Spektroskopia molekularna
65
1 Spektroskopia molekularna Materiały na ćwiczenia do Wykładu 1 Zadanie 1. Wyznaczenie elementów symetrii cząsteczek nadtlenku wodoru H 2 O 2 , formaldehydu CH 2 O, benzenu C 6 H 6 , kwasu solnego HCl i azotu cząsteczkowego N 2 . Wykazać, że trzy osie dwukrotne w grupie benzenu D 6h są ze sobą sprzężone. Zadanie 2. Rozkład reprezentacji grupy C i na reprezentacje nieprzywiedlne. Tablica charakterów: C i E I ------------------------------------------------------------------------------------------- A g 1 1 R x , R y , R z x 2 , y 2 , z 2 xy, xz, yz A u 1 –1 x, y, z ------------------------------------------------------------------------------------------
Transcript of Spektroskopia molekularna

1
Spektroskopia molekularna
Materiały na ćwiczenia do Wykładu 1
Zadanie 1. Wyznaczenie elementów symetrii cząsteczek nadtlenku wodoru H2O2,
formaldehydu CH2O, benzenu C6H6, kwasu solnego HCl i azotu cząsteczkowego
N2. Wykazać, że trzy osie dwukrotne w grupie benzenu D6h są ze sobą sprzężone.
Zadanie 2. Rozkład reprezentacji grupy Ci na reprezentacje nieprzywiedlne.
Tablica charakterów: Ci E I
-------------------------------------------------------------------------------------------
Ag 1 1 Rx, Ry, Rz x2, y
2, z
2
xy, xz, yz
Au 1 –1 x, y, z
------------------------------------------------------------------------------------------

2
Rozwiązanie zadania 1
O O
HH
C2
C2
(2)
C2v
(2mm)
C
H
H
O
v
C2
v
UWAGA: w przypadku cząsteczki H2O2 wygodnie jest przedstawić ją w rzucie
„z góry”
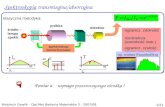
3
C∞v
H Clv
C
C
N Nv
D∞h
I●
C
C
S
S
h
UWAGA:
- płaszczyzn v jest nieskończenie wiele ( v)
- w grupie Dh nie zaznaczono (dla przejrzystości rysunku) nieskończenie wielu
osi C2 prostopadłych do osi cząsteczki C

4
D6h
(6/mmm)
I●
d
C2'(1)
C2'(2)
Rozpatrzmy dwie osie dwukrotne:
- C2’(1) przechodzącą przez węgle (i protony) 2 i 5
- C2’(2) przechodzącą przez węgle (i protony) 1 i 4
Działanie obu transformacjami na węgle i protony benzenu jest następujące:
1 3 1 1
C2’(1) 2 2 C2’(2) 2 6
4 6 3 5
5 5 4 4
Warunek sprzęgania: istnienie w grupie operacji symetrii X takiej, że zachodzi:
C2’(2) = X1
C2’(1)X
X = d płaszczyzna dzieląca na pół wiązania C(1)–C(2) i C(4)–C(5); d1
= d
1 2 1 2 2 1
d 3 6 dC2’(1)d 2 1 3 6
4 5 3 6 4 5
4 5 5 4

5
Rozwiązanie zadania 2.
Jeżeli (x) jest dowolną funkcją zadaną na układzie symetrycznym to zachodzi
rR(Rx) = (x) gdzie operacje symetrii R = E (tożsamość), I (inwersja)
stąd:
rE(Ex) = rE(x) = (x)
rI(Ix) = rI (–x) = (x)
Przyjmijmy więc jako funkcje bazowe dowolnej, przywiedlnej reprezentacji:
f1 = (x) f2 = (–x)
rE f1 = 1 f1 + 0 f2 rE f2 = 0 f1 + 1 f2
rI f1 = 0 f1 + 1 f2 rI f2 = 1 f1 + 0 f2
szukamy macierzy w tej bazie funkcyjnej: rRfi = ji
2
1jj )R(Df∑
czyli: D(E) =
10
01 oraz D(I) =
01
10 to reprezentanci operatorów E
oraz I macierzowo zapisani w bazie f1 i f2 (reprezentacja przywiedlna)
Konstrukcja reprezentacji nieprzywiedlnych:
Dokonujemy transformacji liniowej funkcji bazowych f1, f2 do funkcji f1’, f2’:
f1’ = f1 + f2 f2’ = f1 – f2
czyli fi’ = jij
2
1=j
fM∑ Mij =
1-1
11
rE f1’ = 1 f1’ rI f1’ = 1 f1’ f1’ - funkcja bazowa reprezentacji nieprzywiedlnej Ag
rE f2’ = 1 f2’ rI f2’ = –1 f2’ f2’
-
funkcja bazowa reprezentacji nieprzywiedlnej Au
Macierzowo zapisani reprezentanci operatorów w nowej bazie f1’, f2’:
D’ = M D M–1
M–1
=
2/1-2/1
2/12/1

6
wykonujemy mnożenie macierzy:
D’(E) =
1-1
11
10
01
2/1-2/1
2/12/1 =
10
01 =
u
g
A0
0A
D’(I) =
1-1
11
01
10
2/1-2/1
2/12/1 =
1-0
01 =
u
g
A0
0A
czyli: D’ = Ag Au ; rozkład reprezentacji przywiedlnej na nieprzywiedlne

7
Spektroskopia molekularna
Materiały na ćwiczenia do Wykładu 2
Zadanie 3: Zerowanie całki jednowymiarowej z iloczynu funkcji symetrycznej i
antysymetrycznej wykazane przez zmianę układu współrzędnych x x.
Zadanie 4: Konformacja wokół wiązania w rzucie Newmanna; wyznaczenie
wartości kąta dwuściennego dla fragmentu etanu H1C1C2H2 o
współrzędnych: r(C1) = (0 0 0), r(C2) = (0 1.5 0), r(H1) = (0 0.49 0.87), r(H2) =
(0.75 1.99 0.44).
Do jakiej grupy symetrii można zaliczyć cząsteczkę etanu CH3CH3 zależnie od
kąta ? (do przećwiczenia „w domu”)

8
Rozwiązanie zadania 3.
Rozważmy dwie funkcje jednej zmiennej f(x) i g(x) oraz całkę ich iloczynu
f(x)g(x)dx. Całka taka jest równa powierzchni pod krzywą f(x)g(x).
Niech funkcja f będzie asymetryczna:
f(x) = f(x)
Niech funkcja g będzie symetryczna:
g(x) = g(x)
Dla poglądowego przedstawienia zagadnienia można przyjąć:
f(x) = x
g(x) = x2 1
i narysować wykresy obu funkcji oraz ich iloczynu.
Nie obliczając wartości całki wprowadźmy nowy układ współrzędnych o
przeciwnie do poprzedniego skierowanej osi x (x x), czyli dokonujemy
transformacji symetrii. Całka z iloczynu funkcji f(x) i g(x) w nowym układzie
współrzędnych:
f(x)g(x)dx = f(x)g(x)dx z własności symetrii obu funkcji
musi być równa całce w poprzednim układzie współrzędnych ponieważ pole pod
krzywą nie ulega zmianie przy transformacji:
f(x)g(x)dx = f(x)g(x)dx
a to może zachodzić tylko wtedy gdy f(x)g(x)dx = 0
Rozwiązanie zadania 4.
W rzucie Newmanna dla fragmentu P1ABP2, w którym atomy podstawników
P1 i P2 zmieniają położenie na skutek obrotu wokół wiązania AB, atom A
oznaczamy kropką, atom B koncentrycznym okręgiem (patrzymy wzdłuż wiązania
AB ustawionego prostopadle do płaszczyzny kartki z atomem A ustawinym bliżej
nas) a wiązania podstawników P1 i P2 tworza kąt . Kąt ma wartość dodatnią jeśli
obracamy wiązanie z atomem P2 zgodnie z ruchem wskazówek zegara w stosunku

9
do wiązania P1 a wartość ujemną jeśli obracamy przeciwnie do ruchu wskazówek
zegara.
Rozwiązanie 1.
Korzystamy z rzutu R1 wektora C1H1 i rzutu R2 wektora C2H2 (po jego
przesunięciu do początku układu współrzędnych) na płaszczyznę xz (rzut
Newmanna):
R1 = (0 0 0.87)
R2 = (0.75 0 0.44)
Kat dwuścienny między wektorami R1 i R2 obliczmy z iloczynu skalarnego:
R1 R2 = (0 0 0.87) (0.75 0 0.44) = 0.870.44
|R1| = 0.87
|R2| = 0.87
cos = (0.870.44)/(0.870.87) = 0.5
= 60
Kąt może mieć wartość +60 lub 60 zależnie od kierunku patrzenia wzdłuż
wiązania: C1C2 lub C2C1.
Rozwiązanie 2.
Kąt dwuścienny = 180, gdzie jest tzw. katem torsyjnym między wektorami
normalnymi N1 i N2 do płaszczyzn wyznaczonych przez atomy (punkty) P1AB i
ABP2. Wartość tego kąta uzyskujemy z iloczynu skalarnego N1 i N2:
N1 N2 = |N1||N2|cos
W przypadku fragmentu etanu H1C1C2H2 wektor N1 jest normalny do
płaszczyzny przechodzącej przez H1, C1 i C2, czyli płaszczyzny rozpiętej na
wektorach C1H1 = r(H1) = (0 0.49 0.87) i C1C2 = r(C2) = (0 1.5 0), a wektor
N2 jest normalny do płaszczyzny przechodzacej przez C1, C2 i H2, czyli
płaszczyzny rozpiętej na wektorach C1C2 = r(C2) = (0 1.5 0) i C2H2 = r(H2) po
jego przesunięciu do początku ukladu współrzędnych = (0.75 0.49 0.44).
Współrzędne wektorów N1 i N2 wyznaczamy z iloczynów wektorowych (x)
wektorów rozpinających obie płaszczyzny (patrz ćwiczenia do wykładu Fizyka z
matematyką II; macierze i wyznaczniki):
N1 = (0 0.49 0.87) x (0 1.5 0) = (1.3 0 0)

10
N2 = (0 1.5 0) x (0.75 0.49 0.44) = (0.66 0 1.125)
|N1| = 1.3
|N2| = 1.3
N1 N2 = (1.3)0.66
Stąd:
cos = 0.66/1.3 = 0.5 czyli = 120
a szukany kąt dwuścienny:
= 60
„Praca domowa”
Cząsteczkę etanu można zaliczyć do grupy D3h w konformacji z nakrywającymi się
wiązaniami węgiel-wodór ( = 0), do grupy D3d w konformacji naprzemiennej (
= 60), i do grupy D3 w konformacji o dowolnym różnym od 0 i różnym od 60.

11
Spektroskopia molekularna
Materiały na ćwiczenia do Wykładu 3
Zadanie 5: Wyprowadzenie wzoru na współczynnik Einsteina emisji
spontanicznej z równowagi termodynamicznej układu molekularnego i
promieniowania elektromagnetycznego oraz ze wzoru Plancka na gęstość energii
promieniowania: = 8h(if/c) 3[exp(hif/kT) 1]
1
Zadanie 6: Klasyczne wyprowadzenie wzoru na rozproszenie Ramana dla
cząsteczki dwuatomowej.

12
Rozwiązanie zadania 5.
Rozważmy dwa poziomy energetyczne o energiach Ef > Ei obsadzone przez Nf i Ni
cząsteczek. W warunkach równowagi obowiązuje rozkład Boltzmanna:
Nf/Ni = exp(E/kT) E = Ef Ei T - temperatura
Promieniowanie o częstości if dopasowanej do różnicy energii poziomów:
hif = E
i gęstości wzbudza absorpcję i emisję wymuszoną, tak że ilość cząsteczek
przechodzących na wyższy poziom w jednostce czasu równa się ilości cząsteczek
wracających na niższy poziom; promieniowanie nie zaburza rozkładu obsadzeń.
Ilość cząsteczek wzbudzanych w jednostce czasu w procesie absorpcji wynosi
WfiNf a ilość cząsteczek dezaktywowanych w procesie emisji wymuszonej wynosi
WifNi. Jeśli w warunkach równowagi termodynamicznej proces emisji wymuszonej
byłby jedynym procesem dezaktywacji stanu wzbudzonego f to:
WfiNi = WifNf
Prawdopodobieństwa przejść na jednostkę czasu Wfi = Bfi = Bif = Wif gdzie
współczynnika Einsteina dla absorpcji i emisji wymuszonej są sobie równe
(proporcjonalne do momentu dipolowego przejścia) Bfi = Bif = B. Stąd Nf = Ni co
jest sprzeczne z rozkładem Boltzmanna.
Musi więc istnieć dodatkowy proces dezaktywacji stanu wzbudzonego niezależny
od gęstości promieniowania, z prawdopodobieństwem na jednostkę czasu Afi:
BNf + AfiNf = BNi
Stąd wyznaczamy jego wartość:
Aif = B(Ni/Nf 1)
i korzystając z rozkładu Boltzmanna i dopasowania różnicy energii poziomów do
częstości promieniowania:
Aif = B[exp(hif/kT) 1]
Z wzoru Plancka na gęstość promieniowania w równowadze z ciałem doskonale
czarnym o temperaturze T:
= 8h(if/c) 3[exp(hif/kT) 1]
1
i prawdopodobieństwo emisji spontanicznej na jednostkę czasu:
Afif = 8h(if/c) 3Bif

13
Rozwiązanie zadania 6.
Jeśli na cząsteczkę pada promieniowanie o częstości i to indukowany jest
moment dipolowy (czcionka pogrubiona oznacza wektory i tensory):
d = E gdzie jest tensorem polaryzowalności a E wektorem
natężenia pola elektrycznego
Dla uproszczenia (nie tracąc na ogólności wyprowadzenia) rozpatrzmy czasteczkę
dwuatomową umieszczoną wzdłuż osi z, drgającą z częstością v i rotującą z
częstościa r, na którą pada spolaryzowana liniowo fala płaska. Tensor redukuje
się do skalara zz = i zachodzi:
d = E
E = E0cos(t)
w wyniku oscylacji:
= 0 + 0’cos(vt)
w wyniku rotacji:
= 0 + 0’cos(2rt)
czynnik 2 wynika z faktu, że polaryzowalność osiąga ponownie tę samą wartość po
wykonaniu przez cząsteczkę połowy obrotu.
Podstawiając do wzoru na moment dipolowy:
d = [0 + 0’cos(vt)]E0cos(t) = 0E0cos(t) + 0’E0cos(t) cos(vt)
d = = 0E0cos(t) + 0’E0cos(t) cos(2rt)
Pierwszy człon oznacza rozpraszanie Rayleigha, a drugi można przekształcić
korzystając z tożsamości trygonometrycznych:
cos(a + b) = cos(a)cos(b) sin(a)sin(b)
cos(a b) = cos(a)cos(b) + sin(a)sin(b)
do postaci dla oscylacji:
0’E0cos(t) cos(vt) = (1/2)0’E0{cos[( v)t] + cos[( + v)t]}
i analogicznie dla rotacji:
0’E0cos(t) cos(2rt) = (1/2)0’E0{cos[( 2r)t] + cos[( + 2r)t]}
Pierwszy człon odpowiada stokesowskiemu rozproszeniu Ramana a drugi
antystokesowskiemu rozproszeniu Ramana.

14
Spektroskopia molekularna
Materiały na ćwiczenia do Wykładu 4
Zadanie 7: Wyznaczyć widmo w funkcji częstości dla kwadratowego impulsu
promieniowania o zależności funkcyjnej od czasu t:
f(t) = 1 dla |t| A
0 dla |t| > A
Wskazówka: skorzystać z transformacji Fouriera
F(v) =
dt)ti2exp()t(f
Zadanie 8: Oszacowanie położenia pasma absorpcji IR (w cm1
) dla fragmentu
dwuatomowego C=O; stała siłowa dla drgań rozciągających (według G. Herzberg
„Molecular Spectra and Molecular Structure II”) f = 12.1105 dyn/cm.
Potrzebne wartości stałych fizycznych:
- liczba Avogadro NA = 6.021023
mol1
- prędkość światła c = 31010
cm/s

15
Rozwiązanie zadania 7
Zależności impulsu w funkcji czasu f(t) i w funkcji częstości F() związane są
transformacja Fouriera (podaną we wskazówce):
F() =
dt)ti2exp()t(f =
A
A
dt)ti2exp()t(f
Korzystamy z zależności Eulera: exp(i) = cos isin
F() =
dt)]t2sin(i)t2[cos(
podstawiamy: = 2t dt = [1/(2)]d
[cos(2t) isin(2t)]dt = [cos](1/2)d i[sin](1/2)d
F() = (1/2)[sin(2t)|-A+A
+ icos(2t)|-A+A
] =
)A2sin(
F() jest krzywą “dzwonową”, której przebieg proszę przeanalizować przyjmując
dla uproszczenia A = 1.
Rozwiązanie zadania 8
Częstość oscylacji w przybliżeniu cząsteczki dwuatomowej:
osc = (1/2) /f
masa zredukowana
= MCMO/(MC + MO)

16
Masa węgla MC = 12 j.m.a.; masa tlenu MC = 16 j.m.a.; = 6.86 j.m.a. czyli tyle
wynosi masa w gramach jednego mola, co wyraża się w gramach na jedną
cząsteczkę po podzieleniu przez liczbę Avogadro:
/NA = 1.141023
g
Podstawiamy do wzoru:
osc = (1/6.28) 23-5 10•14.1/10•1.12 g•cm/s•cm•g 2- =
= 0.163.261014
s1
= 0.521014
s1
Położenie przejścia w cm1
obliczamy korzystając z relacji:
= c
Stąd: 1
= osc/c = 0.521014
/31010
cm1
= 0.173104cm
1 = 1730 cm
1

17
Spektroskopia molekularna
Materiały na ćwiczenia do Wykładu 5
Zadanie 9: Analiza struktury rotacyjnej widma absorpcji cząsteczki HCl (widmo z
Alpert, Keiser, Szymanski „Spektroskopia w podczerwieni. Teoria i praktyka”,
PWN 1974); wyznaczenie stałej rotacyjnej B i długości wiązania rHCl.
Zadanie 10: Określenie drgań aktywnych w widmie absorpcyjnym IR i widmie
Ramana cząsteczek CO2 (Dh), i NH3 (C3v) na podstawie własności
transformacyjnych składowych elektrycznego momentu dipolowego i
polaryzowalności (tablice charakterów).
CO2: częstości i symetria drgań: 2349,3cm1
(u+), 1388,3cm
1(g
+), 667,3cm
1(u)
Tablica charakterów:
Dh E 2C
v i 2S
C2
g+
1 1 1 1 1 1 x2 + y
2, z
2
g 1 1 1 1 1 1 Rz
g
2 2cos 0 2 2cos 0 (Rx, Ry) (xz, yz)
g
2 2cos2 0 2 2cos2 0 (x2 y
2, xy)
… ………………………………………………...
u+
1 1 1 1 1 1 z
u 1 1 1 1 1 1
u
2 2cos 0 2 2cos 0 (x, y)
u
2 2cos2 0 2 2cos2 0
… ………………………………………………...

18
NH3: 3336,0cm1
(A1), 950,24cm1
(A1), 3414,9 cm1
(E), 1627,77(E)
Tablica charakterów:
C3v E 2C3 3v
A1
1 1 1 z x2 + y
2, z
2
A2 1 1 1 Rz
E 2 1 0 (x, y) (Rx, Ry) (x2 y
2, xy) (xz, yz)
Wskazówka: funkcja falowa oscylacyjnego stanu podstawowego v = 0 (wielomian
Hermite’a) transformuje się według (tworzy bazę) nieprzywiedlnej reprezentacji
pełnosymetrycznej a funkcja falowa pierwszego stanu wzbudzonego v = 1
transformuje się tak jak współrzędna normalna.
Zadanie 10a: Częstości drgań nieliniowej, symetrycznej cząsteczki trójatomowej
XY2 (H2O) o danych współrzędnych symetrii S1, S2 i S3. Grupy symetrii C2v, dwa
drgania normalne A1 i jedno B1.
C2v E C2 v(xz) v(yz)
A1
1 1 1 1 z x2, y
2, z
2
A2 1 1 1 1 Rz xy
B1 1 1 1 1 x, Ry xz
B2 1 1 1 1 y, Rx yz

19

20

21
Rozwiązanie zadania 9.
Pasmo absorpcji IR cząsteczki HCl dla dowolnego przejścia oscylacyjnego o
częstości
v’0 składa się z dwóch gałęzi struktury rotacyjnej:
gałąź R, J = +1, położenia linii:
(a)
R =
v’0 + 2B’v + (3B’v – B’’v)J + (B’v – B’’v)J2 J = J’’ = 0, 1, 2, 3,...
gałąź P, J = 1, położenia linii:
(b)
P =
v’0 – (B’v + B’’v)J + (B’v – B’’v)J2 J = 1, 2, 3,...
Dla przejścia z poziomu v’’ = 0 na poziom v’ = 1 czyli tonu podstawowego (
10
równe ok. 2885,7 cm1
dla H35
Cl i ok. 2883,6 cm1
dla H37
Cl) rozpatrzmy pasmo
dla cząsteczki H35
Cl o bardziej intensywnych sygnałach (75% zawartości) oraz dla
cząsteczki H37
Cl o mniej intensywnych liniach (25% zawartości); obliczenia są
analogiczne w obu przypadkach.
Niesztywność rotatora nie wpływa w znaczący sposób na położenia linii, natomiast
trzeba uwzględnić sprzężenie osylacji i rotacji, tzn. stała rotacyjna Bv’’
(poziom oscylacyjny v’’ = 0) może być różna od stałej rotacyjnej Bv’ (poziom
oscylacyjny v’ = 1). Przy równych stałych Bv’’ = Bv’= B obie gałęzie składałyby
się z linii równoodległych o 2B. Sprawdzamy dla dwóch pierwszych linii gałęzi P i
gałęzi R dla cząsteczki H35
Cl:
2865,10 2843.62 = 21,48 B = 10,74
2925,90 2906,24 = 19,66 B = 9,83
i dla dwóch ostatnich (oznaczonych) linii obu gałęzi:
2677,73 2651,96 = 25,77 B = 12,88
3059,32 3045,06 = 14,26 B = 7,13

22
Widać, że wartości B są różne; stosunkowo najbliższą wartość rzeczywistej można
otrzymać dla małych J czyli średnia z odstępów dla dwu pierwszych linii w
gałęziach: P (21,48) i R (19,66) daje B = 10,29 cm1
.
Sprawdzamy dla dwóch pierwszych linii gałęzi P i R dla cząsteczki H37
Cl:
2863,02 2841,58 = 21,44 B = 10,72
2923,72 2904,11 = 19,61 B = 9,805
i dla dwóch ostatnich (oznaczonych) linii obu gałęzi:
2675,94 2650,22 = 25,72 B = 12,86
3056,97 3042,73 = 14,24 B = 7,12
Średnia z odstępów dla dwu pierwszych linii w gałęzi P (21,44) i dwu pierwszych
linii w gałęzi R (19,61) daje B = 10,26 cm1
.
Dokładniejsze obliczenie wartości B bez przyjętego założenia (Bv’’ = Bv’ = B)
Położenia dwóch pierwszych linii gałęzi R ze wzoru (a) wprowadzając
upraszczające oznaczenie
10
:
H35
Cl H37
Cl
R(0) = 2906,24 =
+ 2Bv’
R(0) = 2904,11 =
+ 2Bv’
R(1) = 2925,90 =
+ 6Bv’ 2Bv’’
R(1) = 2923,72 =
+ 6Bv’ 2Bv’’
i dla dwóch pierwszych linii gałęzi P ze wzoru (b):
P(1) = 2865,10 =
2Bv’
P(1) = 2863,02 =
2Bv’
P(2) = 2843,62 =
+ 2Bv’ 6Bv’’
P(2) = 2841,58 =
+ 2Bv’ 6Bv’’
Odjęcie od siebie stronami pierwszego i czwartego równania daje wartość Bv’’:
62,62 = 6Bv’’ 62,58 = 6Bv’’
Bv’’ = 10,43 cm1 >
Bv’’ = 10,42 cm1

23
Dodanie do siebie stronami pierwszego i trzeciego równania i podzielenie przez 2
daje częstości przejścia czysto oscylacyjnego
w obu cząsteczkach.
Najdokładniejsze wyznaczenie stałych rotacyjnych otrzymuje się metodą „różnic
kombinacyjnych” na podstawie fitowania prostej do położeń wszystkich linii:
R(J)
P(J) = 4Bv’(J + ½)
R(J 1)
P(J + 1) = 4Bv’’(J + ½)
Wartość tablicowa (bez podania której cząsteczki dotyczy): Bv’’ = 10,5909 cm1
Następnie obliczamy masę zredukowaną (patrz zadanie poprzednie) i korzystamy
ze wzorów na stałą rotacyjną B = h/(82cI) i moment bezwładności I = rHCl
2 :
rHCl = Bc8
h2
(H35
Cl) = (135)/36 = 0.972 Da < (H37
Cl) = (135)/36 = 0.974 Da
i można sprawdzić zgodność stosunku (H35
Cl)/(H37
Cl) ze stosunkiem stałych
rotacyjnych B i stosunkiem częstości drgań
10.
Rozwiązanie zadania 10.
Metodę wyznaczenie symetrii drgań normalnych dowolnej cząsteczki (bez
wyznaczania postaci tych drgań) przez konstrukcję reprezentacji przywiedlnej w
bazie wychyleń kartezjańskich atomów i rozkład jej na reprezentacje
nieprzywiedlne podaje F. Cotton „Teoria grup. Zastosowania w chemii”.
Aktywność przejścia oscylacyjnego w widmie absorpcyjnym IR cząsteczki N-
atomowej wymaga niezerowania przynajmniej jednej składowej wektora
elektrycznego momentu dipolowego przejścia:

24
<n|dk|m> 0 dla k = x, y, z
i analogicznie, aktywność przejścia oscylacyjnego w widmie Ramana wymaga
niezerowania przynajmniej jednej składowej tensora polaryzowalności przejscia:
<n|ij|m> 0 dla i, j = x, y, z
Notacja Diraca:
<n|dx|m> …n*(X1,X2,…XN,Y1,…ZN)dxm(X1,X2,…ZN)dX1dX2 …dZN
i analogicznie dla pozostałych składowych: di i = y, z oraz ij i, j = x, y, z.
Całki te nie znikają, jeśli wyrażenie podcałkowe transformuje się zgodnie z
reprezentacją pełnosymetryczną grupy symetrii danej cząsteczki. Funkcja falowa
oscylacyjnego stanu podstawowego |m> (zgodnie ze wskazówką) jest
pełnosymetryczna, a funkcja stanu jednokrotnie wzbudzonego |n> transformuje się
jak odpowiednie drganie normalne. Składowe elektrycznego momentu dipolowego
transformują się się jak współrzędne x, y i z a składowe polaryzowalności jak ich
iloczyny: x2, y
2, z
2, xy, xz, yz. Stąd, drganie jest aktywne w widmie absorpcji IR
jeśli „należy” do tej samej reprezentacji nieprzywiedlnej co jedna ze
współrzędnych x, y lub z. Drganie jest aktywne w widmie Ramana jeśli należy
do tej samej reprezentacji nieprzywiedlnej co jeden z iloczynów x2, y
2, z
2, xy, xz,
yz, ponieważ iloczyn Kroneckera tych samych reprezentacji zawiera w rozkładzie
na reprezentacje nieprzywiedlne reprezentację pełnosymetryczną.
CO2: aktywne w widmie absorpcji IR są drgania 2349,3cm1
(u+) i
667,3cm1
(u) a aktywne w widmie Ramana jest drganie 1388,3cm1
(g+);
cząsteczka ma środek inwersji co powoduje że aktywność w widmie absorpcji
wyklucza aktywność w widmie Ramana i na odwrót.
NH3: aktywne w widmie absorpcji IR i Ramana są wszystkie drgania.

25
Rozwiązanie zadania 10a.
Energia potencjalna we współrzędnych symetrii:
2V = f11S12 + 2f12S1S2 + f22S2
2 + f33S3
2 f12 = f21
(oczywiście stałe silowe zależą od wyboru układu współrzędnych !!!)
Energia kinetyczna we współrzednych symetrii:
2T = g11
•
S12 + 2g12
•
S 1
•
S 2 + g22
•
S 22 + g33
•
S 32 g12 = g21
Ponieważ energia kinetyczna i potencjalna są niezmiennicze względem operacji
symetrii, fij i gij są równe zeru jeśli Si i Sj mają różną symetrę (zaleta wyboru
współrzędnych symetrii). Przyrównujemy do zera wyznacznik wiekowy:
|f g| = 0 f = [fij] g = [gij]
f11 g11 f12 g12 0
| f12 g12 f22 g22 0 | = 0
0 0 f33 g33
f33 g33 = 0 3 = f33/g33 = 423
2
czyli częstość drgania normalnego Q3 = S3 wynosi 3 = (1/2)33
33
g
f
pozostałe częstości znajdujemy z równania kwadratowego:
(f11 g11)(f22 g22) (f12 g12)2 = 0
1, 2 = )g - g2(g
g2f-gf+gf
2122211
121222111122
)g - g2(g
)g - g)(gf - f4(f -)g2f-gf+gf(
2122211
2122211
2122211
2121222111122

26
po podstawieniu kolejno wartości 1 i 2 do równania wiekowego znajdujemy
wektory własne, które ustawione w kolumnach dają macierz przekształcenia
linowego Lij współrzędnych symetrii S1 i S2 we współrzędne normalne Q1 i Q2:
Q1 = L11S1 + L12S2
Q2 = L21S1 + L22S2
Trzeba jeszcze wyrazić współczynniki gij poprzez masy i parametry przestrzenne
cząsteczki, albo korzystając z tabeli Wilsona, albo (w naszym prostym przypadku)
patrząc na wychylenia atomów położeń równowagi, Y N1: (x1 y1 0), Y N2: (x2
y2 0), X N3: (x1 y1 0)
x1 = S1 S3sin x2 = S1 S3sin x3 = X
Y
m
m2S3sin
ta ostatnia zależność z zasady zachowania pędu wzdłuż osi x:
mXx3 + mYx1 + mYx2 = 0
y1 = S2 S3cos y2 = S2 + S3cos y3 = X
Y
m
m2S2
ta ostatnia zależność z zasady zachowania pędu wzdłuż osi y:
mXy3 + mYy1 + mYy2 = 0
Podstawiając do równania na energię kinetyczną drgań:
T = (1/2)mY(•
x 12 +
•
y 12) + (1/2)mY(
•
x 22 +
•
y 22) + (1/2mX(
•
x 32 +
•
y 32)
i porównując z:
T = (1/2)(g11
•
S 12 + 2g12
•
S 1
•
S 2 + g22
•
S 22 + g33
•
S 32)
otrzymujemy:
g11 = 2mY g22 = 2mY(1 + X
Y
m
m2) g33 = 2mY(1 + 2
X
Ysin
m
m2)
g12 = g13 = g23 = 0

27
Spektroskopia molekularna
Materiały na ćwiczenia do Wykładu 6
Zadanie 11: Wyznaczenie wzoru na długość r wiązania NH na podstawie
obliczenia składowej IZ tensora momentu bezwładności cząsteczki amoniaku NH3 i
składowej IX = IY (wyprowadzenie wzoru dla „ambitnych”):
IX =
)m
m3+1(2
rm3
N
H
2H
[2 (1 N
H
m
m3)sin
2]
gdzie mN i mH są masami azotu i wodoru a jest kątem między wiązaniem NH i
3-krotną osią symetrii cząsteczki.
Zadanie 12:. Oszacowanie stałej rotacyjnej B ze struktury pasma || (częstość 2
PLANSZA 23 z wykładu). Wyznaczenie wartości r z Zadania 11 na podstawie
analizy struktury pasma (częstość 4 PLANSZA 23 z wykładu); A = 6.3 cm1
.

28
29

30
Rozwiązanie zadania 11
Do wyznaczenia długości wiązania NH potrzebna jest znajomość dwóch
składowych tensora momentu bezwładności. Przyjmujemy kierunek osi C3
cząsteczki wzdłuż osi Z układu współrzędnych.
Obliczamy składową Z-tową momentu bezwładności I sumując iloczyny mas przez
kwadraty ich odległości od osi:
IZ = 3mHd2 = 3mHr
2sin
2
Obliczmy skladową IX momentu I przyjmując, że oś x układu współrzędnych
przechodzi przez środek masy cząsteczki i pokrywa z rzutem wiązania NH na
płaszczyznę XY (oś Y jest do niej prostopadła a z symetrii czasteczki IX = IY):
- położenie środka masy na osi Z:
mNu = 3mHw
u + w = rcos
stąd: w =
N
H
m
m3+1
cosr u = (
N
H
m
m3) )
m
m3+1
cosr(
N
H
IX = mNu2 + mHw
2 + 2mH(w
2 + y
2)
gdzie y jest odległością między rzutem H na płaszczyznę XY i osią X;
rozpatrujemy trójkąt równoboczny w płaszczyźnie XY w którym oś X przechodzi
przez środek masy i rzut jednego z wodorów:
y = rsinsin60 = ( 3 /2)rsin
IX = mN2
N
H)
m
m3(
2
N
H)
m
m3+1
cosr(
+mH
2
N
H)
m
m3+1
cosr(
+ 2mH
2
N
H)
m
m3+1
cosr(
+ 2mH(
4
3)r
2sin
2

31
czyli IX =
)m
m3+1(2
rm3
N
H
2H
[2 (1 N
H
m
m3)sin
2]
wyznaczamy długość wiązania NH:
sin2 =
2H
Z
rm3
I
r = ZN
HHX
N
HH I)
m
m3-1)(m6/1(+I)
m
m3+1)(m3/1(
Wartości IX i IZ znajdujemy z analizy struktury rotacyjnej dwóch przejść
oscylacyjnych w następnym zadaniu.
Rozwiązanie zadania 12
Mierzymy odległość na skali widma z pasmem || między punktem 991cm1
i 7601
równą 235 mm; 991760=231cm1
na 235mm czyli 0,983cm1
/mm. Najdokładniej
można wyznaczyć stałą rotacyjną B metodą różnic kombinacyjnych (rozwiązanie
Zadania 8). Szacunkową wartość otrzymujemy z odległości między liniami J = 3 i
J= 4 (lub J= 4 i J=5) przejścia s a, która wynosi 20,3mm, czyli 19,95cm1
;
B = 9,98 cm1
(wartość tablicowa B = 9,941 cm1
)
Korzystamy z podanej stałej rotacyjnej A = 6.3 cm1
, którą można wyznaczyć z
pasma mierząc odstępy między gałęziami Q i znając wartość stałej Coriolisa dla
tego pasma = 0,26. Korzystamy z relacji między stałą rotacyjną i składową
momentu bezwładności (czynnik h/(82c) wstawiamy IX i IZ do wzoru na r w
poprzednim zadaniu. Wartość liczbowa r = 1.01 Å =1.01108
cm.

32
Spektroskopia molekularna
Materiały na ćwiczenia do Wykładu 7
Zadanie 13: Składanie jednoelektronowych orbitali molekularnych cząsteczek
dwuatomowych , ,…, z orbitali atomowych s, p,…; metoda LCAO MO
Zadanie 14:. Elektrony w kolistej studni potencjału V = 0; wyznaczenie energii
przejść elektronowych w benzenie i naftalenie (model FEMO).

33
Rozwiązanie zadania 13
Orbitale atomowe:
1s sferyczniesymetryczny, główna liczba kwantowa n = 1, poboczna liczba l = 0
2s sferyczniesymetryczny, główna liczba kwantowa n = 2, poboczna liczba l = 0
2px, 2py, 2pz wzdłuż osi, odpowiednio x, y i z, o równej energii, n=2, l = 1
Orbitale molekularne LCAO cząsteczek dwuatomowych AB:
(a) kryteria tworzenia:
- energie kombinowanych ze sobą orbitali atomowych powinny być zbliżone;
- kombinowane ze sobą orbitale atomowe muszą mieć jednakową symetrię
względem osi wiązania;
- kombinowane orbitale muszą się nakrywać (przenikać) w dużym stopniu.
(b) cząsteczka ustawiona wzdłuż osi z; układy współrzędnych dla kombinowanych
orbitali atomowych centrowane na atomach A i B; osie z-towe obu układów
skierowane przeciwnie „do siebie”.
(1) Cząsteczki homojądrowe A = B
Jednolelektronowe orbitale molekularne , symetryczne względem obrotu o
dowolny kąt wokół osi wiązania AB:
wiążący: 1s = 1sA + 1sB gęstość elektronowa między atomami
antywiążący: *1s = 1sA 1sB zerowa gęstość elektronowa w środku
między atomami
podobnie 2s i *2s.
wiążący: 2p = 2pAz + 2pBz
antywiążący: *2p = 2pAz 2pBz
jednoelektronowe orbitale molekularne , antysymetryczne względem odbicia w
płaszczyźnie yz (x, x*) lub w płaszczyźnie xz (y, y*):

34
wiążące: x2p = 2pAx + 2pBx y2p = 2pAy + 2pBy
antywiążące: x*2p = 2pAx 2pBx y*2p = 2pAy 2pBy
Energie: 1s < *1s < 2s < *2s < 2p < x2p = y2p < x*2p = y*2p < *2p
Konfiguracja elektronowa cząsteczki N2; obsadzenie każdego poziomu 2g
elektronami o przeciwnie skierowanych spinach (g - stopień degeneracji poziomu):
(1s)2(*1s)
2(2s)
2(*2s)
2(2p)
2(2p)
4 14 elektronów
6 elektronów powłok walencyjnych 2p i 2p tworzą wiązanie potrójne N N.
Kształty orbitali: dowolny podręcznik mechaniki/chemii kwantowej, np. P. W.
Atkins „Molekularna mechanika kwantowa”
(2) Cząsteczki heterojądrowe A B
Orbitale molekularne , wiążące i antywiążące uzyskujemy przez kombinację
zgodnie z podanymi 3 warunkami. UWAGA: mogą mieszać się orbitale atomowe
ns i mpz o głównych liczbach kwantowych n m, zależnie od energii. Dla
cząsteczki C=O orbitale 1s praktycznie się nie mieszają co oznacza się
pozostawiając orbitale zamkniętych powłok wewnętrznych oznaczone np. KC i KO
z dwoma elektronami każdy. Orbitale oraz (wiążące i antywiążące):
k = ck12sC + ck22pzC + ck32sO + ck42pzO k = 1, 2, 3, 4
j = cj12pC + cj22pO j = 1, 2, = x, y
Energie (kolejność) poziomów energetycznych zależy od konkretnej cząsteczki,
Konfiguracja C = O (14 elektronów): KC
2KO
2(1)
2(2)
2(1)
4(3)
2
Inny zapis w postaci kombinacji liniowej atomowych orbitali
zhybrydyzowanych (kombinacji liniowych orbitali 2s, 2px, 2py, 2pz) pokazuje
utworzenie wiązania podwójnego oraz wolnych par elektronowych - orbitale
niewiążące n.

35
Rozwiązanie zadania 14
Elektrony typu cząsteczki benzenu C6H6 przybliżamy jako poruszające się w
jednowymiarowej, kolistej studni potencjału o promieniu r0; długość studni:
2r0 = 6rCC
a potencjał:
V(r) = 0 dla r = r0
V(r) = dla r r0
Równanie Schrödingera dla elektronu o masie me krążącego (zmiana ) w studni:
(ℏ2/2I) d
2/d
2 = E d
2/d
2 = (2EI/ℏ
2) I = mer0
2
rozwiązanie: () = Cexp(iA) = Cexp{i2
IE2
}
sprawdzamy przez wykonanie dwukrotnego różniczkowania i przyrównanie do
siebie obu stron równania (L = P) otrzymując A.
Warunek brzegowy: () = ( + 2) i po podstawieniu otrzymujemy:
exp{2i2
IE2
} = 1 czyli:
2
IE2
= n n = 0, 1, 2, 3,…,
Skwantowane poziomy energetyczne: En = (ℏ2/2I)n
2
6 elektronów benzenu obsadza poziomy n = 0, 1; energia przejścia na n = 2:
E(C6H6) = E2 E1 = 3ℏ2/2I = (
2ℏ
2/6me)(1/rCC
2)
Dla porównania energia przejścia dla naftalenu C10H8 (10 elektronów ) i większej
studni: 10rCC = 2r0, po obsadzeniu poziomów elektronami:
E(C10H8) = (2ℏ
2/10me)(1/rCC
2)
co powoduje batochromowe przesunięcie pasm absorpcyjnych naftalenu w
stosunku do benzenu, zgodnie z doświadczeniem.

36
Spektroskopia molekularna
Materiały na ćwiczenia do Wykładu 8
Zadanie 15: Analiza struktury oscylacyjnej pasma (256 nm) absorpcji gazowego
benzenu; wykluczenie struktury rotacyjnej.
Zadanie 16: Porównanie struktury oscylacyjnej widma absorpcji i fluorescencji
dla roztworu perylenu (w benzenie) i chininy (w 1M H2SO4); analiza efektu
solwatacyjnego.

37

38

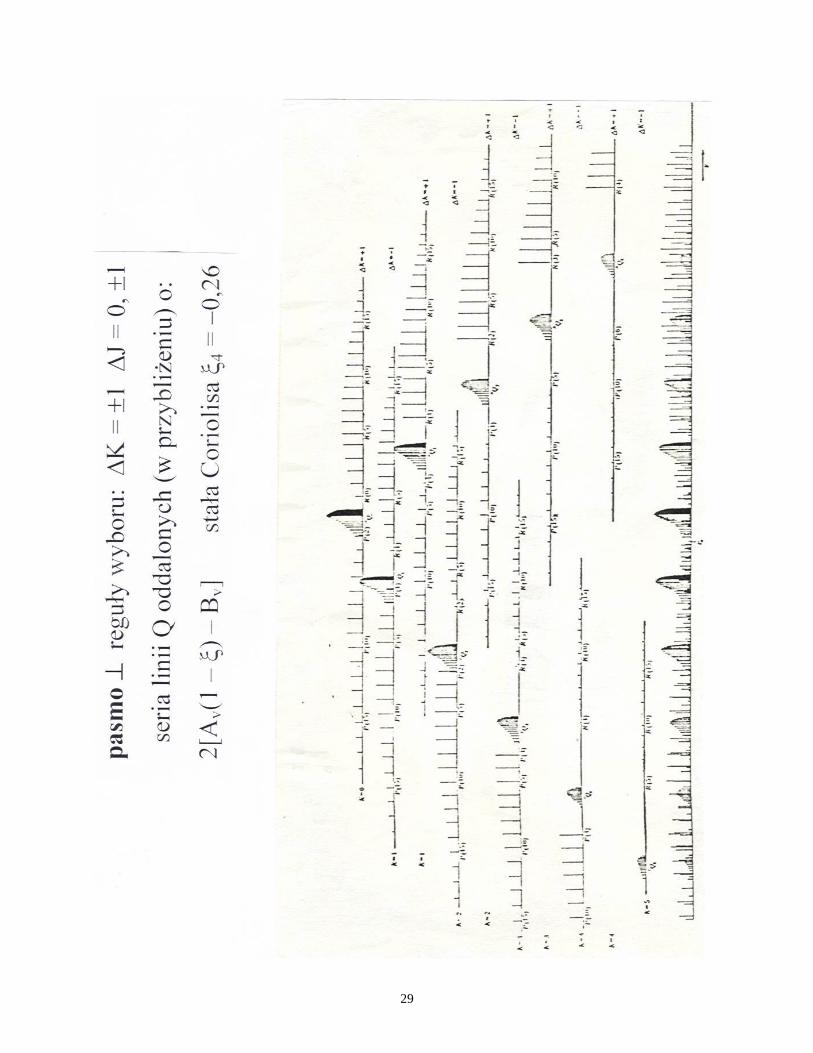
Rozwiązanie zadania 15
Bogata struktura pasma przy 250 nm ma charakter wyłącznie oscylacyjny.
Odległość 272 mm na skali widma odpowiada 46 000 36 000 = 10 000 cm1
,
czyli 36,8 cm1
/mm. Można wyliczyć obie skladowe tensora momentu
bezwładności cząsteczki C6H6 jako rotatora symetrycznego (zadanie do domu).
Wartości B i A nie przekraczają 1 cm1
, a więc odstępy między liniami w
strukturze rotacyjnej nie mogą przekroczyć 2 cm1
. Mierzone na widmie
najmniejsze odstępy są rzędu 1 mm, czyli ok. 30 cm1
, co wyklucza możliwość
obserwacji struktury rotacyjnej.
Dwie główne progresje linii z odstępami ok. 26 mm odpowiadają wzbudzeniu
kolejnych kwantów drgań o częstości ok. 950 cm1
czyli pełnosymetrycznych
drgań a1g. Analiza na podstawie tablicy charakterów grupy D6h grupy symetrii
cząsteczki benzenu pokazuje wzbroniony charakter przejścia elektronowego :
1B2u
1A1g. Zachodzi konieczność jednoczesnego wzbudzenia jednego kwantu
drgania e2g w stanie elektronowym wzbudzonym lub w podstawowym (e2g*), tak
że A1g E1uB2ue2g; „zapożyczenie” z przejścia dozwolonego 1E1u
1A1g.
Schemat przejść z elektronowego poziomu podstawowego 1A1g do
1B2u ze
wzbudzeniami n kwantów drgań a1g (n = 0, 1, 2, 3, 4,…) i jednego kwantu e2g w
stanie elektronowym wzbudzonym (seria najwyższych linii) oraz analogicznych
przejść z pierwszego poziomu oscylacyjnego e2g* poziomu elektronowego 1A1g na
kolejne poziomy oscylacyjne a1g na 1B2u (druga co do intensywności seria)
pokazuje plansza. Pozostałe serie są wzbudzeniami z innych nisko położonych (w
stosunku do poziomu całkowicie podstawowego, vk = 0) poziomów oscylacyjnych.
Nie obserwuje się przejścia v’=0 v’’=0; można jednak oszacować jego
położenie przyjmując jednakowe wartości częstości drgań e2g i e2g*.

Rozwiązanie zadania 16
Symetryczny charakter struktury oscylacyjnej widm absorpcji i fluorescencji
perylenu odzwierciedla jednakowy układ poziomów tej struktury w elektronowym
stanie podstawowym i elektronowym stanie wzbudzonym, w przeciwieństwie do
chininy.
Brak nakrywania linii wspólnego dla absorpcji i fluorescencji przejścia 0 0 dla
(patrz diagram Jabłońskiego) wynika z efektu solwatacyjnego. Przejście
absorpcyjne 0 0 zachodzi nie do równowagowego elektronowego poziomu
wzbudzonego S1, ale do tzw. poziomu Francka-Condona (S1FC
), w którym
wzajemne położenie cząsteczek absorbującej substancji i rozpuszczalnika nie
zdążyło przyjąć najkorzystniejszej energetycznie konfiguracji; jest takie jak w
elektronowym stanie podstawowym - zasada Francka-Condona. Z kolei
fluorescencja zachodzi nie na elektronowy poziom podstawowy o najniższej
energii S0 ale na stan FC, w którym ułożenie cząsteczek jest takie jak w
elektronowym stanie wzbudzonym (S0FC
). Stąd różnice energii spełniają relację:
E(S1FC
) – E(S0) > E(S1) – E(S0FC
)

Spektroskopia molekularna
Materiały na ćwiczenia do Wykładu 9
Zadanie 17: Prawo Lamberta-Beera dla absorpcji i dla emisji.
Zadanie 18: Wyliczenie odległości donor - akceptor z wydajności transferu FRET.

Rozwiązanie zadania 17
Zmniejszenie natężenia promieniowania dI o częstości przy przechodzeniu przez
element l’ l’ +dl’ próbki substancji absorbującej jest zgodnie z prawem Lamberta
- Beera proporcjonalne do molowego stężenia c cząsteczek oraz do natężenia I(l’):
dI(l’) = kI(l')cdl k - stała proporcjonalności
rozwiązujemy równanie różniczkowe:
dI(l’)/dl' = kI(l')c
i w punkcie l' = l na końcu kuwety otrzymujemy I(l) = I
lnI = kcl + C C = lnI0 I0 - natężenie wiązki padającej (dla l’ = 0)
I = I0exp(kcl)
ln(I/I0) = kcl
ln(I/I0) = log(I/I0)ln10
Absorpcja (ekstynkcja) jest zdefiniowana jako:
A = log(I0/I) = ()cl () = (1/ln10)k
() [litrmol1cm
1] jest molowym wpółczynnikiem ekstynkcji.
Wydajność kwantowa emisji (fluorescencji):
F = IE/IA gdzie IE - natężenie promieniowania wyemitowanego
IA - natężenie promieniowania zaabsorbowanego
IE = FIA = F(I0 I) = FI0(1 I/I0) = FI0(1 10()cl)
Dla małych stężęń molowych c możemy rozwinąć 10()cl w szereg Maclaurina
wokół punktu c = 0:

f(x) = f(0) + k
1k
)k(
x!k
)0(f
x c f(x) 10()cl
Zachowując tylko 2 pierwsze wyrazy rozwinięcia:
f(0) = 1; [f(1)
(0)c1/1!] = f
(1)(0)c
f(1)
(0) =d(10()cl)/dc =d(e
()clln10)/dc = () l ln10e
()cl ln10|c=0= ln10()l
otrzymujemy:
10()cl = 1 ln10()cl + …
i po podstawieniu do wzoru na IE mamy prawo Lamberta-Beera dla emisji:
IE = FI0ln10()cl
Rozwiązanie zadania 18
Wydajność E transferu energii FRET z donora D* na akceptor A definiuje się
jako stosunek szybkości tego procesu k = kD*A do całkowitej szybkości
dezaktywacji k + 1
stanu wzbudzonego D*, gdzie jest czasem życia stanu
wzbudzonego przy braku akceptora A:
E = kτ
k
1
Z drugiej strony:
E = 1 FDA/FD
gdzie FDA jest natężeniem fluorescencji donora w obecności akceptora a FD jest
natężeniem fluorescencji donora przy braku akceptora.
Analizując widmo donora indolowego TMA w nieobecności akceptora
dansylowego oraz widmo TUD w obecności tego akceptora (para donor-akceptor)
widać, że wydajność transferu FRET wynosi 90%, gdyż intensywność

fluorescencji w obu przypadkach: FDA ~0.1, FD ~1. DUA jest kontrolnym
widmem znakowanego dansylem kwasu tłuszczowego.
Podstawiając k = τ
1 60 )r
R( , gdzie R0 jest odległościa Förstera a r odległością
donor - akceptor, otrzymujemy:
E = 66
0
60
rR
R
= 0.9 czyli szukana wartość:
r = (0.1)1/6
R0 = 0.68R0
R0 przyjmujemy jako dane; jego wartość można wyznaczyć z nakrywania pasm
emisji donora i absorpcji akceptora:
R06 = (9000ln10
2D)/(128
5NAn
4)F’D()A()
4d
- długość fali [nm]
2/3 dla roztworów; współczynnik orientacji obu dipoli
n 1,4 - współczynnik załamania roztworu
D - wydajność kwantowa donora
NA - liczba Avogadro [mol1
]
F’D() - unormowana intensywność fluorescencji donora
A() - współczynnik ekstynkcji molowej akceptora [litr mol1
cm1
]
Oznaczając: J()= (FD()A()4d)/(FD()d) [litr mol
1cm
1nm
4]:
otrzymujemy po skróceniu [mol1
] i wyrażeniu wszystkich długości w [Å]
R0 = 0.211[2 n
4DJ()]
1/6 [Å]
R0 = 25,9Å
r = 17,6 Å

Spektroskopia molekularna
Materiały na ćwiczenia do Wykładu 10
Zadanie 19: Rozwiązanie równania Schrödingera dla jądra w statycznym polu
magnetycznym B0. Obliczyć częstość rezonansową dla jądra I = ½.
Zadanie 20: Wyliczenie wartości wyrażenia <3cos2 – 1>av opisującego
oddziaływania w cząsteczkach wykonujących szybkie, przypadkowe zmiany
orientacji w przestrzeni.

Rozwiązanie zadania 19
Hamiltonian jądra o spinie I 0 w polu magnetycznym o indukcji B0 skierowanego
wzdłuż osi z układu współrzędnych
H = B0
B0 = B0iz = ħI
równanie Schrödingera:
H = E
H = ħB0Iz
korzystamy z działania na funkcje operatora rzutu spinu na oś z:
Iz = m m = I, I + 1, I + 2,…, I 1, I = |I, m>
lewa strona równania (L): ħB0Iz = ħB0m
prawa strona równania (P): E
po przyrównaniu L = P otrzymujemy położenia skwantowanych poziomów energii
jądra:
Em = ħB0m
Dla jądra I = ½ mamy dwa poziomy:
E1/2 = (1/2)ħB0 = |1/2, 1/2>
E1/2 = (1/2)ħB0 = |1/2,1/2>
i różnica energi obu poziomów:
E = E1/2 E1/2 = ħB0
częstość rezonansowa:
= E/ħ = B0

Rozwiązanie zadania 20
Wartość średnią funkcji f(x) definiuje się jako:
<f(x)>av =
dx)x(P
dx)x(P)x(f = dx)x(P)x(f
dla zadanego i unormowanego prawdopodobieństwa P(x) rozkladu x.
W przypadku cząsteczek przypadkowo zorientowanych w przestrzeni unormowane
prawdopodobieństwo jest dane przez stosunek powierzchni wycinka ds sfery do
powierzchni całej sfery (we współrzędnych sferycznych):
P( ) = 2
2
rπ4
φdθdθsinr = (1/4)sindd
Prawdopodobieństwo jest unormowane (można sprawdzić przez całkowanie).
<3cos2 – 1>av =
π
0
π2
0
(3cos2 – 1) (1/4)sindd
<3cos2 – 1>av = (1/4)
π2
0
d π
0
(3cos2 – 1)sind
<3cos2 – 1>av = (1/2){
π
0
3cos2sind –
π
0
sind}
wartośc drugiej calki wynosi 2
wartość pierwszej całki obliczmy po zastosowaniu podstawienia y = cos i wynosi
ona także 2. Po odjęciu obu całek otrzymujemy:
<3cos2 – 1>av = 0

Spektroskopia molekularna
Materiały na ćwiczenia do Wykładu 11
Zadanie 21: Oznaczanie prostych układów spinowych w cząsteczkach i analiza
odpowiadających im widm NMR.
Zadanie 22: Wyznaczenie wzoru strukturalnego cząsteczki na podstawie
zarejestrowanego widma 1H NMR i wzoru sumarycznego C6H10O2. Uwaga: brak
protonów wymienialnych w wodzie. Dolny rząd przedstawia pełne widmo
protonowe a powyżej przedstawione są rozciągniecia wybranych multipletów: dwa
pojedyncze protony i jedna z grup metylowych. Ciągła linia nad dolnym widmem
oznacza całkowanie, czyli jej skok określa ilość protonów w multiplecie.


Rozwiązanie zadania 21
Przykładowe układy dwuspinowe (sprzęgające się jądra zaznaczone na czerwono):
AX: 2H
13C
15N cjanowodór znakowany (
2H,
13C,
15N)
R1C(1H)=C(
1H)R2 R1, R2 podstawniki o różnych elektroujemnościach
AB: R1C(1H)=C(
1H)R2 R1, R2 podstawniki o bliskich elektroujemnościach
A2 lub X2: R C(1H)=C(
1H)R
R1C(1H)2R2 swobodny obrót wokół wiązań R1C i CR2
Przykładowe układy trójspinowe:
AMX: Br3(13
C2H3)
1H
19F pierscień benzenowy z trzema podstawnikami Br
przy sąsiednich węglach (1,2,3) oraz protonem, grupą metylową znakowaną (13
C,
2H) i fluorem przy pozostałych węglach
ABX: Br3(1H)2
19F pierścień benzenowy z trzema podstawnikami Br przy
sąsiednich węglach (pozycja 1,2,3) oraz dwoma protonami w pozycji orto (pozycja
4,5) i jednym fluorem w pozycji 6
AX2 (A2X): Br3(1H)2
19F pierścień benzenowy z trzema podstawnikami Br
przy sąsiednich węglach (1,2,3), dwoma protonami w pozycji meta (4,6) i jednym
fluorem w pozycji orto (5) do obu protonów.
AB2 (A2B): (CH3)3(1H)3 pierścień benzenowy z trzema podstawnikami
metylowymi przy sąsiednich węglach i trzema protonami
ABC: (CH3)2(CH2OH)(1H)3 pierścień benzenowy z dwoma podstawnikami
metylowymi (1,2), hydroksymetylowym (3) oraz trzema protonami.
Schematyczne widma przedstawia plansza (poniżej); widma układów ABC i A2B
nie są dyskutowane. Dwie linie „kombinacyjne” w układzie ABX, oznaczone
czerwonymi strzałkami, mogą się pojawić lub nie, zależnie od parametrów NMR.

A3 lub X3 (analogicznie jak A2): grupa metylowa z protonami oddalonymi o więcej
niż 4 wiązania od innych jąder I = ½ o obsadzeniu izotopowym bliskim 100%, np.
metyl toluenu deuterowanego w pierścieniu.
Każde kolejne jądro I = ½ o obsadzenie izotopowym bliskie 100% powoduje
dalsze rozszczepienia linii multipletowych jeśli jest odległe od danego jądra o
więcej niż 3 - 4 wiązania, a odległość między rozszczepionymi liniami jest równa
stałej sprzężenia (w przybliżeniu małych stałych sprzężenia w stosunku do różnic
przesunięć chemicznych),
Przykład. Układ multipletowy A2X3 (CH2CH3) uzyskujemy przez kolejne
rozszczepienia każdej linii na dwie, jednakowo odległe (patrz układ AX2). W
konsekwencji uzyskujemy kwartet dla CH2 i tryplet dla CH3. Stosunki
intensywności sygnałów w każdym multiplecie uzyskujemy z trójkąta Pacala:
n = 0 1
n = 1 1 1
n = 2 1 2 1
n = 3 1 3 3 1
gdzie n oznacza ilość sprzężonych jader w sąsiedniej grupie.


Rozwiązanie zadania 22
Poszukiwany związek to:
H3CC(H)=C(H)C(=O)OCH2CH3
zgodnie z widmem, na którym obserwujemy dwa odseparowane układy spinowe:
A3MX A2X3

Spektroskopia molekularna
Materiały na ćwiczenia do Wykładu 12
Zadanie 23: Analiza widma 1H NMR nukleotydu: 7-metylo-GTP w D2O.


Rozwiązanie zadania 23
Sygnał przy 0 ppm - wzorzec
Układ: kwartet 3.66 ppm i tryplet 1.19 ppm - zanieczyszczenia CH2CH3.
Duży sygnał 4.79 ppm - resztka wody H2O w 2H2O (wzbogacenie 99.98%)
Protony w grupach NH2, NH, OH i proton H8 (!!!) ulegają wymianie na deuter.
Grupa N7-CH3 – singlet 4.130 ppm.
Analizując multiplety i strukturę chemiczną widać, że H(1’) może się sprzęgać
tylko z H(2’) dając dublet o przesunięciu chemicznym (środek dubletu) 6.076 ppm
(dokładność ok. 0.005 ppm) i stałej sprzężenia J(1’,2’) = 3.5 Hz.
Protony: H(2’) 4.705 ppm i H(3’) 4.575 ppm. Jest to jedyne możliwe
przyporządkowanie zapewniające zbliżone wartości J(2’,3’) = 5.0 Hz i J(3’,4’) =
5.3 Hz, które można wyliczyć z szerokości multipletów:
H(2’) J(1’,2’) + J(2’,3’) = 8.5 Hz
H(3’) J(2’,3’) + J(3’,4’) = 10.3 Hz
przy wyznaczonej poprzednio stałej J(1’,2’) = 3.5 Hz. W tym przypadku pasmo
4.705 jest pseudotrypletem. Przy odwrotnym założeniu co do położeń H(2’) i H(3’)
duża różnica między sprzężeniami J(2’,3’) i J(3’,4’) (odpowiednio 6.8 Hz i 1.7 Hz)
spowodowałaby powstanie wyraźnego kwartetu przy 4.705 ppm.

Najbardziej sprzężony multiplet to H(4’) 4.411 ppm; ten proton sprzęga się z
H(3’), H(5’) i H(5’’).
Układ AB to H(5’), H(5”) ze stałą sprzężenia J(5’,5’’) = 12 Hz; każdy sprzęga się
dodatkowo z H(4’) i najbliższym fosforem w mostku trifosforanowym (sygnały w
formie kwartetów i pseudotrypletów). Podobnie jak w przypadku H(2’)/H(3’,
zakładając różne kombinacje ze stałymi J(4’,5’), J(4’,5’’), J(5’,P) i J(5’’,P) można,
przy znanej stałej J(3’,4’) = 5.3 Hz, wyznaczyć jednoznacznie ich wartości z
multipletów H(4’), H(5’) i H(5’’), posiłkując się ew. strukturą multipletową na
widmie 31
P NMR. Jednoznaczne przyporządkowanie sygnałów H(5’) i H(5’’) jest
możliwe przy pomocy dodatkowych eksperymentów.
.

Spektroskopia molekularna
Materiały na ćwiczenia do Wykładu 13
Zadanie 24: Analiza widma 13
C NMR nukleozydu w DMSO-d6: 2’-deoxy-
adenozyna
Zadanie 25: Model wektorowy: eksperyment INEPT na przykładzie widma 15
N
NMR 2’,3’,5’-tri-O-metyloadenozyny w CDCl3.

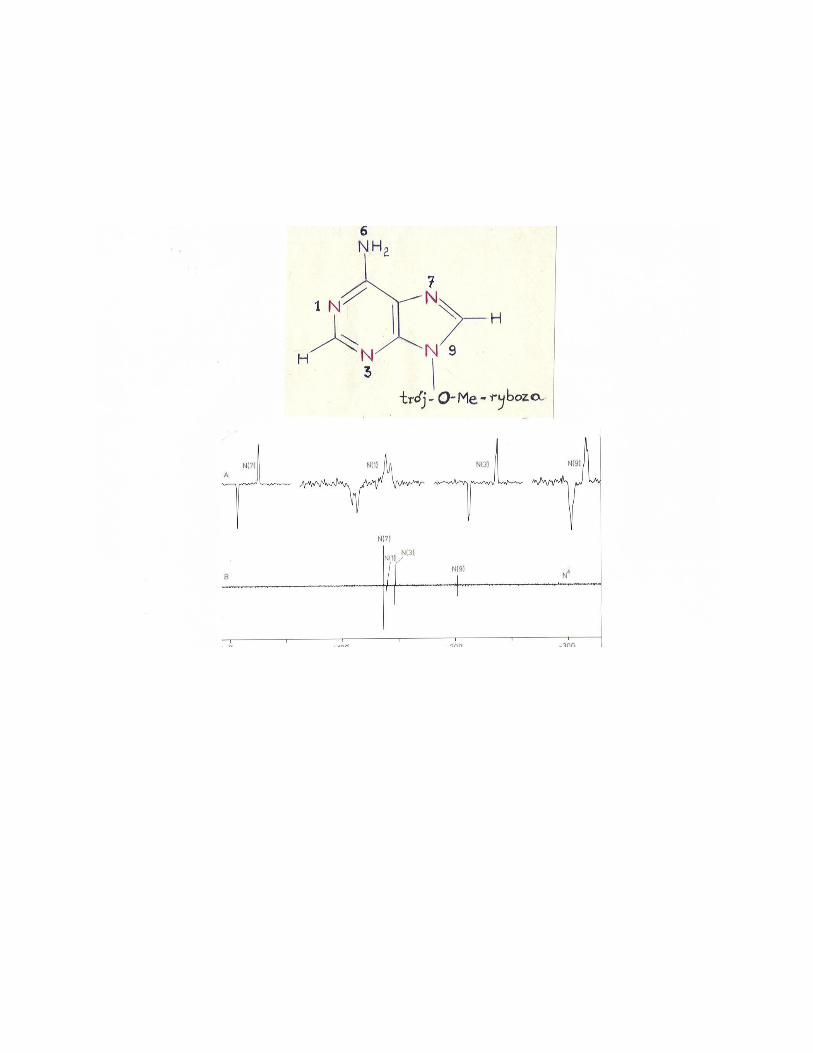
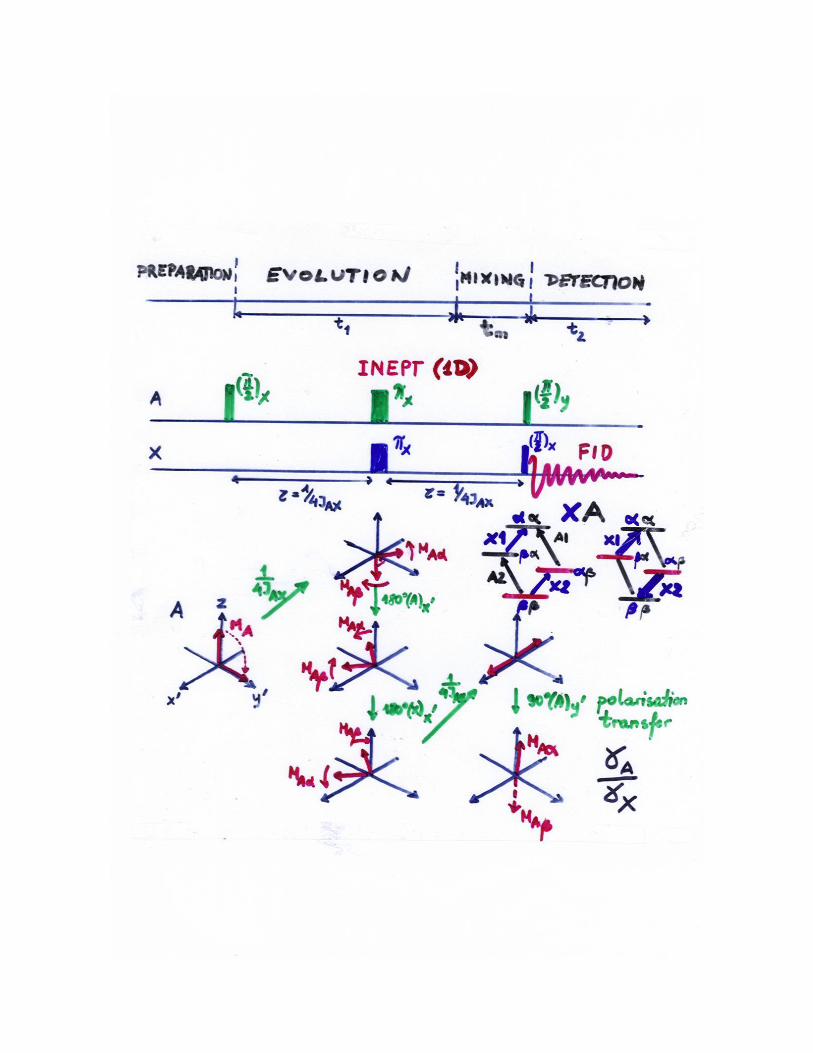

Rozwiązanie zadania 24
Węgle C-13 ze względu na małą zawartość izotopową nie sprzęgają się ze sobą a
jedynie z protonami (dolne widmo). Po odsprzęgnięciu protonów każdy z 5 węgli
cukru 2’-deoxyrybozy i 5 węgli zasady adeninowej daje pojedynczy sygnał -
singlet (górne widmo).
Pierwszy multiplet od prawej na górnym widmie pochodzi od rozpuszczalnika
(C2H3)2S=O (DMSO-d6).
Węgle zasady połączone z pierścieniem aromatycznym mają większe wartości
przesunięć chemicznych, od ok. 53 ppm do ok. 90 ppm. Dwa dublety z odstępami
linii ok. 3 mm to węgle bezpośrednio połączone z protonami: C(2) i C(8), 1JCH
210 Hz.
100 ppm na dolnej skali, czyli 100 x 125,8 Hz, odpowiada 177 mm, a więc 71
Hz/mm, przy częstość podstawowej węgla C-13: SF = 125.769 MHz.
Sprzężone sygnały węgli cukru od ok. 5ppm do 22 ppm pozwalają na
identyfikację trypletów C(2’) i C(5’) (bezpośrednio połączone z dwoma
protonami). Stałe sprzężenia 1JCH 140 Hz (ok. 2 mm) dla wszystkich węgli
cukrowych.
Pełne przyporządkowanie jest możliwe po dokładniejszej analizie sprzężeń w
multipletach i/lub wykonaniu dodatkowych eksperymentów, np. widmo
korelacyjne węgiel-proton 1H,
13C-HSQC, przy znanym przyporządkowaniu widma
protonowego.

Rozwiązanie zadania 25
Załączona plansza przedstawia analizę eksperymentu INEPT w ramach modelu
wektorowego dla układu dwóch jąder: X = 15
N, A = 1H. Widmo
15N NMR
uzyskane w eksperymencie INEPT (załączona plansza) pokazuje ok. 10-krotne
wzmocnienie czterech z pięciu azotów, dla których występują sprzężenia JAX ok. 7
8 Hz z protonami przez 2 i 3 wiązania. Analiza eksperymentu na gruncie modelu
wektorowego oparta jest na:
(1) przypisaniu magnetyzacji prostopadłej do pola B0 odpowiedniego przejścia
rezonansowego;
(2) przypisaniu magnetyzacji równoległej do pola B0 równowagowej różnicy
boltzmannowskich obsadzeń poziomów.
(3) proporcjonalności intensywności rejestrowanych danej linii NMR do różnicy
boltzmannowskich obsadzeń poziomów, między którymi zachodzi przejście.
W trakcie ewolucji magnetyzacji protonowej w układzie wirującym z częstością
rezonansową 1H (dla uproszczenia zaniedbuje się relaksację), zachodzi transfer
polaryzacji, tzn. boltzmannowskiej różnicy obsadzeń poziomów odpowiadających
przejściu protonowemu A2 do poziomów odpowiadających przejściom azotowym.
Zwiększenie tej różnicy powstaje wskutek odwrócenia magnetyzacji MA
(inwersja obsadzeń pozimów ) i daje wzmocnienie linii azotowych.
Efekt zachodzi poprzez sprzężenia skalarne, przy dopasowaniu wartości czasu
między impulsami do wartości stałej sprzężenia JAX, = 1/(4JAX). Dla sprzężenia
JAX = 8 Hz = 30 ms.
Wzmocnienie jest określone przez stosunek współczynników
żyromagnetycznych H/N.

Sygnał N grupy aminowej nie ulega wzmocnieniu ze względu na sprzężenie JAX =
90 Hz tylko z bezpośrednio związanymi protonami i brak inych protonów w
odległości 2 - 3 wiązań. Dla uzyskania analogicznego wzmocnienia trzeba
zastosować eksperyment INEPT z dopasowanym do wartości JAX = 90 Hz, czyli
dla = 3 ms.
Odwrócenie linii w dubletach wynika z inwersji boltzmanowskich obsadzeń
poziomów dla przejścia X2; X = 15
N (magnetyzacja MA „w dół”).