-1N (Foton) oraz BioMax MS (Kodak). W przypadku silnego sygnału (powyżej
50cps) nie stosowano ekranów wzmacniających, natomiast do membran ze słabym
sygnałem (poniżej 10cps) używano filmu BioMax MS wraz z ekranem wzmacniającym
o tej samej nazwie. W przypadku nieco silniejszego sygnału stosowano ekran
wzmacniający Cronex Lightning-Plus (Dupont) wraz z filmem XS-1N. Filmy
wywoływano standardową metodą, a następnie fotografowano cyfrowym aparatem
fotograficznym Olympus C-200.

40
5. Wyniki
1. Aktywność transkrypcyjna promotora genu CTA1 i kinetyka
indukcji
Analizę aktywności transkrypcyjnej genu CTA1 prowadzono wykorzystując
układ reporterowy będący fuzją promotora genu CTA1 i bakteryjnego genu lacZ
kodującego β-galaktozydazę. Konstrukcja została wprowadzona, zarówno do szczepu
kontrolnego (BY4742, dzikiego pod względem badanych czynników transkrypcyjnych),
jak i do szczepów adr1, oaf1 i pip2, na dwóch plazmidach: episomalnym
(występującym w komórce w kilkudziesięciu kopiach) i integracyjnym (występującym
w komórce w jednej kopii). Hodowle będące w stanie derepresji genu CTA1 w
logarytmicznej fazie wzrostu oraz w fazie stacjonarnej przeszczepiano do pożywki z
kwasem olejowym, który indukuje transkrypcję m.in. genu CTA1. Z części hodowli
przygotowywano ekstrakt natychmiast po przeszczepieniu (ekstrakt ten reprezentował
wyjściowy poziom aktywności promotora genu CTA1 w stanie derepresji), natomiast
resztę hodowli w pożywce z kwasem olejowym prowadzono przez 3, 6, 9, 12 i 24
godziny, a następnie sporządzano ekstrakt. Przygotowane ekstrakty wykorzystywano do
oznaczenia aktywności właściwej β-galaktozydazy, która jest pośrednią miarą
aktywności transkrypcyjnej promotora genu CTA1.
Pomiaru dokonywano dla ekstraktów przygotowanych z każdego szczepu
transformowanego odpowiednim plazmidem, w obu fazach wzrostu, w stanie derepresji
genu CTA1 i po upływie odpowiedniego czasu hodowli w warunkach indukcji tego genu.

41
Każdy ekstrakt oznaczano w dwóch powtórzeniach i wyliczano średnią. Dla każdego
szczepu eksperyment powtarzano co najmniej trzy razy. Z nieznanych powodów
wartości aktywności właściwej β-galaktozydazy w powtórzeniach doświadczenia
wykonanych dla jednego szczepu znacznie się różniły. Podobne zjawisko
zaobserwowali także inni badacze [38, 41]. Ponieważ obliczanie wartości średnich z tak
rozbieżnych wyników nie byłoby poprawne, wyniki dotyczące aktywności właściwej
β-galaktozydazy po odpowiednim czasie indukcji genu CTA1 przeliczano w stosunku do
aktywności w warunkach derepresji, a otrzymaną wartość określano jako względny
poziom indukcji (Tab. 5-P). Z niewyjaśnionych również powodów, niektóre wyniki
pomiarów odbiegały znacznie od ogólnej tendencji indukcji lub spójnych powtórzeń
doświadczenia dotyczącego jednego szczepu. W takim przypadku odrzucano ten punkt
jako niewiarygodny i nie brano pod uwagę przy wyliczaniu średniej.
Razem z wykresami pokazującymi względny poziom indukcji w funkcji czasu
hodowli w pożywce z kwasem olejowym, podano także tabele zawierające dane
liczbowe z poszczególnych powtórzeń doświadczenia, na podstawie których obliczano
wartości średnie względnego poziomu indukcji oraz wartości odchylenia standardowego
przedstawione na wykresach (wartości odchylenia standardowego podano również w
tabelach).

42
Bezwzględne wartości aktywności właściwej β-galatozydazy uzyskane w dwóch doświadczeniach
Aktywność właściwa β-galatozydazy [µmol·mg-1·min-1]
Czas hodowli z kw. olej. [godziny]
Symbole Doświadczenie 1 Doświadczenie 2
0 A 0,06 0,043 X3 1,22 0,626 X6 4,97 1,709 X9 4,91 2,0512 X12 6,43 4,3124 X24 11,70 6,17
Względny poziom indukcji
Względny poziom indukcji Czas hodowli z kw. olej. [godziny]
Symbole Doświadczenie 1 Doświadczenie 2
0 A/A 1 13 X3/A 20 176 X6/A 80 469 X9/A 79 5612 X12/A 104 11824 X24/A 189 168
Tab. 5-P Przykład opracowania wyników przedstawionych w postaci bezwzględnych wartości aktywności
β-galatozydazy. Wyniki te wydają się dość rozbieżne, jednak po przeliczeniu na względny poziom indukcji stają się
porównywalne.
Przedstawione w przykładzie wartości pochodzą z wyników uzyskanych dla szczepu oaf1 z plazmidem
integracyjnym z hodowli w logarytmicznej fazie wzrostu.

43
Legenda Wykresy:
BY4742 (szczep kontrolny) adr1 oaf1 pip2
Przedstawiono wykresy z połączonymi linią prostą punktami, odpowiadającymi
wyliczonym wartościom aktywności właściwej β-galaktozydazy. Nie musi to dokładnie
odzwierciedlać rzeczywistego przebiegu procesu, zwłaszcza pomiędzy punktami
przedstawiającymi wartości względnego poziomu indukcji po 12 i 24 godzinach
hodowli. Takie wykresy są jednak bardziej czytelne niż wykresy słupkowe.
Pionowymi liniami oznaczono wartości odchylenia standardowego w danym punkcie.
Wartości odchyleń standardowych umieszczono w tabelach z danymi liczbowymi.
Tabele z danymi liczbowymi:
W poszczególnych kolumnach tabeli umieszczono wartości względnego poziomu
indukcji uzyskane w niezależnych eksperymentach, dotyczące jednego szczepu
transformowanego odpowiednim plazmidem.
Kursywą zaznaczono wartości pominięte przy obliczaniu wartości średnich z powodu
znacznego odstępstwa od ogólnej tendencji indukcji genu CTA1 i spójnych powtórzeń
eksperymentu dotyczącego jednego szczepu.

44
b/p - brak pomiaru
b/d - brak danych spowodowany niemożnością obliczenia wartości odchylenia
standardowego wynikającą ze zbyt małej liczby przeprowadzonych pomiarów
* - symbolem tym oznaczono istotną statystycznie różnicę między poziomem indukcji
w szczepie pozbawionym jednego z trzech badanych czynników transkrypcyjnych w
stosunku do wyniku pomiaru szczepu kontrolnego, na podstawie przeprowadzonego
dwustronnego testu t Studenta różnicy między średnimi przy różnych wariancjach
(poziom istotności p=0,055). Symbol ten umieszczono zarówno w tabelach z danymi
liczbowymi, jak i przy odpowiednich punktach na wykresach.

Numer doświadczenia Szczep
BY4742 1 2 3 4 5 6 Odchylenie standardowe
3h 1 21 26 15 35 127 6
6h 1 136 115 37 115 317 10
9h 5 223 277 76 287 237 27
12h 18 265 493 52 303 550 121
24h 77 815 1059 20 566 776 175
Numer doświadczenia
Szczep adr1 1 2 3 4
Odchylenie standardowe
3h 16 30 25 42 9
6h 96 50 103 268 24
9h* b/p 100 191 149 37
12h b/p b/p b/p 299 b/d
24h* 237 211 272 278 27
Numer doświadczenia
Szczep oaf1 1 2 3
Odchylenie standardowe
3h 15 29 9 8
6h* 62 58 30 14
9h* 63 128 34 14
12h* 77 97 52 18
24h* 227 209 91 9
Numer doświadczenia
Szczep pip2 1 2 3
Odchylenie standardowe
3h 63 33 31 15
6h 72 184 101 47
9h* 143 136 108 15
12h* 232 164 215 29
24h* 289 421 492 84 Tab. 5-1 Wartości względnego poziomu indukcji uzyskane w kolejnych powtórzeniach doświadczenia wraz z
wartościami odchylenia standardowego
Komórki z plazmidem episomalnym Hodowle w logarytmicznej fazie wzrostu
0
200
400
600
800
1000
1200
0 3 6 9 12 15 18 21 24 27
Czas hodowli z kwasem olejowym [godziny]
Wzg
lędn
y po
ziom
indu
kcji
Ryc. 5-1 Przebieg indukcji promotora genu CTA1 pod wpływem kwasu olejowego
1
* *
* * * * *
*
*

Numer doświadczenia Szczep
BY4742 1 2 3 4 5 6 Odchylenie standardowe
3h 4 65 23 34 33 63 17
6h 11 296 141 115 25 223 71
9h 31 468 138 182 160 452 147
12h 73 494 304 176 188 512 145
24h 148 530 395 274 167 550 112
Numer doświadczenia
Szczep adr1 1 2 3 4
Odchylenie standardowe
3h 41 55 26 67 15
6h 73 197 21 109 52
9h b/p 171 50 138 17
12h b/p b/p b/p 120 b/d
24h* 102 172 65 169 45
Numer doświadczenia
Szczep oaf1 1 2 3
Odchylenie standardowe
3h 57 45 28 12
6h* 177 67 63 2
9h 121 126 88 17
12h* 756 96 73 11
24h* 211 88 75 6
Numer doświadczenia
Szczep pip2 1 2 3
Odchylenie standardowe
3h 31 69 48 16
6h 55 161 95 44
9h 67 223 133 33
12h* 89 205 109 10
24h* 102 206 110 4 Tab. 5-2 Wartości względnego poziomu indukcji uzyskane w kolejnych powtórzeniach doświadczenia wraz z
wartościami odchylenia standardowego
Komórki z plazmidem episomalnym Hodowla w fazie stacjonarnej
0
100
200
300
400
500
600
0 3 6 9 12 15 18 21 24 27
Czas hodowli z kwasem olejowym [godziny]
Wzg
lędn
y po
ziom
indu
kcji
Ryc. 5-2 Przebieg indukcji promotora genu CTA1 pod wpływem kwasu olejowego
1
*
* * * *
*

Numer doświadczenia Szczep
BY4742 1 2 3 4 Odchylenie standardowe
3h 72 52 7 69 9
6h 129 228 13 223 45
9h 378 143 49 304 98
12h 414 232 75 394 81
24h 459 294 148 340 69
Numer doświadczenia
Szczep adr1 1 2 3 4
Odchylenie standardowe
3h 49 38 44 52 5
6h 121 98 80 142 24
9h 150 204 193 377 87
12h 214 158 286 335 68
24h 345 b/p b/p 493 74
Numer doświadczenia
Szczep oaf1 1 2 3
Odchylenie standardowe
3h* 20 35 17 8
6h 80 b/p 46 17
9h 79 306 56 12
12h* 104 372 118 7
24h* 189 264 168 10
Numer doświadczenia
Szczep pip2 1 2 3
Odchylenie standardowe
3h 59 58 54 2
6h 128 112 128 8
9h 181 159 169 9
12h 216 238 294 33
24h 292 265 332 28 Tab. 5-3 Wartości względnego poziomu indukcji uzyskane w kolejnych powtórzeniach doświadczenia wraz z
wartościami odchylenia standardowego
Komórki z plazmidem integracyjnym Hodowla w logarytmicznej fazie wzrostu
0
100
200
300
400
500
600
0 3 6 9 12 15 18 21 24 27
Czas hodowli z kwasem olejowym [godziny]
Wzg
lędn
y po
ziom
indu
kcji
Ryc. 5-3 Przebieg indukcji promotora genu CTA1 pod wpływem kwasu olejowego
1 *
*
*
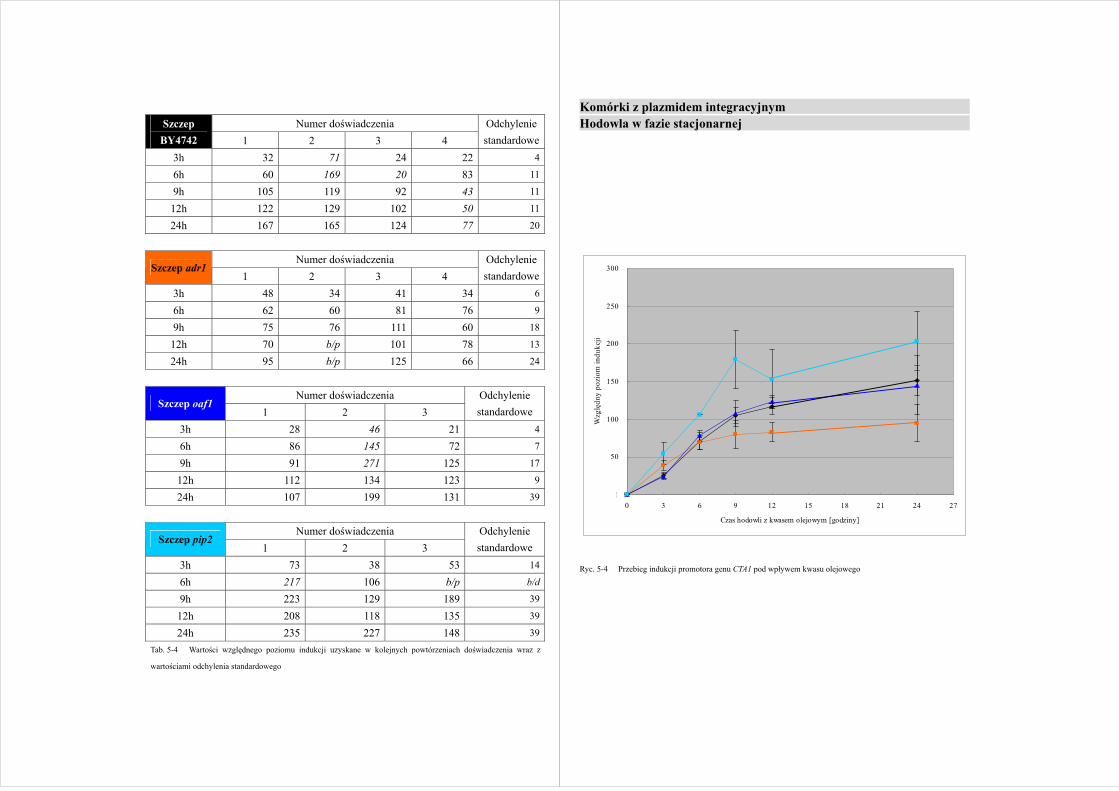
Numer doświadczenia Szczep
BY4742 1 2 3 4 Odchylenie standardowe
3h 32 71 24 22 4
6h 60 169 20 83 11
9h 105 119 92 43 11
12h 122 129 102 50 11
24h 167 165 124 77 20
Numer doświadczenia
Szczep adr1 1 2 3 4
Odchylenie standardowe
3h 48 34 41 34 6
6h 62 60 81 76 9
9h 75 76 111 60 18
12h 70 b/p 101 78 13
24h 95 b/p 125 66 24
Numer doświadczenia
Szczep oaf1 1 2 3
Odchylenie standardowe
3h 28 46 21 4
6h 86 145 72 7
9h 91 271 125 17
12h 112 134 123 9
24h 107 199 131 39
Numer doświadczenia
Szczep pip2 1 2 3
Odchylenie standardowe
3h 73 38 53 14
6h 217 106 b/p b/d
9h 223 129 189 39
12h 208 118 135 39
24h 235 227 148 39 Tab. 5-4 Wartości względnego poziomu indukcji uzyskane w kolejnych powtórzeniach doświadczenia wraz z
wartościami odchylenia standardowego
Komórki z plazmidem integracyjnym Hodowla w fazie stacjonarnej
0
50
100
150
200
250
300
0 3 6 9 12 15 18 21 24 27
Czas hodowli z kwasem olejowym [godziny]
Wzg
lędn
y po
ziom
indu
kcji
Ryc. 5-4 Przebieg indukcji promotora genu CTA1 pod wpływem kwasu olejowego
1

49
Indukcja genu CTA1 w komórkach S.cerevisiae znajdujących się w
logarytmicznej fazie wzrostu pokazuje tendencję wzrostową przez 24 godziny po
przeniesieniu do pożywki z kwasem olejowym, niezależnie od plazmidu, którym
transformowano komórki drożdży (Ryc. 5-1 i Ryc. 5-3). Wyjątkiem jest szczep
kontrolny z plazmidem integracyjnym i szczep adr1 z plazmidem episomalnym, w
których po 12 godzinach hodowli nie obserwuje się wzrostu poziomu indukcji genu
CTA1. Odmienną sytuację można zaobserwować w drożdżach znajdujących się w fazie
stacjonarnej, w których, z wyjątkiem szczepu kontrolnego z plazmidem episomalnym,
wzrost poziomu indukcji genu CTA1 trwa jedynie 9 godzin (Ryc. 5-2 i Ryc. 5-4). Przez
następne 15 godzin hodowli w warunkach indukcji aktywność transkrypcyjna
promotora genu nie wzrasta w sposób znaczący.
W hodowlach znajdujących się w logarytmicznej fazie wzrostu wszystkie
szczepy pozbawione jednego z trzech badanych czynników transkrypcyjnych wykazują
obniżoną zdolność indukcji genu CTA1 w porównaniu do szczepu kontrolnego.
Szczególnie niski poziom indukcji można zaobserwować w szczepie oaf1
transformowanym plazmidem episomalnym (Ryc. 5-1), dla którego już po 6 godzinach
hodowli w pożywce z kwasem olejowym można zauważyć istotne statystycznie
obniżenie stopnia indukcji.
Przebieg indukcji genu CTA1 w komórkach S.cerevisiae transformowanych
plazmidem episomalnym znajdujących się w fazie stacjonarnej (Ryc. 5-2) wyraźnie
różni się od wyników uzyskanych dla hodowli drożdży znajdujących się w
logarytmicznej fazie wzrostu (Ryc. 5-1). W fazie stacjonarnej wszystkie trzy szczepy
pozbawione jednego z trzech czynników transkrypcyjnych wykazują obniżoną w
równym stopniu zdolność indukcji genu CTA1, a szczep oaf1 nie wyróżnia się w żaden

50
sposób na tle dwóch pozostałych badanych szczepów drożdży.
Niespodziewane wyniki uzyskano w hodowlach S.cerevisiae
transformowanych plazmidem integracyjnym, przeniesionych do pożywki z kwasem
olejowym w fazie stacjonarnej (Ryc. 5-4). Nie zaobserwowano znaczącej różnicy
między szczepem kontrolnym i szczepami badanymi (adr1 i oaf1), zaś szczep z
usuniętym genem czynnika transkrypcyjnego Pip2p wykazywał wyższy stopień
indukcji genu CTA1 przez cały czas trwania hodowli w pożywce z kwasem olejowym.
Różnica ta nie była jednak istotna statystycznie.
Warto zaznaczyć, iż nawet w szczepach wykazujących obniżony w stosunku do
szczepu kontrolnego poziom indukcji genu CTA1, w wyniku przeniesienia drożdży do
warunków indukcji aktywność promotora tego genu zwiększała się od 100 do 200 razy.

51
2. Reorganizacja struktury chromatyny w rejonie promotora
genu CTA1
Analizę struktury chromatyny rejonu promotora genu CTA1 prowadzono w
drożdżach hodowanych do wczesnej logarytmicznej fazy wzrostu w trzech warunkach:
represji, derepresji i indukcji. Następnie izolowano jądra komórkowe i analizowano
strukturę chromatyny stosując metodę pośredniego znakowania końców. Podobnie jak
w omawianej wcześniej analizie aktywności transkrypcyjnej promotora genu CTA1
przeprowadzonej przy pomocy genu reporeterowego, strukturę chromatyny badano w
szczepach adr1, oaf1 i pip2, a także w szczepie kontrolnym MHY501α (dzikim pod
względem badanych czynników transkrypcyjnych). Odcinki DNA powstałe w wyniku
trawienia chromatyny poddawano elektroforezie w żelu agarozowym, następnie po
przeniesieniu DNA na membranę przeprowadzano hybrydyzację typu Southern z
odpowiednią sondą znakowaną radioaktywnie i poddawano autoradiografii. Porównując
prążki widoczne na autoradiogramie można uzyskać informacje o rozmieszczeniu
nukleosomów i miejscach wrażliwych na działanie DNazy I w badanym rejonie, a także
o zmianach ich położenia w zależności od warunków w których prowadzono hodowlę i
w zależności od badanego szczepu.
Jednak pełny obraz zmian zachodzących w strukturze chromatyny w rejonie
promotora genu CTA1 można uzyskać dopiero po przeprowadzeniu elektroforezy
preparatów ze wszystkich badanych szczepów we wszystkich warunkach hodowli
(represji, derepresji i indukcji) na jednym żelu agarozowym. Jest to istotne, ponieważ
tylko w ten sposób można dokonać rozdziału fragmentów DNA z równą ruchliwością
umożliwiającą porównanie wykrytych sondą fragmentów DNA na jednym filmie

52
rentgenowskim. Umożliwia to jednoznaczne stwierdzenie wzajemnego położenia
wykrytych fragmentów DNA.
Do chwili obecnej nie przeprowadzono elektroforezy wszystkich preparatów na
jednym żelu agarozowym, tak więc zostaną przedstawione jedynie cząstkowe wyniki
porównań niektórych szczepów i warunków hodowli.
Legenda
Czarne strzałki wskazują prążki wykrywane we wszystkich ścieżkach autoradiogramu,
natomiast czerwone strzałki wskazują prążki pojawiające się lub znikające w niektórych
ścieżkach. Różowe strzałki wskazują prążek o długości 1906pz powstający w wyniku
trawienia endonukleazą restrykcyjną MboI.
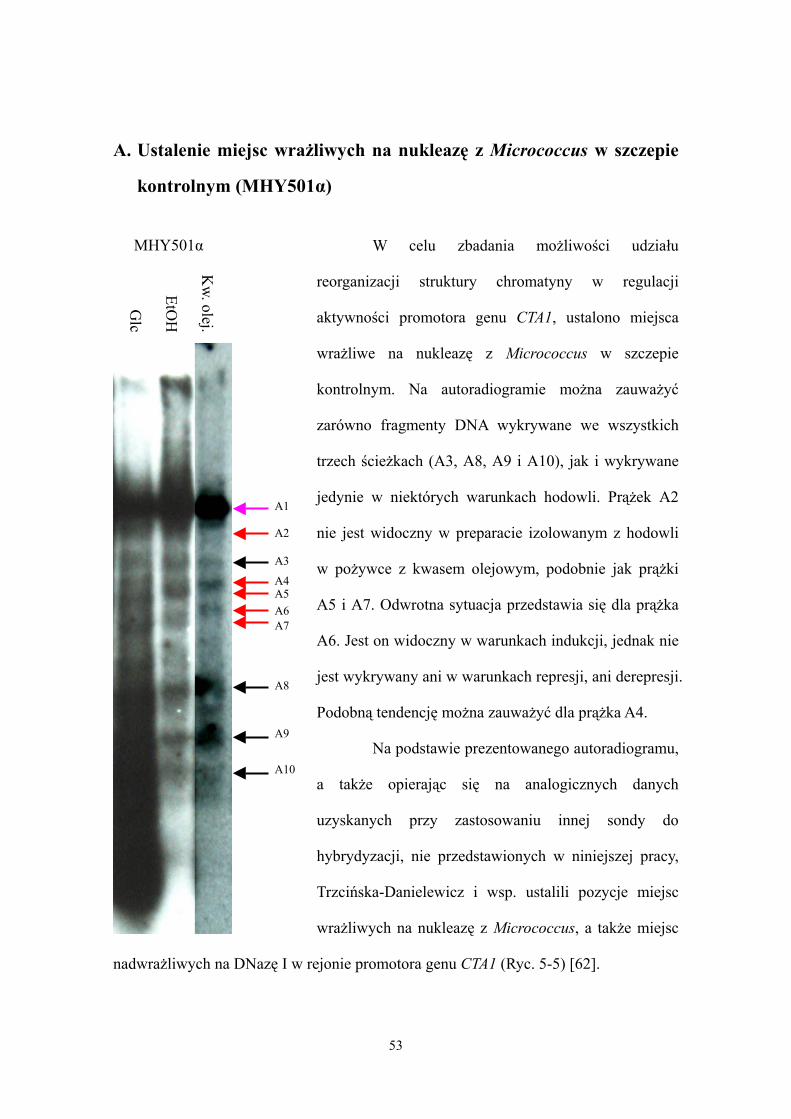
53
A. Ustalenie miejsc wrażliwych na nukleazę z Micrococcus w szczepie
kontrolnym (MHY501α)
W celu zbadania możliwości udziału
reorganizacji struktury chromatyny w regulacji
aktywności promotora genu CTA1, ustalono miejsca
wrażliwe na nukleazę z Micrococcus w szczepie
kontrolnym. Na autoradiogramie można zauważyć
zarówno fragmenty DNA wykrywane we wszystkich
trzech ścieżkach (A3, A8, A9 i A10), jak i wykrywane
jedynie w niektórych warunkach hodowli. Prążek A2
nie jest widoczny w preparacie izolowanym z hodowli
w pożywce z kwasem olejowym, podobnie jak prążki
A5 i A7. Odwrotna sytuacja przedstawia się dla prążka
A6. Jest on widoczny w warunkach indukcji, jednak nie
jest wykrywany ani w warunkach represji, ani derepresji.
Podobną tendencję można zauważyć dla prążka A4.
Na podstawie prezentowanego autoradiogramu,
a także opierając się na analogicznych danych
uzyskanych przy zastosowaniu innej sondy do
hybrydyzacji, nie przedstawionych w niniejszej pracy,
Trzcińska-Danielewicz i wsp. ustalili pozycje miejsc
wrażliwych na nukleazę z Micrococcus, a także miejsc
nadwrażliwych na DNazę I w rejonie promotora genu CTA1 (Ryc. 5-5) [62].
Kw. olej.
EtOH
Glc
A1
A2
A3
A4 A5 A6 A7
A8
A9
A10
MHY501α

54
Ryc. 5-5 Schemat rejonu promotora genu CTA1 wraz z zaznaczonymi miejscami nadwrażliwymi na DNazę I oraz
wrażliwymi na nukleazę z Micrococcus (MNaza). Zaznaczono także położenie sekwencji nukleotydowych sond
molekularnych, którymi wykrywano fragmenty DNA w stosowanej metodzie pośredniego znakowania końców.
Numerem +1 oznaczono pierwszy nukleotyd, który ulega translacji. Zaadaptowano z [62].
Najwyraźniejszy prążek wykrywany we wszystkich trzech ścieżkach (prążek
A1) jest fragmentem DNA o długości 1906pz, który powstaje w wyniku trawienia
endonukleazą restrykcyjną MboI. Fragmenty te są największe wśród wykrywanych
sondą molekularną, ponieważ nie uległy trawieniu nukleazą z Micrococcus lub DNazą I.
Fragment DNA o długości 1906pz jest więc wykrywany we wszystkich
przeprowadzanych eksperymentach niezależnie od badanego szczepu S.cerevisiae i
warunków hodowli, z której zostały wyizolowane jądra komórkowe. Brak na
autoradiogramach prążków większych niż 1906pz świadczy natomiast o tym, że
trawienie endonukleazą restrykcyjną MboI było wyczerpujące. Jest to warunek
konieczny do uzyskania wiarygodnych wyników przy zastosowaniu metody
790990
1 50-80-226-365-490
-669
-645
-1541AUG
-125
+366
DNazaI
--Kw. olej.
-195 -145
DNazaI
-
Glc
-310AUG UGA
-1794 RMS1 CTA1
+1
5’ 3’
MNaza
-156
-515
-546-708
-700
MNaza
Sonda 5’ Sonda 3’
EtOH
MboIMboI

55
pośredniego znakowania końców.
Wszystkie fragmenty DNA migrujące podczas elektroforezy szybciej niż
wspomniany fragment o długości 1906pz powstają w wyniku trawienia z jednej strony
nukleazą z Micrococcus lub DNazą I, z drugiej zaś strony endonukleazą restrykcyjną
MboI.
Dzięki analizie struktury chromatyny w badanych szczepach S.cerevisiae i
ewentualnych jej zmian w stosunku do szczepu kontrolnego będzie możliwe
potwierdzenie lub wykluczenie udziału poszczególnych czynników transkrypcyjnych
Adr1p, Oaf1p i Pip2p w reorganizacji struktury chromatyny rejonu promotora genu
CTA1.

56
B. Analiza miejsc wrażliwych na nukleazę z Micrococcus w szczepie
pip2
Podobnie jak w przypadku szczepu
kontrolnego, w szczepie pip2 również można zauważyć
prążki wykrywane we wszystkich ścieżkach, jak i te
pojawiające się jedynie w niektórych ścieżkach. Do
prążków wykrywanych we wszystkich ścieżkach można
zaliczyć prążki B1 (identyczny z prążkiem A1), B2,
B4-B6, B10-B14, B16-B18. Prążek B3 nie jest
wykrywany w preparacie wyizolowanym z hodowli
prowadzonej w pożywce z kwasem olejowym, podobnie
jak prążek B8. Natomiast prążki B9 i B15 są
wykrywane jedynie w preparacie wyizolowanym z
komórek hodowanych w warunkach indukcji genu
CTA1. Prążek B7 również najwyraźniej widoczny jest w
prepracie wyizolowanym z hodowli w pożywce z
kwasem olejowym, jednak w przypadku tego prążka jest
on widoczny, choć słabiej, także w preparatach
wyizolowanych z hodowli w stanie represji i derepresji
genu CTA1.
Po analizie autoradiogramów (również nie
przedstawionych w niniejszej pracy) ustalono niektóre
miejsca działania nukleazy z Micrococcus w rejonie
Kw. olej.
EtOH
Glc
B1
B2
B3 B4
B5 B6
B7 B8
B9 B10 B11
B12
B13
B14 B15
B16
B17
B18
pip2

57
promotora genu CTA1. Pozycja -220 (prążek B13) i +70 (prążek B17) są miejscami
działania nukleazy z Micrococcus we wszystkich badanych warunkach. Pozycja -150
(prążek B15) jest natomiast miejscem działania nukleazy z Micrococcus występującym
jedynie w warunkach indukcji genu CTA1. Obserwacje te odpowiadają obrazowi ze
szczepu kontrolnego (Ryc. 5-5). Ustalone pozycje miejsc działania nukleazy z
Micrococcus są obarczone stosunkowo dużym błędem. Można jednak stwierdzić, że w
rejonie promotora genu CTA1 zachodzą zmiany w strukturze chromatyny w szczepie
pozbawionym czynnika transkrypcyjnego Pip2p podobne do obserwowanych w
szczepie kontrolnym.

58
C. Analiza miejsc wrażliwych na nukleazę z Micrococcus w szczepach
adr1 i oaf1 w warunkach represji i derepresji genu CTA1
Jak wynika z autoradiogramów
przedstawionych w punktach A i B, układ
wykrywanych fragmentów DNA w preparatach
wyizolowanych z warunków represji i derepresji jest
bardzo podobny. Tak też jest w przypadku
porównania szczepów adr1 i oaf1 w warunkach
represji i derepresji. Wyraźne różnice pojawiają się
jedynie w dwóch miejscach. Są to prążki C6 i C8
wykrywane tylko w szczepie oaf1 w warunkach
represji. Warto także zauważyć, że w szczepie
pozbawionym czynnika transkrypcyjnego Adr1p nie
występują żadne zmiany w układzie wykrywanych
prążków między komórkami hodowanymi w
warunkach represji i derepresji. Pod tym względem
szczep ten różni się od szczepu oaf1, w którym
można zauważyć kilka zmian w układzie
wykrywanych prążków między komórkami hodowanymi w pożywce z glukozą i w
pożywce z etanolem.
Prążki C6 i C8 wykrywane jedynie w szczepie oaf1 w warunkach represji mają
EtOH
Glc
EtOH
Glc
adr1 oaf1
C1 C2
C3
C4 C5 C6 C7
C8 C9
C10
C11
C12
C13
C14

59
długość ok. 1000pz i wskazują na zmiany zachodzące w pobliżu pozycji -650 (licząc od
pierwszego nukleotydu ulegającego translcji). Są to dosyć odległe miejsca od rejonu
promotora genu CTA1 i wydaje się, że nie mają one dużego znaczenia w regulacji
aktywności transkrypcyjnej badanego promotora.

60
D. Próba porównania położenia miejsc wrażliwych na nukleazę z
Micrococcus (MNazę) w szczepach MHY501α, adr1, oaf1 i pip2 w
warunkach derepresji genu CTA1
Porównanie autoradiogramów z
preparatami pochodzącymi ze
wszystkich badanych szczepów
hodowanych w stanie derepresji genu
CTA1 umożliwiło wstępne
zidentyfikowanie prążka obecnego we
wszystkich szczepach z wyjątkiem
szczepu kontrolnego (prążek D1). Inne
prążki zaznaczone kolorem czarnym
wydają się występować we wszystkich
badanych szczepach.
Analiza przeprowadzona w ten
sposób obarczona jest dużym błędem
spowodowanym różną ruchliwością
rozdzielanych fragmentów DNA w
oddzielnych żelach agarozowych, a
także różną intensywnością sygnału
sondy używanej do hybrydyzacji.
Pozwala ona jednak na porównanie
układów wykrywanych prążków i umożliwia wykrycie prążków pojawiających się tylko
w określonym szczepie lub w określonych warunkach hodowli.
MNaza
pip2
oaf1
adr1
MH
Y501α
EtOH
MNaza
D1
D1
MboI-MboI
1906pz

61
E. Analiza miejsc nadwrażliwych na DNazę I w szczepie kontrolnym
(MHY501α) i szczepie oaf1 w warunkach indukcji genu CTA1
Zmiany w układzie wykrywanych fragmentów DNA
są nieliczne. Prążki E3 i E6 są widoczne jedynie w preparacie
wyizolowanym ze szczepu pozbawionego czynnika
transkrypcyjnego Oaf1p. Jednak inne fragmenty DNA są
wykrywane w obu szczepach S.cerevisiae.
Oaf1
MH
Y501α
E1
E2
E3
E4
E5 E6
E7
E8
Kw. olej.

62
Analiza przedstawionych autoradiogramów sugeruje, że w przypadku genu
CTA1 reorganizacja struktury chromatyny nie jest procesem szczególnie ważnym dla
indukcji tego genu. Z przedstawionych w punkcie A oraz B autoradiogramów wynika,
iż w warunkach represji i derepresji struktura chromatyny rejonu promotora genu CTA1
jest bardzo podobna (potwierdza to także autoradiogram przedstawiony w punkcie C),
natomiast drobne, choć wyraźne zmiany zachodzą przy indukcji tego genu.
Jak wcześniej wspomniano, pełny obraz dotyczący reorganizacji struktury
chromatyny badanego rejonu można poznać jedynie przez przeprowadzenie
elektroforezy wszystkich preparatów na jednym żelu agarozowym. Bardzo trudne zatem
jest przypisanie poszczególnych prążków wykrytych przez autoradiografię do miejsc
działania nukleazy z Micrococcus i DNazy I na podstawie kilku oddzielnych
autoradiogramów. Konieczna więc będzie kontynuacja badań w celu jednoznacznego
wyjaśnienia roli czynników transkrypcyjnych Adr1p, Oaf1p i Pip2p w procesie
reorganizacji chromatyny rejonu promotora genu CTA1.
Jednak opierając się na przedstawionych wynikach można stwierdzić, że w
rejonie promotora genu CTA1 zachodzi subtelna reorganizacja struktury chromatyny
przy udziale badanych czynników transkrypcyjnych i ma ona związek m.in. z indukcją
genu CTA1.

63
6. Dyskusja
1. Aktywność transkrypcyjna promotora genu CTA1 i kinetyka
indukcji
Metoda wykorzystująca pomiary aktywności β-galaktozydazy jest metodą
pośrednią i może służyć jedynie do ustalenia przybliżonego poziomu aktywności
transkrypcyjnej promotora genu CTA1. Przeprowadzenie jednak eksperymentu w kilku
niezależnych od siebie powtórzeniach i uzyskanie w nich istotnych statystycznie różnic
w stosunku do szczepu kontrolnego (BY4742) pozwala na formułowanie wniosków
dotyczących udziału czynników transkrypcyjnych Adr1p, Oaf1p i Pip2p oraz ich roli w
indukcji badanego genu.
Kinetyka indukcji genu CTA1 szczepów transformowanych plazmidem
episomalnym znajdujących się w logarytmicznej fazie wzrostu jest liniowa. Tendencję
tę można zauważyć zarówno w szczepie kontrolnym, jak i w szczepach oaf1 i pip2.
Jedynie w szczepie adr1 nie obserwuje się wzrostu poziomu indukcji genu CTA1
między 12 i 24 godziną hodowli w pożywce z kwasem olejowym. Te same szczepy
wykazują nieco inną kinetykę indukcji genu CTA1, kiedy w chwili przenoszenia do
warunków indukcji znajdują się w fazie stacjonarnej. W tym przypadku poziom
indukcji genu CTA1 wzrasta liniowo tylko do 9 godziny hodowli. Osiągnięty poziom
indukcji utrzymuje się aż do 24 godziny hodowli. Wyjątkiem tutaj jest szczep kontrolny,
w którym poziom indukcji genu CTA1 wzrasta przez cały czas trwania hodowli w
warunkach indukcji. Obserwowany przyrost względnego poziomu indukcji z upływem

64
czasu staje się jednak mniejszy. Podobną kinetykę indukcji zaobserwowano już
wcześniej [38]. W tym przypadku również, pomimo znacznie niższej wartości
względnego poziomu indukcji (wynoszącej ok. 70), wzrost względnego poziomu
indukcji był liniowy przez 24 godziny hodowli. Wartości te mogą się różnić z powodu
odmiennego układu doświadczalnego, jednak obserwowane zjawiska mają bardzo
podobną charakterystykę w postaci liniowego wzrostu aktywności genu CTA1.
Indukcja genu CTA1 w szczepach transformowanych plazmidem
integracyjnym przedstawia się podobnie. W logarytmicznej fazie wzrostu, poza
szczepem kontrolnym nie wykazującym wzrostu poziomu indukcji między 12 i 24
godziną hodowli, można uznać, że poziom indukcji wzrasta liniowo przez cały czas
trwania hodowli. Natomiast w fazie stacjonarnej w żadnym z badanych szczepów nie
obserwuje się wyraźnych zmian w poziomie indukcji genu CTA1 po 9 godzinie trwania
hodowli w warunkach indukcji.
Zahamowanie wzrostu poziomu indukcji genu CTA1 w hodowlach będących w
chwili przenoszenia do warunków indukcji w fazie stacjonarnej może wynikać z
ogólnej represji transkrypcji. W komórkach znajdujących się w tej fazie wzrostu
obserwuje się zahamowanie istotnych procesów komórkowych takich jak transkrypcja
[63] i translacja [64, 65]. Możliwe jest więc zahamowanie syntezy białek
odgrywających ważną rolę m.in. w procesie indukcji genu CTA1, a także
β-galaktozydazy będącej kluczowym białkiem w stosowanej metodzie. W przypadku
zahamowania syntezy β-galaktozydazy nie jest możliwe śledzenie rzeczywistej
aktywności transkrypcyjnej promotora genu CTA1.
W szczepie oaf1 transformowanym zarówno plazmidem episomalnym, jak i
plazmidem integracyjnym, w logarytmicznej fazie wzrostu hodowli można zauważyć, iż

65
poziom indukcji genu CTA1 jest wyraźnie niższy w porównaniu do szczepu kontrolnego,
natomiast szczep pip2 transformowany zarówno plazmidem episomalnym, jak i
plazmidem integracyjnym wykazuje podobny do szczepu kontrolnego poziom indukcji
badanego genu. Wyniki te sugerują, iż konieczny jest udział czynnika transkrypcyjnego
Oaf1p do pełnej indukcji genu CTA1. Wysoce prawdopodobne jest więc formowanie
homodimeru (Oaf1p)2 w szczepie pip2, w którym poziom indukcji jest porównywalny
do szczepu kontrolnego. Obserwacje te są zgodne z wcześniej wysuwanymi hipotezami
[39, 45]. Jednak na podstawie przedstawionych wyników nie jest możliwe stwierdzenie,
czy formowanie homodimeru (Oaf1p)2 jest zjawiskiem występującym powszechnie w
indukcji genu CTA1. Istnieje bowiem możliwość, iż homodimer (Oaf1p)2 tworzy się
jedynie w warunkach niedoboru Pip2p. Warto również zauważyć, że pomimo
obniżonego poziomu indukcji genu CTA1 w szczepie oaf1, aktywność promotora tego
genu wzrasta 100-200 razy po trwającej 24 godziny hodowli w pożywce z kwasem
olejowym. Wartość ta wynosi 25-50% w porównaniu do względnego poziomu indukcji
obserwowanego w szczepie kontrolnym. Być może w warunkach braku Oaf1p tworzy
się homodimer (Pip2p)2, który jednak dużo słabiej indukuje gen CTA1 w porównaniu do
homodimeru (Oaf1p)2 lub heterodimeru Oaf1p-Pip2p tworzącego się w szczepie
kontrolnym.
W jednej z przeprowadzonych wcześniej prac [39] zaobserwowano wartość
względnego poziomu indukcji genu CTA1 wynoszącą ok. 4 w szczepie kontrolnym oraz
ok. 2 w szczepie pozbawionym czynnika transkrypcyjnego Oaf1p (obliczenia własne
wg danych z [39]). Wartości te są dużo mniejsze od zaobserwowanych w niniejszej
pracy, jednak stosunek względnego poziomu indukcji w szczepie kontrolnym do
wartości w szczepie pozbawionym czynnika transkrypcyjnego Oaf1p wynosi ok. 2 (4/2).

66
Wartość tę można uznać za porównywalną z obserwowaną w niniejszej pracy (ok. 4
(400/100) w hodowlach, w których zanotowano istotne statystycznie różnice), jeżeli
weźmie się pod uwagę znaczne różnice w stosowanym układzie doświadczalnym [39].
Znaczne różnice w wartościach względnego poziomu indukcji uzyskane w
niniejszej pracy w porównaniu do wcześniejszych prac mogą wynikać nie tylko z
odmiennego układu doświadczalnego (szczepy, plazmidy, metody badania), ale także
niekompletnej derepresji genu CTA1. W celu uzyskania warunków derepresji różne
zespoły badawcze prowadzą hodowlę S.cerevisiae stosując pożywki o stężeniu glukozy
wynoszącym 0% [39], 0,1% [38], 0,3% [42] lub 0,5% [46] uzupełnione zwykle
etanolem lub glicerolem do końcowego stężenia wynoszącego 2%. W pożywce o
stężeniu glukozy wynoszącej 0% komórki S.cerevisiae dzielą się bardzo rzadko,
ponadto często powoduje ona wcześniejsze przejście komórek do fazy stacjonarnej
(wyników nie przedstawiono). Stosowanie pożywki z 0,5% glukozą mogło jednak
opóźnić rozpoczęcie procesu derepresji. Może ona zatem, przy stosowaniu pożywki o
zbyt wysokim stężeniu glukozy, zachodzić dopiero po przeniesieniu komórek drożdży
do pożywki z kwasem olejowym. W takiej sytuacji obserwowany wzrost poziomu
aktywności promotora genu CTA1 jest wynikiem zarówno derepresji, jak i indukcji tego
genu.
Pomimo dużego podobieństwa stosowanych w badanich metod, istnieją także
odmienne wyniki [56] od uzyskanych w niniejszej pracy. W wymienionej pracy szczepy
adr1 i pip2 w logarytmicznej fazie wzrostu charakteryzują się wyższą wartością
względnego poziomu indukcji genu CTA1 w porównaniu do szczepu kontrolnego.
Wyniki te wydają się jednak mało wiarygodne, ponieważ oparte są na jednym
przeprowadzonym doświadczeniu. Natomiast we wspomnianej pracy, podobnie jak w

67
niniejszej, zaobserwowano niższą wartość względnego poziomu indukcji w hodowlach
przeszczepianych w fazie stacjonarnej.
Czynnik transkrypcyjny Adr1p wydaje się nie mieć wpływu na indukcję genu
CTA1 w hodowlach znajdujących się w logarytmicznej fazie wzrostu. Poziom indukcji
badanego genu obserwowany w szczepie adr1 transformowanym zarówno plazmidem
episomalnym, jak i plazmidem integracyjnym nie odbiega zasadniczo od poziomu
indukcji obserwowanego w szczepie kontrolnym. Interesujący jest jednak związek
ekspresji genu PIP2 i Adr1p [66], ponieważ wysoce prawdopodobny jest wpływ Adr1p
na liczbę cząsteczek Pip2p w komórce. Jeżeli jednak za indukację genu CTA1
odpowiedzialny jest homodimer (Oaf1p)2, ani brak Pip2p, ani brak Adr1p nie powinien
mieć wpływu na poziom indukcji genu CTA1. Uzyskane w doświadczeniu wyniki wraz
z prawdopodobnym wpływem Adr1p na ekspresję genu PIP2 przemawiają za
przyjęciem takiej hipotezy, która jest także zgodna z wcześniej wysuwanymi hiptezami
[39, 45].
Stosowany w doświadczeniu plazmid episomalny, jak wcześniej wspomniano,
występuje w komórce w kilkudziesięciu kopiach. Brak wyraźnych różnic w wynikach
uzyskanych w przeprowadzonych doświadczeniach przy użyciu identycznych szczepów
transformowanych plazmidem episomalnym i integracyjnym wskazują więc na dużą
liczbę cząsteczek czynników transkrypcyjnych biorących udział w indukcji genu CTA1
w komórce. Kiedy w komórce jest mała liczba cząsteczek czynników transkrypcyjnych,
w plazmidzie integracyjnym występującym w jednej kopii w komórce wysycenie
sekwencji nukleotydowej promotora genu trwa krócej w porównaniu do plazmidu
episomalnego. Jeśli jednak w komórce jest duży nadmiar liczby cząsteczek czynników
transkrypcyjncyh, w obu plazmidach wysycenie sekwencji nukleotydowej promotora

68
genu CTA1 powinno w przybliżeniu zajmować tyle samo czasu. Uzyskane wyniki, jak
wcześniej wspomniano, sugerują więc występowanie dużego nadmiaru liczby
cząsteczek cznników transkrypcyjnych biorących udział w indukcji genu CTA1.
Obserwacja ta również sugeruje, iż istotnym czynnikiem transkrypcyjnym w
omawianym procesie jest Oaf1p, którego gen wyrażany jest konstytutywnie [45]. Jeśli
czynnik transkrypcyjny Pip2p byłby białkiem koniecznym do indukcji genu CTA1,
wysycenie sekwencji nukleotydowej promotora w plazmidzie episomalnym nastąpiłoby
dużo później w porównaniu do plazmidu integracyjnego, ponieważ gen PIP2
indukowany jest w podobny sposób jak gen CTA1 [45, 66]. Z ostatniej pracy wynika
również, iż ekspresja genu PIP2 jest zależna od czynnika transkrypcyjnego Adr1p [66],
którego gen podlega, podobnie jak gen CTA1 i PIP2, represji glukozowej [68]. Skutki
syntezy cząsteczek Pip2p wcześniej byłyby obserwowane w komórkach z plazmidem
integracyjnym. Brak różnic w kinetyce indukcji genu CTA1 wskazuje zatem na główną
rolę czynnika transkrypcyjnego Oaf1p w omawianym procesie.

69
2. Reorganizacja struktury chromatyny w rejonie promotora
genu CTA1
Obecnie szeroko wykorzystywaną metodą analizy struktury chromatyny jest
metoda oparta na łańcuchowej reakcji polimerazy (PCR) pozwalająca na ustalenie
pozycji nukleosomów z dokładnością do jednego nukleotydu [67]. Wystarczającą
jednak dokładność do wstępnego zbadania struktury chromatyny w celu potwierdzenia
lub wykluczenia udziału czynników transkrypcyjnych Adr1p, Oaf1p i Pip2p w procesie
reorganizacji struktury chromatyny zachodzącym w rejonie promotora genu CTA1
zapewnia stosowana w niniejszej pracy metoda pośredniego znakowania końców [57,
58, 59].
Z wcześniej wykonanej w Zakładzie Biologii Molekularnej pracy wiadomo, że
w rejonie promotora genu CTA1 zachodzi reorganizacja struktury chromatyny zależna
od dostępnego źródła węgla [62]. Uzyskane w niniejszej pracy wyniki potwierdzają tę
obserwację.
Czynnik transkrypcyjny Adr1p jest dobrze poznanym czynnikiem
transkrypcyjnym pod względem reorganizacji struktury chromatyny. Adr1p wiąże się z
sekwencją UAS1 w rejonie promotora genu ADH2 i powoduje destabilizację
nukleosomu uniemożliwiającego dostępu białkom biorącym udział w procesie
transkrypcji do sekwencji TATA-box znajdującej się w rejonie promotora tego genu [23].
Analogiczne działanie Adr1p w rejonie promotora genu CTA1 wydaje się
prawdopodobne, ponieważ sekwencja UAS1 występuje również w rejonie promotora
genu CTA1 [40]. Brak wyraźnych różnic w układzie wykrywanych prążków w stanie

70
represji i derepresji zarówno w szczepie kontrolnym, jak i w szczepie oaf1 i pip2 (w
których funkcjonuje gen ADR1) może jednak świadczyć o tym, że w procesie derepresji
nie zachodzi reorganizacja struktury chromatyny w rejonie promotora genu CTA1. Brak
różnicy w układzie wykrywanych prążków w stanie represji i derepresji w szczepie
pozbawionym Adr1p również potwierdza tę hipotezę.
Podobnie jak w szczepie kontrolnym, w szczepie pip2 wyraźne zmiany
struktury nukleosomowej rejonu promotora zachodzą w warunkach indukcji genu CTA1.
Zmiany te, na podstawie przeprowadzonej wstępnej analizy, wydają się odpowiadać
zmianom obserwowanym w szczepie kontrolnym. Istnieje więc możliwość, iż czynnik
transkrypcyjny Pip2p nie bierze czynnego udziału w reorganizacji struktury chromatyny
rejonu promotora genu CTA1.
Zarówno w świetle analizy aktywności promotora genu CTA1 przeprowadzonej
przy użyciu genu reporterowego, jak i na podstawie wcześniejszych prac [39, 45]
najbardziej interesującym czynnikiem transkrypcyjnym wydaje się Oaf1p. Niestety do
chwili obecnej udało się jedynie przeprowadzić analizę struktury chromatyny w
warunkach represji i derepresji, a więc wtedy, kiedy Oaf1p raczej nie powinien
oddziaływać z promotorem genu CTA1. Podobnie jak w szczepie kontrolnym i szczepie
pip2, nie zaobserwowano wyraźnych zmian w strukturze chromatyny. Możliwe, że
przeprowadzenie analizy preparatu wyizolowanego ze szczepu oaf1 hodowanego w
warunkach indukcji genu CTA1 przyniesie interesujące wyniki dotyczące udziału tego
czynnika transkrypcyjnego w badanym procesie.
Koniec sekwencji nukleotydowej genu RMS1 występującego obok genu CTA1
oddalony jest od pierwszego nukleotydu ulegającego translacji w genie CTA1 jedynie o
310pz (Ryc. 5-5). Część obszaru regulatorowego genu CTA1 może więc leżeć w rejonie

71
sekwencji kodującej genu RMS1. Uzasadniona jest zatem obawa, iż zmiany ekspresji
genu RMS1 mogłyby wpływać na strukturę chromatyny, która następnie może mieć
wpływ na aktywność promotora genu CTA1. Gen RMS1 jest jednak wyrażany
konstytutywnie, a rozluźnienie struktury chromatyny spowodowane brakiem histonu H4
nie wpływa na jego aktywność transkrypcyjną [49]. Zmiany struktury chromatyny
zachodzące w rejonie promotora genu CTA1 są więc z dużym prawdopodobieństwem
związane ze zmianami aktywności promotora genu CTA1. Obserwowane w czasie
indukcji genu CTA1 zmiany struktury chromatyny w obrębie genu RMS1 mogą być
jedynie spowodowane przesunięciem nukleosomu w rejonie promotora genu CTA1.
Może to wymuszać przesunięcia kolejnych nukleosomów, m.in. w obrębie genu RMS1.

72
7. Podsumowanie
W niniejszej pracy ustalono kinetykę indukcji genu CTA1 zarówno w szczepie
kontrolnym drożdży S.cerevisiae, jak i w szczepach pozbawionych jednego z
czynników transkrypcyjnych Adr1p, Oaf1p lub Pip2p. Wzrost wartości względnego
poziomu indukcji jest liniowy we wszystkich badanych szczepach i nie zaobserwowano
żadnych cech wyróżniających któryś ze szczepów na tle innych.
Obserwacje względnego poziomu indukcji sugerują, iż czynnik transkrypcyjny
Oaf1p odgrywa ważną rolę w procesie indukcji genu CTA1. Pozostałe z badanych
czynników transkrypcyjnych wydają się mieć mniejszy wpływ na proces indukcji tego
genu.
Reorganizacja struktury chromatyny jest związana z regulacją aktywności
transkrypcyjnej genu CTA1. Uzyskane wyniki sugerują, iż ten mechanizm jest wyraźnie
związany z indukcją tego genu. W celu zidentyfikowania odpowiedzialnych za ten
proces czynników transkrypcyjnych potrzebne są dalsze badania. Przeprowadzenie
elektroforezy preparatów ze wszystkich szczepów hodowanych w trzech warunkach
regulacji genu CTA1 (represja, derepresja i indukcja) na jednym żelu agarozowym
pozwoli porównać pozycje nukleosomów w poszczególnych szczepach z dużą
wiarygodnością. W ten sposób przeprowadzona analiza najprawdopodobniej
doprowadzi do poznania czynnika transkrypcyjnego związanego z reorganizacją
struktury chromatyny w rejonie promotora genu CTA1.

73
8. Literatura 1. Kornberg RD
Chromatin structure: a repeating unit of histones and DNA. Science 1974 May 24;184(139):868-71
2. Kornberg RD, Thomas JO Chromatin structure; oligomers of the histones. Science 1974 May 24;184(139):865-8
3. Wolffe AP Chromatin: Structure and Function. Academic Press Limited (1995)
4. Arents G, Moudrianakis EN Topography of the histone octamer surface: repeating structural motifs utilized in the docking of nucleosomal DNA. Proc Natl Acad Sci U S A 1993 Nov 15;90(22):10489-93
5. Turner BM, Franchi L, Wallace H Islands of acetylated histone H4 in polytene chromosomes and their relationship to chromatin packaging and transcriptional activity. J Cell Sci 1990 Jun;96 ( Pt 2):335-46
6. Cyert MS, Thorner J Putting it on and taking it off: phosphoprotein phosphatase involvement in cell cycle regulation. Cell 1989 Jun 16;57(6):891-3
7. Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001 Mar 1;410(6824):120-4
8. Goldknopf IL, French MF, Daskal Y, Busch H A reciprocal relationship between contents of free ubiquitin and protein A24, its conjugate with histone 2A, in chromatin fractions obtained by the DNase II, Mg++ procedure. Biochem Biophys Res Commun 1978 Oct 16;84(3):786-93
9. Goldknopf IL, Wilson G, Ballal NR, Busch H Chromatin conjugate protein A24 is cleaved and ubiquitin is lost during chicken erythropoiesis. J Biol Chem 1980 Nov 25;255(22):10555-8

74
10. Hong L, Schroth GP, Matthews HR, Yau P, Bradbury EM Studies of the DNA binding properties of histone H4 amino terminus. Thermal denaturation studies reveal that acetylation markedly reduces the binding constant of the H4 "tail" to DNA. J Biol Chem 1993 Jan 5;268(1):305-14
11. Strahl BD, Allis CD The language of covalent histone modifications. Nature 2000 Jan 6;403(6765):41-5
12. Workman JL, Kingston RE Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu Rev Biochem 1998;67:545-79
13. Wolffe AP, Matzke MA Epigenetics: regulation through repression. Science 1999 Oct 15;286(5439):481-6
14. Jaenisch R, Bird A Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 2003 Mar;33 Suppl:245-54
15. Mohandas T, Shapiro LJ, Sparkes RS Reactivation of an inactive human X chromosome: evidence for X inactivation by DNA methylation. Science 1981 Jan 23;211(4480):393-6
16. Tamaru H, Selker EU A histone H3 methyltransferase controls DNA methylation in Neurospora crassa Nature 2001 Nov 15;414(6861):277-83
17. Irvine RA, Lin IG, Hsieh CL DNA methylation has a local effect on transcription and histone acetylation. Mol Cell Biol 2002 Oct;22(19):6689-96
18. Belotserkovskaya R, Berger SL Interplay between chromatin modifying and remodeling complexes in transcriptional regulation. Crit Rev Eukaryot Gene Expr 1999;9(3-4):221-30
19. Hassan AH, Neely KE, Workman JL Histone acetyltransferase complexes stabilize swi/snf binding to promoter nucleosomes. Cell 2001 Mar 23;104(6):817-27

75
20. Lohr D Chromatin structure and regulation of the eukaryotic regulatory gene GAL80. Proc Natl Acad Sci U S A 1993 Nov 15;90(22):10628-32
21. Lohr D, Lopez J GAL4/GAL80-dependent nucleosome disruption/deposition on the upstream regions of the yeast GAL1-10 and GAL80 genes. J Biol Chem 1995 Nov 17;270(46):27671-8
22. Almer A, Hörz W Nuclease hypersensitive regions with adjacent positioned nucleosomes mark the gene boundaries of the PHO5/PHO3 locus in yeast. EMBO J 1986 Oct;5(10):2681-7
23. Verdone L, Camilloni G, Di Mauro E, Caserta M Chromatin remodeling during Saccharomyces cerevisiae ADH2 gene activation. Mol Cell Biol 1996 May;16(5):1978-88
24. Verdone L, Cesari F, Denis CL, Di Mauro E, Caserta M Factors affecting Saccharomyces cerevisiae ADH2 chromatin remodeling and transcription. J Biol Chem 1997 Dec 5;272(49):30828-34
25. Di Mauro E, Kendrew SG, Caserta M Two distinct nucleosome alterations characterize chromatin remodeling at the Saccharomyces cerevisiae ADH2 promoter. J Biol Chem 2000 Mar 17;275(11):7612-8
26. Rubbi L, Camilloni G, Caserta M, Di Mauro E, Venditti S Chromatin structure of the Saccharomyces cerevisiae DNA topoisomerase I promoter in different growth phases. Biochem J 1997 Dec 1;328 ( Pt 2):401-7
27. Perez-Martin J Chromatin and transcription in Saccharomyces cerevisiae. FEMS Microbiol Rev 1999 Jul;23(4):503-23
28. White CL, Suto RK, Luger K Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions. EMBO J 2001 Sep 17;20(18):5207-18
29. Kunau WH, Dommes V, Schulz H β-oxidation of fatty acids in mitochondria, peroxisomes, and bacteria: a century of continued progress. Prog Lipid Res 1995;34(4):267-342

76
30. Veenhuis M, Mateblowski M, Kunau WH, Harder W Proliferation of microbodies in Saccharomyces cerevisiae. Yeast 1987 Jun;3(2):77-84
31. Gancedo JM Yeast carbon catabolite repression. Microbiol Mol Biol Rev 1998 Jun;62(2):334-61
32. Filipits M, Simon MM, Rapatz W, Hamilton B, Ruis H A Saccharomyces cerevisiae upstream activating sequence mediates induction of peroxisome proliferation by fatty acids. Gene 1993 Sep 30;132(1):49-55
33. Distel B, Erdmann R, Gould SJ, Blobel G, Crane DI, Cregg JM, Dodt G, Fujiki Y, Goodman JM, Just WW, Kiel JA, Kunau WH, Lazarow PB, Mannaerts GP, Moser HW, Osumi T, Rachubinski RA, Roscher A, Subramani S, Tabak HF, Tsukamoto T, Valle D, van der Klei I, van Veldhoven PP, Veenhuis M. A unified nomenclature for peroxisome biogenesis factors. J Cell Biol 1996 Oct;135(1):1-3
34. Brul S, Westerveld A, Strijland A, Wanders RJ, Schram AW, Heymans HS, Schutgens RB, van den Bosch H, Tager JM. Genetic heterogeneity in the cerebrohepatorenal (Zellweger) syndrome and other inherited disorders with a generalized impairment of peroxisomal functions – a study using complementation analysis. J Clin Invest 1988 Jun;81(6):1710-5
35. Cohen G, Rapatz W, Ruis H Sequence of the Saccharomyces cerevisiae CTA1 gene and amino acid sequence of catalase A derived from it. Eur J Biochem 1988 Sep 1;176(1):159-63
36. Gurvitz A, Hamilton B, Ruis H, Hartig A Peroxisomal Degradation of trans-Unsaturated Fatty Acids in the Yeast Saccharomyces
cerevisiae J Biol Chem 2001 Jan 12;276(2):895-903
37. Hortner H, Ammerer G, Hartter E, Hamilton B, Rytka J, Bilinski T, Ruis H Regulation of synthesis of catalases and iso-1-cytochrome c in Saccharomyces cerevisiae by glucose, oxygen and heme. Eur J Biochem 1982 Nov;128(1):179-84

77
38. Skoneczny M, Rytka J Oxygen and haem regulate the synthesis of peroxisomal proteins: catalase A, acyl-CoA oxidase and Pex1p in the yeast Saccharomyces cerevisiae; the regulation of these proteins by oxygen is not mediated by haem. Biochem J 2000 Aug 15;350 Pt 1:313-9
39. Karpichev IV, Small GM Global regulatory functions of Oaf1p and Pip2p (Oaf2p), transcription factors that regulate genes encoding peroxisomal proteins in Saccharomyces cerevisiae. Mol Cell Biol 1998 Nov;18(11):6560-70
40. Cheng C, Kacherovsky N, Dombek KM, Camier S, Thukral SK, Rhim E, Young ET Identification of potential target genes for Adr1p through characterization of essential nucleotides in UAS1. Mol Cell Biol 1994 Jun;14(6):3842-52
41. Simon M, Adam G, Rapatz W, Spevak W, Ruis H The Saccharomyces cerevisiae ADR1 gene is a positive regulator of transcription of genes encoding peroxisomal proteins. Mol Cell Biol 1991 Feb;11(2):699-704
42. Rottensteiner H, Kal AJ, Filipits M, Binder M, Hamilton B, Tabak HF, Ruis H Pip2p: a transcriptional regulator of peroxisome proliferation in the yeast Saccharomyces
cerevisiae. EMBO J 1996 Jun 17;15(12):2924-34
43. Karpichev IV, Luo Y, Marians RC, Small GM A complex containing two transcription factors regulates peroxisome proliferation and the coordinate induction of β-oxidation enzymes in Saccharomyces cerevisiae. Mol Cell Biol 1997 Jan;17(1):69-80
44. Luo Y, Karpichev IV, Kohanski RA, Small GM Purification, identification, and properties of a Saccharomyces cerevisiae oleate-activated upstream activating sequence-binding protein that is involved in the activation of POX1. J Biol Chem 1996 May 17;271(20):12068-75
45. Rottensteiner H, Kal AJ, Hamilton B, Ruis H, Tabak HF A heterodimer of the Zn2Cys6 transcription factors Pip2p and Oaf1p controls induction of genes encoding peroxisomal proteins in Saccharomyces cerevisiae. Eur J Biochem 1997 Aug 1;247(3):776-83

78
46. Baumgartner U, Hamilton B, Piskacek M, Ruis H, Rottensteiner H Functional analysis of the Zn2Cys6 transcription factors Oaf1p and Pip2p. Different roles in fatty acid induction of beta-oxidation in Saccharomyces cerevisiae. J Biol Chem 1999 Aug 6;274(32):22208-16
47. Simon M, Binder M, Adam G, Hartig A, Ruis H Control of peroxisome proliferation in Saccharomyces cerevisiae by ADR1, SNF1 (CAT1, CCR1) and SNF4 (CAT3). Yeast 1992 Apr;8(4):303-9
48. Young ET, Kacherovsky N, Van Riper K Snf1 protein kinase regulates Adr1 binding to chromatin but not transcription activation. J Biol Chem 2002 Oct 11;277(41):38095-103
49. Wyrick JJ, Holstege FC, Jennings EG, Causton HC, Shore D, Grunstein M, Lander ES, Young RA Chromosomal landscape of nucleosome-dependent gene expression and silencing in yeast. Nature 1999 Nov 25;402(6760):418-21
50. Chen P, Johnson P, Sommer T, Jentsch S, Hochstrasser M Multiple ubiquitin-conjugating enzymes participate in the in vivo degradation of the yeast MAT alpha 2 repressor. Cell 1993 Jul 30;74(2):357-69
51. Myers AM, Tzagoloff A, Kinney DM, Lusty CJ Yeast shuttle and integrative vectors with multiple cloning sites suitable for construction of lacZ fusions. Gene 1986;45(3):299-310
52. Sambrook J, Maniatis T, Fritsch EF Molecular Cloning. A laboratory manual. Cold Spring Harbor Laboratory Press (1989)
53. Gietz RD, Woods RA Transformation of yeast by lithium acetate / single-strandedcarrier DNA / polyethylene glycol method. Methods Enzymol 2002;350:87-96
54. Bradford MM A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976 May 7;72:248-54

79
55. Guarente L Yeast promoters and lacZ fusions designed to study expression of cloned genes in yeast. Methods Enzymol 1983;101:181-91
56. Solińska M Analiza aktywności transkrypcyjnej promotora genu CTA1 Saccharomyces cerevisiae. Praca magisterska wykonana w Zakładzie Biologii Molekularnej Wydziału Biologii UW (2002)
57. Wu C The 5' ends of Drosophila heat shock genes in chromatin are hypersensitive to DNase I. Nature 1980 Aug 28;286(5776):854-60
58. Wu C Analysis of hypersensitive sites in chromatin. Methods Enzymol 1989;170:269-89
59. Nedospasov SA, Shakhov AN, Georgiev GP Analysis of nucleosome positioning by indirect end-labeling and molecular cloning. Methods Enzymol 1989;170:408-20
60. Keene MA, Corces V, Lowenhaupt K, Elgin SC DNase I hypersensitive sites in Drosophila chromatin occur at the 5' ends of regions of transcription. Proc Natl Acad Sci U S A. 1981 Jan;78(1):143-6
61. Sledziewski A, Young ET Chromatin conformational changes accompany transcriptional activation of a glucose-repressed gene in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1982 Jan;79(2):253-6
62. Trzcińska-Danielewicz J, Bulkowska U, Fronk J Struktura nukleosomowa promotora genu CTA1 Saccharomyces cerevisiae Plakat prezentowany na XXXVI zjeździe Polskiego Towarzystwa Biochemicznego, Poznań (2000)
63. Choder M A general topoisomerase I-dependent transcriptional repression in the stationary phase in yeast. Genes Dev 1991 Dec;5(12A):2315-26
64. Fuge EK, Braun EL, Werner-Washburne M Protein synthesis in long-term stationary-phase cultures of Saccharomyces cerevisiae. J Bacteriol 1994 Sep;176(18):5802-13

80
65. Boucherie H Protein synthesis during transition and stationary phases under glucose limitation in Saccharomyces cerevisiae. J Bacteriol 1985 Jan;161(1):385-92
66. Rottensteiner H, Wabnegger L, Erdmann R, Hamilton B, Ruis H, Hartig A, Gurvitz A Saccharomyces cerevisiae PIP2 mediating oleic acid induction and peroxisome proliferation is regulated by Adr1p and Pip2p-Oaf1p. J Biol Chem 2003 May 14 (w chwili opracowywania spisu literatury dostępna była jedynie wersja elektroniczna, PMID: 12748191)
67. Venditti P, Costanzo G, Negri R, Camilloni G ABFI contributes to the chromatin organization of Saccharomyces cerevisiae ARS1 B-domain. Biochim Biophys Acta 1994 Nov 22;1219(3):677-89
68. Blumberg H, Hartshorne TA, Young ET Regulation of expression and activity of the yeast transcription factor ADR1. Mol Cell Biol 1988 May;8(5):1868-76

















![Marcin Filipecki - Wykorzystanie miejscowo-specyficznych ...marcin_filipecki.users.sggw.pl/PDFY/Inzynieria...stały opisane w latach osiemdziesiątych XX wieku u drożdży [3, 40].](https://static.fdocuments.pl/doc/165x107/611de75b1b08636f6e439093/marcin-filipecki-wykorzystanie-miejscowo-specyficznych-marcin-stay-opisane.jpg)
