
Skrypt dla liceum ogólnokształcącego Michu Wrocław 2005 wersja 1.4
137
CHEMIA Skrypt dla liceum ogólnokształcącego Michu ® Wrocław 2005 wersja 1.4 Created by Neevia Document Converter trial version http://www.neevia.com
Transcript of Skrypt dla liceum ogólnokształcącego Michu Wrocław 2005 wersja 1.4

Created by Neevia Document Converter trial version
CHEMIASkrypt dla liceum ogólnokształcącego
Michu®
Wrocław 2005wersja 1.4
Created by Neevia Document Converter trial version http://www.neevia.com

1
SPIS TREŚCI
I CHEMIA OGÓLNA1. Budowa atomu:
a) skład i struktura atomu,b) konfiguracja elektronowa, promocje
elektronowe, bloki energetyczne, regułaHunda, teoria wzbudzenia,
c) elektroujemność, związek budowy atomu zukładem okresowym, teoria oktetu,
d) typy wiązań chemicznych,e) hybrydyzacja,f) VSEPR
2. Podstawy chemii:a) podstawowe pojęcia,b) stechiometria,c) termochemia,d) statyka i kinetyka reakcji chemicznych
3. Układy dyspersyjnea) ogólna charakterystyka i podział układów
dyspersyjnych,b) roztwory,c) teorie kwasów i zasad,d) ilościowy opis właściwości roztworów:
- stopień i stała dysocjacji,- prawo rozcieńczeń Ostwalda,- iloczyn jonowy wody,- pH i pOH,- iloczyn rozpuszczalności,
e) reakcje zachodzące w roztworach:- miareczkowanie alkacymetryczne,- amfoteryczność,- zobojętnianie i hydroliza,- wskaźniki,- roztwory buforowe,- związki kompleksowe,
f) koloidy4. Elektrochemia
a) stopnie utlenienia pierwiastkówb) reakcje redox w ujęciu elektronowym,c) najwaŜniejsze utleniacze i reduktory,d) związki chromu i manganu jako utleniacze,e) szereg aktywności metali,f) półogniwa,g) ogniwa galwaniczne,h) energetyka reakcji redox,i) korozja metali i metody jej zwalczania,j) elektroliza.
II CHEMIA ZWIĄZKÓW NIEORGANICZNYCH1. Systematyka związków nieorganicznych:
a) tlenki,b) wodorki,c) wodorotlenki,d) kwasy,e) sole.
2. Litowce,3. Berylowce,4. Borowce,5. Węglowce,6. Azotowce,7. Tlenowce,8. Fluorowce,9. Miedź,10. Srebro,11. Cynk,12. Chrom,13. Mangan,14. śelazo.
III CHEMIA ZWIĄZKÓW ORGANICZNYCH1. Wprowadzenie:
a) skład,b) teoria strukturalna,c) konstytucja i konfiguracja,d) izomeria i homologia,e) efekty elektronowe,f) grupy funkcyjne,g) barwy związków,h) typy i mechanizmy reakcji,
2. Węglowodory:a) podział,b) alkany i metan,c) cykloalkany,d) węglowodory nienasycone,
- alkeny i eten,- alkiny i etin,
e) areny, benzen i jego homologi,3. Jednofunkcyjne pochodne węglowodorów:
a) wstęp,b) fluorowcopochodne węglowodorów:
- fluorowcoalkany,- fluorowcoalkeny,- fluorowcoalkiny,- fluorowcoareny,
Created by Neevia Document Converter trial version http://www.neevia.com

2
c) alkohole i fenole:- nasycone,- polihydroksylowe,- nienasycone,- fenole i fenol,
d) etery,e) związki karbonylowe:
- aldehydy,- ketony,
f) kwasy karboksylowe:- nasycone,- nienasycone,- aromatyczne,- tłuszczowe,- dikarboksylowe,- halogenki kwasowe,- bezwodniki kwasowe,
g) estry i tłuszcze,
h) związki azotowe:- aminy,- amidy,- nitryle i izonitryle,- nitrozwiązki,
i) związki siarkowe:- tiole,- sulfidy,- kwasy sulfonowe,
j) aromatyczne związki heterocykliczne,4. Wielofunkcyjne pochodne węglowodorów:
a) wstęp,b) hydroksykwasy i ketokwasy,c) węglowodany:
- monosacharydy,- oligosacharydy,- polisacharydy,
d) aminokwasy, peptydy, białka,e) kwasy nukleinowef) witaminy.
Created by Neevia Document Converter trial version http://www.neevia.com

3
CHEMIA OGÓLNABUDOWA ATOMU
SKŁAD I STRUKTURA ATOMUBazą chemii jako nauki o przemianach materii jest wiedza o jej budowie. Zachodzące w przyrodzie
zjawiska najlepiej tłumaczy dziś teoria atomistyczna. Opiera się ona na załoŜeniu, iŜ materia zbudowana jest zmikroskopijnych, niewidocznych gołym okiem cząstek, zwanych atomami. Poziom atomu przyjmowano przezdługi czas za kres podzielności materii – zgodnie z tym załoŜeniem kaŜde ciało moŜna dzielić tak długo, aŜdojdzie się do cząstki juŜ dalej niepodzielnej. Współcześnie wiemy, iŜ budowa atomu nie jest jednorodna, leczwyróŜnić moŜna w nim elementy składowe, dalej zresztą takŜe podzielne. Nie zaprzecza to jednak doniosłościteorii atomistycznej ani jej znaczeniu, przeciwnie – stanowi raczej jej uzupełnienie.
Istnieje wiele modeli budowy atomu. Niektóre z nich mają juŜ znaczenie wyłącznie historyczne, niektórezaś są wciąŜ poprawiane i doskonalone. Jednym z pierwszych nowoŜytnych był tzw. model rodzynkowy,wprowadzony przez Josepha Thomsona w 1906r. Autor załoŜył, iŜ atom składa się z fragmentów naładowanychładunkiem elektrostatycznym ujemnym (elektronów), które znajdują się w dodatnio naładowanym ośrodku –niczym rodzynki w cieście. Całość atomu była dzięki temu elektrycznie obojętna. Choć zaletą było zaznaczenieistnienia ładunków dodatnich i ujemnych wewnątrz atomu, to cięŜko wyobrazić sobie dlaczego nie przyciągnąsię one i nie wyrównają potencjałów do zera.
Niedługo później – w 1911r. – Ernest Rutherford przeprowadził doświadczenie obalające modelThomsona. Polegało ono na bombardowaniu naładowanymi cząstkami folii metalowej. Na „królikadoświadczalnego” Rutherford wybrał złoto, gdyŜ moŜna je walcować na bardzo cienkie płytki. Opracowującwyniki, stwierdził on, iŜ większość cząstek przeszła przez złotą folię bez zmiany kierunku, niewielka częśćuległa zakrzywieniu, a nawet odbiciu w stronę przeciwną. Wynikało stąd, iŜ atomy nie są „pełne”, jak tozakładał Thomson, lecz mają wewnątrz bardzo duŜo wolnego miejsca. W ten sposób Thomson odkrył istnienietworu zwanego jądrem atomowym.
Z wyników badań Thomsona korzystamy do dziś, tłumacząc z powodzeniem szereg zjawisk fizycznych ichemicznych. Przyjmujemy, iŜ atom składa się z dodatnio naładowanego jądra oraz z otaczającej je chmuryelektronowej. W skład jądra wchodzą protony i neutrony, określane wspólną nazwą: nukleony, zaś chmurętworzą ujemnie naładowane elektrony. Zestawienie najwaŜniejszych cech tych cząstek przedstawia poniŜszatabela.
Nazwa Symbol Masa spoczynkowa Ładunek Trwałośćelektron e- 9,109.10-31 kg ≈ 0 u - 1,602.10-19 C = - e trwałyproton p+ 1,673.10-27 kg ≈ 1 u + 1,602.10-19 C = + e trwałyneutron n0 1,675.10-27 kg ≈ 1 u 0 T1/2=930s, n0 ---> p+ + e- + 00ΰe
Zbiór atomów posiadających tę samą liczbę protonów w jądrze nazywamy pierwiastkiem. Przy zapisiepierwiastka podajemy najczęściej najwaŜniejsze informacje dotyczące jego składu ilościowego zgodnie zeschematem: AZX. Z to liczba atomowa (porządkowa) – jest ona równa liczbie protonów w jądrze, a tym samymładunkowi jądra oraz liczbie elektronów otaczających jądro. Z kolei A to liczba masowa, równa liczbienukleonów tworzących jądro. Określenie ‘liczba masowa’ bierze się stąd, iŜ, jak łatwo zauwaŜyć, nukleony sąok. 1836 razy cięŜsze niŜ elektron, dlatego w jądrze skupia się ponad 99% masy atomu.
Znając wielkości A i Z moŜemy określić wiele własności jądra. Zakładając, Ŝe ma ono kształt kulisty,moŜemy jego promień wyrazić jako rj=r0
.3√A, gdzie r0=1,23.10-15 m. Liczba neutronów w jądrze jest róŜnicącałkowitej liczby nukleonów i liczby protonów, czyli A – Z. Masa jądra jest mniejsza od sumy mas jegoskładników – róŜnicę tę określamy jako defekt masy i obliczamy następująco: ∆m=Zmp+(A-Z)mn-mj, gdzie mp,mn, mj to odpowiednio masy spoczynkowe protonu i neutronu oraz rzeczywista masa jądra.
Wszystkie identyczne atomy tworzą nuklid. Pierwiastek moŜe składać się z jednego rodzaju nuklidów lub zkilku, bowiem atomy tego samego pierwiastka mogą róŜnić się liczbą neutronów w jądrze. Atomy o tym samymZ lecz róŜnym A nazywamy izotopami. Przykładem jest wodór, składający się z trzech izotopów: protu 11H,deuteru 21H=D oraz trytu 31H=T. Masę średniego składu izotopowego danego pierwiastka wyraŜoną w unitachnazywamy masą atomową. Atomy o tym samym A lecz róŜnym Z nazywamy izobarami – mają one identycznąliczbę nukleonów, np. 36
18Ar i 3616S. Izotony to atomy o identycznej licznie neutronów, czyli o tej samej wartości
róŜnicy A – Z, np. 4020Ca i 39
19K.Zjawisko przekształcania się izotopów danego pierwiastka w izotop trwały lub nietrwały tego samego lub
innego pierwiastka nazywamy promieniotwórczością. WyróŜniamy kilka rodzajów rozpadupromieniotwórczego, z których najwaŜniejsze to:a) Rozpad α polega na wyrzuceniu z jądra cząstki 42α
2+, czyli jądra atomu helu. W wyniku tego liczbaatomowa i masowa zmieniają się w następujący sposób: AZX ---> A-4
Z-2Y + 42α.
Created by Neevia Document Converter trial version http://www.neevia.com

4
b) Rozpad β- polega na rozpadzie neutronu w jądrze na proton, elektron oraz antyneutrino elektronowe,zgodnie z równaniem: 10n ---> 11p + 0-1e
- + 00ΰe – powstały elektron wyrzucany jest z jądra. Zmianęilościową składu atomu ilustruje równanie: AZX ---> AZ+1Y + 0-1β
- + 00ΰe.c) Rozpad β+ zachodzi w przyrodzie bardzo rzadko i w przeciwieństwie do pozostałych jest rozpadem
sztucznym. Polega na rozpadzie protonu w jądrze na neutron, pozyton oraz neutrino elektronowe, zgodniezrównaniem: 11p ---> 10n + 0+1e
+ + 00νe. Pozyton jest antycząstką w stosunku do elektronu – posiada takąsamą masę i wartość bezwzględną ładunku, róŜni się natomiast jego znakiem. Po rozpadzie pozytonwyrzucany jest z jądra. Zmianę składu atomu przedstawia schemat: AZX ---> AZ-1Y + 0+1β
+ + 00νe.d) Przemiana γ powiązana jest z wyrzutem wysokoenergetycznego fotonu z jądra. Najczęściej jest
następstwem rozpadu α, po którym wzbudzone jądro przechodzi do stanu trwałego przez pozbycie się częścienergii właśnie w postaci fotonu.Rozpadem promieniotwórczym rządzą określone prawa fizyczne. Istotne jest, iŜ rozpadowi w jednostce
czasu nie ulega określona masa materiału, lecz pewna jego część. Przykładowo jeŜeli promieniowanie pochodziz 1 kg substancji promieniotwórczej, to po czasie t rozpadnie się 0,5 kg, ale jeŜeli weźmiemy 2 kg, to po tymsamym czasie rozpadnie się 1 kg! Wynika stąd, iŜ szybkość rozpadu zaleŜy wprost proporcjonalnie od liczbyatomów w próbce; po wprowadzeniu współczynnika proporcjonalności – stałej rozpadu λ otrzymujemy:
τλλ
λλλλ
ttt
N
N
N
N
eNeNNeN
N
tN
Ndt
N
dNdt
N
dNN
dt
dN
−−− ==⇒=⇒
⇒−=⇒−=⇒−=⇒=− ∫∫
000
0
ln00 ,
gdzie τ=1/λ to średni czas Ŝycia jąder. Z prawa rozpadu wynika, iŜ połowa próbki rozpadnie się po czasie:
λλλλλ 693,02ln
2lnln 21
0021
021 ==⇒=⇒−=⇒=⇒= − TTTeNNNN T ,
zwanym czasem połowicznego zaniku (okresem półtrwania). Na podstawie tej wielkości dzieli się izotopy natrwałe i nietrwałe. Izotop przyjmuje się za trwały, gdy jego okres półtrwania wynosi ponad miliard lat. Cociekawe, niektóre pierwiastki mają ponad trzydzieści izotopów, jednak Ŝaden z nich nie naleŜy do trwałych.
Jak łatwo się domyślić, poglądy na temat budowy atomu nie zatrzymały się na modelu Rutherforda.Problemem do rozwiązania pozostawał opis ruchu elektronów wokół jądra. KrąŜący elektron powinien bowiemstopniowo tracić energie kinetyczną, a po jej wyczerpaniu zostać przyciągnięty przez jądro. Rozwiązanie podałw 1913r. Niels Bohr w postaci teorii orbit stacjonarnych. Zgodnie z nią elektron nie traci energii, gdy porusza się
po orbicie dozwolonej, czyli gdy moment pędu elektronu jest całkowitą wielokrotnością stałej π2h=η , gdzie
h oznacza stałą Plancka (I postulat). Dodatkowo przejście elektronu w wyŜszej orbity dozwolonej na niŜsząodpowiada emisji fotonu o energii równej róŜnicy energii elektronu na tych orbitach: ∆En→k=En-Ek=hν (IIpostulat).
RozwaŜania te pozwoliły na dokładny opis ruchu elektronu, zgonie z determinizmem fizycznym. Nowościąnatomiast okazała się udowodniona przez Wernera Heisenberga (1927r.) zasada nieoznaczoności, zgodnie zktórą nie moŜna jednocześnie i z dowolną dokładnością zmierzyć dwóch wielkości fizycznych kanoniczniesprzęŜonych tj. połoŜenia i pędu, połoŜenia kątowego i momentu pędu, energii i czasu itd. Dodatkowo iloczynniepewności pary wielkości fizycznych kanonicznie sprzęŜonych jest nie mniejszy od stałej Plancka.SpostrzeŜenie to ugodziło w przewidywalność ruchu elektronu i zasugerowało, iŜ brak dokładności nie jestkonsekwencją niedoskonałości przyrządów czy technik, lecz wypływa z samej natury elektronu.
Kolejnych informacji dostarczyła hipoteza, jaką wysunął w 1924r. Ludwik de Broglie. Stwierdził on, iŜ zruchem cząstki skojarzona jest fala materii (tzw. fala de Broglie’a) o określonej długości λ = h / p, gdzie hoznacza stałą Plancka, a p – pęd cząstki. PowyŜszy wzór moŜna przedstawić w kilku alternatywnych formach,np. wiedząc, Ŝe pęd jest iloczynem masy i prędkości: λ = h / (mv). Z kolei energia ciała E = p2 / 2m, gdzie m tomasa ciała, stąd p = √2mE, czyli λ = h / √2mE. Energia spoczywającej cząstki o ładunku q rozpędzonej przezróŜnicę potencjałów U wynosi E = e U, stąd λ = h / √2meU.
Powiązanie dotychczasowych zdobyczy fizyki pozwoliło Erwinowi Schrödingerowi podać równanie ruchu
elektronu (1926r.) w postaci: t
iVm ∂
Ψ∂=Ψ
+∆− ηη
2. Jego rozwiązaniami są funkcje falowe Ψ, opisujące
orbitale atomowe. Orbital atomowy jest fragmentem przestrzeni wokółjądrowej, w której istnieje duŜe (≥90%)prawdopodobieństwo znalezienia elektronu. Miarą tego prawdopodobieństwa jest kwadrat modułu funkcjifalowej |Ψ|2. Na stan kwantowy elektronu składa się jego energia, zachowanie w polu magnetycznym, orientacjawektora momentu pędu oraz spin, czyli ruch wirowy wokół własnej osi. Wielkości te opisywane są przy pomocyliczb kwantowych, wynikających z równania falowego ruchu elektronu.
Created by Neevia Document Converter trial version http://www.neevia.com

5
a) Główna liczba kwantowa (n) kwantuje energię elektronu i decyduje o wielkości obszaru orbitalnego.Związana jest z powłoką elektronową (poziomem energetycznym), czyli zbiorem elektronów o zbliŜonychwartościach energii. Elektron a n-tej powłoce posiada energię En=-E1/n
2, gdzie E1 jest energią na powłocepierwszej; przyjmuje się ją za wartość stałą dla danego atomu, np. dla atomu wodoru E1 = 13,6 eV. Głównaliczba kwantowa przyjmuje wartości kolejnych liczb naturalnych: 1, 2, 3 itd. teoretycznie aŜ donieskończoności (w praktyce do 7, gdyŜ atomy znanych pierwiastków mają od 1do 7 powłokelektronowych). Kolejne powłoki oznacza się wielkimi literami alfabetu:
n 1 2 3 4 5 6 7nazwa powłoki K L M N O P Q
b) Poboczna (orbitalna, azymutalna) liczba kwantowa (l) kwantuje orbitalny moment pędu elektronu idecyduje o kształcie obszaru orbitalnego. Związana jest z podpowłoką elektronową (podpoziomemenergetycznym), czyli zbiorem elektronów o takich samych wartościach energii. Liczba ta przyjmujewartości od 0 do n - 1, gdzie n to główna liczba kwantowa. Stanom tym odpowiada połoŜenie elektronu naokreślonych podpowłokach:
l 0 1 2 3 4nazwa podpowłoki s p d f g
Danej wartości pobocznej liczby kwantowej odpowiada określony kształt orbitalu.
Orbital typu s ma kształt kulisty, co oznacza, iŜ prawdopodobieństwo napotkania elektronu w kaŜdymkierunku, począwszy od jądra, jest jednakowe (rysunek lewy). Orbital typu p ma kształt ósemki obrotowej, apołoŜony jest na jednej z osi układu współrzędnych (x, y, z – m = -1, 0, 1), co oznacza, iŜ wzdłuŜ tej osiwystępuje największe prawdopodobieństwo napotkania elektronu (rysunek środkowy). Orbital typu d makształt dwóch ósemek obrotowych połoŜonych róŜnie względem osi układu współrzędnych (np. rysunekprawy).
c) Magnetyczna liczba kwantowa (m) kwantuje rzut orbitalnego momentu pędu elektronu na wyróŜnionykierunek w przestrzeni i decyduje o orientacji obszarów orbitalnych w polu magnetycznym. Związana jest zpoziomem orbitalnym. Przyjmuje wartości od –l do l, gdzie l to poboczna liczba kwantowa, czyli razem2n+1 róŜnych wartości. Dzięki temu warunkowi moŜna określić, ile istnieje moŜliwych ustawień orbitalu,czyli ile orbitali mieści powłoka:
l=0 l=1 l=2 l=3 l=41 orbital s 3 orbitale p 5 orbitali d 7 orbitali f 9 orbitali g
Podpoziomy p, d i dalsze nazywamy zdegenerowanymi, gdyŜ występuje na nich więcej orbitali o tej samejenergii, róŜniących się tylko orientacją przestrzenną.
d) Spinowa liczb kwantowa (s) kwantuje własny moment pędu elektronu. Ma ona wartość stałą, równa ½.e) Magnetyczna spinowa liczba kwantowa (ms) kwantuje rzut własnego momentu pędu
elektronu na wyróŜniony kierunek w przestrzeni. Przyjmuje ona dwie wartości: ms=½(tzw. spin dodatni) oraz ms=-½ (tzw. spin ujemny).
Stanem kwantowym elektronu nazywamy jego opis za pomocą czterech liczbkwantowych. Np. (3,1,1,½) oznacza powłokę M, podpowłokę 3p, orbital 3p oraz spin dodatni.Elektrony objęte są zakazem Pauliego: nie istnieją w atomie elektrony mające takie same wartości wszystkichliczb kwantowych – muszą się róŜnić co najmniej jedną z nich. Wynika stąd, iŜ na jednym orbitalu mogąwystępować maksymalnie dwa elektrony o przeciwnych liczbach magnetycznych spinowych. Pojemnościpodpowłok wynoszą zatem:
s – 1 . 2 =2 e-
p – 3 . 2 = 6 e-
d – 5 . 2 = 10 e-
f – 7 . 2 = 14 e-
Teoretyczną pojemność n-tej powłoki otrzymamy obliczając sumę:2 + 6 + 10 + 14 + ... + 2 . (2m+1) = 2 . [ 1 + 3 + 5 + 7 + 2m+1 ] = 2 . n2,gdzie n jest główną liczba kwantową – jest to tzw. wzór Bohra.
Created by Neevia Document Converter trial version http://www.neevia.com

6
KONFIGURACJA ELEKTRONOWA, PROMOCJE ELEKTRONOWE, BLOKI ENERGETYCZNE, REGUŁAHUNDA, TEORIA WZBUDZENIA
Konfiguracja elektronowa pierwiastka to rozmieszczenie jego elektronów na powłokach ipodpowłokach atomu. Kolejność obsadzania elektronami poszczególnych stanów kwantowychw atomach wieloelektronowych określa schemat zwany „złotym deszczem” (rysunek obok).Podczas zapisu konfiguracji elektronowej atomu lub jonu stosuje się schemat: numer powłoki –symbol podpowłoki – liczba elektronów na podpowłoce (w wykładniku symbolu). Dlauproszczenia zapisu, coraz dłuŜszego w przypadku dalszych pierwiastków, podaje się wnawiasie symbol poprzedzającego pierwiastek helowca oraz pozostałą część konfiguracji.Ilustrują to poniŜsze przykłady:
20Ca 1s22s22p63s22p64s225Mn2+ [18Ar] 4s25d3
20Ca [18Ar] 4s226Fe3+ [18Ar] 3d5
26Fe [18Ar] 4s23d616S
2- [10Ne] 3s23p6
82Pb [54Xe] 4f145d106s22p220Ca2+ [18Ar]
11Na+ [10Ne] 13Al3+ [10Ne]Powłoką walencyjną nazywamy powłokę o najwyŜszej w danym atomie wartości energii, wypełniającej sięelektronami jako ostatnia. Znajdujące się na niej elektrony nazywamy walencyjnymi. One właśnie biorą udział wreakcjach chemicznym i wiązaniach chemicznych, są ponadto odpowiedzialne za wartościowość pierwiastka.Dla pierwiastków grup głównych układu okresowego liczba elektronów walencyjnych jest równa numerowigrupy. Pozostałe elektrony nazywamy elektronami rdzenia. Te ostatnie wraz z jądrem atomu tworzą zrąbatomowy.
Powszechnie występującym zjawiskiem są promocje elektronowe. Promocja polega na tym, Ŝekonfiguracja elektronowa ulega zmianie ze względu na większą trwałość nowo powstałego układu. Najczęściejdotyczy ona jednego elektronu, który przeniesiony zostaje na wyŜszą podpowłokę powłoki niŜszej. Jest tokonsekwencją faktu, iŜ kolejna powłoka zapełnia się szybciej niŜ dalsze podpowłoki powłoki niŜszej. Wynika toz przyjmowanych przez znajdujące się na nich elektrony wartości energii, a ilustruje to najlepiej „złoty deszcz”.Typowymi układami wyjściowymi do promocji są: d4s2 przekształcający się w d5s1 oraz d9s2 – w d10s1. Wynikastąd, iŜ promocjom powinny ulegać pierwiastki grupy 6 i 11 i jest tak na prawdę. Przykładowo z połoŜenia złotaw układzie okresowym wynika konfiguracja: 79Au [54Xe] 4f145d96s2, jednak w rzeczywistości elektronywalencyjne są ułoŜone następująco: 4f145d106s1. Na uwagę zasługuje równieŜ pallad, gdzie konfiguracja 4d85s2
zmienia się w 4d10, czyli promocji ulegają aŜ dwa elektrony, pustosząc całkowicie podpoziom s. Opróczwymienionych, promocji ulegają pierwiastki takie jak: 24Cr, 29Cu, 41Nb, 42Mo, 43Tc, 44Ru, 45Rh, 47Ag, 78Pt.
Ze względu na konfigurację elektronową w układzie okresowym wyróŜnia się tzw. bloki energetyczne.Forma długa (układ Wernera) grupuje pierwiastki według symbolu podpoziomu energetycznego. Dziękiznajomości konfiguracji w poszczególnych blokach moŜna szybko ustalić budowę pierwiastka bez korzystania z„deszczu”.
s: ns1-2 d: (n-1)d1-10ns2
p: ns2np1-6 f: (n-2)f1-14(n-1)d1ns2
Ze względu na potrzebę czytelnego przedstawienia konfiguracji elektronów walencyjnych wprowadza siętzw. zapis klatkowy. KaŜda klatka oznacza jeden orbital będący w stanie pomieścić dwa elektrony o przeciwniezorientowanych spinach. Zbiór odpowiedniej ilości klatek daje podpoziom walencyjny. Ilustruje to poniŜszyrysunek:
W ustalaniu właściwej konfiguracji pomocna jest reguła Hunda, zwana teŜ regułą maksymalnejróŜnorodności. Składa się nań kilka zasad:a) liczba niesparowanych elektronów na podpoziomie zdegenerowanym powinna być
moŜliwie największa,b) pary elektronowe tworzą się dopiero po zapełnieniu wszystkich orbitali danego
podpoziomu przez elektrony niesparowane,c) elektrony niesparowane danego podpoziomu mają ten sam kierunek spinu.
Pod wpływem rozmaitych czynników (zderzenie z elektronem, absorpcja fotonu, działanie termiczne)atomy mogą ulegać wzbudzeniu. Wówczas ich elektrony przechodzą na wyŜsze podpoziomy energetyczne, cozwiązane jest ze zmianą konfiguracji oraz wzrostem wartościowości.
Created by Neevia Document Converter trial version http://www.neevia.com

7
ELEKTROUJEMNOŚĆ, ZWIĄZEK BUDOWY ATOMU Z UKŁADEM OKRESOWYM, TEORIA OKTETU
Elektroujemnością nazywamy zdolność pierwiastka do wiązania własnych i obcych elektronów walencyjnych.Elektroujemność pierwiastków maleje w grupach a roŜnie w okresach wraz ze wzrostem liczby atomowejpierwiastka. Metale mają niską elektroujemność (są elektrododatnie). Aktywność metali rośnie z grupach imaleje w okresach wraz ze wzrostem liczby atomowej, ogólnie – wzrasta ze spadkiem elektroujemności. Z koleiaktywność niemetali wzrasta ze wzrostem elektroujemności , malaje więc ona w grupach a rośnie w okresachwraz ze wzrostem liczby atomowej.
Efektem ekranowania jądra nazywamy zjawisko osłabienia oddziaływania pomiędzy jądrem atomu aelektronami walencyjnymi, powstające wskutek obecności między nimi elektronów rdzenia. Siła efektu rośniewraz ze wzrostem numeru okresu – liczby powłok ekranujących. Jest to jedna z przyczyn róŜnicelektroujemności pomiędzy pierwiastkami.
Związek budowy układu okresowego z właściwościami pierwiastkówWielkość Kierunek wzrostu
elektroujemnośćenergia jonizacjipowinowactwo elektronowepromień atomuaktywność metalizasadowość tlenków i wodorkówmetalicznośćaktywność niemetalikwasowość tlenków i wodorkówniemetalicznośćmoc kwasów tlenowychmoc kwasów beztlenowychmaksymalna wartościowość względem tlenu →maksymalna wartościowość względem wodoru →IV←
KaŜdy układ w przyrodzie dąŜy do moŜliwie najstabilniejszego stanu energetycznego. W przypadku atomukonsekwencje tego dąŜenia opisuje teoria oktetu – pierwiastki dąŜą do uzyskania trwałej, ośmioelektronowej(ew. dwuelektronowej) powłoki zewnętrznej (tzw. oktet lub dublet elektronowy) – takiej, jaką mają gazyszlachetne. Mogą tego dokonać przez oddawanie, przyjmowanie lub uwspólnianie elektronów w wiązaniachchemicznych.
2He 1s2 K2 dublet10Ne 1s22s22p6 K2L8 oktet18Ar 1s22s22p63s23p6 K2L8M8 oktet
a) Metale mają niską elektroujemność i liczbę elektronów walencyjnych, dlatego dąŜą do trwałości poprzezoddawanie elektronów, tworząc kationy. Np.:11Na 1s22s22p63s1 ---> 11Na+ 1s22s22p6 (oktet) XNa=0,9
Created by Neevia Document Converter trial version http://www.neevia.com

8
20Ca 1s22s22p63s23p64s2 ---> 20Ca2+ 1s22s22p63s23p6 (oktet) XCa=1,0Metale grup pobocznych mogą tworzyć kationy poprzez oddawanie elektronów z podpoziomu s ostatniejpowłoki oraz z podpoziomu d przedostatniej powłoki. Np.:26Fe 1s22s22p63s23p63d64s2
26Fe2+ 1s22s22p63s23p63d6
26Fe3+ 1s22s22p63s23p63d5
b) Niemetale mają wysoką elektroujemność i liczbę elektronów walencyjnych, dlatego dąŜą do trwałościpoprzez przyjmowanie elektronów, tworząc aniony. Np.:17Cl 1s22s22p63s23p5 ---> 17Cl- 1s22s22p63s23p6 (oktet) XCl=3,08O 1s22s22p4 ---> 8O
2- 1s22s22p6 (oktet) XO=3,5
TYPY WIĄZAŃ CHEMICZNYCH
a) Wiązanie jonowe tworzy się pomiędzy pierwiastkami skrajnie róŜniącymi się elektroujemnością (∆X=|XA-XB|>1,7), czyli pomiędzy metalami i niemetalami. Występuje najczęściej w tlenkach metali, wodorotlenkachi solach. Najbardziej typowe wiązanie tworzy się pomiędzy pierwiastkami grup I i II a VI i VII. Wiązaniejonowe polega na oddaniu elektronów walencyjnych przez np. metal i włączeniu ich do powłokiwalencyjnej np. niemetalu. W ten sposób tworzą się kationy i aniony, które budują sieć krystaliczną związkujonowego.Właściwości związków jonowych:
- trwałość sieci krystalicznej,- stały stan skupienia i wysokie temperatury topnienia,- przewodnictwo prądu elektrycznego po stopieniu lub rozpuszczeniu w wodzie.
Dla przykładu rozpatrzmy proces tworzenia chlorku potasu:
Aby oderwać elektron od atomu potasu naleŜy dostarczyć energii jonizacji E1. Przyłączeniu elektronu doatomu chloru towarzyszy wydzielenie energii powinowactwa elektronowego E2. Aby reakcja zaszła, naleŜydo układu dostarczyć róŜnicę wartości tych energii, czyli deficyt energetyczny, gdyŜ E1 > E2. Warunkiemtrwałości powstałego związku jest aby suma energii sieci krystalicznej oraz powinowactwa elektronowegobyła większa od energii jonizacji. Zazwyczaj energia sieci krystalicznej jest bardzo duŜa, co powodujewłasności takie jak stały stan skupienia oraz wysokie temperatury topnienia.
XK=0,9 XCl=3,0 ∆X=2,1>1,7Nie istnieją cząsteczki chlorku potasu – tworzy on sieć krystaliczną. KCl jest jedynie wzorem empirycznym,wyraŜającym stosunek liczby jonów w sieci. O tym, jaka ilość jonów przeciwnego znaku otacza dany jon wsieci krystalicznej, decyduje tzw. liczba koordynacyjna. Wyznacza się ją na podstawie znajomościpromienia kationu i anionu, co przedstawia poniŜsza tabela.
Stosunek promienia jonu metalu do niemetalu 0,15-0,23 0,23-0,41 0,41-0,73 0,73-1,00 ≥1,00Liczba koordynacyjna jonu metalu 2 4 6 8 12
Wartościowość jest liczbą wiązań, jakie moŜe wytworzyć atom danego pierwiastka. Jest ona zawszedodatnia i w grupach głównych równa jest numerowi grupy. Stopień utlenienia pierwiastka to liczbaładunków elementarnych, które pojawiłby się na atomie danego pierwiastka, gdyby wszystkie wiązania wdanym związku były czysto jonowe. W przeciwieństwie do wartościowości moŜe przyjmować takŜewartości ujemne.
b) Wiązanie kowalencyjne powstaje pomiędzy atomami o takiej samej lub zbliŜonej wartości elektroujemności(∆X=0÷0,4). Jego powstanie polega na uwspólnieniu elektronów walencyjnych brakujących do oktetu (ew.
Created by Neevia Document Converter trial version http://www.neevia.com
9
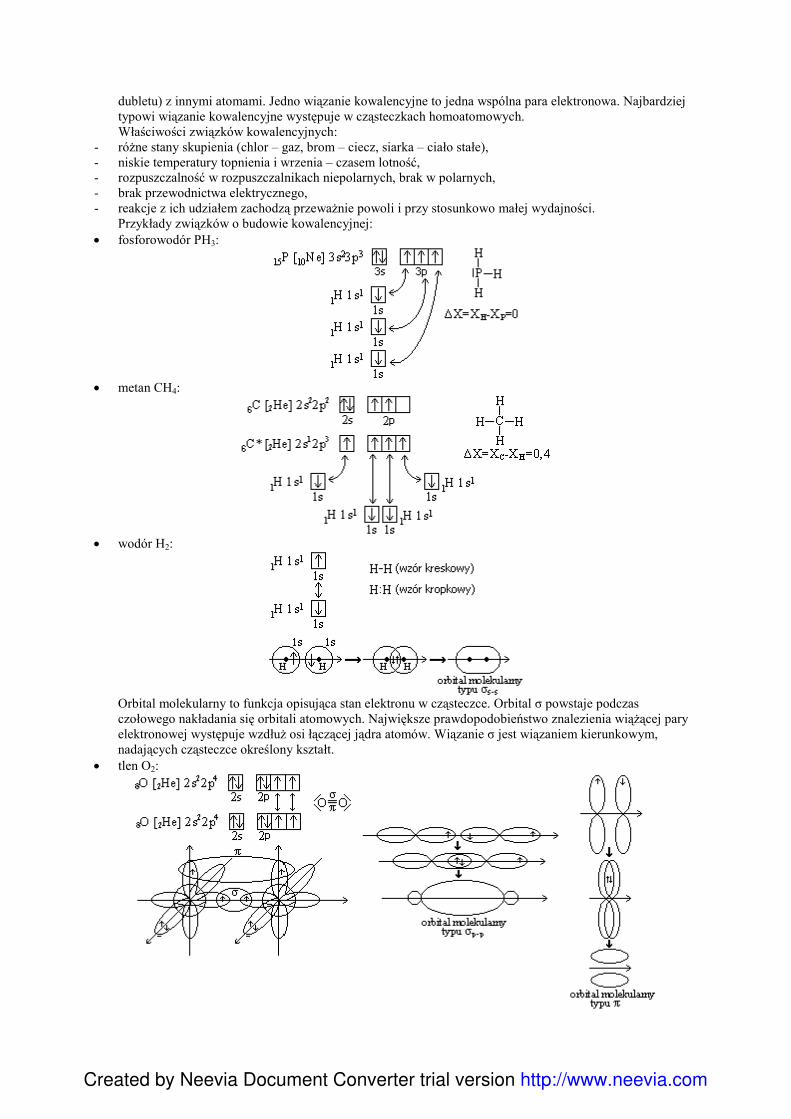
dubletu) z innymi atomami. Jedno wiązanie kowalencyjne to jedna wspólna para elektronowa. Najbardziejtypowi wiązanie kowalencyjne występuje w cząsteczkach homoatomowych.Właściwości związków kowalencyjnych:
- róŜne stany skupienia (chlor – gaz, brom – ciecz, siarka – ciało stałe),- niskie temperatury topnienia i wrzenia – czasem lotność,- rozpuszczalność w rozpuszczalnikach niepolarnych, brak w polarnych,- brak przewodnictwa elektrycznego,- reakcje z ich udziałem zachodzą przewaŜnie powoli i przy stosunkowo małej wydajności.
Przykłady związków o budowie kowalencyjnej:• fosforowodór PH3:
• metan CH4:
• wodór H2:
Orbital molekularny to funkcja opisująca stan elektronu w cząsteczce. Orbital σ powstaje podczasczołowego nakładania się orbitali atomowych. Największe prawdopodobieństwo znalezienia wiąŜącej paryelektronowej występuje wzdłuŜ osi łączącej jądra atomów. Wiązanie σ jest wiązaniem kierunkowym,nadających cząsteczce określony kształt.
• tlen O2:
Created by Neevia Document Converter trial version http://www.neevia.com

10
Orbital molekularny π powstaje podczas bocznego nakładania się orbitali atomowych p. Jest onzorientowany równolegle do osi wiązania σ. Składa się z dwóch jednakowych części, znajdujących się poobu stronach prostej przechodzącej przez jądra atomów tworzących wiązanie. Para elektronowa zajmującaobszar molekularny π tworzy wiązanie π. Nie występuje ono nigdy samodzielnie – pary elektronowe π wrazz parą elektronów σ tworzą wiązania wielokrotne, tj. podwójne lub potrójne.
c) Wiązanie kowalencyjne spolaryzowane (polarne = biegunowe) powstaje między atomami znacznieróŜniącymi się elektroujemnością. Polega ono na uwspólnieniu elektronów walencyjnych, przy czymwiąŜąca para elektronowa znajduje się bliŜej atomu o wyŜszej elektroujemności. Atom ten ładuje sięujemnie, a cała cząsteczka staje się dipolem. Miarą przesunięcia pary elektronowej jest moment dipolowy
wiązania: lqρρ⋅=µ . Naturalną jednostką jest kulombometr [C.m], poniewaŜ
jednak niewygodnie jest posługiwać się tak małymi liczbami, przyjmuje siędebje [D] (czyt. ‘debaj’). 1 D = 3,33.10-30 C.m.Właściwości związków polarnych:
- rozpuszczalność w wodzie i w innych rozpuszczalnikach o budowie jonowej,- dysocjacja jonowa w środowisku wodnym – moŜliwość przewodzenia prądu elektrycznego,- niskie temperatury topnienia,- w ciałach stałych tworzenie nietrwałych sieci krystalicznych – kryształy są miękkie i nietrwałe.
Przykłady związków o budowie polarnej:• dwutlenek węgla CO2:
• woda H2O:
• amoniak NH3:
• czterochlorek węgla CCl4:
d) Wiązanie koordynacyjne (donorowo – akceptorowe) jest wiązaniem typu kowalencyjnego. Polega nauwspólnieniu elektronów pochodzących od jednego atomu, zwanego donorem. Na schemacie zaznacza sięje strzałką z grotem skierowanym do donora do akceptora.Przykłady cząstek zawierających wiązanie koordynacyjne:
• kation amonowy NH4+:
Created by Neevia Document Converter trial version http://www.neevia.com

11
• kation hydroniowy H3O+:
e) Wiązanie metaliczne (struktura metaliczna) występuje w kryształach metali lub ich stopów. Atomy metalizostają przekształcone w elementy sieci krystalicznej: jądra atomowe obsadzają węzły sieci, zaś elektronywalencyjne tworzą chmurę elektronową, która scala całą sieć. Węzły są wierzchołkami komórekelementarnych. Dzięki bliskości węzłów, elektrony mogą swobodnie poruszać się we wnętrzu metalu, cojest warunkiem łatwego przewodzenia prądu elektrycznego.Cechy metali wynikające z ich budowy:
- duŜa gęstość,- wysokie temperatury topnienia i wrzenia,- twardość, kowalność i ciągliwość,- dobre przewodnictwo cieplne i elektryczne.f) Wiązanie wodorowe polega na oddziaływaniu typu elektrostatycznego. Występuje ono pomiędzy atomem o
duŜej elektroujemności a atomem wodoru związanym spolaryzowanym wiązaniem kowalencyjnym z innymsilnie elektroujemnym atomem. Tym ostatnim jest najczęściej azot, tlen lub fluorowiec. Wstępowaniewiązań wodorowych nie pozostaje bez wpływu na właściwości substancji. Najczęściej objawia się większągęstością, lepkością i lotnością, czasem innym stanem skupienia. RóŜnice widać na przykładzieizomerycznych alkoholi i eterów – pomimo braku róŜnic w masie cząsteczkowej niŜsze alkohole są ciekłe,zaś etery – gazowe. Do ciekawych wniosków prowadzi równieŜ obserwacja stanu skupienia wodorkówpierwiastków grupy 16 – wszystkie one są gazami wyjątkiem najlŜejszego – wody. Ten pozorny paradokstłumaczy duŜa róŜnica elektroujemności pomiędzy wodorem a tlenem, tak iŜ występujące pomiędzycząsteczkami wody wiązania wodorowe utrzymują ją w stanie ciekłym (zjawisko asocjacji).Przykłady substancji zawierających wiązania wodorowe:
- stały cyjanowodór (struktura liniowa):.....H-C≡N|.....H-C≡N|.....H-C≡N|.....H-C≡N|.....H-C≡N|.....
- stały fluorowodór (struktura zygzakowata):
- woda – asocjat (H2O)n:
HYBRYDYZACJA
Hybrydyzacja orbitali atomowych to metoda wyjaśniania geometrycznego kształtu cząsteczek. Hybrydyzacjajest operacją matematyczną polegającą na uśrednieniu kształtu i energii orbitali walencyjnych atomu centralnegocząsteczki. WyróŜnia się kilka podstawowych typów hybrydyzacji.a) Hybrydyzacja tetraedryczna (sp3) polega na uśrednieniu energii i kształtu jednego orbitalu s i trzech orbitali
p. Orbitale zhybrydyzowane mają kształt zdeformowanej ósemki obrotowej, z której kaŜda skierowana jestku naroŜnikowi czworościanu foremnego. Atom węgla w stanie hybrydyzacji tetragonalnej otacza sięwyłącznie wiązaniami pojedynczymi.Przykłady związków z hybrydyzacją sp3:
• metan CH4:
Cząsteczka metanu ma kształt czworościanu foremnego – tetraedru. W kaŜdym z naroŜy znajduje się takisam element, stąd cząsteczka cechuje się regularnością i zerowym momentem dipolowym. Pomiędzysymetralnymi orbitali s-p (wiązaniami węgiel – wodór) występuje kąt 109o28’, będący kątemcharakterystycznym tego typu hybrydyzacji.
Created by Neevia Document Converter trial version http://www.neevia.com

12
• amoniak NH3:
W cząsteczce amoniaku występuje wolna para elektronowa. Fakt ten sprawia, iŜ budowa przestrzenna niejest idealnie regularna, gdyŜ wolna para odpycha orbitale z większą siłą, niŜ one siebie nawzajem. Następujelekkie wygięcie orbitali w stronę przeciwną, a kąt pomiędzy wiązaniami odbiega od wartościcharakterystycznej – wynosi 107o.
• woda H2O:
W cząsteczce wody występują dwie wolne pary elektronowe. Z tego powodu wiązania ulegają odgięciu, akąt pomiędzy nimi – zmniejszeniu (104o27’). Zgięta budowa cząsteczki wody pozwala na traktowanie ejjako dipola elektrostatycznego.
b) Hybrydyzacja trygonalna (sp2) polega na uśrednieniu energii i kształtu jednego orbitalu s i dwóch orbitali p.Zhybrydyzowane orbitale skierowane są ku wierzchołkom trójkąta równobocznego. Kąt charakterystycznyhybrydyzacji wynosi zatem 120o.Przykładem takiej cząsteczki jest fluorek boru BF3:
c) Hybrydyzacja digonalna polega na uśrednieniu energii i kształtu jednego orbitalu s i jednego p. Atomytworzące cząsteczkę są ułoŜone liniowo, a kat charakterystyczny hybrydyzacji wynosi 180o.Przykładem takiej cząsteczki jest chlorek berylu BeCl2:
d) Istnieją równieŜ inne typy hybrydyzacji – pokaŜemy to na przykładzie sześciofluorku siarki SF6, któregocząsteczka ma kształt bipiramidy tetragonalnej.
Created by Neevia Document Converter trial version http://www.neevia.com

13
VSEPR
VSEPR (ang. vallence shell electron pair repulsion – dosł. odpychanie się par elektronów powłok walencyjnych)jest metodą określania kształtu przestrzennego drobin chemicznych. MoŜe być stosowana pod kilkomawarunkami:- drobina składa się wyłącznie z atomów grup głównych (bloki energetyczne s i p),- drobina posiada tylko jeden atom centralny,- podstawniki (ligandy) są atomami prostymi – nie mogą to być grupy złoŜone.Etapy postępowania przy posługiwaniu się metodą VSEPR:a) zapis cząstki w postaci EAnHm, gdzie E to atom centralny, A – ligandy niewodorowe, H – ligandy
wodorowe i odczytanie wartości n i m,b) obliczenie całkowitej liczby elektronów walencyjnych w cząsteczce lub jonie (Lwal),c) obliczenie liczby powstałych par elektronowych (Lpar = ½ . Lwal),d) obliczenie ilości własnych par elektronowych atomu centralnego (LwpE = Lpar - 4n - m),e) obliczenie liczby przestrzennej drobiny (Lp = LwpE + n + m),f) wykonanie rysunku odpowiednio do uzyskanej wartości liczby przestrzennej.
Wartość Lp Kształt cząsteczki2 budowa liniowa3 trójkąt równoboczny4 tetraedr5 bipiramida trygonalna6 bipiramida tetragonalna7 bipiramida pentagonalna
Ponadto zachodzą następujące relacje:a) liczba wiązań σ łączących atom centralny z ligandami: Lσ = n + m, czyli Lp = LwpE + Lσ,b) liczba wiązań π w drobinie (dla grup IV-VIII): Lπ = 4 - Lp; wartość ujemna oznacza brak wiązań π.
PODSTAWY CHEMII
PODSTAWOWE POJĘCIA
a) Zjawisko fizyczne to zjawisko, podczas którego następuje zmiana właściwości fizycznych substancji, np.stanu skupienia, gęstości, lepkości, spręŜystości czy kształtu.
b) Zjawisko chemiczne (reakcja chemiczna) to zjawisko prowadzące do zmiany składu chemicznego substancjioraz ich właściwości fizycznych.
c) Atom to najmniejsza część pierwiastka zachowująca jeszcze jego właściwości chemiczne.d) Cząsteczka to zespół co najmniej dwóch atomów (mogą być one takie same bądź róŜne). Cząsteczki
dzielimy na homoatomowe (cząsteczki pierwiastków) oraz heteroatomowe (cząsteczki związkówchemicznych).
e) Substancja chemiczna to rodzaj materii o ściśle określonych właściwościach fizycznych i chemicznych,posiadającej określoną masę. Substancje dzielimy na proste (pierwiastki) oraz złoŜone (związki chemiczne).
f) Pierwiastek chemiczny (pojęcie to wprowadził Robert Boyle) to najprostsza substancja, której nie da się juŜdalej rozłoŜyć metodami chemicznymi.
g) Symbol chemiczny pierwiastka to skrót łacińskiej nazwy pierwiastka (jedno- lub dwuliterowy).h) Związek chemiczny to substancja składająca się z co najmniej dwóch pierwiastków, połączonych ze sobą w
charakterystyczny, określony i uporządkowany sposób.i) Wzór chemiczny to przedstawienie składu ilościowego i jakościowego związku chemicznego przy pomocy
symboli chemicznych oraz indeksów stechiometrycznych.
Created by Neevia Document Converter trial version http://www.neevia.com

14
j) Postulaty Johna Daltona (1804r.):- KaŜdy pierwiastek chemiczny jest zbiorem małych cząstek zwanych atomami. Atomy tego samego
pierwiastka mają identyczne właściwości chemiczne. (Postulat ten nie uwzględnia występowania izotopów.)- Atomy róŜnych pierwiastków róŜnią się od siebie właściwościami fizycznymi i chemicznymi. Istnieje tyle
rodzajów atomów o określonych właściwościach, ile jest rodzajów pierwiastków. (TakŜe ten postulat nieuwzględnia występowania izotopów.)
- Atom danego pierwiastka nie moŜe ulec przekształceniu w atom innego pierwiastka na drodze chemicznej.(MoŜliwe jest to natomiast na drodze fizycznej w sposób naturalny – przez rozpad promieniotwórczy bądźteŜ w sposób sztuczny – przez fuzję jąder.)
- Łączenie się pierwiastków w związki chemiczne polega na łączeniu się ich atomów w większe zespołyzwane cząsteczkami.
- Związek chemiczny jest zbiorem cząsteczek. Wszystkie cząsteczki danego związku mają taki sam składatomowy pierwiastków i identyczne właściwości chemiczne.
- Rozkład związku chemicznego na pierwiastki polega na rozpadzie cząsteczek tego związku na atomyposzczególnych pierwiastków. MoŜe to być proces wieloetapowy.
- Atomy tego samego pierwiastka mogą równieŜ łączyć się ze sobą w cząsteczki.k) Wartościowość pierwiastka w związku chemicznym to ilość wiązań chemicznych tworzonych przez atom
tego pierwiastka w cząsteczce związku chemicznego.l) Postulaty teorii wartościowości (J. Wislicenus, 1868r.):- Pierwiastki w wstanie czystym nie posiadają wartościowości.- Dany pierwiastek moŜe mieć więcej wartościowości niŜ jedną.- Wartościowość wodoru jest stała i wynosi I.- Wartościowość tlenu jest równieŜ stała i wynosi II.- Pierwiastki leŜące w grupach nieparzystych mają na ogół wartościowości nieparzyste, zaś pierwiastki grup
parzystych – wartościowości parzyste. („Na ogół”, poniewaŜ istnieją od tej reguły wyjątki, np.czterowartościowy atom chloru.)
m) Prawo stosunków stałych (prawo stałości składu, J. L. Proust, 1799r.): stosunek wagowy pierwiastków wdanym związku chemicznym jest zawsze stały, charakterystyczny dla danego związku i nie zaleŜy odsposobu otrzymywania tego związku.
n) Prawo stosunków wielokrotnych (J. Dalton): jeŜeli dwa pierwiastki tworzą ze sobą kilka związkówchemicznych, to ilości wagowe jednego pierwiastka, przypadające na tę samą ilość drugiego pierwiastka,pozostają do siebie w stosunku niewielkich liczb naturalnych.
o) Równanie chemiczne to zapis przebiegu reakcji chemicznej przy pomocy symboli i wzorów chemicznych.KaŜde równanie chemiczne musi spełniać prawo stałości składu oraz prawo zachowania masy.
p) Prawo zachowania masy (A. Lavoisser, M. Łomonosow): masa substratów biorących udział w reakcji jestrówna masie produktów otrzymywanych w jej wyniku.
q) Reakcja syntezy to reakcja, podczas przebiegu której przynajmniej dwa substraty łączą się w jeden produkto bardziej złoŜonej budowie.
r) Reakcja analizy to reakcja, podczas przebiegu której jeden złoŜony substrat rozpada się na kilka produktówo prostszej budowie.
s) Reakcja wymiany pojedynczej (prostej) to reakcja, podczas przebiegu której z pierwiastka i związkuchemicznego powstaje inny pierwiastek i inny związek chemiczny.
t) Reakcja wymiany podwójnej to reakcja, podczas przebiegu której z dwóch związków chemicznych tworząsię dwa inne związki chemiczne o innych właściwościach.
STECHIOMETRIAa) Międzynarodowa jednostka masy atomowej (1 unit) to masa odpowiadająca 1/12 masy izotopu węgla 12C.
1 u = 1,66.10-24 g 1 g = 6,022.1023 ub) Masa atomowa to masa atomu pierwiastka wyraŜona w unitach.c) Masa cząsteczkowa to masa cząsteczki pierwiastka wyraŜona w unitach. Oblicza się ją sumując masy
poszczególnych atomów wchodzących w skład danego związku.d) Mol to liczba cząstek równa ilości atomów węgla zawartych w 12 g jego izotopu 12C. Liczba ta nosi nazwę
liczby Avogadra i wynosi: NA=6,022.1023 mol-1.e) Masa molowa (M) to masa jednego mola cząstek. Masa molowa danej substancji równa się liczbowo jej
masie atomowej lub cząsteczkowej, wyraŜonej w g / mol.f) Objętość molowa (VM) to objętość jednego mola cząstek. Objętość molowa gazu doskonałego jest stała i
wynosi V0=22,4 dm3/mol. Najczęściej gazy rzeczywiste przybliŜa się do gazu doskonałego, stąd moŜnaprzyjąć, Ŝe objętość mola dowolnego gazu wynosi V0.
g) Warunki normalne: T0=273,16K=0oC, p0=101325 Pa=1 atm.h) Warunki standardowe: T0=292K=25oC, p0=101325 Pa=1 atm.
Created by Neevia Document Converter trial version http://www.neevia.com

15
i) Prawo Avogadra (1811r.): jednakowe objętości róŜnych gazów zawierają w tych samych warunkachciśnienia i temperatury jednakowe liczby cząsteczek tych gazów. W 22,4 dm3 dowolnego gazu znajduje sięzatem, 6,022.1023 cząsteczek tego gazu.
j) Prawo Gay – Lussaca (1809r.): objętości reagujących ze sobą gazów i powstających w wyniku reakcjiproduktów gazowych mają się do siebie tak jak niewielkie liczby naturalne (w tych samych warunkachciśnienia i temperatury).
k) W równaniu reakcji chemicznej często zestawia się ze sobą ilości reagujących substratów, jak równieŜ ilościpowstałych w wyniku przemiany produktów. Porównywać moŜemy albo masy albo ilości moli (atomów lubcząsteczek) albo objętości wybranych reagentów. Dzieląc przez siebie odpowiednie masy otrzymujemystosunek wagowy (masowy), odpowiednie ilości moli – stosunek molowy, zaś odpowiednie objętości –stosunek objętościowy. Zgodnie z matematyczną definicją równość dwóch stosunków nazywamy proporcją.Proporcje reagentów (proste bądź mieszane) wykorzystywane są w zadaniach w celu szybkiego obliczenianieznanych wartości mas, ilości moli czy objętości.
TERMOCHEMIAa) Układ chemiczny to ogół substancji chemicznych biorących udział w reakcji. Układy chemiczne moŜna
podzielić na otwarte, zamknięte oraz izolowane.b) Układ otwarty to układ, który ma moŜliwość wymiany z otoczeniem zarówno masy jak i energii, np.
otwarty cylinder.c) Układ zamknięty to układ, który ma moŜliwość wymiany energii z otoczeniem, ale nie ma moŜliwości
wymiany masy, np. zamknięty cylinder.d) Układ izolowany to układ, który nie ma moŜliwości wymiany z otoczeniem ani masy ani energii. Układy
izolowane istnieją wyłączenie hipotetycznie, gdyŜ nie znamy doskonałych izolatorów cieplnych. Wymianęenergii z otoczeniem moŜne pominąć przy bardzo szybkim zachodzeniu procesu, np. w podczas przemianyadiabatycznej.
e) Energia wewnętrzna układu chemicznego (Ew, U) to całkowita ilość energii zawartej wewnątrz danegoukładu. Jej wartość jest niemoŜliwa do określenia, gdyŜ składa się na nią bardzo wiele rodzajów energii, zktórych pewne nie zostały jeszcze dokładnie poznane.
f) I zasada termodynamiki: zmiana energii wewnętrznej moŜe odbywać się na sposób ciepła lub pracyobjętościowej, z czego wynika zaleŜność: ∆U=±Q±Vobj.
g) Entalpia reakcji (H) to ciepło reakcji zachodzącej pod stałym ciśnieniem. Na podstawie I zasadytermodynamiki zmiana entalpii wyraŜa się wzorem: ∆H=∆U+p∆V (p=const), gdzie drugi składnik sumyokreśla wartość pracy objętościowej. Zmiana objętości, wynikająca ze zmiany liczby moli gazu o jeden, podciśnieniem normalnym, równowaŜna jest z wykonaniem pracy równej co do wartości:W=Vmp0=0,0224m3/mol.101325N/m2=2271J/mol≈2,5kJ/mol.
h) Zmiana entalpii (entalpia reakcji) to róŜnica sumy entalpii tworzenia produktów oraz sumy entalpiitworzenia substratów; rozwaŜając odpowiednie entalpie spalania składniki występują w odwrotnejkolejności. KaŜdy układ w przyrodzie dąŜy do zgromadzenia jak najmniejszej ilości energii, gdyŜ jest tosytuacja najstabilniejsza i najbardziej prawdopodobna, a więc dąŜy do zmniejszenia swojej entalpii. Pojęcieto tłumaczy zatem zachodzenie procesów egzoergicznych.
i) Entalpia standardowa (H0) to entalpia liczona w odniesieniu do jednego mola reagentu.j) Reakcja egzoenergetyczna (egzoergiczna) to reakcja, podczas przebiegu której energia wydzielana jest do
otoczenia. Entalpia takiego procesu jest ujemna. Reakcje tego typu są samorzutne – po zainicjowaniuzachodzą dalej samodzielnie.
k) Reakcja endoenergetyczna (endoergiczna) to reakcja, podczas przebiegu której energia jest pobierana zotoczenia, np. na sposób ciepła. Entalpia takiego procesu jest dodatnia. Reakcje tego typy są wymuszone, ado podtrzymania przebiegu wymagają stałego dopływu energii.
l) Entropia (S) jest miarą rozkładu energii w układzie chemicznym. Jest funkcję stanu, więc jej wartość zaleŜywyłącznie od początkowego i końcowego stanu układu. Jest wielkością addytywną, co oznacza, iŜrozpatrywany proces moŜna podzielić na kilka etapów i obliczyć entropie cząstkowe, które po dodaniudadzą entropię całkowitą. W termodynamice fenomenologicznej entropia określana jest róŜniczką zupełną:dQ=TdS, stąd dS=dQ/T, czyli S=∫dQ/T + const,gdzie dQ oznacza nieskończenie małą ilość energii wymienionej na sposób ciepła w procesie odwracalnym,a T – temperaturę bezwzględną. Wynika stąd, iŜ w układach izolowanych, wobec braku wymiany ciepła zotoczeniem, entropia nie zmienia się. Podobnie kaŜdy cykl odwracalny moŜna podzielić na nieskończonąliczbę cząstkowych procesów adiabatycznych, zatem podczas przemiany odwracalnej entropia nie ulegazmianie. W termodynamice statystycznej przyjmuje się: S=k.lnW, gdzie k oznacza stałą Boltzmanna, zaśW – liczbę sposobów podziału energii w układzie, dla układów makroskopowych jest ona olbrzymia wporównaniu z liczbą Avogadra.
Created by Neevia Document Converter trial version http://www.neevia.com

16
m) Entropia reakcji to róŜnica sumy entropii produktów oraz sumy entropii substratów. KaŜdy układ wprzyrodzie dąŜy do stanu równowagi poprzez obniŜenie energii wewnętrznej oraz zwiększenie entropii.Układy dąŜą zatem do zapewnienia moŜliwie największej róŜnorodności sposobów podziału energii międzydrobiny oraz poszczególne rodzaje form jej gromadzenia. Entropia rośnie ze wzrostem temperatury, oobniŜaniem ciśnienia oraz przy rozpadzie wiązań chemicznych. Pojęcie entropii tłumaczy takie procesy jakzachodzenie przemian endoergicznych (czynnik entropowy przewaŜa nad energetycznym), zachodzeniereakcji w układach izolowanych oraz samorzutne mieszanie się gazów. Samorzutnie zachodzą wyłącznieprocesy nieodwracalne, zwiększające entropię środowiska. Wszechświat dąŜy do stanu równowagi poprzezcoraz większe nieuporządkowane (chaos) materii. Za jedyny prawdziwie izolowany układ moŜna uznaćjedynie cały Wszechświat (wobec braku otoczenia) – zachodzą więc w nim nieodwracalne procesyzwiększania entropii. Na tej podstawie moŜna wnioskować, iŜ w odległej przyszłości procesy te doprowadządo ustania wszelkiego ruchu i osiągnięcia przez Wszechświat stanu równowagi termodynamicznej (tzw. stanśmierci cieplnej Wszechświata). Teoria ta opisuje jednak zdarzenie bardzo mało prawdopodobne.
n) Entropia standardowa (S0) to entropia liczona w odniesieniu do jednego mola reagentu.o) Entalpia swobodna (G) to funkcja stanu określająca samorzutność przebiegu oraz stan równowagi reakcji
chemicznej. Jej zmianę podaje równania Gibbsa – Helmholtza postaci ∆G= ∆H-T∆S. KaŜdy układ dąŜy dozmniejszenia energii wewnętrznej (∆H<0) oraz do wzrostu entropii (∆S>0), co w efekcie daje ∆G<0 – stantaki oznacza reakcję samorzutną. Analogicznie ∆G=0 oznacza ustalenie się stanu równowagi w układzie,zaś ∆G<0 – zachodzenie reakcji wymuszonej. Entalpia swobodna reakcji to róŜnica sum entalpiiswobodnych produktów oraz substratów.
p) Prawo Hessa: efekt cieplny reakcji nie zaleŜy od drogi reakcji (pod warunkiem, Ŝe p,V=const), a pracaobjętościowa jest jedyną formą przemiany energii. Efekt cieplny reakcji zaleŜy zatem wyłącznie odpoczątkowego oraz końcowego stanu układu.
q) Prawo Lavoissier’a – Laplace’a: efekt cieplny reakcji odwrotnej do danej ma taką samą wartość jak reakcjidanej, lecz przeciwny znak. Jest to swoista konsekwencja zasady zachowania energii.
STATYKA I KINETYKA REAKCJI CHEMICZNYCHa) Reakcja nieodwracalna to reakcja, podczas której w środowisku powstają wyłącznie produkty. Przyjmuje
się, Ŝe tego typu reakcje zachodzą do końca. NaleŜą do tej grupy reakcje zobojętniania oraz te, w wynikuktórych wytrąca się nierozpuszczalny osad, wydziela się gaz lub powstaje słaby elektrolit.
b) Reakcja odwracalna to reakcja, podczas której gromadzące się produkty reagują ze sobą i odtwarzająsubstraty. W środowisku reakcji oprócz produktów występują nie przereagowane substraty. Reakcje tegotypu nie zachodzą do końca. NaleŜy tu np. dysocjacja słabych elektrolitów lub hydroliza soli. Większośćreakcji naleŜy właśnie do tej grupy.
c) Równowaga to ustalony stan np. reakcji chemicznej.d) Równowaga statyczna to stan, w którym procesy chemiczne nie zachodzą, a stęŜenia reagentów nie
zmieniają się w czasie.e) Równowaga dynamiczna to stan, w którym zachodzą przemiany chemiczne, jednak tyle samo substratów
przechodzi w produkty, co produktów w substraty. W stanie równowagi dynamicznej szybkość reakcji danejoraz reakcji do niej przeciwnej są sobie równe, a stęŜenia reagentów ustalają się wówczas na stałympoziomie.
f) Miarą szybkości reakcji jest szybkość pojawianie się produktów (i ubytku substratów) w środowisku, stądv=dc/dt. Szybkość reakcji zaleŜy proporcjonalnie od stęŜenia początkowego substratów, więc v=k.c0, gdziek oznacza współczynnik proporcjonalności, zwaną stałą szybkości reakcji.
g) Reguła van’t Hoffa to empiryczna i orientacyjna reguła pozwalająca na określenie zmian szybkości reakcjipod wpływem zmian temperatury. W jej myśl szybkość reakcji wzrasta od dwóch do czterech razy przypodwyŜszeniu temperatury o 10oC. Liczba określająca, ile razy wzrasta szybkość reakcji, nazywana jestwspółczynnikiem temperaturowym: Θ=kT+10/kT=kT/kT-10. Reguły tej nie stosuje się do reakcji wybuchowychi przebiegających w obecności stałych katalizatorów.
h) Dla większych ilości reagentów wyraŜenia opisujące szybkość reakcji powiększają się o iloczynodpowiednich stęŜeń.A <=v1=> B, v1=k1[A],A + A <=v1=> B, v1= k1[A][A]=k1[A]2,aA <=v1=> B, v1=k1[A]a itd.Ogólnie dla modelowej reakcji: aA + bB <=v1
v2=> cC + dD wyraŜenia na stałe szybkości przyjmują postać:v1=k1[A]a[B]b, v2=k2[C]c[D]d. PoniewaŜ w stanie równowagi prędkości obu przeciwstawnych reakcji sąrówne, otrzymujemy: v1=v2 => k1[A]a[B]b=k2[C]c[D]d => k1/k2=[C]c[D]d/[A]a[B]b=const. Interpretacjąotrzymanej zaleŜności jest podaje prawo działania mas.
i) Prawo działania mas (prawo Guldberga – Waagego): w stanie równowagi chemicznej iloczyn stęŜeń(ciśnień) reagentów w wykładnikach potęgowych ich współczynników stechiometrycznych jest wielkością
Created by Neevia Document Converter trial version http://www.neevia.com
17
stałą dla danej reakcji i w określonej temperaturze i określaną stałą równowagi reakcji (stęŜeniową Kc lubciśnieniową Kp – w praktyce najczęściej posługujemy się tą pierwszą). Pomiędzy stałymi równowagi tejsamej reakcji zachodzi zaleŜność: Kp=Kc(RT)∆n, gdzie R oznacza stałą gazową, T – temperaturębezwzględną, zaś ∆n – przyrost liczby moli produktów gazowych podczas reakcji. Stała stęŜeniowa iciśnieniowa są więc sobie równe, gdy podczas przebiegu reakcji nie zmienia się liczba moli produktówgazowych. Stała równowagi jest wielkością chemiczną charakteryzującą liczbowo stan równowagidynamicznej reakcji odwracalnej. Jej wartość nie zaleŜy od stęŜeń reagentów, a jedynie od temperatury,zmieniając się w sposób zaleŜny od efektu energetycznego reakcji.
j) Reguła przekory Le Chateliera – Browna: kaŜdy układ w przyrodzie dąŜy do osiągnięcia stanu równowagi, agdy na układ znajdujący się w stanie równowagi zadziała jakiś czynnik zewnętrzny, wówczas w układzietym zachodzą zmiany zmniejszające skutki działania tego czynnika. Pod pojęciem czynników zewnętrznychrozumiemy zmianę temperatury, ciśnienia bądź stęŜenia jednego z reagentów.
- wpływ temperatury na stan równowagi – stała równowagi zaleŜy tylko i wyłącznie od temperatury:• dla reakcji egzoergicznych (∆H<0) maleje ze wzrostem temperatury,• dla reakcji endoergicznych (∆H>0) rośnie ze wzrostem temperatury,- wpływ ciśnienia na stan równowagi:• dla reakcji, w których objętość produktów jest większa od objętości substratów, wzrost ciśnienia powoduje
przesunięcie równowagi reakcji w lewo, zaś spadek ciśnienia – w prawo,• dla reakcji, w których objętość produktów jest mniejsza od objętości substratów, wzrost ciśnienia powoduje
przesunięcie równowagi reakcji w prawo, zaś spadek ciśnienia – w lewo,- wpływ zmiany stęŜenia jednego z reagentów:
przesunięcie równowagi zmniejszenie stęŜenia zwiększenie stęŜeniasubstraty w lewo w prawoprodukty w prawo w lewo
UKŁADY DYSPERSYJNE
OGÓLNA CHARAKTERYSTYKA I PODZIAŁ UKŁADÓW DYSPERSYJNYCH
Układ dyspersyjny (rozproszony) to układ złoŜony z co najmniej dwóch składników, zwanych fazami, z którychjeden jest rozproszony w drugim. Fazy zmieszane są ze sobą w określonej (ogólnie jednak dowolnej) proporcjiilościowej, nie są połączone chemicznie, zachowują swoje indywidualne właściwości fizyczne i chemiczne,moŜne je rozdzielić metodami fizycznymi. Do tych ostatnich naleŜą m. in.: dekantacja, sedymentacja,segregacja, sączenie, destylacja, ekstrakcja, adsorpcja, chromatografia, odparowanie, krystalizacja, wymroŜenieitd.KaŜdy układ dyspersyjny składa się z fazy rozpraszającej, czyli ośrodka dyspersyjnego oraz z fazy rozproszonej,czyli substancji zdyspergowanej. Zarówno jedną jak i drugą fazę mogą stanowić pierwiastki lub związkichemiczne we wszystkich stanach skupienia. W zaleŜności od stanu skupienia układy dyspersyjne dzielimy wnastępujący sposób:
rozproszona \ rozpraszająca gaz ciecz ciało stałegaz roztwory gazowe (np.
powietrze)piany (np. woda sodowa) piany stałe (np. pumeks)
ciecz aerozole i mgły (np.deszcz)
roztwory właściwe (np.ocet) i emulsje (np. mleko)
(np. mokra gąbka)
ciało stałe dymy (np. kurz) roztwory właściwe, koloidyi zawiesiny
stopy metali (np. brąz)
Układy dyspersyjne dzielimy na jednorodne (homogeniczne) oraz niejednorodne (heterogeniczne).a) Układy homogeniczne charakteryzuje rozproszenie molekularne, a rozmiary cząsteczek rozproszonych są
wielkości mniejszych od 1 nm. Nazywa się je roztworami właściwymi. Są one jednorodne fizycznie, tzn.kaŜda próbka, pobrana z dowolnego miejsca układu, ma taki sam skład jakościowy i ilościowy.
b) Układy heterogeniczne cechuje rozproszenie koloidalne bądź makroskopowe. W pierwszy przypadkunazywamy je koloidami, a rozmiary cząstek rozproszonych zawierają się pomiędzy 1 nm a 200 nm. Wdrugim przypadku są to zawiesiny, gdzie rozmiary cząstek rozproszonych przewyŜszają 200 nm. Te ostatniesą zazwyczaj układami nietrwałymi, złoŜonymi z cząsteczek substancji stałej w cieczy lub gazie.
Created by Neevia Document Converter trial version http://www.neevia.com

18
Określanie składu mieszanin:a) procent wagowy i objętościowy:
pkw=mk/mm
.100%, pko=Vk/Vm
.100%,(indeks k dotyczy składnika numer k, zaś indeks m – mieszaniny),
b) przeliczanie procentu wagowego na procent objętościowy lub odwrotnie dla dwuskładnikowej mieszaninygazowej:p1
o=p1w.M2
.100%/(p1w.M2+p2
w.M1),p1
w=p1o.M1
.100%/(p1o.M1+p2
o.M2),c) ułamek wagowy, objętościowy i molowy:
ukw=mk/mm, ∑uw=1,
uko=Vk/Vm, ∑uo=1,
ukmol=nk/nm, ∑umol=1,
d) przeliczanie ułamków ilościowych dla mieszanin dwuskładnikowych:umol uw uo
umol -molmol
ww
wmol
uu
MuMu
Muu
12
1221
211
1−=
+=
molmol
oo
omol
uu
MduMdu
Mduu
12
122211
2111
1−=
+=
uw
ww
molmol
molw
uu
MuMu
Muu
12
2211
111
1−=
+=
-ww
oo
ow
uu
dudu
duu
12
2211
111
1−=
+=
uo
oo
molmol
molo
uu
dMudMu
dMuu
12
122211
2111
1−=
+=
oo
ww
wo
uu
dudu
duu
12
1221
211
1−=
+=
-
ROZTWORY
Roztworem nazywamy układ złoŜony z substancji rozpuszczonej (stałej, ciekłej bądź gazowej) w ciekłymrozpuszczalniku. Roztwory podzielić moŜna na właściwe (rzeczywiste, homogeniczne) oraz koloidalne(heterogeniczne). Rozpuszczalność ciał stałych i cieczy silnie zaleŜy od temperatury (zazwyczaj rośnie z jejwzrostem, gdyŜ proces rozpuszczania jest endoergiczny) i prawie nie zaleŜy od ciśnienia (substancje te sąpraktycznie nieściśliwe). Na rozpuszczalność gazów widoczny wpływ mają oba wymienione czynniki – malajeona ze wzrostem temperatury i rośnie ze wzrostem ciśnienia. W roztworach substancji stałych istnieje stanrównowagi dynamicznej pomiędzy osadem a substancją rozpuszczoną, ilościowo opisywany przez iloczynrozpuszczalności.
roztwórosadnierozpuszcza
cjakrystaliza⇔
Ze względu na stopień nasycenia roztwór moŜe być:a) nasycony – kiedy po dalszym wprowadzaniu substancji rozpuszczonej stęŜenie nie ulega zmianie (nie da się
dalej rozpuszczać danej substancji),b) nienasycony – kiedy po wprowadzeniu dodatkowej ilości substancji rozpuszczonej stęŜenie wzrasta (moŜna
w nim dalej rozpuszczać daną substancję),c) przesycony – o stęŜeniu większym od stęŜenia roztworu nasyconego w danej temperaturze. MoŜe on
powstać podczas obniŜania temperatury roztworu nasyconego w warunkach utrudnionego powstawaniazarodków krystalizacji. Wstrząśnięcie lub dodanie substancji rozpuszczonej powoduje gwałtowanąkrystalizację jej nadmiaru i powstanie roztworu nasyconego.
Krystalizacja to proces tworzenia się i wzrostu kryształów z cieczy, roztworu bądź fazy gazowej.Rozpuszczalność to maksymalna ilość gramów danej substancji, jaką w danych warunkach ciśnienia itemperatury moŜna rozpuścić w 100g rozpuszczalnika, otrzymując roztwór nasycony.
Created by Neevia Document Converter trial version http://www.neevia.com

19
Przy opisie właściwości roztworu największe znaczenie obok składu jakościowego ma podanie jego stęŜenia.a) stęŜenie procentowe to ilość gramów substancji zawarta w 100g roztworu:
%100%100 ×+
=×=lnikarozpuszczasubstancji
substancji
roztworu
substancji
pmm
m
m
mC ,
b) stęŜenie molowe to ilość moli substancji rozpuszczonej w 1dm3 rozpuszczalnika; jest wielkością zaleŜną odtemperatury:
×==
mol
dm
VM
m
V
nc
roztworusubstancji
substancji
roztworu
substancji
mol
3
,
c) stęŜenie molalne – ilość moli substancji rozpuszczonej zawarta w 1kg roztworu; jest wielkością niezaleŜnąod temperatury:
=
kg
mol
m
nc
roztworu
substancji
a .
Przeliczanie stęŜeń (M – masa molowa substancji rozpuszczonej)Cp cmol ca
Cp -r
molp
d
McC
%100⋅=
Mc
McC
a
ap +
⋅=
1
%100
cmol%100⋅
=M
dcc
rp
mol-
Mc
dcc
a
ramol +=
1
caMC
Cc
p
p
a )%100( −=
molr
mola
Mcd
cc
−= -
Reguła mieszania roztworów o znanych stęŜeniach procentowych:
)( 122
1
1
2ppxp
pxp
ppxCCC
CC
CC
m
m>>
−
−= .
Reguła mieszania roztworów o znanych stęŜeniach molowych:
molxmol
molmolx
CC
CC
V
V
−−
=2
1
2
1
12( molmolxmol CCC >> i przy pominięciu kontrakcji – załoŜeniu addytywności objętości)
Niektóre właściwości fizyczne roztworów:a) Kontrakcja to efekt towarzyszący mieszaniu roztworów, polegający na otrzymaniu roztworu o objętości
mniejszej od sumy objętości roztworów wyjściowych. Podobnie jak przy mieszaniu kamieni z piaskiem tenostatni zajmuje wole przestrzenie pomiędzy większymi okruchami, tak samo zachowują się cząsteczkirozpuszczalnika i substancji rozpuszczonej. Przy mieszaniu roztworów wodnych o niewielkiej róŜnicystęŜeń kontrakcja praktycznie nie występuje. Przy mieszaniu roztworu o duŜym stęŜeniu z czystymrozpuszczalnikiem kontrakcja wynosi nie więcej niŜ kilka procent. Np. przy zmieszaniu 480cm3 wody z20cm3 96% kwasu siarkowego (d=1,84g/cm3) otrzymujemy 495cm3, a nie przewidywane 500cm3;kontrakcja w tym przypadku wynosi: ∆V/V=5/500=1%. W typowych obliczeniach laboratoryjnychdotyczących stęŜeń molowych na ogół nie zachodzi potrzeba uwzględniania kontrakcji. W celu uzyskaniabardzo dokładnego wyniku naleŜy odpowiednie stęŜenia molowe zamienić na stęŜenia procentowe izastosować regułę mieszania roztworów procentowych.
b) Zmiana temperatury topnienia i wrzenia roztworu w stosunku do czystego rozpuszczalnika: roztwory wrą wwyŜszych i zamarzają w niŜszych temperaturach niŜ występujące w nich rozpuszczalniki. PodwyŜszenietemperatury wrzenia, podobnie jak obniŜenie temperatury topnienia, to wielkości koligatywne –charakterystyczne dla roztworu, a zaleŜne wyłącznie od liczby niezaleŜnych cząsteczek w danej objętościroztworu. Są one wprost proporcjonalne do stęŜenia molalnego roztworu i dla substancji nie dysocjującejwynoszą: ∆Tk=Ek
.ma, ∆Tw=Eb.ma. Występujące w powyŜszych równaniach współczynniki
Created by Neevia Document Converter trial version http://www.neevia.com

20
proporcjonalności noszą nazwy stałej krioskopowej oraz stałej ebulioskopowej. Wartości ich moŜnaobliczyć z równań prawa Raoulta: Ek=RTk
2M/Lt, Eb=RTw2M/Lp, gdzie R oznacza uniwersalną stałą gazową,
M – masę molową rozpuszczalnika, Tk, Tw – temperatury bezwzględne krzepnięcia i wrzeniarozpuszczalnika, Lt, Lp – ciepła molowe topnienia i parowania rozpuszczalnika. Przykładowo dla wodywartości te wynoszą: Ek=-1,87oC, Eb=0,51oC.
c) Ciśnienie osmotyczne: obecność w roztworze substancji rozpuszczonych powoduje powstawaniedodatkowego ciśnienia, zwanego ciśnieniem osmotycznym. Aby roztwór pozostał w równowadzeosmotycznej z czystym rozpuszczalnikiem, naleŜy nań wywrzeć nadwyŜkę ciśnienia równą ciśnieniuosmotycznemu. Przez równowagę osmotyczną rozumiemy, iŜ rozpuszczalnik, oddzielony od roztworuprzegrodą półprzepuszczalną, przenikliwą dla rozpuszczalnika a nieprzenikliwą dla substancjirozpuszczonej, nie wnika osmotycznie do roztworu ani teŜ nie jest z niego wyciskany nadwyŜką ciśnienia wroztworze. Zgodnie z prawem van’t Hoffa ciśnienie osmotyczne moŜna interpretować jako ciśnienie równetakiemu, jakie wywierałaby substancja rozpuszczona, jeśli znajdowałaby się w danej temperaturze w staniegazowym i zajmowała objętość równą objętości roztworu. Dla rozcieńczonych roztworów nieelektrolitówmoŜna tą wartość wyrazić przekształcając równanie Clapeyrona w równanie van’t Hoffa:
cRTV
nRTpnRTpV ==⇒= ,
gdzie c oznacza stęŜenie molowe roztworu, R – uniwersalną stałą gazową, a T – temperaturę bezwzględną.
TEORIE KWASÓW I ZASAD
Teorie kwasów i zasad są próbami wyjaśnienia zespołu cech wspólnych, charakterystycznych dla kwasów iprzeciwstawnego mu zespół cech zasad. Spośród kilkunastu zaproponowanych, największe uznanie zyskały:teoria Arrheniusa (1887r.), teoria Brönsteda – Lowry’ego (1923r.) oraz teoria Lewisa.
Teoria Arrheniusa sformułowana została w postaci trzech postulatów.Postulat I: Niektóre substancje, zwane elektrolitami, ulegają pod wpływem wody rozpadowi na dwa rodzajecząstek: jony dodatnie (kationy) oraz jony ujemne (aniony). Proces ten nazywamy dysocjacją jonową lubelektrolityczną.Z I postulatu wynika, iŜ wszystkie substancje moŜna podzielić na dwie duŜe grupy:- elektrolity, będące związkami o budowie jonowej bądź kowalencyjnej silnie spolaryzowanej (kwasy,
zasady, sole, wodorki kwasowe), których roztwory dobrze przewodzą prąd elektryczny dzięki obecności wnich cząstek obdarzonych ładunkiem (jonów), które mogą się przemieszczać w polu elektrycznym,
- nieelektrolity, będące związkami o budowie kowalencyjnej wcale lub bardzo słabo spolaryzowanej(cząsteczki pierwiastków oraz wiele związków organicznych), które nie dysocjują i nie przewodzą prądu.
Postulat II: Liczba wytworzonych podczas dysocjacji ładunków dodatnich musi być równa liczbiewytworzonych ładunków ujemnych. Jest to konsekwencją zasady zachowania ładunku. Zgodnie z tymzałoŜeniem przyjmuje się następujące definicje:- kwasy to substancje, które w procesie dysocjacji odłączają katony wodoru oraz aniony reszt kwasowych,
ogólnie: HnR ---> nH+ + Rn-;- zasady to substancje, które w procesie dysocjacji odłączają katony metali oraz aniony wodorotlenkowe,
ogólnie: M(OH)n ---> Mn+ + nOH-;- sole to substancje, które w procesie dysocjacji odłączają katony metali oraz aniony reszt kwasowych,
ogólnie: MnRm ---> nMm+ + mRn-.Postulat III: Niektóre elektrolity w swoich roztworach zawierają takŜe niezdysocjowane cząstki – są to tzw.elektrolity słabe.Z III postulatu wynika, Ŝe wszystkie elektrolity moŜna podzielić na:- elektrolity mocne, całkowicie rozpadające się na jony, których roztwory nie zawierają niezdysocjowanych
cząstek; do grupy tej naleŜą: wszystkie sole, kwasy: HNO3, H2SO4, HClO4, HCl, HBr, HI, wodorotlenkilitowców i berylowców za wyjątkiem berylu i magnezu,
- elektrolity słabe, rozpadające się na jony tylko częściowo, których roztwory zawierają duŜe ilościniezdysocjowanych cząstek.
Teoria Brönsteda – Lowry’ego zwana jest takŜe teorią protonową. W jej myśl kwasami nazywamy związki lubjony zdolne do oddawania kationów wodoru, zaś zasadami – związki lub jony zdolne do ich przyjmowania.PoniewaŜ kation wodoru to nic innego jak proton, toteŜ kwasy nazywamy donorami protonów(protonodawcami), a zasady – akceptorami protonów (protonobiorcami). KaŜda reakcja kwasu z zasadąprowadzi do wytworzenia nowej, sprzęŜonej pary kwas – zasada. Związki tworzące parę sprzęŜoną róŜnią się wskładzie cząsteczek co najwyŜej o proton (kation wodoru). Im mocniejszy kwas, tym słabsza jest sprzęŜona znim zasada. Według teorii protonowej kwasy moŜna podzielić na:
Created by Neevia Document Converter trial version http://www.neevia.com

21
- kwasy cząsteczkowe (HCl, H2SO4),- kwasy kationowe (NH4
+),- kwasy anionowe (HSO4
-),zaś zasady na:- zasady cząsteczkowe (NH3),- zasady anionowe (HSO4
-).Teoria protonowa rozszerza zatem pojęcie kwasu i zasady o nowe grupy związków. Niektóre cząsteczki mogąmieć ponadto charakter amfoteryczny, czyli wykazywać w zaleŜności od warunków właściwości kwasowe bądźzasadowe – np. HSO4
- moŜe zarówno przyłączyć, jak i odszczepić kation wodoru.
Teoria Lewisa równieŜ poszerza pojęcie kwasów i zasad. Zgodnie z jej załoŜeniami, kwasem jest związek lubjon posiadający deficyt elektronowy – kwasami Lewisa są zatem zarówno wszystkie kationy metali, jak iniektóre cząsteczki obojętne, np. BF3. Atom boru posiada w tym związku sześć elektronów walencyjnych, azgodnie z regułą oktetu dąŜy do uzyskania ośmiu – posiada zatem deficyt, gdyŜ 6<8. Analogicznie – zasadą jestzwiązek posiadający wolną parę elektronową, np. Cl- czy NH3. Dzięki swym właściwościom kwasy Lewisawykorzystywane są jako katalizatory w niektórych syntezach organicznych, np. w reakcjach Friedla – Craftsa.
ILOŚCIOWY OPIS WŁAŚCIWOŚCI ROZTWORÓW
W celu ilościowego opisu procesu dysocjacji wprowadza się wielkości chemiczne: stopień oraz stałą dysocjacji.a) Stopień dysocjacji (α) wyraŜa się stosunkiem ilości cząstek zdysocjowanych do ilości cząstek
wprowadzonych do roztworu: α=n/n0=c/c0. Np. dla reakcji AB <==> A+ + B-: α=[A+]/[AB]=[B-]/[AB](symbole w nawiasach kwadratowych oznaczają odpowiednie stęŜenia molowe). Dla elektrolitówdysocjujących wielostopniowo wyróŜnia się kolejne stopnie dysocjacji, przy czym α1>α2>α3 itd. Stopieńdysocjacji zaleŜy od:
- rodzaju elektrolitu,- temperatury – zasadniczo wzrasta z jej wzrostem,- obecności innych substancji,
- stęŜenia substancji wprowadzonej
∝
0
1c
α .
b) Stała dysocjacji (K), czyli stała równowagi reakcji dysocjacji, np. dla reakcji AB <==> A+ + B-:K=[A+][B-]/[AB]. Dla elektrolitów dysocjujących wielostopniowo wyróŜnia się kolejne stałe dysocjacji,przy czym K1>K2>K3 itd. Stała dysocjacji zaleŜy tylko i wyłącznie od temperatury.
RozwaŜmy następujący prosty przykład procesu dysocjacji:równanie reakcji AB <==> A+ + B-
stęŜenie początkowe c0 - -stęŜenie reagujące c c cstęŜenie równowagowe c0 - c c c
αα
αα
ααα
−=
−/=
−
⋅=
−⋅
==/−+
1)1(][
]][[ 20
0
20
2
00
00
0
c
c
c
cc
cc
cc
cc
AB
BAK
PowyŜszy związek to tzw. prawo rozcieńczeń Ostwalda. MoŜna za jego pomocą wyznaczyć stopień dysocjacji:
0
02
20
02
1
02
20
20
20
20
2
40
2
4
4
0
)1()1(/1
c
KcKK
c
KcKK
KcK
KKccKK
cKc
K
++−=<
+−−=
+=∆
=−+⇒=−⇒
⇒=−⇒−⋅−
=
αα
αααα
ααααα
Pierwsze rozwiązanie nie ma sensu chemicznego – stopień dysocjacji nie moŜe przyjmować wartości ujemnych.W przypadku, gdy α<5% lub gdy c0/K≥400, wówczas:
0
2010
cKc =⇒≈Κ ⇒ ≈ −1 ⇒ ≈ αααα .
Created by Neevia Document Converter trial version http://www.neevia.com

22
Autodysocjacja wody to rozpad cząsteczki wody pod wpływem innej cząsteczki wody; sumarycznie proces tenmoŜna przedstawić jako: H2O + H2O <==> H3O
+ + OH-.
Eksperymentalnie wyznaczona stała dysocjacji powyŜszej reakcji wynosi: K=1,8.10-16. W wyraŜeniu opisującymstałą pojawia się stęŜenie molowe wody, przyjmowane za wartość stałą i obliczane następująco:
3
3
25,55
18
1000][ 2 dm
mol
molg
dm
g
OmolHMMV
m
V
ncOH ======
ρ.
( )( )214
21416162
22
3
3
10]][[
10109,995,55108,1][
]][[][][
]][[
moldm
moldm
OHH
OHK
OHHOHKOH
OHHK
−−+
−−−
−+−+
=
≈⋅=⋅⋅=
=⇒=
PowyŜsza wartość to tzw. iloczyn jonowy wody – jest ona zawsze stała dla wszystkich roztworów wodnych wdanej temperaturze. Pozwala ona na obliczenie stęŜenie H+ lub OH- w danym roztworze, gdy dana jest jedna ztych wielkości. Wartości powyŜszych stęŜeń są bardzo małe, do rzędu czternastu miejsc po przecinku włącznie.Dla uniknięcia stosowania niewygodnych liczb podaje się nie samą ich wartość, lecz potęgę, do jakiej naleŜypodnieść podstawę równą 10, aby tę wartość otrzymać. PoniewaŜ są to najczęściej liczby ujemne, poprzedza sięje znakiem minus. W ten sposób określa się pojęcie pH oraz pOH:
]log[]log[10][
]log[]log[10][−−−−
++−+
−=⇒=−⇒=
−=⇒=−⇒=
OHpOHOHpOHOH
HpHHpHH
pOH
pH
.
Z powyŜszych definicji wynika następująca własność:
1410log])][log([]log[]log[ 14 =−=−=−−=+ −−+−+ OHHOHHpOHpH .
Ze względu na wartości stęŜeń H+ i OH- wprowadza się pojęcia odczynu roztworu:odczyn roztworu proporcje stęŜeń wartości stęŜeń pH i pOH
obojętny [H+]=[OH-] [H+]2=[OH-]2=10-14 => [H+]=[OH-]=10-7 mol/dm3 pH=pOH=7kwasowy [H+]>[OH-] [H+]>10-7 mol/dm3 [OH-]<10-7 mol/dm3 pH<7 pOH>7zasadowy [H+]<[OH-] [H+]<10-7 mol/dm3 [OH-]>10-7 mol/dm3 pH>7 pOH<7
Iloczyn rozpuszczalności to pojęcie wprowadzone dla roztworów nasyconych soli trudno rozpuszczalnych. GdystęŜenie osadu przyjmuje się za stałe, wówczas iloczyn stałej dysocjacji i stęŜenia osadu takŜe jest niezmienny, awięc iloczyn stęŜeń jonów w wykładnikach potęgowych ich współczynników stechiometrycznych takŜe będziestały i określany jako iloczyn rozpuszczalności (Ir). Jest to wielkość stała dla danej soli w danej temperaturze –charakteryzuje ona sól pod względem rozpuszczalności: im niŜsza wartość Ir, tym słabiej rozpuszczalna jest danasól. Gdy do roztworu nasyconego dodaje się jonów trudno rozpuszczalnej soli, wówczas iloczyn stęŜeń jonów wroztworze przekracza wartość Ir, co powoduje wytrącenie osadu; dodanie wody do wyjściowego roztworupowoduje zmniejszenie stęŜenia i przejście części osadu w postaci jonów do roztworu. JeŜeli w roztworzeznajdują się jony dwóch soli o róŜnych Ir, to jako pierwsza wytrąci się z roztworu sól o mniejszej wartości Ir.
REAKCJE ZACHODZĄCE W ROZTWORACH
Miareczkowanie alkacymetryczneAnaliza chemiczne to zespół czynności zmierzających do ustalenia jakościowego i ilościowego składu próbki.Jednym z jej działów jest analiza miareczkowa (objętościowa), polegająca na oznaczaniu składnika roztworu napodstawie pomiaru objętości substancji zuŜytej w celu ilościowego przeprowadzenia reakcji z tym składnikiem(titrantu). Titrant (roztwór mianowany) musi być roztworem o znanym z duŜą dokładnością stęŜeniuokreślonego składnika. Czynność w analizie miareczkowej, polegająca na dodawaniu z biurety titrantu doroztworu substancji oznaczanej do momentu zakończenia reakcji (osiągnięcia punktu równowaŜnikowego)nazywamy miareczkowaniem. Często w ramach analizy miareczkowej stosuje się alkacymetrię (miareczkowaniealkacymetryczne), polegającą na oznaczaniu stęŜenia kwasu poprzez miareczkowanie zasadą (alkalimetria) bądźzasady poprzez miareczkowanie kwasem (acydymetria). Punktem równowaŜnikowym (PR) nazywamy moment,
Created by Neevia Document Converter trial version http://www.neevia.com

23
w którym oznaczany składnik przereagował ilościowo z titrantem. Określa się go za pomocą wskaźników lubinnymi metodami, np. elektrochemiczną. Roztwór znajdujący się w PR jest całkowicie zobojętniony, co nieoznacza zawsze jednak odczynu obojętnego – sole hydrolizujące mogą mieć inny odczyn. Określa się równieŜpunkt końcowy miareczkowania (PK), na ogół nieznacznie róŜniący się od PR.Do ustalenia PR wykorzystuje się często metodę pomiaru przewodnictwa elektrycznego (η) roztworumiareczkowanego. Przewodnictwo roztworu w PR ma wartość minimalną, gdyŜ obojętny roztwór w bardzomałym stopniu przewodzi prąd elektryczny. Ilustrując zobojętnianie obserwujemy najpierw liniowy spadekprzewodnictwa, następnie linia wykresu ostro zaznacza PR, po czym przewodnictwo rośnie, gdyŜ przy dalszymdodawaniu titrantu zaczyna on dysocjować, zwiększając liczbę jonów w roztworze. W przypadku wytrącania sięnierozpuszczalnego osadu przewodnictwo w PR spada do zera – np. przy miareczkowaniu roztworu kwasusiarkowego wodorotlenkiem baru. Dla sformalizowania przewodnictwa elektrycznego roztworów wprowadza sięnastępujące wielkości chemiczne:- przewodność elektrolityczną: χ=1/ς [m2/Ω] (ς=RS/l),- przewodność molowa elektrolitu to stosunek przewodności elektrolityczne do stęŜenia molowego:
Λ=χ/c [m5/Ω/mol]; jeŜeli stęŜenie wyraŜone jest w mol/dm3, wówczas Λ=1000χ/c.
Amfoteryczność to zdolność niektórych związków chemicznych, najczęściej tlenków i wodorotlenków, dowykazywania zarówno właściwości kwasowych, jak i zasadowych. Związki takie nazywamy amfoterycznymilub amfoterami. Pierwiastki amfoteryczne leŜą na przekątnej układu okresowego i mają elektroujemnośćzbliŜoną do wodoru (ok. 1,5-2). W wodorotlenkach amfoterycznych wiązania pomiędzy atomami wodoru i tlenuoraz tlenu i metalu mają podobną moc. Tlenki i wodorotlenki amfoteryczne z reguły nie rozpuszczają się wwodzie. W reakcjach z mocnymi kwasami zachowują się jak zasady, a z zasadami – jak kwasy.NajwaŜniejsze amfotery to związki następujących pierwiastków: Be+2, Al+3, V+4, V+5, Cr+3, Mn+4, Fe+2, Fe+3,Co+2, Cu+1, Cu+2, Zn+2, Ga+3, Ge+4, As+3, Zr+4, Nb+5, Pd+4, Ag+1, Cd+2, In+3, Sn+2, Sn+4, Sb+3, Sb+5, Te+4, Hf+4,Ta+5, Re+4, Ir+3, Pt+4, Au+3, Pb+2, Pb+4, Ce+4, U
+6. Związki pierwiastków bloku d posiadających nie zapełnioneorbitale walencyjne są na ogół barwne. Wśród związków organicznych charakter amfoteryczny wykazują przedewszystkim aminokwasy z racji posiadania dwóch grup funkcyjnych – karboksylowej (kwasowej) oraz aminowej(zasadowej).
Zobojętnianie to reakcja łączenia się kwasowych kationów wodorowych z zasadowymi anionamiwodorotlenkowymi w obojętne cząsteczki wody: H+ + OH- ---> H2O.Hydroliza to reakcja odwrotna do zobojętniania – jest to reakcja jonów soli z cząsteczkami wody, w wynikuktórej roztwory wodne soli mogą mieć odczyn róŜny od obojętnego.
Rodzaj soli Typ hydrolizy Odczyn środowiskasól mocnego kwasu i słabej zasady hydroliza kwasowa (kationowa) kwasowysól słabego kwasu i mocnej zasady hydroliza anionowa (zasadowa) zasadowysól słabego kwasu i słabej zasady kwasowo-zasadowa (kationowo-anionowa) zbliŜony do obojętnego
sól mocnego kwasu i mocnej zasady brak hydrolizy obojętnyOdczyn środowiska, w którym hydrolizuje sól słabego kwasu i słabej zasady, nie jest idealnie obojętny,poniewaŜ stałe dysocjacji tworzących sól kwasu i zasady na ogół nieznacznie róŜnią się od siebie. O odczynieroztworu decyduje zatem relacja KK i KZ.
Wskaźniki (indykatory) to najczęściej słabe kwasy lub zasady organiczne, występujące w dwóch formachbarwnych. Przykładami są: oranŜ metylowy (na kwasy), błękit tymolowy, fenoloftaleina (na zasady) orazwskaźnik uniwersalny, będący odpowiednio spreparowaną mieszaniną innych wskaźników.
Roztwór buforowy (bufor = zderzak) to roztwór mający zdolność przeciwdziałania zmianom pH zarówno przyrozcieńczaniu, jak i dodawaniu do nich niewielkich ilości mocnych kwasów lub zasad. Roztworem buforowymmoŜe być:- roztwór słabego kwasu i jego soli z mocna zasadą,- roztwór słabej zasady i jej soli z mocnym kwasem,- roztwór dwóch soli kwasu wieloprotonowego.Działanie buforujące przedstawione zostanie na przykładzie buforu octanowego (CH3COOH + CH3COONa).protonodawca: CH3COOH <==> CH3COO- + H+ [CH3COOH]≈ckwasu
protonobiorca: CH3COONa <==> CH3COO- + Na+ [CH3COO-]≈csoli
a) protonodawca chroni bufor przed zmianami pH podczas dodawania do niego zasady, gdyŜ zobojętnia ją:CH3COOH + OH- ---> CH3COO- + H2O
b) protonobiorca chroni bufor przed zmianami pH podczas dodawania do niego kwasu, gdyŜ zobojętnia go:CH3COO- + H+ ---> CH3COOH
Created by Neevia Document Converter trial version http://www.neevia.com

24
Dla buforu słabego kwasu moŜna zapisać:
KKKsolikwasu
soli
kwasuK
soli
kwasuK
soli
kwasuK
kwasu
soliK
pKpKpKpHcc
c
cK
c
cKHpH
c
cKH
c
HcK
=−=−=⇒=
−−=
⋅−=−=
⋅=⇒⋅
=
+
++
01log
logloglog]log[
][][
Analogicznie dla buforu słabej zasady:
ZZZsolizasady
soli
zasady
Z
soli
zasady
Z
soli
zasady
Z
zasady
soliZ
pKpKpKpOHcc
c
cK
c
cKOHpOH
c
cKOH
c
OHcK
=−=−=⇒=
−−=
⋅−=−=
⋅=⇒⋅
=
−
−−
01log
logloglog]log[
][][
W przypadku, gdy objętości kwasu (zasady) i soli są róŜne, w miejsce stosunków stęŜeń naleŜy wstawićodpowiednie stosunki molowe.
Związki kompleksowe to połączenia tworzące się jako trwalsze formy obecności cząsteczek lub jonów wroztworach. Ich powstawanie jest przyczyną pogłębienia barwy roztworów. Kompleksy zbudowane są zcentralnie połoŜonego atomu lub jonu oraz otaczających go ligandów. Kompleksy mogą zawierać jeden lubwięcej jonów (atomów) centralnych, zwanych rdzeniami, stąd wyróŜnia się kompleksy jedno- orazwielordzeniowe. Ligandem moŜe być cząsteczka lub jon posiadające wolną parę elektronową. Liczbę ligandówotaczających atom centralny określa tzw. liczba koordynacyjna; zazwyczaj jest ona liczbą parzystą. Ligandypodzielić moŜna na jednokleszczowe (monodentne – koordynujące jedną parę elektronową) orazwielokleszczowe (multidentne – koordynujące dwoma lub więcej parami elektronowymi). Ze względu naładunek, kompleksy moŜna podzielić na kationowe, anionowe oraz obojętne. Przy zapisie kompleksu w postaciwzoru chemicznego kompleks ujmuje się w nawias kwadratowy.Najczęściej występujące ligandy oraz skojarzone z nimi przedrostki:
Wzór Nazwa Wzór NazwaH2O akwa- S2O3
2- tiosiarczano-NH3 amino- SCN- tiocyjaniano-CO karbonylo- NO nitrozylo-Cl- chloro- azotyno-CH3COO- octano- nitro-
W celu ilościowego ujęcia mocy kompleksu się następujące wielkości chemiczne:a) stała nietrwałości (rozpadu) kompleksu to stała równowagi hipotetycznej reakcji ELn <==> En+ + nL-, czyli:
][]][[
EL
LEK
nn
N
−+
= ; im wyŜsza wartość stałej nietrwałości, tym dany kompleks jest mniej trwały;
b) stała trwałości (tworzenia kompleksu) to odwrotność stałej nietrwałości, a zarazem stała równowagi reakcji
En+ + nL- <==> ELn, czyli: nn
N
TLE
EL
KK
]][[
][1−+== ; im wyŜsza wartość stałej trwałości, tym trwalszy
jest dany związek kompleksowy.
KOLOIDY
Koloidy to układy dwu- lub wieloskładnikowe, heterogeniczne, w których cząstki fazy rozproszonej mająśrednic rzędu 1-200nm. Cząstki koloidalne nie są widoczne gołym okiem ani pod mikroskopem, przechodzątakŜe przez sączki filtracyjne. NajwaŜniejsze układy koloidowe to:
Created by Neevia Document Converter trial version http://www.neevia.com

25
Nazwa Stan skupienia faz Przykładyzole ciało stałe w cieczy lub w gazie smog, dym w powietrzu, roztwór białka, skrobi, krochmal,
kleje pochodzenia roślinnego, budynie i kisielemgły ciecz w gazie mgła atmosferycznaaerozoledymy ciało stałe w gazie dym z komina, kurz domowy
emulsje ciecz w cieczy mleko, majonez, masło, margaryna, kremyciekłe gaz w cieczy piana mydlanapianystałe gaz w ciele stałym pumeks, styropian, materace piankowe
Koloidy moŜna podzielić na:a) koloidy cząsteczkowe – fizycznie jednorodne, których składnikami są makrocząsteczki, po wprowadzeniu
do wody zawsze otrzymujemy roztwór koloidowy, np. koloid białka, skrobi, celulozy, glikogenu, kauczuku;b) koloidy fazowe – fizycznie niejednorodne, z nieciągłą powierzchnią podziału obu faz, na której zachodzi
adsorpcja elektrolitu, warunkująca powstanie ładunku elektrycznego, np. zol złota (Au)x (tzw. purpuraKasjusza), koloid srebra (Ag)y, koloid chlorku srebra (AgCl)z (x, y, z > 100).
Zasadniczo koloidy moŜna otrzymać na dwa sposoby:a) metoda dyspersyjna polega na rozpraszaniu większych cząstek do rozmiarów koloidowych – do tego celu
wykorzystuje się specjalne urządzenia, zwane młynkami koloidowymi (dzielą się na termiczne, elektryczneitd., w zaleŜności od rodzaju czynnika rozpraszającego),
b) metoda kondensacyjna i polimeryzacyjna polega na łączeniu atomów lub cząsteczek w większe zespoły orozmiarach odpowiadających cząsteczkom koloidowym (np. metoda strąceniowa).
Ze względu na stosunek do cząsteczek rozpuszczalnika koloidy dzielimy na:a) liofilowe (gdy rozpuszczalnikiem jest woda – hydrofilowe), których cząsteczki otaczają się zwartym
płaszczem cząsteczek rozpuszczalnika, dzięki czemu stają się bardziej odporne na działanie czynnikówścinających; są podatne na pęcznienie, tworzenia galaret i Ŝelów (np. koloid białka);
b) liofobowe (ew. hydrofobowe), w których oddziaływanie z cząsteczkami rozpuszczalnika jest bardzo słabe,przez co są one bardziej podatne na ścinanie od liofilowych (np. koloid srebra czy złota).
Właściwości roztworów koloidowych:a) efekt Tyndalla – specyficzny sposób rozpraszania wiązki światła w charakterystyczny stoŜek Tyndalla,b) barwa najczęściej biała; niektóre koloidy posiadają barwę zaleŜną od rozmiarów cząstek koloidowych,
grubości warstwy, przez którą przechodzi światło oraz od sposobu obserwacji,c) opalizowanie na świetle – obserwacja tęczowych refleksów,
d) zdolność do koagulacji, czyli łączenia się w większe zespoły, np.: koagulatkoloidkoagulacja
peptyzacja⇔ :
- koagulacja odwracalna: )()( 3
osadbialkowysolonebiałiakoloidNaNOsolirr
adestylowanwoda
−
⇔ ,
- koagulacja nieodwracalna (denaturacja): bialkoanezdenaturowbiałiakoloidcydenaturujączynnik
⇔ ;
do czynników denaturujących zaliczamy: wysoką temperaturę, jony metali cięŜkich, alkohol, kwasy, zasady,formalinę,
e) tendencja do adsorpcji cząsteczek rozpuszczalnika na swojej powierzchni i wytwarzanie dzięki temuładunku elektrycznego o określonym znaku oraz otaczania się jonami o znaku przeciwnym na skutekprzyciągania elektrostatycznego.
ELEKTROCHEMIA
STOPNIE UTLENIENIA PIERWIASTKÓW
Stopień utlenienia pierwiastka w związku o budowie jonowej jest równy ładunkowi jonu, jaki tworzy atom tegopierwiastka w związku. Stopień utlenienia pierwiastka w związku o budowie kowalencyjnej jest równy liczbiewszystkich ładunków dodatnich lub ujemnych, które przypisalibyśmy atomowi pierwiastka, gdyby związek miałbudowę jonową.Zasady ustalania stopnia utlenienia pierwiastka:a) pierwiastki w stanie wolnym mają zawsze stopień utlenienia równy zero,
Created by Neevia Document Converter trial version http://www.neevia.com

26
b) wodór przyjmuje z związkach chemicznych zawsze stopień utlenienia równy +1, z wyjątkiem wodorkówmetali, gdzie posiada –1,
c) tlen przyjmuje w związkach chemicznych zawsze stopień utlenienia równy –2, z wyjątkiem tlenku fluoru(+2) oraz nadtlenków (–1),
d) metale w związkach chemicznych mają zawsze dodatnie stopnie utlenienia, liczbowo równewartościowości,
e) suma algebraiczna wszystkich stopni utlenienia w obojętnym związku chemicznym jest równa zero, a wjonie złoŜonym równa jest ładunkowi tego jonu.
REAKCJE REDOX W UJĘCIU ELEKTRONOWYM
a) Utleniania (dezelektronacja) to reakcja oddawania elektronów i podwyŜszania stopnia utlenieniapierwiastka.
b) Redukcja (elektronacja) to reakcja przyjmowania elektronów i obniŜania stopnia utlenienia pierwiastka.c) Utleniacz (dezelektronator, elektronobiorca) to atom pierwiastka, który przyjmując elektrony powoduje
utlenienie atomu innego pierwiastka, a sam się redukuje. Typowe utleniacze chętnie przyjmują elektrony isą „na minusie”.
d) Reduktor (elektronator, elektronodawca) to atom pierwiastka, który oddając elektrony powoduje redukcjęatomu innego pierwiastka, a sam się utlenia. Typowe reduktory chętnie oddają elektrony i są „na minusie”.
e) Prawo zachowania ładunku dla reakcji redox: liczba elektronów oddanych w procesie utleniania musi byćrówna liczbie elektronów przyjętych w procesie redukcji.
NAJWAśNIEJSZE UTLENIACZE I REDUKTORY
Utleniaczem moŜe być kaŜdy atom pierwiastka, który ma moŜliwość przyjęcia elektronów i obniŜenia swegostopnia utlenienia. W związku z powyŜszym utleniaczami mogą być:a) atomy niemetali w stanie wolnym (np. O2, Cl2, S8),b) jony lub atomy metali występujących na swoich wyŜszych stopniach utlenienia (Fe3+, Sn4+, Pb4+, Cr6+,
Mn7+),c) atomy niemetali występujące na swoich wyŜszych stopniach utlenienia (N5+, S6+, Cl7+).Utleniaczami natomiast nie mogą być (są jednocześnie dobrymi reduktorami):a) atomy metali w stanie wolnym (Mg0, Fe0),b) atomy niemetali występujące na swoich najniŜszych stopniach utlenienia (Cl-, Br-, I-, F-, S2-, N3-).
ZWIĄZKI CHROMU I MANGANU JAKO UTLENIACZE
a) Chrom jest pierwiastkiem bloku d układu okresowego. Z połoŜenia wynika, iŜ powinien on miećnastępującą konfigurację: 25Cr: [18Ar]4s23d4, jednak w wyniku promocji elektrony walencyjne przyjmująukład 4s15d5. Pozwala to atomowi chromu na przyjmowanie stopni utlenienia +2, +3 oraz +6. W tymostatnim przypadku tlenek chromu CrO3 wykazuje właściwości kwasowe. Chrom tworzy dwa głównekwasy tlenowe: chromowy (VI) K2CrO4 o barwie Ŝółtej oraz dwuchromowy (VI) K2Cr2O7 o barwiepomarańczowej. W roztworze wodnym pomiędzy jonami chromianowymi (VI) i dwuchromianowymi (VI)wytwarza się stan równowagi opisany równaniem: 2CrO4
- + H+ <==> Cr2O72- + OH-; wynika z niego, iŜ w
środowisku kwaśnym przewaŜa dwuchromian (VI), a w zasadowym – chromian (VI).Badanie doświadczalne zdolności utleniających związków chromu w zaleŜności od odczynu środowiskamoŜna przeprowadzić w następujący sposób. Do dwóch probówek z roztworem dwuchromianu (VI) potasudodaje się: do jednej kwasu siarkowego (VI), a do drugiego zasady potasowej, a następnie do obu wkraplasię jodek potasu. Obserwujemy wydzielanie się pęcherzyków gazu (jod) w obu probówkach. Ponadto wprobówce z zasadą kolor roztworu zmienia się z pomarańczowego na zielony, co świadczy o pojawieniu sięjonów Cr3+ (zmiana o trzy stopnie utlenienia). W drugiej probówce zmiana następuje aŜ o cztery stopnieutlenienia, dając jony Cr2+, łączące się w siarczan (VI) chromu (II), strącający się w postaci bezbarwnegoosadu. Z takiego przebiegu doświadczenia wnioskujemy, iŜ lepsze własności utleniające związku chromuwykazują w środowisku kwaśnym, w obojętnym zapewne są one słabsze, a najsłabsze w zasadowym.Chromiany znalazły równieŜ zastosowanie w alkometrach, gdyŜ reagują z alkoholami i utleniają je dozwiązków karbonylowych (aldehydów lub ketonów), np. izopropanol do acetonu:3 CH3CH(OH)CH3 + K2Cr2O7 + 4H2SO4 ---> 3 CH3COCH3 + Cr2(SO4)3 + K2SO4 + 7H2O.Wizualnym efektem zachodzenia reakcji jest zmiana barwy środowiska z pomarańczowego na zielony.
b) Mangan jest równieŜ pierwiastkiem bloku energetycznego d. Posiada on konfigurację 25Mn: [18Ar]4s23d5, copozwala mu na przyjmowanie wartościowości od I do VII włącznie. Mangan bierze udział w róŜnychreakcjach redox, np.:
Created by Neevia Document Converter trial version http://www.neevia.com

27
MnO2 + 4 HCl ---> MnCl2 + Cl2, + H2O,MnO2 + H2O2 + H2SO4 ---> MnSO4 + O2 + 2 H2O,2 KMnO4 + 16 HCl ---> 2MnCl2 + 2KCl + 5Cl2 + 8H2O.Doświadczalne badanie zdolności utleniających związków manganu w zaleŜności od odczynu środowiskareakcji wykonuje się podobnie jak w przypadku związków chromu. Do trzech probówek zawierającychroztwór manganianu (VII) potasu KMnO4 dodaje się: kwasu siarkowego (VI), wody destylowanej orazwodorotlenku potasu, a następnie – roztwór siarczanu (IV) sodu. W probówce pierwszej (kwas) fioletowyroztwór odbarwia się do przezroczystego, w drugiej (woda) staje się czarno-brunatny, a w trzeciej (zasada) –zielony. Tłumaczy się to zachodzeniem następujących reakcji:I (kwaśne) MnO4
- ---H+---> Mn2+
2 KMnO4 + 5 Na2SO3 + 3 H2SO4 ---> 2 MnSO4 + 5 Na2SO4 + K2SO4 + 3 H2O,II (obojętne) MnO4
- ---H2O---> MnO2↓2 KMnO4 + 3 Na2SO3 + H2O ---> 3 Na2SO4 + 2 MnO2↓ + 2 KOHIII (zasadowe) MnO4
- ---OH----> MnO42-
2 KMnO4 + Na2SO3 + 2 KOH ---> Na2SO4 + 2 K2MnO4 + H2O.Ostatnia reakcja wykazuje ogromną czułość – moŜna za jej pomocą wykryć obecność manganu w ilości 10-6
mg. Podobnie jak przy chromie, najsilniejsze własności utleniające nadmanganian potasu posiada wśrodowisku kwaśnym, słabsze w obojętnym, najsłabsze zaś w środowisku zasadowym. Miarą mocyutleniającej jest liczba elektronów wymienionych (tu: przyjętych) przez atomy magnezu równa liczbieutlenionych cząsteczek siarczanu (IV) sodu (odpowiednio: 5, 3, 1).
SZEREG AKTYWNOŚCI METALI
Szereg aktywności metali to ułoŜenie metali od najbardziej aktywnego do najmniej aktywnego. W szeregu tymwystępuje równieŜ wodór, który dzieli wszystkie metale na nieszlachetne (poprzedzające go w szeregu) iszlachetne (występujące za nim w szeregu). Metale nieszlachetne wydzielają wodór z rozcieńczonych kwasównieorganicznych, podczas gdy metale szlachetne nie dają takiej reakcji. Dowolny metal występujący w szeregumoŜe być reduktorem kationów wszystkich metali występujących za nim i nie moŜe być reduktorem kationówmetali występujących przed nim. Odwracając powyŜszą regułę – dowolny metal moŜe utlenić do kationu kaŜdyinny metal występujący w szeregu przed nim i nie moŜe utlenić metalu występującego za nim. Wynika z tego, iŜnajlepsze reduktory znajdują się na początku szeregu aktywności (przed nimi prawie nic juŜ nie ma), zaśnajlepsze utleniacze znajdują się na końcu szeregu (za nimi prawie nic juŜ nie ma).Skrócony szereg aktywności ma następującą postać:Li, K, Ba, Sr, Ca, Na, Mg, Be, Al, Mn, Zn, Cr, Fe, Cd, Co, Ni, Sn, Pb, H2, Bi, Sb, As, Cu, Ag, Hg, Pt, Au
PÓŁOGNIWA
a) definicjaPółogniwem (zwyczajowo – elektrodą) nazywamy układ złoŜony z co najmniej dwóch kontaktujących sięze sobą faz: przewodnika metalicznego (I rodzaju, elektrody) zanurzonego w przewodniku jonowym (IIrodzaju). Liczba faz półogniwa moŜe być większa.
b) podział i przykładyKlasyfikacja półogniw ma charakter umowny.Ze względu na budowę (liczbę i rodzaj faz) rozróŜnia się kilka rodzajów półogniw.
- Półogniwo I rodzaju zbudowane jest z przewodnika metalicznego zanurzonego w roztworze elektrolitu.Przykładem jest półogniwo miedziowe: Cu | Cu2+.
- Półogniwo II rodzaju zbudowane jest z przewodnika metalicznego, pokrytego solą trudno rozpuszczalnątego metalu i zanurzonego w roztworze elektrolitu o anionie identycznym z anionem soli trudnorozpuszczalnej. Obecne są dwie granice faz: roztwór – sól oraz sól – metal, na granicach których ustala sięrównowaga. Przykładem jest półogniwo chlorkowo – srebrowe: Ag, AgCl | Cl-(aq)
jako K(+): Ag+ + e- ---> Ag0, AgCl (rozpuszczanie) ---> Ag+ + Cl-
jako A(-): Ag0 ---> Ag+ + e-, Ag+ + Cl- ---> AgCl (osadzanie)ogólnie: Ag + Cl- <==> AgCl + e-
StęŜenie roztworu zaleŜy od anionu, a dzięki stałemu stęŜeniu potencjał równieŜ jest stabilny.Półogniwem II rodzaju jest takŜe półogniwo kalomelowe: Hg(c), Hg2Cl2(s) | Cl-(aq)
2 Hg + 2 Cl- <==> Hg2Cl2 + 2e-
- Półogniwo III rodzaju zbudowane jest z przewodnika metalicznego, pokrytego solą trudno rozpuszczalnątego metalu, która z kolei jest pokryta drugą solą trudno rozpuszczalną o anionie wspólnym z pierwszą izanurzonego w roztworze elektrolitu zawierającego kation metalu występującego w drugiej z tych soli.
Created by Neevia Document Converter trial version http://www.neevia.com

28
Przykładem jest półogniwo ołowiowo – szczawianowo – wapniowe: Pb(s), PbC2O4(s), CaC2O4(s) | Ca2+(aq).
Działanie jest analogiczne jak w przypadku półogniw II rodzaju.Ze względu na typ reakcji elektrodowej wyróŜnia się:
- Półogniwo metaliczne (elektroda metaliczna) – kiedy faza metaliczna bierze udział w reakcji elektrodowej.Zbudowane jest z przewodnika metalicznego zanurzonego w roztworze zawierającym jego jony. Zachodzina nim redukcja (elektronacja) lub utlenianie (dezelektronacja). Przykładem jest półogniwo cynkowe:Zn | Zn2+.
- Półogniwo redox – kiedy faza metaliczna nie bierze udziału w reakcji elektrodowej, lecz jedynie spełnia rolęprzekaźnika elektronów. Do tego typu zaliczamy półogniwa gazowe – zbudowane z przewodnikametalicznego zanurzonego w roztworze nasyconym gazem i zawierającym jony powstające w wynikureakcji elektrodowej tego gazu, np.: półogniwo wodorowe Pt, H2(g) | H
+(aq), chlorowe Pt, Cl(g) | Cl-(aq),
tlenowe Pt, O2(g) | OH-(aq).
c) odwracalność półogniwPółogniwem odwracalnym nazywamy półogniwo znajdujące się w stanie równowagi termodynamicznej.Półogniwa I rodzaju są odwracalna względem kationu (reakcja potencjałotwórcza: M0 <=> Mn+ + n e-), zaśpółogniwa II rodzaju – względem anionu (reakcja potencjałotwórcza: Xn- <=> X + n e-).
d) potencjał półogniwaRozwaŜmy granicę faz: przewodnik metaliczny – roztwór zawierający jego jony(rysunek obok). Na granicy tej obecne są róŜnoimienne ładunki, występuje zatem skokpotencjału. Wartość bezwzględna potencjału półogniwa jest niemoŜliwa dozmierzenia, podobnie jak całkowita energia wewnętrzna substancji, moŜna natomiastmierzyć go metodą porównawczą – względem innego półogniwa o znanympotencjale. Pomiaru tego dokonuje się względem normalnej (standardowej) elektrodywodorowej, której potencjał przyjęto umownie za równy zeru w kaŜdej temperaturze.NEW jest elektrodą gazową odwracalną względem kationu.NEW: Pt, H2(g) | H
+(aq)
p=1013hPa, pH=0 => [H+]=1M
VE HH 0|0
2 =+ (w kaŜdej temperaturze)
- pomiar dla cynku:zestawienie ogniwa: A (-) Zn | Zn2+ || H+ | H2, Pt (+) K i odczytanie z woltomierzawartości SEM
SEME
ESEM
EESEM
ZnZn
ZnZn
ZnZnHH
−=
−=
−=
+
+
++
2
2
22
|0
|0
|0
|0
0
- pomiar dla miedzi:zestawienie ogniwa: A (-) Pt, H2 | H
+ || Cu2+ | Cu (+) K i odczytanie z woltomierza wartości SEM
SEME
ESEM
EESEM
CuCu
CuCu
HHCuCu
=
−=
−=
+
+
++
2
2
22
|0
|0
|0
|0
0
Potencjał standardowy (E0) to potencjał danej elektrody zanurzonej w 1-molowym roztworze kationów tegometalu względem NEW. Potencjały standardowe elektrod metalicznych ułoŜone od najniŜszego donajwyŜszego tworzą tzw. szereg napięciowy elektrod metalicznych – szereg elektrochemiczny. Kolejnośćelektrod w szeregu napięciowym pokrywa się z kolejnością metali w szeregu aktywności.
e) równanie NernstaPotencjał półogniwa zaleŜy od wielu czynników, m. in. od temperatury, stęŜenia, a w przypadku gazów –takŜe od ciśnienia. Dla półogniwa odwracalnego zmiany potencjału ilustruje równanie Nernsta postaci:
r
u
a
a
zF
RTEE ln0 ±= , gdzie:
E – potencjał ogniwa w nowych warunkach,E0 – potencjał standardowy półogniwa,R – stała gazowa (R=8,31 J/mol/K),F – stała Faradaya (F=86500 C),z – liczba ładunkowa reakcji (liczba moli wymienionych elektronów w odniesieniu do jednego molareagenta),
Created by Neevia Document Converter trial version http://www.neevia.com

29
T – temperatura bezwzględna (w skali Kelvina),au, ar – aktywności odpowiednio: formy utlenionej i zredukowanej.Równanie to dotyczy półogniw I i II rodzaju (dla półogniw gazowych dotyczy ciśnienia normalnego). Znakwystępujący we wzorze zaleŜy od kierunku odwracalności procesu: + dla odwracalności względem kationu(I rodzaj), – dla odwracalności względem anionu (II rodzaj). Aktywność obu form moŜna przedstawić jakoa=f.c, gdzie c oznacza stęŜenie reagentu, a f to współczynnik proporcjonalności, zwany współczynnikiemaktywności: w roztworach rozcieńczonych jego wartość zbliŜona jest do jedności, podczas gdy w stęŜonychprzyjmuje wartość 0<f<1. Dla półogniw redox w miejscu stęŜenia pojawia się odpowiedni iloczyn stęŜeń wwykładnikach współczynników stechiometrycznych (stała równowagi).Dla półogniwa metalicznego (odwracalnego względem kationu) w warunkach normalnych moŜna przyjąćaktywność formy zredukowanej za jednostkową, a po wstawieniu odpowiednich stałych i wielkościcharakteryzujących warunki środowiska oraz zamianie logarytmu naturalnego na dziesiętny, wzór Nernsta
przyjmuje postać: ucn
EE log059,00 += .
OGNIWA GALWANICZNE
Ogniwem nazywamy zestawienie dwóch półogniw. Aby popłynął prąd, musi wystąpić róŜnica potencjałówmiedzy elektrodami, a zatem musza one zawierać albo róŜne metale albo elektrolity muszą róŜnić się stęŜeniem(tzw. ogniwo stęŜeniowe). Ogniwo metaliczne jest zestawieniem dwóch róŜnych metali połączonychprzewodnikiem i zanurzonych do roztworu elektrolitu. Pod wpływem róŜnicy potencjałów następuje
wytworzenie pola elektrycznego (dxdVgradE −= ) , którego siły powodują ruch elektronów od metalu
aktywniejszego (niŜszy potencjał) do metalu mniej aktywnego (wyŜszy potencjał). Oba półogniwa nazywamyelektrodami: anoda to elektroda, na której zachodzi utlenianie (w ogniwach ma ona znak –), zaś katoda to ta, naktórej zachodzi redukcja (w ogniwach ma ona znak +). Istnieje bardzo wiele rodzajów ogniw – tu podamyjedynie kilka najpopularniejszych przykładów.a) ogniwo Volty
Gdy do wodnego roztworu kwasu siarkowego (VI) zanurzymy dwiepłytki: cynkową oraz miedzianą, wówczas na pierwszej z nich zaczniewydzielać się wodór zgodnie z równaniem reakcji:Zn + H2SO4 ---> ZnSO4 + H2,natomiast miedź jest metalem półszlachetnym i nie daje podobnegoefektu (rysunek lewy). Układ ten nie wnosi nic ciekawego. JeŜelijednak obie płytki połączymy przewodnikiem, a w tak zmontowanyobwód włączymy Ŝarówkę, to zacznie ona świecić (rysunek prawy). W przewodniku odbywa się przepływelektronów wytworzonych w procesie utleniania na anodzie do katody, gdzie teraz redukowany jest wodór.Ten ostatni wydziela się na płytce miedzianej w postaci pęcherzyków, które blokują przystęp nowychkationów wodorowych, wobec czego praca ogniwa ustaje – zjawisko to nosi nazwę polaryzacji elektrody(tu: katody). Dla ogniwa Volty moŜna zapisać:A(-): Zn0 ---> Zn2+ + 2e- (utlenianie)K(+): 2H+ + 2e- ---> H2↑ (redukcja)sumarycznie: Zn + H2SO4 ---> ZnSO4 + H2↑
Zn + 2H+ + SO42- ---> Zn2+ + SO4
2- + H2↑Zn + 2H+ ---> Zn2+ + H2↑
W roztworze powstaje siarczan (VI) cynku, zaś miedź spełnia wyłącznie rolę przewodnika. Ogniwo moŜnaprzedstawić schematycznie: A (-) Zn | H2SO4 | Cu (+) K, gdzie kreski pionowe oznaczają granice faz.
b) Ogniwo Daniella, podobnie jak ogniwo Volty, jest ogniwem cynkowo – miedziowym,posiada jednak tę zaletę, iŜ nie występuje w nim kłopotliwa polaryzacja katody.Problem ten rozwiązano stosując zasadnicze zmiany w konstrukcji ogniwa. OgniwoDaniella składa się z dwóch oddzielnych półogniw – cynkowego i miedziowego –połączonych przewodnikiem oraz kluczem elektrolitycznym (aby w obwodzie płynąłprąd, musi on być obwodem zamkniętym). Półogniwem cynkowym jest płytkacynkowa zanurzona w roztworze zawierającym kationy cynkowe, podobniepółogniwem miedziowym jest płytka miedziana zanurzona do roztworu z kationami miedziowymi. Kluczemelektrolitycznym jest rurka szklana wypełniona ośrodkiem Ŝelującym (np. koloidowym roztworemkrzemionkowym), w którym rozpuszczony jest chlorek lub azotan (V) potasu.schematycznie: A (-) Zn | ZnSO4 || CuSO4 | Cu (+) K, gdzie || oznacza kluczzapis uproszczony: Zn | Zn2+ || Cu2+ | Cu
Created by Neevia Document Converter trial version http://www.neevia.com

30
A(-): Zn0 ---> Zn2+ + 2e-
K(+): Cu2+ + 2e- ---> Cu0
Zn + Cu2+ ---> Zn2+ + Cu
VSEM
EESEM
CuZn
ZnZnCuCuCuZn
1,1)76,0(34,0/
/0
/0
/22
=−−=
−= ++
c) Ogniwo Lenclanchégo jest ogniwem nie podlegającym regeneracji. Generalnie zbudowane jest z dwóchelektrod: cynkowej oraz węglowej, otoczonej workiem z dwutlenkiem manganu, zanurzonych w roztworzechlorku amonu (salmiaku). W ogniwie „suchym” elektrodą ujemną jest cynkowa obudowa ogniwa. Teostatnie to popularne baterie R6 o SEM=1,5V.
ogniwo mokre: A (-) Zn | NH4Cl | MnO2, C (+) Kogniwo suche: A (-) Zn | ZnCl2(aq), NH4Cl | MnO2, C (+) KW ogniwie Lenclanchégo zachodzi cały szereg reakcji chemicznych, warunkujących jego działanie. Naanodzie zachodzi utlenianie metalicznego cynku (reakcja pierwotna):A(-): Zn0 ---> Zn2+ + 2 e-.Jednocześnie dysocjuje salmiak:NH4Cl <==> NH4+ + Cl-,a powstałe aniony chlorkowe łączą się z kationami cynkowymi (wtórna reakcja anodowa):A(-): Zn2+ + 2 Cl- ---> ZnCl2.Na katodzie zachodzi redukcja powstałych z dysocjacji kationów amonowych:K(+): 2 NH4+ + 2 e- ---> H2 + 2 NH3.Wydzielony amoniak kompleksuje z chlorkiem cynku, dając chlorek dwuaminocynku:ZnCl2 + 2 NH3 ---> [Zn(NH3)2]Cl2,zaś wodór łączy się z dwutlenkiem manganu, depolaryzując katodę:2 MnO2 + H2 ---> 2 MnO(OH);w wyniku reakcji tworzy się zasadowy tlenek manganu.Sumaryczna reakcja ogniwa Lenclanchégo ma postać:2 Zn + 4 NH4Cl + 4 MnO2 ---> ZnCl2 + [Zn(NH3)2]Cl2 + 4 MnO(OH).
d) Ogniwo Clarka to ogniwo cynkowo – rtęciowe o następującym schemacie:A (-) Zn(amalgamat) | ZnSO4 || HgSO4, Hg (+) KA(-): Zn ---> Zn2+ + 2 e-
K(+): Hg2+ + 2 e- ---> HgZn + Hg2+ ---> Zn2+ + HgSEM=1,433V
e) Ogniwo Westona jest ogniwem kadmowo – rtęciowym:A (-) Cd(amalgamat 12%) | nas. 3 CdSO4
. 8 H2O || HgSO4, Hg (+) KA(-): Cd ---> Cd2+ + 2 e-
K(+): Hg2+ + 2 e- ---> HgCd + Hg2+ ---> Cd2+ + HgOgniwo Westona określa się mianem ogniwa normalnego, gdyŜ utrzymuje ono przez długi czas stałąwartość SEM=1,018V (w temperaturze 20oC). Ma ono zastosowanie w laboratoriach jako wzorzec napięcia.
f) Ogniwo Bunsena jest ogniwem cynkowo – azotowym:A (-) Zn | ZnSO4(aq) (8-10%) || HNO3 (65-70%), C (+) KSEM=1,88V
g) Ogniwo Greneta / Poggendorffa zbudowane jest z elektrody cynkowej oraz grafitowej, zanurzonych wroztworze wodnym dwuchromianu (VI) potasu oraz kwasu siarkowego (VI):A (-) Zn | K2Cr2O7, H2SO4 (aq) | C (+) KSEM=2V
h) Ogniwo paliwowe – najprostszym przykładem jest ogniwo wodorowo – tlenowe,w którym elektrolitem jest 50% roztwór wodorotlenku potasu, zaś elektrodami –porowate płyty niklowe.
Created by Neevia Document Converter trial version http://www.neevia.com

31
A (-) Ni, H2 | KOH(aq) | O2, Ni (+) KA(-): H2 + 2OH- ---> 2 H2O + 2 e-
K(+): ½ O2 + H2O + 2 e- ---> 2 OH-
H2 + ½ O2 ---> H2OW ogniwie następuje synteza wody z pierwiastków. Ogniwa paliwowe są powszechnie stosowane – np. wpojazdach kosmicznych.
i) Akumulator ołowiowy jest ogniwem regenerowalnym. Akumulatorsamochodowy złoŜony jest z sześciu takich półogniw połączonych szeregowo.A (-) Pb(ołów gąbczasty) | H2SO4(aq) | PbO2, Pb(płyta ołowiana pokryta tlenkiem) (+) KH2SO4: C≈37%, d=1,27-1,29g/cm3
JeŜeli gęstość kwasu spadnie poniŜej wartości krytycznej: 1,19g/cm3, to ogniwanie będzie moŜna powtórnie naładować.A(-): Pb ---> Pb2+ + 2 e- (reakcja pierwotna)Pb2+ + SO4
2- ---> PbSO4↓ (reakcja wtórna)Pb + SO4
2- ---> PbSO4 + 2 e- (reakcja sumaryczna anody)K(+): PbO2 + 4 H+ + 2 e- ---> Pb2+ + 2 H2O (reakcja pierwotna)Pb2+ + SO4
2- ---> PbSO4↓ (reakcja wtórna)PbO2 + SO4
2- + 4 H+ + 2 e- ---> PbSO4 + 2 H2O (reakcja sumaryczna katody)Sumaryczne równanie ilustrujące zasadę działania akumulatora ma postać:Pb + PbO2 + 2 H2SO4 <====> 2 PbSO4 + 2 H2O(równanie przedstawia stan pracy; stan ładowania ilustruje równanie czytane od prawej strony).SEM=2,3V (1,8V oznacza wartość krytyczną)SEMbaterii= 6 . SEM=6 . 2,2V=13,2V (napięcie akumulatora)Trwałość akumulatorów ołowiowych (kwasowych) jest większa niŜ alkalicznych, chociaŜ wadą jeststosunkowo duŜa masa. Z drugiej strony – w porównaniu z innymi częściami pojazdu akumulator niestanowi powaŜnego obciąŜenia.
j) Akumulator Edisona jest przykładem akumulatora zasadowego (alkalicznego). Anodę stanowi Ŝelazo, zaśkatodę mieszanina niklu i jego stałych związków. Elektrolitem jest 10% roztwór wodny wodorotlenkupotasu.A (-) Fe, Fe(OH)2 | KOH(aq) | Ni(OH)2, Ni(OH)3, Ni (+) KA(-): Fe + 2 OH- ---> Fe(OH)2 + 2 e-
K(+): 2 Ni(OH)3 + 2 e- ---> 2 Ni(OH)2 + 2 OH-
Fe + 2 Ni(OH)3 <===> 2 Ni(OH)2 + Fe(OH)2
Ze względu na relatywnie małą masę oraz większą trwałość w stanie rozładowania akumulator Edisonaznalazł szerokie zastosowanie w telekomunikacji, do zasilania przenośnej aparatury elektrycznej orazpojazdów elektrycznych.
k) Akumulator srebrowo – cynkowy jest równieŜ przykładem ogniwa alkalicznego. Anodę stanowią związkucynku, katodę – związku srebra, zaś elektrolitem jest wodorotlenek potasu.A (-) Zn, ZnO | KOH(aq) | AgO, Ag2O, Ag (+) KA(-): Zn + 2 OH- ---> ZnO + H2O + 2 e-
K(+): AgO + H2O + 2 e- ---> Ag + 2 OH-
Zn + AgO <===> Ag + ZnOAkumulator ten jest wprawdzie drogi, lecz stosowany jest wszędzie tam, gdzie wymagana jest duŜaniezawodność aparatury, np. w samolotach i rakietach, do zasilania precyzyjnej aparatury pomiarowe, wtelewizji do silnego oświetlenia studia, a takŜe w lampach górniczych.
l) Ogniwo rtęciowo – cynkowe znane jest równieŜ pod nazwą ogniwapinezkowego. Jest ono powszechnie stosowane w zegarkachelektronicznych oraz miniaturowych kalkulatorach.A (-) Zn, ZnO, Zn(OH)2 | KOH(aq) | HgO, Hg (+) KA(-): 2 Zn + 4 OH- ---> ZnO + Zn(OH)2 + H2O + 4 e-
K(+): 2 HgO + 2 H2O + 4 e- ---> 2 Hg + 4 OH-
2 Zn + 2 HgO + H2O ---> ZnO + Zn(OH)2 + 2 Hg
ENERGETYKA REAKCJI REDOX
a) Entalpia swobodna reakcji redox wyraŜa się wzorem ∆G=-zF∆E, gdzie z to liczba ładunkowa reakcji, F –stała Faradaya, a ∆E – siła elektromotoryczna ogniwa. JeŜeli SEM jest dodatnie, wówczas entalpiaswobodna reakcji jest ujemna, co gwarantuje samorzutność zachodzenia procesu. Aby uniknąć ujemnychwartości SEM anodę ustawia się po lewej stronie, a katodę po prawej, gdyŜ zgodnie z konwencjąsztokholmską: ∆E=Ep-El, gdzie Ep, El to potencjały odpowiednio prawego i lewego półogniwa.
Created by Neevia Document Converter trial version http://www.neevia.com

32
b) Przewidywanie kierunku przebiegu reakcji redox w oparciu o potencjałystandardowe półogniw – tzw. metoda zegara. Niech dane będą dwie reakcjeelektrodowe:R1 <==> U1
n+ + n e- (E1),R2 <==> U2
m+ + m e- (E2)oraz niech E2>E1. Wówczas przemiany zachodzące w tak zestawionymogniwie ilustruje wykres – „zegar” (rysunek obok). W półogniwie oniŜszym potencjale reakcja redox przebiega w stronę utleniania (półogniwo to staje się anodą), zaś wpółogniwie o wyŜszym potencjale reakcje redox przebiega w stronę redukcji (półogniwo to staje się katodą).Przebieg reakcji sumarycznej moŜna zatem zilustrować następująco:R1 ---> U1
n+ + n e- / . mU2
m+ + m e- ---> R2 / . n
m R1 + n U2m+ + nm e- ---> n R2 + m U1
n+ + nm e-
m R1 + n U2m+ ---> n R2 + m U1
n+
Warto zwrócić uwagę, Ŝe liczba ładunkowa sumarycznej reakcji wynosi z=n.m.c) Stała równowagi reakcji redox. Kontynuując rozwaŜanie reakcji z poprzedniego podpunktu, moŜna wyrazić
potencjały półogniw następująco:
][
][ln,
][
][ln
2
2022
1
1011
R
U
mF
RTEE
R
U
nF
RTEE
mn ++
+=+= .
Siła elektromotoryczna powstałego ogniwa to róŜnica potencjałów półogniw, czyli:
KzF
RTE
RU
RU
zF
RTE
R
U
R
U
zF
RTE
R
Um
R
Un
nmF
RTE
R
U
nmF
mRT
R
U
nmF
nRTE
R
U
nF
RT
R
U
mF
RTEE
R
U
nF
RTE
R
U
mF
RTEEEE
nmn
mnm
m
mn
n
nmnm
nmnm
nm
ln][][
][][ln
][
][ln
][
][ln
][
][ln
][
][ln
][
][ln
][
][ln
][
][ln
][
][ln
][
][ln
][
][ln
0
1
21
120
1
1
2
20
1
1
2
20
1
1
2
20
1
1
2
201
02
1
101
2
20212
−∆=
+∆=
=
−+∆=
−+∆=
=−+∆=−+−=
=
+−
+=−=∆
−
+
+
++++
++++
++
Gdy w środowisku reakcji ustali się stan równowagi, wówczas ogniwo nie pracuje, a jego siłaelektromotoryczna wynosi zero.
RT
EzF
RT
EzF
KKe
KK
eKRT
EzFKEK
zF
RTK
zF
RTEE
3,2
000
0
0
10log3,2log
logln
lnln0ln0
∆
∆
=⇒==
=⇒∆
=⇒∆=⇒=−∆⇒=∆
Znając zatem róŜnicę potencjałów reakcji połówkowych oraz warunki środowiska, moŜemy określić stałąrównowagi reakcji redox. Dodatkowo wprowadzając: zF∆E0=-∆G, otrzymujemy:
RT
G
RT
G
eK 3,210∆−∆−
== .Z ostatniego równania wynika, iŜ:
- dla reakcji samorzutnej (∆G<0) K>1,- w stanie równowagi (∆G=0) K=1,- dla reakcji wymuszonej (∆G>0) K<1.
KOROZJA METALI I METODY JEJ ZWALCZANIA
Korozja jest procesem niszczenia metali na skutek wpływu czynników środowiska. Ze względu na przebiegwyróŜniamy dwa typy korozji.a) Korozja chemiczna zachodzi w środowisku suchym pod wpływem tlenu lub działania agresywnych gazów
przemysłowych. Prowadzi do powstania ochronnej warstewki tlenku (czasem takŜe wodorotlenku lub soli).Ten typ korozji ma znaczenie pozytywne, gdyŜ pasywna warstwa chroni przed dalszą korozją. Np.śniedzenie miedzi, czernienie srebra, pasywacja Ŝelaza, chromu, glinu.
Created by Neevia Document Converter trial version http://www.neevia.com

33
b) Korozja elektrochemiczna zachodzi w środowisku wilgotnympod wpływem tlenu atmosferycznego. Przyspiesza ją kwaśneśrodowisko oraz obecność gazów przemysłowych, podczas gdyzasadowość środowiska – hamuje. Istota korozjielektrochemicznej polega na tworzeniu się tzw. mikroogniwlokalnych, w których Ŝelazo stanowi anodę. Ulegającsystematycznemu utlenianiu, przechodzi w postaci jonów zmetalu do środowiska i w ten sposób niszczeje.
c) W mikroogniwie zachodzą następujące reakcje:A(-): Fe ---> Fe2+ + 2 e- (reakcja pierwotna)Fe2+ + 2 OH- ---> Fe(OH)2↓ (biały) (reakcja wtórna)4 Fe(OH)2 + 2 H2O + O2 ---> 4 Fe(OH)3↓ (brązowy) (reakcja wtórna)K(+): ½ O2 + H2O + 2 e- ---> 2 OH-
Efektem korozji jest powstanie rdzy: Fe2O3 . n H2O (n=1, 2, 3...), np. dla n=3 Fe2O3
. 3 H2O = 2 Fe(OH)3.Metody walki z korozją:a) wytwarzanie powłok ochronnych z farb, lakierów i tworzyw sztucznych,b) wytwarzanie powłok ochronnych z innych metali- Powłoki czynne wytwarzane są z metali o potencjale standardowym niŜszym
niŜ Ŝelaza, np. z cynku czy magnezu. Po uszkodzeniu nadal chronią oneprzed korozją do czasu, gdy ulegną całkowitemu utlenienie. W powstałymmikroogniwie Ŝelazo stanowi katodę, a więc nie niszczeje.A(-): Zn ---> Zn2+ + 2 e- (reakcja pierwotna)Zn2+ + 2 OH- ---> Zn(OH)2 (reakcja wtórna)K(+): H2O + ½ O2 + 2 e- ---> 2 OH-
- Powłoki bierne wytwarzane są z metali o potencjale standardowym wyŜszymniŜ Ŝelaza, np. z niklu, miedzi, srebra, złota. Po uszkodzeniu znacznieprzyspieszają one korozję, gdyŜ w powstałym mikroogniwie Ŝelazo stanowianodę i bardzo szybko ulega degradacji.A(-): Fe ---> Fe2+ + 2 e- (reakcja pierwotna)Fe2+ + 2 OH- ---> Fe(OH)2↓ (reakcja wtórna)4 Fe(OH)2 + 2 H2O + O2 ---> 4 Fe(OH)3↓ (reakcja wtórna)K(+): ½ O2 + H2O + 2 e- ---> 2 OH-
O wyborze powłoki nie zawsze decyduje jej skuteczność, ale równieŜ względy estetyczne – powłoki biernesą bowiem o wiele ładniejsze od powłok czynnych.
c) osłabianie agresywności środowiska i / lub jego alkalizacja,d) stosowanie ochrony protektorowej: protektor to metal aktywniejszy od Ŝelaza, przytwierdzany do
powierzchni wyrobu stalowego, np. kadłuba okrętu bądź rurociągu, działający jako anoda w powstałymogniwie; rolę protektora spełniają często płyty magnezowe,
e) stosowanie ochrony katodowej, czyli nadawanie niŜszego potencjału poprzez łączenie z ujemnym biegunemogniwa; metoda ta jest często praktykowana w odniesieniu do kotłów przemysłowych.
ELEKTROLIZA
a) istota procesu elektrolizyRozwaŜmy procesy zachodzące w ogniwiecynkowo – miedziowym. Zgodnie zzasadami energetycznymi ∆E>0 => ∆G<0,czyli procesy zachodzą samorzutnie wprzedstawionym kierunku. Zobaczmy co sięstanie, gdy do biegunów ogniwa przyłoŜymyodwrotnie spolaryzowane napięcie owartości większej od siły elektromotorycznejwytwarzanej prze to ogniwo. Obserwujemydokładne odwrócenie przebiegu samorzutniezachodzących reakcji redox. Zespółwymuszonych reakcji chemicznychzachodzących na elektrodach i w elektrolitach towarzyszących przepływowi przezeń prądu stałegonazywamy elektrolizą. Termin ten oznacza dosłownie „rozkład pod wpływem prądu”. Elektrolizę moŜnaprzeprowadzać zarówno w stopionych elektrolitach (związkach o budowie jonowej – np. sole, tlenki metali iwodorotlenki – które pod wpływem wysokiej temperatury ulegają termodysocjacji) jak i w roztworach
Created by Neevia Document Converter trial version http://www.neevia.com

34
wodnych elektrolitów (np. w roztworach kwasów, zasad i soli, gdzie pod wpływem wody zachodzidysocjacja elektrolityczna). Aparat, w którym przeprowadzamy elektrolizę, nazywamy elektrolizerem. Wprzypadku zamiany ogniwa w elektrolizer anoda staje się katodą i odwrotnie, zmieniają się równieŜ ichznaki: anoda jest dodatnia, a katoda ujemna. Niezmiennym natomiast pozostaje fakt, iŜ na anodzie zachodziproces utleniania, a na katodzie – redukcji. Aby zaszła elektroliza, naleŜy do biegunów ogniwa przyłoŜyćnapięcie wyŜsze od jego SEM. Najmniejszą wartość zewnętrznego napięcia, przy którym obserwujemyciągłą reakcję elektrodową, nazywamy napięciem rozkładowym (Ur). Jest ono charakterystyczne dla danegoukładu: przewodniki metaliczne – przewodnik jonowy. W praktyce przyjmuje się, iŜ Ur to napięcieuzyskane przez ekstrapolację krzywej I=f(U) do I=0 (wykres). RóŜnicę pomiędzySEM ogniwa a Ur elektrolizera zeń uzyskanego nazywamy nadnapięciemelektrolizy: η=Ur-E. RozróŜnia się nadnapięcie procesu katodowego (ηK) ianodowego (ηA). Nadnapięcie zaleŜy od naboju powierzchni katody, gęstościprądu zewnętrznego oraz od temperatury, co związane jest z energią i ruchliwościąjonów.ZaleŜności pomiędzy przedstawionymi wielkościami ilustruje schemat:
pwK = EK - ηK pw
A = EA - ηA
η = ηA + ηK
Ur = pwA – pw
K = EA – EK + ηA + ηK
Ur = SEM + ηη > 0 => Ur > SEM
gdzie: pw – potencjały wydzielania, E – potencjały półogniw, η – nadnapięcie.b) kolejność wydzielania produktów w elektrolizerze
Na anodzie zachodzi utlenianie reduktorów. Najlepsze reduktory mają najmniejszy potencjał standardowy,dlatego na anodzie produkty wydzielają się w kolejności od najniŜszego potencjału począwszy.Analogicznie na katodzie zachodzi redukcja utleniaczy. Najlepsze utleniacze mają największy potencjałstandardowy, dlatego na katodzie produkty wydzielają się w kolejności od najwyŜszego potencjałupocząwszy. Bardzo często produktami elektrolizy są wodór i / lub tlen. Korzystając z równania NernstamoŜna określić jaki odczyn środowiska sprzyja redukcji kationów wodorowych do wolnego wodoru, a jakiutlenianiu anionów wodorotlenkowych do wolnego tlenu.
WODÓR TLENK(-): 2 H2O + 2 e- ---> H2↑ + 2 OH- (alkalizacja) A(+): 2 H2O ---> O2↑ + 4 H+ + 4 e- (zakwaszenie)pH=0 E = E0 = 0 V E = E0 - 0,059 . log 10-14 = 1,24 V pOH=14pH=7 E = E0 + 0,059 . log 10-7 = -0,41 V E = E0 - 0,059 . log 10-7 = 0,82 V pOH=7pH=14 E = E0 + 0,059 . log 10-14 = -0,83 V E = E0 = 0,41 V pOH=0
Wodór wydziela się na katodzie, zatem najkorzystniejszym jest potencjał najmniejszy, czyli redukcjiwodoru sprzyja środowisko zasadowe. Z kolei tlen wydziela się na anodzie, zatem najkorzystniejszy jestpotencjał największy, czyli utlenianiu tlenu sprzyja środowisko kwaśne. PowyŜsze wartości potencjałówwydzielania wodoru i tlenu w zestawieniu z szeregiem napięciowym metali oraz z potencjałami normalnymireakcji redox informują nas o kolejności wydzielania się produktów.Na anodzie utleniają się kolejno:- aniony kwasów beztlenowych od najniŜszego potencjału (np. S2- ---> S0 + 2 e-),- aniony wodorotlenkowe: 4 OH- ---> O2↑ + 2 H2O + 4 e-,- woda: 2 H2O ---> O2↑ + 4 H+ + 4 e-.Na katodzie redukują się kolejno:- kationy metali od najwyŜszego potencjału (np. Cu2+ + 2 e- ---> Cu0),- kationy wodorowe (gdy odczyn jest kwaśny): 2 H+ + 2 e- ---> H2↑,- woda (gdy w roztworze znajdują się kationy metali aktywnych): 2 H2O + 2 e- ---> 2 OH- + H2↑.
c) produkty elektrolizy poszczególnych grup związków chemicznych – jakościowe ujęcie elektrolizy- stopione elektrolity – substancje wyjściowe• tlenek metalu ---> metal + tlen
Al2O3 ---∆---> 2 Al3+ + 3 O2-
K(-): 2 Al3+ + 6 e- ---> 2 AlA(+): 3 O2- ---> 3/2 O2↑ + 6 e-
Al2O3 ---> 2 Al + 3/2 O2
• wodorotlenek ---> metal + tlen + wodaNaOH ---∆---> Na+ + OH-
K(-): Na+ + e- ---> NaA(+): OH- ---> ½ O2↑ + ½ H2O + e-
NaOH ---> Na + ½ O2↑ + ½ H2O
Created by Neevia Document Converter trial version http://www.neevia.com

35
• sól ---> metal + zredukowana reszta kwasowaFe2S3 ---∆---> 2 Fe3+ + 3 S2-
K(-): 2 Fe3+ + 6 e- ---> 2 FeA(+): 3 S2- ---> 3 S + 6 e-
Fe2S3 ---> 2 Fe + 3 S- roztwory wodne – według kolejności wynikającej z potencjałów; w przypadku identycznych jonów
pochodzących z róŜnych źródeł wydzielają się one stamtąd, gdzie jest ich najwięcej• zasada ---> wodór z wody + tlen z zasady
NaOH ---> Na+ + OH-
H2O <==> H+ + OH-
K(-): 2 H+ + 2 e- ---> H2↑A(+): 2 OH- ---> H2O + ½ O2↑ + 2 e-
H2O ---> H2↑ + ½ O2↑Produkty wskazują na elektrolizę wody; w roztworze pozostaje zasada – środowisko ulega alkalizacji.
• kwas beztlenowy ---> wodór z kwasu + niemetalHBr ---> H+ + Br-
K(-): 2 H+ + 2 e- ---> H2↑A(+): 2 Br- ---> Br2↑ + 2 e-
2 HBr ---> H2↑ + Br2↑• kwas tlenowy ---> wodór z kwasu + tlen z wody
H2SO4 ---> 2 H+ + SO42-
H2O <==> H+ + OH-
K(-): 2 H+ + 2 e- ---> H2↑A(+): 2 OH- ---> ½ O2↑ + H2O + 2 e-
H2O ---> ½ O2↑ + H2↑Produkty wskazują na elektrolizę wody; w roztworze pozostaje kwas – środowisko ulega zakwaszeniu.
• sól beztlenowa metalu lekkiego ---> wodór z wody + niemetalKCl ---> K+ + Cl-
H2O <==> H+ + OH-
K(-): 2 H+ + 2 e- ---> H2↑A(+): 2 Cl- ---> Cl2↑ + 2 e-
W roztworze pozostaje zasada – środowisko ulega alkalizacji.• sól beztlenowa metalu cięŜkiego ---> metal + niemetal
CuS ---> Cu2+ + S2-
K(-): Cu2+ + 2 e- ---> CuA(+): S2- ---> S + 2 e-
CuS ---> Cu + S• sól tlenowa metalu lekkiego ---> wodór z wody + tlen z wody
Li2SO4 ---> 2 Li+ + SO42-
H2O <==> H+ + OH-
K(-): 2 H+ + 2 e- ---> H2↑A(+): 2 OH- ---> ½ O2↑ + H2O + 2 e-
H2O ---> ½ O2↑ + H2↑Jest to właściwie elektroliza wody – jony soli spełniają jedynie rolę nośników ładunku elektrycznego.
• sól tlenowa metalu cięŜkiego ---> metal + tlen z wodyCu(NO3)2 ---> Cu2+ + 2 NO3
-
H2O <==> H+ + OH-
K(-): Cu2+ + 2 e- ---> CuA(+): 2 OH- ---> ½ O2↑ + H2O + 2 e-W roztworze powstaje kwas azotowy – środowisko ulega zakwaszeniu.
d) prawa elektrolizy Faradaya – ilościowe ujęcie elektrolizyRozwaŜmy następującą reakcję połówkową:
K(-): Un+ + n e- ---> R1 cząstka n elektronów
mcz n . em q
Created by Neevia Document Converter trial version http://www.neevia.com

36
Z porównania otrzymujemy:
⋅=⋅
=⋅⋅=⋅=⋅⋅
=⇒
⇒⋅
=⇒⋅⋅
=⋅
⇒⋅
=⇒⋅
=
tIqFn
MktIkqk
Fn
qMm
Fn
q
M
m
Nen
q
Nm
m
en
q
m
m
q
en
m
m
AAczcz
cz
,
Pierwsze prawo elektrolizy stwierdza, iŜ masa wydzielonego produktu jest wprost proporcjonalna doładunku, jaki przepłynął przez elektrolizer, czyli iloczynowi wartości prądu stałego oraz czasu trwaniaprocesu. Współczynnik proporcjonalności k zwany jest równowaŜnikiem elektrochemicznym; jest onilościowo równy masie produktu wydzielonego w jednostkowym przedziale czasu w wyniku przepływuprądu o jednostkowym natęŜeniu. Drugie prawo elektrolizy podaje wyraŜenie opisujące równowaŜnik – jeston wprost proporcjonalny do masy molowej produktu, a odwrotnie proporcjonalny do liczby ładunkowejreakcji. Stała F zwana jest stałą Faradaya; liczbowo równa jest ona iloczynowi liczby Avogadra i ładunkuelementarnego:F = NA . e = 6,02 . 1023 mol-1 . 1,6 . 10-19 C ≈ 96 500 C / mol.
e) praktyczne zastosowanie elektrolizy:- Otrzymywanie metali lekkich (sodu, wapnia, glinu) z ich stopionych elektrolitów. W przypadku glinu nie
moŜna stosować redukcji tlenku ze względu na zbyt kosztowny nakład energetyczny, dlatego stosuje siętermoelektrolizę. Surowcem wyjściowym jest boksyt: Al2O3
. x H2O, do którego dodaje się topiku – kriolitu:
Na3AlF6.Al2O3 ---∆---> 2 Al3+ + 3 O2-
K(-): 2 Al3+ + 6 e- ---> 2 AlA(+): 3 O2- ---> 3/2 O2↑ + 6 e-
Al2O3 ---> 2 Al + 3/2 O2
Rolę katody spełnia ciekły glin pozostały po poprzedniejelektrolizie (nigdy nie spuszcza się go do końca), anodą zaśszereg elektrod węglowych (grafitowych). Pod wpływemwydzielającego się tlenu te ostatnie utleniają się do tlenku i dwutlenku węgla. Jest to zjawisko niepoŜądane,lepiej jednak tracić grafit niŜ pozwalać na ponowne utlenianie glinu. Ostatecznymi produktami są zatem:glin oraz tlenki węgla.
- pokrywanie metali mniej szlachetnych powłokami galwanicznymi z metali szlachetniejszych (np. cynk,nikiel, miedź, srebro) – silniki, ramy rowerowe itp.
- rozdzielanie metali z mieszanin przy wykorzystaniu róŜnicy potencjałów- produkcja przemysłowa wodorotlenku sodu• metoda przeponowa – elektroliza roztworu chlorku sodu
NaCl ---> Na+ + Cl-
H2O ---> H+ + OH-
K(-): 2 H2O + 2 e- ---> H2↑ + 2 OH-
A(+): 2 Cl- ---> Cl2↑ + 2 e-
2 NaCl + 2 H2O ---> 2 NaOH + H2↑ + Cl2↑Otrzymywany w ten sposób wodorotlenek jest pozostałością po elektrolizie, przez co jest silniezanieczyszczony.
• metoda rtęciowaPoniewaŜ nadnapięcie wydzielania wodoru na katodzie rtęciowej jest bardzo wysokie (ηH = 1,2 - 1,3 V),redukcji ulega sód. Tworzy on z rtęcią amalgamat, po czym jest wypłukiwany. Otrzymany w ten sposóbwodorotlenek jest zadowalająco czysty.NaCl ---> Na+ + Cl-
K(-): 2 Na+ + 2 e- ---> NaHg + 2 Na ---> 2 Na|Hg
A(+): 2 Cl- ---> Cl2↑ + 2 e-
2 NaCl + Hg ---> 2 Na|Hg + Cl2↑2 Na|Hg + 2 H2O ---> 2 NaOH + H2↑ + Hg
- elektrorafinacja miedzi – jeŜeli anoda wykonana jest z metalu, którego kationy znajdują się w roztworze, toprocesem anodowym jest utleniania metalu anodyK(-): Cu2+ + 2 e- ---> CuA(+): Cu ---> Cu2+ + 2 e-
Cu (zanieczyszczona z huty) ---elektrorafinacja---> Cu (czysta do uŜytku)
Created by Neevia Document Converter trial version http://www.neevia.com
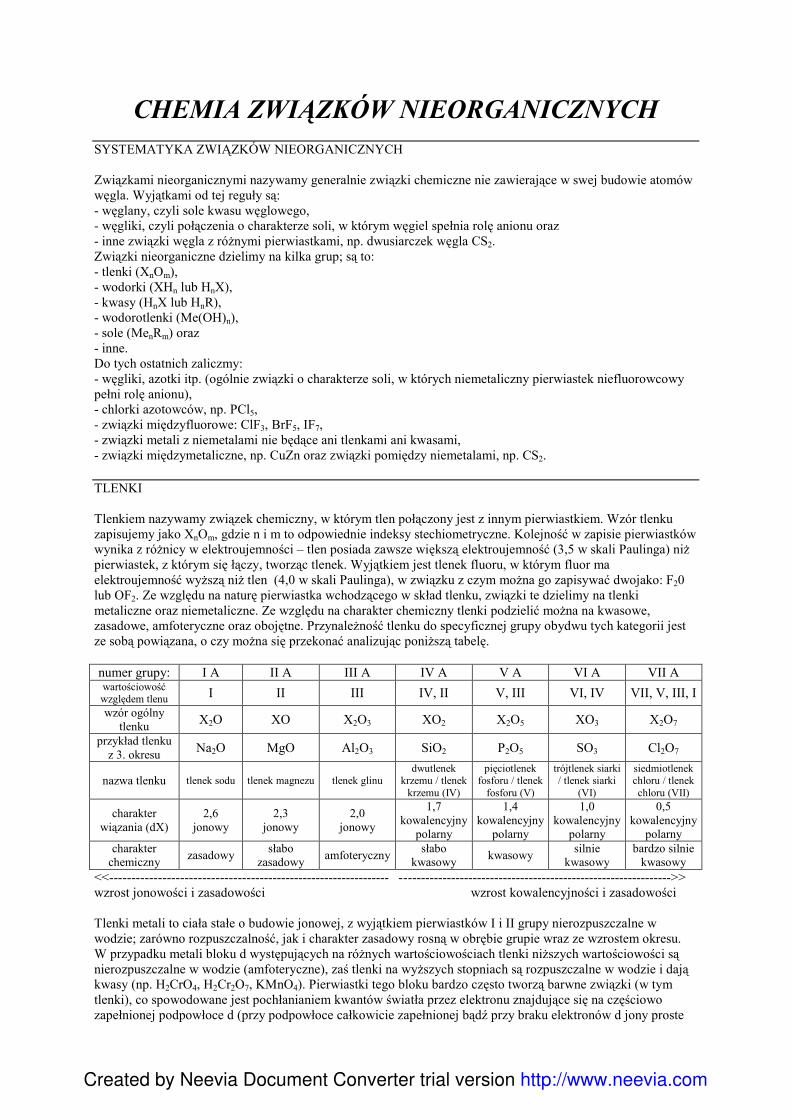
37
CHEMIA ZWIĄZKÓW NIEORGANICZNYCHSYSTEMATYKA ZWIĄZKÓW NIEORGANICZNYCH
Związkami nieorganicznymi nazywamy generalnie związki chemiczne nie zawierające w swej budowie atomówwęgla. Wyjątkami od tej reguły są:- węglany, czyli sole kwasu węglowego,- węgliki, czyli połączenia o charakterze soli, w którym węgiel spełnia rolę anionu oraz- inne związki węgla z róŜnymi pierwiastkami, np. dwusiarczek węgla CS2.Związki nieorganiczne dzielimy na kilka grup; są to:- tlenki (XnOm),- wodorki (XHn lub HnX),- kwasy (HnX lub HnR),- wodorotlenki (Me(OH)n),- sole (MenRm) oraz- inne.Do tych ostatnich zaliczmy:- węgliki, azotki itp. (ogólnie związki o charakterze soli, w których niemetaliczny pierwiastek niefluorowcowypełni rolę anionu),- chlorki azotowców, np. PCl5,- związki międzyfluorowe: ClF3, BrF5, IF7,- związki metali z niemetalami nie będące ani tlenkami ani kwasami,- związki międzymetaliczne, np. CuZn oraz związki pomiędzy niemetalami, np. CS2.
TLENKI
Tlenkiem nazywamy związek chemiczny, w którym tlen połączony jest z innym pierwiastkiem. Wzór tlenkuzapisujemy jako XnOm, gdzie n i m to odpowiednie indeksy stechiometryczne. Kolejność w zapisie pierwiastkówwynika z róŜnicy w elektroujemności – tlen posiada zawsze większą elektroujemność (3,5 w skali Paulinga) niŜpierwiastek, z którym się łączy, tworząc tlenek. Wyjątkiem jest tlenek fluoru, w którym fluor maelektroujemność wyŜszą niŜ tlen (4,0 w skali Paulinga), w związku z czym moŜna go zapisywać dwojako: F20lub OF2. Ze względu na naturę pierwiastka wchodzącego w skład tlenku, związki te dzielimy na tlenkimetaliczne oraz niemetaliczne. Ze względu na charakter chemiczny tlenki podzielić moŜna na kwasowe,zasadowe, amfoteryczne oraz obojętne. PrzynaleŜność tlenku do specyficznej grupy obydwu tych kategorii jestze sobą powiązana, o czy moŜna się przekonać analizując poniŜszą tabelę.
numer grupy: I A II A III A IV A V A VI A VII Awartościowośćwzględem tlenu I II III IV, II V, III VI, IV VII, V, III, I
wzór ogólnytlenku X2O XO X2O3 XO2 X2O5 XO3 X2O7
przykład tlenkuz 3. okresu Na2O MgO Al2O3 SiO2 P2O5 SO3 Cl2O7
nazwa tlenku tlenek sodu tlenek magnezu tlenek glinudwutlenek
krzemu / tlenekkrzemu (IV)
pięciotlenekfosforu / tlenek
fosforu (V)
trójtlenek siarki/ tlenek siarki
(VI)
siedmiotlenekchloru / tlenekchloru (VII)
charakterwiązania (dX)
2,6jonowy
2,3jonowy
2,0jonowy
1,7kowalencyjny
polarny
1,4kowalencyjny
polarny
1,0kowalencyjny
polarny
0,5kowalencyjny
polarnycharakter
chemicznyzasadowy
słabozasadowy
amfoterycznysłabo
kwasowykwasowy
silniekwasowy
bardzo silniekwasowy
<<---------------------------------------------------------------- --------------------------------------------------------------->>wzrost jonowości i zasadowości wzrost kowalencyjności i zasadowości
Tlenki metali to ciała stałe o budowie jonowej, z wyjątkiem pierwiastków I i II grupy nierozpuszczalne wwodzie; zarówno rozpuszczalność, jak i charakter zasadowy rosną w obrębie grupie wraz ze wzrostem okresu.W przypadku metali bloku d występujących na róŜnych wartościowościach tlenki niŜszych wartościowości sąnierozpuszczalne w wodzie (amfoteryczne), zaś tlenki na wyŜszych stopniach są rozpuszczalne w wodzie i dająkwasy (np. H2CrO4, H2Cr2O7, KMnO4). Pierwiastki tego bloku bardzo często tworzą barwne związki (w tymtlenki), co spowodowane jest pochłanianiem kwantów światła przez elektronu znajdujące się na częściowozapełnionej podpowłoce d (przy podpowłoce całkowicie zapełnionej bądź przy braku elektronów d jony proste
Created by Neevia Document Converter trial version http://www.neevia.com

38
są bezbarwne).Tlenki niemetali to zazwyczaj gazy o budowie kowalencyjnej polarnej; w obrębie ich cząsteczek sumamomentów dipolowych wynosi przewaŜnie zero.
Tlenki kwasowe reagują z wodą dając kwasy oraz z zasadami, zobojętniając je, np.:SO2 + H2O ---> H2SO3,SO2 + 2NaOH ---> Na2SO3 + H2O.Podobnie tlenki zasadowe reagują z wodą dając zasady oraz z kwasami, takŜe je zobojętniając, np.:Na2O + H2O ---> 2NaOH,Na2O + H2SO3 ---> Na2SO3 + H2O.Tlenki obojętne to takie, które nie reagują ani z kwasami ani z zasadami, np.: CO, NO, N2O, SiO.Tlenki amfoteryczne są tlenkami pierwiastków o stosunkowo wysokiej elektroujemności (1,5 – 2), np.: Al2O3,PbO, ZnO, Cr2O3, FeO, Fe2O3, CuO, Cu2O, Ag2O, Au2O3. Tlenki amfoteryczne nie rozpuszczają się w wodzie,reagują z kwasami i bardzo mocnymi zasadami, dają przez to dwa rodzaje soli: kiedy pierwiastek z tlenkuspełnia w soli rolę kationu (klasyczna reakcja z kwasem) oraz kiedy ów pierwiastek wchodzi w skład resztykwasowej (reakcja z mocną zasadą). W reakcji z zasadami jon nie zmienia stopnia utlenienia) nie jest to reakcjaredox). Dla glinu i cynku reakcje wyglądają następująco:Al2O3 + 6HCl ---> 2AlCl3 + 3H2O,Al2O3 + 2NaOH + 3H2O ---> 2NaAl(OH)4 ---T---> 2NaAlO2 + 2H2O, czterohydroksoglinian soduZnO + 2HCl ---> ZnCl2 + H2O,ZnO + 2NaOH + H2O ---> Na2Zn(OH)4 ---T---> Na2ZnO2 + 2H2O. czterohydroksocynkan sodu
Tlenki moŜna otrzymywać róŜnorodnymi metodami. Najczęściej stosowanymi z nich są:- spalanie pierwiastków, np.:S + O2 ---> SO2,4Na + O2 ---> 2Na2O,N2 + O2 ---> 2NO (w łuku elektrycznym – metoda Mościckiego),- spalanie związków chemicznych, np.:ZnS + 3/2 O2 ---> ZnO + SO2, (sfaleryt – blenda cynkowa)2NH3 + 5/2 O2 ---> 2NO + 3H2O,2NH3 + 7/2 O2 ---> 2NO2 + 3H2O,- rozkład związków chemicznych (głównie soli i wodorotlenków), np.:CaCO3 ---T---> CaO + CO2,NH4NO3 ---T---> N2O + 2H2O,Cu(OH)2 ---T---> CuO + H2O,- utlenianie niŜszych tlenków do wyŜszych, np.:2NO + O2 ---> 2NO2,2SO2 + O2 ---V2O5+Pt/Pd--->2SO3
- redukcja wyŜszych tlenków do niŜszych, np.:Fe2O3 + C ---> 2FeO + CO,Fe2O3 + CO ---> 2FeO + CO2.
WODORKI
numer grupy: I A II A III A IV A V A VI A VII Awartościowośćwzględemwodoru
I II III IV III II I
wzór ogólnywodorku
XH XH2 XH3 XH4 XH3 H2X HX
przykład z3. okresu
NaH MgH2 AlH3 SiH4 PH3 H2S HCl
nazwawodorku
wodoreksodu
wodorekmagnezu
wodorekglinu
krzemowodór fosforowodór siarkowodór chlorowodór
charakterwiązania (dX)
1,2spolaryzowany
0,9spolaryzowany
0,6spolaryzowany
0,3kowalencyjny
0,0kowalencyjny
0,4spolaryzowany
0,9spolaryzowany
charakterchemiczny
zasadowy zasadowy słabozasadowy
obojętny obojętny słabokwasowy
kwasowy
Created by Neevia Document Converter trial version http://www.neevia.com

39
Wodorkami nazywamy dwuskładnikowe połączenia pierwiastków z wodorem. Dzielimy je, podobnie jak tlenki,według dwóch kategorii: na kwasowe, zasadowe i obojętne oraz na wodorki metali i niemetali. PomiędzyprzynaleŜnością do obu grup istnieje ścisła zaleŜność.Wodorki metali (umownie grupy I – V) to ciała stałe o małych gęstościach. Mają budowę polarną i charakterzasadowy. Powstają w reakcjach wymuszonych, są bardzo aktywne chemicznie i łatwo się rozkładają zwydzielenia ciepła. Reagują z wodą, w wyniku czego powstają zasady oraz gazowy wodór, np.:NaH + H2O ---> NaOH + H2.Wodorki niemetali (grupy VI i VII) to bezbarwne trujące gazy o charakterystycznych zapachach. Cechują sięroŜną rozpuszczalnością w wodzie, co wiąŜe się z charakterem chemicznych. Nierozpuszczalne i obojętne są:CH4, BH3, PH3, SiH4, GeH4, natomiast rozpuszczalne są: NH3, H2S, H2Se, H2Te, HF, HCl, HBr, HI; opróczzasadowego amoniaku pozostałe są kwasowe. Charakter kwasowy, podobnie jak moc, rośnie w grupie i wokresie (wzrost promienia jonowego osłabia wiązanie z wodorem, co ułatwia jego odłączanie i zwiększa stopieńdysocjacji). Rozpuszczalne wodorki reagują z wodą, w wyniku czego powstają kwasy beztlenowe (ew. zasadaamonowa: NH3 + H2O --> NH4OH).
Metody otrzymywania wodorków:- synteza z pierwiastków, np.:2Na + H2 ---> 2NaH,H2 + Cl2 ---p,T--> 2HCl, \ specjalneN2 + 3H2 ---kat--> 2NH3, / warunki- z soli w reakcjach jonowych:• amoniak w reakcji soli amonowej z roztworem mocnej zasady, np.:
(NH4)2SO4 + 2NaOH ---> 2NH4OH + Na2SO4 ---> 2NH3 + 2H2O + Na2SO4,• wodorki kwasowe w reakcjach odpowiednich soli beztlenowych i mocnego kwasu, np.:
2NaCl + H2SO4 ---> 2HCl + Na2SO4.
WODOROTLENKI
1) Wodorotlenkami nazywamy połączenia metalu z jednowartościowymi grupami hydroksylowymi(wodorotlenowymi). Zapisać je moŜna wzorem ogólnym M(OH)n, gdzie M to atom metalu, a –OH to grupawodorotlenowa. Wiązanie O-H jest wiązaniem kowalencyjnym silnie spolaryzowanym, zaś w wiązanie M-O jestwiązaniem jonowym.2) Wodorotlenki to krystaliczne ciała stałe o róŜnej barwie i rozpuszczalności w wodzie; dobrze rozpuszczalnesą wodorotlenki litowców i Ba, słabo rozpuszczają się wodorotlenki Be, Mg, Ca i Sr, zaś pozostałe sąpraktycznie nierozpuszczalne. Roztwory rozpuszczalnych wodorotlenków nazywamy zasadami. Zasady barwiąindykatory (wskaźniki) chemiczne – fenoloftaleinę na malinowo, lakmus na niebiesko, wskaźnik uniwersalny naniebiesko bądź zielono. Wodorotlenki litowców są silnie higroskopijne, tzn. pochłaniają parę wodną z powietrza,dlatego przechowuje się je w formie pastylek (mała powierzchnia kontaktowa); mają ponadto właściwości Ŝrącei są śliskie w dotyku – dlatego równieŜ śliskie jest mydło, gdyŜ podczas jego kontaktu z wodą wydziela sięzasada sodowa jako produkt hydrolizy. Zasady mocnych wodorotlenków nazywamy ługami; niektóre z nichmają specyficzne nazwy zwyczajowe, np.: roztwór NaOH to soda kaustyczna, a KOH to potaŜ Ŝrący.3) Wodorotlenki moŜna podzielić na zasadowe oraz amfoteryczne. Wodorotlenki zasadowe rozpuszczają się wwodzie i reagują z kwasami, np.: NaOH, KOH, CaOH. Wiązanie M-O jest o wiele słabsze wobec O-H i przykontakcie z wodą ulega degradacji, co daje kation metalu i anion wodorotlenowy.Wodorotlenki amfoteryczne nie rozpuszczają się w wodzie, reagują zarówno z kwasami, jak i z mocnymizasadami. Właściwości wodorotlenków amfoterycznych tłumaczy się wysoką elektroujemnością wchodzącegow ich skład pierwiastka, co prowadzi do upodobnienia siły wiązań M-O oraz O-H, w efekcie Ŝadne z nich nierozpada się i rozpuszczenie nie jest moŜliwe. Produkt reakcji zaleŜy dodatkowo od ilości uŜytych substratów.Np.: Al(OH)3 + 3HCl --> AlCl3 + 3H2O,Al(OH)3 + NaOH --> NaAlO2 + 2H2O,Al(OH)3 + 3NaOH + --> Na3AlO3 + 3H2O.4) Ze względu na zdolność do dysocjacji wodorotlenki dzielimy na mocne i słabe. Do pierwszej grupy zaliczmywodorotlenki litowców, Ca, Ba, Sr, zaś do drugiej – Mg, Be i resztę.5) Najpopularniejsze metody otrzymywania wodorotlenków to:- reakcja tlenków litowców i berylowców z wodą, np.:K2O + H2O --> 2KOHCaO + H2O --> Ca(OH)2,
- reakcja bezpośrednio litowców i berylowców z wodą (Be i Mg wyłącznie w wyŜszych temperaturach), np.:2Na + 2H2O --> 2NaOH + H2,Ca + 2H2O --> Ca (OH)2 + H2,
Created by Neevia Document Converter trial version http://www.neevia.com

40
- reakcje strącania pomiędzy solami i zasadami, np.:CuCl2 + 2KOH --> 2KCl + Cu(OH)2,FeCl3 + 3NaOH --> 3NaCl + Fe(OH)3.
KWASY
1) Kwasy dzielimy na beztlenowe oraz tlenowe. Kwasami beztlenowymi nazywamy roztwory wodne wodorkówkwasowych. Kwasy tlenowe to połączenia podobne w budowie chemicznej do zasad, składające się zpierwiastka i jednej bądź więcej grupy –OH. Kwasy cechuje budowa kowalencyjna polarna. Istotną róŜnicą wporównaniu do zasad jest moc wiązań M-O i O-H. W kwasach bowiem to pierwsze jest zdecydowaniemocniejsze, w związku z czym przy kontakcie z wodą uwalnia się kation wodoru i reszta kwasowa.Ze względu na ilość atomów wodoru w cząsteczce kwasy moŜna podzielić na jedno- (np. HNO3) orazwieloprotonowe, te ostatnie zaś na dwu- (H2SO4), trzy- (H3PO4), cztero- (H4SiO4), pięcio- (H5IO6),sześcioprotonowe (H6TeO6) itd. Kwasy występują w stanie stałym (krzemowe, fosforowe, borowe) bądźciekłym (ogromna większość, np. HNO3). Kwasy cechują się róŜną rozpuszczalnością w wodzie – większość jestrozpuszczalna (np. HCl), ale np. kwasy krzemowe są praktycznie nierozpuszczalne. Proces rozpuszczanianiektórych kwasów jest bardzo silnie egzotermiczny i moŜe doprowadzić do wrzenia rozpuszczalnika; tak dziejesię dla kwasu siarkowego, dlatego teŜ podczas rozcieńczania naleŜy wkraplać go do wady, a nie wlewać dońwody.2) Kwasy beztlenowe otrzymuje się w reakcjach rozpuszczania kwasowych wodorków niemetali w wodzie, np.:HCl -------H2O------> H+ + Cl-,HF -------H2O------> H+ + F-,H2S -------H2O------> H+ + HS- -------H2O------> 2H+ + S+.Z kolei kwasy tlenowe powstają w reakcjach bezwodnika kwasowego z wodą, np.:SO2 + H2O ----> H2SO3,SO3 + H2O ----> H2SO4,N2O5 + H2O ----> 2HNO3.Wyjątkowo kwasy krzemowe otrzymuje się w reakcjach strąceniowych mocnego kwasu z krzemianem:2H+ + SiO3
2- ----> H2SiO3,np.: H2SO4 + Na2SiO3 ----> H2SiO3 + Na2SO4.3) Niektóre kwasy (węglowy, siarkawy) rozkładają się pod wpływem temperatury na wodę oraz odpowiednibezwodnik:H2CO3 ----T----> H2O + CO2,H2SO3 ----T----> H2O + SO2.4) Moc kwasów tlenowych zaleŜy od elektroujemności wchodzącego do nich niemetalu i wzrasta z jej wzrostem.W związku z tym w okresach wzrasta na prawo, zaś w grupach malaje w dół. W przypadku kilku kwasówtlenowych tego samego pierwiastka o róŜnych wartościowościach moc kwasu wzrasta ze wzrostemwartościowości. Przy ogólnym porównywaniu mocy kwasów róŜnych pierwiastków naleŜy brać pod uwagę ichnajwyŜsze wartościowości. Moc kwasów beztlenowych wzrasta w okresach ze wzrostem elektroujemności (naprawo) i rośnie w grupach ze wzrostem promienia jonowego niemetalu (w dół).Ogólnie kwasy moŜemy podzielić na mocne (dysocjujące w znacznym stopniu) oraz słabe (przewaga cząstekniezdysocjowanych). NaleŜy zaznaczyć, iŜ tak określona moc kwasu nie pokrywa się zawsze ze skutkamidziałania; przykładowo kwas fluorowodorowy jest kwasem słabym, jednak ze względu na duŜą agresywność jeston jedynym kwasem uŜywanym do trawienia szkła.5) Kwasy reagują z odczynnikami zasadowymi, tj. zasadami, tlenkami zasadowymi i amfoterycznymi, dającsole. Reagują równieŜ z metalami nieszlachetnymi (o ujemnych potencjałach standardowych) dając sole i wodór.Nie reagują natomiast z metalami szlachetnymi (o dodatnich potencjałach standardowych). Np.:2H3PO4 + 3Ca(OH)2 ---> Ca3(PO4)3 + 6H2O,2H3PO4 + 3BaO ---> Ba3(PO4)3 + 3H2O,3H2SO4 + Al2O3 ---> Al2(SO4)3 +3H2O,Fe + H2SO4 ---> FeSO4 + H2,Ag + H2SO4 --X--> nie zachodzi.6) Istnieje grupa kwasów reagujących z tymi ostatnimi, jednak dzieje się to drogą reakcji redox. Ze względu narolę tych kwasów w takiej reakcji nazwano je kwasami utleniającymi. NaleŜą do nich: HNO3, stęŜony i gorącyH2SO4 oraz tlenowe kwasy chlorowe. Kwasy utleniające reagują z metalami szlachetnymi w ten sposób, Ŝeutleniają pierwiastek na wyŜszy stopień utlenienia, po czym tworzą z nim sól. Jednocześnie część uŜytego doreakcji kwasu redukuje się; ściślej mówiąc redukowany jest atom centralny występujący w kwasie, co powodujepowstawanie obok wspomnianej soli równieŜ cząsteczki tlenku zredukowanego atomu centralnego oraz wody.W przypadku niektórych kwasów produkty zaleŜą dodatkowo od ich stęŜenia.
Created by Neevia Document Converter trial version http://www.neevia.com

41
Np.:Cu + 4HNO3 -> Cu(NO3)2 + 2NO2 + 2H2O (>15%) niebieski rudobrązowyCu + 8HNO3 -> 3Cu(NO3)2 + 2NO + H2O (<15%) niebieski bezbarwnyCu + HNO3 -X-> nie zachodzi (<1%)Cu + 2H2SO4 -> CuSO4 + SO2 + 2H2O (stęŜony i gorący)7) Do reakcji z metalami wybitnie szlachetnymi, złotem oraz platyną, zdolna jest wyłącznie mieszaninastęŜonych kwasów – solnego oraz azotowego (V) – w stosunku objętościowym 3:1 (molowym 4:1). Reakcja tazłoŜony przebieg: wpierw kwas solny reaguje z kwasem azotowym, wytwarzając chlorek nitrozylu, którynastępnie atakuje metal szlachetny. Dla złota produktem reakcji jest kwas czterochlorozłotowy, co moŜnazapisać następująco:HNO3 + 3HCl -> NOCl + Cl2 + 2H2O,Au + HCl + NOCl +Cl2 -> HAuCl4 + NO.8) Natomiast kwasy utleniające nie reagują z niektórymi metalami nieszlachetnymi, np.: glinem, chromem czyŜelazem na zimno. Przyczyną jest powstawanie na ich powierzchni ochronnej warstwy pasywnej tlenku, couniemoŜliwia dalszy przebieg reakcji. Proces powstawania takiej powłoki nazywamy pasywacją.9) Kwasy utleniające mają takŜe moŜliwość utleniania niektórych niemetali, np. węgla, siarki czy fosforu.Ilustrują to równania:3C + 4HNO3 -> 3CO2 + 4NO + 2H2O,C + 2H2SO4 -> 2SO2 + CO2 + 2H2O,3S + 4HNO3 -> 3SO2 + 4NO + 2H2O,S + 2H2SO4 -> 3SO2 + 2H2O,3P + 5HNO3 -> 3H3PO4 + 5NO.10) W reakcjach kwasów utleniających z niektórymi metalami nieszlachetnymi będącymi silnymi reduktoraminie obserwuje wydzielania wodoru. Pierwiastki te powodują znaczną redukcję atomu centralnego kwasu (naweto osiem stopni utlenienia!), co przyczynia się do powstawania innych produktów w środowisku reakcji, tj.tlenków bądź soli, np.:4Zn + 10HNO3 -> 4Zn(NO3)2 + NH4NO3 + 4H2O,4Mg + 10HNO3 -> 4Mg(NO3)2 + N2O + 5H2O.11) StęŜone kwasu utleniające praktycznie nie dysocjują, poniewaŜ w niemal stuprocentowym roztworze prawienie występuje rozpuszczalnik (wówczas trudno taki układ nazwać roztworem). Ponadto w przypadku kwasusiarkowego (VI) moŜna obserwować ciekawy paradoks stęŜeniowy; otóŜ moŜna w nim rozpuścić dodatkowoduŜe ilości bezwodnika - SO3, przez co otrzymuje się tzw. dymiący kwas siarkowy bądź oleum o wzorzeumownym H2SO4
. SO3. Podczas dolewania do oleum wody zwiększamy objętość układu, natomiast niezmieniamy stęŜenia, poniewaŜ rozpuszczany bezwodnik wciąŜ daje nowe porcje kwasu i stęŜenie się niezmniejsza. Gdyby bezwzględnie przyjąć, iŜ dodawanie rozpuszczalnika zmniejsza stęŜenie roztworu, wówczasokazałoby się, iŜ kwas siarkowy moŜe przyjmować wartości stęŜeń wyŜsze o stu.12) Kwas azotowy (V) daje reakcję charakterystyczną z białkami. W wyniku nitrowania aminokwasówaromatycznych powstają Ŝółte nitrozwiązki i całe środowisko przyjmuje barwę Ŝółtą. Reakcja ta nosi nazwęreakcji ksantoproteinowej (ksantos – Ŝółty, proteina – białko). Po zalkalizowaniu środowiska przyjmuje onobarwę pomarańczową. Skóra ludzka po zetknięciu z kwasem azotowym równieŜ przyjmuje zabarwienie Ŝółte.
SOLE
1) Solami nazywamy połączenia metali z resztami kwasowymi, skąd natychmiast wynika wzór ogólny MenRm.W zaleŜności od typu kwasu, którego reszta wchodzi w skład soli, wyróŜniamy sole tlenowe oraz beztlenowe. Wprzypadku tych pierwszych końcówkę kwasu –owy zamieniamy na –an i dopisujemy nazwę metalu wdopełniaczu, natomiast w przypadku drugich zamieniamy końcówkę na –ek i postępujemy podobnie.2) Sole są krystalicznymi ciałami stałymi o róŜnej barwie i rozpuszczalności w wodzie. Proces rozpuszczaniasoli w wodzie jest endotermiczny, gdyŜ wiązania jonowe są tak silne, Ŝe energia pochłaniana do ich rozbicia niemoŜe zostać pokryta przez energię uzyskaną podczas hydratacji powstałych jonów. W związku z powyŜszymistnieje generalna prawidłowość, iŜ ze wzrostem temperatury rozpuszczalność soli rośnie, choć zaleŜności nie sąproporcjonalne, o czym świadczą zróŜnicowane krzywe rozpuszczalności. Choć większość soli jest bezbarwna,wiele z nic ma określony kolor; zabarwienie moŜe pochodzić od kationu (barwa odkationowa) bądź o anionu(barwa odanionowa). Przykłady: Cu2+ – niebieskie, Ni2+ – jasnozielone, Cr3+ - zielone, Fe2+ - brązowe, Fe3+ -słomkowoŜółte; I- - Ŝółte, Br- - beŜowe, Se2- - róŜowe (szkło), MnO4
- - fioletowe, CrO42- - cytrynowo Ŝółte,
Cr2O72- - pomarańczowe. Metale nadające solom barwę leŜą w bloku energetycznym d, co zostało wytłumaczone
Created by Neevia Document Converter trial version http://www.neevia.com
42
przy okazji barwy tlenków. Wskutek hydratacji jonów bądź tworzenia związków koordynacyjnych barwa moŜeulec pogłębieniu.3) - Sole są mocnymi elektrolitami, co oznacza, iŜ w roztworach prawie całkowicie występują jony.- Uczestniczą w reakcjach wymiany w roztworach wodnych; wymiana moŜe być pojedyncza (typu metal + sóllub sól beztlenowa + niemetal) bądź podwójna (typu sól + sól, sól + mocna zasada lub sól + mocny kwas).- Sole pochodzące od słabych kwasów i / lub słabych zasad ulegają hydrolizie, który to proces szczegółowowytłumaczono w dziale Chemia ogólna.– Wiele soli podatnych jest na rozkład pod wpływem czynników zewnętrznych (temperatury, światła), np.:CaCO3 ---T---> CaO + CO2,Na2SO3 ---T---> Na2O + SO2,2KNO3 ---T---> 2KNO2 + O2,2KClO3 ---T---> 2KCl + 3O2,2KMnO4 ---T---> K2MnO4 + MnO2 + O2,2AgCl ---hv---> 2Ag + Cl2,2AgBr ---hv---> 2Ag + Br2.4) Aby otrzymać sól naleŜy uŜyć odpowiednich odczynników, z których jeden jest dawcą kationu, drugi zaśdawcą anionu. W ogólnym przypadku daje to bardzo szeroki wachlarz moŜliwości, co ilustruje poniŜsza tabela:
Metody otrzymywania soliniemetal tlenek niemetalu kwas sól
metal2Na + Cl2 --> 2NaCl 2Na + 2CO2 -->
--> Na2CO3 + COZn + 2HCl -->--> ZnCl2 + H2
Fe + CuSO4 -->--> FeSO4 + Cu
tlenek niemetaluNa2O + Cl2 -->--> NaCl + NaOCl
Na2O + CO2 -->--> Na2CO3
CaO + 2HNO3 -->--> Ca(NO3)2 + H2O
Na2O + 2NH4Cl -->--> 2NaCl + 2NH3 + H2O
wodorotlenek2NaOH + Cl2 -->--> NaCl + NaOCl + H2O
Ca(OH)2 + CO2 -->--> CaCO3 + H2O
NaOH + HCl -->--> NaCl + H2O
Ca(OH)2 + 2NH4Cl -->--> CaCl2 + 2NH4OH
sól2KBr + Cl2 -->--> 2KCl + Br2
3Na2CO3 + P2O5 -->2Na3PO4 + 2HCl
CaCl2 + H2CO3 -->--> 2HCl
AgNO3 + NaCl -->--> AgCl + NaNO3
LITOWCE
Litowce są pierwszą grupą w układzie okresowym, co oznacza się I A (po staremu) lub 1 (według nowegosystemu zalecanego przez IUPAC). Posiadają konfigurację elektronową ns1
- posiadają zatem jeden elektronwalencyjny. Poza tym cechuje je mała energia jonizacji, co owocuje łatwym uleganiem wzbudzeniu i jonizacji.Wzbudzone atomy pierwiastków dają widmo w zakresie fal widzialnych, co jako tzw. próba płomienia słuŜy doidentyfikacji pierwiastków; i tak: lit daje barwę karminową (czerwono - róŜową), sód – Ŝółtą (moŜna się o tymprzekonać rzucając szczyptę soli na palnik kuchenki gazowej), potas – liliową, rubid – fiołkowo-róŜową, a cez –błękitną. Litowce są bardzo dobrymi przewodnikami elektryczności. Wszystkie mają nieduŜą gęstość, którarośnie w grupie wraz ze wzrostem liczby atomowej (czyli lit jest najlŜejszym pierwiastkiem w przyrodzie). Sąmetalami miękkimi, np. sód moŜna kroić noŜem. Niskie są równieŜ temperatury topnienia i wrzenia, któremaleją w grupie w dół (wyjątkowo cez wrze w temperaturze wyŜszej niŜ rubid). Unikalną cechą litowców jestrozpuszczalność w ciekłym amoniaku. Litowce z powodu higroskopijności i duŜej aktywności łatwo tworzą tlenki (X2O), a czasem równieŜ nadtlenki(XO - jeden atom tlenu więcej) i ponadtlenki (XO2 - dwa atomy tlenu więcej). Mówiąc o ponadtlenkach naleŜywymienić głównie nadtlenek potasu KO2 – pozostałe nie są tak powszechne. Tlenki litowców wykazującharakter silnie zasadowy. Nadtlenki i ponadtlenki są silnymi utleniaczami; te ostatnie mają charakterrodnikowy, co oznacza, iŜ w cząsteczce ich obecny jest nie sparowany elektron, a sumaryczna liczba elektronówjest nieparzysta. Spin nie sparowanego elektronu nadaje atomom właściwości magnetyczne – stąd wniosek, iŜponadtlenki są paramagnetykami.Sole litowców najczęściej dobrze rozpuszczają się w wodzie. Nie mają one barwy odkationowej, łatwo ulegająhydratacji, szczególnie sodu i litu. Oznacza to, iŜ w ich cząsteczkach naleŜy spodziewać się obecności wodykrystalizacyjnej (np.: Na2SO4
. 10H2O - sól glauberska, Na2CO3 . 10H2O - soda krystaliczna czy Na2B4O7
.
10H2O - boraks). Sole litu rozpuszczają się stosunkowo najtrudniej – albo występują one w formie hydratówalbo są nierozpuszczalne. Częściej spotykane są sole sodu niŜ potasu. Ten ostatni wykorzystywany jest przezrośliny, natomiast sód powoduje niszczenie roślin; wykorzystywano to niegdyś podczas wojny – posypującziemię solą wróg sprawiał, iŜ długo nie nadawała się ona do ponownej uprawy. W organizmach zwierzęcych sódi potas mają niebanalne znaczenie w regulacji właściwości błon komórkowych; utrzymywanie jonów soduwewnątrz, a potasu – na zewnątrz komórki, warunkuje jej polaryzację, a przez to pośrednio jej moŜliwość doprzekazywania impulsów drugiej komórce.
Created by Neevia Document Converter trial version http://www.neevia.com

43
Ze względu na układ elektronów do grupy tej naleŜy wodór. Od pozostałych litowców odróŜnia go wielewłaściwości – od stanu skupienia (gaz o gęstości 0,071 g/cm3) do elektroujemności (2,1, podczas gdy pozostałe:0,7 – 1). Wodór jest pierwiastkiem reaktywnym: gwałtownie reaguje z fluorem, dając fluorowodór:H2 + F2 ---> 2HF,z tlenem tworzy mieszaninę wybuchową i po zapaleniu gwałtownie reaguje, dając wodę:2H2 + O2 ---> 2H2O,zaś pod działaniem promieni świetlnych reaguje z chlorem:H2 + Cl2 ---> 2HCl.Wodór moŜna otrzymać na wiele sposobów, m. in.:a) z gazu wodnego:C + H2O --1000oC--> CO + H2,gaz wodny gaz syntezowyb) w reakcji metanu z wodą:CH4 + H2O ---Ni/1100oC---> CO + 3H2,c) w reakcji metali nieszlachetnych z kwasami, np.:Zn + 2HCl ---> ZnCl2 + H2,d) poprzez elektrolizę wody:2H2O -----> 2H2 + O2.
1) Do najwaŜniejszych związków litowców naleŜy wodorotlenek sodu NaOH. Jest to bardzo silna zasada, zwanaługiem sodowym, atakująca nawet krzemionkę SiO2 (dlatego nie przechowuje się jej w szklanych butelkach).Czysty NaOH jest białym, silnie higroskopijnym proszkiem, który w wyniku hydratacji „rozpływa się” napowietrzu (aby temu zapobiec przechowuje się w pastylkach). MoŜna go otrzymać w reakcji kaustyfikacji:Na2CO3 + Ca(OH)2 ---> 2NaOH + CaCO3.Reaguje z dwutlenkiem węgla, dając wodorowęglan sodu, który ogrzany dalej daje sodę:NaOH + CO2 ---> NaHCO3,2NaHCO3 --T--> Na2CO3 + CO2 + H2O.Znalazł zastosowanie w produkcji mydła, wypłukiwania celulozy drzewnej, fabrykacji barwników oraz środkówczyszczących i piorących.2) Wodorotlenek potasu KOH jest silniejsza zasadą od ługu sodowego. W podobny sposób reaguje zdwutlenkiem węgla, dając w efekcie węglan potasu:KOH + CO2 ---> KHCO3,2KHCO3 --T--> K2CO3 + CO2 + H2O.Jedyna róŜnica jest taka, iŜ wodorowęglan potasu łatwiej rozpuszcza się w wodzie od tworzącego zawiesinęwodorowęglanu sodu. Jest on wykorzystywany do produkcji miękkich, szarych mydeł.3) Sole sodowe i potasowe wykorzystywane są pospolicie w rolnictwie jako nawozy sztuczne. Najczęściej są toazotany, znane pod nazwą saletr: NaNO3 – saletra chilijska, KNO3 – saletra indyjska.4) Węglan sodu, zwany zwyczajowo sodą kalcynowaną, znajduje zastosowanie w produkcji szkła, mydła orazśrodków zapobiegających zawilgoceniu (ze względu na właściwości higroskopijne). podobnie zresztą moŜnauŜywać węglanu potasu. Na skalę przemysłową sodę otrzymuje się metodą Solvaya, która zostanie tuszczegółowo opisana.Surowcami jest sól kamienna NaCl oraz skała wapienna CaCO3. Proces technologiczny odbywa się dwomarównoległymi szlakami, które łączą się ze sobą we współzaleŜny cykl. Po oczyszczeniu sól kamiennadostarczana jest do karbonizatora, który ma na celu przekształcenie chlorku sodu w jego wodorowęglan. Obecnew karbonizatorze: woda i dwutlenek węgla reagują ze sobą, dając kwas węglowy, który łączy się z amoniakiem ipowstaje wodorowęglan amonu. Ostatni produkt wchodzi w reakcję wymiany podwójnej z chlorkiem sodu,dając poŜądany wodorowęglan sodu oraz chlorek amonu (salmiak). Sumarycznie:H2O + CO2 + NH3 +NaCl ----> NaHCO3 + NH4Cl.Powstały wodorowęglan, znany po zwyczajową nazwą sody oczyszczonej, tworzy wpierw zawiesinę, po czymwytrąca się w postaci delikatnego osadu. Półprodukt ten uŜywany jest jako środek spulchniający w przemyślepiekarskim, a pospolicie stanowi główny składnik proszku do pieczenia. Tak wytrącony osad podgrzewa się wprocesie kalcynacji, skutkiem czego wodorowęglan przekształca się w węglan:2NaHCO3 ----T----> Na2CO3 + CO2 +H2O.W ostatniej reakcji otrzymujemy końcowy produkt – sodę. Łatwo ulega ona uwodnieniu, tworząc hydrat znanyjako soda krystaliczna: Na2CO3
. 10H2O. Łatwość pochłaniania wody zadecydowała o zastosowaniuosuszającym.Równolegle przebiega proces przetwarzania drugiego substratu, tj. węglanu wapnia. Związek ten poddaje siędziałaniu wysokiej temperatury w procesie praŜenia, skutkiem czego otrzymuje się tlenek wapnia (wapnopalone):CaCO3 ---1000oC---> CaO + CO2,
Created by Neevia Document Converter trial version http://www.neevia.com

44
które następnie wprowadza się do wody (gaszenie). Reakcja przebiega burzliwie, dając jako główny produktwodorotlenek wapnia (wapno gaszone):CaO + H2O ----> Ca(OH)2.Wówczas następuje proces regeneracji, w którym powstałe wapno gaszone łączy się z salmiakiem jakoproduktem karbonizacji, który nie wytrącił się z roztworu:Ca(OH)2 + 2NH4Cl ----> CaCl2 + 2NH3 + H2O.Powstały amoniak jest powtórnie uŜywany w procesie karbonizacji, zaś chlorek wapnia jest zbędnym produktemubocznym.Sumarycznie zapisany przebieg metody Solvaya przedstawia się następująco:2NaCl + CaCO3 + H2O ---> Na2CO3 +CaCl2,skąd wnioskować moŜna o jej substratach (sól kamienna i skała wapienna) i produktach (soda i chlorek wapnia).Na uwagę zasługuje brak w równaniu reakcji amoniaku; substrat ten spełnia w całym obiegu rolę jakbykatalizatora – reaguje z jednym z substratów, po czym jest odzyskiwany w procesie regeneracji. W praktycewystępuje potrzeba uzupełniania zapasów amoniaku ze względu na nie stuprocentową wydajność reakcji.
BERYLOWCE
Berylowce to druga grupa główna w układzie okresowym – II A lub 2. Posiadają konfigurację elektronową ns2 -
dwa elektrony walencyjne na podpowłoce s. W wyniku wzbudzenia jeden z elektronów s przechodzi napodpowłokę p; tak powstały układ ulega (teoretycznie!) hybrydyzacji digonalnej sp, co doprowadza dopowstania dwóch jednakowych orbitali zachowujących układ liniowy (znajdują się pod wzajemnym kątem180o). Kształt liniowy posiadają zatem równieŜ cząsteczki oparte w budowie na atomie berylowca. Wporównaniu do litowców mają mniejszą objętość (większy ładunek jądra silniej przyciąga elektrony), większągęstość (co wynika z poprzedniego), wyŜsze temperatury topnienia i wrzenia oraz większą twardość. Promieńatomowy oraz związana z nim reaktywność rosną w grupie w dół, wraz ze wzrostem liczby atomowej. Wprzyrodzie berylowce występują głównie w postaci soli, a zwłaszcza węglanów (CaCO3 - kamień wapienny,kreda, kalcyt, aragonit, CaCO3
. MgCO3 – dolomit). Związki berylowców równieŜ wykazują tendencję dotworzenia hydratów, która maleje w grupie w dół (np.: CuSO4
. 5H2O - kamień siny, CaSO4 . 2H2O – gips lub
alabaster, MgSO4 . 7H2O - sól gorzka, 3MgO . 4SiO2
. H2O – talk). Wszystkie berylowce posiadają ujemnepotencjały normalne, co stanowi o ich aktywności chemicznej. DuŜa aktywność wymusza stosowanie metodyelektrolizy ze stopionych soli podczas otrzymywania tych pierwiastków. Berylowce są silnymi reduktorami,przez co łatwo reagują z kwasami w wypierają z nich wodór. Począwszy od wapnia i idąc dalej w dół grupy,związki berylowców ulegają wzbudzeniu i emitują energię w postaci kwantów światła widzialnego. Oznacza to,iŜ ich obecność badać moŜna testem płomieniowym, gdyŜ barwią płomień na określony kolor: wapń –ceglastoczerwony (szybko znika), bar – jasnozielony, stront – purpurowy, rad – karminowy. W grupie tejwystępuje izomorfizm (równopostaciowość); np. BaSO4 i CaSO4 mają identyczny typ komórki elementarnej,zbliŜone rozmiary komórek, taki sam typ wzoru chemicznego, podobną postać krystalograficzną i analogicznąstrukturę – warunkuje to zdolność do tworzenia kryształów mieszanych i naprzemienne narastanie kryształówobu substancji
BERYLBeryl jest najlŜejszy z całej grupy. W wilgotnym powietrzu pokrywa się warstwą tlenku. Nie reaguje z wodą aniw temperaturze pokojowej, ani nawet po podgrzaniu. Podobnie jak tlenek, posiada właściwości amfoteryczne. Wkontakcie ze stęŜonym kwasem azotowym pasywuje, pokrywając się ochronna warstwą tlenku. Jest toksycznydla człowieka.
MAGNEZMagnez jest najmniej aktywny z całej grupy. Pozostawiony na powietrzu pokrywa się warstwą tlenku orazbardzo trudno rozpuszczalnego wodorotlenku, w małym stopniu wpływając na alkalizację środowiska. Reaguje zwodą na gorąco, tj. w temperaturze >70oC. Silnie ogrzany (>760oC) zapala się oślepiającym płomieniem,wydzielając duŜe ilości ciepła, co zadecydowało o wykorzystaniu magnezu jako materiału zapalnego do lampbłyskowych, głównie podczas fotografowania nocą. W reakcji tej powstaje trudno rozpuszczalny tlenekmagnezu MgO, stosowany jako składnik materiałów ogniotrwałych. Twardość i odporność na niekorzystneczynniki zadecydowały o technicznym zastosowaniu magnezu w budownictwie. Stosowany jest równieŜ jakoprotektor do pokrywania kadłubów okrętów i ochrony ich przed korozją. Jonów magnezowych Mg2+ nie moŜnawykryć w obecności jonów amonowych NH4
+, które znacznie szybciej wiąŜą aniony wodorotlenowe.
WAPŃWapń występuje w przyrodzie gównie wchodząc w skład soli – siarczanów oraz wapieni; te ostatnie jakowęglany najłatwiej wykryć działając kwasem, np. solnym:
Created by Neevia Document Converter trial version http://www.neevia.com

45
CaCO3 + 2HCl ---> CaCl2 + H2O + CO2;reakcji towarzyszy silne pienienie roztworu spowodowane wydzielaniem pęcherzyków gazu.Obecność jonów Ca2+ w roztworze moŜna stwierdzić dodając doń rozpuszczalnego szczawianu. Z roztworustrąci się wówczas nierozpuszczalny biały osad szczawianu wapnia CaC2O4.Z jonową budową węglanu wapnia związana jest jego budowa krystaliczna. Ze względu na sposób ułoŜeniajonów w sieci krystalicznej wyróŜniamy dwie formy kryształów: kalcyt (forma podstawowa) oraz aragonit.Drobnokrystaliczną odmianą kalcytu jest marmur. Czyste marmury są białe bądź przezroczyste (tzw. marmurykarryjskie z włoskiego kamieniołomu w Carras), natomiast drobne zanieczyszczenia nadają im określone barwy.Kryształy kalcytu są dwójłomne, co oznacza, iŜ podające nań światło załamuje się pod dwoma róŜnymi kątami.Powstają dzięki temu dwa promienie: zwyczajny, załamujący się zgodnie z prawem Snelliusa oraznadzwyczajny, nie spełniający powyŜszego warunku. Promienie te posiadają ponadto waŜną właściwość,mianowicie oba są całkowicie spolaryzowane, a płaszczyzny polaryzacji są do siebie prostopadłe. Otrzymaneświatło spolaryzowane słuŜyć moŜe do określania stęŜenia substancji optycznie czynnej w jej roztworze (np.zawartości sacharozy w soku buraczanym).Występujący w przyrodzie wapień poddaje się rozkładowi termicznemu w wapiennikach, gdzie zachodzireakcja:CaCO3 ---1000oC---> CaO +CO2,w której powstaje aktywny i trudno rozpuszczalny tlenek wapnia, zwany wapnem palonym. Tak powstałyprodukt zalewa się wodą:CaO + H2O ----> Ca(OH)2.Reakcja jest silnie egzotermiczne i prowadzi do powstania wapna gaszonego. Wodorotlenek wapnia jest słaborozpuszczalny w wodzie i tworzy tzw. mleko wapienne, jednak pomimo tego jest bardzo silną zasadą. Wlaboratorium słuŜy np. do wykrywania dwutlenku węgla – wpierw pojawia się zmętnienie, zauwaŜa się strąceniebiałego osadu, który następnie rozpuszcza się (wskutek przemiany w wodorowęglan, o czym później):Ca(OH)2 + CO2 ---> CaCO3 + H2O.Reakcja ta znalazła zastosowanie w budownictwie: składniki zaprawy (wapno) pod wpływem atmosferycznegodwutlenku węgla ulegają stwardnieniu i łączą elementy budowlane. Z powodu emisji do atmosferyzwiększonych ilości dwutlenku węgla, skały wapienne oraz budowle są niszczone wskutek zachodzenianastępującego procesu:2CaCO3 + CO2 + H2O <---> Ca(HCO3)2.Kwasowość środowiska wpływa na przemianę węglanów w wodorowęglany charakteryzujące się o wielewiększą rozpuszczalnością w wodzie. Te ostatnie są po prostu wymywane – na tej samej zasadzie działająprocesy krasowe, jak i destrukcyjne skutki kwaśnych opadów (przykładowo zniszczenie rzeźb krakowskiejStarówki). Wzrost temperatury sprzyja przebieganiu reakcji w drugą stronę – węglan wytrąca się, a lotneskładniki opuszczają środowisko.Przy produkcji cukru zasadę wapniową wprowadza się do soku buraczanego celem oczyszczenia (defekacji).Wiązaniu legają wszelkie zanieczyszczenia kwasowe, jak równieŜ dwutlenek węgla (saturacja), które pozobojętnieniu strącają się w postaci osadu, podczas gdy sacharoza pozostaje w roztworze.Wapń wchodzi równieŜ w skład gipsów. Miękki i łupliwy kamień gipsowy wydobywany w kamieniołomach tonic innego jak uwodniony siarczan (VI) wapnia CaSO4
. 2H2O. W stosunkowo niewysokiej temperaturze (zmałym nakładem energetycznym) moŜna przeprowadzić dehydratację skały gipsowej:2[CaSO4
. 2H2O] ---110oC---> 2CaSO4 . H2O + 3H2O,
i otrzymać popularny gips. Ten cechuje się łatwym wchłanianiem wody, któremu towarzyszy wzrost objętości(powyŜsza reakcja zachodzi wówczas w drugą stronę). Ze względu na ostatnią właściwość odlewy gipsowerzeźb są niezwykle dokładne, gdyŜ w przeciwieństwie do gipsu beton kurczy się podczas twardnienia. Mostycementowe muszą być wylewane stopniowo wskutek dylatacji.Drobnokrystaliczną odmianą gipsu jest alabaster (w przeciwieństwie do pierwszego jest bezwodny). ZbliŜona domarmuru struktura pozwoliła na zastosowanie go do celów dekoracyjnych juŜ w czasach sumeryjskich.Szczególnie trudno rozpuszczalnymi solami są fluorki ze względu na duŜe róŜnice elektroujemności i energiewiązań. Sprawia to, iŜ fluorek wapnia CaF2 buduje szkliwo zębowe – element skrajnie nierozpuszczalny.
BARBar jest najaktywniejszym berylowcem - posiada aktywność porównywalną z potasem, dzięki czemu z łatwościąwypiera wodór z wody. Podczas otrzymywania baru z jego tlenku stosuje się metodę aluminotermiczną.Siarczany berylowców są solami trudno rozpuszczalnymi, a rozpuszczalność maleje w grupie w dół:siarczan (VI) baru jest nierozpuszczalny nawet w stęŜonych kwasach, co pozwoliło na zastosowanie go wdiagnostyce medycznej. Pacjent zjada papkę zawierającą BaSO4, po czym tworzy się zdjęcie rentgenowskiejamy brzusznej. Ze względu na duŜą liczbę masową bar pochłania promieniowanie, co uwidocznione jest nazdjęciu jako białe plamy. Analizując wynik specjalista moŜe stwierdzić chorobę wrzodową lub inne schorzenia.Reakcja, w której strąca się biały osad nierozpuszczalny w kwasach, jest reakcją charakterystyczną na jony Ba2+.
Created by Neevia Document Converter trial version http://www.neevia.com

46
Przy okazji berylowców omówimy pojęcie twardości wody. W naturalnej wodzie zawsze znajdują się pewneilości jonów wapniowych i magnezowych. W przypadku wód mineralnych jest to zjawisko poŜądane, poniewaŜwystępujące w formie słabo wysyconych i rozpuszczalnych wodorowęglanów makroelementy są dobrzeprzyswajalne przez organizm ludzki (nawet 20 – 40%). Natomiast obecność takich jonów w wodzie uŜywanej doprania, zwana twardością wody, jest wysoce niewskazane, gdyŜ tworzą one z mydłami nierozpuszczalne sole. Wpraniu pojawia się kłaczkowaty osad a tkanina staje się sztywna. TakŜe włosy umyte szarym mydłem będąmatowe (w celu uzyskania połysku moŜna dodać kwasku cytrynowego). W zaleŜności od czynnikapowodującego twardość moŜna podzielić ją na:a) twardość przemijającą (węglanową), spowodowaną obecnością w wodzie wodorowęglanów wapnia imagnezu. Podczas gotowania wody pod wpływem temperatury powstaje lotny z nich dwutlenek węgla oraznierozpuszczalne węglany, strącające się w postaci kamienia kotłowego, np.:MgCO3 + H2O ---> Mg(OH)2 + CO2.Powstający kamień kotłowy, będący mieszaniną węglanu wapnia i magnezu, osadza się na elementachgrzewczych i powoduje ich nadmierne ogrzewanie – łatwiej się wtedy topią i niszczą, nie mówiąc juŜ ododatkowym zuŜyciu energii.Twardość przemijającą moŜna usunąć dodając do wody określone substancje, a następnie odsączając powstałyosad, np.:Ca(OH)2 + Ca(HCO3)2 ---> 2CaCO3 + 2H2O,2Ca(OH)2 + Mg(HCO3)2 ---> 2CaCO3 + Mg(OH)2 + 2H2ONa2CO3 + Ca(HCO3)2 ---> CaCO3 + 2NaHCO3.Usuwaniu twardości słuŜą równieŜ jonity. Są to specjalne substancje, które dzięki posiadanemu na powierzchniładunkowi elektrycznemu osadzają na niej niepoŜądane jony – w tym przypadku Ca2+ i Mg2+.b) twardość nieprzemijającą (siarczanową), spowodowaną obecnością w wodzie siarczanów wapnia i magnezu.W usuwaniu twardości pomocne są detergenty, czyli syntetyczne środki piorące. Oprócz zmniejszania napięciapowierzchniowego roztworu posiadają bardzo często naturalny charakter zasadowy.Twardość wody jest wielkością mierzalną. Określa się ją zawartością soli utwardzających w przeliczeniu na CaOi oznacza w stopniach. 1o niemiecki odpowiada zawartości 10 mg CaO w 1 dm3 wody. 1o niemiecki = 1,25o
angielskiego = 1,79o francuskiego. Wodę o twardości 1-7o niemieckich uznaje się za wodę miękką, zaś 7-14o –za wodę twardą.
BOROWCE
Borowce to trzecia grupa główna w układzie okresowym – III A lub 13. Posiadają konfigurację elektronowąns2np1
- zatem trzy elektrony walencyjne rozmieszczone na podpoziomach s i p.. W wyniku wzbudzenia jeden zelektronów s przechodzi na podpowłokę p; tak powstały układ ulega (teoretycznie!) hybrydyzacji trygonalnejsp2, co doprowadza do powstania trzech jednakowych orbitali zachowujących układ trójkątny (orbitaleskierowane w wierzchołki trójkąta równobocznego znajdują się pod wzajemnym kątem 120o). Cząsteczki opartew budowie na atomie borowca posiadają zatem budowę płaską, trójkątną. Borowce mają mniejsza aktywnośćchemiczną w porównaniu z pierwiastkami grupy I i II. WiąŜąc się z innymi pierwiastkami trzema wiązaniamizapewniają sobie układ sześciu elektronów walencyjnych. Jest to ilość mniejsza od potrzebnego do uzyskaniatrwałości oktetu - wobec deficytu elektronowego zgodnie z teorią Lewisa związki takie są kwasami. Dzięki tejwłaściwości związki borowców z fluorowcami, a zwłaszcza halogenki glinu (AlCl3, AlBr3, AlF3), pełnią rolękatalizatorów w reakcjach syntez organicznych, m. in. w reakcjach Friedla – Craftsa, których mechanizmzostanie omówiony później. Borowce łatwo tworzą tlenki, mogą nawet odbierać tlen związany z innymipierwiastkami, przez co redukują środowisko.
BORPierwszym pierwiastkiem w grupie, od którego zresztą bierze się jej nazwa, jest bor. Jest to pierwiastekpółmetaliczny, występujący w związkach z wartościowością III. W przyrodzie występuje on w wielu formach.Czysty bor występuje w dwóch postaciach alotropowych: forma krystaliczna jest bardzo twarda, odporna nadziałanie tlenu stęŜonego kwasu azotowego, zaś forma bezpostaciowa jest miękka i łatwo utlenia się napowietrzu w podwyŜszonej temperaturze. W warunkach takich bor moŜe równieŜ redukować parę wodną,dwutlenek węgla oraz krzemionkę. Identyfikacja boru opiera się na analizie widmowej i teście płomieniowym:wprowadzony w ogień bor daje barwę zieloną, jednak nie tak jaskrawą jak bar.Naturalnym związkiem o duŜej zawartości boru jest boraks, czyli uwodniony czteroboran sodu Na2B4O7
.
10H2O. Stopiony boraks rozpuszcza tlenki metali, dając charakterystycznie zabarwione szkliwo, tzw. perłęboraksową. PoniewaŜ jej kolor zaleŜy od uŜytego pierwiastka, moŜe być wykorzystywana do identyfikacjipierwiastków przy dostępie tlenu (jakościowa analiza chemiczna). I tak: Mn2O7 stopiony z boraksem ma kolorfioletowy, CaO – ciemnoniebieski lub granatowy, zaś CrO3 – zielony. Rozpuszczanie tlenków na powierzchni
Created by Neevia Document Converter trial version http://www.neevia.com

47
metali pozwoliło uŜyć boraksu jako środka odrdzewiającego. Perły boraksowej uŜywa się ponadto przyprodukcji płytek ceramicznych, gdyŜ zmniejsza to wymaganą temperaturę. Znajduje takŜe zastosowanie jakotopik przy lutowaniu. Dodawany jest do gorących emalii, gdyŜ ze względu na duŜy współczynnik załamaniaświatła nadaje im połysk. UŜywany jest do produkcji szkła Ŝaroodpornego, w połączeniu z ołowiem stanowiskładnik szkła kryształowego, zaś stopiony z berylem – składnik szkła uŜywanego w mikrofalówkach. Boraksreaguje z nadtlenkiem wodoru, dając nadtlenoboran o silnych właściwościach wybielających. Oprócz wybielaniatkanin ten ostatni słuŜy do barwienia jedwabiu oraz drogich wyrobów skórzanych.Bor występujący w postaci tlenku B2O3 występuje w towarzystwie krzemianów i glinokrzemianów. Tlenek borujest ciałem stałym, rozpuszczającym się w wodzie ze nieznacznym wydzieleniem ciepła (jest to procesniewymuszony). Reakcja prowadzi do powstania kwasu ortoborowego:B2O3 + 3H2O ---> 2H3BO3.Związek ten ma właściwości antyseptyczne, dlatego jest wykorzystywany jako środek dezynfekcyjny w postacisilnie rozcieńczonego roztworu pod nazwą wody borowej. Podczas ogrzewania kwas ortoborowy traci jednącząsteczkę wody i przechodzi w kwas metaborowy:H3BO3 ---T---> HBO2 + H2O.Kwasy borowe posiadają słaby charakter kwasowy.Bor tworzy z niektórymi pierwiastkami nietrwałe borki, np. borek magnezu Mg3B2. Są to na ogół związkinietrwałe, łatwo rozkładające się pod działaniem wilgoci i kwasów. Wyjątkowo węglik boru CB4 posiadaogromną twardość, co zadecydowało o zastosowaniu go do produkcji materiałów ściernych. W naturalnychzłoŜach jest zanieczyszczony grafitem, obok którego występuje. Czysty węglik boru, dzięki duŜej zdolnościpochłaniania neutronów, uŜywany był do budowy reaktorów jądrowych oraz schronów atomowych.W połączeniu w wodorem bor tworzy borowodory. Najprostszym byłby BH3 – byłby, gdyŜ naturalnie związektaki nie występuje. Spotykany jest jego dimer B2H6 charakteryzujący się deficytem elektronowym, co ułatwiareakcję ze związkami posiadającymi wolną parę elektronową (np. z amoniakiem). Dimery i polimeryborowodoru aŜ do B6H10 włącznie są gazami o bardzo nieprzyjemnym zapachu, wydzielającymi się podczasgnicia substancji organicznych.
GLINRównie waŜnym pierwiastkiem w grupie borowców jak sam bor jest glin. Pierwiastek ten występuje pospoliciew skorupie ziemskiej w postaci glinokrzemianów, np. skalenia czy miki. Praktycznie wykorzystywana jestglinka porcelanowa, czyli kaolin (uwodniony krzemian glinu – [Al2(OH)4][Si2O5] – w zapisie tlenkowym:Al2O3
. 2SiO2 . 2H2O), z której wypala się cegły. Następstwem ruchów geologicznych jest występowanie w
kaolinie zanieczyszczeń, np. czerwonych związków Ŝelaza Fe2O3, co nadaje cegłom kolor czerwony. Tlenekglinu Al2O3 występuje w dwóch alotropowych odmianach: krystalicznej, jako korund, minerał bardzo twardy iwykorzystywany w produkcji materiałów ściernych oraz ziemistej, takŜe uŜywanej w materiałach szlifierskich.Następstwem jonowej budowy tlenku glinu jest wysoka temperatura topnienia. Siła wiązań sprawia, iŜ tlenkuglinu nie redukują typowe reduktory jak węgiel czy dwutlenek węgla. Cechuje go ponadto wysoki współczynnikzałamania światła. Chemicznie czysty minerał jest bezbarwny, natomiast zanieczyszczenia pierwiastkami blokud nadają mu piękną barwę. Powstają w ten sposób kamienie szlachetne: ciemnoczerwony rubin przydomieszkowaniu jonami Cr3+ oraz niebieski szafir przy domieszkowaniu jonami Ti3+. Związki glinu wykazującharakter amfoteryczny, stąd mówi się o kwasach: ortoglinowym H3AlO3 oraz metaglinowym HAlO2. Teostatnie występują jednak wyłącznie w postaci anionów, wchodząc w skład odpowiednich soli. Związki glinuczęsto biorą udział w tworzeniu połączeń koordynacyjnych z liczbą koordynacyjną zaleŜną od stopnia utlenienia– odpowiednio 4 dla –1 oraz 6 dla –3.WaŜnym z punktu widzenia praktyki związkiem glinu jest kriolit Na3AlF6, który dzięki obniŜaniu temperaturytopnienia znalazł zastosowanie w elektrolitycznym otrzymywaniu glinu na skalę przemysłową. Innym istotnymzwiązkiem jest chlorek glinu AlCl3, ciało stałe dobrze rozpuszczalne w wodzie. Swe znaczenie zawdzięczaistnieniu deficytu elektronowego, dzięki czego moŜe być jako kwas wykorzystywany w syntezie organicznej, np.jako katalizator przy bromowaniu benzenu. Wodorek litowo – glinowy Li[AlH4] lub LiH . AlH3 posiada bardzosilne właściwości redukujące, gdyŜ występujący w nim wodór -1 dąŜy do utlenienia się na stopień 0 bądź +1.Zadecydowało to o jego wykorzystaniu w chemii organicznej w reakcjach hydrogenacji, czyli uwodornienia.
Niektóre metale jednowartościowe (potas, sód, rubid, cez) oraz kation amonowy tworzą z metalamidwuwartościowymi (Al, Cr, Fe, Co, Ga, In, Ti, V) tzw. sole podwójne typu MIMIII(SO4)2 . 12H2O, co moŜnaprzedstawić alternatywnym wzorem MI
2SO4 . MIII
2(SO4)3 . 24 H2O. Sole te, zwane ałunami, tworzą dorodna
barwne kryształy. Ałun chromowo – sodowy stosowany jest w garbarstwie do wyprawiania skór, podobnie jakałun glinowo – potasowy, który znalazł teŜ zastosowanie w farbiarstwie , jako wypełniacz przy produkcjipapieru oraz do tamowania krwawień.
Created by Neevia Document Converter trial version http://www.neevia.com

48
WĘGLOWCE
Węglowce to czwarta grupa główna w układzie okresowym – IV A lub 14. Posiadają konfigurację elektronowąns2np2
- zatem cztery elektrony walencyjne rozmieszczone po równo na podpoziomach s i p. W wynikuwzbudzenia jeden z elektronów s przechodzi na podpowłokę p; tak powstały układ ulega hybrydyzacjitetragonalnej sp3, co doprowadza do powstania czterech jednakowych orbitali skierowanych w naroŜaczworościanu foremnego; shybrydyzowane orbitale znajdują się pod wzajemnym kątem 109o. Cząsteczki opartew budowie na atomie węglowca posiadają zatem budowę czworościanu.
WĘGIELPierwiastkiem rozpoczynającym grupę jest węgiel. Występuje on w kilku odmianach alotropowych: diament,grafit oraz fulleren. Istotne róŜnice pomiędzy dwoma pierwszymi wynikają z występującego w obrębie ichcząsteczek typu hybrydyzacji. W diamencie występuje hybrydyzacja sp3, co oznacza, iŜ atomy są ułoŜone bliskosiebie i kaŜdy połączony jest z czterema innymi obecnymi dookoła. Powoduje to relatywnie duŜą gęstość itwardość – jest to najtwardszy minerał w przyrodzie. Brak swobodnych elektronów sprawia, iŜ diament jestizolatorem. O wysokim współczynniku załamania światła w diamencie przekonać się moŜna obserwując barwnerefleksy powstające podczas przejścia światła przez brylant. Z kolei w graficiewystępuje hybrydyzacja sp2, co oznacza, iŜ kaŜdy atom połączony jest płasko ztrzema dookoła. Grafit ma zatem budowę warstwową, a poszczególnepłaszczyzny atomów mogą się względem siebie przesuwać. Z tego powodugrafit jest miękki i ma mniejszą gęstość od grafitu. Czwarty elektron, niezwiązany z Ŝadnym innym atomem, jest wolny i moŜe być nośnikiem prąduelektrycznego – dlatego teŜ grafit posiada doskonałe właściwości przewodzące.Odmianą grafitu jest węgiel aktywny; cechuje go bardzo duŜa powierzchniawchłaniania i sorpcji, dzięki czemu stosowany jest do zatrzymywaniaszkodliwych substancji. Podawany jako lekarstwo zatrzymuje biegunkę, zaśstosowany w maskach gazowych oczyszcza zatrute powietrze.Fullereny to specyficzna odmiana węgla; naleŜą do nowych, intensywnie badanych materiałów. Najprostszyfulleren - C60 jest zamkniętą klatką zbudowaną z 60 atomów węgla w kształcie piłki noŜnej, złoŜoną z 12pięciokątów i 20 sześciokątów foremnych (patrz rysunek). MoŜna go otrzymać przez laserowe odparowanie gra-fitu w atmosferze helu. Fullereny znalazły juŜ zastosowanie jako doskonałe smary. Z tych wielkich cząsteczekC60 moŜna zbudować kryształ molekularny, który nazywa się fullerytem i który jest półprzewodnikiem o prostejprzerwie energetycznej równej 1,9 eV. Istnieją takŜe sferyczne obiekty z wielu współśrodkowych fullerenów(tzw. struktury cebulowe): C60, C240, C540, C960, które nazywane są hiperfullerenami. Ponadto moŜnadomieszkować fullereny „zapakowując" obce atomy w C60 i tworzyć tzw. fullerydki: np. fullerydek z metalemalkalicznym K3C60. Wykazuje on nadprzewodnictwo wysokotemperaturowe z temperaturą przejścia 33 K.Metodą wyładowania łukowego moŜna tworzyć rurki fullerenowe (tzw. nanorurki kwantowe) - współosiowecylindry odległe o 0,34 nm i długości ok. l µm, które są bardzo trwałymi włóknami, stosowanymi jakoświatłowody dla promieniowania gamma. Interesująca właściwość „samozasklepiania" rurek fullerenowych dajeich duŜą odporność na uszkodzenia. MoŜna wreszcie „patroszyć" fullereny; np. dodając do sześciokątówsiedmiokąty, wewnętrzną stronę odwracać na zewnątrz. Dokonując „mariaŜu topologii z węglem" moŜnaprowadzić geometryczne zabawy budowania róŜnych obiektów przestrzennych z atomów węgla otrzymującnowe struktury o nieoczekiwanych właściwościach.Ogromną rolę w przyrodzie i gospodarce odgrywają związki węgla. Związki organiczne, ze względu na ichmnogość, omówione będą osobno. Wśród nieorganicznych związków węgla waŜną rolę pełnią tlenki.1) Najprostszym tlenkiem węgla jest CO, znany jako czad. Jest to bezbarwny i bezwonny gaz, nieco lŜejszy odpowietrza, bardzo słabo rozpuszczalny w wodzie i nie reagujący z nią. Na uwagę zasługuje budowa cząsteczki:otóŜ po dwa niesparowane elektrony z tlenu i węgla tworzą dwa wiązania koordynacyjne spolaryzowane, na obuatomach pozostają wolne pary, a dodatkowo jedna para z atomu tlenu tworzy trzecie, koordynacyjne wiązanie zatomem węgla. Cząsteczka wykazuje słaby moment dipolowy. Taka sama budowa strukturalna, identyczna masamolowa oraz liczba elektronów walencyjnych upodabnia cząsteczkę CO do N2 – mówimy, iŜ są izosteryczne.Dzięki dysponowaniu wolnymi parami w wyŜszych temperaturach tworzy lotne związki z metalami, zwanekarbonylkami, np.: Ni(CO)4, Fe(CO)5. Wiązanie jest potrójne, czyli silnie nienasycone. Dzięki temu tlenekwęgla bardzo łatwo wchodzi w reakcje, jest gazem palnym, a w wyŜszych temperaturach moŜe odbierać tleninnym związkom. Redukcyjne właściwości sprzyjają zastosowaniu go przy otrzymywaniu metali. W procesiewielkopiecowym najpierw jest on otrzymywany w wyniku przepuszczanie strumienia powietrza przezrozŜarzony koks (reakcja Braundarda):C + O2 ---> CO2, CO2 + C --->2CO,
Created by Neevia Document Converter trial version http://www.neevia.com

49
a następnie wchodzi w reakcję z rudą:Fe2O3 + 3CO ---T---> 3CO2 + 2Fe.Podobnie przebiega proces otrzymywania cynku; wpierw ruda siarczkowa (sfaleryt lub wurtzyt) jest spalana dotlenku:2ZnS + 3O2 --->2ZnO + 2SO2,który następnie poddawany jest redukcji:ZnO + CO ---> Zn + CO2.W odpowiednio wysokiej temperaturze tlenek węgla odtlenia wodę:CO + H2O <---1100oC---> CO2 + H2.W przedstawionym równaniem układzie ustala się stan równowagi. Jest to przykład reakcji konwersji, o którejmówimy, gdy jeden gaz lub mieszanina przekształca się w drugą (w powyŜszej reakcji ze względu natemperaturę wszystkie substraty występują w stanie gazowym).Czad tworzy się podczas półspalania związków organicznych, jak równieŜ przy kontakcie metanu z wodą:CH4 + H2O ---T---> CO + 3H2.MoŜna go takŜe otrzymać działając stęŜonym kwasem siarkowym na kwas mrówkowy. Ten pierwszy odbieradrugiemu cząsteczkę wody, w wyniku czego pozostaje tlenek węgla:HCOOH ---st. H2SO4 ---> H2O (natychmiast pochłaniana przez higroskopijny kwas) + CO.MoŜe on wchodzić w reakcję z zasadą sodową, dając mrówczan sodu:CO + NaOH ---T, p---> HCOONa.W wyniku reakcji koksu z wodą w wysokiej temperaturze powstaje mieszanina wodoru i tlenku węgla, zwanagazem syntezowym (generatorowym, wodnym):C + H2O ---1000oC---> H2 + CO.Odpowiednia proporcja składników oraz właściwe warunki termiczno – ciśnieniowe pozwalają na otrzymanie zgazu syntezowego róŜnorodnych związków organicznych.Reaguje równieŜ z siarką, dając ostatecznie dwusiarczek węgla:2CO + 2S ---> 2COS (nietrwały) ---> CO2 + CS2.Z chlorem tworzy fosgen, który wykorzystywany był jako gaz bojowy:CO + Cl2 ---> COCl2.Fosgen ma jednak równieŜ bardziej cywilizowane zastosowania – przykładowo w reakcji z amoniakiem dajemocznik, wykorzystywany w produkcji tworzyw sztucznych (np. Ŝywic):COCl2 + 2NH3 ----> NH2CONH2 + 2HCl.Czad jest gazem silnie trującym. Śmiertelne jest dla człowieka juŜ stęŜenie 0,3% w powietrzu. Czad łączy się zhemoglobiną krwi 250 razy trwalej niŜ czyni to tlen, co blokuje funkcję transportującą tego związku, gdyŜpowstała karboksyhemoglobina HbCO nie przenosi tlenu.2) Najbardziej znanym jest dwutlenek węgla, biorący udział w procesach biologicznych. Powstaje przez pełnespalanie związków organicznych (przy bogatym dostępie tlenu). Otrzymać go moŜna przez rozkład węglanówmetodą termiczną bądź przez działanie nań mocnym kwasem np.:CaCO3 ---1000oC---> CaO + CO2,CaCO3 + 2HCl ---> CaCl2 + H2O + CO2.Ostatnia reakcja moŜe być wykorzystywana do identyfikacji węglanów, gdyŜ towarzyszy jej charakterystycznesilne pienienie roztworu spowodowane wydzielaniem pęcherzyków gazu.Dwutlenek węgla jest bezbarwnym i bezwonnym gazem, cięŜszym od powietrza. Przez krótki czas moŜna goprzechowywać w naczyniach otwartych. Nie jest gazem palnym ani nie podtrzymuje palenia. Jest związkiemniepolarnym. W obrębie jego cząsteczki zachodzi hybrydyzacja digonalna sp. Cząsteczka CO2 ma budowęliniową, a sumaryczny moment dipolowy wynosi zero. Rozpuszcza się w wodzie, przy czym rozpuszczalnośćmaleje ze wzrostem temperatury i rośnie ze wzrostem ciśnienia. Przykładowo – w butelce wody i w małymstopniu reaguje z nią, dając kwas węglowy:H2O + CO2 <---> H2CO3.Reakcja jest silnie odwracalna, powstały produkt jest bardzo nietrwały i gdy stęŜenie jego przekroczy setnączęść procenta, rozkłada się. Kwas węglowy jest kwasem dwuprotonowym, przechodzi więc dysocjacjędwustopniową i moŜe tworzyć dwa rodzaje soli: obojętne węglany oraz kwaśne wodorowęglany. Wodorosolerozpuszczają się lepiej od soli obojętnych, o czym mowa była w temacie poświęconym berylowcom. Węglanylitowców (nie dotyczy litu i amonu) są dobrze rozpuszczalne, podobnie wodorowęglany litowców, trochę gorzejw przypadku berylowców. Dwutlenek węgla moŜna skroplić pod zwiększonym ciśnieniem oraz zestalić wtemperaturze –78,5oC – stosunkowo wysokiej w porównaniu z innymi naturalnymi gazami. Zestalony znany jestpod nazwą suchego lodu; sublimuje on, a wrzucony do wody unosi jej cząsteczki ponad powierzchnię, co dajeefekt „dymu”. Obecnie wykorzystuje się go w produkcji środków chłodzących. W małych ilościach dwutlenekwęgla nie wywiera szkodliwego działania na organizm ludzki, jednak stęŜenie jego nie moŜe być całkowiciedowolne. Za dopuszczalne stęŜenie, tzw. wskaźnik Pettenkofera, przyjmuje się 0,1%.
Created by Neevia Document Converter trial version http://www.neevia.com

50
Substancje o bardzo duŜym powinowactwie do tlenu mogą go zredukować, np.:Mg + CO2 ---> 2MgO + C.Dwutlenek węgla wykorzystywany jest w analizie jakościowej. Wpuszczony do roztworu zasady wapiennejpowoduje zmętnienie i wytrącenie białego osadu węglanu wapnia:Ca(OH)2 + CO2 ---> CaCO3 + H2O.Przy nieustannym dostarczaniu nowych porcji zaczyna się reakcja w wodą:H2O + CO2 <---> H2CO3.Powstający kwas węglowy reaguje z węglanem i przekształca go w bardziej rozpuszczalny wodorowęglan:2CaCO3 + H2CO3 <---> Ca(HCO3)2.Skutek jest taki, Ŝe powstały osad ulega rozpuszczeniu; taki cykl przemian pozwala jednoznacznie stwierdzićobecność w środowisku wymienionych powyŜej substancji.Dwutlenku węgla uŜywa się ponadto do produkcji sody metodą Solvaya, produkcji wód gazowanych,oczyszczania soku buraczanego w procesie saturacji, wiązania zanieczyszczeń zasadowych oraz do wypełnianiagaśnic.3) Dwa opisane powyŜej tlenki wymieniane są zawsze gdy mowa o związkach węgla. Ale oprócz nich istniejąrównieŜ inne tlenki. Podczas ogrzewania związku organicznego jakim jest kwas malonowy HOOC-CH2-COOHw obecności pięciotlenku fosforu otrzymuje się jego bezwodnik:HOOC-CH2-COOH<------>C3O2 + 2H2O.MoŜna mówić zatem o kolejnym tlenku węgla o wzorze C3O2. Zaproponować moŜna dla niego liniowy wzórcząsteczki, w którym wszystkie wiązania są podwójne: O=C=C=C=O. W przedstawionej reakcji związek fosforuze względu na duŜą higroskopijność pełni rolę osuszacza.Naukowo dowiedziono równieŜ istnienia kolejnych tlenków węgla o wzorach sumarycznych C6O9 oraz C12O9,przy czym dla tego ostatniego zaproponowano budowę cykliczną.4) W wyniku trzykrotnego chlorowania metanu otrzymuje się trichlorometan CHCl3, znany pod zwyczajowąnazwą chloroform. Wykorzystywany był do narkozy podczas zabiegów medycznych. W wyniku całkowitegochlorowania metanu otrzymuje się waŜnym technicznie związek - czterochlorek węgla CCl4. Związek ten jestcieczą wielokrotnie cięŜszą od wody. Choć trujący dla człowieka, znalazł zastosowanie dzięki skutecznemurozpuszczaniu tłuszczy.5) Węgiel wchodzi w skład reszt kwasowych kwasów: cyjanowodorowego, piorunowego oraz rodanowego.Kwas cyjanowodorowy HCN występuje w dwóch odmianach mezomerycznych, pomiędzy którymi ustala sięstan równowagi dynamicznej: HCN <=>HNC. Posiada charakterystyczny zapach gorzkich migdałów i jestbardzo silną trucizną: 50 mg czystego HCN powoduje śmierć w przeciągu kilku sekund. Jego sole, cyjanki, sąrównieŜ bardzo silnymi truciznami o krótkim czasie efektu śmiertelnego. Najgroźniejsze są sole rozpuszczalne wwodzie, a więc cyjanki litowców i berylowców, np. cyjanek potasu KCN. Jony cyjankowe biorą udział wtworzeniu związków kompleksowych rozpuszczalnych w wodzie, np.:AgCN + CN- ---> Ag(CN)2
-.(osad) (rozpuszczalny)W cyjankach rozpuszczają się metale szlachetne, takie jak srebro czy złoto, stąd w wielu rejonach światawykorzystuje się je do wypłukiwania tych metali ze zmielonych skał. śelazocyjanek potasu K4[Fe(CN)6] znanyjest jako błękit pruski i słuŜył niegdyś do barwienia tkanin na niebiesko. Jego hydrat K4[Fe(CN)6]
. 3H2O tworzyŜółtawe kryształy uŜywane do cementacji stali.Kwas piorunowy (fulminowy) HOCN jest nietrwały i istnieje tylko w roztworach wodnych. Jest aktywnychemicznie, dając reakcje przyłączania i polimeryzacji. Istotną rolę spełniają jego sole – pioruniany. Piorunianrtęci Hg(OCN)2 to biała substancja stała wybuchająca przy uderzeniu. Właściwości te stanowią o jejzastosowaniu w spłonkach jako detonator. Otrzymuje się ją jako produkt reakcji azotanu (V) rtęci z etanolem.Kwas rodanowy (tiocyjanowy) HSCN jest mocnym, nietrwałym kwasem beztlenowym. Jest bezbarwną ciecząmieszającą się z wodą. Otrzymuje się go przez gotowanie siarki w wodnym roztworze cyjanków. Zastosowaniemają jego sole, głównie w analizie chemicznej, gdyŜ jony rodankowe tworzą barwne kompleksy z niektórymikationami, np. z Cu2+ szafirowy a z Fe3+ krwistoczerwony.
KRZEMKrzem półmetalem występującym w dwóch odmianach alotropowych. Pierwsza - metaliczna ma budowękrystaliczną. Cechuje się wyŜszą od grafitu twardością i potrafi rysować szkło. Jest przewodnikiem, aprzewodnictwo to rośnie ze wzrostem temperatury; przypomnijmy, iŜ dla metali obowiązuje odwrotna zaleŜność– wraz ze wzrostem temperatury rośnie ich opór. Na zimno krzem jest bierny chemicznie, natomiast w wyŜszychtemperaturach łączy się z tlenem, fluorem i azotem (tworzy azotki). W łuku elektrycznym reaguje z wodoremtworząc wodorokrzemiany (silany). Na gorąco rozpuszcza się w wielu metalach, a z niektórymi z nich tworzykrzemki o teoretycznym charakterze kwasowym, np. krzemek magnezu Mg2Si. Niewielkimi ilościami krzemudomieszkuje się stale o podwyŜszonych właściwościach spręŜystych. Druga – bezpostaciowa odmiana posiadawiększą powierzchnię czynną, czemu zawdzięcza szybsze wchodzenie w reakcje chemiczne. Rozpuszcza się w
Created by Neevia Document Converter trial version http://www.neevia.com

51
bardzo mocnych zasadach, tworząc krzemiany z wydzieleniem wodoru:Si + 2OH- + H2O ---> SiO3
2- + 2H2.W przyrodzie krzem występuje ponadto w postaci związanej. Przykładem jest węglik krzemu SiC, znany jakokarborund; minerał ten jest bardzo twardy, dzięki czemu uŜywany jest jako materiał do produkcji wyrobówszlifierskich.1) Wśród związków krzemu waŜne miejsce zajmują jego połączenia z wodorem. Wspomniano juŜ wcześniej, iŜreagując z wodorem w łuku elektrycznym, krzem daje silany. MoŜna mówić tu o całym szereguhomologicznym, podobnie jak przy związkach organicznych: SiH4, Si2H6, Si3H8, Si4H10 itd. Przedstawionezwiązki są gazami o zróŜnicowanej gęstości. Są bardzo nietrwałe, zapalają się na powietrzu i ulegają działaniuzasad:SiH4 + 2OH- + H2O ----> SiO3
2- +4H2.Mogą polimeryzować dając łatwopalne polisilany (SiH2)n. Gdy atom wodoru jest w nich zastępowany przezgrupy alkilowe, wówczas mówimy o alkilosilanach.2) Krzem tworzy związki równieŜ z fluorowcami. NajwaŜniejszym w tej grupie jest fluorek krzemu SiF4,pozostałe nie odgrywają istotnej roli. Fluorek krzemu powstaje podczas trawienia szkła z wykorzystaniem kwasufluorowodorowego:SiO2 + 4HF <==> 2H2O + SiF4.Z powodu trujących właściwości tego ostatniego coraz częściej proces ten przeprowadza się za pomocą reakcjialternatywnej:SiO2 + 2CaF2 + 2H2SO4 ---> 2CaSO4 + 2H2O + SiF4.Fluorek krzemu reaguje równieŜ z samym kwasem fluorowodorowym:2 SiF4 + 4HF ---> 2H2[SiF6].Produkt ostatniej reakcji to kwas fluorokrzemowy; posiada relatywnie dosyć silne właściwości kwasowe idobrze rozpuszcza się w wodzie.3) Bardzo powszechną formą występowania krzemu w przyrodzie jest krzemionka, czyli tlenek krzemu SiO2.Jest to krystaliczne ciało stałe o bardzo uporządkowanej strukturze, w której występują wyłącznie wiązaniapojedyncze, a kaŜdy atom tlenu związany jest z dwoma atomami krzemu. Konstrukcja oparta jest naczworościanach o duŜym stopniu upakowania, co gwarantuje duŜą gęstość i twardość. Największe ilościkrzemionki znajdują się w piasku. W przyrodzie występuje w postaci amorficznej, skrytokrystalicznej, azabarwiony przez zanieczyszczenia zyskuje barwę (np. kamienie – opal, agat, krwawnik). Czysto krystalicznaodmiana nosi nazwę kwarcu, a dokładnie kryształu górskiego. Ze względu na czynność optyczną wyróŜniamykwarc prawo- oraz lewoskrętny. Zabarwiony tlenkami metali daje kamienie półszlachetne: fioletowy ametyst,czarny morion czy Ŝółty cytryn. Kwarc jest kryształem dwójłomnym, co oznacza iŜ posiada zdolnośćpodwójnego załamania światła spolaryzowanego. Na tym zjawisku oparta jest budowa nikoli, czyli przyrządymierzące zawartość cukrów w ich roztworach. Stopiona krzemionka zastyga na bezpostaciowe szkło kwarcowe,przepuszczające promieniowaniu ultrafioletowe. Lampy kwarcowe wykorzystywane są zarówno w szpitalach docelów dezynfekcyjnych, jak i w solariach do opalania. Bezpostaciowa odmiana krzemionki to tzw. ziemiaokrzemkowa. Jest to materiał wybitnie porowaty o duŜych zdolnościach sorpcyjnych, uŜywany do produkcjidynamitu przez wysycenie go nitrogliceryną. Ze względu na swe właściwości wchłania ona trzykrotnie więcejmateriału wybuchowego niŜ sama waŜy.Krzemionki uŜywa się do produkcji szkła. Pomimo odporności na działanie wielu chemikaliów wchodzi wreakcję podczas stapiania z wodorotlenkiem sodu:SiO2 + 2NaOH ---> Na2SiO3 + H2O;w rzeczywistości uŜywa się do tego celu sody:Na2CO3 + SiO2 ---> Na2SiO3 + CO2.Oczywiście moŜna stosować odpowiednie sole potasu. W wyniku powyŜszych reakcji otrzymuje sięrozpuszczalne w wodzie krzemiany, tzw. szkło wodne. Następnie w celu stabilizacji przeprowadza się podobnyszereg reakcji dla węglanu wapnia. Powstały metakrzemian wapnia nie jest juŜ rozpuszczalny w wodzie, przezco zwiększa odporność i wytrzymałość nowo powstałego szkła. Gotowy wyrób jest zatem stopioną mieszaninąkrzemianu sodu (ew. potasu), krzemianu wapnia i krzemionki. W zaleŜności od uŜytych składników cechują gookreślone właściwości. I tak: szkło potasowo – wapniowe charakteryzuje się duŜym połyskiem iwspółczynnikiem załamania światła, przez co stosowane jest do wyrobu soczewek. z kolei szkło sodowe jesttańsze, bardziej wraŜliwe na zmiany temperatury i słabiej załamuje światło. Szkło dekoracyjne z dodatkiemołowiu posiada piękny połysk; wykonanych z niego „kryształów” nie moŜna jednak uŜywać do podawaniaproduktów spoŜywczych, gdyŜ łatwo pękają pod wpływem ciepła. Typowe szkło ołowiowe stosowane jest woptyce do wyrobu soczewek okularowych i mikroskopowych. W naczyniach szklanych nie wolnoprzechowywać wodorotlenku sodu, który powoduje nadŜerki i przecieki; z tego powodu pastylki wodorotlenkutrzyma się w pojemnikach z tworzyw sztucznych.Krzemian sodu moŜe reagować z kwasami, np. z kwasem solnym:Na2SiO3 + 2HCl ---> H2SiO3 + 2NaCl.
Created by Neevia Document Converter trial version http://www.neevia.com

52
Powstały kwas metakrzemowy posiada właściwości kwasowe słabsze od (bardzo słabego przecieŜ) kwasuwęglowego, ponadto słabo rozpuszcza się w wodzie i strąca się w postaci białego galaretowatego osadu.Wytwarza się faza koloidowa SiO2
. H2O. Dodatek wody powoduje przejście kwasu metakrzemowego w kwasortokrzemowy:H2SiO3 + H2O ---> H2SiO4,który równieŜ jest kwasem bardzo słabym i źle rozpuszczalnym w wodzie, strącając się w postacigalaretowatego osadu.
CYNACyna jest metalem występującym na dwóch stopniach utlenienia: +2 i +4, w związku z czym tworzy dwa tlenki:zasadowy SnO i amfoteryczny SnO2. Pierwszy, dąŜąc do stopnia utlenienia +4 wykazuje właściwościredukcyjne. Drugi jako amfoter ulega reakcji z mocnymi zasadami:SnO2 + 2NaOH ---> Na2SnO3 + H2O.W roztworze powstaje oczywiście forma bogatsza w cząsteczki wody, sześciohydroksocynian soduNa2[Sn(OH)6], który następnie przechodzi w powyŜszy cynian sodu.Cyna jest odporna na czynniki atmosferyczne, nie ulega działaniu słabych kwasów ani słabych zasad. UŜywanajest do lutowania, jak równieŜ do produkcji blachy białej, z której powstają puszki do konserw. Jest plastyczna imoŜe być rozwałkowywania na cienkie paski. Powstała folia cynowa słuŜy do opakowania wyrobówcukierniczych. Prawdziwość tego moŜna łatwo zbadać, gdyŜ dobrej jakości cyna dzięki ziarnistej budowiechrzęści w charakterystyczny sposób, zaś folia aluminiowa nie daje takiego efektu. Podobnie sprawdzić moŜnaczystość cienkich profili cynowych, gdyŜ zanieczyszczenia równieŜ pozbawiają cynę efektu szelestu.
OŁÓWMetal ten występuje w przyrodzie w postaci związanej jako róŜnorodne minerały, np.: PbO – glejta, PbS –galena, PbCO3 – cerusyt, PbSO4 – anglezyt, Pb3(PO4)2 – piromorfit. Jest to metal cięŜki, pozostawiony napowietrzu pokrywa się warstwą zasadowego węglanu ołowiu PbO . Pb(OH)2
. PbCO3, co zabezpiecza go przedkorozją. W obecności stęŜonych kwasów utleniających (siarkowego, azotowego) ulega pasywacji, tworzącnierozpuszczalne sole, np. PbSO4. Jest giętki, ciągliwy i kowalny; posiada niską temperaturę topnienia, dziękiczemu wykorzystuje się go do wykonywania odlewów. Dla organizmów Ŝywych jest silnie trujący, kumulującsię głównie w mózgu. Dzięki duŜej liczbie masowej uŜywany jest do zatrzymywania promieniowania –przykładowo przed działaniem promieniowania gamma chroni dopiero gruba warstwa ołowiu. Przypomnijmytylko, iŜ zanik natęŜenia promieniowania w warstwie absorbującej ma charakter wykładniczy: I=I0e
-kx, gdzie x togrubość płytki, a k – współczynnik proporcjonalności. Tworzy bardzo trwałe związki na +2 stopniu utlenienia,natomiast te na +4 wykazują pewien charakter amfoteryczny. Bardzo łatwo łączy się z siarką, co znalazłozastosowanie przy identyfikacji białek (choć siarka występuje w trzech tylko aminokwasach – cystynie, cysteiniei metioninie – to zawiera ją praktycznie kaŜde naturalne białko). Podczas ogrzewania białka z octanem ołowiu(CH3COO)2Pb wydziela się powoli czarny osad siarczku ołowiu PbS, co jest charakterystyczne dla produktówzawierających siarkę.
AZOTOWCE
Azotowce to piąta grupa główna w układzie okresowym – V A lub 15. Posiadają konfigurację elektronowąns2np3
- zatem pięć elektronów walencyjnych rozmieszczonych na podpoziomach s i p. W grupie tej występujązarówno niemetale (azot, fosfor), półmetale (arsen, antymon) jak i metale (bizmut). Azotowce niemetalicznewystępują na wyŜszych stopniach utlenienia, zaś metaliczne na niŜszych.
AZOTPierwiastkiem rozpoczynającym grupę jest azot. Jest to gaz stanowiący główny składnik (78%) powietrza i niecood niego lŜejszy. Spotykamy z się z nim najczęściej w postaci związanej, np. saletr: NaNO3 – saletra chilijska,KNO3 – saletra indyjska, Ca(NO3)2 – saletra norweska, NH4NO3 – saletra amonowa (dawniej pospolity środeksłuŜący do konserwacji mięsa). Wchodzi w skład aminokwasów, a przez to równieŜ peptydów i białek. Na skalęprzemysłową otrzymywany jest przez frakcjonowaną destylację powietrza w temperaturze –210oC. Wlaboratorium otrzymać go moŜna przez rozkład termiczny wybuchowego azotanu (III) amonu:NH4NO3 ---T---> N2 + 2H2O.Ciekły azot jest powszechnie wykorzystywany do szeroko rozumianego chłodzenia: od transplantowanychorganów medycznych do procesów przemysłowych. W cząsteczce azotu występują trzy wiązania kowalencyjne,co nadaje jej ogromną trwałość i energię jonizacji (947 kJ/mol). Z tego powodu temperaturze pokojowej jestbierny, zaś w temperaturach wyŜszych tworzy z wieloma metalami (Ca, Mg, Si) związki zwane azotkami. Łączysię z wodorem dając amoniak oraz w wysokiej temperaturze z tlenem dając tlenki. Zapach świeŜościwystępujący w powietrzu po burzy to efekt powstawania tlenków azotu w łuku elektrycznym piorunu.
Created by Neevia Document Converter trial version http://www.neevia.com

53
1) Amoniak NH3 powstaje w wyniku reakcji azotu z wodorem:N2 + 3H2 ---(war)--> 2NH3.Aby taka reakcja zaszła, wymagane są odpowiednie warunki: ze względu na zmniejszanie objętości stosuje sięwysokie ciśnienie, ze względu na endotermiczność – wysoką temperaturę (reguła przekory). Oprócz tego dodajesię odpowiednich katalizatorów: najbardziej optymalnymi są osm, iryd bądź platyna, moŜna równieŜ stosowaćtańszą, ale zdecydowanie mniej wydajną mieszaninę Ŝelaza, tlenku glinu i wodorotlenku potasu.Amoniak jest lŜejszym od powietrza gazem o intensywnym zapachu. Łatwo się skrapla, a dzięki pochłanianiuznacznych ilości ciepła znalazł zastosowanie w chłodziarkach. W stanie skroplonym przewodzi prąd (ale bardzosłabo) wskutek autodysocjacji na kation amonowy i anion amidowy:2NH3 ---> NH4+ + NH2-.MoŜe rozpuszczać metale i z niektórymi z nich tworzyć amidki, np.: amidek sodu NaNH2, wapnia Ca(NH2)2 czyglinu Al(NH2)3. W powietrzu spala się z wydzieleniem azotu:4NH3 + 3O2 ---> 2N2 + 6H2O.Spalany w tlenie wobec katalizatora daje mieszaninę tlenków azotu:4NH3 + 5O2 ---> 6H2O + 4NO (reakcja główna),4NH3 + 7O2 ---> 6H2O + 4NO2 (reakcja uboczna).W obrębie cząsteczki występują wiązania kowalencyjne. Azot jest w stanie hybrydyzacji tetragonalnej sp3,dzięki czemu cząsteczka ma budowę zgiętą. W trzech naroŜach czworościanu foremnego znajdują się jądrawodoru, zaś w czwartym – wolna para elektronowa. Ta cecha budowy dalece wpływa na właściwości amoniaku;są to przede wszystkim: słaby charakter zasadowy oraz zdolność do tworzenia związków kompleksowych.Omówimy teraz kolejno obie te właściwości.W wodzie amoniak łączy się z kationami wodorowymi dzięki przekazaniu im swej wolnej pary:NH3 + H+ ---> NH4+,co jako reakcja z wodą przedstawia się następująco:NH3 + H2O <==> NH4OH <==> NH4+ + OH-.Powstaje więc związek o charakterze zasadowym – zasada amonowa. Jej właściwości zasadowe są słabe, alebardzo łatwo reaguje z wieloma kwasami. Wszystkie sole amonowe przy ogrzewaniu bądź działaniumocniejszymi zasadami wydzielają wonny amoniak, co słuŜy identyfikacji tych soli. Przykładowo jeśli wnachylonej do poziomu rurce ogrzewać będziemy biały proszek salmiaku NH4Cl, a na obu końcach umieścimyzwilŜone papierki lakmusowe, to dolny zabarwi się na czerwono, a górny na niebiesko. Mechanizmdoświadczenia jest następujący: w wyniku rozkładu termicznego soli amonowej wydzielają się lotne gazy –amoniak oraz chlorowodór:NH4Cl ---T---> NH3 + HCl.Amoniak jako lŜejszy od powietrza unosi się w górę, gdzie rozpuszcza się w wodzie i daje anionywodorotlenowe barwiące papierek na niebiesko:NH3 + H2O <==> NH4OH <==> NH4+ + OH-.Z kolei chlorowodór jako cięŜszy od powietrza spływa w dół rurki, rozpuszcza się w wodzie i daje kationywodorowe, barwiące papierek na czerwono.HCl + H2O ---> H3O
+ + Cl-.Amoniak tworzy wiele związków kompleksowych z nierozpuszczalnymi bądź trudno rozpuszczalnymi solami(np. halogenkami), w które to połączenia wchodzi z liczbą koordynacyjną 2, np.:AgCl + 2NH3 ---> [Ag(NH3)2]Cl,AgBr + 2NH3 ---> [Ag(NH3)2]Br.Istotne jest, iŜ powstałe związki kompleksowe są juŜ rozpuszczalne. Natomiast nie jest w stanie rozpuścić wpodobny sposób jodku srebra AgI, którego wiązanie jest zbyt silne. Z jonami pierwiastków bloku d (miedź,mangan, Ŝelazo, nikiel, kobalt) tworzy kationy kompleksowe o charakterystycznych barwach: Cu2+ - niebieski,Fe2+ - zółty, Fe3+ - brunatny, Ni2+ - Ŝółty, Co2+ - niebieski. MoŜna to wytłumaczyć dwustopniowo: pierwiastkibloku d posiadające niesparowane elektrony d powodują rozpraszanie się na nich światła, zaś dodatek amoniakuwprowadzającego wolne pary elektronowe znacznie ten efekt pogłębia. Przykładowo jon miedzi Cu2+ otacza sięw wodzie czterema cząsteczkami wody (na kaŜdej występują dwie pary elektronowe), tworząc kationtetraakwamiedzi (II), zaś po dodaniu amoniaku otacza się czterema jego cząsteczkami, dając kationtetraamionomiedzi.Amoniak słuŜy do produkcji mocznika:COCl2 + 2NH3 ----> NH2CONH2 + 2HCloraz kwasu azotowego i nawozów sztucznych:
Created by Neevia Document Converter trial version http://www.neevia.com

54
Pochodnymi amoniaku są: hydrazyna NH2NH2 oraz hydroksyloamina NH2OH, o których zastosowaniachpowiemy później.2) WaŜne znaczenie mają tlenki azotu. Tlenkiem o najniŜszej wartościowości jest podtlenek azotu N2O.Otrzymać go moŜna przez termiczny rozkład azotanu (V) amonu:NH4NO3 ---T---> N2O + 2H2O.W temperaturze pokojowej jest to bezbarwny gaz o przyjemnym, słodkawym zapachu. Wdychany działa naorganizm ludzki oszałamiająco, dlatego stosowano go jako gaz rozweselający, np. do krótkotrwałegoznieczulenia przy zabiegach dentystycznych. Tlenek azotu (I) posiada parzystą liczbę elektronów, z którychwszystkie są sparowane, co przyczynia się zapewne do stosunkowo duŜej bierności chemicznej w zwykłejtemperaturze.3) Tlenek azotu (II) powstaje podczas reakcji tlenu z azotem przy dostarczeniu duŜych ilości energii. MoŜe to sięodbywać na sposób katalityczny (termiczny):N2 + O2 ---1500oC---> 2NO,bądź w łuku elektrycznym, np. podczas wyładowań atmosferycznych lub w specjalnym reaktorze (metodaMościckiego). Tlenek azotu wydziela się równieŜ podczas reakcji niezbyt stęŜonego (1-15%) kwasu azotowegoz metalami szlachetnymi, np.:Cu + 8HNO3 ---> 3Cu(NO3)2 + 2NO + H2O.W warunkach normalnych jest to bezbarwny gaz nieco cięŜszy od powietrza, bardzo słabo rozpuszczalny wwodzie. Pierwiastki o duŜym powinowactwie do tlenu (jak magnez czy glin) po zakończeniu reakcji spalania wpowietrzu mogą palić się dalej w atmosferze tlenku azotu. Posiada on nieparzystą liczbę elektronów (11),dlatego wykazuje właściwości paramagnetyczne i charakter rodnikowy. W dąŜeniu do uzyskania sparowanychelektronów reaguje egzotermicznie z powietrzem:2NO + O2 ---> 2NO2.Efektem ostatniej reakcji jest czerwonobrunatny, cięŜszy od powietrza dwutlenek azotu. Omawiany tlenektworzy połączenia kompleksowe z fluorowcami – nitrozyle, np. chlorek nitrozylu NOCl czy bromek nitrozyluNOBr. Dla analizy jakościowej istotna jest reakcja tworzenia kompleksu z jonami Ŝelaza Fe2+:FeSO4 + NO ---> Fe(NO)SO4.Powstały siarczan nitrozyloŜelaza (II) tworzy w środowisku reakcji ciemnobrunatną obrączkę.4) Tlenek azotu (III) N2O3 jest cieczą o barwie niebieskawej. Trwałość zapewnia temperatura poniŜej –10oC, wwyŜszych bowiem ulega dysproporcjonowaniu na tlenki azotu (II) i (IV). Jest bezwodnikiem kwasuazotowego (III):N2O3 + H2O ---> 2HNO2,który równieŜ jest związkiem nietrwałym i dysproporcjonuje:3HNO2 ---> HNO3 + 2NO + H2O.Nieco bardziej trwałe są przynajmniej jego sole – azotany (III). Ze względu na pośredni stopień utlenienia moŜepełnić zarówno funkcję utleniacza, jak i reduktora.5) Tlenek azotu (IV) NO2 jest czerwonobrunatnym, cięŜszym od powietrza i silnie trującym gazem. Powstajeprzez utlenianie tlenku azotu (II). Ze względu na nieparzystą liczbę elektronów walencyjnych samorzutnie iegzotermicznie dimeryzuje:2NO2 <==> N2O4.Powstały dimer jest produktem bezbarwnym. Przesunięcie równowagi reakcji w którąś stronę jest odpowiedziąna zmianę temperatury – w niŜszych występuje raczej N2O4, zaś w wyŜszych NO2. Dwutlenek azotu łatworównieŜ ulega dysproporcjonowaniu:2NO + H2O ---> HNO2 + HNO3.Powstaje w reakcjach kwasu azotowego (V) z metalami o dodatnich potencjałach normalnych, np.:Cu + 4HNO3 ---> Cu(NO3)2 + 2NO2 + 2H2O.Jest silnym utleniaczem; wiele pierwiastków o duŜym powinowactwie do tlenu świetnie pali się w jegoatmosferze.6) Pięciotlenek azotu N2O5 jest ciałem stałym. Bezwodnik kwasu azotowego (V), jest związkiem nietrwałym ijuŜ w temperaturze pokojowej rozkłada się z wydzieleniem tlenu:2N2O5 ---> 2N2O4 + O2,przez co staje się niebezpieczny w przechowywaniu.7) Kwas azotowy (V) HNO3 jest bardzo waŜnym produktem przemysłowym. Na skalę przemysłową otrzymujesię go poprzez utlenianie amoniaku. Miesza się z wodą w kaŜdym stosunku. Jest związkiem nietrwałym, aprzechowywany na świetle brunatnieje wskutek wydzielania się tlenku azotu (IV):2HNO3 ---> 2NO2 + ½O2 + H2O.Bardzo rozcieńczony (<1%) kwas azotowy zachowuje się jak klasyczny kwas: przewodzi prąd elektryczny,reaguje tylko z metalami o ujemnych potencjałach standardowych i wydziela w takich reakcjach wodór. StęŜonynatomiast nie dysocjuje, dlatego nie przewodzi prądu i zachowuje się jak typowy środek utleniający. Jest bardzosilnym utleniaczem, utlenia niemetale w stanie czystym (fosfor, siarkę, węgiel) do ich tlenków i kwasów z
Created by Neevia Document Converter trial version http://www.neevia.com

55
wydzieleniem tlenku azotu(II):3P + 5HNO3 -> 3H3PO4 + 5NO,3S + 4HNO3 -> 3SO2 + 4NO + 2H2O,3C + 4HNO3 -> 3CO2 + 4NO + 2H2O.Działaniu stęŜonego kwasu azotowego ulegają metale wybitnie szlachetne, np. iryd, platyna czy złoto. Procesy tesą jednak bardziej złoŜone. Do reakcji z metalami wybitnie szlachetnymi zdolna jest wyłącznie mieszaninastęŜonych kwasów – solnego oraz azotowego (V) – w stosunku objętościowym 3:1 (molowym 4:1). Reakcja taprzebieg dwuetapowy: wpierw kwas solny reaguje z kwasem azotowym, wytwarzając chlorek nitrozylu, którynastępnie atakuje metal szlachetny. Dla złota produktem reakcji jest kwas czterochlorozłotowy, co moŜnazapisać następująco:HNO3 + 3HCl -> NOCl + Cl2 + 2H2O,Au + HCl + NOCl +Cl2 -> HAuCl4 + NO.Metale o niŜszych potencjałach, będące silnymi reduktorami mogą go zredukować nawet o 8 stopni utlenienia:4Zn + 10HNO3 -> 4Zn(NO3)2 + NH4NO3 + 4H2O,4Mg + 10HNO3 -> 4Mg(NO3)2 + N2O + 5H2O.Niektóre metale (Ŝelazo, chrom, glin) pokrywaj się w jego obecności pasywną warstwą tlenku; jest to czasamizjawisko poŜądane – przykładowo moŜna dzięki temu przewozić stęŜony kwas azotowy w stalowych cysternach.Kwas azotowy reaguje z dymiącym kwasem siarkowym (oleum, roztwór SO3 w H2SO4), odszczepiając kationnitroniowy:2H2SO4 + HNO3 ---> 2HSO4
- + NO+ + H3O+,
który bardzo agresywnie atakuje inne związki, np. benzen czy toluen. Z tego powodu do nitrowania(wprowadzania grupy nitrowej do) związków chemicznych nie stosuje się czystego kwasu azotowego, lecz tzw.mieszaninę nitrującą, złoŜonej z HNO3 i H2SO4. W reakcjach tych powstają Ŝółte nitrozwiązki, często cechującesię właściwościami wybuchowymi, np. sym-trinitrotoluen (TNT) znany powszechnie pod nazwą trotylu.Obecność kwasu azotowego wykrywa się na podstawie reakcji z siarczanem (VI) Ŝelaza (II) FeSO4, w której towytwarza się wspomniana juŜ wcześniej ciemnobrunatna obrączka siarczanu (VI) nitrozyloŜelaza (II).Jego sole – azotany(V) – są nośnikami duŜych ilości tlenu, dlatego znalazły zastosowanie jako składnikimateriałów wybuchowych. Zmieszane z substancjami łatwopalnymi ulegają samozapłonowi; przykładowoazotanu amonu nie naleŜy mieszać z węglem drzewnym, trocinami, papierem ani nie rozbijać metalowymiprzedmiotami.8) Rzadko wymieniany jest związek zwany kwasem azotowodorowym HN3 (H-N=N=>N|). Istotne znaczeniemają jedynie jego sole, azydki. Są bardzo nietrwałe i pobudzenie mechaniczne wyzwala w nich wybuchowereakcje rozkładu. Z tego powodu wiele azydków stanowi główny składnik detonatorów, np.: azydek ołowiuPb(N3)2, azydek rtęci Hg2(N3)2 czy azydek srebra AgN3.
FOSFORNastępnym w grupie azotowców pierwiastkiem jest fosfor. W przyrodzie występuje on w postaci związanej -pięciotlenku fosforu P2O5, fosforanów (np. jednym z głównych składników kości jest fosforan wapnia) bądź wpostaci czystej – jednej z kilku odmian alotropowych.a) Fosfor biały jest bezbarwną substancją stałą, bardzo miękką i plastyczną. Nie rozpuszcza się w wodzie ani walkoholach, za to rozpuszcza się w PCl3, SCl2 i CS2. Posiada niską temperaturę topnienia (44oC) i wrzenia(280oC). Występuje w postaci tetraedrycznych cząsteczek P4. Jest bardzo aktywny chemicznie: w atmosferzeczystego chloru samorzutnie się zapala, łatwo reaguje z bromem i siarką. Rozdrobniony lub podgrzany do 60oCzapala się na powietrzu:4P + 5O2 ---> P4O10 <==> 2P2O5.Naświetlony świeci w ciemności, stąd wyrabia się z niego elementy świecące w zegarkach nocnych. Jest bardzosilną trucizną, dawka śmiertelna czystego fosforu białego wynosi 0,1 g. Z powodu zagroŜenia samozapłonem iwybuchem przechowywany jest pod wodą.b) Fosfor czerwony moŜna otrzymać z odmiany białej przez powolne ogrzewanie bez dostępu powietrza. WpowyŜszych warunkach tetraedry polimeryzują, wytwarzając długą, liniową cząsteczkę Pn (n>>1). Z róŜnicy wstrukturze wynikają róŜnice we właściwościach: forma czerwona jest trwalsza, nietrująca, mało aktywnachemicznie, nierozpuszczalna w chlorkach i siarczkach, posiada wyŜsze temperatury zmian fazy, zapala się wtemperaturze powyŜej 240oC. Wykorzystywany jest do produkcji zapałek: główka i draska wykonane są zmieszaniny fosforu czerwonego i chloranu, będącego nośnikiem tlenu.c) Pozostałe odmiany alotropowe – fosfor czarny i fioletowy – nie są szczególnie wykorzystywane.1) W połączeniu z wodorem fosfor tworzy fosforowodór PH3. W przeciwieństwie do podobnego w budowieamoniaku nie posiada on właściwości zasadowych. Jest związkiem silnie trującym, o przykrym zapachuzgniłych ryb.2) Fosfor tworzy kilka tlenków: P2O3, P2O5 oraz nietrwały P2O4. Dwa pierwsze z nich są bezwodnikamikwasowymi. Pięciotlenek fosforu P2O5 jest białą substancją stałą o konsystencji sypkiej; łatwo sublimuje i jest
Created by Neevia Document Converter trial version http://www.neevia.com

56
bardzo higroskopijny, dzięki czemu moŜe być wykorzystywany jako środek osuszający substancji o charakterzeobojętnym lub kwasowy.3) Fosfor tworzy dwa szeregi kwasów – w jednym występuje na stopniu utlenienia +3 , a w drugim na +5.Powstanie konkretnego kwasu uzaleŜnione jest od ilości dostępnej w środowisku reakcji wody:P2O3 + H2O ---> 2HPO2 (kwas metafosforowy (III)),P2O3 + 2H2O ---> H4P2O5 (kwas pirofosforowy (III) = difosforowy (III)),P2O3 + 3H2O ---> 2H3PO3 (kwas ortofosforowy (III)),P2O5 + H2O ---> 2HPO3 (kwas metafosforowy (V)),P2O5 + 2H2O ---> H4P2O7 (kwas pirofosforowy (V) = difosforowy (V)),P2O5 + 3H2O ---> 2H3PO4 (kwas ortofosforowy (V)).Interesującym z pewnego względu jest kwasortofosforowy (III). Jako kwas trzyprotonowy powinientworzyć on trzy rodzaje soli, a tworzy tylko dwie.MoŜna stwierdzić, iŜ dysocjuje on dwustopniowo, aostateczny anion ma postać HPO3
2- (patrz rysunek).Dzieje się tak, poniewaŜ jeden z atomów wodorupołączony jest bezpośrednio z atomem fosforu i nie ma przez to moŜliwości dysocjacji.Kwas ortofosforowy (V) tworzy trzy szeregi soli: sole obojętne, wodorosole oraz diwodorosole. Te dwa ostatnierozpuszczają się w wodzie lepiej niŜ sole obojętne. Z tego powodu nawozy fosforowe mają postać diwodorosoli,np. superfosforat Ca(H2PO4)2. Wśród soli obojętnych rozpuszczalne są ortofosforany litowców ( z wyjątkiemlitu) oraz amonu, pozostałe są nierozpuszczalne. Fosforany i polifosforany stosowane były jako składnikiśrodków piorących i zmiękczających. Zostały jednak wycofane ze względu na skutki, jakie wywoływały ściekifosforowe, m. in. nadmierny rozwój roślin, obrastanie zbiorników wodnych i kadłubów statków oraz deficyttlenowy i śmierć zwierząt wodnych.4) Połączenia fosforu z fluorowcami, np. PCl5, stosowane są w przemyśle chemicznym do chlorowaniaaldehydów.
ARSENArsen jest półmetalem występującym równieŜ w odmianach alotropowych: arsen metaliczny As4 oraz arsenŜółty. Jest to pierwiastek o aktywności mniejszej od fosforu. Wszystkie jego związki są silnie trujące.1) Arsenowodór AsH3 posiada zapach zbliŜony do czosnku. Jest silną trucizną. Podczas ogrzewania rozkłada sięna pierwiastki, co w kryminalistyce funkcjonuje jako próba lustra arsenowego (wykrywanie otrucia związkamiarsenu juŜ w niewielkich stęŜeniach):2AsH3 ---T---> 2As + 3H2. 2) Tlenek arsenu (III) As2O3, znany jako arszenik, jest równieŜ silną trucizną. Bardzo źle rozpuszcza się wwodzie, z którą tworzy kwas arsenowy (III):As2O3 + 3H2O ---> 2H3AsO3.Powstały produkt jest bardzo słabym kwasem, wykazującym pewien charakter amfoteryczny.
ANTYMONAntymon jest pierwiastkiem półmetalicznym o właściwościach zbliŜonych do arsenu. Występuje w odmianachalotropowych. Nie ulega działaniu nawet stęŜonych kwasów, jedynie na gorąco roztwarza się w wodziekrólewskiej – jest zatem metalem szlachetnym. Antymonowodór SbH3 posiada właściwości trująceporównywalne z arsenowodorem. RównieŜ podobnie jak on rozkłada się w wyŜszych temperaturach napierwiastki, dając lustro antymonowe (próba na antymon juŜ w małych stęŜeniach).
BIZMUTBizmut jest jedynym w grupie azotowców pierwiastkiem o właściwościach metalicznych. Wprowadzony dostopu znacznie zmniejsza jego temperaturę topnienia.
TLENOWCE
Tlenowce to szósta grupa główna w układzie okresowym – VI A lub 16. Posiadają konfigurację elektronowąns2np4
- zatem sześć elektronów walencyjnych rozmieszczonych na podpoziomach s i p. W stanie podstawowymatom tlenowca posiada dwie pary elektronowe – jedną s i jedną p – oraz dwa niesparowane elektrony walencyjnep, które mogą brać udział w wiązaniach. Ogranicza to wartościowość do II – taka sytuacja występuje wprzypadku tlenu. JeŜeli tlenowiec dysponuje wolnym orbitalem walencyjnym d (okres trzeci i poniŜsze), to wwyniku wzbudzenia mogą nań przemieszczać się elektrony z par s i p. Rozszerza to zakres wartościowości o IV(wzbudzenie pojedyncze) i VI (wzbudzenie podwójne). Oznacza to równieŜ, iŜ mogą pojawić się rzadko
Created by Neevia Document Converter trial version http://www.neevia.com

57
wymieniane typy hybrydyzacji, np. sp3d2 w sześciofluorku siarki SF6. W grupie tej występują zarówno niemetale(tlen, siarka, selen), półmetale (tellur) jak i metale (polon).
TLENPierwiastkiem rozpoczynającym grupę jest tlen. W warunkach normalnych jest to gaz o gęstości 1,43 g/cm3, azatem nieco cięŜszy od powietrza. Stanowi około jedną piątą (21%) składu powietrza. Występuje w dwóchodmianach alotropowych: zwykły tlen (O2) oraz ozon (O3), którego największe ilości występują w górnejwarstwie atmosfery zwanej ozonosferą. Występuje pomiędzy nimi stan równowagi:2O3 <==> 3O2.Jest pierwiastkiem bardzo aktywnym (przy czym ozon jest aktywniejszy), łączy się praktycznie z wszystkimipierwiastkami i daje tlenki. Występuje na –2 stopniu utlenienia, wyjątkowo w nadtlenkach na –1 ze względu naistnienie ugrupowania -O-O- oraz w tlenku fluoru OF2 na +2 ze względu na wyŜszą elektroujemność fluoru. Wprzeciwieństwie do pozostałych tlenowców nie przyjmuje wyŜszych stopni utlenienia, co zostało wyjaśnionewcześniej.Tlen występuje w postaci trzech izotopów: 16O, 17O oraz 18O. Ostatni izotop jest promieniotwórczy, dlategowykorzystywany jest do badania mechanizmów reakcji metodą tzw. tlenu znaczonego. Przykłady zastosowaniatej metody podane będą później, np. przy reakcjach estryfikacji.
SIARKASiarka jest Ŝółtym ciałem stałym występującym w kilku odmianach alotropowych. W kaŜdej z nich występującząsteczki siarki S8, róŜniące się jedynie konfiguracją przestrzenną, a ściślej typem sieci krystalicznej.a) Odmiana alfa (rombowa) jest trwała w temperaturze do 95,6oC. Ma barwę Ŝółtą, nie rozpuszcza się w wodzie,bardzo słabo zaś w substancjach organicznych. Ogrzana powyŜej punktu trwałości topi się i przechodzi w formębeta.b) Odmiana beta (jednoskośna) w zwykłej temperaturze jest nieaktywna chemicznie, ulega działaniu fluorowcówz wyjątkiem jodu. W wysokich temperaturach łączy się niemal z wszystkimi pierwiastkami, z wyjątkiem azotu,złota, platynowców i helowców. W powietrzu spala się do dwutlenku siarki SO2. Jako pierwiastek elektroujemny(X=2,5) łatwo reaguje z metalami tworząc siarczki; jest to przyczyną czernienia wyrobów z miedzi i srebra.PowyŜsze reakcje są egzotermiczne i wymagają jedynie inicjacji. Z wodorem siarka łączy się tylko w wysokichtemperaturach, ze względu na małą róŜnicę elektroujemności. Siarka ma wiele zastosowań. W lecznictwiewykorzystuje się ją w postaci maści siarkowej do leczenia chorób skóry, gdyŜ niszczy drobnoustroje trądzikowe.UŜywana jest do konserwacji tanich wyrobów alkoholowych (np. win, na których etykiecie widniejedopuszczalna zawartość dwutlenku siarki), równieŜ soków i przecierów owocowych (jagodzianki po przetarciupoddaje się działaniu dwutlenku siarki). Przy wulkanizacji w wysokiej temperaturze dodana siarka tworzymostki siarczkowe pomiędzy łańcuchami polimerów, np. kauczuku czy gumy, przez co zmniejsza płynność tychwyrobów. Siaka wykorzystywana jest ponadto do produkcji barwników, prochu strzelniczego oraz w przemyślechemicznym do produkcji kwasy siarkowego (VI).1) Jak powiedziano, w wyŜszych temperaturach siarka łączy się z wodorem, dając siarkowodór H2S. Jestbezbarwny gaz o bardzo przykrej woni zgniłych jaj, silnie trujący o działaniu duszącym z powodu redukcjioksyhemoglobiny. W warunkach laboratoryjnych otrzymać go moŜna działając na siarczek metalu kwasemsolnym, np.:Na2S + 2HCl ---> H2S + NaCl.Siarkowodór jest wraŜliwy na wysokie temperatury i rozkłada się wówczas na pierwiastki:H2S ---T---> H2 + S.Jest gazem palnym, przy czym produkt zaleŜy od dostępności tlenu w środowisku:2H2S + 3O2 ---> 2H2O + 2SO2,H2S + ½O2 ---> H2O + S.Wykazuje dosyć silne właściwości redukcyjne, gdyŜ potencjał reakcji S2- -->S0 wynosi –0,51V. Ponadto dobrzerozpuszcza się w wodzie i dysocjuje dwustopniowo:H2S <==> H+ + HS- <==> 2H+ + S2-.Jest słabym kwasem, moŜe tworzyć dwa szeregi soli: obojętne siarczki oraz kwaśne wodorosiarczki. Siarczki sąbardzo trudno rozpuszczalne w wodzie i mają ciemne (czarne bądź brunatne) zabarwienie; wyjątek stanowiąsiarczki litowców i amonu. Siarczki litowców mogą rozpuścić w sobie znaczne ilości siarki, a w wynikuzachodzącej reakcji tworzą wielosiarczki. Siarczki rozpuszczalne w wodzie hydrolizują zasadowo, np. (NH4)2Sjest solą słabego kwasu i słabej zasady, lecz ostatecznie jego wodny roztwór ma odczyn zasadowy, gdyŜ zasadaamonowa dysocjuje lepiej niŜ siarkowodór. Niektóre siarczki wskutek hydrolizy nie istnieją w roztworachwodnych, np. siarczek magnezu MgS:MgS ---H2O---> Mg2+ + S2-,2H2O <==> 2H+ + 2OH-,
Created by Neevia Document Converter trial version http://www.neevia.com

58
Mg2+ + 2OH- --> Mg(OH)2,S2- + 2H+ ---> H2S.Produkty hydrolizy to ulatniający się gaz (H2S) oraz strącający się osad (Mg(OH)2). Ze względu na słabąrozpuszczalność wiele siarczków metali to rudy tych metali, np.: Cu2S – chalkozyn, CuS – kowelin, CuS . FeS –chalkopiryt, PbS – galena ołowiowa, FeS2 – piryt lub markosyt, HgS – cynober, Ag2S – argentyt, ZnS – sfalerytlub wurtzyt, MoS2 – molibdenit. Istnieje generalna prawidłowość, iŜ wodorosiarczki rozpuszczają się w wodzielepiej od siarczków obojętnych.2) Tlenek siarki SO2 jest związkiem trwałym. Występuje w przyrodzie jako składnik wyziewów wulkanicznych.W warunkach domowych otrzymać go moŜna przez spalanie zapałek, o czym świadczy charakterystycznyzapach. W przemyśle powstaje podczas utleniania siarczków (proces technologiczny polega na przeprowadzeniusiarczku w tlenek, a następnie jest redukcji koksem) np.:4FeS2 + 11O2 --> 2Fe2O3 + 8SO2.Tlenek siarki powstaje równieŜ jako produkt reakcji stęŜonego kwasu siarkowego z metalami o dodatnimpotencjale normalnym, np.:Cu + 2H2SO4 -> CuSO4 + SO2 + 2H2O.Wydziela się, gdy działamy stęŜonym kwasem siarkowym na niemetale, np.:S + 2H2SO4 -> 3SO2 + 2H2O,C + 2H2SO4 -> 2SO2 + CO2 + 2H2O.Tlenek siarki jest gazem bezbarwnym o ostrym zapachu, dwukrotnie cięŜszym od powietrza. Jest szkodliwyzarówno dla człowieka, jak i dla mikroorganizmów. Cząsteczka ma budowę zgiętą ( |O=S=>O| ), posiadaniezerowy moment dipolowy, a w związku z tym budowę polarną. Łatwo się skrapla; w ciekłym dwutlenkusiarki rozpuszcza się wiele soli, np. PCl3. Jest pochłaniany przez wodę i jako bezwodnik tworzy niezbyt mocnykwas siarkowa (IV):SO2 + H2O ---> H2SO3.Dzięki pośredniemu stopniowi utlenienia siarczany (IV) mogą być utleniaczami i reduktorami. Rozpuszczalne wwodzie siarczany (IV) stosowane były jako środki bielące, np. Na2SO3 utleniając się redukuje niepoŜądanezwiązki barwne w tkaninie, zaś zredukowane produkty są łatwo rozpuszczalne i mogą zostać usunięte przezpocieranie. Preparaty siarkowe miały przewagę nad wybielaczami chlorowymi dzięki miłemu zapachowi.3) Trójtlenek siarki SO3 otrzymywany jest katalitycznie przez utlenianie dwutlenku w aparatach kontaktowych:SO2 + ½O2 ---(war)---> SO3.Ze względu na zmniejszanie objętości podczas przebiegu reakcji stosuje się wysokie ciśnienie, a ze względu naendotermiczność – temperaturę 400oC (reguła przekory). Dodatkowo obecny jest zazwyczaj katalizatorwanadowy, którym najczęściej jest pięciotlenek wanadu V2O5. W typowych warunkach trójtlenek siarki jestciałem stałym, sublimującym w biały dym. UŜywany jest głównie do produkcji kwasu siarkowego (VI), moŜesię w nim rozpuszczać i tworzyć tzw. dymiący kwas siarkowy (oleum). Reakcja z wodą jest tak egzotermiczna,iŜ powstający wrzący kwas mógłby rozprysnąć się i stanowić zagroŜenie, dlatego teŜ SO3 uŜywane jest wnadmiarze. Pełniąc rolę inhibitora sprawia, iŜ reakcja jest w pewien sposób kontrolowana.4) Kwas siarkowy (VI) rozpuszcza się w wodzie w kaŜdym stosunku. Tworzenie wiązań wodorowych isamorzutna hydratacja są procesami bardzo egzotermicznymi, stąd rozpuszczaniu towarzyszy wydzielenieduŜych ilości ciepła. Występuje równieŜ zjawisko kontrakcji – objętość mieszaniny maleje. Dzięki ogromnemupowinowactwu do wody oleum uŜywane jest do osuszania gazów, a nawet odbiera wodę substancjom, w którychjest ona związana chemicznie, np.:C2H5OH ---st. H2SO4/120-130oC---> C2H4 + H2O (pochłaniana),2C2H5OH ---st. H2SO4---> C2H5OC2H5 + H2O (pochłaniana), eter dietylowyCnH2nOn = Cn(H2O)n ---st. H2SO4---> nC + nH2O (pochłaniana).Ostatnia reakcja ilustruje zwęglanie cukrów przez stęŜony kwas siarkowy; moŜe to być zarówno cukier domowy,jak i na drewno (celuloza). Skóra poparzona kwasem siarkowym pokrywa się czarnymi strupami. StęŜony kwassiarkowy jest silnym utleniaczem: utlenia niemetale (węgiel, siarkę, fosfor), a na gorąco równieŜ metaleszlachetne. Metale takie jak Ŝelazo, glin czy chrom pasywują w jego obecności i pokrywają się ochronnąwarstwą tlenków. Rozcieńczony kwas siarkowy w temperaturze pokojowej na ma właściwości utleniających izachowuję się jak zwykły kwas dwuzasadowy. Tworzy dwa szeregi kwasów – obojętne siarczany (VI) i kwaśnewodorosiarczany (VI). Większość tych pierwszych jest dobrze rozpuszczalna w wodzie, wyjątkiem są siarczanywapniowców, np. BaSO4 lub innych – PbSO4.5) Gdyby w cząsteczce kwasu siarkowego (VI)jeden z atomów tlenu wymienić na atom siarkina stopniu utlenienia –2, to otrzymalibyśmykwas tiosiarkowy H2S2O3. Jest on związkiemnietrwałym i w środowisku kwaśnym powolisię rozkłada:
Created by Neevia Document Converter trial version http://www.neevia.com

59
H2S2O3 ---H+---> S + SO2 + H2O.
Jego sole, tiosiarczany, są dobrze rozpuszczalne w wodzie i przede wszystkim – trwałe. Anion tiosiarczanowyS2O3
2- posiada duŜo (10) par elektronowych, dzięki czemu moŜe być donorem par w połączeniachkoordynacyjnych, które z łatwością tworzy. Przykładowo w fotografii tiosiarczan sodu stosowany jest jakoutrwalacz – wypłukuje i usuwa z negatywu i pozytywu nie rozłoŜone halogenki srebra:AgBr + 2Na2S2O3 ---> Na3[Ag(S2O3)2] + NaBr.osad rozpuszczalnyPowstałe związki: tiosrebrzan sodu i bromek sodu mogą być łatwo wypłukane. Tiosiarczan soduwykorzystywany jest w przemyśle papierniczym i włókienniczym do usuwania nadmiaru chloru uŜytego dobielenia, stąd nazwa techniczna: antychlor. W reakcji aniony tiosiarczanowe działają redukująco na chlor, któryzjonizowany tworzy NaCl, podczas gdy sam tiosiarczan utlenia się do siarczanu (VI):Na2S2O3 + 4Cl2 + 10NaOH --> 2Na2SO4 +8NaCl + H2O.6) Niektóre związki siarki warte są uwagi ze względu na udział w reakcjach substytucji w chemii organicznej; sąto np.: dwuchlorek siarki SCl2 i jego dimer S2Cl4 oraz chlorotlenek siarki SO2Cl2.7) Jak juŜ wcześniej wspomniano, siarka uŜywana była przez wiele wieków do produkcji prochu strzelniczego.W najprostszym przypadku naleŜy zmieszać siarkę (zapalnik) z materiałem palnym (węgiel drzewny) wobecności utleniacza (np. saletry indyjskiej) w proporcjach 1:1,5:7,5. Po zainicjowaniu przebiega reakcja:S + 3C + 2KNO3 --> 3CO2 + N2 + K2S.Dwa pierwsze produkty są gazami, trzeci natomiast – ciałem stałym, dlatego teŜ pisany proch spala się zwydzieleniem dymu. Głównym składnikiem prochu bezdymnego jest triazotan (V) celulozy, opisany dokładniejw dziale poświęconym chemii organicznej. Jako zapalniki inicjujące reakcję uŜywane są np. piorunian rtęciHg(OCN)2 bądź azydek ołowiu Pb(N3)2.8) W przyrodzie siarka współtworzy wiele naturalnych związków dezynfekujących. Na przykład podczaskrojenia cebuli uwalnia się z niej propantiol C3H6SO, który reagując z wodą znajdującą się na powierzchni okadaje kwas siarkowy, będący przyczyną łzawienia. Z tego teŜ powodu krojenie cebuli pod wodą nie powodujemęczącego „płaczu”. Znanym warzywem o działaniu dezynfekującym jest czosnek; zawiera on szereg związkówsiarkowych o działaniu odkaŜającym, co ilustruje poniŜszy schemat:
.
SELENSelen jest pierwiastkiem rozpoczynającym w grupie tlenowców przejście niemetali w metale. Występuje wodmianach alotropowych. W związkach przyjmuje róŜne stopnie utlenienia, jednak najtrwalsze są związki wktórych ma +4. Nie przewodzi prądu elektrycznego – jest izolatorem. Natomiast pod wpływem kwantówpromieniowania świetlnego generuje impulsy elektryczne, dzięki czemu moŜe być stosowany jako zamiennikimpulsów świetlnych na sygnały w postaci prądu. Ze względu na tą ostatnią właściwość znalazł zastosowanie wurządzeniach alarmowych, fotokomórkach, telewizorach i urządzeniach kserograficznych. Selen jestmikroelementem, niezbędnym w śladowych ilościach do prawidłowego funkcjonowania organizmu ludzkiego(erytrocyty). Dodatek selenu barwi szkło na kolor róŜowy lub czerwono – rubinowy, przez co neutralizujezieloną barwę pochodzącą od zanieczyszczeń krzemionki związkami Ŝelaza. W połączeniu z wodorem selentworzy selenowodór H2Se, który wprowadzony do wody wykazuje mocniejsze właściwości kwasowe niŜsiarkowodór. Tworzy dwa szeregi soli: selenki i wodoroselenki.
Tellur i polon tworzą związki o typowej wartościowości +4. Porównując selen, tellur i polon pod względemelektrycznym stwierdzamy, iŜ potencjał normalny maleje w grupie wraz ze wzrostem liczby atomowej.
FLUOROWCE
Fluorowce to siódma grupa główna w układzie okresowym – VII A lub 17. Posiadają konfigurację elektronowąns2np5
- zatem siedem elektronów walencyjnych rozmieszczonych na podpoziomach s i p. W staniepodstawowym atom fluorowca posiada trzy pary elektronowe – jedną s i dwie p – oraz jeden niesparowanyelektron walencyjny p, który moŜe brać udział w wiązaniach. Ogranicza to wartościowość do I – taka sytuacjawystępuje w przypadku fluoru. JeŜeli fluorowiec dysponuje wolnym orbitalem walencyjnym d (okres trzeci iponiŜsze), to w wyniku wzbudzenia mogą nań przemieszczać się elektrony z par s i p. Rozszerza to zakres
Created by Neevia Document Converter trial version http://www.neevia.com

60
wartościowości tak, Ŝe pozostałe fluorowce przyjmują wartościowości I, III, V, i VII. Oznacza to równieŜ, iŜmogą pojawić się niestandardowe typy hybrydyzacji, np. sp3d3 w siedmiofluorku jodu IF7. W grupie tejdominują niemetale; półmetaliczny astat występuje w warunkach naturalnych w śladowych ilościach 10-24% iaktualnie nie ma zastosowania. Fluorowce cechuje zróŜnicowany stan skupienia: fluor i chlor to gazy ocząsteczkach dwuatomowych, brom jest cieczą a jod – łatwo sublimujących ciałem stałym. Elektroujemnośćmaleje w grupie wraz ze wzrostem liczby atomowej. Fluorowce tworzą wodorki kwasowe, które wprowadzonedo wody dają kwasy beztlenowe. Tak powstałe roztwory mają stosunkowo silne właściwości kwasowe, przyczym moc kwasu wzrasta w grupie wraz ze wzrostem liczby atomowej. Tłumaczyć to naleŜy wzrastaniempromienia jonowego, a tym samym odległości pomiędzy jądrami fluorowca i wodoru – to z kolei znacznieosłabia działającą między nimi siłę; w efekcie łatwiej wodór moŜe być odszczepiany łatwiej, a ze wzrostemstopnia dysocjacji rośnie pH oraz moc kwasu. KaŜdy fluorowiec wypiera z soli sąsiada leŜącego poniŜej siebie wgrupie (fluor wypiera wszystkie pozostałe, chlor wypiera brom i jod, brom wypiera jod, a jod moŜe byćwyłącznie wypierany przez inne fluorowce ze swoich soli).
FLUORPierwiastkiem rozpoczynającym grupę jest fluor. W warunkach normalnych jest gazem o barwie Ŝółtozielonej ointensywnym, nieco chlorowym zapachu. Atakuje drogi oddechowe człowieka. Ma najwyŜszą elektroujemnośćw układzie okresowym (X=4,0), jest w związku z tym najaktywniejszym pierwiastkiem. Bezpośrednio nie łączysię jedynie z tlenem, chlorem, azotem i helowcami. W wysokiej temperaturze działaniu chloru ulegają: diament,złoto i platynowce. Bardzo energicznie reaguje z wodorem nawet w ujemnych temperaturach i bez udziałuświatła. Odbiera wodór innym związkom chemicznym, np. wodzie.W wyniku reakcji fluoru z wodorem powstaje fluorowodór:F2 + H2 ---> 2HF.MoŜna go równieŜ otrzymać przez działanie stęŜonym kwasem siarkowym (VI) na fluorek wapnia:CaF2 + H2SO4 ---> CaSO4 + H2F2.Ze względu na bardzo duŜą róŜnicę elektroujemności cząsteczki fluorowodoru wytwarzają bardzo silne wiązania
wodorowe, skutkiem czego wytwarza się struktura liniowa: .Skutek jest taki, iŜ forma dimeru H2F2 jest trwalsza od formy monomeru. Czasami tworzone są długie łańcuchypolimeru (HF)n o budowie liniowej. Kwas fluorowodorowy w stosunkowo małym stopniu ulega dysocjacji i jestkwasem słabym, choć agresywnym. Jego sole – fluorki – cechują się zróŜnicowaną rozpuszczalnością w wodzie:rozpuszczalne są fluorki litowców i srebra, natomiast reszta jest nierozpuszczalna. Wybitnym przykładem złejrozpuszczalności fluorków jest fluorek wapnia CaF2, wchodzący w skład szkliwa zębowego. Fluorowodórniszczy substancje organiczne, przez co stosowany jest jako środek dezynfekcyjny i bakteriobójczy. Wykazujeodporność na działanie czynników utleniających. UŜywany jest do trawienia szkła:Na2SiO3 + 6HF ---> 2NaF + SiF4 + 3H2O,CaSiO3 + 6HF ---> CaF2 + SiF4 + 3H2O.Powstały w powyŜszych reakcjach fluorek krzemu reaguje równieŜ z samym kwasem fluorowodorowym:2 SiF4 + 4HF ---> 2H2[SiF6].Produkt ostatniej reakcji to kwas fluorokrzemowy; jest bardzo mocnym kwasem i dobrze rozpuszcza się wwodzie.
CHLORChlor występuje w przyrodzie w postaci minerałów, np.: NaCl – halit, KCl – sylwin. W warunkachlaboratoryjnych moŜna go otrzymać przez elektrolizę chlorków bądź odpowiednią reakcją, np. działanie kwasemsolnym na braunsztyn:4HCl + MnO2 ---> MnCl2 + Cl2 + 2H2O.W warunkach normalnych chlor jest Ŝółtozielonym gazem o duszącej woni. Nazwa jego wzięła się od barwy,gdyŜ gr. chloros oznacza zielony. Chlor działa draŜniąco na błony śluzowe człowieka, podczas pierwszej wojnyświatowej uŜywany był jako gaz bojowy. Jest wielokrotnie cięŜszy od powietrza, dzięki czemu „pełznie” popodłoŜu. Rozpuszcza się w wodzie i wchodzi z nią w reakcję dysproporcjonowania:H2O + Cl2 ---> HCl + HClO.Jest pierwiastkiem bardzo czynnym chemicznie i silnym utleniaczem, a bezpośrednie reakcje z chlorem mogąprzebiegać bardzo gwałtownie. Jest mniej aktywny od fluoru, gdyŜ reakcja z wodorem nie przebiega wciemności:H2 + Cl2 ---> 2HCl.Natomiast przy dostępie światła reakcja przebiega bardzo gwałtownie, gdyŜ powstałe pod wpływem światłarodniki natychmiast dąŜą do sparowania elektronów walencyjnych. Działa utleniająco na substancje organiczne,stosowany jest do bielenia tkanin i papieru. Nadmiaru jego moŜna się pozbyć za pomocą „antychloru”, czylitiosiarczanu sodowego:Na2S2O3 + 4Cl2 + 10NaOH --> 2Na2SO4 +8NaCl + H2O.
Created by Neevia Document Converter trial version http://www.neevia.com

61
1) W wyniku łączenia się chloru z wodorem otrzymujemy chlorowodór HCl. Nie dysponując powyŜszymiodczynnikami moŜna go otrzymać np. działając mocnym kwasem na chlorek:2NaCl + H2SO4 ---> Na2SO4 + 2HCl,albo przez hydrolizę fluorku fosforu (III):PCl3 + 3H2O ---> H3PO3 + 3HCl.Chlorowodór jest gazem bezbarwnym, cięŜszym od powietrza. Rozpuszcza się w wodzie i dysocjuje, dziękiczemu roztwór przewodzi prąd elektryczny. Łączy się z wodą egzotermicznie w wyniku automatycznejhydratacji. Jest mocnym kwasem , zdysocjowanym niemal stuprocentowo. Jest związkiem trwałym, a rozkład napierwiastki stanowi problem. Wchodzi w skład wody królewskiej wraz z kwasem azotowym (V). Kwaschlorowodorowy jest kwasem jednozasadowym, dlatego tworzy tylko jeden szereg soli obojętnych. Większośćchlorków jest dobrze rozpuszczalna w wodzie, z wyjątkiem AgCl, PbCl2 oraz Hg2Cl2. Pierwszy z nich, chloreksrebra AgCl, wchodzi w postaci kompleksu z amoniakiem i cyjanowodorem w skład materiałów światłoczułychna kliszach fotograficznych.2) Chlor tworzy cały szereg tlenków, co spowodowane jest mnogością stopni utlenienia. Są to: Cl2O, Cl2O3,ClO2, Cl2O5, Cl2O6 (dimer ClO3) oraz Cl2O7. Jak widać, chlor tworzy związki zarówno na parzystych, jak i nanieparzystych stopniach utlenienia. Tlenki chloru są związkami nietrwałymi, są nośnikami tlenu i mają silnewłaściwości utleniające. W reakcjach z wodą tworzą cały szereg tlenowych kwasów chlorowych:3) HClO (kwas podchlorowy, kwas chlorowy (I)) jest związkiem nietrwałym, występuje wyłącznie w duŜymrozcieńczeniu. Powstaje w wyniku reakcji dysproporcjonowania chloru w wodzie:H2O + Cl2 ---> HCl + HClO.Jego sole, podchloryny, uŜywane były jako środki bielące.4) HClO2 (kwas chlorowy (III)) jest nietrwałym związkiem, słabym kwasem, ale za to silnym utleniaczem.5) HClO3 (kwas chlorowy (V)) jest w porównaniu z poprzednimi trwały. RównieŜ posiada właściwościutleniające6) HClO4 (kwas chlorowy (VII)) jest najtrwalszym i najmocniejszym kwasem z grupy. Jest bardzo silnymutleniaczem, a aniony chloranowe (VII) rozkładają się wybuchowo.Moc kwasów chlorowych rośnie wraz ze wzrostem stopnia utlenienia, czyli kwas chlorowy (VII) jest mocniejszyod chlorowego (V), ten z kolei jest mocniejszy od chlorowego (III), zaś chlorowy (I) jest najsłabszy.
BROMW warunkach normalnych brom jest ciemnobrunatną cieczą. Słabo rozpuszcza się w wodzie, a powstała wodabromowa ma kolor jasnobrązowy; o jej znaczeniu dla wykrywania nienasyconego charakteru związkóworganicznych powiemy później. Brom rozpuszcza się dobrze w rozpuszczalnikach organicznych. Brom działaniszcząco na substancje organiczne (choć wolniej niŜ chlor) i szkodliwie na organizmy Ŝywe; pary bromu mająsilnie duszący zapach.1) Podczas łączenia się bromu z wodorem na świetle powstaje bromowodór. Jest to gaz cięŜszy od powietrza, amocnym zapachu. Rozpuszcza się w wodzie i daje kwas bromowodorowy, silniejszy od chlorowodorowego.Bromki są związkami barwnymi, o kolorze jasnoŜółtym bądź jasnobeŜowym.2) Bromek srebra AgBr o barwie jasnoŜółtej stanowi element masy światłoczułej na kliszach fotograficznych.3) Podobnie jak chlor, równieŜ brom tworzy kwasy tlenowe: kwas bromowy (I) HBrO i kwas bromowy (III)HBrO3. Są to związki nietrwałe, o charakterze utleniającym słabszym od kwasów chlorowych. Ze względu narozkład przechowuje się je w formie 50% roztworów.
JODW warunkach normalnych jod jest ciałem stałym, występującym w postaci szarych metalicznych łusek. Bardzołatwo sublimuje, a jego pary są fiołkowe. Słabo rozpuszcza się w wodzie, rozpuszczalność tą podnosi obecnośćanionów jodkowych, gdyŜ tworzone są wówczas jony trójjodkowe:J2 + J- ---> J3
-.Roztwór przyjmuje wówczas barwę ciemnobrunatną. Jod stosunkowo dobrze rozpuszcza się wrozpuszczalnikach organicznych, np. 3% roztwór jodu z dodatkiem jodku potasu KI w etanolu to jodyna,uŜywana niegdyś powszechnie do dezynfekcji ran. Spośród fluorowców jest najmniej aktywny, posiadanajwiększy potencjał normalny. Ze skrobią tworzy barwę ciemnogranatową, przez co stosowany jest do jejwykrywania, np. margarynę poddaje się próbie z jodem, czy nie zawiera nawet niewielkich ilości skrobi. Wanalizie chemicznej stosowany jest podczas jodometrii, opartej na reakcjach redox, np.Cr2O7
2- + 6J- + 14H+ ---> 2Cr3+ + 3J2 + 3H2O.1) Przez syntezę jodu z wodorem przy uŜyciu nakładu energetycznego bądź przez reakcję jodków z mocnymikwasami moŜna otrzymać jodowodór. Jest to gaz cięŜszy od powietrza o przenikliwym zapachu. Rozpuszcza sięw wodzie i daje kwas jodowodorowy. Powstałe w roztworze jony jodkowe I- są czynnikiem redukującym. Jodkisą na ogół solami rozpuszczalnymi, jednak np. AgI, PbI2 i Hg2I2 to związki bardzo trudno rozpuszczalne.Dodając do roztworu amoniaku nie moŜna rozpuścić jodków przez ukompleksowienie, tak jak to wychodzi z
Created by Neevia Document Converter trial version http://www.neevia.com

62
nierozpuszczalnymi chlorkami. Wiele jodków to substancje barwne: AgI jest Ŝółty, HgI2 – ceglastoczerwony,zaś Hg2I2 – szarozielony.2) Podobnie jak poprzednicy, jod tworzy kwasy tlenowe. Kwas jodowy (I) HIO jest kwasem nietrwałym isłabszym od sąsiadów.3) Kwas jodowy (V) HJO3 jest dosyć silnym utleniaczem i relatywnie trwałym związkiem, gdyŜ moŜna gowydzielić z roztworu w postaci kryształów.
MIEDŹ
Miedź jest metalem połoŜonym w grupie I B lub 11 i w 4 okresie. Rozpoczyna grupę pierwiastków zwanychmiedziowcami, do której naleŜy równieŜ srebro i złoto. Znajduje się w grupie pobocznej, w blokuenergetycznym d. Powinna posiadać konfigurację nd9(n-1)s2, jednak w wyniku promocji wszystkie miedziowcezyskują konfigurację nd10(n-1)s1. Atom miedzi posiada zatem jeden niesparowany elektron, co wskazywałoby najej jednowartościowość. Istnieje teoretyczna moŜliwość wzbudzenia atomu i przeniesienia elektronów na orbitalp; dzięki temu miedź moŜe być dwu-, a w rzadkich przypadkach nawet trójwartościowa. Elektroujemność tegopierwiastka wynosi X=1,9 w skali Paulinga, przez co niewiele róŜni się od wodoru. NaleŜałoby spodziewać sięamfoteryczności i rzeczywiście ona występuje. Na tym przykładzie potwierdza się prawidłowość, iŜ pierwiastkiamfoteryczne leŜą na umownej przekątnej układu okresowego. W atomie miedzi odległości pomiędzy jądrem aelektronami walencyjnymi nie są zbyt duŜe. Miedź jest bardzo dobrym przewodnikiem ciepła i elektryczności –posiada drugi w kolejności najmniejszy opór właściwy, dzięki czemu stosuje się ją w przemyśleelektrotechnicznym do produkcji przewodów (najmniejszy opór właściwy posiada srebro, którego rzadkość iwysoka cena wyklucza podobne zastosowanie). Jest bardzo ciągliwa i kowalna, co umoŜliwia obróbkęplastyczną. Wprowadzona w płomień palnika, zabarwia go na kolor zielony.Cechuje się bardzo niewielką aktywnością chemiczną. Podobnie zachowuje się reszta miedziowców, przy czymszlachetność rośnie w dół – świadczą o tym dodatnie i rosnące wartości potencjałów normalnych. Miedź reagujez silnymi kwasami utleniającymi, doprowadzając do redukcji atomu centralnego, np.:Cu + 4HNO3 -> Cu(NO3)2 + 2NO2 + 2H2O (>15%) niebieski rudobrązowyCu + 8HNO3 -> 3Cu(NO3)2 + 2NO + H2O (<15%) niebieski bezbarwnyCu + HNO3 -X-> nie zachodzi (<1%)Cu + 2H2SO4 -> CuSO4 + SO2 + 2H2O (stęŜony i gorący).Natomiast Ŝaden z miedziowców nie reaguje z kwasami beztlenowymi. Miedź tworzy związki, w którychwystępuje na stopniu utlenienia +1 oraz +2, przy czym te drugie są trwalsze. Ze względu na amfoterycznycharakter tlenki i wodorotlenki miedzi są trudno rozpuszczalne. Wodorotlenek miedzi (I) jest silniejszą zasadąniŜ miedzi (II).W przyrodzie miedź wstępuje w postaci minerałów siarczkowych oraz, w małych ilościach, tlenowych, np.:Cu2O – kupryt, CuO – melakonit, Cu2S – chalkozyn, CuS – kowelin, CuS . FeS (CuFeS2) – chalkopiryt,Cu(OH)2
. CuCO3 - malachit, CuSO4 . 5H2O - kamień siny. W procesie przetwarzania rud miedzi wpierw
siarczki przeprowadza się w tlenki, np.:Cu2S + 2O2 ---> 2CuO + SO2,a następnie redukuje koksem:CuO + C ---> Cu + CO.Wyroby miedziane pokrywają się warstwą czerwonego tlenku miedzi (I) Cu2O, dlatego mają charakterystycznyodcień. W wyniku łączenia się miedzi z siarką, występującą w niewielkich ilościach w powietrzu, przyjmują onebarwę czarno-brunatną. Nie ulega działaniu suchego tlenu, natomiast w wilgotnym powietrzu pokrywa sięzasadowym węglanem miedzi Cu(OH)2
. CuCO3 = [Cu(OH)]2CO3, czyli patyną – z tego powodu miedzianedachy z czasem zielenieją. Bezwodne sole miedzi są bezbarwne, natomiast uwodnione mają kolor niebieski lubniebieskozielony. Solidarnie z resztą związków miedzi, sole ulegają działaniu kwasów utleniających. Miedź maszerokie zastosowanie: wymieniono juŜ produkcję przewodów elektrycznych, poza tym blachy miedzioweuŜywane są w róŜnych konstrukcjach, wyrabia się z niej kotły parowe, aparaty destylacyjne, kryje się nią dachy.Miedź wchodzi w skład licznych stopów, np. z cyną tworzy brązy, z cynkiem – mosiądze, z glinem – brązaluminiowy (brązal); stop uŜywany do bicia monet to połączenie miedzi i niklu oraz glinu i cynku.
Tlenek miedzi (I) występuje w przyrodzie jako minerał kupryt. Tlenek ma barwę ceglastoczerwoną. Powstaje wwyniku próby Trommera, stosowanej do wykrywania grupy aldehydowej – zawarty w odczynniku Fehlinganiebieski wodorotlenek miedzi (II) redukuje się do czerwonego tlenku miedzi (I), a aldehyd utlenia się do kwasukarboksylowego. Dzięki tworzeniu połączeń kompleksowych nierozpuszczalne związki miedzi rozpuszczają się
Created by Neevia Document Converter trial version http://www.neevia.com

63
w nadmiarze amoniaku, natomiast ze stęŜonym kwasem solnym łączą się koordynacyjnie. Szereg soli kwasówbeztlenowych miedzi (I) jest trwały (np.: CuCl, CuCN, CuSCN), natomiast sole kwasów tlenowych wroztworach wodnych dysproporcjonują z wydzieleniem czystej miedzi oraz soli miedzi (II).
Tlenek miedzi (II) ma barwę czarną, nie rozpuszcza się w wodzie ze względu na amfoteryczny charakter. Jestutleniaczem i redukuje inne substancje. Bezwodne sole są białe, zaś hydraty przyjmują barwę niebieską bądźzielonkawą (np. CuSO4
. 5H2O). Większość soli miedzi jest rozpuszczalna w wodzie – CuCl2, CuBr, CuI2 itd.Sole miedzi moŜna rozpuszczać w stęŜonym kwasie solnym, roztworze amoniaku czy mocnych wodorotlenkach.Miedź w postaci kationów Cu2+ jest mikroelementem, stanowi składnik enzymów oddechowych u wielu grupzwierząt oraz wchodzi w skład enzymów. W nadmiernych ilościach jej sole wywierają szkodliwe działanie naorganizmy Ŝywe za względu na denaturację białek – szkodliwe jony moŜna wykryć a pomocą amoniaku(tworzenie kolorowych kompleksów – patrz tekst o amoniaku).
SREBRO
Srebro występuje w samorodkach, lecz procentowy udział molowy metalu jest niewielki. Czyste srebrowydobywa się przez ługowania amoniakiem, amalgamację rtęcią bądź metodą elektrolityczną. Jest to metal obarwie srebrzystej, bardzo kowalny i ciągliwy, świetny przewodnik ciepła i elektryczności, dzięki czemuuŜywany jest do wyrobu specjalistycznych obwodów elektrycznych. Jest nieaktywny chemicznie i odporny nadziałanie tlenu cząsteczkowego, natomiast z ozonem reaguje juŜ w temperaturze pokojowej. Reakcje srebra zfluorowcami zachodzą bardzo powoli. Natomiast łatwo następuje łączenie z siarką z wydzieleniemnierozpuszczalnego czarnego siarczku srebra AgS – jest to przyczyną czernienia wyrobów srebrnych, np.sztućców. Ze względu na duŜy dodatni potencjał srebro naleŜy do silnych utleniaczy i reaguje jedynie z silnymikwasami utleniającymi. Trwałością odznaczają się związki srebra za +1 stopniu utlenienia. Srebro tworzypołączenia kompleksowe z amoniakiem i cyjanowodorem, w których występuje z liczbą koordynacyjną 2.Czyste srebro jest miękkie, dlatego zazwyczaj do uŜytku wprowadza się jego stopy z miedzią. Srebro tworzystopy z wieloma metalami, z wyjątkiem Ŝelaza, manganu i chromu. Stopy z duŜa zawartością srebra uŜywane sądo produkcji biŜuterii; zawartość srebra w wyrobie oznacza się przez podanie próby: I – 916, II – 875, III – 800(ułamki masowe w promilach). Dla porównania stopy złota stosowane w dentystyce maja próby: I – 750, II –575, III – 573. Tradycyjnie odsetek złota w wyrobie podaje się w karatach: 24 karaty odpowiadają 100%, zaśzazwyczaj uŜywa się złota 14-karatowego, co odpowiada 58,3%. Karat to równieŜ uŜywana w jubilerstwiejednostka masy odpowiadająca 2,5g. Srebro moŜna odróŜnić od złota poprzez reakcję z kwasem azotowym (V) –w przypadku srebra powstanie niebieska plama. Srebro jest metalem szlachetnym szkodliwym dla organizmówŜywych (np. lapis AgNO3 zwany jest kamieniem piekielnym). Ze względu na bakteriobójcze działanie jonówAg+ srebro stanowi składnik środków dezynfekcyjnych. Ze względu na połysk było wykorzystywane doprodukcji luster.Wiele związków srebra jest barwnych.1) Tlenek srebra (I) Ag2O jest czerwonobrunatnym proszkiem. Rozpuszcza się w wodzie i w wyniku hydrolizynadaje jej odczyn zasadowy.2) Azotan AgNO3 i chloran (V) AgClO3 srebra są nośnikami tlenu, zaś fluorek AgF ma właściwościwybuchowe.3) Tiosiarczan srebra Ag2S2O3 w reakcji z wodą i mocniejszym kwasem daje trudno rozpuszczalny czarny osadsiarczku srebra oraz kwas siarkowy (VI),np.:2Ag2S2O3 + 2HCl + 2H2O ---> Ag2S + 2AgCl + 2H2SO4.4) Węglan Ag2CO3 i fosforan (V) Ag3PO4 srebra maja barwę Ŝółtą, a chromian (VI) Ag2CrO4 – ciemnobrunatną.
CYNK
Cynk jest metalem połoŜonym w grupie II B lub 12 i w 4 okresie. Ze względu na rozpoczynania grupy, nazwanoją cynkowcami (cynk, kadm, rtęć). Cynk posiada konfigurację elektronów walencyjnych 3d104s2 i występujewyłącznie na +2 stopniu utlenienia. Posiada niski potencjał standardowy i jest bardzo silnym reduktorem:4Zn + 10HNO3 -> 4Zn(NO3)2 + NH4NO3 + 4H2O.W powyŜszej reakcji cynk powoduje zmianę stopnia utlenienia azotu z +5 na –3, a więc o 8 jednostek, a moŜnato powiedzieć o niewielu reakcjach. Jest metalem amfoterycznym, a produkt reakcji z kwasami zaleŜy odwarunków środowiska reakcji:Zn + 4HNO3 -> Zn(NO3)2 + 2NO2 + 2H2O,Zn + 8HNO3 -> 3Zn(NO3)2 + 2NO + H2O.
Created by Neevia Document Converter trial version http://www.neevia.com

64
Reaguje z mocnymi zasadami, np.:ZnO + 2NaOH + H2O ---> Na2Zn(OH)4 ---T---> Na2ZnO2 + 2H2O; czterohydroksocynkan sodu[Zn(OH)4]
2- ----2H2O---> [ZnO2]
2-.MoŜe tworzyć zasadowe hydroksysole oraz sole obojętne. Sole cynku w roztworach wodnych wskutek hydrolizykationowej wykazują odczyn kwasowy, np. ZnCl2.Do trudno rozpuszczalnych soli cynku zaliczmy: ZnS, ZnCO3,Zn(PO4)3 i ZnF2. Wodorotlenek Zn(OH)2 równieŜ posiada charakter amfoteryczny – nie rozpuszcza się wwodzie, wytrącając się w formie galaretowatego osadu. Rozpuszcza się w nadmiarze mocnych zasad, słabo zaśw amoniaku, z którym tworzy połączenie kompleksowe [Zn(NH3)4]
2+.Cynk występuje w przyrodzie w postaci minerałów, np.: ZnS – sfaleryt lub wurtzyt, ZnCO3 – galman. Wszystkierudy doprowadza się do formy tlenkowej, którą następnie redukuje się koksem lub tlenkiem węgla. Tak powstałycynk poddaje się elektrolitycznej rafinacji (elektrorafinacji). Czysty metal ma niebieskawy połysk, jest ciągliwy ikowalny oraz łatwo topliwy (ok. 400oC). Ogrzany do 100oC staje się kruchy. Stosowany jest do produkcji blachycynkowej oraz do cynkowania blach stalowych – jest to ochrona przeciwkorozyjna czynna (aktywna), gdyŜ wrazie uszkodzenia w powstałym ogniwie galwanicznym cynk utlenia się aŜ do całkowitego rozpuszczenia. Cynkjest równieŜ składnikiem wielu stopów: z miedzią tworzy mosiądze, z miedzią i srebrem – tzw. nowe srebro,uŜywane do produkcji biŜuterii i sztućców, stop miedzi z dodatkiem do 10% cynku daje tombak imitujący złoto,stopiony z glinem daje znal.1) Tlenek glinu ZnO jest jedną z form występowania tego pierwiastka. Czysty tlenek uŜywany jest jako białafarba o duŜych zdolnościach kryjących, znana pod nazwą bieli cynkowej. Cynk jako mikroelement niezbędnyjest człowiekowi w procesie regeneracji naskórka; dzięki dodatkowym właściwościom antyseptycznym idealnienadaje się do produkcji leków naskórnych, tzw. maści cynkowej. W kosmetyce uŜywany jest jako składnikpudrów. W przemyśle chemicznym katalizuje reakcję syntezy metanolu z gazu syntezowego:CO + H2 ---ZnO---> CH3OH.2) Siarczek cynku ZnS to występująca jako minerał blenda cynkowa. W przeciwieństwie do większościsiarczków, których barwa jest czarna lub brązowa, siarczek cynku jest biały. Zmieszany z siarczanem (VI) barudaje białą farbę znaną jako litopan.3) Siarczan (VI) cynku ZnSO4 jest solą dobrze rozpuszczalną w wodzie. Roztwór ZnSO4 słuŜy do impregnacjidrewna – zmniejsza się przez to jego palność, a dzięki antyseptycznym właściwościom jonów Zn2+ zabezpieczaje przed rozkładem.
CHROM
Chrom połoŜony jest w grupie VI B lub 6 i w 4 okresie. Rozpoczyna grupę pierwiastków zwanychchromowcami, do której naleŜy równieŜ molibden i wanad. Znajduje się w grupie pobocznej, w blokuenergetycznym d. Z połoŜenia w układzie okresowym wynika konfiguracja nd4(n-1)s2, jednak w wynikupromocji wszystkie chrom i molibden zyskują konfigurację nd5(n-1)s2 - atom chromu posiada zatem jedenniesparowany elektron. Ogólnie chromowce są metalami bardzo trwałym i trudno topliwymi, wykazująodporność na czynniki atmosferyczne i silne środki utleniające. Ulegają pasywacji poddane działaniu stęŜonychkwasów, a nie wskutek ujemnego potencjału normalnego. Chrom występuje na kilku stopniach utlenienia (+2,+3, +6), z czego wynikają róŜnice we właściwościach. Ze względu na duŜą liczbę elektronów walencyjnychzwiązki chromu są barwne: Cr2+ jest niebieskie w roztworze wodnym, Cr3+ - niebieskie, CrO4
2- - Ŝółte, a Cr2O72-
- pomarańczowe.1) Tlenek chromu (III) Cr2O3 ma barwę zieloną. Jest związkiem nierozpuszczalnym w wodzie, wykazującymcharakter amfoteryczny. Tworzy połączenia koordynacyjne, w które wchodzi z liczbą koordynacyjną +6, np.:Na3[Cr(OH)6] ---
-3H2O---> Na3CrO3.
W wyniku słabej hydratacji dole chromu przyjmują czasem barwę fioletową.2) Tlenek chromu (VI) CrO3 posiada sam w sobie barwę czerwoną. Jest substancją bardzo higroskopijną.Reaguje z wodą, dając kwas chromowy (VI):CrO3 + H2O ---> H2CrO4.Roztwór przyjmuje wówczas barwę od Ŝółtopomarańczowej do czerwonej, gdyŜ wytworzone jonychromianowe (VI) bardzo łatwo ulegają dimeryzacji do jonów dwuchromianowych (VI); w roztworze wytwarzasię stan równowagi dynamicznej pomiędzy dwiema formami jonów:2CrO4
2- + H+ <====> Cr2O72- + OH-.
Ŝółty pomarańczowoczerwony3) Chromiany (VI) i dwuchromiany (VI) to silne utleniacze, redukujące się podczas reakcji do zielonych Cr3+.Dwuchromiany utleniają: S(+4)->S(+6), S(-2)->S(0), a w środowisku silnie kwaśnym: Cl(-1)->Cl(0), Br(-1)->Br(0) oraz I(-1)->I(0). Dwuchromiany rozpuszczają się w wodzie lepiej od chromianów. Rozpuszczalne sąchromiany litowców, amonu, magnezu, wapnia i strontu.
Created by Neevia Document Converter trial version http://www.neevia.com

65
MANGAN
Mangan połoŜony jest w grupie VII B lub 7 i w 4 okresie. Rozpoczyna grupę pierwiastków zwanychmanganowcami, do której naleŜy równieŜ technet i ren. Znajduje się w grupie pobocznej, w blokuenergetycznym d. Posiada konfigurację elektronów walencyjnych 4s23d5 oraz wolny do zagospodarowaniaorbital p. Pozwala to manganowi na przyjmowanie wartościowości od II do VII, przy czym najtrwalsze sązwiązki o wartościowości II. Mangan jest metalem aktywnych chemicznie, łatwo reaguje z kwasami irozpuszcza się w nich. Ulega działaniu chlorowców juŜ w zwykłej temperaturze, dając halogenki manganu(MnCl2, MnBr2). Łączy się chętnie z tlenem i daje wiele tlenków: MnO (zasadowy), Mn2O3 (amfoteryczny),Mn2O7 (kwasowy).1) Tlenek manganu (II) MnO jest w warunkach pokojowych szarozielonym proszkiem, bardzo słaborozpuszczalnym w wodzie. Związek ten łatwo się utlenia, podobnie jak Mn(OH)2 utlenia się na powietrzu ipowstaje wodorotlenek manganu (IV) MnO(OH)2. Jon Mn2+ tworzy połączenia kompleksowe, w które wchodziz liczbą koordynacyjną 6, np. jon sześcioamionomanganowy (II) [Mn(NH3)6]
2+. W roztworze wodnym jon Mn2+
jest prawie bezbarwny, moŜe lekko róŜowy, dlatego trwały zanik zabarwienia utleniających związków manganuoznacza koniec reakcji rodox.2) Związki manganu na +3 stopniu utlenienia są nietrwałe i szybko dysproporcjonują.3) Tlenek manganu (IV) MnO2 jest czarnym proszkiem. Jest nierozpuszczalny w wodzie i wykazuje charakteramfoteryczny. Ze względu na pośredni stopień utlenienia moŜe być zarówno utleniaczem jak i reduktorem.Związki manganu na +4 stopniu utlenienia są trwałe w środowisku zasadowym.4) Manganiany (VI) są trwałe w środowisku zasadowym.5) Tlenek manganu (VII) Mn2O7 to w warunkach normalnych oleista ciecz. Związek ten jest tlenkiemkwasowym. Istniejący teoretycznie kwas manganowy (VII) znany jest tylko w rozcieńczonych roztworach. Jonmanganowy (VII) jest w roztworze wodnym fioletowy. Manganiany (VII) to silne utleniacze, utleniające m. in.:S(+4)->S(+6) i S(-2)->S(0). Procesy redox zaleŜą od pH środowiska: najsilniejsze właściwości utleniająceosiąga się w środowisku kwasowym (mangan przechodzi ze stopnia utlenienia +7 na +2, a fioletowy roztwórstaje się bezbarwny), słabsze w obojętnym (+7 na +4, powstaje czarny brunatny osad MnO2), zaś najsłabsze wzasadowym (+7 na +6, roztwór staje się trawiastozielony). Manganiany są przenośnikami tlenu i mogą utleniaćwiele substancji, zaś wymieszane z materiałami palnymi (papier) powodują samozapłon.
śELAZO
W układzie okresowym Ŝelazo jest pierwiastkiem grupy VIII B lub 8, w okresie 4. Posiada konfiguracjęelektronów walencyjnych 3d64s2. Tworzy związki na +2 i +3 stopniu utlenienia. Przykładem tego są tlenki:Ŝelaza (II) FeO, nieco zasadowy oraz Ŝelaza (III) Fe2O3, bardziej amfoteryczny. Rzadko spotykane i nietrwałesą związki Ŝelaza na stopniu +6 – są to Ŝelaziany o pozornie kwasowym charakterze. Rozpuszczone sole Ŝelaza(II) hydrolizują kationowo i są prawie bezbarwne, bardziej stęŜone mają zabarwienie zielonkawoŜółte. Z koleisole Ŝelaza (III) rozpuszczone w wodzie wskutek hydrolizy tworzą po pewnym czasie czerwonobrunatny osad.W wyniku reakcji Ŝelaza z kwasami powstają sole Ŝelaza (II), co wynika z szeregu napięciowego (róŜnicapotencjałów wodoru i Ŝelaza (III) jest niewystarczająca do wydzielenia wodoru, a potrzeba 0,20); pozostawionyna powietrzu roztwór powoli brunatnieje wskutek przemiany jonów Fe2+ w Fe3+. Wobec stęŜonych kwasówutleniających Ŝelazo pasywuje, czyli pokrywa się ochronną warstwą tlenku; czasami jest to zjawisko poŜądane –pozwala to przykładowo na przewóz stęŜonego kwasu siarkowego (VI) w stalowych cysternach. śelazowystawione na działanie czynników atmosferycznych lub zanurzone w wodzie koroduje. Korozji sprzyjanasycenie środowiska reakcji tlenem, rozpuszczenie w nim soli oraz zakwaszenie, podczas gdy środowiskozasadowe hamuje postęp korozji. śelazo, nikiel i kobalt posiadają właściwości ferromagnetyczne, jednak wprzypadku Ŝelaza są one największe.W przyrodzie Ŝelazo występuje w postaci rud, np.: Fe2O3 – hematyt, FeO . Fe2O3 – magnetyt, FeS2 – piryt lubmarkosyt, FeCO3 – syderyt, Fe(OH)3 – limonit. Rudę Ŝelazową poddaje się obróbce w wielkim piecu: w wynikuredukcji dwutlenku węgla rozŜarzonym koksem powstaje tlenek węgla:C + CO2 --> 2CO,który stopniowo redukuje Ŝelazo – wpierw do tlenków:3Fe2O3 + CO --> 2Fe3O4 + CO2,Fe3O4 + CO --> 3FeO + CO2,a następnie do czystego metalu:FeO + CO --> Fe + CO2.W procesie wielkopiecowym następuje wytapianie surówki, którą następnie wzbogaca się i powstaje stal. Istotnyfakt, to iŜ czyste Ŝelazo jest kruche i raczej nie nadaje się do praktycznego zastosowania. Dodatek 0,5 - 1,5%węgla tak dalece zmienia właściwości stali, szczególnej jej wytrzymałość mechaniczną, twardość i kruchość, iŜ
Created by Neevia Document Converter trial version http://www.neevia.com

66
moŜe być wszechstronnie wykorzystywana w konstrukcjach technicznych. Dalsze dodawanie węgla do 2,5 - 3%powoduje powstanie kruchego Ŝeliwa.Domieszki hematytu w naturalnej krzemionce powodują, iŜ najtańsze szkło jest Ŝółtawe bądź zielonkawe (tzw.szkło luxver, np. w starych przychodniach). Neutralizację tego koloru uzyskuje się przez dodanie selenu,barwiącego szkło na róŜowo lub czerwono – rubinowo. Wytworzone domieszkami barwy znoszą się i szkło stajesię przezroczyste.
Created by Neevia Document Converter trial version http://www.neevia.com

67
CHEMIA ZWIĄZKÓW ORGANICZNYCHWPROWADZENIE
Związkami organicznymi nazywamy ogromną większość związków węgla; wyjątki od powyŜszego przepisupodane zostały w dziale poprzednim. Oprócz węgla pierwiastkami tworzącymi najpopularniejsze związkiorganiczne są: wodór, tlen, azot, siarka, fosfor, fluor, chlor, brom oraz jod.Zasady rządzące budową związków organicznych zebrane zostały w tzw. teorię strukturalną, którą tworzy kilkareguł:a) Atomy węgla w związkach organicznych są zawsze czterowartościowe. W wyniku hybrydyzacji
tetragonalnej sp3 następuje uśrednienie funkcji falowych opisujących orbitale elektronowe węgla, skutkiemczego wszystkie wiązania są równocenne.
b) Atomy węgla mogą łączyć się między sobą w łańcuchy o dowolnej długości – proste lub rozgałęzione, atakŜe w pierścienie o dowolnej wielkości. W tym miejscu zaznaczymy tylko, iŜ od ilości atomów węgla wpierścieniu zaleŜy jego trwałość oraz moŜliwość przyjmowania róŜnych pozycji w przestrzeni.
c) Atomy węgla mogą łączyć się ze sobą wiązaniami pojedynczymi, podwójnymi oraz potrójnymi. Oczywiściez rodzajem wiązania nierozerwalnie sprzęŜony jest typ hybrydyzacji, co stanowi uzupełnienie punktu a). Odkrotności wiązania zaleŜy równieŜ jego sztywność i zdolność rotacji.
d) Elektrony atomu węgla nie biorące udziału we wiązaniach węgiel-węgiel biorą udział we wiązaniach zinnymi atomami (np. wodoru, tlenu itd.).
Cechą charakterystyczną kaŜdego związku organicznego jest jego skład jakościowy i ilościowy oraz kolejnośćpowiązania atomów, czyli konstytucja oraz ich rozmieszczenie w przestrzeni, czyli konfiguracja. Bardzo częstonatrafiamy na związki o takim samym składzie chemicznym, a w związku z tym – wzorze sumarycznym, jednakróŜniące się wzorem strukturalnym; nazywamy je izomerami, a zjawisko ich występowania – izomerią. IzomeryróŜnią się od siebie właściwościami fizycznymi i / lub aktywnością chemiczną. Znane są róŜne rodzaje izomerii,zaleŜne od róŜnic pomiędzy izomerami. Odnosząc się do wymienionych wyŜej cech związków, wyróŜniamyizomerię konstytucyjną (szkieletową i podstawnikową) oraz konfiguracyjną (geometryczną i optyczną).a) Izomeria szkieletowa polega na tym, Ŝe dwa związki organiczne zawierają taką samą liczbę atomów węgla,
mają taki sam wzór sumaryczny, jednak róŜnią się szkieletem węglowym. WyróŜniamy w jej obrębieszczególne przypadki, np. izomerię połoŜenia wiązania podwójnego.
b) Izomeria połoŜenia podstawnika polega na identyczności wzorów sumarycznych i szkieletów węglowychizomerów, zaś przyłączeniem niewodorowego podstawnika do innego atomu węgla.
c) Izomeria geometryczna (cis-trans, Z-E) występuje w obrębie związków z wiązaniami wielokrotnymi –wiązania pi uniemoŜliwiają rotację wokół wiązania, dlatego teŜ najbliŜsze podstawniki nie mogą zamieniaćsię miejscami; jeŜeli występują po jednej stronie, mówimy o układzie cis (zuzammen – niem. razem ), wprzeciwnym wypadku – o układzie trans (entgegen – niem. oddzielnie).
d) Izomeria optyczna związana jest z występowaniem w cząsteczce atomu węgla połączonego z czteremaróŜnymi podstawnikami (tzw. asymetrycznego atomu węgla, oznaczany gwiazdką). Mogą to byćstereoizomery (enancjomery, lustrzane odbicia) bądź diastereoizomery (gdy obecny jest więcej niŜ jedenatom *C).
Równie często jak izomery spotykamy związki o takiej samej budowie zasadniczej, lecz róŜniące się liczbą grup-CH2-; nazywamy je homologami. Homologi ułoŜone według wzrastającej stopniowo liczby grup –CH2–tworzą tzw. szereg homologiczny. Mówimy zatem o szeregu homologicznym alkanów, alkadienów,cykloalkanów itd. Porównując dwa rysunki związków chemicznych moŜemy stwierdzić, iŜ są to izomery,homologi, bądź związki toŜsame (identyczne).W wielu przypadkach obecność określonego elementu budowy związku (np. ugrupowania atomów) daje efekt wpostaci z góry znanych właściwości. Omówimy to na przykładach:a) efekt indukcyjny obserwuje się w cząsteczkach z elektroujemnym podstawnikiem przy atomie węgla; układtaki powoduje wytworzenie cząstkowego ładunku – ujemnego na podstawniku i dodatniego na węglu, poniewaŜten pierwszy ściąga elektrony z swe sąsiedztwo,b) efekt mezomeryczny polega na przemieszczeniu elektronów wewnątrz cząsteczki tak, iŜ trudno jest wskazaćich dokładne połoŜenie – moŜna wskazać dwie struktury graniczne, zaś w rzeczywistości obecna jest formapośrednia (tzw. wiązanie przechodnie); przykładowo takie przemieszczenie następuje po dysocjacji grupykarboksylowej:
Created by Neevia Document Converter trial version http://www.neevia.com

68
Wiele związków oganicznych wykazuje podobieństwo przez obecność charakterystycznej grupy atomów wokreślonym układzie, co ułatwia wydatnie ich klasyfikację. Grupy takie nazywamy grupami funkcyjnymi. WponiŜszej tabeli zebrano najwaŜniejsze z nich.
Grupa funkcyjnaKlasa związków
budowa nazwafluorowcopochodne -X (F, Cl, Br, I) fluorowiecalkohole, fenole -OH hydroksylowakwasy karboksylowe -COOH karboksylowaaldehydy -CHO aldehydowaketony -CO- karbonylowa (ketonowa)estry -COO- estrowaetery -C-O-C- eterowanitrozwiązki -NO2 nitrowazwiązki azowe -N=N- azowanitryle -CN cyjanowa (nitrylowa)aminy -NH2 aminowaamidy -CONH2 amidowatiole -SH tiolowasulfozwiązki -SO3H sulfonowa
Na podstawie znajomości wzoru strukturalnego substancji moŜna dociekać, czy będzie ona barwna. Kolorzwiązku zaleŜy do obecności w jego cząsteczce chromoforu oraz auksochromu.a) Chromoforem (grupą chromoforową) nazywamy grupę funkcyjną, której obecność nadaje barwę. Grupy
takie zawierają wiązanie podwójne, gdyŜ na powstałym skupisku elektronów łatwo rozszczepia się światło.Do najpopularniejszych chromoforów naleŜą grupy: etylenowa: –CH=CH–, karbonylowa: –CO–, nitrowa: –NO2, nitrozowa: –NO, azowa: –N=N–, siarczkowa: –CS–.
b) Auksochromem (grupą auksochromową) nazywamy grupę, która wprowadzona do cząsteczki barwnikaprzesuwa pasmo absorpcyjne w jego widmie, czyli zmienia barwę. Pozwala to niekiedy na przesunięciewidma w zakres widzialny dla oka ludzkiego. Najczęściej spotykane auksochromy to: –N=R, –NHR, –NH2,–OH, –O–CH3, –Br, –Cl.
Dla przykładu – związkiem barwnym i wskaźnikiem chemicznym jest oranŜ metylowy: p,p-dimetyloaminoazobenzenosulfonian sodu: (H3C)2-N-C6H4-N=N-C6H4-SO3Na. Posiada on rdzeń (chromogen)nadający barwę, złoŜony z chromoforowej grupy azowej oraz dwóch pierścieni aromatycznych, a takŜeauksochromowe grupy: metylowe, aminowe i sulfonowe. Sumowanie efektu powyŜszych składników daje barwępomarańczową (w środowisku obojętnym).Związki organiczne ulegają rozmaitym reakcjom, które klasyfikuje się nieco odmiennie niŜ w przypadkuzwiązków nieorganicznych. Ze względu na relacje między substratami i produktami wyróŜniamy:a) reakcje podstawienia (substytucji), kiedy atom lub grupa atomów zostaje zastępowana przez inną, np.:CH3Cl + NaOH ---H2O---> NaCl + CH3OH,b) reakcje przyłączenia (addycji), kiedy do wiązania wielokrotnego przyłącza się cząsteczka powodując jegodegradację, np.:CH2CH2 + HCl ---> CH3CH2Cl,c) reakcje odszczepienia (eliminacji), kiedy od substratu oderwane zostają dwa podstawniki, dające popołączeniu wydzielany produkt, np.:CH3CH2Cl ---NaOH+C2H5OH---> CH2CH2 + NaCl + H2O,CH3CH(OH)CH3 ---Cu+CrO3---> CH3COCH3 + H2 (2CrO3 + 3H2 ---> 3H2O + Cr2O3).Reakcje moŜna równieŜ podzielić ze względu na charakter biorących w niej udział reagentów. Tu wyróŜniamy:a) reakcje rodnikowe (homolityczne), kiedy biorą w niej udział substancje z niesparowanym elektronem, czylirodniki (paramagnetyczne związki niepolarne): AooB ---> Ao + oB (występujące po prawej stronie elektrony nienadają cząsteczce ładunku, tylko sygnalizują niesparowanie),b) reakcje jonowe (heterolityczne), kiedy biorą w niej udział substancje o budowie polarnej bądź jonowej:AooB ---> Aoo(+) + B(-); zachodzi między związkami o podobnej budowie (zasada wspomagania).Reakcje mogą zachodzić według jednego z dwóch charakterystycznych mechanizmów, co równieŜ moŜestanowić kryterium ich podziału:a) mechanizm elektrofilowy (ze skłonnością do elektronów) ma miejsce, gdy czynnikiem atakującym jest układz niedoborem (deficytem, luką) elektronów, np. kation, kwas Lewisa czy spolaryzowany dipol z biegunemdodatnim,b) mechanizm nukleofilowy (ze skłonnością do jądra) ma miejsce, gdy czynnikiem atakującym jest układ zwolnymi parami elektronowymi, np. aniony, zasady Lewisa czy spolaryzowany dipol z biegunem ujemnym.
Created by Neevia Document Converter trial version http://www.neevia.com

69
WĘGLOWODORY
Węglowodorami, jak sama nazwa wskazuje, nazywamy związki węgla z wodorem. Nie występuje tu Ŝadnacharakterystyczna grupa funkcyjna. Zbiór węglowodorów jest niezwykle liczny, dlatego dla ułatwieniapodzielono go na szereg klas:
Dalszy podział przedstawiony zostanie podczas omawiania poszczególnych klas.
ALKANY
Alkany są nasyconymi węglowodorami alifatycznymi. Nasycony charakter oznacza występowanie wcząsteczkach wyłącznie wiązań pojedynczych. Zgodnie z załoŜeniami teorii strukturalnej wzór ogólny alkanówto CnH2n+2, gdzie n to liczba atomów węgla w cząsteczce. Alkany tworzą zatem własny szereg homologiczny,którego pierwszym przedstawicielem jest metan – CH4. Cząsteczki o więcej niŜ trzech atomach węgla mogąwystępować w postaci izomerów strukturalnych – w praktyce oznacza to moŜliwość rozgałęzień w obrębiecząsteczki. Alkany prostołańcuchowe mają wyŜsze temperatury wrzenia niŜ ich izomery rozgałęzione o takiejsamej ilości atomów węgla, co wynika z kumulacji oddziaływań wzdłuŜ długiej osi cząsteczki. Rozgałęzieniaoznaczają równieŜ występowanie atomów węgla o róŜnej rzędowości. Rzędowość atomów węgla określa liczbainnych atomów węgla, z którą atom wyjściowy jest połączony: i tak węgiel 1-rzędowy połączony jest tylko zjednym atomem węgla, 2-rzędowy – z dwoma itd. Z teorii strukturalnej wynika, iŜ maksymalna rzędowośćatomu węgla wynosi cztery. Czyste alkany są bezbarwnymi związkami nierozpuszczalnymi w wodzie, za torozpuszczalnymi w substancjach organicznych. Ich stan skupienia silnie zaleŜy od długości i rozgałęzieniałańcucha. N-alkany (łańcuch prosty) z 1-4 atomami węgla są gazami, z 5-15 – cieczami, zaś powyŜej 15 –ciałami stałymi.Alkany otrzymać moŜna bardzo róŜnorodnymi metodami:a) poprzez uwodornienie (hydrogenację) węglowodorów nienasyconych w warunkach zwiększonego ciśnienia,
np.:C2H4 + H2 ---p---> C2H6,
b) poprzez uwodornienie fluoropochodnych alkanów, np.: RCl + H2 ---> RH + HCl,
c) w wyniku reakcji Würtza, czyli traktowania chlorowcopochodnych alkanów sodem metalicznym, np.:CH3CH2Cl + CH3CH2Cl + 2Na ---> CH3CH2CH2CH3 + 2NaCl;naleŜy zauwaŜyć, iŜ jeśli poddaje się takiej reakcji dwa róŜne alkany, to otrzymuje się aŜ cztery pochodna,co jest wynikiem losowego łączenia się powstałych rodników,
d) przez dekarboksylację kwasów organicznych, co w praktyce jest trudne do przeprowadzenia ze względu nastopień utlenienia węgla (+3), np.:CH3COONa + NaOH ---T, CaO---> CH4 + Na2CO3,
e) przez działanie woda na węglik; gdy jest to węglik z nieparzystą liczbą atomów węgla, produktem reakcjijest równieŜ węglowodór o nieparzystej liczbie atomów węgla w łańcuchu, np.:Al4C3 + 12H2O ---> 4Al(OH)3 + 3CH4,
f) w wyniku reakcji Kolbego; polega ona na elektrolizie soli kwasów karboksylowych, np.:2CH3COONa <==> 2CH3COO- + 2Na+,A(+): 2CH3COO- - 2e- ---> CH3CH3 + 2CO2,
g) metodą Bergiusa, czyli bezpośrednio z pyłu węglowego i wodoru:nC + mH2 ---T,p---> CnH2m,
h) metodą Fischera – Tropscha, w której najpierw działając wodą na rozŜarzony koks otrzymujemy gazwodny:C + H2O ---> CO + H2,
Created by Neevia Document Converter trial version http://www.neevia.com

70
a następnie, spręŜając go w odpowiednich proporcjach, otrzymujemy Ŝądany węglowodór:nCO + mH2 ---> CnH2m-2 + H2O,
i) wykorzystując związki Grignarda, czyli magnezoorganiczne – takie, w których występuje atom magnezuzwiązany z atomem węgla; wpierw redukuje się chlorowcopochodną alkanu magnezem:RCl + Mg ---> RMgCl,a następnie działając na produkt reakcji wodą z wydzieleniem hydroksychlorku magnezu:RMgCl + H2O ---> RH + Mg(OH)Cl.
Alkany z racji nasyconego charakteru są mało reaktywne, mimo to ulegają róŜnorodnym reakcjom:a) Spalanie ich jest procesem wybuchowym, więc lotne alkany bądź opary ciekłych tworzą z powietrzemmieszaniny wybuchowe. Ogólne wzory na trzy rodzaje spalania podana są poniŜej:
( )
( )
( ) OHnnCOn
HC
OHnnCOOn
HC
OHnnCOOn
HC
nn
nn
nn
2222
2222
22222
12
1
12
12
12
13
++→
++
++→
++
++→
++
+
+
+
b) Halogenowanie na świetle wg mechanizmu wyjaśnionego przy okazji metanu:RH + X2 ---> RX + HX.Atom fluorowca moŜe przyłączyć się do kaŜdego miejsca w cząsteczce, stąd teŜ nagminnie występuje izomeriapołoŜenia podstawnika. Z elektrostatycznej dyslokacji chmury elektronowej wynika, iŜ głównym produktemreakcji jest ten, który powstał przez przyłączenie się halogenku do atomu węgla o wyŜszej rzędowości. Np.chlorowanie 2-metylopropanu prowadzi do otrzymania 1-chloro-2-metylopropanu oraz 2-chloro-2-metylopropanu, ten drugi jednak występuje w zdecydowanej przewadze.c) Nitrowanie zachodzi tu dosyć trudno, nie jest jednak niemoŜliwe:RH + HONO2 ---> RNO2 + H2O.d) Sulfonowanie jest reakcją z oleum, w wyniku której powstaje odpowiedni kwas sulfonowy. Sole tychostatnich znane są jako detergenty.RH + H2SO4 ---> RSO3 + H2O.e) Chlorosulfonowanie to reakcja z tlenochlorkiem siarki; produktem tej reakcji jest odpowiedni sulfochlorek:RH + SO2Cl2 ---> RSO2Cl + HCl.f) Izomeryzacja to przekształcanie n-alkanów w ich rozgałęzione izomery, np.:CH3CH2CH2CH3 ---war---> CH3CH(CH3)CH3.g) Kraking polega na katalitycznym ogrzewaniu mieszaniny alkanów, które pod wpływem tych czynnikówpękają na krótsze alkany i odpowiednie alkeny, np.:CH3(CH2)5CH3 ---war---> CH3(CH2)2CH3 + CH2=CHCH3.h) Aromatyzacja to przekształcanie węglowodoru alifatycznego w aromatyczny, np.:CH3CH2CH3 ---war---> ∆ + H2.
METANNajprostszym alkanem i – jak się wydaje – najprostszym węglowodorem jest metan. Jego wzór sumaryczny:CH4. W obrębie atomu węgla zachodzi umownie hybrydyzacja, w wyniku której powstają cztery równocenneorbitale. Wzajemne oddziaływanie pomiędzy tymi ostatnimi powoduje, iŜ cząsteczka metanu ma kształtczworościanu foremnego, zaś atomy wodoru skierowane są w jego naroŜa. Z tetraedrycznej budowy wynika, iŜwiązania węgiel – wodór skierowane są względem siebie pod kątem 109o28’. Takie ustawienie przestrzennewiązań powoduje kompensację ich momentów dipolowych. W efekcie cząsteczka jest niepolarna, wobec czegonie ulega rozpuszczenie w substancjach polarnych (np. wodzie), rozpuszcza się zaś z rozpuszczalnikachorganicznych. RóŜnica elektroujemności pomiędzy atomem węgla i wodoru wynosi 0,4, skąd wnioskujemy, iŜwystępują tu wiązania kowalencyjne. Długość wspomnianego wiązania wynosi 109pm, zaś energia 425 kJ/mol.Metan otrzymać moŜna na wiele sposobów. Na skalę przemysłową wykorzystuje się gaz ziemny, którego metanjest głównym składnikiem (stanowi 80-90%). W laboratorium dokonuje się to np. działając wodą na węglikglinu:Al4C3 + 12H2O ---> 4Al(OH)3 + 3CH4,bądź teŜ, aby przyspieszyć reakcję, stosuje się zamiast wody kwas solny:Al4C3 + 12HCl ---> 4AlCl3 + 3CH4.Inną popularną metodą jest dekarboksylacja octanu sodu przez działanie nań tlenkiem wapnia w zasadziesodowej:CH3COONa + NaOH ---T,CaO---> CH4 + Na2CO3.
Created by Neevia Document Converter trial version http://www.neevia.com

71
W normalnych warunkach metan jest bezbarwnym i bezwonnym gazem, pozbawionym smaku. Jest lŜejszy odpowietrza, nie rozpuszcza się w wodzie z powodu odmiennego charakteru wiązań w obu cząsteczkach. Metanwykazuje relatywnie małą aktywność chemiczną, głównie z powodu nasyconego charakteru, mimo tego jednakulega reakcjom spalania:spalanie całkowite: CH4 + 2O2 ---> CO2 + 2H2O + E1,półspalanie: CH4 + 3/2O2 ---> CO + 2H2O + E2,spalanie niecałkowite: CH4 + O2 ---> C + 2H2O + E3.Porównując powyŜsze typy spalania widzimy, iŜ róŜnią się one ilością potrzebnego do ich zajścia tlenu,produktem końcowym oraz ilością wydzielonej energii. Proces spalania metanu zachodzi z maksymalnymwykorzystaniem dostępnego w środowisku reakcji tlenu, stąd teŜ ilość ta decyduje o rodzaju spalania. Zwróćmyjeszcze uwagę na produkty końcowe – powstający podczas całkowitego spalania dwutlenek węgla nie jestszczególnie groźny, natomiast wydzielający się przy półspalaniu tlenku węgla jest niebezpieczną trucizną. Z tegopowodu tak waŜne jest odpowiednie przewietrzenie pomieszczeń, w których pracują wszelkiego rodzaju piecegazowe, gdyŜ niedostatek tlenu powoduje pośrednio wydzielanie toksycznego czadu.Metan ulega równieŜ reakcjom z fluorowcami. Reakcje te zachodzą w obecności światła, które rozbijadwuatomowe cząsteczki na wolne rodniki. Jeden z tych ostatnich przyłącza się do cząsteczki metanu, powodującoderwanie atomu wodoru (powstaje obojętna cząsteczka kwasu beztlenowego). Powstały rodnik metanowybłyskawicznie łączy się z drugim fluorowcowym i tak powstaje cząsteczka fluorowcopochodnej metanu, np.:CH4 + Cl2 ---> CH3Cl + HCl.Proces ten moŜna prowadzić dalej, iŜ do zastąpienia wszystkich atomów wodoru atomami fluorowców, np.:CH3Cl + Cl2 ---> + HCl,CH2Cl2 + Cl2 ---> CHCl3 + HCl,CHCl3 + Cl2 ---> CCl4 + HCl.W wyniku powyŜszego chlorowania metanu otrzymujemy kolejno: mono-, di-, tri- i tetrachlorometan.Fluorowanie metanu jest przykładem reakcji podstawienia, czyli substytucji, gdyŜ kolejne atomy wodorusukcesywnie zastępowane są innymi. Reakcje podstawienia charakterystyczne są dla węglowodorównasyconych.
CYKLOALKANY
Cykloalkany to nasycone węglowodory pierścieniowe (cykliczne). Nasycony charakter oznacza występowanie wcząsteczkach wyłącznie wiązań pojedynczych. Nierozgałęzione cylkoalkany mają wzór ogólny postaci CnH2n,gdzie n oznacza liczbę atomów węgla w cząsteczce. PoniewaŜ nie istnieje wielokąt o mniejszej liczbiewierzchołków niŜ trójkąt, toteŜ analogicznie n nie moŜe być mniejsze od trzech. Cykloalkany tworzą szereghomologiczny, którego pierwszym przedstawicielem jest cyklopropan.Cyklopropan (∆) posiada pierścień płaski. Godne uwagi jest, iŜ kąt pomiędzy sąsiadującymi wiązaniami węgiel– węgiel wynosi 60o, zaś charakterystyczny kąt hybrydyzacji tetragonalnej – w przybliŜeniu 109o. DuŜa róŜnicawartości tych kątów ma swoje odzwierciedlenie w silnych napręŜeniach działających na cząsteczkę odwewnątrz. Powodują one swoiste wygięcie wiązań, stąd teŜ mówi się o „bananowych” wiązaniach π.W cząsteczce cyklobutanu () nie mamy do czynienia z tak duŜymi wartościami napręŜeń, gdyŜ kąt pomiędzywiązaniami C-C zbliŜony jest do kąta charakterystycznego hybrydyzacji (90≈109). Związek ten, podobnie jakpoprzedni, posiada pierścień płaski.Nie moŜna tego natomiast powiedzieć o cząsteczce cyklopentanu () i dalszych przedstawicieli. Tutaj sytuacja zkątami między wiązaniami przedstawia się odwrotnie: są one porównywalne bądź większe od kątacharakterystycznego, więc plaski do tej pory pierścień ulega pofałdowaniu. W przypadku cyklopentanu istniejetylko jedna moŜliwość ułoŜenia wiązań.Jednak juŜ cykloheksan występuje w dwóch alternatywnych postaciach, zwanych konformacjami: łódkowej (zosią symetrii, występują większe napięcia) bądź krzesłowej (ze środkiem symetrii, mniej napięciowa i trwalszaod poprzedniej). Ze względu na oddziaływania naprzeciwległych atomów wodoru konformacja łódkowaprzyjmuje postać łódki skręconej.Izomeria geometryczna występuje równieŜ w przypadku dekaliny – dziesięciowęglowego związku o dwóchsześcioczłonowych skondensowanych pierścieniach. Występuje ona w dwóch formacjach: równoległej (cis) oraznaprzemianległej (trans). Wyznacznikiem jest połoŜenie atomów wodoru po tej samej bądź po róŜnych stronachpierścienia.Cykloalkany otrzymać moŜna na róŜne sposoby:a) przez cyklizację dihalogenkoalkanów, które zawierają atomy fluorowca przy pierwszym i ostatnim atomiewęgla (tzw. wiązanie termalne), np.:X-CH2-CH2-CH2-X + Zn ---> ZnX2 + ∆,
Created by Neevia Document Converter trial version http://www.neevia.com

72
b) przez cyklizację soli kwasów karboksylowych pod wpływem temperatury, przy czym najłatwiej ulegają temuprocesowi sole wapniowe; w przykładowej, przedstawionej poniŜej reakcji najpierw zachodzi zobojętnianie, anastępnie odszczepienie cząsteczki węglanu wapnia, w wyniku czego powstaje cykliczny keton:
,c) poprzez uwodornienie związków aromatycznych, np.:
.Same cykloalkany podlegają równieŜ wielu reakcjom (choć pomimo tego wszystkie odporne są na działanieczynników utleniających). Przemiany te mogą polegać na rozerwanie wiązań węgiel – węgiel (w praktyceoznacza to addycję) bądź węgiel – wodór (jest to substytucja). Addycja zachodzi najczęściej przy działaniuczynników jonowych bądź polarnych, zaś substytucja podczas działania fluorowcami na świetle. W przypadkucyklopropanu większość katalizowanych reakcji to addycje:∆ + HJ ---> CH3CH2CH2J (1-jodopropan),∆ + H2O ---H+---> CH3CH2CH2OH (propanol),∆ + H2SO4 ---> CH3CH2CH2SO3H (kwas propanosulfonowy),∆ + H2 ---Ni---> CH3CH2CH3 (propan).
Ogólnie rzecz biorąc istnieje kilka moŜliwości wzajemnego ustawienia pierścieni:
Na lewym rysunku widzimy pierścienie izolowane, tj. rozdzielone atomem węgla nie naleŜącym do Ŝadnego znich. W tej sytuacji wzajemne oddziaływanie chmury elektronowej jest minimalne i w nieznacznym stopniuwpływa na właściwości cząsteczki. Przykładem związku zwierającego p. i. jest trifenylometan.Dalej przedstawione są pierścienie połączone tylko jednym wiązaniem pojedynczym. Występuje tu nałoŜenie sięchmury elektronowej sąsiadujących pierścieni, choć wciąŜ jest ono jeszcze relatywnie niewielkie. Wiązaniepojedyncze umoŜliwia rotację, zaś wiązania wielokrotne usztywniają pierścień, co powoduje występowanieizomerii geometrycznej. Przykładem związku z takim układem jest bifenyl.Trzeci rysunek przedstawia sytuację, w której jeden atom węgla jest wspólny dla dwóch pierścieni. Związkizawierające w swojej budowie takie ugrupowanie nazywamy spiranami. PoniewaŜ wiąŜący atom węgla jestczterowartościowy i tetraedryczny, zatem oba pierścienie leŜą w płaszczyznach wzajemnie prostopadłych.Wynika stąd, iŜ cząsteczka nie moŜe posiadać środka symetrii, zatem jest optycznie czynna i występuje wpostaci izomerów optycznych. Najprostszym przykładem takich związków jest spiropentan, zaś przedstawionyna powyŜszym rysunku związek to spiro-4,5-dekan.Rysunek z prawej strony ilustruje pierścienie skondensowane, czyli posiadające dwa wspólne atomy węgla. Wtakiej sytuacji układ jest płaski, bez jakiejkolwiek moŜliwości rotacji. Ponadto następuje zagęszczenie chmuryelektronowej na odcinku łączącym oba cykliczne fragmenty, co w praktyce objawia się to zwiększeniemprawdopodobieństwa przyłączenia podstawnika w tym właśnie miejscu. Przykładami związków zawierającychp. s. jest dekalina oraz naftalen (w tym drugim przypadku do problemu jeszcze wrócimy).
WĘGLOWODORY NIENASYCONE
Nienasycony charakter związków naleŜących do tej grupy wynika z obecności w ich budowie wiązańwielokrotnych, tj. podwójnych i potrójnych. Powstają one przez boczne nałoŜenie się orbitali, czyliwystępowanie obok wiązań σ równieŜ wiązań π. Te ostatnie odczytywane są przez zbliŜającą się cząsteczkę jakoskupisko chmury elektronowej, stad teŜ chętnie przyłączają się w tym miejscu odczynniki elektrofilowe (addycjado wiązania). Widzimy juŜ zatem, w jak istotny sposób obecność wiązań wielokrotnych modyfikuje właściwościzwiązków chemicznych.Typowe reakcje addycji zachodzą pod wpływem czynników jonowych bądź polarnych. Reakcja przebiega przezkompleks π. Wpierw zgodnie z mechanizmem elektrofilowym do jednego wiązania przyłącza się atomobdarzony ładunkiem dodatnim, w wyniku czego powstaje wspomniany kompleks, a następnie czynniknukleofilowy, obdarzony ładunkiem ujemnym, atakuje drugi atom węgla. Ilustruje to przedstawiony rysunek:
Created by Neevia Document Converter trial version http://www.neevia.com

73
Powstały w przykładzie związek zawiera juŜ tylko wiązanie pojedyncze, umoŜliwiające swobodną rotację.Gdyby atakować wiązanie potrójne, powstałby izomer trans, gdyŜ czynnik nukleofilowy atakuje cząsteczkę zprzeciwnej strony niŜ elektrofilowy (do problemu powrócimy przy omawianiu alkinów).Energie kolejnych wiązań σ, π, π stopniowo maleją – wynoszą odpowiednio: 348 kJ/mol, 264 kJ/mol i 226kJ/mol. Pomimo to całkowita energia wiązania rośnie wraz ze wzrostem krotności i o ile dla wiązaniapojedynczego wynosi 348 kJ/mol, to dla podwójnego: 612 kJ/mol, zaś dla potrójnego: 838 kJ/mol. Bez względuna moŜliwość łatwej addycji, wiązanie potrójne jest najtrudniejsze do rozerwania. Ze wzrostem energii idzie wparze skrócenie długości. I tak: wiązanie pojedyncze ma 154 pm, podwójne: 134 pm, zaś potrójne: 120 pm.Boczne nałoŜenie orbitali powoduje usztywnienie wiązania, co oznacza, iŜ atomy węgla połączone za pomocąniego, jak równieŜ wszystkie bezpośrednio sąsiadujące atomy znajdują się w jednej płaszczyźnie. Dodatkowonie ma moŜliwości zmiany zaistniałego stanu, gdyŜ wiązanie π blokuje rotację wokół całego wiązania. Oznaczato, iŜ atom, znalazłszy się w jednym połoŜeniu (np. po tej samej stronie wiązania co inny), nie moŜe juŜ gozmienić. Efektem tego jest występowanie izomerii geometrycznej cis-trans (Z-E) w związkach o róŜnychpodstawnikach przy atomach węgla połączonych wiązaniem wielokrotnym. Jest to rodzaj izomerii przestrzennej,czyli stereoizometrii.PrzynaleŜność wybranego związku do szeregu cis lub trans pociąga za sobą róŜnice we właściwościachfizycznych i chemicznych. W przypadku izomerów trans, zwłaszcza symetrycznych, moŜna mówić o istnieniupewnego środka symetrii, gdzie znoszą się momenty dipolowe naprzemianległych fragmentów. W związku zpowyŜszym wypadkowy moment dipolowy cząsteczki ma niewielką wartość i z dobrym przybliŜeniem moŜnago uznać za zerowy. Zupełnie inaczej rzecz ma się w przypadku izomerów cis. Tu składowe momentówdipolowych prostopadłe do długiej osi cząsteczki dodają się, w związku z czym moment wypadkowy zawsze mawartość niezerową. Sprawia to, iŜ oddziaływanie pomiędzy sąsiadującymi cząsteczkami jest większe, więc tebardziej się do siebie zbliŜają. W efekcie izomery cis mają większą gęstość oraz wyŜsze temperatury wrzenia niŜizomery trans tego samego związku.W przypadku dwóch i więcej wiązań wielokrotnych nie bez znaczenia jest ich wzajemna odległość, mierzonailością dzielących je atomów węgla:a) Gdy wiązania wielokrotne znajdują się obok siebie (–C=C=C–), mówimy o układzie skumulowanym.
Występuje w tym przypadku znaczne zagęszczenie chmury elektronowej w pobliŜu wiązania, co powoduje,iŜ atakujący czynnik elektrofilowy z duŜym prawdopodobieństwem uderzy w to właśnie miejsce.
b) Gdy wiązania wielokrotne przedzielone są jednym wiązaniem pojedynczym (–C=C–C=C–), wówczasmówimy o układzie sprzęŜonym. Charakterystyczna jest tu delokalizacja elektronów π, które w wynikuprzyciągania elektrostatycznego zbliŜają się w stronę środkowych atomów węgla. Powoduje to wyrównaniedługości wiązań, które mierzą pomiędzy długością wiązania pojedynczego i podwójnego (np. w cząsteczcebutadienu: CH2=CH–CH=CH2). Oznacza to równieŜ moŜliwość otrzymania rozmaitych produktów addycjido wiązania. Delokalizacja odpowiedniej liczby elektronów π w cyklicznych związkach nienasyconych jestwarunkiem o ich aromatyczności.
c) Gdy wiązania wielokrotne przedzielone są więcej niŜ jednym wiązaniem pojedynczym (–C=C–C–C=C–),mówimy o układzie izolowanym. W takim przypadku wzajemne oddziaływanie wiązań jest minimalne imaleje wraz z ilością dzielących atomów.
Znane nam alifatyczne związki nienasycone moŜemy podzielić ze względu na krotność wiązania oraz zewzględu na liczbę wiązań danego typu. Związki alifatyczne o jednym wiązaniu podwójnym nazywamyalkenami, o dwóch – alkadienami, o trzech – alkartienami itd. Analogicznie związki z jednym wiązaniupotrójnym to alkiny, o dwóch – alkadiiny, o trzech – alkatriiny itd. Wiązania wielokrotne mogą równieŜwystępować w związkach cyklicznych – mówimy wówczas o cykloalkenach, cykloalkadienach, cykloalkinach,cykloalkadiinach itd.
ALKENYAlkeny to związki alifatyczne zawierające w cząsteczce jedno wiązanie podwójne. W związku z tym ostatnimposiadają mniej atomów wodoru niŜ alkany o takich samych długościach łańcuchów. Ich wzór ogólny ma postaćCnH2n, gdzie n oznacza ilość atomów w cząsteczce. PoniewaŜ nie istnieje związek o wzorze CH2, zatem n niemoŜe być mniejsze od dwóch. Ostatni przypadek odpowiada cząsteczce etenu. Alkeny tworzą szereghomologiczny, w którym kaŜdy reprezentant ma podwójną nazwę: eten – etylen, propen – propylen, buten –butylen itd. Druga opcja nazwy jest uŜywana częściej zwyczajowo.
Created by Neevia Document Converter trial version http://www.neevia.com

74
Alkeny otrzymać moŜna róŜnymi sposobami:a) przez eliminację fluorowcowodorów z halogenopochodnych,b) przez eliminację z difluorowcopochodnych, w których podstawniki halogenowe leŜą przy sąsiadujących
atomach węgla, np.:-CH2-CH(X)-CH(X)-CH2- + Zn ---> -CH2-CH=CH-CH2- + ZnX2,
c) przez eliminację wody z alkoholi:-CH2-CH2-CH2-OH ---Al2O3/H2SO4---> -CH2-CH=CH2.
Alkeny ulegają reakcjom charakterystycznym dla związków nienasyconych:a) Są palne – w zaleŜności od zasobności środowiska w tlen występuje spalanie całkowite, połowiczne bądźniecałkowite. Mogą tworzyć z tlenem i z powietrzem mieszaniny wybuchowe,b) Dają reakcje addycji do wiązania podwójnego. Mogą być dzięki temu wykrywane za pomocą próbyodbarwienia wody bromowej oraz roztworu nadmanganianu potasu. Reagują ponadto:- z wodorem na katalizatorze, dając alkany,- z fluorowcami, dając difluorowcoalkany,- z fluorowcowodorami, dając fluorowcoalkany,- z wodą w środowisku kwasowym, dając alkohole,- z kwasami, np. siarkowym (VI), dając kwas alkilosiarkowy, lub chlorowym (I), dając chlorowe pochodne
alkoholi.Ponadto alkeny łatwo ulegają polimeryzacji. Proces ten zachodzić moŜe według mechanizmu jonowego bądźrodnikowego. Produktami są znane z codziennego Ŝycia substancje: polietylen, polipropylen itp.MoŜliwe addycje do wiązania podwójnego alkenów przedstawia poniŜszy schemat:
Jak łatwo zauwaŜyć, podczas addycji cząsteczek asymetrycznych (HX, H2O) do wiązania podwójnego wdłuŜszych alkenach występują róŜne moŜliwości przebiegu reakcji, od tego zaś zaleŜy rodzaj otrzymanegoproduktu. O tym, do którego atomu węgla przyłączy się która część asymetrycznej cząsteczki, decyduje rozkładchmury elektronowej. Gęstość tej ostatniej jest większa w okolicy atomu węgla o niŜszej rzędowości. Nie ma wtym nic dziwnego – przecieŜ im więcej atomów wodoru dookoła atomu węgla, tym bardziej chmura ulegarozproszeniu. W takiej sytuacji głównym produktem reakcji jest związek powstały przez przyłączenie atomuwodoru do atomu węgla o niŜszej rzędowości (tam, gdzie jest „więcej wodorów”). PowyŜsze stwierdzenie nosinazwę reguły Markownikowa. Przykładem jest przedstawiony poniŜej proces hydratacji etylenu:
.Skoro atom węgla o niŜszej rzędowości ma większe powinowactwo do wodoru, to oprócz tego, Ŝe łatwiej goprzyłącza, równieŜ trudniej go oddaje. Oznacza to, iŜ podczas eliminacji cząsteczki niesymetrycznej z pochodnejgłównym produktem reakcji będzie ten, który powstanie w wyniku oderwania atomu wodoru od atomu węgla owyŜszej rzędowości (stamtąd, gdzie jest „mniej wodorów”). Stanowi to treść reguły Zajcewa. Np.:
.
Created by Neevia Document Converter trial version http://www.neevia.com

75
ETENEten, czyli etylen, jest najprostszym przedstawicielem szeregu homologicznego alkenów. Posiada wzórsumaryczny C2H4. W jego cząsteczce obecne jest jedno wiązanie pojedyncze.W warunkach domowych otrzymać go moŜna przez ogrzewanie folii polietylenowej. Następuje wówczasdepolimeryzacja i ulatniają się cząsteczki monomeru:(-CH2-CH2-)n ---T---> n CH2=CH2.Oprócz tego eten powstaje w wielu reakcjach eliminacji, np.:a) z etanolu przez dehydratację: CH3-CH2-OH ---Al2O3,T---> CH2=CH2 + H2O,b) z difluoropochodnej na pyle cynkowym: Cl-CH2-CH2-Cl + Zn ---> CH2=CH2 + ZnCl2,c) z fluoropochodnej: CH3-CH2-Cl + NaOH ---C2H5OH,T≈80oC---> CH2=CH2 + NaCl + H2O,d) z etanu (na katalizatorze Raneya): CH3-CH3 ---Ni---> CH2=CH2 + H2.W warunkach normalnych etylen jest bezbarwnym gazem pozbawionym smaku i zapachu, nierozpuszczalnym wwodzie, za to rozpuszczalnym w ciekłych alkoholach, benzenie oraz toluenie. ZauwaŜmy, iŜ pod tymiwzględami niewiele róŜni się od nasyconego bliźniaka – etanu, stąd niemoŜliwe jest ich rozróŜnienie napodstawie obserwacji właściwości fizycznych.Eten ulega reakcjom spalania, podobnie jak większość węglowodorów. Z powietrzem i tlenem tworzymieszaniny wybuchowe.Dzięki obecności w cząsteczce wiązania podwójnego etylen ma charakter nienasycony. Słabsze energetyczniewiązanie π łatwo ulega degradacji, pozwalając na addycję. Przyłączanie jest reakcją charakterystyczną dlawęglowodorów (i innych związków) nienasyconych. Do etenu przyłączać się mogą róŜne cząsteczki:a) fluorowce:CH2=CH2 + X2 ---> X-CH2-CH2-XW wyniku powyŜszej reakcji powstaje odpowiednia difluoropochodna. Ma to znaczenie przy wykrywaniunienasyconego charakteru wiązań. Rozpuszczając brom w wodzie otrzymujemy wodę bromową opomarańczowym zabarwieniu; gdy ta zetknie się z etenem, odbarwia się. Syntezę tę wykorzystujemy zatem doodróŜniania związków nasyconych od nienasyconych (np. etanu od etenu). Do identyfikacji wiązańwielokrotnych moŜe równieŜ słuŜyć rozcieńczony nadmanganian potasu, który w kontakcie ze związkaminienasyconymi odbarwia się z fioletowego na brunatny.b) fluorowcowodory:CH2=CH2 + HX ---> CH3-CH2-X,c) wodór:CH2=CH2 + H2 ---> CH3-CH3,d) woda (reakcja katalizowana odczynem kwasowym):CH2=CH2 + H2O ---H+---> CH3-CH2-OHW przypadku reakcji etylenu z wodą w środowisku nadmanganianu potasu powstaje glikol etylenowy:3CH2=CH2 + 4H2O + 2KMnO4 ---> 3HO-CH2-CH2-OH+ 2MnO2 + 2KOH.Ten najprostszy diol jest cząsteczką polarną i łatwo tworzy wiązania wodorowe. Dzięki znacznemu obniŜaniutemperatury krzepnięcia wody znalazł zastosowanie w płynach niezamarzających do chłodnic.e) inna cząsteczka etylenu – mówimy wówczas o addycji wzajemnej, czyli polimeryzacji; pod wpływemodpowiednich warunków cząsteczki monomeru łączą się w długie łańcuchy polimeru:n CH2=CH2 ---p, T---> (-CH2-CH2-)n.Taki polimer posiada na końcach fragmenty metylowe, które wykazują wraŜliwość na światło.
ALKINYAlkiny to węglowodory alifatyczne nienasycone, zawierające jedno wiązanie potrójne w cząsteczce. Tworzą oneszereg homologiczny, którego pierwszym przedstawicielem jest etin. Ich wzór ogólny ma postać CnH2n-2, gdzie noznacza ilość atomów w cząsteczce. KaŜdy przedstawiciel alkinów posiada dwie nazwy – właściwą orazspolszczoną, powstała przez zamianę litery „i” na „y”, np.: etin – etyn, propin – propyn, butin – butyn itd.Alkiny są związkami palnymi, a dzięki nienasyconemu charterowi dają addycje do wiązania potrójnego. Wprzypadku dłuŜszych cząsteczek produkty addycji mogą być róŜnorodne i występować w większej liczbie –przykładowo całkowita addycja fluorowcowodoru do propinu daje teoretycznie cztery, praktycznie zaś trzyróŜne produkty. Gdy dołączymy do tego jeszcze moŜliwości wewnątrzcząsteczkowych przegrupowań, liczbapochodnych alkinów stanie się imponująca. Co więcej w grupie tej występują, oprócz wcześniej poznanychzwiązków, aldehydy i ketony.Reakcje alkinów przedstawione zostaną na przykładzie propinu. Reaguje on:a) z wodorem na katalizatorze:
H-C≡C-CH3 + H2 ---> H2C=CH-CH3 + H2 ---> CH3-CH2-CH3
b) z fluorowcami:H-C≡C-CH3 + X2 ---> HXC=CX-CH3 + X2 ---> HX2C-CX2-CH3
Created by Neevia Document Converter trial version http://www.neevia.com

76
c) z fluorowodorami:H-C≡C-CH3 + HX ---> H2C=CX-CH3 (główny) + HX ---> CH3-CX2-CH3 (główny)
H2XC-CHX-CH3 (poboczny)HXC=CH-CH3 (poboczny) + HX ---> H2XC-CHX-CH3 (główny)
HX2C-CH2-CH3 (poboczny)d) z wodą:
H-C≡C-CH3 + H2O ---> H2C=C(OH)-CH3 (główny) ---> H3C-CO-CH3 (propanon)H(OH)C=CH-CH3 (poboczny) ---> CH3-CH2-CHO (propanal)
Spośród wszystkich węglowodorów alkiny wykazują najbardziej kwasowy charakter, poniewaŜ mogą reagowaćz metalami i dawać sole. Dzieje się tak, poniewaŜ potrójne wiązanie miedzy atomami węgla jest wielokrotniesilniejsze niŜ pojedyncze wiązanie węgla z wodorem. Z tego powodu ten ostatni moŜe zostać wymieniony naatom metalu.Chcąc otrzymać w wyniku uwodornienia wiązania potrójnego izomer trans alkanu, przeprowadza się reakcjęalkinu z metalicznym sodem w atmosferze ciekłego amoniaku, otrzymując Ŝądany produkt oraz amidek sodu:R1-C≡C-R2 + 2Na + 2NH3 ---> R1-
HC=CH-R2 + 2NaNH2.
ETINEtin (spolszczone: etyn), czyli acetylen jest pierwszym przedstawicielem szeregu homologicznego alkinów.Dzięki obecności wiązania potrójnego cząsteczka ma kształt liniowy. Odpowiada to kątowi charakterystycznemuhybrydyzacji digonalnej – 180o. Czterowartościowy atom węgla wytwarza dwa wiązania σ, zaś nałoŜenie boczneorbitali powoduje powstanie dwóch wiązań π.Acetylen moŜna otrzymać na kilka sposobów:a) przez traktowanie wodą karbidu. Karbid, czyli węglik wapnia (a właściwie jego acetylenek), powstaje przez
spiekanie wapna palonego produkowanego z kamieni wapiennych:CaCO3 ---T---> CaO + CO2
z koksem:CaO + 3C ---> CaC2 + CO.Następnie tak powstały karbid hydrolizuje:CaC2 + 2H2O ---> Ca(OH)2 + C2H2.Reakcja jest silnie egzotermiczna, co wykorzystywane było w lampkach tzw. karbidówkach.
b) przez pirolityczną eliminację wodoru z alkenów, np. ogrzewając metan do wysokich temperaturpowodujemy jego rozkład z wydzieleniem etynu:2CH4 ---T≈1500oC---> C2H2 + 3H2.
Acetylen posiada nieswoiste właściwości fizyczne, nie wyróŜnia się zatem z grona innych węglowodorów. Jestto gaz bezbarwny, bez smaku i zapachu, nie rozpuszcza się w wodzie. SpręŜony do ciśnienia 1,5 atm. wybucha.Acetylen jest gazem palnym. Spalanie całkowite zachodzi z wydzieleniem bardzo duŜych ilości ciepła(T≈3000oC, ∆H=-1300 kJ/mol), co zadecydowało o zastosowaniu go w palnikach do cięcia metali. Dziękinienasyconemu charakterowi etyn daje reakcje addycji do wiązanie potrójnego (najczęściej moŜna je prowadzićdalej, aŜ do całkowitego wysycenia wiązań). Acetylen reaguje:a) z wodorem na katalizatorze:
H-C≡C-H + H2 ---> H2C=CH2 + H2 ---> CH3-CH3
b) z halogenkami, odbarwiając m. in. wodę bromową:H-C≡C-H + X2 ---> HXC=CXH + X2 ---> HX2C-CX2H
c) z halogenkowodorami:H-C≡C-H + HX ---> H2C=CXH + HX ---> CH3-CX2H (główny) oraz H2XC-CXH2 (poboczny)JeŜeli prowadzimy reakcję dla chloru, to w pierwszym etapie reakcji powstaje chloroetan, czyli chlorekwinylu, będący waŜnym produktem w syntezie organicznej.
d) z wodą wobec katalizatora siarkowego (kwas siarkowy (VI) oraz siarczan (VI) rtęci) (reakcja Kuczerowa):H-C≡C-H + H2O ---HgSO4/H2SO4---> H2C=CHOH ---> CH3-CHOW pierwszym etapie powyŜszej reakcji powstaje forma przejściowa, zwana enolem. Zawiera ona bardzonietrwały układ polegający na występowaniu grupy hydroksylowej przy atomie węgla połączonym z drugimwiązaniem podwójnym. Formacja taka szybko izomeryzuje, dając aldehyd octowy.
e) z metalami, dając acetylenki i wydzielając wodór:2 H-C≡C-H + 2Na ---> 2 H-C≡C-Na + H2
Acetylenki metali lekkich reagują z wodą, np. karbid CaC2, zaś metali cięŜkich – są nierozpuszczalne wwodzie, np. Ag HC2 lub Cu(HC2)2. Suche acetylenki są związkami wybuchowymi.
f) z cyjanowodorem, dając akrylonitryl:H-C≡C-H + HCN ---> CH2=CH-CN,który kopolimeryzowany z monomerami dienowymi daje rodzaj kauczuku syntetycznego.
Created by Neevia Document Converter trial version http://www.neevia.com

77
g) z kwasem octowym, dając octan winylu:H-C≡C-H + CH3COOH ---> CH2=CH-COO=CH3
Ten ostatni w wyniku hydrolizy daje etenol, czyli alkohol winylowy, który w wyniku izomeryzacjiprzekształca się w aldehyd etylowy. Natomiast polimeryzowany daje poli(octan winylu), uŜywany doprodukcji błon fotograficznych.
h) z drugą cząsteczką acetylenu w obecności odpowiednich katalizatorów:2 H-C≡C-H ---Cu2Cl2, NH4Cl---> CH2=CH-C≡CHProduktem tej dimeryzacji jest winyloacetylen (acetylen z wodorem podstawionym grupą winylową:CH2=CH-). Związek ten poddany działalności chlorowodoru daje chloropren (2-chloro-1,3-butadien):CH2=CH-C≡CH + HCl ---> CH2=CH-CCl=CH2,który słuŜy do produkcji kauczuku chloroprenowego, odpornego na oleje i smary. Z kolei bliźniaczy izopren(cis-2-metylo-1,3-butadien) jest monomerem kauczuku naturalnego, który wykazuje wraŜliwość narozpuszczalniki organiczne. Syntetyczny kauczuk butadienowy cechuje duŜa elastyczność. Grupaizoprenowa występuje w wielu związkach organicznych: terpenach, olejkach eterycznych, witaminie A orazβ-karotenie.
i) W wyniku przepuszczania acetylenu przez węgiel aktywny w warunkach wysokiej temperatury następujejego trimeryzacja, której produktem jest benzen:3 H-C≡C-H ---T, C---> C6H6.
W wyniku polimeryzacji otrzymanego wcześniej chlorku winylu powstaje poli(chlorek winylu) – waŜnetworzywo sztuczne (oznaczenia: PCW, PCV, PVC).n H2C=CHCl ---p, T---> [-CH2-CHCl-]n
Postać końcowego produktu reakcji zaleŜy ściśle od warunków, zwłaszcza ciśnienia. MoŜna zatem otrzymaćtwardy winidur bądź miękki igielit. Z winiduru wytwarzało się m. in. obudowy telefonów domowych, zaś zigielitu np. obsadki piór.
WĘGLOWODORY AROMATYCZNE – ARENY
Węglowodory aromatyczne, czyli areny, są szczególnym przypadkiem związków cyklicznych. PokaŜemy to naprzykładzie najprostszego z nich, czyli benzenu C6H6. W świetle przedstawionego wcześniej podziału icharakterystyki węglowodorów powinien to być cyklo-1,3,5-heksatrien, zaś cząsteczka powinna mieć wyglądjak na rysunku z lewej strony. Obecne są w niej trzy wiązania podwójne sprzęŜone parami. MoŜna jednakpostawić pytanie: dlaczego wiązania podwójne mają taki właśnie układ, a nie wprost przeciwny? Nie ma Ŝadnejprzyczyny, dla której połoŜenie wiązań nie miałoby być odwrócone, więc alternatywny układ jest równiepoprawny. Oba przedstawione warianty są skrajnymi strukturami mezomerycznymi (rezonansowymi). Kekulèzaproponował, iŜ obie formy wzajemnie w siebie przechodzą, co obrazuje rysunek środkowy.
Taki model cząsteczki benzenu rozwiązuje wiele problemów, jednak powstaje szereg innych. Wiemy bowiem, iŜwiązania pojedyncze i podwójne róŜnią się długością – jak zatem „złoŜyć” sześciokąt z takich odcinków? Podrugie sygnalizowałby to, Ŝe jedne wiązania róŜnią się od drugich, czego doświadczalnie nie wykazano.Ostatecznie – wiązania podwójne cechuje łatwość addycji, zaś cząsteczka benzenu takiej cechy nie posiada.Wszystkie te fakty prowadza do hipotezy, iŜ benzen cechuje jednorodna budowa w kaŜdym wierzchołku, cojednak trudno pogodzić z poznanymi dotychczas informacjami nt. wiązań chemicznych. Wobec tego przyjmujesię, iŜ w pierścieniu aromatycznym występuje wiązanie zdelokalizowane, któremu nie moŜna jednoznacznieprzypisać miejsca występowania. Wzajemne sprzęŜenie wiązań powoduje, iŜ elektrony tworzące wiązania πzbiegają się w sekstet elektronowy, oznaczany na rysunku prawym przez okrąg wewnątrz pierścienia.Charakterystycznymi cechami związku aromatycznego są zatem: płaska budowa pierścienia i połoŜenie w jednejpłaszczyźnie oraz obecność wieloelektronowego zdelokalizowanego wiązania typu π. Liczba elektronówtworzących wiązanie zdelokalizowane nie moŜe być dowolna – ma ona postać 4 n + 2, gdzie n przyjmujewartości dodatnie i całkowite (reguła Hückla), a dodatkowo wszystkie pochodzą z tego samego poziomuenergetycznego. Areny są związkami niepolarnymi bądź bardzo słabo polarnymi. Wiązanie π otaczającecząsteczkę z góry i z dołu powoduje, iŜ moŜe ona zostać odczytana jako naładowana ujemnie i / lub przyciągaćelementy elektrofilowe (typowe są reakcje substytucji elektrofilowej).Areny moŜna otrzymać w wyniku intensywnej pirolizy (1000oC) paliw kopalnych: ropy naftowej oraz węglakamiennego. Wszystkie one mają szkodliwy wpływ na funkcjonowanie organizmu ludzkiego.
Created by Neevia Document Converter trial version http://www.neevia.com

78
Obecność związków aromatycznych moŜna wykryć przez ogrzewanie próbki w roztworze chloroformu zchlorkiem glinu (lub Ŝelaza) – powstający produkt ma intensywnie Ŝółtą, niebieską bądź czerwoną barwę dziękireakcji:
.
BENZENBenzen jest rozpuszczalnikiem organicznym. Ma postać bezbarwnej cieczy o charakterystycznym zapachu,lotnej i łatwo parującej, co świadczy o niskiej temperaturze wrzenia. Nie rozpuszcza się w wodzie, za to złatwością w alkanach. Otrzymać go moŜna ze smoły pogazowej pozostałej po koksowaniu węgla, bądź wwyniku trimeryzacji acetylenu:3 C2H2 ---p, T---> C6H6.Benzen ulega róŜnorodnym reakcjom:a) Jest to związek palny, a ze względu na uwolnienie energii rezonansu proces jest silnie egzotermiczny (∆H=-
3268 kJ/mol):C6H6 + 15/2 O2 ---> 6CO2 + 3H2O.Daje reakcje halogenowania w obecności odpowiednich kwasów Lewisa. Chcąc wprowadzić do pierścieniaaromatycznego podstawnik halogenowy X, naleŜy zastosować związek Ŝelaza (III) bądź glinu z tympierwiastkiem, np.:C6H6 + Cl2 ---FeCl3/AlCl3---> C6H5Cl + HCl.Produktem powyŜszej reakcji jest chlorobenzen, czyli chlorek fenylu. Mechanizm tak katalizowanej reakcjijest następujący: kwas Lewisa, dąŜąc do uzyskania oktetu elektronowego, przyłącza cząsteczkę chloru ipowstaje kompleks AlCl4
-Cl+. Część zakończona dodatnim atomem chloru atakuje pierścień aromatyczny iprzyłącza się doń. Powstaje wówczas dodatni karbokation oraz jon AlCl4
-. Ten ostatni przyłącza się dowodoru w miejscu ataku fluorowca, powodując powstanie chlorobenzenu oraz nietrwałego połączeniaAlCl3-ClH, które rozpada się na wyjściową cząsteczkę katalizatora oraz cząsteczkę chlorowodoru.W podobny sposób moŜna dołączyć do pierścienia aromatycznego łańcuch alifatyczny. Do syntezy potrzebahalogenową pochodną łańcucha podstawioną w miejscu przyszłego połączenia z pierścieniem. W reakcjitworzy się kompleks łańcuch-katalizator, który przyłącza się do pierścienia. Następnie tworzy sięzamierzony produkt reakcji, a kompleks katalizator-halogenowodór rozpada się. Metoda ta znana jest jakoreakcje Friedla – Craftsa. Np.:C6H6 + RBr ---FeBr3/AlBr3---> C6H5R + HBr.W przypadku alkilowych pochodnych benzenu temperatura wrzenia rośnie wraz ze wzrostem liczby grup-CH2- w łańcuchu.
b) Łatwiej niŜ związki alifatyczne ulega działaniu mieszaniny nitrującej:C6H6 + HNO3 ---H2SO4---> C6H5NO2 + H2O,dając nitrobenzen. Łatwa reakcja nitrowania jest charakterystyczna wyłącznie dla związków aromatycznych,a powstałe nitropochodne mają zawsze barwę Ŝółtą.
c) Benzen reaguje z oleum, dając kwas benzenosulfonowy:C6H6 + H2SO4 ---SO3---> C6H5SO3H + H2O.
d) MoŜe równieŜ dawać addycje do pierścienia aromatycznego, np.:C6H6 + 3H2 ---Ni|T≈200oC|2,5Mpa---> C6H12 (cykloheksan),C6H6 + 3Cl2 ---hν--> C6H6Cl6 (1,2,3,4,5,6-heksachlorocykloheksan).
HOMOLOGI BENZENUZnane są liczne homologi benzenu, głównie jego pochodne powstałe przez podstawienie wodorów metylami,etylami itd. Najprostsze z nich to:- metylobenzen, czyli toluen: C6H5-CH3,- etylobenzen: C6H5-CH2-CH3,- dimetylobenzeny, czyli ksyleny: C6H4(CH3)2 – występują trzy izomery.W przypadku pochodnej benzenu z dwoma podstawnikami przy pierścieniu aromatycznym występuje izomeriaorto-meta-para. Jest ona wynikiem róŜnych moŜliwości ustawienia podstawników. Oczywiście w przypadku
Created by Neevia Document Converter trial version http://www.neevia.com
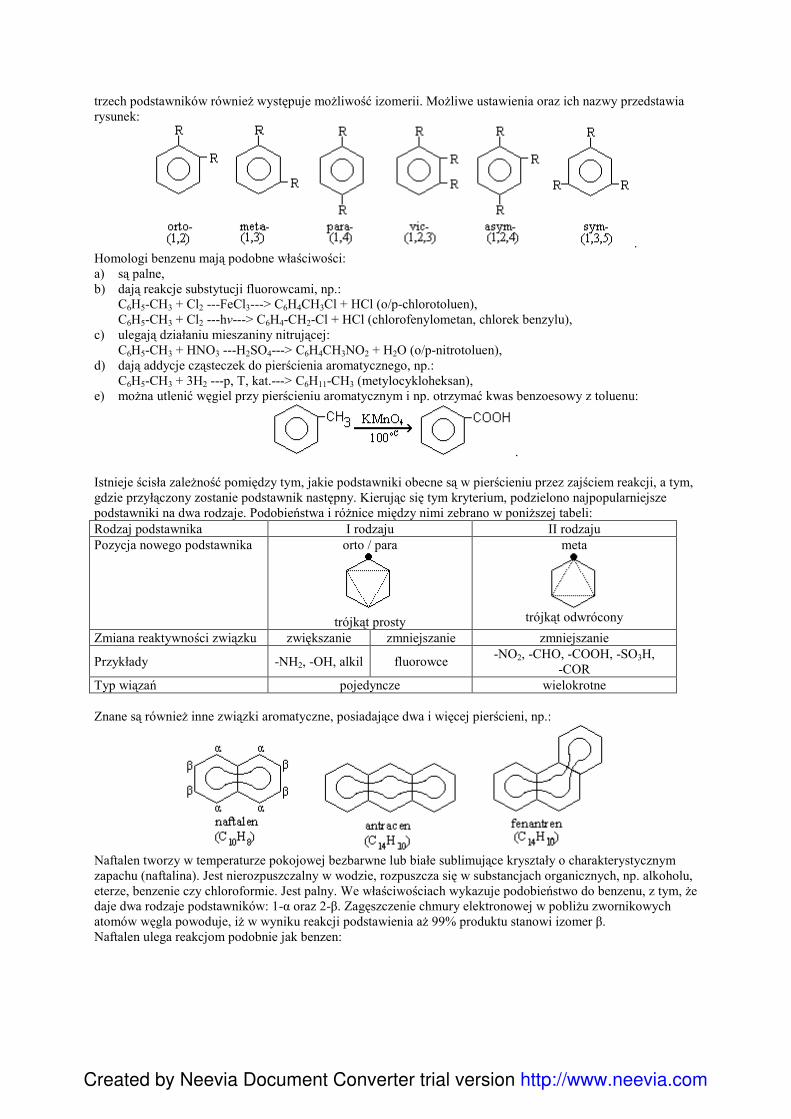
79
trzech podstawników równieŜ występuje moŜliwość izomerii. MoŜliwe ustawienia oraz ich nazwy przedstawiarysunek:
.Homologi benzenu mają podobne właściwości:a) są palne,b) dają reakcje substytucji fluorowcami, np.:
C6H5-CH3 + Cl2 ---FeCl3---> C6H4CH3Cl + HCl (o/p-chlorotoluen),C6H5-CH3 + Cl2 ---hν---> C6H4-CH2-Cl + HCl (chlorofenylometan, chlorek benzylu),
c) ulegają działaniu mieszaniny nitrującej:C6H5-CH3 + HNO3 ---H2SO4---> C6H4CH3NO2 + H2O (o/p-nitrotoluen),
d) dają addycje cząsteczek do pierścienia aromatycznego, np.:C6H5-CH3 + 3H2 ---p, T, kat.---> C6H11-CH3 (metylocykloheksan),
e) moŜna utlenić węgiel przy pierścieniu aromatycznym i np. otrzymać kwas benzoesowy z toluenu:
.
Istnieje ścisła zaleŜność pomiędzy tym, jakie podstawniki obecne są w pierścieniu przez zajściem reakcji, a tym,gdzie przyłączony zostanie podstawnik następny. Kierując się tym kryterium, podzielono najpopularniejszepodstawniki na dwa rodzaje. Podobieństwa i róŜnice między nimi zebrano w poniŜszej tabeli:Rodzaj podstawnika I rodzaju II rodzajuPozycja nowego podstawnika orto / para
trójkąt prosty
meta
trójkąt odwrócony
Zmiana reaktywności związku zwiększanie zmniejszanie zmniejszanie
Przykłady -NH2, -OH, alkil fluorowce-NO2, -CHO, -COOH, -SO3H,
-CORTyp wiązań pojedyncze wielokrotne
Znane są równieŜ inne związki aromatyczne, posiadające dwa i więcej pierścieni, np.:
Naftalen tworzy w temperaturze pokojowej bezbarwne lub białe sublimujące kryształy o charakterystycznymzapachu (naftalina). Jest nierozpuszczalny w wodzie, rozpuszcza się w substancjach organicznych, np. alkoholu,eterze, benzenie czy chloroformie. Jest palny. We właściwościach wykazuje podobieństwo do benzenu, z tym, Ŝedaje dwa rodzaje podstawników: 1-α oraz 2-β. Zagęszczenie chmury elektronowej w pobliŜu zwornikowychatomów węgla powoduje, iŜ w wyniku reakcji podstawienia aŜ 99% produktu stanowi izomer β.Naftalen ulega reakcjom podobnie jak benzen:
Created by Neevia Document Converter trial version http://www.neevia.com

80
a) bromowanie – powstaje mieszanina 1-bromonaftalenu i 2-bromonaftalenu:
,b) nitrowanie – powstaje mieszanina 1-nitronaftalenu i 2-nitronaftalenu:
,c) uwodornienie – powstaje dekalina:
.
JEDNOFUNKCYJNE POCHODNE WĘGLOWODORÓW
PowyŜsze związki są produktami reakcji węglowodorów z róŜnorodnymi odczynnikami, w wyniku którychpowstają połączenia typu R-X, gdzie R oznacza rodnik węglowodorowy, zaś X niewęglowodorową grupęfunkcyjną. Rodnik moŜe być dowolnego rodzaju – alkil, cykloalkil, alkenyl, alkinyl czy aryl. Z kolei spiswiększości grup funkcyjnych podany został we wprowadzeniu do chemii organicznej.
FLUOROWCOPOCHODNE WĘGLOWODORÓWZe związkami tymi spotkaliśmy się juŜ wcześniej, przy omawianiu produktów substytucji i addycji dowęglowodorów. Fluorowcopochodne mają postać RX, gdzie R jest rodnikiem, zaś X to atom fluoru, chloru,bromu bądź jodu. Oczywiście w cząsteczce moŜe znajdować się więcej niŜ jeden atom fluorowca, jednakrozwaŜając róŜnorodność przypadków nie moŜna by podać wzoru ogólnego.Fluorowcopochodne moŜna klasyfikować na wiele sposobów:- ze względu na liczbę atomów fluorowca w cząsteczce: mono-, di-, trifluorowcopochodne itd.,- ze względu na rzędowość węgla, z którym połączony jest fluorowiec: I-, II- i III-rzędowe,- ze względu na rodzaj grupy węglowodorowej, z którą związany jest fluorowiec: fluorowcoalkany, -alkeny, -
alkiny, -areny.
FLUOROWCOALKANYFluorowcoalkany tworzą szereg homologiczny, w którym temperatura wrzenia rośnie wraz ze wzrostem masyatomowej chlorowca, liczby atomów chlorowca, długości łańcucha węglowego oraz rzędowości pochodnej (I-rzędowe cechuje największa temperatura wrzenia). Zazwyczaj są to związki nierozpuszczalne w wodzie. Wgrupie tej moŜe występować czynność optyczna, czyli zdolność do skręcania płaszczyzny światłaspolaryzowanego liniowo. Podstawowym warunkiem izomerii optycznej jest obecność w cząsteczce atomuwęgla połączonego z czterema róŜnymi podstawnikami – tzw. asymetrycznego atomu węgla. Izomery optyczne(enancjomery, stereoizomery) mają się do siebie jak swoje lustrzane odbicia, dlatego cząsteczki takie nazywamychiralnymi. Do rozróŜnienia konkretnego przypadku słuŜy system Ingolda. W metodzie tej wpierw numeruje siępodstawniki od najcięŜszego do najlŜejszego; następnie patrzy się na cząsteczkę tak, by najlŜejszy podstawnik(bardzo często jest nim atom wodoru) znajdował się z tyłu, aŜ wreszcie sprawdza się kierunek, w którymnaleŜałoby się poruszać, aby przejść przez podstawniki zgodnie z rosnącą numeracją. JeŜeli naleŜałoby sięporuszać zgodnie z kierunkiem ruchu wskazówek zegara, to mówimy o odmianie prawoskrętnej (R – retrus), wprzeciwnym wypadku – o lewoskrętnej (S – sinister).O właściwościach chemicznych fluorowcoalkanów decyduje polaryzacja wiązania węgiel – podstawnik.Zazwyczaj cząsteczka jest i tak mniej lub bardziej polarna, gdyŜ bardziej elektroujemny atom halogenku ściągachmurę elektronową w swoje sąsiedztwo (przykład opisanego we wprowadzeniu efektu indukcyjnego).Powoduje to pojawienie się na najbliŜszym atomie węgla cząstkowego ładunku dodatniego, zaś na samymatomie fluorowca – ujemnego. Sprawia to, iŜ reakcje zachodzą głównie według mechanizmu nukleofilowego(typowe są substytucje nukleofilowe). W ich wyniku atom fluorowca moŜe zostać wymieniony na inną grupęfunkcyjną: -OH (otrzymamy alkohol), -SH (tiol), -NH2 (aminę), -CN (nitryl) itd.1. Wśród substytucji nukleofilowych wyróŜniamy reakcje I rzędu – gdy ich szybkość zaleŜy wyłącznie od
stęŜenia jednego z reagentów oraz II rzędu – gdy ich szybkość zaleŜy od stęŜenia wszystkich substratów.
Created by Neevia Document Converter trial version http://www.neevia.com

81
a) I rzędu:
W pierwszym przypadku prawdopodobieństwo ataku z góry i z dołu jest takie samo, powstaje zatem mieszaninaracemiczna, w której obecne są zarówno izomery R i S. Natomiast w drugim przypadku mamy zawsze doczynienia z inwersją konfiguracji, czyli zmianą typu skręcalności optycznej (przeciwieństwem inwersji jestretencja – zachowanie konfiguracji wokół centrum chiralności).b) Większość substytucji nukleofilowych II rzędu zachodzi z inwersją, np.:
RX + Mg ---> RMgX (związek Grignarda),RX + 2NH3 ---> RNH2 + NH4X (amina),RX + OH- ---> ROH + X- (alkohol),RX + CN- ---> RCN + X- (nitryl),RX + HC≡C- ---> R-C≡CH (alkin),R-CH2X + R’-ONa ---> R-CH2-O-R’ + NaX (eter).
2. Atomy halogenów mogą być eliminowane z fluorowcopochodnych. Do czynników szczególniesprzyjających eliminacji zaliczamy: wyŜszą rzędowość podstawionego atomu węgla oraz wysokązasadowość atakującego czynnika, który łatwiej łączy się z protonem niŜ proton wymienia.
a) Rzędowość atomu węgla zawierającego podstawnik ma wpływ na łatwość zachodzenia tego procesu. Wwyniku efektu indukcyjnego na fluorowcu gromadzi się cząstkowy ładunek ujemny, a na najbliŜszym węglu– dodatni. Im wyŜsza jest rzędowość tego ostatniego, tym cząstkowy ładunek ma większą wartość. Zatemłatwiej jest oderwać podstawnik od atomu węgla o wyŜszej rzędowości.
b) O tym, jak zasadowość atakujących czynników ma wpływ na przebieg reakcji, łatwo przekonać się naprzykładzie traktowania monofluorowcopochodnej wodorotlenkiem sodu. W celu eliminacji cząsteczki HXreakcję prowadzi się w etanolu. Ten ostatni reaguje z zasadą, dając etanolan sodu, który w wynikudysocjacji daje jony etanolanowe C2H5O
-. Te z kolei są bardziej nukleofilowe niŜ aniony OH- ze względu nawiększe zagęszczenie chmury elektronowej (efekt indukcyjny). Ostatecznie anion etanolanowy odłącza odfluoropochodnej atom wodoru, zaś jon Na+ - atom fluorowca. Ostatecznie otrzymuje się alken, eliminowanyprodukt oraz pomocnicze substancje wyjściowe (lewa strona rysunku). JeŜeli natomiast reakcję prowadzi siębez obecności etanolu, wówczas jedynymi czynnikami atakującymi są aniony wodorotlenkowe. Gdy kationNa+ odszczepia atom fluorowca, grupa OH- wchodzi na jego miejsce. W wyniku przemiany otrzymuje sięetanol oraz fluorowiec sodu (sama reakcja jest substytucją, a nie eliminacją) (prawa strona rysunku). Wpraktyce obie reakcje zachodzą konkurencyjnie. Na rysunku zakreślono produkty końcowe obu przemian:
.3. Jak wspomniano przy reakcjach Friedla – Craftsa, halogenowe pochodne węglowodorów łatwiej jest
wprowadzić do pierścienia aromatycznego niŜ surowy łańcuch. UmoŜliwia to uzyskanie pochodnych typuAr-R, czyli połączenia aromatyczno – alifatycznego.
4. Fluorowcopochodne moŜna redukować w amalgamacie sodowym, np.:CH3-CH2-CH2-I ---Na(Hg)---> CH3-CH2-CH3 + HI.
Do najwaŜniejszych fluorowcoalkanów zaliczamy:- CH3Cl – przemysł chemiczny, chłodnictwo,- CHCl3 – chloroform (rozpuszczalnik Ŝywic, tłuszczów, olejów, wywabiacz plam, oczyszczanie
antybiotyków, dawniej narkoza),- CCl4 – tetra(chlorek węgla) – gaśnice tetrowe,- CCl2F2 – freon 12, uŜywany w lodówkach,- CHI3 – środek antyseptyczny,
Created by Neevia Document Converter trial version http://www.neevia.com

82
- C2H5Cl – do znieczuleń miejscowych.
FLUOROWCE ALIFATYCZNYCH WĘGLOWODORÓW NIENASYCONYCHW przypadku fluorowcoalkenów i fluorowcoalkinów występuje izomeria geometryczna cis-trans. Jest topowodem róŜnic w wartościach momentów dipolowych, a w związku z tym – w gęstości i temperaturze wrzenia.MoŜna je otrzymać na róŜne sposoby:a) przez addycję halogenowodoru do alkinu (zgodnie z r. Markownikowa), np.:
HC≡C-CH3 + HX ---> H2C=CX-CH3,b) przez halogenowanie i eliminację (izomer przeciwny do zgodnego z r. M.), np.:
CH3-CH=CH2 + Br2 ---> CH3-CHBr-CH2Br ---C2H5ONa---> CH3-CBr=CH2 + HBr,c) przez addycję halogenku i eliminację:
HC≡CH + Cl2 ---> CHCl2-CHCl2 ---C2H5ONa---> CHCl=CCl2.Fluorując węglowodory nienasycone w wysokiej temperaturze moŜna wymusić rodnikowy mechanizm reakcji.Wówczas powstający produkt jest zaprzeczeniem reguły Markownikowa, gdyŜ zachodzi substytucja przy atomiewęgla o hybrydyzacji sp3, a nie addycja do wiązania podwójnego, np.:CH2=CH-CH3 + Cl2 ---T≈600oC---> CH2=CH-CH2Cl + HCl.Właściwości pochodnych nienasyconych węglowodorów zaleŜą od wzajemnego połoŜenia atomu chlorowcaoraz wiązania podwójnego:a) Gdy chlorowiec znajduje się przy atomie węgla z wiązaniem podwójnym, wówczas na skutek efektu
mezomerycznego chmury elektronowe (wiązania wielokrotnego oraz chlorowca) ulegają ściągnięciu na tenwłaśnie atom węgla. Wskutek tego układ alkanowy (C-C-Cl) posiada większy moment dipolowy i jestreaktywniejszy niŜ układ nienasycony (C=C-Cl). Ten drugi staje się niepodatny na czynniki nukleofilowe.
b) Gdy atom chlorowca i wiązanie wielokrotne są rozdzielone przestrzennie, wówczas większa reaktywnośćcechuje naturalnie związek nienasycony. W tym przypadku układ nienasycony (C=C-C-Cl) jestreaktywniejszy od alkanowego (C-C-C-Cl).
Do najwaŜniejszych fluoropochodnych nienasyconych naleŜą:- CH2=CHCl – chlorek winylu (produkcja PVC),- CF2=CF2 – tetrafluoroeten (monomer teflonu),- CHCl=CCl2 – tri (uniwersalny rozpuszczalnik, synteza organiczna).
FLUOROWCOARENYSą to związki aromatyczne, w których atom wodoru zastąpiony został atomem fluorowca. Właściwościfluorowcoarenów prześledzimy na przykładzie chlorobenzenu C6H5Cl. Chlor cechuje się znacznąelektroujemnością, co w połączeniu z bogactwem elektronowym pierścienia aromatycznego daje silnezagęszczenie chmury elektronowej. Wiązanie pomiędzy atomem chloru a atomem węgla w pierścieniuaromatycznym ulega skróceniu, a orbitale obydwu pierwiastków nakładają się jeszcze bardziej. Sprawia to, iŜwymiana chloru na inne pierwiastki zachodzi jedynie w drastycznych warunkach, np.:C6H5Cl + NaOH ---T≈350oC|p≈6.106Pa---> C6H5OH + NaCl.Ponadto chlor naleŜy do grupy podstawników pierwszego rodzaju zmniejszających reaktywność związku. Wrezultacie łatwiej jest chlorować benzen niŜ chlorobenzen. Z racji rodzaju nowy podstawnik kierowany jest wpozycje orto- oraz para-. Jednak ze względów sterycznych pozycja orto- jest mniej uprzywilejowana.
ALKOHOLE I FENOLE
ALKOHOLE NASYCONE MONOHYDROKSYLOWEAlkoholami nazywamy pochodne węglowodorów, w których co najmniej jeden atom wodoru zastąpiony zostałgrupą hydroksylową –OH. Wynika stąd wzór ogólny postaci ROH. Dodatkowo istotne jest, aby grupawodorotlenkowa znajdowała się przy atomie węgla o konfiguracji sp3, czyli połączonego z innymi atomamiwyłącznie wiązaniami pojedynczymi. W przeciwnym wypadku mówimy o enolach, które są nietrwałe i jakoszczególny przypadek omówione zostaną później. Występujący w cząsteczce alkoholu rodnik węglowodorowynie moŜe być arylem, gdyŜ grupa hydroksylowa połączona z pierścieniem aromatycznym ma odmiennewłaściwości, stąd całą klasę tych związków nazywamy fenolami (fenole są „bliźniakami” alkoholi).Alkohole moŜna klasyfikować na róŜne sposoby, np. ze względu na rzędowość atomu węgla, przy którymwystępuje grupa hydroksylowa (I-, II-, III-rzędowe) czy na ilość grup –OH w cząsteczce (mono-, di-,trihydroksylowe itd.). Generalnie alkohole mają wyŜsze temperatury wrzenia i lepiej rozpuszczają się w wodzieniŜ alkany o podobnej masie cząsteczkowej, np. metan jest gazem, metanol – cieczą. Przyczyną tego jestwystępowanie wiązań wodorowych pomiędzy atomami tlenu a atomami wodoru z grup hydroksylowych. Nie sąone wprawdzie silne, jednak ze względu na liczność obniŜają lotność i podwyŜszają temperaturę wrzenia.Cząsteczka kaŜdego alkoholu składa się z węglowodorowej części hydrofobowej oraz hydroksylowej –hydrofilowej. Ich wzajemna proporcja, zaleŜna od długości łańcucha węglowego, decyduje o właściwościach
Created by Neevia Document Converter trial version http://www.neevia.com

83
fizycznych. Alkohole odalkanowe o długości łańcucha 1-3 rozpuszczają się nieograniczenie w wodzie (mieszająsię z nią w kaŜdym stosunku), o długości 4-8 – rozpuszczają się w wodzie z tendencją malejącą, zaś dłuŜsze nierozpuszczają się w wodzie. Jednocześnie rośnie rozpuszczalność w substancjach organicznych: benzenie,benzynie, toluenie, tłuszczach czy nafcie. Ze wzrostem długości łańcucha zmienia się równieŜ stan skupienia:dla 1-10 atomów węgla alkohole są cieczami o rosnącej gęstości, zaś dłuŜsze są ciałami stałymi,przypominającymi w wyglądzie i w dotyku woski. NiezaleŜnie od stanu skupienia alkohole są bezbarwne. Wprzypadku alkoholi II- i III-rzędowych moŜe występować izomeria optyczna.Alkohole moŜna otrzymać na wiele sposobów:a) w reakcji zasady z chlorkiem alkanu:
RX + KOH ---> ROH + KX,b) przez addycję cząsteczki wody do alkenu, np.:
CH2=CH2 + H2O ---kat.---> C2H5OH,c) przez działanie kwasem siarkowym i hydrolizę, np.:
CH3-CH=CH2 + H2SO4 ---> CH3-CH(OSO3H)-CH3 ---H2O---> CH3-CH(OH)-CH3,d) przez hydrogenację (redukcję) aldehydów lub ketonów:
R-CHO ---[H]---> ROH,R1-CO-R2 ---[H]---> R1-CH(OH)-R2,
e) w wyniku hydrolizy estrów:R1-COO-R2 + H2O ---H+---> R1-COOH + R2-OH,
f) działając kwasem azotowym (III) na aminy (metoda rozpoznawania amin I-rzędowych), np.:CH3-CH2-CH2-NH2 + HNO2 ---> CH3-CH2-CH2-OH + N2 + H2O,
g) przez hydrolizę związków Grignarda:R1-CHO + R2-MgX ---> R1-CH(OMgX)-R2 ---H2O---> R1-CH(OH)-R2 + Mg(OH)X,
h) przez fermentację cukrów, np.:C6H12O6 ---> 2C2H5OH + 2CO2.
Pomimo obecności grupy –OH, charakterystycznej dla wodorotlenków nieorganicznych, alkohole nie dysocjująjonowo. Spowodowane jest to zbliŜeniem róŜnic elektroujemności wiązań węgiel – tlen (∆X=1) oraz tlen –wodór (∆X=1,4), co czyni taki układ nierozróŜnialny dla atakujących go cząsteczek wody. Wobec tego wwodzie alkohole dają odczyn obojętny, będąc swoistymi związkami amfoterycznymi.a) Reagują z metalami alkalicznymi, wymieniając wodór na metal, dając sole – alkoholany, np.:
CH3-CH2-CH2-OH + Na ---> CH3-CH2-CH2-ONa + ½H2 (1-propanolan sodu),które w wodzie dysocjują anionowo:CH3-CH2-CH2-ONa + H2O ---> CH3-CH2-CH2-OH + NaOH,CH3-CH2-CH2-O
- + H2O ---> CH3-CH2-CH2-OH + OH-.
Alkoholany magnezu lub glinu słuŜą do otrzymywania absolutnie czystych alkoholi (100%), czego nie dasię zrobić przez destylację.
b) Reagują z wodorkami zasadowymi oraz z amidkami:ROH + NaH ---> RONa + H2,ROH + NaNH2 ---> RONa + NH3.
c) W środowisku kwaśnym mogą wymienić grupę hydroksylową. Wpierw kation wodoru przyłącza się do paryelektronowej atomu tlenu, dając jon alkilooksoniowy R-O+(H)-H, od którego następnie moŜe odłączyć sięobojętna cząsteczka wody, naraŜając dodatni łańcuch na działanie czynniku nukleofilowego, np. anionu.Dlatego wchodzą w reakcję z mocnymi kwasami beztlenowymi:ROH + HX ---H2SO4---> RX + H2O.
d) Reagują z chlorkami azotowców (PCl3, PCl5, POCl3), np.:ROH + PCl3 ---> RCl + POCl3 + HCl.
e) W podwyŜszonej temperaturze i w obecności osuszaczy (Al2O3, H2SO4) ulegają eliminacji cząsteczki wody,dając alkeny bądź etery:-CH2-CH(OH)- ---T,Al2O3---> -CH=CH-,2ROH ---kat.,H2SO4---> R-O-R + H2O.
f) W reakcji z kwasami karboksylowymi dają estry:R1-COOH + R2-OH ---H2SO4---> R1-COO-R2 + H2O.W przypadku kwasów nieorganicznych proces estryfikacji zachodzi o wiele łatwiej, np.:CH3OH + HNO3 ---> CH3-O-NO2 + H2O (azotan etylu),CH3OH + H2SO4 ---> CH3-O-SO3H + H2O (siarczan (VI) etylu).
g) Ulegają utlenieniu (odwodornieniu, dehydrogenacji) wobec silnych utleniaczy (KMnO4, K2Cr2O7), dającaldehydy i ketony, np.:CH3-CH2-OH ---[O],Cu,T≈300oC---> CH3-CHO + H2O (etanal),CH3-CH(OH)-CH3 ---[O]---> CH3-CO-CH3 + H2O (propanon).Podczas reakcji utleniania odszczepiany jest wodór z grupy hydroksylowej oraz z następnego atomu węgla.
Created by Neevia Document Converter trial version http://www.neevia.com

84
JeŜeli natomiast na najbliŜszym atomie węgla wodorów brak, a w takiej sytuacji znajdują się alkohole III-rzędowe, wówczas bezpośrednie utlenienie nie jest moŜliwe.
h) Dają pozytywny wynik próby jodoformowej – pod wypływem ogrzewania alkoholu z zasadowymroztworem jodu (fioletowy) pojawia się Ŝółte zabarwienie jodoformu:C2H5OH + 4I2 + 5KOH ---> CHI3 + HCOOH + 5KI + 4H2O.Reakcja ta zachodzi dla 2-hydroksozwiązków oraz metyloketonów. Pozwala odróŜnić alkohole od fenolioraz ketony od aldehydów.
Do najwaŜniejszych alkoholi odalkanowych naleŜą:a) Metanol czyli spirytus drzewny – otrzymywany jest przez bezpośrednią syntezę w obecności katalizatorów
(ZnO, MnO2, Cr2O3):CO + 2H2 ---p,T≈350oc,kat.---> CH3OH.Jest bardzo silną trucizną, wywołującą po spoŜyciu ślepotę i mogącą doprowadzić do śmierci. SłuŜy dootrzymywania aldehydu mrówkowego, do produkcji tworzyw sztucznych i włókien syntetycznych, leków,barwników, jako paliwo i rozpuszczalnik.
b) Etanol – na skalę przemysłową otrzymywany przez fermentację enzymatyczną cukrów (Polska),katalityczną hydratację etylenu (Irak), bądź hydrolizę zasadową halogenków etylu. W warunkachpokojowych jest to bezbarwna ciecz o charakterystycznym zapachu. Ma małą gęstość (0,79 g/cm3) itemperaturę wrzenia (78oC), jest lotny. Miesza się z wodą w kaŜdym stosunku, przy próbie destylacji wrzejako mieszanina azeotropowa (95,5% etanolu i 4,5% wody – jest to maksymalne stęŜenie procentowedestylowanego etanolu, tzw. spirytus rektyfikowany). Reaguje z metalami alkalicznymi dając alkoholanyoraz z kwasami beztlenowymi dając halogenki alkilowe. W podwyŜszonej temperaturze ulega dehydratacjina tlenku glinu. Wykorzystywany jest jako rozpuszczalnik i surowiec w syntezie organicznej.
c) Dalsi przedstawiciele szeregu homologicznego alkanoli są składnikami rozpuszczalników.d) Alkohol benzylowy C6H5-CH2-OH stanowi składnik substancji zapachowych. Jego estry są
wykorzystywane w przemyśle perfumeryjnym.
ALKOHOLE POLIHYDROKSYLOWESą to alkohole posiadające więcej niŜ jedną grupę wodorotlenową, np. diole – dwie, triole – trzy itd. Wszystkiemają postać lepkich cieczy dzięki wiązaniu wody przez grupy hydroksylowe, dlatego np. do kremów i pastdodaje się gliceryny. Alkohole polihydroksylowe identyfikuje się przez reakcję z wodorotlenkiem miedzi (II): wwodzie tworzy on niebieski osad, zaś wiąŜąc się z blisko połoŜonymi grupami –OH tworzy rozpuszczalnykompleks, zaś barwa roztworu ulega pogłębieniu i staje się ciemnoniebieska:
.Diole często nazywa się je glikolami. Otrzymuje się je przez hydratację i utlenianie alkenów, np.:
.Powstały w wyniku tej reakcji glikol etylenowy (1,2-etanodiol) jest bardzo silną trucizną. Ze względu nawiększą liczbę grup hydroksylowych glikole lepiej od alkoholi rozpuszczają się w wodzie. ObniŜają ponadto jejtemperaturę krzepnięcia, co zadecydowało o ich zastosowaniu jako składników płynów niezamarzających. Diolemogą ulegać dehydratacji – powstają wówczas enole, które w wyniku izomeryzacji dają ketony, np.:CH3-CH(OH)-CH(OH)-CH3 ---> CH3-CH=CH(OH)-CH3 ---> CH3-CH2-CO-CH3.Gliceryna (1,2,3-propantriol) jest związkiem nietrującym, wytwarzanym przez przyrodę – jest składnikiemkaŜdego tłuszczu. Ponadto stanowi waŜny składnik procesów technologicznych i surowiec do produkcjipropylenu. MoŜna ją otrzymać na róŜne sposoby, z których przedstawimy dwa najwaŜniejsze.a) Surowcem wyjściowym jest propen. Najpierw ulega on utlenieniu wg mechanizmu rodnikowego:
CH2=CH-CH3 ---T,[O],CuO---> CH2=CH-CHO,w wyniku czego otrzymujemy najprostszy aldehyd nienasycony – akroleinę. Tą poddaje się działaniunadtlenku wodoru, otrzymując 2,3-dihydroksypropanal:CH2=CH-CHO + H2O2 ---> CH2(OH)-CH(OH)-CHO,który po uwodornieniu na katalizatorze daje glicerynę:CH2(OH)-CH(OH)-CHO + H2 ---kat.---> CH2(OH)-CH(OH)-CH2(OH).
b) Wymuszając wysoką temperaturą rodnikowy przebieg reakcji chlorujemy propen, otrzymując produktniezgodny z regułą Markownikowa:CH2=CH-CH3 + Cl2 ---T≈500oC---> CH2=CH-CH2-Cl + HCl.
Created by Neevia Document Converter trial version http://www.neevia.com

85
Następnie poddajemy go działaniu kwasu chlorowego (I):CH2=CH-CH2-Cl + HClO ---> CH2(OH)-CHCl-CHCloraz wodorotlenku sodu:CH2(OH)-CHCl-CHCl + 2NaOH ---> CH2(OH)-CH(OH)-CH2(OH) + 2NaCl.
Przez działanie na glicerynę kwasem azotowym (V) otrzymuje się nitroglicerynę. UŜywana jest ona do produkcjileków nasercowych. Z kolei zmieszana w stosunku 3:1 z ziemią okrzemkową daje dynamit. W wyniku inicjacjizachodzi silnie wybuchowa reakcja:4C3H5(NO3)3---->12CO2 + 10H2O + 6N2 + O2,której wszystkie produkty są gazami. W układzie ograniczonym przestrzennie oznacza to powstanie duŜychciśnień, zdolnych do rozsadzania konstrukcji.
ALKOHOLE NIENASYCONEEnole są związkami chemicznymi zawierającymi charakterystyczny układ atomów – grypę hydroksylowąpołączoną z atomem węgla przy, którym obecne jest wiązanie podwójne. W miejsce to nie przyłączają się innecząsteczki, dlatego alkohole nienasycone nie odbarwiają wody bromowej. Są związkami nietrwałymi,ulegającymi samoistnemu przegrupowaniu. Pomiędzy substratem a produktem wytwarza się pewien stanrównowagi, przesunięty na korzyść odmiany ketonowej. Sytuację taką nazywamy tautomerią, w tym przypadku– ketonowo – enolową (jest to jej najpopularniejszy rodzaj) – rysunek:
.Tautomeria ketonowo – enolowa ma znaczenie w biochemii, np. wzajemne przechodzenie w siebie formaminokwasu cytruliny jest waŜnym etapem cyklu mocznikowego (ornitynowego).Niektóre alkohole nienasycone stanowią surowiec w syntezie organicznej. Prowadząc reakcje odwrotne moŜnawięc otrzymać niektóre z nich. Przykładowo depolimeryzując poli(octan winylu) otrzymujemy jego monomer:CH3-COO-CH=CH2, który w hydrolizując w środowisku kwasowym daje kwas octowy oraz nienasyconyalkohol winylowy:CH3-COO-CH=CH2 + H2O ---H+---> CH2=CH-OH + CH3COOH.W wyniku izomeryzacji ten ostatni przekształca się w aldehyd etylowy.
FENOLEFenole są związkami chemicznymi zbliŜonymi pod względem budowy do alkoholi, zawierają bowiem grupęhydroksylową –OH. Istnieje jednak zasadnicza róŜnica – w fenolach grupa ta związana jest bezpośrednio zpierścieniem aromatycznym. Wynika z tego wzór ogólny fenoli postaci Ar-OH, gdzie Ar jest rodnikiemarylowym. Zastępując we wzorach znanych węglowodorów aromatycznych jeden z atomów wodoru grupąwodorotlenkową, otrzymujemy kolejne fenole. Najprostszym z nich jest benzenol, popularnie określany jakofenol. Do grupy tej naleŜą równieŜ niektóre pochodne naftalenu. Izomeryczne odmiany metylobenzenoli(metylofenoli) znane są pod wspólną nazwą krezoli. W pierścieniu aromatycznym moŜe występować więcejgrup hydroksylowych – dwie, trzy itd. Wzory wymienionych fenoli przedstawia poniŜszy rysunek:
Fenole moŜna otrzymać:a) przez suchą destylację węgla kamiennego – głównie krezole,b) ze smoły pogazowej przez ekstrakcję:
C6H5-OH (w smole) + NaOH(aq) ---> C6H5-ONa + H2O,C6H5-ONa + H2O + CO2 ---> C6H5-OH + NaHCO3.
Created by Neevia Document Converter trial version http://www.neevia.com

86
c) przez syntezę organiczną z chlorobenzenu, co wymaga jednak dodatkowych warunków:C6H5Cl + 2NaOH(aq) ---T≈370oC,p,kat.---> C6H5-ONa + NaCl + H2O,C6H5-ONa + H2O + CO2 ---> C6H5-OH + NaHCO3,
d) przez utlenianie izopropylobenzenu:
,e) z amin I-rzędowych.Wszystkie fenole dają reakcje barwne z kationami Ŝelaza Fe3+, co ułatwia ich identyfikację. Kompleksowyprodukt jest najczęściej fioletowy, niebieski bądź czerwony – zaleŜnie od konkretnego związku. Bardziejkwasowe nitrofenole reagują z węglanem i wodorowęglanem sodu, dając czerwone, pomarańczowe bądź Ŝółtefenolany.
FENOLW temperaturze pokojowej jest to higroskopijne, krystaliczne ciało stałe, początkowo bezbarwne, róŜowiejące napowietrzu. Słabo rozpuszcza się w zimnej wodzie, za to dosyć dobrze w ciepłej. Fenol jest trucizną bardzoszkodliwą dla zdrowia, ma właściwości parzące i dezynfekujące, długi kontakt niekorzystnie wpływa na układkostny. Kategorycznie nie moŜe on występować w wodzie pitnej.Fenol posiada charakter polarny ze względu na obecność grupy hydroksylowej. Z tego powodu moŜna bywnioskować, iŜ będzie on dysocjował i to zasadowo. I rzeczywiście – dysocjuje, lecz kwasowo. Zjawiskokwasowości grupy -OH występuje u fenoli właśnie z powodu połączenia tej grupy z aromatycznym atomemwęgla. Sześcioelektronowe, zdelokalizowane wiązanie π bardzo silnie oddziałuje na wolne pary elektronowetlenu, powodując ich ściągnięcie w stronę pierścienia (efekt indukcyjny). Tym samym w obrębie podstawnikazachodzi rozsunięcie chmury elektronowej i osłabienie wiązania pomiędzy atomem tlenu a wodoru. Ten ostatnistosukowo łatwo ulega odszczepieniu, dając odczyn kwasowy:C6H5-OH <===H2O===> C6H5-O
- + H+.Fenol jest jednak bardzo słabym kwasem, o czym świadczy stała równowagi powyŜszej reakcji: K≈10-10. ObniŜaon odczyn środowiska do pH≈6. Dzięki kwasowemu charakterowi nie reaguje bezpośrednio z kwasami ani nietworzy estrów.Kwasowość fenolu moŜna zwiększyć dzięki podstawnikom przy pierścieniu aromatycznym – podstawniki IIrodzaju zdolne są przyciągać chmurę elektronową z pierścienia, ściągając jeszcze większy ładunek cząstkowy zatomu tlenu. Ponadto najsilniejszy wpływ mają grupy w połoŜeniu orto- oraz para-, najsłabszy zaś w połoŜeniumeta-. Najczęściej wykorzystuje się nitrowanie – i faktycznie nitrofenol jest bardziej kwasowy od fenolu.Prowadząc reakcję dalej moŜna otrzymać wybuchowy kwas pikrynowy – 2,4,6-trinitrofenol C6H2(NO2)3OH,którego moc zbliŜona jest do mocy kwasu siarkowego (VI).1) Z racji kwasowego charakteru reaguje on z metalami alkalicznymi i zasadami, zobojętniając je i dając sole
zwane fenolanami, np.:C6H5-OH + Na ---> C6H5-ONa + ½H2,C6H5-OH + NaOH ---> C6H5-ONa + H2O.Oznacza to, iŜ ma silniejsze właściwości kwasowe od alkoholi.
2) Fenolany z łatwością hydrolizują anionowo, dając wyjściowy fenol, np.:C6H5-ONa + H2O <==> C6H5-OH + NaOH,C6H5-O
- + H2O <==> C6H5-OH + OH-.3) Fenolany mogą stanowić surowiec do otrzymywania kwasu salicylowego:
C6H5-ONa + CO2 ---p,T≈125oC---> C6H4(OH)(COONa) ---H+---> C6H4(OH)(COOH) (izomer orto).4) Fenol z fenolanów moŜna równieŜ wypierać kwasami mocniejszymi, np.:
C6H5-O- + H2O + CO2 ---> C6H5-OH + NaHCO3.
5) Fenol ulega bardzo łatwo reakcjom nitrowania, dając mieszaninę o- i p-nitrofenolu:C6H5-OH + HNO3 ---> C6H5-NO2 + H2O.
6) Daje równieŜ reakcje substytucji z fluorowcami, np.:C6H5-OH + 3Br2 ---> C6H2Br3OH + 3HBr.Reakcję tą wykorzystuje się do oznaczania stęŜenia fenolu w ściekach.
7) Ulega równieŜ uwodornieniu na katalizatorze, dając cykloheksanol:C6H5-OH + 3H2 ---p≈2,5MPa,T≈200oC,Ni---> C6H11-OH.
Created by Neevia Document Converter trial version http://www.neevia.com

87
8) MoŜna go utlenić przez działanie kwasem chromowym (VI):
.Wpierw powstaje hydrochinon, a następnie p-benzochinon (lub po prostu p-chinon), cykliczny ketonnienasycony. Chinon jest składnikiem witamin z grupy K.
9) Fenol kondensowany z aldehydem mrówkowym daje polikondensat w postaci Ŝywicy lub bakelitu:
.
PORÓWNANIE ALKOHOLI I FENOLICecha Alkohole Fenole
grupa funkcyjna hydroksylowa -OH hydroksylowa -OHatom węgla przy grupie funkcyjnej tetraedryczny (sp3) aromatyczny (sp2)podstawienie grupy funkcyjnej tak niewzór ogólny R-(OH)n Ar-(OH)n
dysocjacja i odczyn brak dysocjacji – odczyn obojętny dysocjacja kwasowa – odczyn kwasowyz metalami alkalicznymi produkty – alkoholany produkty – fenolanyz wodorotlenkiem sodu nie takz fluorowcem \ nitrowanie nie takz fluorowcowodorem tak niez wodorotlenkiem miedzi tylko polihydroksylowe takz kationem Ŝelaza (FeCl3) nie tak - barwnaz Na2CO3 lub NaHCO3 nie nitrofenole – barwne
reak
cja
z kwasem chromowym (VI) tak – utlenianie do aldehydu,ketonu bądź kwasu
nie
próba jodoformowa tak nie
ETERY
Etery to związki organiczne złoŜone w ogólnym przypadku z dwóch fragmentów węglowodorowychpołączonych atomem tlenu. Wynika stąd ich wzór ogólny postaci R1-O-R2. W pewnym sensie etery nazwaćmoŜna izomerami alkoholi – tak jakby atom tlenu z grupy hydroksylowej przemieścił się do wnętrza łańcucha.W kwestii nazewnictwa istnieją dwie tendencje: albo podaje się po słowie „eter” nazwy obu fragmentów wformach przymiotnikowych (np. eter metylowo-etylowy) albo zaznacza obecność tlenu elementem „oksy” (np.etoksymetan). Czasem, gdy jednym z fragmentów jest rodnik metylowy, zupełnie się go pomija.Etery nie tworzą wiązań wodorowych, dlatego mają mniejsze gęstości i większą lotność od alkoholi – eterdimetylowy i metylowo-etylowy są gazami. Z reguły nie rozpuszczają się w wodzie, ale za to dobrze wrozpuszczalnikach organicznych. Mają zazwyczaj przyjemny zapach.1) Etery wykazują bardzo małą aktywność chemiczną. Między innymi dzięki temu znalazły zastosowanie jako
rozpuszczalniki substancji organicznych – są niepolarne i nie ulegają hydrolizie
Created by Neevia Document Converter trial version http://www.neevia.com

88
2) Ulegają działaniu bardzo mocnych kwasów beztlenowych (acydolizie), np.:C6H5-O-CH3 + HI ---> C6H5-OH + CH3I.
3) Etery alifatyczne łatwo tworzą nietrwałe i wybuchowe nadtlenki, np. oksydietylowy:C2H5-O-C2H5 + O2 ---> C2H5-O-CH(CH3)-O-OH.
Fragmenty węglowodorowe mogą tworzyć zamknięty łańcuch – wówczas mówimy o eterach cyklicznych, czylioksiranach. Najprostszym z nich jest eter etylowy, zwany od nazwy klasy oksiranem; słuŜy on do otrzymywaniaglikolu etylowego. Dzięki występującym wewnątrz pierścienia napręŜeniom oksirany są zdecydowanie bardziejaktywne chemicznie niŜ etery niecykliczne.
ZWIĄZKI KARBONYLOWE – ALDEHYDY I KETONY
Wymienione grupy związków są ze sobą mocno spokrewnione, przede wszystkim dzięki obecności w obu grupykarbonylowej –(C=O)–. RóŜnica dotyczy właściwie jej sąsiedztwa – w aldehydach jest ona połączona z atomemwodoru, zaś w ketonach – z fragmentem węglowodorowym. Występujący w niej węgiel, z racji wiązaniapodwójnego z tlenem, znajduje się w stanie hybrydyzacji sp2, zatem zarówno sama grupa, jak i wszystkieokoliczne atomy leŜą w jednej płaszczyźnie. Grupa ma budowę polarną, co decyduje o przebiegu reakcji. Anialdehydy ani ketony nie dysocjują, posiadają zatem odczyn obojętny.Aktywność związków karbonylowych zaleŜy od otoczenia grupy – przykładowo metanal jest reaktywniejszy odaldehydu o dłuŜszym łańcuchu, a ten z kolei – od ketonu. Problem leŜy w rozkładzie chmury elektronowej –elektroujemny atom tlenu ściąga w swoje sąsiedztwo elektrony, przez co na atomie węgla pojawia się cząstkowyładunek dodatni (metanal). Jednak juŜ obecność tetraedrycznego atomu węgla połączonego z karbonylowymzmniejsza na tym ostatnim ładunek cząstkowy przez odpychanie elektronów (dalsze aldehydy). Efekt potęgujesię, gdy zamiast obu atomów wodoru obecne są grupy węglowodorowe (ketony).Nie występują tu jednak ugrupowania hydroksylowe. Brak międzycząsteczkowych wiązań wodorowych pozwalana większą lotność od alkoholi. Wiązanie podwójne oznacza, przynajmniej teoretycznie, moŜliwość addycjinukleofilowej do wiązania. Występująca pomiędzy atomami węgla i tlenu róŜnica elektroujemności sprawia, iŜwęgiel karbonylowy jest miejscem aktywnym chemicznie, choć najczęściej obserwuje się aktywność węgla wpozycji α. Występuje ponadto zjawisko tautomerii ketonowo – enolowej, polegające na izomeryzacjiugrupowania typu –CH2–CHO do –CH=CH-OH. Powstające enole łatwo jednak powracają do stanu trwalszego.Związki karbonylowe moŜna otrzymać na wiele sposobów:a) przez formylowanie alkenów – w wyniku reakcji z tlenkiem węgla następuje wytworzenie wiązania
termalnego i wydłuŜenie łańcucha:R-CH=CH2 + CO + H2 ---p---> R-CH2-CH2-CHO,
b) przez przyłączanie wody do węglowodorów nienasyconych:R-C≡CH + H2O ---H2SO4/HgSO4---> R-C(OH)=CH2 ---> R-CO-CH3,w wyniku czego powstaje keton (wyjątkowo dla acetylenu – metanal),
c) przez utlenianie węglowodorów aromatycznych, np.:C6H5-CH3 ---[O]---> C6H5-CHO + H2O,
d) przez hydrolizę geminalnych dichlorków i dibromków, np.:C6H5-CHCl2 + H2O ---> C6H5-CHO + 2HCl,
e) przez utlenianie alkoholi I- i II-rzędowych:R-CH2-OH ---[O]---> R-CHO + H2O,R1-CH(OH)-R2 ---[O]---> R1-CO-R2 + H2O,
f) przez dehydrogenację alkoholi (stąd nazwa: aldehyd = alkohol dehydrogenatus, łac. alkohol odwodorniony):R-CH2-OH ---Cu|T≈300oC---> CH3-CHO + H2,
g) przez utlenianie geminalnych dioli w obecności kwasu jodowego (VII) lub octanu ołowiu (IV), np.:CH3-CH(OH)-C(OH)(CH3)-CH3 ---HIO4/Pb(CH3COO)4---> CH3-CHO + CH3-CO-CH3,
h) przez termiczny rozkład soli kwasów karboksylowych, np.:(CH3COO)2Ca ---T---> CH3-CO-CH3 + CaCO3.
Grupa karbonylowa wchodzi w szereg reakcji nukleofilowych:a) z bardzo słabymi kwasami, np. cyjanowodorowym, dając cyjanohydrazynę aldehydu bądź ketonu:
R-CHO + HCN ---> R-CH(CN)(OH),b) z amoniakiem, dając aldehydoamoniak:
R-CHO + NH3 ---> R-CH(NH2)(OH)W reakcji metanalu z amoniakiem otrzymujemy urotropinę – lek stosowany przy infekcjach drógmoczowych:
Created by Neevia Document Converter trial version http://www.neevia.com

89
,c) z pochodnymi amoniaku (aminami, hydrazyną, hydroksyaminą i fenylohydrazyną), przy czym aldehydy
wchodzą w reakcję łatwiej – np. z aminami:R1-CHO + R2-NH2 ---H
+---> R1-CH=N-R2 + H2O,R1-CO-R2 + R3-NH2 ---H
+---> R1-(C=N-R3)-R2 + H2OPowyŜsza reakcja jest addycją – eliminacją nukleofilową. Powstają w niej tzw. zasady Schiffa, czyli aldo-lub ketoiminy. Dodatkowo ma tu miejsce tautomeria aminowo – iminowa, gdyŜ powstałe związki mogąizomeryzować przez przeniesienie atomu wodoru z najbliŜszego atomu węgla na atom azotu, w ten sposóbzmienia się połoŜenie wiązania podwójnego. W reakcji z hydrazyną powstaje hydrazon:R-CHO + NH2-NH2 ---> R-CH=N-NH2 + H2O,zaś z hydroksyaminą – oksyny aldehydu lub ketonu (odpowiednio – aldooksyny lub ketooksyny):R-CHO + NH2-OH ---> CH3-CH=N-OH + H2O.Reakcja z fenylohydrazyną daje fenylohydrazon, który wydziela się z roztworu w formie stałej:R-CHO + C6H5-NH-NH2 --->R-CH=N-NH-C6H5 + H2O.Reakcja ta ma znaczenie w identyfikacji cukrów.
d) z wodorosiarczanem sodu, dając sól sodową kwasu α-hydroksosulfonowego:R-CHO + NaHSO3 ---> R-CH(OH)(SO3Na)Powstała substancja jest krystaliczną solą, zatem łatwo ją wydzielić, co jest przydatne w analiziechemicznej. PowyŜsza reakcja ma znaczenie przy identyfikacji cukrów, zachodzi bowiem z aldehydami iketonami metyloalkilowymi, zaś nie zachodzi z ketonami aromatycznymi i alifatyczno - aromatycznymi.
e) z alkoholami w środowisku polarnym (kwaśnym bądź zasadowym), dając hemiacetale bądź hemiketale R3:R1-CHO + R2-OH ---H+/OH----> R1-CH(OH)-O-R2,które okazują się na tyle nietrwałe, iŜ nie moŜna ich wydzielić z roztworu, mogą jednak reagować znastępną cząsteczką alkoholu, tym razem wyłącznie w środowisku kwasowym (pomaga to wydzielićcząsteczkę wody), dając acetale bądź ketale R4:R1-CH(OH)-O-R2 + R3-OH ---H+---> R1-CH(-O-R2)-O-R3 + H2OTe ostatnie są trwałe w środowisku zasadowym, zaś w kwaśnym hydrolizują do hemiacetali i alkoholi. Zpodobnymi reakcjami mamy do czynienia przy badaniu właściwości cukrów, głównie hydrolizydisacharydów.
f) Do niŜszych aldehydów moŜe przyłączyć się cząsteczka wody, dając hydrat aldehydu, np.:HCHO + H2O ---> HO-CH2-OH.Dzięki moŜliwości występowania jako hydrat formaldehyd dobrze rozpuszcza się w wodzie. Jego 37%roztwór – formalina – znalazł zastosowanie jako środek konserwujący. Wraz ze wzrostem długości łańcuchawęglowego rosną problemy związane z hydratacją – i tak np. juŜ etanal ulega hydratacji tylko w 50%. Wcelu ułatwienia hydratacji moŜna podstawić atomy wodoru halogenkami – np. chloral CCl3-CHO hydratujew 100%. Im bardziej elektroujemny jest podstawnik, tym większy ma on wpływ na reaktywność związku.
Do wielu reakcji włącza się równieŜ węgiel w pozycji α, zawierający atomy wodoru. Często w bezpośredniąreakcję wchodzi odmiana enolowa.a) Dimeryzacja aldolowa umoŜliwia przekształcenie aldehydów w ich dimery, np.:
2 CH3-CHO ---OH----> CH3-CH(OH)-CH2-CHO,w wyniku czego powstaje aldehydoalkohol (aldol), β-hydroksyaldehyd (3-hydroksyaldehyd).
b) Związki karbonylowe moŜna halogenkować, a zachodzące podstawienie jest raczej elektrofilowe, np.:CH3-CHO + Br2 ---HBr---> Br-CH2-CHO + HBr.W efekcie powstaje α-bromoaldehyd octowy (α-bromoetanal).
c) W środowisku wybitnie zasadowym zachodzi reakcja haloformowa, której ulegają wyłącznie metyloketonyoraz 2-hydroksyalkany (alkohole II-rzędowe) – jej szczególnym przypadkiem jest reakcja jodoformowa,np.:CH3-CH2-CO-CH3 + I2 ---NaOH---> CHI3 + CH3-CH2-CO-OH,której charakterystycznym objawem jest Ŝółty osad jodoformu.
d) Ze względu na pośredni stopień utlenienia związki karbonylowe powinny zarówno łatwo dać się utlenić, jaki zredukować. Produktami reakcji są alkohole: w przypadku aldehydów – I-rzędowe, zaś w przypadkuketonów – II-rzędowe, np.:CH3-CHO + H2 ---Ni, Pt---> CH3-CH2-OH,
Created by Neevia Document Converter trial version http://www.neevia.com

90
CH3-CO-CH3 + H2 ---Ni, Pt---> CH3-CH(OH)-CH3.Utlenianie ketonów mogłoby zachodzić jedynie w bardzo drastycznych warunkach i połączone byłoby zrozerwaniem istniejących wiązań. Natomiast aldehydy dosyć łatwo ulegają utlenianiu do kwasówkarboksylowych, np.:CH3-CHO ---[O]---> CH3-COOH.Wobec powyŜszego do identyfikacji aldehydów stosuje się reakcje polegające na ich utlenianiu, z którychnajpopularniejszymi są: próba Tollensa oraz próba Trommera.
e) Próba Tollensa polega na działaniu na aldehyd amoniakalnym roztworem tlenku srebra. Do jegosporządzenia uŜywa się azotanu (V) srebra, wodorotlenku sodu oraz amoniaku (tworzy się złoŜony związekkompleksowy). W wyniku procesu redox powstaje odpowiedni kwas oraz metaliczne srebro, które pokrywacienką warstwą ścianki naczynia, stąd reakcję tą nazywa się próbą lustra srebrnego (uzyskanie efektu lustrazachodzi jedynie w bardzo czystych (odtłuszczonych) naczyniach). Schematycznie:2 [Ag(NH3)2]OH + HCHO ---> HCOOH + 2 Ag + 4 NH3 + H2O.Do obliczeń stosuje się uproszczoną wersję powyŜszego równania:Ag2O + HCHO ---> 2Ag + HCOOH.
f) Próba Trommera polega na ogrzewaniu aldehydu z roztworem wodorotlenku miedzi (II) w środowiskuzasadowym. Przed przeprowadzeniem reakcji ten ostatni naleŜy otrzymać „na świeŜo”, np. z chlorku miedzii wodorotlenku sodu – wytrąca się wówczas niebieski, galaretowaty osad. Dodatkowo dla większegokontaktu rozpuszcza się go w winianie sodowo – potasowym, otrzymując odczynnik Fehlinga.HCHO + 2Cu(OH)2 ---> HCOOH + Cu2O + 2H2O.Reakcja ta jest bardzo uŜyteczna przy wykrywaniu cukrów – wchodzą w nią jedynie redukującemonosacharydy, zaś cukry nieredukujące dają w efekcie czarny osad tlenku miedzi (II), powstały przeztermiczny rozkład wodorotlenku:Cu(OH)2 ---T---> CuO + H2O.
MoŜe się równieŜ zdarzyć, iŜ w połoŜeniu α brak atomu wodoru. Wówczas zachodzą reakcje innego rodzaju:a) W środowisku silnie zasadowym aldehydy ulegają dysproporcjonowaniu (reakcja Cannizzaro), dając
odpowiedni kwas w postaci soli oraz odpowiedni alkohol np.:2 C6H5-CHO ---NaOH---> C6H5-COOH + C6H5-CH2-OH.
b) Aldehydy aromatyczne i alifatyczne nie zawierające wodoru w połoŜeniu α w obecności cyjanków ulegająkondensacji benzoinowej, dając α-hydroksyketony, np.:2 C6H5-CHO ---NaCN---> C6H5-CH(OH)-CO-C6H5.
c) Karbonylowe związki aromatyczne ulegają w środowisku zasadowym kondensacji do ketonównienasyconych (reakcja Kleisena), np.:C6H5-CHO + C6H5-CO-CH3 ---NaOH---> C6H5-CH=CH-CO-C6H5 + H2O.
Spotykamy równieŜ tendencję do tworzenia połączeń pomiędzy poszczególnymi cząsteczkami.a) Aldehyd mrówkowy polimeryzuje, tworząc łańcuchy liniowe (n dochodzi do 100):
n HCHO ---> (-CH2-O-)n.W ten sposób powstaje paraformaldehyd (paraform), wytrącający się jako biały, kłaczkowaty osad. Wpostaci kostek uŜywany jest jako paliwo do kuchenek turystycznych.
b) W odpowiednich warunkach metanal trimeryzuje, dając cykliczny trioksan:
.c) Cząsteczki etenalu łączą się ze sobą w trimery (ciekły paraldehyd, po lewej) bądź tetramery (stały
metaldehyd, po prawej):
.Do najwaŜniejszych związków karbonylowych naleŜą:a) Metanal – lotny gaz o intensywnym zapachu, dobrze rozpuszczalny w wodzie. Otrzymuje się go przez
utlenianie (dehydrogenację) metanolu. Wodnym roztworem 37% – formaliną – konserwuje się preparatyanatomiczne. Metanal stanowi surowiec do produkcji Ŝywic fenolowo – formaldehydowych.
Created by Neevia Document Converter trial version http://www.neevia.com

91
b) Etanal – gaz o ostrym zapachu, dobrze rozpuszczalny w wodzie. Otrzymywany z etylenu przez hydratację idehydrogenację lub w reakcji Kuczerowa na katalizatorze (PdCl2, CuCl2). Wykorzystywany jest doprodukcji kwasu octowego, polimerów oraz Ŝywic fenolowych.
c) Aldehyd benzoesowy jest cieczą o zapachu migdałów. Otrzymuje się go z toluenu, produktu suchejdestylacji węgla kamiennego. W wyniku chlorowania na świetle powstaje dichlorofenylometan, który wwyniku hydrolizy daje szukany produkt oraz chlorowodór. Jest to składnik esencji zapachowych do ciast.Jego pochodna – wanilina jest równieŜ środkiem zapachowym.
d) Propanon (aceton ) jest lotną cieczą o charakterystycznym zapachu. Otrzymywany jest z propenu przezhydratację do 2-propanolu i dehydrogenację. Posiada w obrębie cząsteczki zarówno fragment polarny, jak iniepolarny, dlatego rozpuszcza się dosyć dobrze zarówno w wodzie jak i związkach organicznych. Mieszasię z wodą w kaŜdym stosunku, nie dysocjuje. Jest palny i z powietrzem tworzy mieszaniny wybuchowe.Jest składnikiem zmywaczy kosmetycznych i wielu rozpuszczalników, np. odtłuszczaczy galwanicznych.
Nienasycone związki karbonylowe mogą być bardzo reaktywne chemicznie, jeŜeli wiązanie podwójne występujeprzy węglu karbonylowym. Bardzo łatwo zachodzą wówczas addycje, których produktami mogą być:a) kwasy: R-CH=C=O + H2O ---> R-CH2-COOH,b) estry: R1-CH=C=O + R2-OH ---> R1-CH2-COO-R2,c) amidy kwasowe: R1-CH=C=O + R2NH2 ---> R1-CH2-CO-NH-R2.Przykładem aldehydu nienasyconego jest akroleina (propenal): CH2=CH-CHO. Otrzymać ją moŜna z propylenuw wyniku reakcji:CH2=CH-CH3 ---CuO, T---> CH2=CH-CHO.Powstaje równieŜ z gliceryny podczas smaŜenia i jełczenia tłuszczów. W laboratorium reakcję tą moŜnaprzyspieszyć przez dodanie katalizatora – wodorosiarczanu (VI) potasu:HO-CH2-CH(OH)-CH2-OH ---T,KHSO4---> CH2=CH-CHO + 2H2O.W wyniku działania na akroleinę nadtlenkiem wodoru powstaje aldehyd glicerynowy – najprostszy cukier:CH2=CH-CHO + H2O2 ---> HO-CH2-CH(OH)-CHO.
Pochodne związków karbonylowych – hydroksyaldehydy i hydroksyketony – to typowe cukry, czyliwęglowodany. Najprostszym z nich jest aldehyd glicerynowy, 2,3-dihydroksypropanal.
KWASY KARBOKSYLOWE
KWASY NASYCONEKwasami karboksylowymi nazywamy związki posiadające grupę karboksylową. Jest ona połączeniem grupykarbonylowej –CO– oraz hydroksylowej –OH, co daje razem –COOH. Atom węgla występuje w niej namaksymalnym stopniu utlenienia (+3), co ma wpływ na właściwości. W obrębie grupy funkcyjnej ma miejsceefekt mezomeryczny:
,następuje wyrównanie długości wiązań węgiel – tlen oraz delokalizacja elektronów wiązania podwójnego.Energia ta jest niemniejsza od energii dysocjacji kationu wodorowego, dlatego odszczepienie faktyczniezachodzi. Im jednak łańcuch rodnikowy jest dłuŜszy, tym mocniejsze wiązanie pomiędzy atomem tlenu awodoru a dysocjacja zachodzi trudniej. Moc kwasów zaleŜy wówczas od obecności podstawnikówelektroujemnych oraz ich odległości od grupy funkcyjnej. Do tych ostatnich zaliczają się: fluorowce, grupahydroksylowa oraz podstawniki II rodzaju. W przypadku kwasów aromatycznych wiązanie zdelokalizowaneściąga elektrony grupy funkcyjnej do wewnątrz pierścienia, a osłabienie wiązania tlen – wodór ułatwiadysocjację (kwas benzoesowy jest silniejszy od kwasu octowego, choć słabszy od mrówkowego). Kwasowośćzwiększa równieŜ obecność kolejnych grup karboksylowych. Z obecności w cząsteczce grupy karboksylowej wynika szereg własności, np. to, Ŝe kwasy karboksylowe sącieczami (wyŜsze – ciałami stałymi). NiŜsze kwasy alifatyczne (do kwasu masłowego) są nieograniczenierozpuszczalne w wodzie i mają ostry, draŜniący zapach. Rozpuszczalność i stan skupienia wynikają z silnejasocjacji wiązaniami wodorowymi. Z tego teŜ powodu częstą formą występowania są dimery, które mają czasemnawet w postać pary (ta ostatnia właściwość zwiększa dwukrotnie masę cząsteczkową). Dzięki słabymwiązaniom wodorowym (20 razy słabsze od tradycyjnych) niektóre kwasy, np. octowy, występują w postacinietrwałych polimerów. Wraz ze wzrostem długości łańcucha węglowodorowego wpływ grupy karboksylowejna własności sukcesywnie maleje. Kwasy niŜsze, rozpuszczalne w wodzie są dla człowieka trujące, wyŜszenatomiast są nieszkodliwe.Związki te moŜna nazywać w róŜny sposób:- podając nazwę rodnika i dodatek „karboksylowy” (kwas wodorokarboksylowy, metanokarboksylowy itd.),
Created by Neevia Document Converter trial version http://www.neevia.com

92
- podając przymiotnikową formę nazwy alkanu o tej samej liczbie atomów węgla (kwas metanowy, etanowyitd.),
- uŜywając nazwy zwyczajowej (kwas mrówkowy, octowy itd.).Po odrzuceniu od cząsteczki kwasu grupy hydroksylowej powstaje rodnik acylowy o wzorze ogólnym R-CO-.Rodniki acylowe pochodzące od poszczególnych kwasów mają odpowiednie nazwy: formyl –CHO, acetyl –CO-CH3, propionyl –CO-C2H5, butyryl –CO-C3H7, benzoil –CO-C6H5 itd.Kwasy karboksylowe moŜna otrzymać:a) przez utlenianie węglowodorów,b) przez hydrolizę pochodnych kwasowych – halogenków, estrów kwasowych, amidów,c) przez utlenianie alkoholi i aldehydów,d) przez utlenianie ketonów (zachodzi z rozerwaniem łańcucha),e) przez przekształcanie ketonów metylowych (reakcja chloroformowa), np.:
CH3-CO-CH2-CH3 + Cl2 ---OH----> CHCl3 + C2H5COOH,f) przez karboksylowanie soli fenolowych, np.:
C6H5-ONa + CO2 ---T,p,H2O---> C6H5(OH)-COONa,g) przez hydrolizę nitryli, np.:
CH3-C≡N ---T,H2O/H2SO4---> CH3-COOH + NH3
Nitryle otrzymać moŜna z halogenopochodnych węglowodorów w reakcji z cyjankami metali, np.:RX + NaCN ---> R-C≡N + R-N=C + NaX.Powstaje wówczas mieszanina nitrylu i izonitrylu oraz sól nieorganiczna. JeŜeli jako metal w powyŜszejreakcji zastosujemy np. srebro, to powstająca sól będzie wytrącać się ze środowiska reakcji, zwiększająctym samym jej wydajność.
h) z wykorzystaniem związków Grignarda, np.:R-CH2-Cl + Mg ---eter---> R-CH2-Mg-Cl,R-CH2-Mg-Cl + CO2 ---H
+---> R-CH2-COOMgCl,R-CH2-COOMgCl + H2O ---> R-CH2-COOH + Mg(OH)Cl.
Kwasy karboksylowe ulegają róŜnorodnym reakcjom:a) Kwasy organiczne są kwasami słabymi, jednak mocniejszymi od kwasu węglowego, dlatego wypierają go z
soli:CaCO3 + 2 CH3COOH ---> (CH3COO)2Ca + H2O + CO2.Ponadto reagują z tlenkami metali, wodorotlenkami i metalami aktywnymi. Dla porównania – alkoholewchodzą w reakcję tylko z tymi ostatnimi.
b) W obecności mocnych kwasów związki karboksylowe zachowują się jak zasady, tj. przyłączają kationwodoru, w wyniku czego powstaje karbokation:R-COOH + H+ ---> R-C(+)(OH)-OH.
c) Podczas elektrolizy do anody dąŜą aniony reszt kwasowych. Na elektrodzie zachodzi reakcja Kolbego –reszta kwasowa rozkłada się na cząsteczkę dwutlenku węgla oraz rodnik. Po połączeniu tych ostatnichpowstaje odpowiedni węglowodór o parzystej licznie atomów węgla:A(+): 2 R-COO- ---> R-R + 2 CO2.
d) W wyniku reakcji kwasów z halogenkami powstają α-halogenopochodne:-CH2-COOH + Br2 ---p---> -C-CH(Br)-COOH + HBr,które moŜna następnie podstawić innymi grupami (np. aminową – otrzymujemy aminokwas).
e) Pod wpływem chlorków azotowców (SOCl2, PCl3, PCl5) kwasy karboksylowe mogą przekształcać się whalogenki kwasowe:R-COOH + SOCl2 ---> R-CHCl + SO2 + HCl,które są o wiele bardziej reaktywne od kwasów i estrów.
f) Ze względu na najwyŜszy stopień utlenienia, kwasy karboksylowe są związkami trudnymi do utlenienia (sązłymi reduktorami, dają negatywny wynik prób redukcyjnych – Tollensa i Trommera). Wyjątkiem jest kwasmrówkowy, który posiada jakgdyby grupę aldehydową:HCOOH + Ag2O ---> CO2 + H2O + 2Ag.RównieŜ kwas pirogronowy (oksopropionowy) i szczawiowy (etanodiowy) mogą być utlenione zgodnie zrównaniami:CH3-CO-COOH ---[O]---> CH3-CHO + CO2,HOOC-COOH ---[O]---> 2CO2 + H2O.Ostatnia reakcja potwierdza skuteczność kwasu szczawiowego w wywabianiu plam z nadmanganianupotasu.
g) Kwasy karboksylowe bardzo trudno ulegają redukcji. Pozwala na to jedynie obecność szczególnie silnychutleniaczy, np. wodorku litowo – glinowego LiAlH4 czy dimeru borowodoru B2H6:R-COOH ---LiAlH4/H2O---> R-CH2-OH + Al(OH)3 + LiOH.
Created by Neevia Document Converter trial version http://www.neevia.com

93
h) W niektórych reakcjach od kwasu oderwana zostaje grupa hydroksylowa (wykryć to moŜna przezoznaczenie jej radioaktywnym izotopem tlenu 18O). Przykładem jest estryfikacja, polega na łączeniu siękwasu z alkoholem, w wyniku czego powstaje ester oraz cząsteczka wody. Reakcja ma przebiegnukleofilowy, katalizowana jest najczęściej przez kwas siarkowy:R1-COOH + R2-OH ---H2SO4---> R1-COO-R2 + H2O.Powstała cząsteczka wody jest pochłaniana przez higroskopijny kwas, zaś ester z łatwością się ulatnia.Kwas siarkowy pełni ponadto rolę czynnika atakującego.
i) Trudnym zabiegiem jest dekarboksylacja kwasów. Wyjątkiem jest tu kwas octowy, który ogrzewany zwodorotlenkiem sodu i tlenkiem wapnia wydziela metan:CH3-COOH + NaOH + CaO ---T≈300oC ---> CH4 + Na2CO3 + CaCO3.Dekarboksylacji o wiele łatwiej ulegają np. sole sodowe:CH3-COOH + NaOH ---CaO---> CH4 + Na2CO3,C6H5-COONa + NaOH ---> C6H6 + Na2CO3.Sytuacja zmienia się równieŜ, gdy w połoŜeniu α bądź β znajduje się elektroujemny podstawnik, np. atomchlorowca lub druga grupa karboksylowa. Wówczas dekarboksylacja zachodzi o temperaturze rzędu 100oC,np.:C6H4(Cl)COOH ---H2O,T≈100oC---> C6H5-Cl + CO2,Cl-CH2-COOH + NaOH --- ---> CH3Cl + Na2CO3,CH3-CH(OH)-CH2-COOH ---T≈100oC---> CH3-CO-CH3 + CO2,HOOC-CH2-COOH ---T≈100oC ---> CH3-COOH + CO2.W przypadku kwasu pirogronowego proces ten moŜe zachodzić juŜ w temperaturze pokojowej.
Do waŜniejszych kwasów karboksylowych naleŜą:a) Kwas mrówkowy – bezbarwna Ŝrąca ciecz o ostrym zapachu. Miesza się w kaŜdym stosunku z wodą,
alkoholem i eterem. MoŜna go otrzymać przez katalizowaną reakcję wodorotlenku sodu z tlenkiem węgla:CO + NaOH ---H2O,p≈15MPa,T≈200oC---> HCOONa,a następnie działając na mieszaninę poreakcyjną kwasem siarkowym (VI):HCOONa + H2SO4 ---> HCOOH + NaHSO4.Mrówczan sodu słuŜy do otrzymywania kwasu szczawiowego:2 HCOONa ---T≈400oC---> NaOOC-COONa,NaOOC-COONa + H2 ---> HOOC-COOH.Działając nań kwasem siarkowym doprowadza się do rozkładu na czad i wodę, pochłanianą przez kwas:HCOOH ---T,H2SO4---> CO + H2O.Kwas metanowy wykorzystywany jest w przemyśle farbiarskim, garbarstwie, jako środek dezynfekcyjny,zewnętrzny lek przeciwreumatyczny oraz w syntezie organicznej.
b) Kwas octowy – stuprocentowy i bezwodny zwany jest lodowatym ze względu na wygląd zewnętrzny.Otrzymać go moŜna: z acetylenu w reakcji Kuczerowa, z aldehydu octowego, przez zakładową destylacjędrewna oraz przez utlenianie etanolu pod wpływem bakterii octowych Mycoderma aceti (metodaprzeciwprądu). Ostatnia reakcja odpowiada kwaśnieniu alkoholu oraz soków owocowych - do celówspoŜywczych nadaje się ocet spirytusowy. Kwasu etanowego uŜywa się do produkcji octanu celulozy,octanu winylu, w przemyśle spoŜywczym oraz do produkcji środków zapachowych (estry).
c) Kwas propinowy uŜywany jest jako składnik środków ochrony roślin.
KWASY NIENASYCONEKwasy nienasycone zawierają w cząsteczce wiązanie wielokrotne (jedno lub więcej). Stwarza to moŜliwośćaddycji doń innych cząsteczek. Dzięki temu kwasy nienasycone odbarwiają wodę bromową, a w reakcji zwodorem na katalizatorze ulegają utwardzeniu.NajwaŜniejszym z kwasów nienasyconych jest kwas propenowy (akrylowy): CH2=CH-COOH oraz jegohomolog – kwas α-metakrylowy: CH2=C(CH3)-COOH. W wyniku estryfikacji powyŜszych kwasów otrzymujesię półprodukty słuŜące do produkcji polimerów, z których najpopularniejszym jest tzw. szkło organiczne –pleksiglas, czyli poli(metakrylan metylu).
KWASY AROMATYCZNEW tym przypadku rodnikiem jest aryl. Kwasy aromatyczne dają reakcje charakterystyczne dla związkówaromatycznych, tj. nitrowanie i podstawienie fluorowcem, np.:C6H5-COOH + HNO3 ---> C6H5(NO2)-COOH + H2O (kwas m-nitrobenzoesowy),C6H5-COOH + Cl2 ---> C6H5(Cl)-COOH + HCl (kwas m-chlorobenzoesowy).NajwaŜniejszym z powyŜszych jest kwas benzoesowy (benzenokarboksylowy): C6H5-COOH. Otrzymać gomoŜna z toluenu, który najpierw poddaje się chlorowaniu do trichlorofenylometanu, a następnie w środowiskuzasadowym przekształca w poŜądany kwas. Związek ten jest bardzo trudno rozpuszczalny w wodzie. UŜywanyjest do produkcji barwników, a jego sole znalazły zastosowanie jako środki konserwujące – np. benzoesan sodu.
Created by Neevia Document Converter trial version http://www.neevia.com

94
KWASY TŁUSZCZOWEKwasy odalkanowe nazywamy kwasami tłuszczowymi ze względu na ich występowanie tłuszczach. Dzielą sięna nasycone (CnH2n+1-COOH) oraz nienasycone, te drugie dalej na jednonienasycone (CnH2n-1-COOH) orazwielonienasycone (CnH2n-x-COOH). Nazwy oraz występowanie najwaŜniejszych kwasów tłuszczowychprzedstawia poniŜsza tabela:
Wzór Nazwa zwyczajowa (systematyczna) WystępowanieNasycone kwasy tłuszczowe (NKT)
C4H9-COOH kwas walerianowy olej krotonowyC5H11-COOH kwas kapronowy olej kokosowy i palmowyC7H15-COOH kwas kaprylowy olej palmowyC9H19-COOH kwas kaprynowy olej kokosowy i palmowyC11H23-COOH kwas laurynowy olej kokosowy i palmowyC13H27-COOH kwas mirystynowy olej kokosowyC15H31-COOH kwas palmitynowy powszechnie w tłuszczachC16H33-COOH kwas margarynowy margarynaC17H35-COOH kwas stearynowy powszechnie w tłuszczachC19H39-COOH kwas arachinowy olej arachidowy
Nienasycone kwasy tłuszczowe (NNKT)C17H33-COOH kwas olejowy, oleinowy (9-oktadecenowy)C17H31-COOH kwas linolowy (9,12-oktadecenodienowy)C17H29-COOH kwas linolenowy (9,12,15-oktadecenotrienowy)C19H31-COOH kwas arachidonowy (5,8,11,14-ejkozanotetraenowy)
róŜne oleje roślinne
Jak łatwo zauwaŜyć, ogromna większość kwasów tłuszczowych posiada w łańcuchu parzystą liczbę atomówwęgla. Prawidłowość ta wynika z biologicznego mechanizmu ich syntezy, polegającej na łączeniu kolejnychpodjednostek dwuwęglowych. Ponadto wszystkie nienasycone kwasy tłuszczowe są izomerami cis, dzięki czemuistniejące między łańcuchami oddziaływania pozwalają na utrzymanie właściwej lepkości.Sole sodowe i potasowe wyŜszych kwasów tłuszczowych są dobrze rozpuszczalne w wodzie, gdyŜ energiahydratacji równowaŜy energię odpychania wody przez element hydrofobowy. Łańcuchy węglowodorowe kierująsię do środka, tworząc układy hydrofobowe, tzw. micele. Rozcieńczone roztwory tych soli są w zasadzieroztworami rzeczywistymi, ulegającymi stuprocentowej hydratacji, zaś roztwory stęŜone mają charakterkoloidalny.
KWASY DIKARBOKSYLOWESą to związki posiadające w cząsteczce dwie grupy karboksylowe. Mogą być nasycone, nienasycone bądźaromatyczne. Nasycone kwasy alifatyczne tworzą szereg homologiczny:HOOC-COOH - kwas etanodiowy (dikarboksylowy, szczawiowy),HOOC-CH2-COOH - kwas propanodiowy (metanodikarboksylowy, malonowy),HOOC-(CH2)2-COOH - kwas butanodiowy (1,2-etanodikarboksylowy, bursztynowy),HOOC-(CH2)3-COOH - kwas pentanodiowy (1,3-propanodikarboksylowy, glutarowy),HOOC-(CH2)4-COOH - kwas heksanodiowy (1,4-butanodikarboksylowy, adypinowy) itd.W przypadku kwasów nienasyconych występuje izomeria geometryczna cis-trans, a poszczególne formy mogąmieć odmienne nazwy zwyczajowe, np. kwas cis-etenodikarboksylowy nazywamy maleinowym, zaś jegoizomer trans – kwasem fumarowym. Wśród kwasów aromatycznych występuje izomeria orto-meta-para, np.kwas o-benzenodikarboksylowy to kwas ftalowy, izomer meta – izoftalowy, a para – tereftalowy.Z racji posiadania dwóch grup mogących odszczepić kation wodoru, związki te charakteryzują dwie stałedysocjacji. Pierwsza z nich jest zawsze większa od drugiej, gdyŜ zgodnie z regułą przekory trudniej wprowadzićdo środowiska kwasowego dodatkowy czynnik kwasotwórczy. Dla porównania – dla kwasu szczawiowego:K1=5,4.10-2, a K2=5,4.10-5. Kwasy te są mocniejsze od analogicznych kwasów monokarboksylowych, gdyŜwzajemne odpychanie blisko połoŜonych grup funkcyjnych ułatwia dysocjację. Wśród kwasów nienasyconychizomery cis są zdecydowanie mocniejsze od bliźniaków trans ze względu na sumowanie się momentówdipolowych: dla porównania pierwsza stała dysocjacji kwasu maleinowego wynosi 1170.10-5, a kwasufumarowego – 93.10-5. Kwasy aromatyczne dysocjują tym lepiej, im bliŜej siebie połoŜone są grupy funkcyjne.JeŜeli grupy karboksylowe połoŜone są w bezpośrednim sąsiedztwie (α) lub przedzielone są pojedynczymatomem węgla (β), to w trakcie ogrzewania ulegają dekarboksylacji, np.:HOOC-COOH ---T≈160-180oC---> HOOC + CO2,HOOC-CH2-COOH ---T≈150-160oC---> CH3COOH + CO2.
Created by Neevia Document Converter trial version http://www.neevia.com

95
W przypadku większych odległości (γ, δ) ogrzewanie prowadzi do powstania cyklicznych bezwodników iwydzielenia wody, np.:
,lub do powstania cyklicznych ketonów:
.W przypadku kwasów nienasyconych ze względów przestrzennych bezwodniki tworzą jedynie izomery cis.Zastąpienie grup hydroksylowych jedną grupą iminową prowadzi do powstania imidów, np. ftalimidu:
.Ten ostatni wykorzystuje się do syntezy amin, aminokwasów (np. kwasu antranilowego), związkówheterocyklicznych i barwników, np. indygo.Do waŜniejszych kwasów dikarboksylowych zaliczamy:a) Kwas szczawiowy, występujący w przyrodzie (rabarbar, szczaw), jest swoistym reduktorem. W obecności
nadmanganianu potasu i w środowisku kwaśnym ulega dekarboksylacji do dwutlenku węgla, dzięki temustosuje się go jako wywabiacz plam spowodowanych KMnO4. SłuŜy ponadto do oznaczanie jego stęŜenia wanalizie chemicznej. Z kationami wapnia tworzy nierozpuszczalne szczawiany, a w organizmie ludzkimpowoduje niebezpieczne dla zdrowia odwapnienie. Jest składnikiem kamieni szczawianowych tworzącychsię w moczu. Osobom zagroŜonym powyŜszą dolegliwością nie wolno jeść produktów zawierających duŜoszczawianów, tj. czarnych porzeczek, aronii, szczawiu, rabarbaru, kakao i czekolady.
b) Kwas adypinowy słuŜy do produkcji włókien poliamidowych.c) Kwas ftalowy podczas ogrzewania tworzy bezwodnik ftalowy, uŜywany do produkcji Ŝywic poliestrowych
(kwas tereftalowy – do produkcji elany), zaś po podstawieniu fenolem daje indykator chemiczny –fenoloftaleinę:
.
HALOGENKI KWASOWESą to pochodne kwasów karboksylowych, w których grupa hydroksylowa zastąpiona została atomem chlorowca.Wynika stąd ich wzór ogólny postaci R-C(O)-X. Nie tworzą one wiązań wodorowych, mają formę lotnychcieczy o ostrym zapachu. Otrzymać je moŜna w reakcji chlorków azotowców ew. tlenowców z kwasami, np.:3 R-COOH + PBr3 ---> 3 R-C(O)-Br + H3PO3,R-COOH + SOCl2 ---> R-C(O)-Cl + SO2 + HCl.Halogenki kwasowe są związkami bardzo aktywnymi chemicznie – reaktywniejszymi od odpowiadających imkwasów dzięki zwiększeniu cząstkowego ładunku na atomie węgla. W reakcji z wodą odtwarzają kwasy, odktórych pochodzą. Bardzo łatwo reagują z alkoholami:R1-C(O)-Cl + R2-OH ---> R1-COO-R2 + HCl (ester),R-C(O)-Cl + NH3 ---> R-CO-NH2 + HCl (amid kwasowy).
Created by Neevia Document Converter trial version http://www.neevia.com

96
BEZWODNIKI KWASOWETeoretycznie są to produkty kondensacji dwóch cząsteczek tego samego kwasu. W najprostszy sposób moŜna jeotrzymać w reakcji chlorków kwasowych z solą sodową lub potasową kwasu, np.:R-C(O)-Cl + R-COONa ---> R-COO-CO-R + NaCl.Wzajemne oddziaływanie powoduje większą reaktywność bezwodników od kwasów, z których powstały,chociaŜ nie tak jak w przypadku halogenków. Bezwodniki bardzo łatwo reagują z czynnikami nukleofilowymi:a) z wodą, wówczas odtwarzają kwasy,b) z alkoholami, dając estry i kwasy,c) z amoniakiem lub aminami, dając amidy kwasowe.
ESTRY
Estrami nazywamy produkty kondensacji kwasów karboksylowych z alkoholami, czyli tzw. reakcji estryfikacji.Zachodzi ona w środowisku kwaśnym, polega na umownym odrzuceniu grupy hydroksylowej kwasu oraz atomuwodoru od alkoholu, w wyniku czego powstaje ester:R1-COOH + R2-OH ---H+---> R1-COO-R2 + H2O.Wydziela się woda, w której grupa wodorotlenkowa pochodzi od kwasu, co moŜna sprawdzić metodą atomówznaczonych (wykorzystuje się radioaktywny izotop węgla 18O). Reakcja estryfikacji jest na ogół mało wydajna,dlatego zamiast kwasów stosuje się halogenki lub bezwodniki kwasowe. Wówczas w środowisku reakcji obokestru powstaje kwas, a nie woda, nie ma zatem warunków do typowej hydrolizy.Estry posiadają grupę funkcyjną zwaną estrową: -C(O)O-. W jej obrębie zachodzi sprzęŜenie par elektronowychtlenu z elektronami π wiązania podwójnego. Wiązania przesuwają się nieznacznie ku sobie, następujewyrównanie ich długości i delokalizacja. Efekt indukcyjny powoduje zmniejszenie bezwzględnej wartościładunku na atomie węgla, wskutek czego staje się on mniej aktywny. Miejscami aktywnymi w cząsteczce są:grupa karbonylowa oraz węgiel w połoŜeniu α. Reaktywność tej pierwszej jest jednak mniejsza niŜ waldehydach, ketonach czy kwasach.Z powodu ekranowania grupy hydrofilowej przez rodniki hydrofobowe estry nie tworzą wiązań wodorowych animiędzy sobą, ani z cząsteczkami wody (dlatego są w niej nierozpuszczalne i cechują się duŜą lotnością).Rozpuszczają się natomiast dobrze w substancjach organicznych. NiŜsze estry są cieczami o przyjemnychzapachach, stąd wykorzystywane są w produktach spoŜywczych i kosmetycznych: mrówczan etylu – rum, octanetylu – gruszki, maślan etylu – ananasy, octan butylu lub amylu – banany.JeŜeli estryfikacja zachodzi pomiędzy alkoholami a kwasami mineralnymi, to równieŜ powstają estry, np.triazotan (V) gliceryny. Do estrów zaliczamy takŜe cykliczne laktydy i laktony, powstające w wyniku termicznejdehydratacji hydroksykwasów.a) Wg teorii Brönsteda estry są zasadami mogącymi przyłączyć kation wodoru, tworząc układ –C(OH)=O-C-.
Jednak przyłączony wodór łatwo dysocjuje.b) Estry mogą być zarówno bardzo słabymi kwasami jak i zasadami, dlatego ulegają dwojakiego rodzaju
hydrolizie:- kwasowej (odwracalnej):
R1-COO-R2 + H+ + H2O ---> R1-COOH + R2-OH,- zasadowej (nieodwracalnej):
R1-COO-R2 + OH- + H2O ---> R1-COO- + R2-OH.JeŜeli do ostatniej reakcji uŜyjemy zasady sodowej lub potasowej, to produktem reakcji będzie mydło,dlatego hydrolizę zasadową nazywamy niekiedy zmydlaniem estrów.
c) Estry ulegają reakcji alkoholizy zachodzącej w środowisku polarnym, polegającej na wymianie resztyalkoholowej:R1-COO-R2 + R3-OH <==> R1-COO-R3 + R2-OH.
d) W wyniku działania amoniakiem otrzymuje się amidy I-rzędowe:R1-COO-R2 + NH3 ---H2O,T≈0oC---> R1-CONH2 + R2-OH.MoŜna równieŜ działać pochodnymi amoniaku, np.:R1-COO-R2 + NH2NH2 ---> R1-CONHNH2 + R2-OH,R1-COO-R2 + NH2OH ---> R1-CONHOH + R2-OH.
e) Reakcja estrów z aminami prowadzi do otrzymania amidów II-rzędowych, np.:R1-COO-R2 + R3-NH2 ---> R1-CONH-R3 + R2-OH.
f) Estry mogą ulegać kondensacji z ketonami, jednak musi to zachodzić w obecności silnej zasady, np.:R1-COO-R2 + CH3-CO-CH3 ---C2H5ONa---> CH3-CO-CH2-CO-R + R2-OH.Powstaje wówczas β-diketon.
g) W środowisku zasadowym estry ulegają kondensacji (reakcja Claissena), podobnie jak kwasy zawierająceatom wodoru w połoŜeniu α.
Created by Neevia Document Converter trial version http://www.neevia.com

97
h) W podwyŜszonej temperaturze (ok. 200oC) estry moŜna zredukować do alkoholi. Reakcja zachodzi łatwiejniŜ analogiczna dla kwasów.
Z biologicznego punktu widzenia szczególne znaczenie mają fosfoglicerydy, czyli estry kwasu fosfatydowego.Ten ostatni powstaje przez estryfikację jednego z I-rzędowych atomów węgla w glicerolu kwasemortofosforowym H3PO4:HO-CH2-CH(OH)-CH2-OH + H3PO4 ---> HO-CH2-CH(OH)-CH2-O-P(O)(OH)-OH + H2O.W miejscu podstawienia obecna jest grupa hydroksylowa, którą moŜna estryfikować alkoholami. I tak:a) w wyniku estryfikacji kolaminą (etanoloaminą, 2-aminoetanolem) HO-CH2-CH2-NH2 otrzymuje się
kefalinę:
b) przez kondensację z choliną otrzymuje się lecytynę:
.
TŁUSZCZETłuszcze są estrami gliceryny i wyŜszych kwasów tłuszczowych (za wyŜsze uznajemy na mniejsze niŜ kwasmasłowy). Ich ogólny wzór ma postać:
.W tłuszczach występujących w przyrodzie rodniki R1, R2, R3 są alifatyczne, nierozgałęzione, a liczba atomówwęgla (wliczając węgiel karboksylowy) jest parzysta. Proste łańcuchy węglowodorowe są równoległe i gęstoupakowane, dlatego tłuszcze są ciałami stałymi. Tłuszcze są bardziej charakterystyczne dla świata zwierzęcego.Tłuszcze nienasycone mogą być półpłynne lub płynne i występują w roślinach (wyjątkiem jest tran). Wprzypadku naturalnych nienasyconych kwasów tłuszczowych wiązanie wielokrotne występuje zazwyczaj naśrodku łańcucha, np. w kwasie oleinowym C17H33-COOH – pomiędzy 9 i 10 atomem węgla. Kwasywielonienasycone cechują się równomiernym rozłoŜeniem wiązań, np. kwas arachidonowy – przy 5, 8, 11, 14atomie węgla (stałe odległości pomiędzy wiązaniami). W przyrodzie występują raczej izomery cis, co wpływana odległości pomiędzy poszczególnymi łańcuchami. Większa swoboda pozwala na obniŜenie temperaturytopnienia i zachowanie stanu ciekłego. Tłuszcze nienasycone moŜna uwodorniać na katalizatorze, otrzymująctłuszcze mniej płynne:
.Ich nienasycony charakter wykrywać moŜna reakcją z wodą bromową, którą odbarwiają. Nienasycenie tłuszczumoŜna przedstawić w sposób wymierny za pomocą liczby jodowej – jest to liczba gramów jodu przyłączającegosię do nienasyconych kwasów tłuszczowych zawartych w 100g danego tłuszczu.Pod wpływem niekorzystnych czynników środowiska tłuszcze ulegają jełczeniu. W początkowej fazie procespolega na hydrolizie glicerydów do gliceryny i kwasów tłuszczowych. Te ostatnie zakwaszają środowiska,przyspieszając niekorzystne przemiany, a ponadto mogą wydawać odraŜającą woń, np. kwas masłowy.
Created by Neevia Document Converter trial version http://www.neevia.com

98
Ostatecznie kwasy nienasycone ulegają jełczeniu aldehydowemu, a kwasy nasycone – ketonowemu np. kwaslaurynowy do metylononyloketonu CH3-CO-C9H19).Tłuszcze są związkami palnymi, co wykorzystywano w staroŜytności w lampkach oliwnych i łojowych.Z racji estrowej natury tłuszcze ulegają hydrolizie kwasowej i zasadowej. Ta ostatnia, przeprowadzana wobecności wodorotlenku sodu lub potasu, daje sole zwane mydłami, np.:
.Ilość miligramów wodorotlenku potasu potrzebną do zobojętnienia kwasów tłuszczowych powstałych zhydrolizy 1g tłuszczu nazywamy liczbą zmydlania. Mydła są rozpuszczalne w wodzie z wyjątkiemnierozpuszczalnych mydeł wapniowych i magnezowych, te jednak nie słuŜą do prania, a ich powstawanie jestzazwyczaj procesem niepoŜądanym. Rozcieńczone roztwory mydeł moŜna traktować jako roztwory rzeczywiste,zaś roztwory bardziej stęŜone tworzą emulsje. Mydła, jako anionowe środki piorące, są wraŜliwe na działanietwardej wody – z kationami Ca2+ i Mg2+ tworzą nierozpuszczalne osady.Inne środki anionowe to alkiloarylosiarczany ArR-O-SO3Na lub alkilosiarczany sodowe (mersolany) R-O-SO3Na. Powstające sole wapniowe i magnezowe nie strącają się, lecz ulegają rozpuszczeniu.Kationowe środki piorące to głównie sole amin IV-rzędowych. Jedna z reszt węglowodorowych posiada długiłańcuch, dzięki czemu cząsteczka staje się środkiem powierzchniowo czynnym. Dodatkowym atutem środkówkationowych jest ich niewraŜliwość na twardą wodę.Środki niejonowe uŜywane są do celów technologicznych. Nie ulegają one dysocjacji, jednak posiadają grupyhydroksylowe umoŜliwiające rozpuszczalność oraz fragmenty węglowodorowe, wykazujące powinowactwo dotłuszczów. Wykorzystuje się je do flotacji, tj. oddzielania i wzbogacania rud.
ZWIĄZKI AZOTOWE
AMINYAminy to azotowe związki organiczne, pochodne amoniaku, w którego cząsteczce jeden lub więcej atomówwodoru zastąpiono rodnikiem alkilowym lub arylowym. RozróŜniamy aminy: I-rzędowe: R-NH2, II-rzędowe:R1-NH-R2, III-rzędowe: R1-N(R2)-R3 oraz IV-rzędowe: [R1-(R2)N(R3)-R4]X (w postaci soli). Rzędowość aminłatwo rozpoznać na podstawie wyniku reakcji z kwasem azotowym (III):a) aminy I-rzędowe dają alkohole oraz cząsteczkowy azot, wydzielający się w postaci pęcherzyków:
R-CH2-NH2 + HNO2 ---> R-CH2-OH + N2 + H2O,b) aminy II-rzędowe dają N-nitrozoaminy – oleiste ciecze trudno rozpuszczalne w wodzie o barwie Ŝółtej lub
Ŝółtoczerwonej:R1-NH-R2 + HNO2 ---> R1-N(R2)-N=O + H2O,
c) aminy III-rzędowe praktycznie nie reagują z kwasem azotowym (III).Atom azotu w aminach występuje w stanie konfiguracji sp3 – fragment cząsteczki cechuje budowa zgięta zwyeksponowaną wolną parą elektronową, co decyduje o właściwościach. Elektroujemność azotu jest mniejszaod elektroujemności tlenu, dlatego wiązanie azot – wodór jest mniej polarne niŜ tlen – wodór. Z tego powoduaminy I- i II-rzędowe tworzą bardzo mało słabych wiązań wodorowych. Aminy III-rzędowe nie mogą tworzyćwiązań wodorowych i nie ulegają asocjacji. Stąd etyloamina oraz aminy zawierające grupy metylowe są wtemperaturze pokojowej gazami, podczas gdy analogiczne alkohole dzięki oddziaływaniommiędzycząsteczkowym są cieczami. Metylowe i etylowe aminy I- i II-rzędowe są związkami dobrzerozpuszczalnymi w wodzie, zaś aminy aromatyczne, np. anilina, bardzo trudno lub w ogóle nie rozpuszczają sięw wodzie. Spowodowane jest to brakiem moŜliwości tworzenia wiązań wodorowych spowodowanymosłabieniem wiązania azot – wodór przez sprzęŜenie sekstetu aromatycznego z parą elektronową atomu azotu.Metylowe i etylowe pochodne azotu (mała masa cząsteczkowa) mają nieprzyjemny zapach zgnilizny.Aminy otrzymać moŜna:a) przez działanie amoniakiem na halogenopochodne węglowodorów (aminy alifatyczne):
RX + 2 NH3 ---> R-NH2 + NH4X,b) przez działanie aminami na halogenopochodne węglowodorów (aminy alifatyczne):
R1-NH2 + R2X ---> R1-NH-R2 + HX,c) w reakcji alkoholi z amoniakiem w odpowiednich warunkach, np.:
CH3OH + NH3 ---p,T≈300oC,Al2O3---> CH3NH2 + H2O (pochłaniana),
Created by Neevia Document Converter trial version http://www.neevia.com

99
d) przez redukcję amidów (tzw. przegrupowanie Hofmanna):R-CO-NH2 + Br2 + 4NaOH ---> R-NH2 + Na2CO3 + 2NaBr + 2H2O,
e) przez redukcję nitryli w obecności bardzo mocnego reduktora (LiAlH4, B2H6):RCN ---LiAlH4---> R-CH2-NH2,
f) przez redukcję oksymów, np.:CH3-CO-CH3 + NH2OH ---> CH3-CH(=NOH)-CH3 (oksym),CH3-CH(=NOH)-CH3 ---+2H---> CH3-CH(NH2)-CH3,
g) przez redukcję nitrozwiązków (aminy aromatyczne):R-NO2 + 3H2 ---> R-NH2 + 2H2O,
h) ogólnie – ze wszystkich pochodnych aminowych silnymi reduktorami(aldehyd / keton + amoniak ---redukcja---> amina).
Podobnie jak amoniak dysocjuje w wodzie na kation amonowy i anion wodorotlenkowy, tak i aminyrozpuszczalne w wodzie dysocjują i dają odczyn zasadowy:RNH2 + H2O ---> RNH3
+ + OH-.Aminy są słabszymi zasadami od wodorotlenków. Zasadowość amin kształtuje się bardzo rozmaicie wzaleŜności od rodzaju i ilości podstawników. I tak: z powodu sprzęŜenia pary elektronowej atomu azotu zsekstetem aromatycznym fenyloamina jest zasadą słabszą od amoniaku. Dzięki efektowi indukcyjnemu grupametylowa połączona z aminową zwiększa cząstkowy ładunek na atomie azotu, dlatego metyloamina jestsilniejsza od amoniaku. Podstawienie kolejnych atomów wodoru fragmentami węglowodorowymi potęgujepowyŜszy efekt, stąd trimetyloamina jest zasadą o wiele silniejszą niŜ amoniak. Schematycznie zasadowośćwymienionych związków przedstawia się następująco:C6H5-NH2 < NH3 < CH3NH2 < (CH3)2NH < (CH3)3N.Aminy są odczynnikami nukleofilowymi, co pozwala im na tworzenie soli amoniowych z halogenkamikwasowymi, np.:CH3-CH2-NH2 + HCl ---> CH3-CH2-NH3
+Cl-.Powstałe związki są kryształami jonowymi i posiadają wszystkie cechy typowych soli. IV-rzędowe wodorotlenkiamoniowe to bardzo mocne zasady o mocy zbliŜonej do mocy zasad nieorganicznych – moŜna je otrzymać z soliamin IV-rzędowych:2 [R1-(R2)N(R3)-R4]
+X- + Ag2O + H2O ---> 2 [R1-(R2)N(R3)-R4]+OH- + 2 AgX.
W wyniku reakcji amoniaku z chlorkami sulfonowymi powstają sulfonoamidy, np.:CH3-C6H4-SO2Cl + 2 NH3 ---> CH3-C6H4-SO2-NH2 + NH4Cl.Przez podstawienie przyazotowych atomów wodoru w tych ostatnich otrzymuje się pochodne o właściwościachdezynfekcyjnych.
AMIDY KWASOWEAmidami kwasowymi nazywamy pochodne kwasów karboksylowych, w których grupa hydroksylowazastąpiona została grupą aminową –NH2. W ten sposób powstaje nowa grupa – amidowa: -CO-NH2, będącapołączeniem grupy karbonylowej i aminowej. Obecne w tej ostatniej atomy wodoru mogą być podstawioneinnymi rodnikami, przez co wyróŜniamy amidy: I-rzędowe: R-CO-NH2, II-rzędowe: R1-CO-NH-R2, III-rzędowe: R1-CO-N(R2)-R3 oraz IV-rzędowe: R1-CO-N(R2)(R3)-R4. Istnienie amidów IV-rzędowych umoŜliwiawolna para elektronowa znajdująca się na atomie azotu – wytwarza ona koordynacyjne wiązanie z czwartymrodnikiem. Do powstałego kationu przyłącza się anion, dlatego istnieją one w postaci soli: [R1-CO-N(+)(R2)(R3)-R4]X
(–).Amidy otrzymać moŜna w róŜnoraki sposób:a) działając na chlorki kwasowe lub estry amoniakiem lub aminami, np.:
R1-COCl + 2 R2-NH-R3 ---> R1-CO-N(R2)-R3 + R1-NH3+Cl--R2,
R1-COO-R2 + R3-NH-R4 ---> R1-CO-NH(R3)-R4 + R2-OH,b) przez hydrolizę nitryli:
R-CN + H2O ---> R-CO-NH2,c) przez silne ogrzewanie soli amonowych lub amoniowych kwasów karboksylowych:
R-COOH + NH3 <==> R-COO-NH4+ ---T---> R-CO-NH2 + H2O (I-rzędowa),
R1-COOH + R2-NH2 <==> R1-COO-R2NH3+ ---T---> R1-CO-NH-R2 + H2O (II-rzędowa),
R1-COOH + R2-NH-R3 <==> R1-COO-R2R3NH2+ ---T---> R1-CO-N(R2)-R3 + H2O (III-rzędowa),
metody tej jednak nie stasuje się ze względu na znikomą wydajność i duŜy nakład energetyczny.W budowie samej grupy amidowej wyróŜnia się dwie struktury graniczne: -C(+)(=O)-N= oraz –C(-O-)=N(+)=. Wrzeczywistości występuje układ pośredni (efekt mezomeryczny). W obu formach wiązanie podwójne znajdujesię przy atomie węgla (sp2), więc układ jest w przybliŜeniu płaski (atakujący wiązanie reagent moŜe zbliŜać się zdowolnej strony). W przypadku drugiej formy moŜna mówić o izomerii geometrycznej cis-trans względematomu tlenu. Grupa amidowa jako całość jest polarna – biegun dodatni znajduje się w pobliŜu atomu węgla, aujemny pomiędzy atomami tlenu i azotu. Obecność wolnych par elektronowych na tych ostatnich (w
Created by Neevia Document Converter trial version http://www.neevia.com

100
szczególności azotu) umoŜliwia tworzenie wiązań wodorowych. Stąd amidy mają stosunkowo wysokietemperatury wrzenia i małą lotność. NiŜsze amidy (do czterech atomów węgla w cząsteczce) są rozpuszczalne wwodzie i w substancjach, z którymi mogą tworzyć wiązania wodorowe. Mocznik, podobnie jak sole amonowe,rozpuszcza się endoergicznie, co wykorzystuje się w mieszaninach oziębiających. W przypadku amin III-rzędowych (brak atomów wodoru przy azocie) następuje asocjacja wskutek polarności dipoli. Najprostsze amidykwasu mrówkowego, np. formamid HCO-NH2, są cieczami, zaś pozostałe to krystaliczne ciała stałe o barwiebiałej.a) Miejscami aktywnymi w cząsteczce amidu są atomy węgla i azotu; w niektórych reakcjach bierze udział
atom tlenu. To, od którego atomu rozpocznie się reakcja, zaleŜy od środowiska – np. w kwasowym zachodzinastępująca tautomeria:-C(=O)-N= + H+ <==> -C(=O(+)H)-N= <==> -C(-OH)=N=.
b) Amidy rozpuszczalne w wodzie dają odczyn obojętny. Pewien charakter kwasowy wykazują natomiastimidy (R1-CO-NH-CO-R2) – następuje osłabienie wiązania pomiędzy atomami azotu i wodoru, coumoŜliwia dysocjację.
c) Amidy i imidy ulegają hydrolizie kwasowej i zasadowej. Są to reakcje substytucji nukleofilowej z udziałemkarbokationu C(+).
- hydroliza kwasowa – odwracalna:R-CO-NH2 + H+ + H2O <==> R-COOH + NH4
+
- hydroliza zasadowa – nieodwracalna:R-CO-NH2 + OH- + H2O <==> R-COO- + NH3
Szczególnym przypadkiem jest hydroliza mocznika, zachodząca wg równań:NH2-CO-NH2 + H2SO4 + H2O ---> CO2 + (NH4)2SO4,NH2-CO-NH2 + 2 NaOH ---> Na2CO3 + 2NH3.
d) Amidy są mniej reaktywne od estrów i gorzej ulegają hydrolizie.e) Amidy I-rzędowe w obecności kwasu azotowego (III) wydzielają azot cząsteczkowy, np.:
CH3-CH2-CH2-NH2 + HNO2 ---> CH3-CH2-CH2-OH + N2 + H2O,f) Amidy I-rzędowe w podwyŜszonej temperaturze i w obecności czynników odwadniających (P2O5, SOCl2)
ulegają dehydratacji:R-CH-NH2 ---T---> R-C≡N| + H2O (pochłaniana).
g) W obecności silnych reduktorów (LiAlH4, B2H6) moŜna przeprowadzić amidy w aminy:R-CO-NH2 ---+2H---> R-CH2-NH2 + H2O,R1-CO-NH-R2 ---+2H---> R1-CH2-N-R2 + H2O.
AMIDY KWASU WĘGLOWEGOKwas węglowy H2CO3 jest kwasem nieorganicznym. Cechuje się duŜa nietrwałością i juŜ w niewielkichstęŜeniach rozkłada się na dwutlenek węgla i wodę. Przez zastąpienie pojedynczej grupy hydroksylowej grupąaminową otrzymujemy monoamid k. w. – kwas karbaminowy HO-CO-NH2, zaś przez zastąpienie obu grup –diamid k. w., czyli mocznik NH2-CO-NH2. Estry kwasu karbaminowego to uretany o wzorze ogólnym R-O-CO-NH2.Mocznik otrzymać moŜna na róŜne sposoby:a) w laboratorium – przez ogrzewanie izocyjanianu amonu – pod wpływem temperatury zachodzi
izomeryzacja:NH4OCN ---T---> NH2CONH2,
b) przez reakcję fosgenu z amoniakiem:COCl2 + 2 NH3 ---> NH2CONH2 + 2HCl,
c) bezpośrednio z amoniaku i tlenku węgla przy uŜyciu siarki:2 NH3 + CO + S ---> NH2CONH2 + H2S,
d) na skalę przemysłową – z amoniaku i dwutlenku węgla:2 NH3 + CO2 ---p≈2-2,5atm,T≈180-200oC---> NH2CONH2 + H2O.
Jest to związek rozpuszczalny w wodzie, dający odczyn obojętny. Wolne pary elektronowe atomów azotu mogąprzyłączać kation wodorowy, co daje pewne właściwości zasadowe. Pozwala to mocznikowi na reakcję zkwasem azotowym (V), w wyniku czego powstaje azotan (V) mocznika: CO(NH2)2
.HNO3. Związek ten nierozpuszcza się w wodzie i strąca się w postaci osadu, co umoŜliwia oznaczenie stęŜenia mocznika np. w moczu.Wolne pary elektronowe zwiększają ponadto ogólną reaktywność mocznika. Reaguje on np. z formaldehydem,dając hydroksymetylomocznik (tworzywo polikondensacyjne):2 NH2-CO-NH2 + HCHO ---> NH2-CO-NH-CH2-NH-CO-NH2 + H2O.Mocznik podczas ogrzewania przekształca się w dwumocznik, czyli biuret:2 NH2CONH2 ---T---> NH2-CO-NH-CO-NH2 + NH3.
Created by Neevia Document Converter trial version http://www.neevia.com

101
Biuret jest o tyle istotny, Ŝe zawiera w cząsteczce charakterystyczne dla białek wiązanie peptydowe. Wkontakcie z jonami miedzi (II) biuret daje intensywnie fioletowe zabarwienie. Reakcja ta, zwana biuretową,słuŜy do wykrywania oligopeptydów od tripeptydów wzwyŜ.Pochodnymi mocznika są: tiomocznik NH2-CS-NH2 oraz guanidyna NH2-CNH-NH2. Pochodne tej ostatniejmają szerokie zastosowanie w medycynie, np. przeciwbiegunkowa sufloguanidyna czy znany antybiotyk –streptomycyna.
NITRYLENitryle to pochodne kwasu cyjanowodorowego, w cząsteczce którego atom wodoru zastąpiono rodnikiemalkilowym bądź arylowym. Ich wzór ogólny ma postać: R-C≡N|. Nazwy tych związków tworzy się przezdodanie do rodnika końcówki „–karbonitryl” lub przez poprzedzenie nazwy rodnika wyrazem „cyjanek”.Nitryle otrzymuje się głównie w reakcji wymiany podwójnej pomiędzy chlorowcopochodnymi węglowodorów icyjankami nieorganicznymi, np.:RX + KCN ---> RCN + KX.Produktem głównym reakcji jest poŜądany nitryl, zaś ubocznym – izonitryl.Cząsteczki nitryli są silnie polarne, przez co charakteryzują się duŜym momentem dipolowym. Konsekwencjątego jest wzrost aktywności chemicznej; dzięki łatwości przyłączania protonu nitryle pełnią funkcjeprzenośników wodoru w przemianach chemicznych. Ponadto moŜna z nich otrzymać wiele innych grupzwiązków.WaŜnym surowcem w syntezie organicznej jest nienasycony akrylonitryl (etanokarbonitryl): CH2=CH-CN. Wwyniku jego polimeryzacji otrzymuje się włókno syntetyczne – anilanę:n CH2=CH-CN ---war.---> -[-CH2-CH(CN)-]n-.
IZONITRYLEIzonitryle róŜnią się kolejnością przyłączenia atomów węgla i azotu do fragmentu węglowodorowego. Ich wzórogólny ma postać: R-N=C. Są to związki toksyczne o bardzo przykrym zapachu.Ich powstanie słuŜy do wykrywania amin I-rzędowych w reakcji z chloroformem:R-NH2 + CHCl3 + 2 NaOH ---> RNC + 3 NaCl + 3H2O.W wyniku ogrzewania zachodzi samorzutna izomeryzacja do nitryli.W środowisku kwasowym izonitryle ulegają hydrolizie do amin I-rzędowych i kwasu mrówkowego:RNC + 2H2O ---H+---> RNH2 + HCOOH.
NITROZWIĄZKIZwiązki nitrowe są pochodnymi węglowodorów, w których atom wodoru zastąpiony został grupą –NO2. Istniejezasadnicza róŜnica pomiędzy nimi a azotanami (III), które posiadają grupę funkcyjną o identycznym wzorzesumarycznym – w nitrozwiązkach atom azotu połączony jest bezpośrednio z atomem węgla, natomiast wazotanach obecne jest połączenie przez atom tlenu. Ponadto w nitrozwiązkach jeden z atomów tlenu połączonyjest koordynacyjnie, zaś w azotanach wszystkie wiązania są typu kowalencyjnego. W wyniku oddziaływaniaelektrostatycznego pomiędzy wolnymi parami elektronowymi obu atomów tlenu i elektronami wiązaniapodwójnego następuje w obrębie grupy nitrowej delokalizacja wiązań i efekt mezomeryczny. Grupa funkcyjnawykazuje charakter polarny. NiŜsze nitroalkany są cieczami, zaś wyŜsze – ciałami stałymi.Nitrozwiązki alkilowe są produktami reakcji węglowodorów z mieszaniną nitrującą – stęŜonymi kwasami:siarkowym (VI) i azotowym (V):RH + HNO3 ---H2SO4---> R-NO2 + H2O.Aromatyczne nitrozwiązki otrzymuje się w reakcjach nitrowania, zachodzącej tu bardzo łatwo (nawet bez uŜyciakwasu siarkowego). Mają ona barwę Ŝółtą lub pomarańczową, niektóre wydają przyjemną woń, np. nitrobenzen– zapach gorzkich migdałów. Redukcja tego ostatniego daje anilinę:C6H5-NO2 ---+6H---> C6H5-NH2 + 2H2O.Znane są aromatyczne związki polinitrowe, np. trotyl (2,4,6-trinitrotoluen) czy kwas pikrynowy (2,4,6-trinitrofenol). Z reguły posiadają one właściwości wybuchowe. Grupy nitrowe są podstawnikami II rodzaju, stądjednoczesna obecność kilku znacznie zubaŜa pierścień w elektrony. Rekompensata następuje przez ściąganieelektronów z atomów tlenu, co objawia się ułatwieniem ich dysocjacji.
ZWIĄZKI SIARKOWE
TIOLETiole, zwanie dawniej merkaptanami, są siarkowymi odpowiednikami alkoholi – atom tlenu zastąpiony jest wnich atomem siarki. Wynika stąd wzór R-SH. Siarka jest pierwiastkiem o mniejszej elektroujemności od tlenu(leŜy w trzecim okresie, a tlen w pierwszym), dlatego wiązanie S-H jest słabsze od bliźniaczego O-H. Atomwodoru moŜe być łatwiej odszczepiany, zatem tiole posiadają bardziej kwasowy charakter od alkoholi.
Created by Neevia Document Converter trial version http://www.neevia.com

102
Potwierdzeniem tego jest następujące porównanie: alkohole reaguje z halogenkiem:R-OH + HBr ---> RBr + H2O,lecz analogiczne tiole – nie:R-SH + HBr ---> nie zachodzi,gdyŜ oba reagenty mają kwasowy charakter. Słabsze są równieŜ oddziaływania pomiędzy cząsteczkami, dlategocechują je niŜsze temperatury wrzenia oraz większa lotność. Pod wpływem utleniaczy moŜna przekształcićgrupę tiolową, otrzymując nietrwałe disulfidy:2 R-SH ---[O]---> R-S-S-R + H2O,obecne nagminnie w procesach biologicznych, bądź inne związki, np. kwasy sulfonowe:R-SH ---b. silne utleniacze---> R-SO3H.Pochodne tych ostatnich stosowane są jako środki bakteriobójcze. Zalicza się tu takŜe sulfonamidy, jaknajprostszy pod względem budowy sulfanilamid. Podstawienie atomów wodoru daje inne pochodne, np.sulfotiosol czy przeciwbiegunkową sulfoguanidynę:
.
SULFIDYSulfidy, dawniej tioetery, są siarkowymi odpowiednikami estrów. Mają wzór ogólny R1-S-R2, gdzie R oznaczafragmenty węglowodorowe. Sulfidy są cieczami o nieprzyjemnym zapachu, pod względem chemicznym bardziejreaktywne niŜ odpowiadające im etery. W wyniku selektywnego chlorowania sulfidu dietylowego otrzymuje sięsiarczek β, β-dichloroetylowy – gaz bojowy zwany musztardowym lub iperytem (od zastosowania go po razpierwszy pod Ypres).
KWASY SULFONOWEKwasami sulfonowymi nazywamy związki postaci R-SO3H, gdzie R jest rodnikiem węglowodorowym. Są tokwasy mocne, tworzące z kationami trwałe sole. Występującą w nich grupę hydroksylową moŜna zastąpić,otrzymując np. chlorki sulfonowe R-SO2-Cl, estry kwasów sulfonowych R1-SO2-O-R2 czy sulfonamidy R-SO2-NH2. Te ostatnie są ciałami stałymi i wykazują pewien charakter zasadowy objawiający się moŜliwościąprzyłączenia wodoru. Wykazują działanie bakteriobójcze, np. para-aminobenzenosulfoamid H2N-SO2-C6H4-NH2, którego pochodne podaje się przy biegunkach. Jego pochodną jest sacharyna, związek około 500 razysłodszy od sacharozy, choć dla człowieka nieprzyswajalny. Stanowi ona składnik słodzików dietetycznych.
AROMATYCZNE ZWIĄZKI HETEROCYKLICZNE
Są to związki o budowie cyklicznej, w których atom węgla zastąpiony został „obcym”, stąd przedrostek„hetero”. Niewęglowy heteroatom moŜe wnieść własny elektron do pierścienia, który dzięki temu moŜe nabraćcech aromatyczności (zatem nie jest to wyłączny atrybut wybranych pierścieni węglowych). Najczęściejspotykamy pierścienie pięcio- oraz sześcioczłonowe, gdyŜ w tych najłatwiej spełnić warunek aromatyczności, tj.regułę Hückla: 2 + 4 n elektronów zdelokalizowanych. Charakter aromatyczny, mierzony zdolnością dooddawanie elektronów do pierścienia, malaje w szeregu: furan – pirol – tiofen. Następstwem aromatycznegocharakteru jest wyrównanie długości wiązań oraz zmniejszenie wartości momentów dipolowychRoboczo moŜna podzielić związki heterocykliczne na:a) z pierścieniem pięcioczłonowym:- Furan ma waŜne znaczenie przy nazewnictwie cukrów. Jego pochodną jest aldehyd furylowy (furfural):
Created by Neevia Document Converter trial version http://www.neevia.com

103
.Produktem dehydratacji w środowisku kwaśnym cukrów z grupy pentoz jest furfural, który tworzy
barwne (wiśnia) połączenie z floroglucyną (s-trihydroksobenzen) oraz z orcyną (kolor błękitno-zielony). Zkolei produktem dehydratacji heksoz jest 5-hydroksymetylofurfural, tworzący z rezorcyną (m-dihydroksybenzen) i kwasem solnym barwę pomarańczowoczerwoną.
- Pirol wykazuje słaby charakter kwasowy, gdyŜ jako nieliczny posiada w cząsteczce atom wodoru połączonyw heteroatomem. Ów atom wodoru odszczepia się tym łatwiej, iŜ wiązanie z atomem azotu osłabione jestprzez ubytek elektronów wciągniętych do sekstetu. Cztery pierścienie pirolowe połączone przez czterymostki metinowe tworzą płaski pierścień porfirynowy. W wyniku tworzenia połączeń kompleksowych zróŜnorodnymi jonami (Mg2+, Fe2+) powstają porfiryny, stanowiące składnik chlorofilu, hemoglobiny,witaminy B12 i innych związków biochemicznych.
b) z pierścieniem sześcioczłonowym:- Piran jest tlenowym związkiem sześcioczłonowym. Jego kation ma charakter aromatyczny i jest stosunkowo
trwały. Sam piran nie występuje w przyrodzie w stanie wolnym, znane są jego liczne pochodne. Układpiranowy spotykany jest w wielu barwnikach naturalnych – antocyjanach.
Czerwone antocyjany są najpiękniejszymi naturalnymi barwnikami. W wyniku utlenienia dają flawonyo silnym działaniu bakteriobójczym. Te ostatnie wchodzą w skład wielu związków, np. kwercetyna w składrutyny (witamina P).
- Pirydyna (azyna) ma ostry smak i zapach (pieprz). Cząsteczka posiada wolną parę elektronową na atomieazotu, co zwiększa jej moment dipolowy i pozwala na rozpuszczalność w wodzie praktycznie w kaŜdymstosunku. Wolna para nie bierze udziału w sekstecie elektronowym, przez co pirydyna jest mocniejszązasadą od pirolu. Charakter słabej zasady ujawnia się w reakcji z kwasami, wynikiem której są związki ocharakterze soli. Podstawienie elektrofilowe zachodzi trudniej niŜ w benzenie, gdyŜ wolna para odciągaelektrony i osłabia sekstet. Pirydyna ulega uwodornieniu na katalizatorze, w wyniku czego powstajepiperydyna.WaŜne z biologicznego punktu widzenia są pochodne pirydyny: nikotyna, kwas izonikotynowy, amid kwasunikotynowego (witamina PP), kardiamid i koramina.
c) z dwoma heteroatomami:- Imidazol jest składnikiem zasad purynowych. Zamiast jednego z atomów azotu moŜe występować atom
tlenu bądź siarki (tiazol – składnik katalizujący dekarboksylację).
- Pirymidyna (1,3-diazyna) jest bardzo słabą zasadą pomimo obecności wolnych dwóch par elektronowych.Bardzo istotne są pochodne pirymidyny zawierające grupy hydroksylowe, gdyŜ ulegają procesowitautomerii: cytozyna, tymina, uracyl, kwas barbiturowy.
- W wyniku kondensacji pierścienia imidazolu i pirymidyny powstaje puryna, stanowiąca szkielet dlaniektórych zasad azotowych (adenina czy guanina).
WIELOFUNKCYJNE POCHODNE WĘGLOWODORÓW
W odróŜnieniu od pochodnych jednofunkcyjnych, omawiane związki zawierają w cząsteczce dwie lub więcejgrup funkcyjnych (patrz wprowadzenie). Oznacza to ogromną róŜnorodność zarówno samej budowy, jak imoŜliwości oraz wynikających nich zastosowań.
Created by Neevia Document Converter trial version http://www.neevia.com

104
HYDROKSYKWASYHydroksykwasy są dwufunkcyjnymi pochodnymi węglowodorów, zawierającymi obok grupy karboksylowejrównieŜ grupę hydroksylową. Ich najbardziej ogólny wzór ma postać: (HO)n-R-(COOH)m. Obecnośćelektroujemnej grupy zwiększa moc tych kwasów, największy zaś wpływ na właściwości ma ona w połoŜeniu α.Wiele hydroksykwasów posiada asymetryczny atom węgla, w związku z czym są one chiralne. Dzięki temuwystępuje czynność optyczna, polegająca na skręcaniu płaszczyzny światła spolaryzowanego liniowo. Izomery iszeregi D i L wyróŜnia się na podstawie konfiguracji ostatniego asymetrycznego atomu węgla – jeŜeli grupahydroksylowa znajduje się w rzucie Fischera po prawej stronie, to cząsteczka ma konfigurację D, gdy zaś polewej – L. Oznaczenie skręcalności +/- opiera się na eksperymentalnie wyznaczonym kierunku skręcania –izomer skręcający płaszczyznę światła w prawo oznacza się „+”, a w lewo „–” . Teoretyczna liczbastereoizomerów cząsteczki o n asymetrycznych atomach węgla wynosi 2n. Wyjątkiem są cząsteczki posiadającewewnętrzną oś symetrii, a przez to nieczynne optycznie – nazywamy je odmianami mezo-.PowyŜsze sytuacje zaprezentować moŜna na przykładzie kwasu winowego:
.Hydroksykwasy dają róŜne reakcje chemiczne:a) reagują z odczynnikami zasadowymi, np. wodorotlenkami metali alkalicznych i zobojętnieją je, dając
hydroksysole, np.:CH3-CH(OH)-COOH + NaOH ---> CH3-CH(OH)-COONa + H2O,
b) ulegają estryfikacji z kwasami, alkoholami i fenolami, np. estryfikacja kwasu salicylowego kwasemoctowym daje kwas acetylosalicylowy, czyli aspirynę:C6H4(COOH)-OH + CH3COOH ---> C6H4(COOH)-O-CO-CH3 + H2O,z kolei podstawienie w pozycji para- grupy aminowej daje kwas p-aminosalicylowy (PAS) – lekprzeciwgruźliczy,
c) w wyniku ogrzewania α-hydroksykwasy tworzą cykliczne estry zwane laktydami, np.:
,d) w wyniku ogrzewania w środowisku kwaśnym β-hydroksykwasy ulegają dehydratacji, np.:
HO-CH2-COOH ---T,H2O---> CH2=CH-COOH + H2O,CH3-CH(OH)-CH2-COOH ---T,H2O---> CH3-CH=CH-COOH + H2O.Gdy istnieje moŜliwość otrzymania dwóch produktów, obowiązuje reguła Zajcewa.
e) juŜ słabe ogrzewanie γ- lub δ-hydroksykwasów powoduje powstawanie cyklicznych estrów wewnętrznych,zwanych laktonami, np.:
.
Hydroksykwasy występują powszechnie w przyrodzie – najczęściej występujące zebrano w tabeli:Wzór Nazwa zwyczajowa Nazwa systematyczna
HO-CH2-COOH kwas glikolowy,hydroksyoctowy
kwas hydroksyetanowy
CH3-CH(OH)-COOH kwas mlekowy kwas α-hydroksypropionowyCH3-CH(OH)-CH2-COOH kwas β-hydroksymasłowy kwas β-hydroksybutanowyHOOC-CH2-CH(OH)-COOH kwas jabłkowy,
hydroksybursztynowykwas α-hydroksybutanodiowy
HOOC-CH(OH)-CH(OH)-COOH kwas winowy, kwas 2,3-dihydoksybutanodiowy
Created by Neevia Document Converter trial version http://www.neevia.com
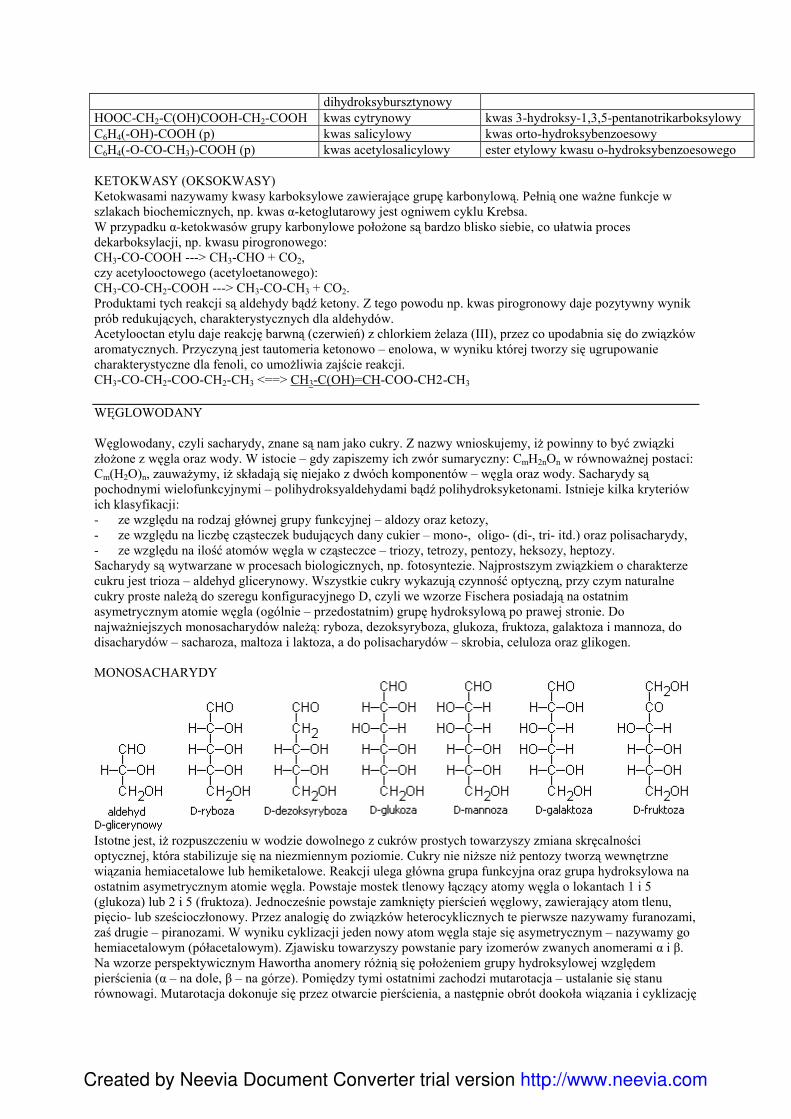
105
dihydroksybursztynowyHOOC-CH2-C(OH)COOH-CH2-COOH kwas cytrynowy kwas 3-hydroksy-1,3,5-pentanotrikarboksylowyC6H4(-OH)-COOH (p) kwas salicylowy kwas orto-hydroksybenzoesowyC6H4(-O-CO-CH3)-COOH (p) kwas acetylosalicylowy ester etylowy kwasu o-hydroksybenzoesowego
KETOKWASY (OKSOKWASY)Ketokwasami nazywamy kwasy karboksylowe zawierające grupę karbonylową. Pełnią one waŜne funkcje wszlakach biochemicznych, np. kwas α-ketoglutarowy jest ogniwem cyklu Krebsa.W przypadku α-ketokwasów grupy karbonylowe połoŜone są bardzo blisko siebie, co ułatwia procesdekarboksylacji, np. kwasu pirogronowego:CH3-CO-COOH ---> CH3-CHO + CO2,czy acetylooctowego (acetyloetanowego):CH3-CO-CH2-COOH ---> CH3-CO-CH3 + CO2.Produktami tych reakcji są aldehydy bądź ketony. Z tego powodu np. kwas pirogronowy daje pozytywny wynikprób redukujących, charakterystycznych dla aldehydów.Acetylooctan etylu daje reakcję barwną (czerwień) z chlorkiem Ŝelaza (III), przez co upodabnia się do związkówaromatycznych. Przyczyną jest tautomeria ketonowo – enolowa, w wyniku której tworzy się ugrupowaniecharakterystyczne dla fenoli, co umoŜliwia zajście reakcji.CH3-CO-CH2-COO-CH2-CH3 <==> CH3-C(OH)=CH-COO-CH2-CH3
WĘGLOWODANY
Węglowodany, czyli sacharydy, znane są nam jako cukry. Z nazwy wnioskujemy, iŜ powinny to być związkizłoŜone z węgla oraz wody. W istocie – gdy zapiszemy ich zwór sumaryczny: CmH2nOn w równowaŜnej postaci:Cm(H2O)n, zauwaŜymy, iŜ składają się niejako z dwóch komponentów – węgla oraz wody. Sacharydy sąpochodnymi wielofunkcyjnymi – polihydroksyaldehydami bądź polihydroksyketonami. Istnieje kilka kryteriówich klasyfikacji:- ze względu na rodzaj głównej grupy funkcyjnej – aldozy oraz ketozy,- ze względu na liczbę cząsteczek budujących dany cukier – mono-, oligo- (di-, tri- itd.) oraz polisacharydy,- ze względu na ilość atomów węgla w cząsteczce – triozy, tetrozy, pentozy, heksozy, heptozy.Sacharydy są wytwarzane w procesach biologicznych, np. fotosyntezie. Najprostszym związkiem o charakterzecukru jest trioza – aldehyd glicerynowy. Wszystkie cukry wykazują czynność optyczną, przy czym naturalnecukry proste naleŜą do szeregu konfiguracyjnego D, czyli we wzorze Fischera posiadają na ostatnimasymetrycznym atomie węgla (ogólnie – przedostatnim) grupę hydroksylową po prawej stronie. DonajwaŜniejszych monosacharydów naleŜą: ryboza, dezoksyryboza, glukoza, fruktoza, galaktoza i mannoza, dodisacharydów – sacharoza, maltoza i laktoza, a do polisacharydów – skrobia, celuloza oraz glikogen.
MONOSACHARYDY
Istotne jest, iŜ rozpuszczeniu w wodzie dowolnego z cukrów prostych towarzyszy zmiana skręcalnościoptycznej, która stabilizuje się na niezmiennym poziomie. Cukry nie niŜsze niŜ pentozy tworzą wewnętrznewiązania hemiacetalowe lub hemiketalowe. Reakcji ulega główna grupa funkcyjna oraz grupa hydroksylowa naostatnim asymetrycznym atomie węgla. Powstaje mostek tlenowy łączący atomy węgla o lokantach 1 i 5(glukoza) lub 2 i 5 (fruktoza). Jednocześnie powstaje zamknięty pierścień węglowy, zawierający atom tlenu,pięcio- lub sześcioczłonowy. Przez analogię do związków heterocyklicznych te pierwsze nazywamy furanozami,zaś drugie – piranozami. W wyniku cyklizacji jeden nowy atom węgla staje się asymetrycznym – nazywamy gohemiacetalowym (półacetalowym). Zjawisku towarzyszy powstanie pary izomerów zwanych anomerami α i β.Na wzorze perspektywicznym Hawortha anomery róŜnią się połoŜeniem grupy hydroksylowej względempierścienia (α – na dole, β – na górze). Pomiędzy tymi ostatnimi zachodzi mutarotacja – ustalanie się stanurównowagi. Mutarotacja dokonuje się przez otwarcie pierścienia, a następnie obrót dookoła wiązania i cyklizację
Created by Neevia Document Converter trial version http://www.neevia.com
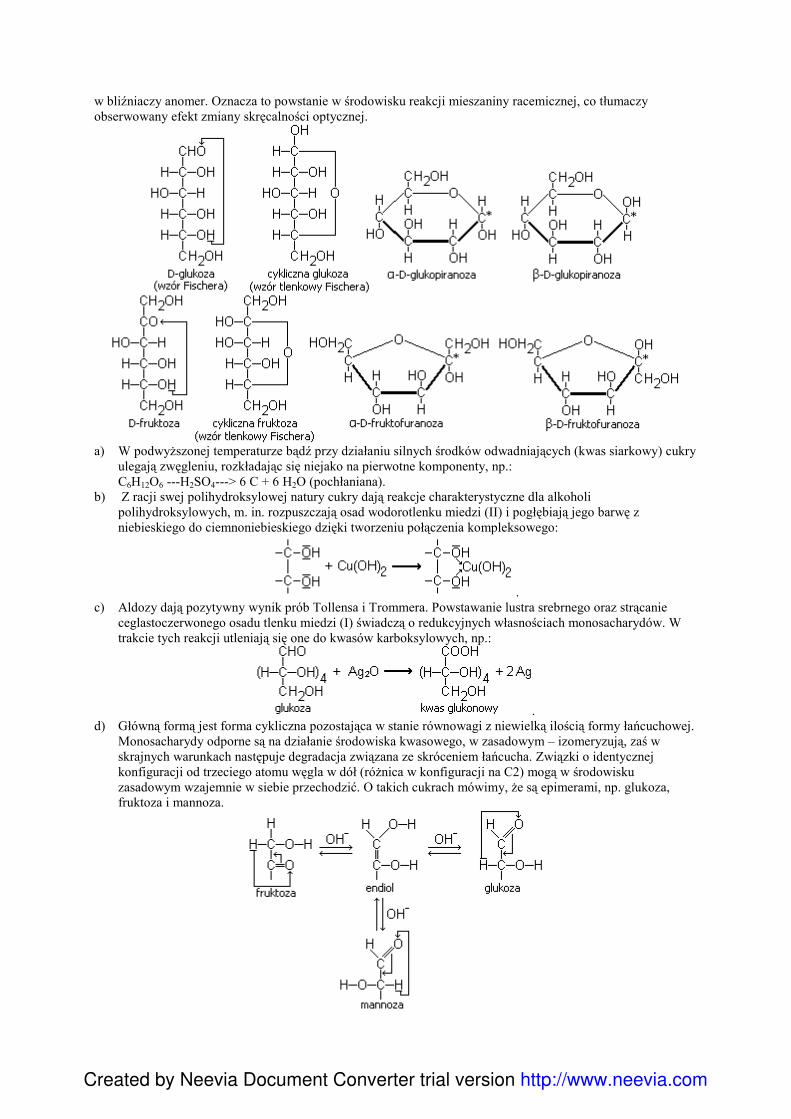
106
w bliźniaczy anomer. Oznacza to powstanie w środowisku reakcji mieszaniny racemicznej, co tłumaczyobserwowany efekt zmiany skręcalności optycznej.
a) W podwyŜszonej temperaturze bądź przy działaniu silnych środków odwadniających (kwas siarkowy) cukryulegają zwęgleniu, rozkładając się niejako na pierwotne komponenty, np.:C6H12O6 ---H2SO4---> 6 C + 6 H2O (pochłaniana).
b) Z racji swej polihydroksylowej natury cukry dają reakcje charakterystyczne dla alkoholipolihydroksylowych, m. in. rozpuszczają osad wodorotlenku miedzi (II) i pogłębiają jego barwę zniebieskiego do ciemnoniebieskiego dzięki tworzeniu połączenia kompleksowego:
.c) Aldozy dają pozytywny wynik prób Tollensa i Trommera. Powstawanie lustra srebrnego oraz strącanie
ceglastoczerwonego osadu tlenku miedzi (I) świadczą o redukcyjnych własnościach monosacharydów. Wtrakcie tych reakcji utleniają się one do kwasów karboksylowych, np.:
.d) Główną formą jest forma cykliczna pozostająca w stanie równowagi z niewielką ilością formy łańcuchowej.
Monosacharydy odporne są na działanie środowiska kwasowego, w zasadowym – izomeryzują, zaś wskrajnych warunkach następuje degradacja związana ze skróceniem łańcucha. Związki o identycznejkonfiguracji od trzeciego atomu węgla w dół (róŜnica w konfiguracji na C2) mogą w środowiskuzasadowym wzajemnie w siebie przechodzić. O takich cukrach mówimy, Ŝe są epimerami, np. glukoza,fruktoza i mannoza.
Created by Neevia Document Converter trial version http://www.neevia.com

107
e) W środowisku silnie kwaśnym i pod wpływem podwyŜszonej temperatury monosacharydy ulegajądehydratacji, dając charakterystyczne produkty (reakcja Seliwanowa). Z pentoz powstaje aldehyd furylowy,czyli furfural, który w reakcji z floroglucyną daje barwę wiśniową, a z orcyną (odczynnik Biala) – błękitno-zieloną. Z kolei z heksoz powstaje 5-hydroksymetylofurfural, który z rezorcyną (odczynnik Seliwanowa)daje zabarwienie pomarańczowoczerwone.
f) Przy udziale bardzo silnych środków redukujących (NaBH4, B2H6, Na|Hg) moŜna zredukować cukry dopolihydroksyzwiązków. Produktem redukcji glukozy i fruktozy jest sorbitol:
.g) Monosacharydy mogą równieŜ ulegać utlenianiu. W przypadku ketoz prowadzi to do rozerwania łańcucha
węglowego, np.:
.h) Z kolei produkt utleniania aldoz zaleŜy silnie od warunków przemiany i tak:- reakcja z wodą bromową zachodzi w obecności wodorosiarczanu (VI) sodu i prowadzi do utlenienia grup
aldehydowych – powstają kwasy aldonowe (np. dla glukozy – kwas glukonowy),- reakcja z kwasem azotowym (V) prowadzi do utlenienia początkowej i ostatniej grupy hydroksylowej –
powstają kwasy aldarowe (np. dla glukozy – kwas glukarowy),- reakcja katalizowana enzymatycznie prowadzi do utlenienia ostatniej grupy hydroksylowej – powstają
kwasy alduronowe (np. dla glukozy – kwas glukuronowy – znaczenie przy detoksykacji organizmu).i) W przeciwieństwie do aldehydów aldozy nie tworzą połączenia z wodorosiarczanem sodu.j) Cukry reagują z pochodnymi amoniaku. W reakcji z hydroksyloaminą powstają oksymy, zaś z
fenylohydrazyną – osazony:
Traktowanie epimerów fenylohydrazyną daje w efekcie ten sam osazon. DostrzeŜenie tej prawidłowościstanowiło waŜny krok w pracach Fischera nad ustaleniem budowy cukrów.
k) W reakcje wchodzić moŜe równieŜ hemiacetalowa grupa hydroksylowa, a produktami są glikozydy. Są topochodne cukrów, w których fragment sacharydowy połączony jest za pośrednictwem atomu tlenu (O-glikozydy) bądź azotu (N-glikozydy) z fragmentem innego związku. Przykładowo glukoza daje O-glikozydw reakcji z metanolem:
.Reakcja analogiczna do powyŜszej, zachodząca pomiędzy dwoma cząsteczkami α-D-glukozy, prowadzi dopowstania wiązania α(1,4)-glikozydowego i daje w wyniku dwucukier – maltozę (cukier słodowy). N-glikozydy rybozy i dezoksyrybozy z zasadami azotowymi (purynowymi i pirymidynowymi) to nukleozydywchodzące w skład kwasów nukleinowych, np.:
Created by Neevia Document Converter trial version http://www.neevia.com

108
.
OLIGOSACHARYDYOligosacharydy są produktami kondensacji 2 do 10 cząsteczek cukrów prostych. W grupie tej niewątpliwienajwaŜniejsze są disacharydy – sacharoza, maltoza, laktoza i celobioza. Disacharydy powstają dziękiwytworzeniu wiązania O-glikozydowego pomiędzy atomem węgla hemiacetalowym bądź hemiketalowym orazgrupą hydroksylową drugiej cząsteczki cukru prostego. Wiązanie to moŜe powstać między dwoma atomamiwęgla hemi, wówczas jednak produkt pozbawiony jest własności redukcyjnych. Wiązanie acetalowe (mostektlenowy) jest trwałe w środowisku zasadowym, w kwaśnym zaś ulega hydrolizie, odtwarzając wyjściowe cukryproste.
a) Sacharoza (cukier buraczany, trzcinowy) powstaje w wyniku kondensacji α-D-glukozy z β-D-fruktozą.Powstałe wiązanie 1,4-glikozydowe blokuje węgle hemi, co uniemoŜliwia powtórne otwarcie pierścienia.Cząsteczka nie ulega mutarotacji i nie wykazuje właściwości redukujących (negatywny wynik prób Tollensai Trommera).Sacharozę otrzymuje się w cukrowniach z buraków cukrowych. Zszatkowane buraki (krajanka) poddawanesą dyfuzji – działaniu gorącej pary wodnej w celu przyspieszenia rozpuszczania sacharozy. Otrzymany wten sposób sok dyfuzyjny (rzadki) ulega defekacji – oczyszczaniu przez działanie mlekiem wapiennym.Sacharoza tworzy wówczas rozpuszczalne w wodzie cukrzany wapnia. Kolejnym etapem jest saturacja,polegająca na działaniu dwutlenkiem węgla (lub siarki gdy buraki są przymarznięte). Powoduje to stracenienadmiaru jonów wapniowych oraz zanieczyszczeń, które nie związały się z zasadą. W wyniku odparowaniawody otrzymuje się sok gesty. Dalsze zagęszczenie i zaszczepienie zarodkami krystalizacji powodujepowstanie kryształów, które ulegają odwirowaniu. Powtórne zagęszczenie daje cukier przemysłowy, zaśostatni (trzeci) odciek – melasę.Roztwór sacharozy cechuje dodatnia skręcalność optyczna, która po hydrolizie zmienia się na ujemną.Zjawisko to nosi nazwę inwersji, dlatego sacharozę nazywa się cukrem inwertowanym, a enzymkatalizujący jej hydrolizę – inwertazą. Na skalę techniczną inwersję sacharozy stosuje się do produkcjisztucznego miodu (po dodaniu karmelu i esencji). Sacharoza ogrzana powyŜej 150-160oC topi się,przyjmując barwę brunatną i przekształcając się w karmel. Ten ostatni jest naturalnym barwnikiemdodawanym do produktów spoŜywczych, np. wyrobów cukierniczych czy alkoholowych.
Created by Neevia Document Converter trial version http://www.neevia.com

109
b) Maltoza (cukier słodowy) jest produktem kondensacji dwóch cząsteczek α-D-glukozy. Wytwarza sięwiązanie 1,4-glikozydowe, co oznacza pozostawienie wolnego węgla hemi. UmoŜliwia to otwarciepierścienia, mutarotację i właściwości redukcyjne.
c) Celobioza jest bliźniaczką maltozy, gdyŜ tworzą ją dwie cząsteczki β-D-glukozy. Wiązanie wytwarza siętakŜe pomiędzy atomami C1 i C4.
d) Laktoza (cukier mleczny) składa się z β-D-glukozy oraz β-D-galaktozy. Tworzące się wiązanie łączy atomywęgla 1 (ze strony galaktozy) oraz 4 (ze strony glukozy).
POLISACHARYDYPolisacharydy (wielocukry) są wielkocząsteczkowymi polimerami utworzonymi z fragmentów heksoz. KaŜdymer łączy się z sąsiednimi za pośrednictwem wiązań glikozydowych. Powoduje to oddzielenie od niego atomuwodoru z jednej oraz grupy hydroksylowej z drugiej strony. Wynika stąd wzór ogólny postaci: (C6H10O5)n, gdzien oznacza liczbę merów, wahającą się w szerokich granicach – od kilkuset do wielu tysięcy. Ze względu narodzaj merów wyróŜniamy homopolisacharydy (utworzone z jednego rodzaju cukru prostego) orazheteropolisacharydy (utworzone z róŜnych reszt cukrowych). W całej cząsteczce obecny jest tylko jeden wolnyatom węgla hemi, co tłumaczy brak właściwości redukujących. śaden z polisacharydów nie jest w smaku słodki.Do najwaŜniejszych wielocukrów zaliczamy: skrobię, celulozę, glikogen oraz pektyny.a) Skrobia utworzona jest z podjednostek glukozowych połączonych głównie wiązaniami α-1,4-
glikozydowymi (tak jak w cząsteczce maltozy). Amyloza jest cząsteczką o budowie liniowej, w której niewystępują inne wiązania. Natomiast w amylopektynie obok powyŜszych występują co 15 - 20 merówwiązania α-1,6, przez co cząsteczka ma budowę rozgałęzioną. Amyloza w obecności jodu barwi się na kolorniebieski, co jest wynikiem specyficznego ustawiania się łańcuchów wokół cząsteczki jodu; barwa znika popodgrzaniu środowiska. Dla zwiększenia kontaktu w praktyce uŜywa się roztworu jodu w jodku potasu (tzw.płyn Lugola). Reakcja jest bardzo czuła, dlatego wskaźnik skrobiowy często stosuje się w jodometrii.Skrobia niechętnie rozpuszcza się w zimnej wodzie, w ciepłej natomiast rozpuszczanie połączone jest ztworzeniem zolu (kleiku skrobiowego). W środowisku kwaśnym lub przy udziale enzymów skrobia ulegahydrolizie, dając mieszaninę cukrów niskich (np. tworzenie syropu konfiturowego). Dalsze etapy hydrolizyprowadzą do otrzymania maltozy, a ostatecznie – glukozy. Skrobia jest materiałem zapasowym dla świataroślinnego.
b) Glikogen jest węglowodanem zapasowym w świecie zwierzęcym, pełni podobne funkcje co skrobia uroślin. Budową zbliŜony jest do amylopektyny: składa się z merów glukozowych połączonych wiązaniamiα-1,4, zaś w znacznych odstępach pojawiają się wiązania α-1,6. W organizmie ludzkim ulega hydrolizieenzymatycznej, zwiększając stęŜenie glukozy we krwi.
c) Celuloza jest związkiem istotnym zwłaszcza dla roślin, buduje bowiem ich ściany komórkowe. Wcząsteczce występują wyłącznie wiązania β-1,4, dzięki czemu ma ona budowę liniową. Długie polimery sąpodstawowym składnikiem włókien roślinnych, np. lnu czy konopi. Choć dla człowieka nieprzyswajalna,moŜe być trawiona symbiotycznie w Ŝołądkach przeŜuwaczy.Z drzewa lub słomy wyodrębnia się krótkie włókna, oddzielane od pozostałych składników. W tym celudziała się na surowiec wodorosiarczanem (IV) wapnia, który rozpuszcza substancje niecelulozowe. Wformie czystej celuloza słuŜy do produkcji papieru, a po przetworzeniu – surowców włókienniczych. Dalszaobróbka pozwala otrzymać tworzywa sztuczne (modyfikowane), np. włókna wiskozowe czy octanowe oraznitrocelulozowe lakiery i materiały wybuchowe. Octan celulozy otrzymuje się działając na substratbezwodnikiem octowym, w wyniku czego otrzymuje się acetylocelulozę. Ta wykorzystywane jest doprodukcji błon fotograficznych oraz jedwabiu octanowego.Wiskoza jest zregenerowana celulozą. W celu jej otrzymania poddaje się celulozę działaniu zasady sodowej.Rozpuszczaniu towarzyszy powstanie płynnej alkalicelulozy. Produktem reakcji tej ostatniej zdwusiarczkiem węgla jest ksantogenian celulozy, który w formie zgęstniałej i stęŜonej tłoczy się przezmikrodysze do gorącej kąpieli w kwasie siarkowym (VI). Następuje zobojętnienie środowiska i odtworzeniecelulozy. Nowa postać – jedwab celulozowy jest bardzo gładką tkaniną. W ten sposób moŜna równieŜotrzymać celofan, uŜywany do opakowywania wyrobów cukierniczych, kwiatów itp.
d) Pektyny są polimerami kwasu α-D-galaktouronowego. Wcząsteczce obecne są wiązania pomiędzy C1 i C4. Wpektyny obfitują niektóre owoce, np. czarna porzeczka,dzięki czemu łatwo zrobić z nich galaretki (dodane dodŜemu pęcznieją w kontakcie z wodą).
Created by Neevia Document Converter trial version http://www.neevia.com

110
AMINOKWASY
Aminokwasy są dwufunkcyjnymi pochodnymi węglowodorów, zawierającymi w cząsteczce grupękarboksylowa oraz aminową. Wynika stąd ich wzór ogólny postaci: (H2N)n-R-(COOH)m, gdzie R jestfragmentem węglowodorowym. W warunkach naturalnych mają postać krystalicznych ciał stałych o barwiebiałej. Ogromna większość z nich wykazuje czynność optyczną, tworząc szeregi konfiguracyjne D i L(wyjątkiem jest np. glicyna). Wszystkie występujące w przyrodzie aminokwasy są α-aminokwasami okonfiguracji L. Oznacza to, iŜ główne grupy funkcyjne przyłączone są do jednego, I-rzędowego atomu węglaoraz Ŝe numeracja podstawników według kryterium masy rośnie podczas obrotu przeciwnie do wskazówekzegara.Aminokwasy otrzymać moŜna:a) przez hydrolizę peptydów i białek,b) przez działanie amoniakiem na α-halogenopochodne kwasów:
R-CH(Cl)-COOH + 2 NH3 ---> R-CH(NH2)-COOH + NH4Cl,c) przez redukcję aldehydów z cyjankiem amonowym:
R-CHO + NH4CN ---> R-CH(NH2)-CN ---H2O---> R-CH(NH2)-COOH + HCN.Większość aminokwasów dobrze rozpuszcza się w wodzie, tworząc roztwory, w których wykazują na ogółcharakter obojętny. Dzięki jednoczesnej obecności w cząsteczce grupy o charakterze kwasowym i zasadowym,tworzą one sole wewnętrzne, np.:H2N-CH2-COOH <==> H3N
+-CH2-COO-.Pozwala to występować cząsteczkom aminokwasów w postaci jonów obojnaczych i tworzyć kryształy jonowe.Oddziaływania jonowe tłumaczą polarną i krystaliczną strukturę aminokwasów. Związane są z tym wysokietemperatury topnienia; dalsze ogrzewanie prowadzi do rozkładu.a) Z racji amfoterycznego charakteru moŜliwa jest reakcja zarówno z kwasami, jak i z zasadami. W
środowisku zasadowym występują w formie anionowej:H2N-CH2-COOH + NaOH ---> H2N-CH2-COONa + H2O, (glicynian sodu)H3N
+-CH2-COO- + OH- ---> H2N-CH2-COO- + H2O,zaś w kwasowym – w formie kationowej:H2N-CH2-COOH + HCl ---> Cl-H3N
+-CH2-COOH = HCl.H2N-CH2-COOH, (chlorowodorek glicyny)H3N
+-CH2-COO- + H+ ---> H3N+-CH2-COOH + H2O.
b) Oznacza to, iŜ – w zaleŜności od odczynu środowiska – cząsteczka występuje w jednej z trzech form:H3N
+-CH2-COO- <==OH-==> H3N+-CH2-COO- <==H+==> H3N
+-CH2-COOH.Dla kaŜdego aminokwasu moŜna wskazać taką wartość pH, przy której stęŜenie jonu obojnaczego jestmaksymalne, a stęŜenia formy kationowej i anionowej są równe. Wartość taką nazywamy punktemizoelektrycznym – dla większości aminokwasów mieści się on w granicach 5 – 6, dla kwasowych jestmniejszy od 4, a dla zasadowych – większy od 9. Z racji najmniejszego stęŜenia form jonowych izwiązanego z tym minimum hydratacji, w punkcie izoelektrycznym aminokwasy wykazują najmniejsząrozpuszczalność.
c) Reakcja aminokwasów z kwasami karboksylowymi prowadzi do związków kompleksowych.d) Aminokwasy reagują z alkoholami, tworząc estry:
H2N-CH(R1)-COOH + R2-OH ---> H2N-CH(R1)-COO-R2.e) JeŜeli grupa aminowa jest w dalekim połoŜeniu w stosunku do grupy karboksylowej (4, 5, 6), to w wyniku
ogrzewania następuje wewnętrzna cyklizacja i powstają laktamy (cykliczne amidy). Przykładowo kwas 6-aminoheksanowy tworzy kaprolaktam, będący surowcem do produkcji tworzyw syntetycznych –poliamidów.
f) W odpowiednich warunkach moŜna pozbawić aminokwasy jednej z grup funkcyjnych. W wynikudekarboksylacji otrzymuje się tzw. aminy biogenne, zaś dezaminacja prowadzi do powstania ketokwasów:H2N-CH(R)-COOH ---war.---> R-CO-COOH + NH3.
g) Aminokwasy bezbarwne stają się widoczne po reakcji z ninhydryną. Produktami reakcji ninhydrynowej sąaldehydy odaminokwasowe oraz fioletowy barwnik:
Created by Neevia Document Converter trial version http://www.neevia.com

111
AMINOKWASY BIAŁKOWEZnanych jest około 170 aminokwasów, spośród których nieco ponad 20 występuje w białkach. RóŜnorodnośćich budowy i właściwości jest podstawą rozmaitych klasyfikacji. Najczęściej dzieli się je:a) ze względu na polarność rodnika na:- z R niepolarnym (n),- z R polarnym niejonizującym (p),- z R polarnym jonizującym – kwaśne (k) i zasadowe (z),b) ze względu na budowę rodnika na:- z łańcuchem bocznym alifatycznym (a),- z łańcuchem bocznym zawierającym grupy hydroksylowe (h),- z łańcuchem bocznym zawierającym atomy siarki (s),- z łańcuchem bocznym zawierającym grupy kwasowe lub ich amidy (k),- z łańcuchem bocznym zawierającym grupy zasadowe (z),- zawierające pierścienie aromatyczne (r),- iminokwasy (i),c) ze względu na moŜliwość syntezy przez organizm ludzki na:- egzogenne (niezbędne, niezastąpione) – nie są syntezowane w organizmie ludzkim, a ich obecność i
odpowiednie stęŜenie w białkach spoŜywczych decyduje o wartości odŜywczej; wyjątkowo histydyna jestniezbędna dla dzieci do lat 12, ale nie jest niezbędna dla dorosłych (g),
- endogenne – są syntezowane w organizmie ludzkim (przez hepatocyty – komórki wątroby) (n),d) ze względu na produkty metabolizmu na:- glikogenne, których metabolizm prowadzi do wytwarzania sacharydów (g),- ketogenne, których metabolizm prowadzi do wytworzenia związków ketonowych – kwasu β-
hydroksymasłowego, kwasu acetomasłowego, acetonu (k).Oryginalne badania „Ŝywieniowe” pozwoliły zaklasyfikować tryptofan jako glikogenny, a lizynę jakoglikogenną i ketogenną. Natomiast doświadczenia pośrednie nad metabolizmem aminokwasów wykazały,Ŝe lizyna jest zdecydowanie ketogenna, a tryptofan głównie ketogenny i tylko słabo glikogenny.
Zestawienie aminokwasów naturalnych przedstawiają poniŜsze tabele.
Lp. SymbolNazwa
zwyczajowa
Pola
rnoś
ć ro
dnik
a
Bud
owa
rodn
ika
Egz
o- /
endo
genn
ość
Prod
ukty
met
abol
izm
u
Punk
t izo
elek
tryc
zny
Występowanie
1. Gly glicyna n a n g 5,97 kolagen, elastyna, Ŝelatyna (25%)
2. Ala alanina n a n g 6,00 fibroina jedwabiu (30%), w kaŜdym białku małe ilości
3. Val walina n a g g 5,96 białka nasion (13%), większość białek w małych ilościach
4. Leu leucyna n a g k 6,02 zeina (21%), wszystkie białka
5. Ile izoleucyna n a g gk 5,98 wszystkie białka
6 Pro prolina n i n g 6,30 głównie białka tkanki łącznej
7. Phe fenyloalanina n r g gk 5,48 we wszystkich białkach (3-5%), białko wirusa mozaikitytoniowej (10%)
Created by Neevia Document Converter trial version http://www.neevia.com
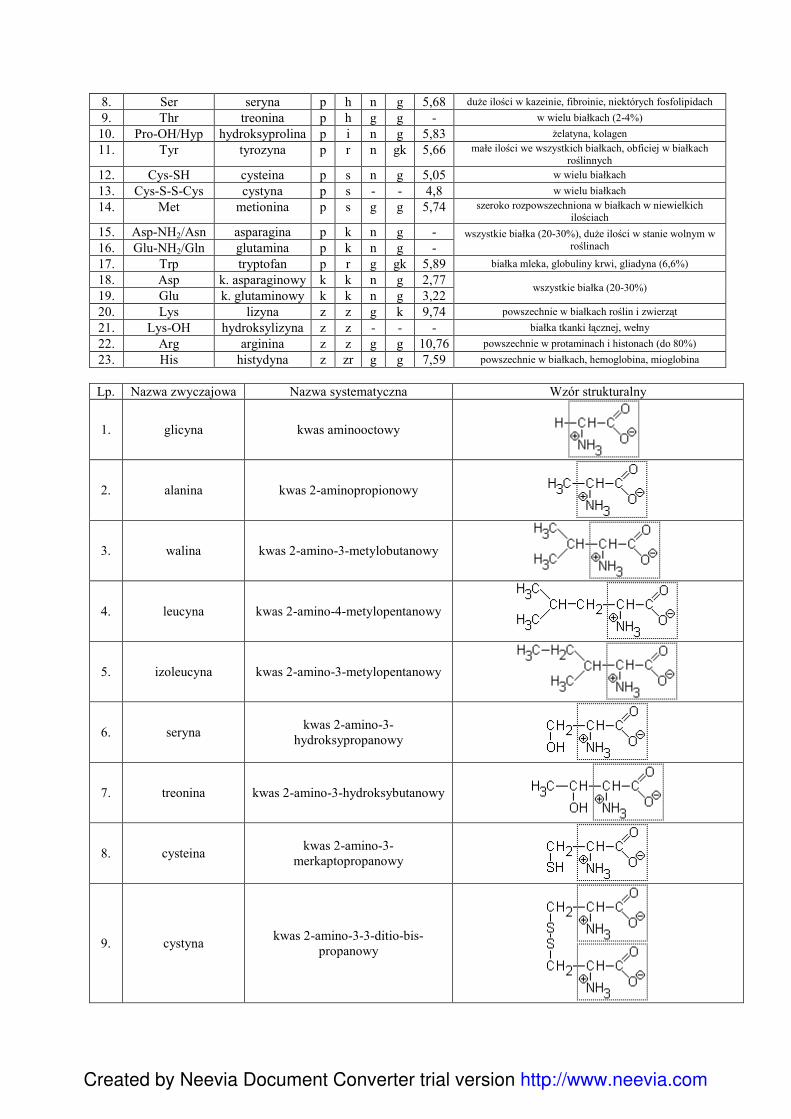
112
8. Ser seryna p h n g 5,68 duŜe ilości w kazeinie, fibroinie, niektórych fosfolipidach
9. Thr treonina p h g g - w wielu białkach (2-4%)
10. Pro-OH/Hyp hydroksyprolina p i n g 5,83 Ŝelatyna, kolagen
11. Tyr tyrozyna p r n gk 5,66 małe ilości we wszystkich białkach, obficiej w białkachroślinnych
12. Cys-SH cysteina p s n g 5,05 w wielu białkach
13. Cys-S-S-Cys cystyna p s - - 4,8 w wielu białkach
14. Met metionina p s g g 5,74 szeroko rozpowszechniona w białkach w niewielkichilościach
15. Asp-NH2/Asn asparagina p k n g -16. Glu-NH2/Gln glutamina p k n g -
wszystkie białka (20-30%), duŜe ilości w stanie wolnym wroślinach
17. Trp tryptofan p r g gk 5,89 białka mleka, globuliny krwi, gliadyna (6,6%)
18. Asp k. asparaginowy k k n g 2,7719. Glu k. glutaminowy k k n g 3,22
wszystkie białka (20-30%)
20. Lys lizyna z z g k 9,74 powszechnie w białkach roślin i zwierząt
21. Lys-OH hydroksylizyna z z - - - białka tkanki łącznej, wełny
22. Arg arginina z z g g 10,76 powszechnie w protaminach i histonach (do 80%)
23. His histydyna z zr g g 7,59 powszechnie w białkach, hemoglobina, mioglobina
Lp. Nazwa zwyczajowa Nazwa systematyczna Wzór strukturalny
1. glicyna kwas aminooctowy
2. alanina kwas 2-aminopropionowy
3. walina kwas 2-amino-3-metylobutanowy
4. leucyna kwas 2-amino-4-metylopentanowy
5. izoleucyna kwas 2-amino-3-metylopentanowy
6. serynakwas 2-amino-3-
hydroksypropanowy
7. treonina kwas 2-amino-3-hydroksybutanowy
8. cysteinakwas 2-amino-3-
merkaptopropanowy
9. cystynakwas 2-amino-3-3-ditio-bis-
propanowy
Created by Neevia Document Converter trial version http://www.neevia.com
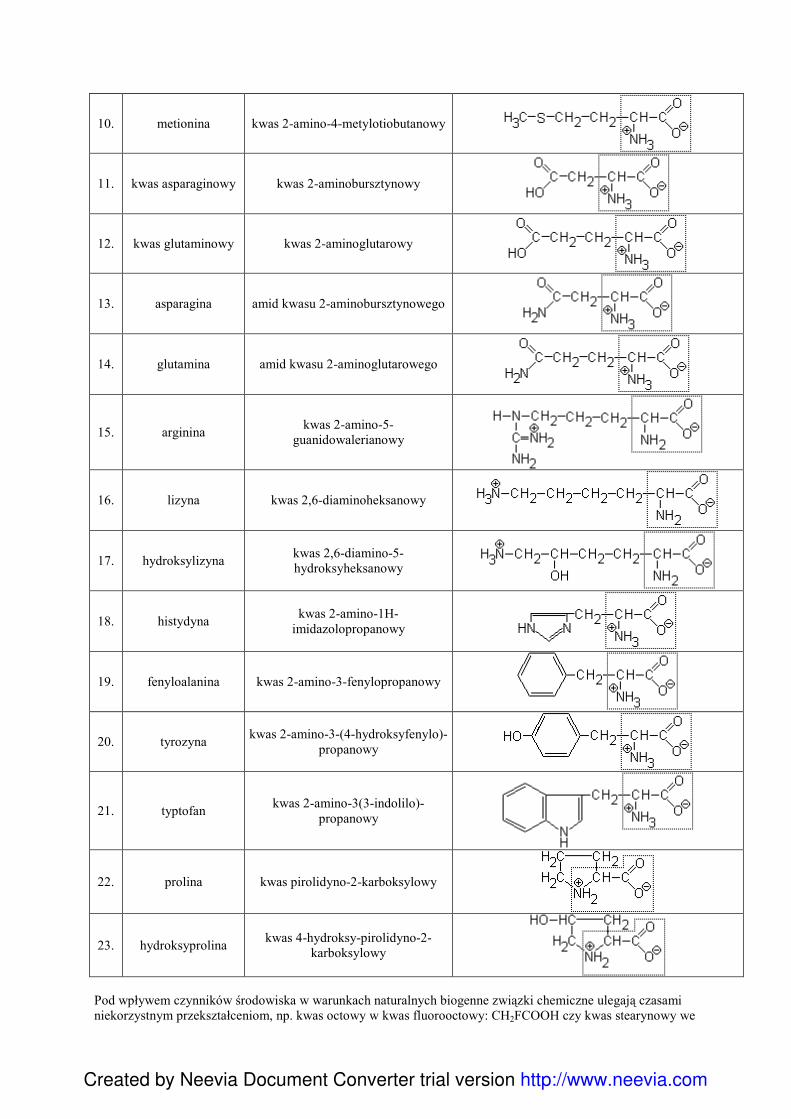
113
10. metionina kwas 2-amino-4-metylotiobutanowy
11. kwas asparaginowy kwas 2-aminobursztynowy
12. kwas glutaminowy kwas 2-aminoglutarowy
13. asparagina amid kwasu 2-aminobursztynowego
14. glutamina amid kwasu 2-aminoglutarowego
15. argininakwas 2-amino-5-
guanidowalerianowy
16. lizyna kwas 2,6-diaminoheksanowy
17. hydroksylizynakwas 2,6-diamino-5-hydroksyheksanowy
18. histydynakwas 2-amino-1H-
imidazolopropanowy
19. fenyloalanina kwas 2-amino-3-fenylopropanowy
20. tyrozynakwas 2-amino-3-(4-hydroksyfenylo)-
propanowy
21. typtofankwas 2-amino-3(3-indolilo)-
propanowy
22. prolina kwas pirolidyno-2-karboksylowy
23. hydroksyprolinakwas 4-hydroksy-pirolidyno-2-
karboksylowy
Pod wpływem czynników środowiska w warunkach naturalnych biogenne związki chemiczne ulegają czasaminiekorzystnym przekształceniom, np. kwas octowy w kwas fluorooctowy: CH2FCOOH czy kwas stearynowy we
Created by Neevia Document Converter trial version http://www.neevia.com

114
fluorostearynowy: CH2F(CH2)16COOH. Aminokwasy nie są pod tym względem wyjątkiem – przykładowoatomy siarki w cystynie bywają zastąpione podobnymi atomami selenu, dając selenocystynę: HOOC-CH(NH2)-CH2-Se-CH2-CH(NH2)-COOH. Często obserwuje się przemianę histydyny w histaminę(rysunek obok), co ma miejsce przy ukąszeniach owadów i obrzękach. Np. jad pszczółzawiera 30% roztwór dwóch enzymów oraz trzech peptydów zasadowych (MCD,mellityna). Te ostatnie powodują rozpad komórek, szczególnie erytrocytów. Poukąszeniu z komórek tucznych uwalnia się związania dotychczas histamina. Podobny efekt obserwujemy przyoparzeniach i odmroŜeniach. Z kolei pokrzywa pospolita (Urtica dioica) wydziela parzącą substancjęzawierającą histaminę oraz acetylocholinę, które powodują wydzielania esterazy cholinowej niszczącej komórki.Z aminokwasów: waliny i cysteiny następuje biogeneza penicylin. Penicylinytworzą szeroką grupę związków – znalazły zastosowanie jako antybiotyki, czylisubstancje hamujące wzrost i rozmnaŜania się bakterii oraz innychdrobnoustrojów. Ogólny wzór penicylin przedstawia rysunek. MoŜna na nimzauwaŜyć czteroczłonowy (a więc wykazujący wewnętrzne napięcia) pierścieńlaktamowy, stąd moŜna je zaliczyć do laktamów. RóŜnorodność w grupiepenicylin wynika z tego, iŜ posiadają one róŜne rodniki, a wodór kwasowy moŜe ulec wymianie na metale lubzasady organiczne. Najczęściej spotykane rodniki przedstawia poniŜsza tabela:
Symbol Nazwa penicyliny WzórF pentylopenicylina -CH2-CH=CH-CH2-CH3
G benzylopenicylina -CH2-C6H5
K heptylopenicylina -(CH2)6-CH3
V fenoksymetylopenicylna -CH2-O-C6H5
X hydroksybenzylopenicylina -CH2-C6H4-OHW praktyce najczęściej uŜywa się penicyliny G. Sole sodowe i potasowe penicyliny są dobrze rozpuszczalne wwodzie, dzięki czemu działają na organizm krotko i szybko. Z kolei sole trudno rozpuszczalne przedłuŜają jejdziałanie w organizmie.
PEPTYDYPeptydy powstają w wyniku reakcji kondensacji zachodzącej pomiędzy aminokwasami, np.:2 H2N-CH2-COOH ---T / enzymy---> H2N-CH2-CO-HN-CH2-COOH. (N-glicyloglicyna: Gly-Gly)W kaŜdym peptydzie wyróŜniamy dwa końce – aminowy N oraz karboksylowy C, przy czym obowiązuje zasadapodawania kolejności aminokwasów od N końca do C końca. Peptydy traktuje się jako acyloaminokwasy, więckońcówkę nazwy aminokwasu, którego grupa karboksylowa jest podstawiona zmienia się na „-ylo”. Jedynieaminokwas znajdujący się na końcu łańcucha, w wolną grupą karboksylową, zachowuje nie zmienioną nazwę.KaŜdy peptyd posiada charakterystyczne ugrupowanie atomów, zwane wiązaniem peptydowym. Jego obecnośćod tripeptydów w górę wykrywać moŜna za pomocą reakcji biuretowej. Z racji występowania wiązaniawielokrotnego obecne jest zjawisko izomerii geometrycznej cis-trans względem atomu tlenu. W obrębiewiązania następuje przyciąganie elektrostatyczne pomiędzy wolną parą elektronową atomu azotu orazwiązaniem podwójnym węgiel – tlen, co prowadzi do przegrupowania układu – atom wodoru zasila atom tlenu,a wiązanie podwójne przemieszcza się pomiędzy atomy azotu i węgla. Powstała struktura enolowa jestnietrwała, a reakcja jej tworzenia – łatwo odwracalna. NiezaleŜnie od formy tautomerycznej wiązanie podwójneobecne jest przy atomie węgla, co sugeruje hybrydyzację sp2. Dzięki temu wszystkie okoliczne atomy połoŜonesą w jednej płaszczyźnie, co na wpływ na dalsze właściwości.Liczność rodziny peptydów wymusza potrzebę ich klasyfikacji. Ze względu na liczbę aminokwasów moŜnapodzielić je na: oligopeptydy (2-10), polipeptydy (10-100) oraz białka (>100). Inny podział oparty jest na masiecząsteczkowej – za polipeptydy uwaŜa się połączenia do 104u, zaś struktury cięŜsze – za białka.
BIAŁKAWraz ze wzrostem masy i rozmiarów cząsteczek komplikuje się struktura dwu- i trójwymiarowa. Pewien wyrazrozmiarów cząsteczek białek dają ich wzory sumaryczne: albumina (białko jaja kurzego): C237H386O78N58S2,pojedynczy łańcuch hemoglobiny: C738H1160O208N203S2Fe, cała hemoglobina: C3032H4816O872N789S8Fe4, jedno zbiałek w mleku: C1864H3012O576N468S21. Do opisu budowy białek stosuje się pojęcie rzędowości struktur.a) Struktura I-rzędowa określa sekwencję aminokwasów w łańcuchu polipeptydowym.b) Struktura II-rzędowa podaje sposób skręcenia łańcucha w przestrzeni. Oddziaływania wodorowe pomiędzy
ugrupowaniami C-O i N-H mogą ułoŜyć łańcuch na jeden z dwóch sposobów.- Struktura α (α-helisa) ma kształt prawoskrętnej spirali. Kierunek skrętu zdeterminowany jest przez fakt
budowy cząsteczki z enancjomerów L: w wyniku przyciągania lŜejsza grupa N-H przychyla się do cięŜszejgrupy C-O właśnie w prawo. W przypadku dwóch łańcuchów oba rodzaje grup układają się naprzeciwsiebie.
Created by Neevia Document Converter trial version http://www.neevia.com

115
- Struktura β ma postać złoŜonej harmonijki, w której płaszczyzny wiązań peptydowych oraz łańcuchówbocznych są do siebie wzajemnie prostopadłe. Powodem są proste wiązania peptydowe.
c) Struktura III-rzędowa opisuje sposób ułoŜenia α-helisy lub struktury β w przestrzeni. W tworzeniu jej biorąudział róŜnorodne oddziaływania (wodorowe, jonowe, van der Waalsa, mostki disiarczkowe).
d) Strukturę IV-rzędową tworzą wzajemnie oddziałujące ze sobą skręcone łańcuchy polipeptydowe.Zbudowana jest z podjednostek białkowych na bazie ich oddziaływań. Za jej utrzymanie odpowiedzialne sągłównie: mostki disiarczkowe, siły van der Waalsa oraz wiązania wodorowe pomiędzy bocznymiodgałęzieniami. Strukturę tę wykryto tylko w niektórych typach białek, cechuje się największą złoŜonościąa zarazem została najsłabiej poznana.
Zniszczenie struktury III- i IV-rzędowej (rozerwanie wiązań wodorowych) powoduje utratę aktywności białka,czyli denaturację. Czynnikami denaturującymi są: wysoka temperatura, silnie polarne (kwaśne lub zasadowe)środowisko, alkohol, formalina, rozpuszczalne sole metali cięŜkich (głównie ołowiu, rtęci i srebra). Czasaminastępuje odwracalna utrata postaci białka – mówimy wówczas o koagulacji (wysoleniu - działa się tu solamimetali lekkich, np. NaCl, NH4Cl). W wyniku odciągnięcia pewnej ilości wody półpłynny zol białkowy zmieniasię w półstały Ŝel. Dodanie do koagulatu wody powoduje przywrócenie poprzedniej konsystencji – proces tennazywamy peptyzacją.Obecność białek z środowisku wykryć moŜna za pomocą reakcji ksantoproteinowej lub biuretowej. Pierwsza znich polega na działaniu stęŜonym kwasem azotowym (V). Daje ona pozytywny rezultat przy kontakcie zrodnikami aromatycznymi, które w wyniku nitrowania uzyskują barwę Ŝółtą. Reakcja biuretowa wykrywaobecność wiązań peptydowych, które z jonami miedzi (II) dają intensywnie fioletowe zabarwienie. Pozytywnemiano próby nie oznacza jednak koniecznie obecności związków aminokwasowych.Podobnie jak kaŜdemu aminokwasowi przyporządkować moŜna punkt izoelektryczny, tak kaŜdej cząsteczcebiałka odpowiada punkt izojonowy. Własność ta dotyczy czystych polipeptydów, dla danego związku jest stała icharakterystyczna. Określa się ją jako wartość pH, przy której liczba protonów związanych z grupamizasadowymi jest równa liczbie protonów odszczepionych przez grupy kwasowe (wyrównanie liczby ładunków).Białka ulegają dwojakiego rodzaju hydrolizie. Niestety takŜe niektóre aminokwasy rozkładają się przy hydroliziekwasowej (tryptofan) lub zasadowej (cysteina, arginina). Nie istnieje rozkład idealny, oczywiście z wyjątkiemprocesu enzymatycznego. Hydroliza jest uŜytecznym narzędziem biochemicznym – pozwala na ustalenieproporcji występowania aminokwasów w badanym białku, choć nie daje informacji nawet o strukturze I-rzędowej.Białka moŜna róŜnorodnie klasyfikować:a) ze względu na budowę cząsteczki na:- białka proste (proteiny), zbudowane tylko i wyłącznie z aminokwasów:• albuminy (osocze krwi, mleko, zboŜa, jaja),• globuliny (mleko, mięso, jaja, owoce, nasiona),• histony (leukocyty),• protaminy (ikra, ryby),• skleroproteiny (paznokcie, włosy, Ŝyły, tętnice),- białka złoŜone (proteidy), zawierające składniki niebiałkowe, np.:• fosfoproteidy (reszta kwasu fosforowego) (mleko – kazeina),• glikoproteidy (sacharyd) (ślina),• lipoproteidy (lipid),• metaloproteidy (metal),• chromoproteidy (cząsteczka barwnika) (barwniki oddechowe, np. hemoglobina),• nukleoproteidy (kwas nukleinowy) (jądro komórkowe),b) ze względu na kształt cząsteczki na:- białka globularne, których cząsteczka ma w przybliŜeniu kształt kulisty, są rozpuszczalne w wodzie,- białka fibrylarne, włókienkowe, których cząsteczka ma kształt włókna, są nierozpuszczalne w wodzie,c) ze względu na stan skupienia na:- białka stałe (kolagen, elastyna),- białka półpłynne (występujące w cytoplazmie),- białka płynne (występujące w osoczu krwi),d) ze względu na przydatność dla organizmu:- białka pełnowartościowe (zawierają aminokwasy egzogenne),- białka niepełnowartościowe (składają się z aminokwasów endogennych).
Created by Neevia Document Converter trial version http://www.neevia.com

116
KWASY NUKLEINOWE
Kwasy nukleinowe są polimerami mniejszych jednostekzwanych nukleotydami, które z kolei moŜna rozszczepić nakwas ortofosforowy i nukleozydy, a te ostatnie na cukry(pentozy) oraz zasady azotowe (purynowe lub pirymidynowe).
Całkowity rozkład moŜna przeprowadzić chemiczniedziałaniem ługów lub kwasów (0,3 n NaOH w 37o, 6 n HCl w100o lub 12 n HClO4 w 100o). Oba rodzaje kwasów róŜniezachowują się pod hydrolitycznym działaniem tych związków;na ogół trwalszy okazuje się DNA. MoŜna tez przeprowadzićcałkowitą hydrolizę działaniem specyficznych enzymów:nukleodepolimeraz (rybonukleazy i dezoksyrybonukleazy),nukleotydaz i nukleozydaz. Ostatecznie całkowitą hydrolizękwasów nukleinowych moŜna ująć tak jak przedstawiono nazałączonych obok schemacie.
Zasady purynowe. JuŜ w zeszłym stuleciu zauwaŜono, Ŝe w skład kwasów nukleinowych wchodząheterocykliczne związki dwupierścieniowe. Fischer nazwał te związki „ciałami purynowymi” – po niemieckuPurinkörper od łacińskiego purum uricum (acidum uricum oznacza kwas moczowy). W purynie, podstawowymtego typu związku, moŜna wyróŜnić pierścień pirymidynowy i imidazolowy. Często dla przejrzystości wzoru niewpisuje się do pierścienia purynowego atomów węgla i związanych z nimi atomów wodoru.
Wolnej puryny w przyrodzie nie znaleziono, znane są natomiast jej liczne pochodne, przewaŜnie amino- ihydroksypuryny. W jednej i drugiej odmianie występują dwa związki purynowe: 6-aminopuryna zwana adeniną,otrzymana po raz pierwszy z trzustki przez Kossela oraz 2-amino-6-hydroksypuryna nosząca nazwę guaniny,znalezione juŜ w roku 1844 w guanie, naturalnym nawozie powstałym głownie z ptasich ekskrementów (stądpochodzi nazwa guaniny). Aminowe grupy adeniny i guaniny zachowują się podobnie jak grupy aminoweaminokwasów i mogą przechodzić w postać kationową, przyłączając jon wodorowy H+. JednakŜe aromatycznycharakter pierścienia purynowego zmniejsza w pewnej mierze zasadowe własności tych związków.
Innymi występującymi w przyrodzie pochodnymi puryn są: hipoksantyna (6-hydroksypuryna), ksantyna(2,6-dihydroksypuryna) i kwas moczowy (2,6,8-trihydroksypuryna). Są to między innymi produkty przemianadeniny i guaniny powstające w procesie dezaminacji (odszczepiania grup aminowych) i utleniania tychzwiązków. Zarówno zasady purynowe, jak i kwas moczowy mogą występować w dwóch tautomerycznychodmianach: ketonowej i enolowej.
Kwas moczowy był pierwszą z odkrytych pochodnych puryny. Otrzymał go K. W. Sokeele w roku 1776 zkamieni i osadów moczowych. Jest to równieŜ główny azotowy składnik wydalin zwierząt urykotelicznych:ptaków, owadów i węŜy. Kwas moczowy jest bardzo słabo rozpuszczalny w wodzie (1:15500 w 37o), natomiastw środowisku alkalicznym przechodzi w jedno- lub dwuzasadowy, rozpuszczalny moczan (postać enolowa).
W świecie roślinnym występują metylowe pochodne puryn. Do najbardziej znanych naleŜą: teofilina (1,3-dimetyloksantyna), znaleziona w liściach herbaty, teobromina (3,7-dimetyloksantyna), otrzymana z owocówkakaowych oraz kofeina (1,3,7-trimetyloksantyna), zwana równieŜ teiną, występująca w liściach i ziarnachkawy, w liściach herbaty, w orzeszkach kola i innych. Metylowe pochodne puryn wykryto w niewielkich
Created by Neevia Document Converter trial version http://www.neevia.com
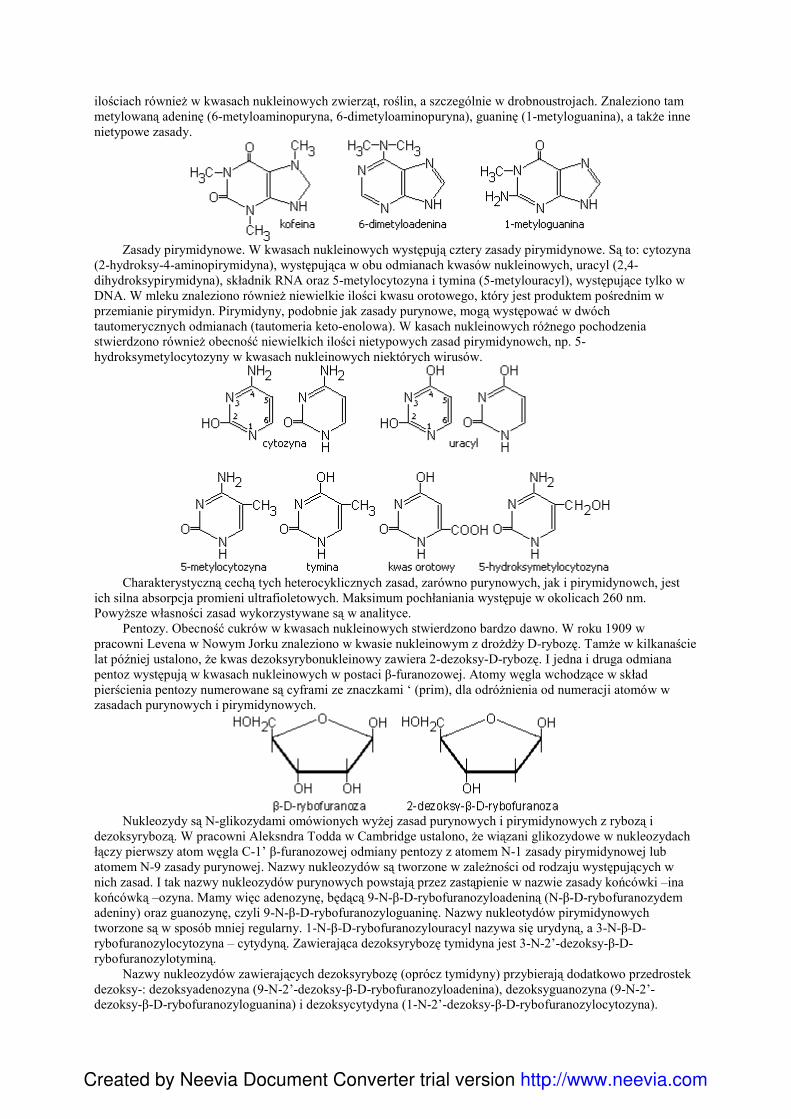
117
ilościach równieŜ w kwasach nukleinowych zwierząt, roślin, a szczególnie w drobnoustrojach. Znaleziono tammetylowaną adeninę (6-metyloaminopuryna, 6-dimetyloaminopuryna), guaninę (1-metyloguanina), a takŜe innenietypowe zasady.
Zasady pirymidynowe. W kwasach nukleinowych występują cztery zasady pirymidynowe. Są to: cytozyna(2-hydroksy-4-aminopirymidyna), występująca w obu odmianach kwasów nukleinowych, uracyl (2,4-dihydroksypirymidyna), składnik RNA oraz 5-metylocytozyna i tymina (5-metylouracyl), występujące tylko wDNA. W mleku znaleziono równieŜ niewielkie ilości kwasu orotowego, który jest produktem pośrednim wprzemianie pirymidyn. Pirymidyny, podobnie jak zasady purynowe, mogą występować w dwóchtautomerycznych odmianach (tautomeria keto-enolowa). W kasach nukleinowych róŜnego pochodzeniastwierdzono równieŜ obecność niewielkich ilości nietypowych zasad pirymidynowch, np. 5-hydroksymetylocytozyny w kwasach nukleinowych niektórych wirusów.
Charakterystyczną cechą tych heterocyklicznych zasad, zarówno purynowych, jak i pirymidynowch, jestich silna absorpcja promieni ultrafioletowych. Maksimum pochłaniania występuje w okolicach 260 nm.PowyŜsze własności zasad wykorzystywane są w analityce.
Pentozy. Obecność cukrów w kwasach nukleinowych stwierdzono bardzo dawno. W roku 1909 wpracowni Levena w Nowym Jorku znaleziono w kwasie nukleinowym z droŜdŜy D-rybozę. TamŜe w kilkanaścielat później ustalono, Ŝe kwas dezoksyrybonukleinowy zawiera 2-dezoksy-D-rybozę. I jedna i druga odmianapentoz występują w kwasach nukleinowych w postaci β-furanozowej. Atomy węgla wchodzące w składpierścienia pentozy numerowane są cyframi ze znaczkami ‘ (prim), dla odróŜnienia od numeracji atomów wzasadach purynowych i pirymidynowych.
Nukleozydy są N-glikozydami omówionych wyŜej zasad purynowych i pirymidynowych z rybozą idezoksyrybozą. W pracowni Aleksndra Todda w Cambridge ustalono, Ŝe wiązani glikozydowe w nukleozydachłączy pierwszy atom węgla C-1’ β-furanozowej odmiany pentozy z atomem N-1 zasady pirymidynowej lubatomem N-9 zasady purynowej. Nazwy nukleozydów są tworzone w zaleŜności od rodzaju występujących wnich zasad. I tak nazwy nukleozydów purynowych powstają przez zastąpienie w nazwie zasady końcówki –inakońcówką –ozyna. Mamy więc adenozynę, będącą 9-N-β-D-rybofuranozyloadeniną (N-β-D-rybofuranozydemadeniny) oraz guanozynę, czyli 9-N-β-D-rybofuranozyloguaninę. Nazwy nukleotydów pirymidynowychtworzone są w sposób mniej regularny. 1-N-β-D-rybofuranozylouracyl nazywa się urydyną, a 3-N-β-D-rybofuranozylocytozyna – cytydyną. Zawierająca dezoksyrybozę tymidyna jest 3-N-2’-dezoksy-β-D-rybofuranozylotyminą.
Nazwy nukleozydów zawierających dezoksyrybozę (oprócz tymidyny) przybierają dodatkowo przedrostekdezoksy-: dezoksyadenozyna (9-N-2’-dezoksy-β-D-rybofuranozyloadenina), dezoksyguanozyna (9-N-2’-dezoksy-β-D-rybofuranozyloguanina) i dezoksycytydyna (1-N-2’-dezoksy-β-D-rybofuranozylocytozyna).
Created by Neevia Document Converter trial version http://www.neevia.com
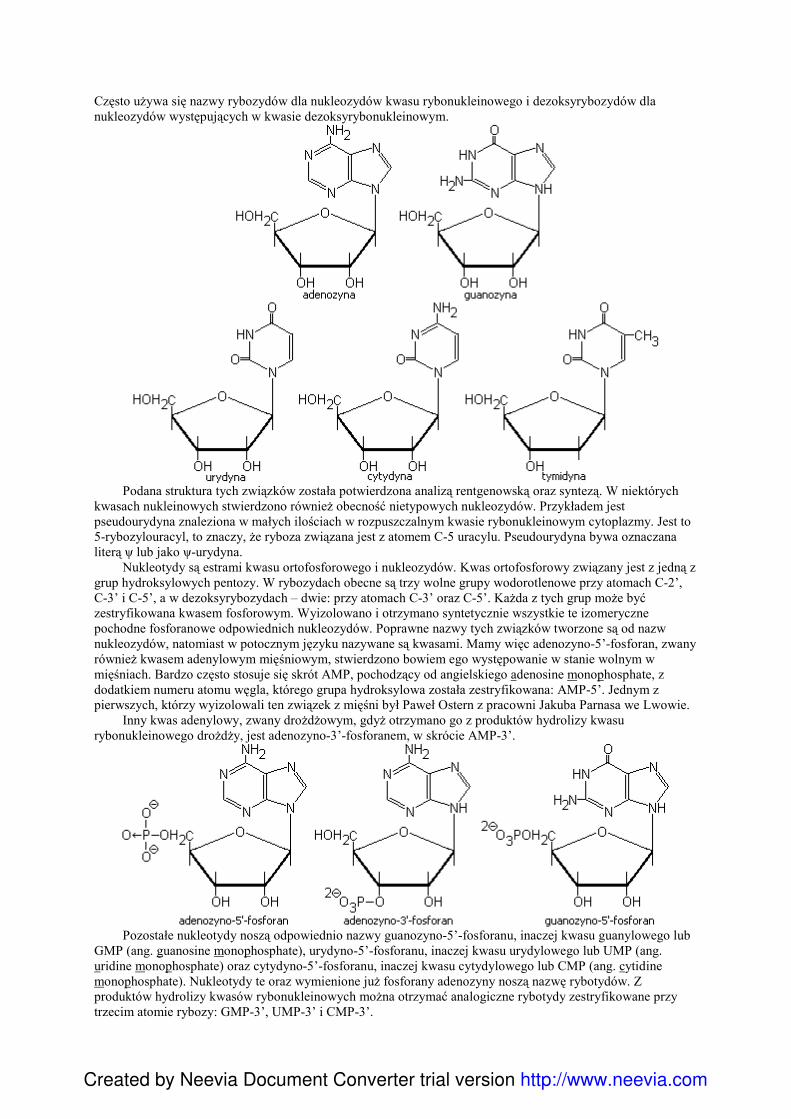
118
Często uŜywa się nazwy rybozydów dla nukleozydów kwasu rybonukleinowego i dezoksyrybozydów dlanukleozydów występujących w kwasie dezoksyrybonukleinowym.
Podana struktura tych związków została potwierdzona analizą rentgenowską oraz syntezą. W niektórychkwasach nukleinowych stwierdzono równieŜ obecność nietypowych nukleozydów. Przykładem jestpseudourydyna znaleziona w małych ilościach w rozpuszczalnym kwasie rybonukleinowym cytoplazmy. Jest to5-rybozylouracyl, to znaczy, Ŝe ryboza związana jest z atomem C-5 uracylu. Pseudourydyna bywa oznaczanaliterą ψ lub jako ψ-urydyna.
Nukleotydy są estrami kwasu ortofosforowego i nukleozydów. Kwas ortofosforowy związany jest z jedną zgrup hydroksylowych pentozy. W rybozydach obecne są trzy wolne grupy wodorotlenowe przy atomach C-2’,C-3’ i C-5’, a w dezoksyrybozydach – dwie: przy atomach C-3’ oraz C-5’. KaŜda z tych grup moŜe byćzestryfikowana kwasem fosforowym. Wyizolowano i otrzymano syntetycznie wszystkie te izomerycznepochodne fosforanowe odpowiednich nukleozydów. Poprawne nazwy tych związków tworzone są od nazwnukleozydów, natomiast w potocznym języku nazywane są kwasami. Mamy więc adenozyno-5’-fosforan, zwanyrównieŜ kwasem adenylowym mięśniowym, stwierdzono bowiem ego występowanie w stanie wolnym wmięśniach. Bardzo często stosuje się skrót AMP, pochodzący od angielskiego adenosine monophosphate, zdodatkiem numeru atomu węgla, którego grupa hydroksylowa została zestryfikowana: AMP-5’. Jednym zpierwszych, którzy wyizolowali ten związek z mięśni był Paweł Ostern z pracowni Jakuba Parnasa we Lwowie.
Inny kwas adenylowy, zwany droŜdŜowym, gdyŜ otrzymano go z produktów hydrolizy kwasurybonukleinowego droŜdŜy, jest adenozyno-3’-fosforanem, w skrócie AMP-3’.
Pozostałe nukleotydy noszą odpowiednio nazwy guanozyno-5’-fosforanu, inaczej kwasu guanylowego lubGMP (ang. guanosine monophosphate), urydyno-5’-fosforanu, inaczej kwasu urydylowego lub UMP (ang.uridine monophosphate) oraz cytydyno-5’-fosforanu, inaczej kwasu cytydylowego lub CMP (ang. cytidinemonophosphate). Nukleotydy te oraz wymienione juŜ fosforany adenozyny noszą nazwę rybotydów. Zproduktów hydrolizy kwasów rybonukleinowych moŜna otrzymać analogiczne rybotydy zestryfikowane przytrzecim atomie rybozy: GMP-3’, UMP-3’ i CMP-3’.
Created by Neevia Document Converter trial version http://www.neevia.com
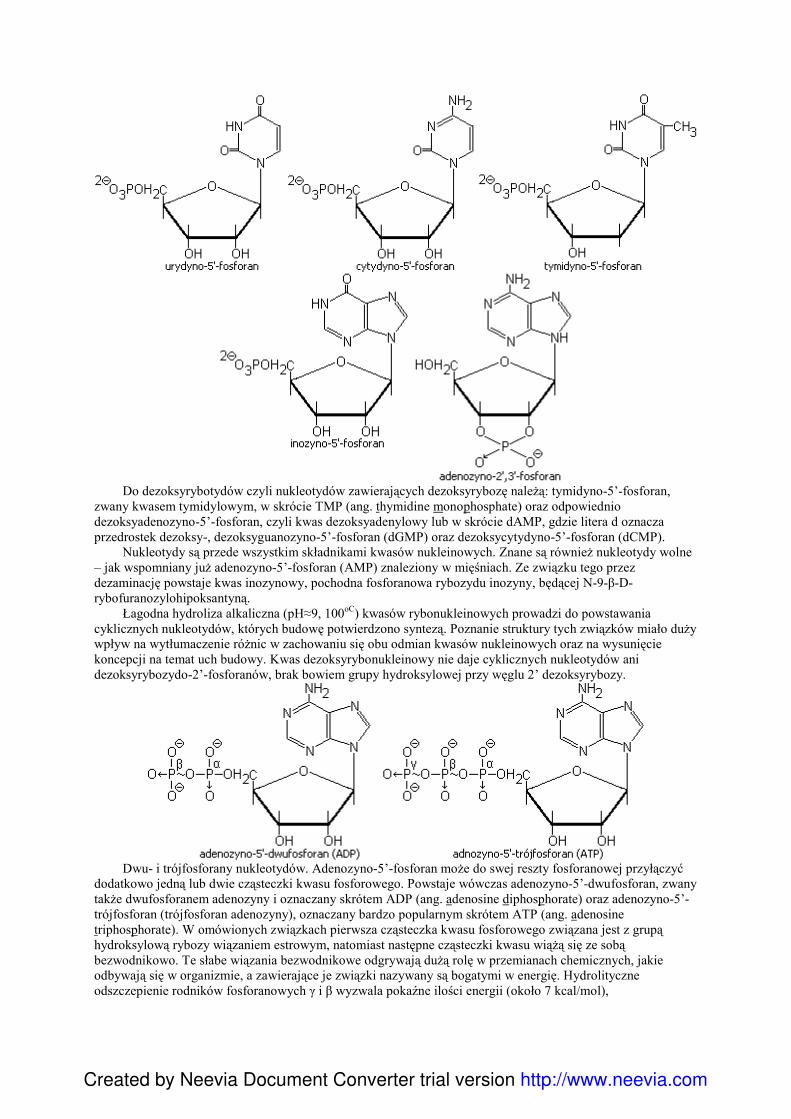
119
Do dezoksyrybotydów czyli nukleotydów zawierających dezoksyrybozę naleŜą: tymidyno-5’-fosforan,zwany kwasem tymidylowym, w skrócie TMP (ang. thymidine monophosphate) oraz odpowiedniodezoksyadenozyno-5’-fosforan, czyli kwas dezoksyadenylowy lub w skrócie dAMP, gdzie litera d oznaczaprzedrostek dezoksy-, dezoksyguanozyno-5’-fosforan (dGMP) oraz dezoksycytydyno-5’-fosforan (dCMP).
Nukleotydy są przede wszystkim składnikami kwasów nukleinowych. Znane są równieŜ nukleotydy wolne– jak wspomniany juŜ adenozyno-5’-fosforan (AMP) znaleziony w mięśniach. Ze związku tego przezdezaminację powstaje kwas inozynowy, pochodna fosforanowa rybozydu inozyny, będącej N-9-β-D-rybofuranozylohipoksantyną.
Łagodna hydroliza alkaliczna (pH≈9, 100oC) kwasów rybonukleinowych prowadzi do powstawaniacyklicznych nukleotydów, których budowę potwierdzono syntezą. Poznanie struktury tych związków miało duŜywpływ na wytłumaczenie róŜnic w zachowaniu się obu odmian kwasów nukleinowych oraz na wysunięciekoncepcji na temat uch budowy. Kwas dezoksyrybonukleinowy nie daje cyklicznych nukleotydów anidezoksyrybozydo-2’-fosforanów, brak bowiem grupy hydroksylowej przy węglu 2’ dezoksyrybozy.
Dwu- i trójfosforany nukleotydów. Adenozyno-5’-fosforan moŜe do swej reszty fosforanowej przyłączyćdodatkowo jedną lub dwie cząsteczki kwasu fosforowego. Powstaje wówczas adenozyno-5’-dwufosforan, zwanytakŜe dwufosforanem adenozyny i oznaczany skrótem ADP (ang. adenosine diphosphorate) oraz adenozyno-5’-trójfosforan (trójfosforan adenozyny), oznaczany bardzo popularnym skrótem ATP (ang. adenosinetriphosphorate). W omówionych związkach pierwsza cząsteczka kwasu fosforowego związana jest z grupąhydroksylową rybozy wiązaniem estrowym, natomiast następne cząsteczki kwasu wiąŜą się ze sobąbezwodnikowo. Te słabe wiązania bezwodnikowe odgrywają duŜą rolę w przemianach chemicznych, jakieodbywają się w organizmie, a zawierające je związki nazywany są bogatymi w energię. Hydrolityczneodszczepienie rodników fosforanowych γ i β wyzwala pokaźne ilości energii (około 7 kcal/mol),
Created by Neevia Document Converter trial version http://www.neevia.com
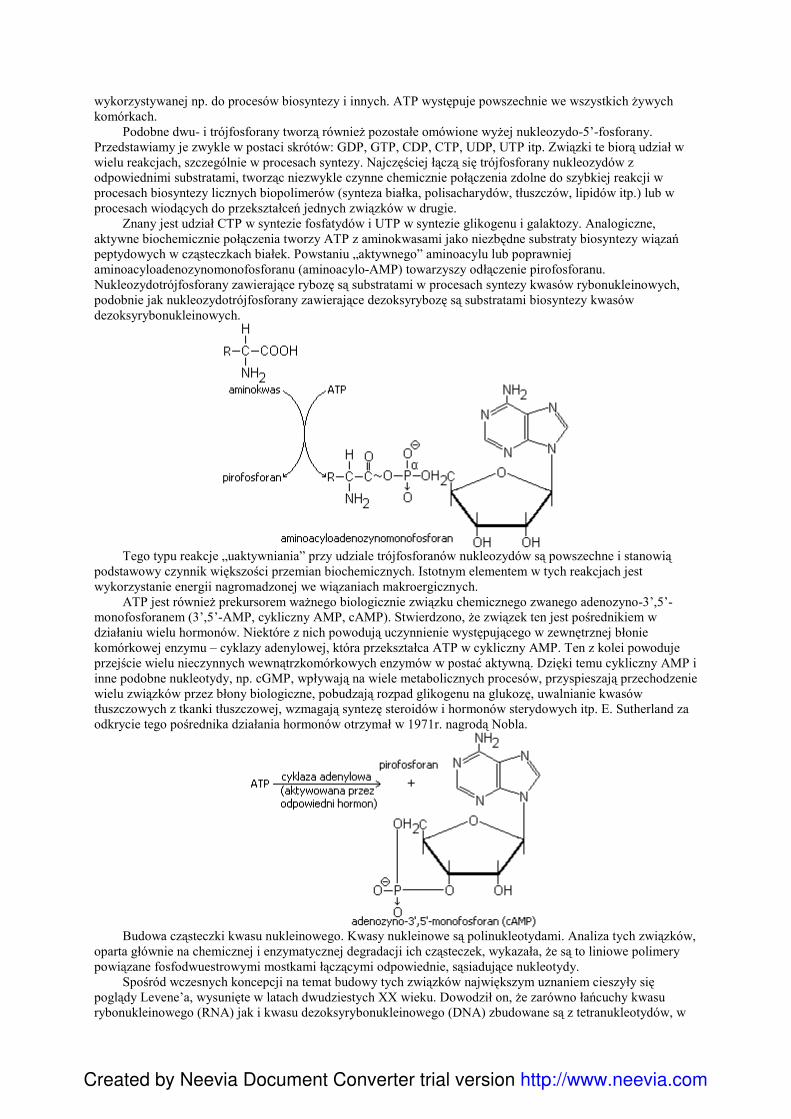
120
wykorzystywanej np. do procesów biosyntezy i innych. ATP występuje powszechnie we wszystkich Ŝywychkomórkach.
Podobne dwu- i trójfosforany tworzą równieŜ pozostałe omówione wyŜej nukleozydo-5’-fosforany.Przedstawiamy je zwykle w postaci skrótów: GDP, GTP, CDP, CTP, UDP, UTP itp. Związki te biorą udział wwielu reakcjach, szczególnie w procesach syntezy. Najczęściej łączą się trójfosforany nukleozydów zodpowiednimi substratami, tworząc niezwykle czynne chemicznie połączenia zdolne do szybkiej reakcji wprocesach biosyntezy licznych biopolimerów (synteza białka, polisacharydów, tłuszczów, lipidów itp.) lub wprocesach wiodących do przekształceń jednych związków w drugie.
Znany jest udział CTP w syntezie fosfatydów i UTP w syntezie glikogenu i galaktozy. Analogiczne,aktywne biochemicznie połączenia tworzy ATP z aminokwasami jako niezbędne substraty biosyntezy wiązańpeptydowych w cząsteczkach białek. Powstaniu „aktywnego” aminoacylu lub poprawniejaminoacyloadenozynomonofosforanu (aminoacylo-AMP) towarzyszy odłączenie pirofosforanu.Nukleozydotrójfosforany zawierające rybozę są substratami w procesach syntezy kwasów rybonukleinowych,podobnie jak nukleozydotrójfosforany zawierające dezoksyrybozę są substratami biosyntezy kwasówdezoksyrybonukleinowych.
Tego typu reakcje „uaktywniania” przy udziale trójfosforanów nukleozydów są powszechne i stanowiąpodstawowy czynnik większości przemian biochemicznych. Istotnym elementem w tych reakcjach jestwykorzystanie energii nagromadzonej we wiązaniach makroergicznych.
ATP jest równieŜ prekursorem waŜnego biologicznie związku chemicznego zwanego adenozyno-3’,5’-monofosforanem (3’,5’-AMP, cykliczny AMP, cAMP). Stwierdzono, Ŝe związek ten jest pośrednikiem wdziałaniu wielu hormonów. Niektóre z nich powodują uczynnienie występującego w zewnętrznej błoniekomórkowej enzymu – cyklazy adenylowej, która przekształca ATP w cykliczny AMP. Ten z kolei powodujeprzejście wielu nieczynnych wewnątrzkomórkowych enzymów w postać aktywną. Dzięki temu cykliczny AMP iinne podobne nukleotydy, np. cGMP, wpływają na wiele metabolicznych procesów, przyspieszają przechodzeniewielu związków przez błony biologiczne, pobudzają rozpad glikogenu na glukozę, uwalnianie kwasówtłuszczowych z tkanki tłuszczowej, wzmagają syntezę steroidów i hormonów sterydowych itp. E. Sutherland zaodkrycie tego pośrednika działania hormonów otrzymał w 1971r. nagrodą Nobla.
Budowa cząsteczki kwasu nukleinowego. Kwasy nukleinowe są polinukleotydami. Analiza tych związków,oparta głównie na chemicznej i enzymatycznej degradacji ich cząsteczek, wykazała, Ŝe są to liniowe polimerypowiązane fosfodwuestrowymi mostkami łączącymi odpowiednie, sąsiadujące nukleotydy.
Spośród wczesnych koncepcji na temat budowy tych związków największym uznaniem cieszyły siępoglądy Levene’a, wysunięte w latach dwudziestych XX wieku. Dowodził on, Ŝe zarówno łańcuchy kwasurybonukleinowego (RNA) jak i kwasu dezoksyrybonukleinowego (DNA) zbudowane są z tetranukleotydów, w
Created by Neevia Document Converter trial version http://www.neevia.com
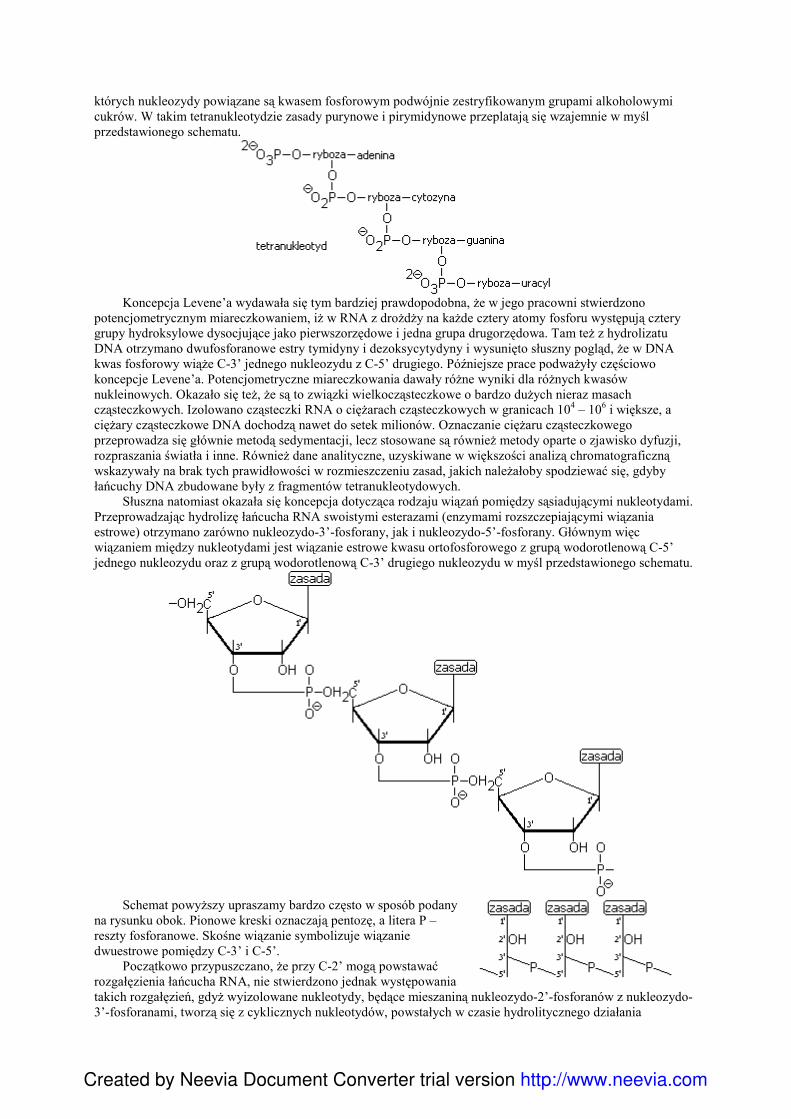
121
których nukleozydy powiązane są kwasem fosforowym podwójnie zestryfikowanym grupami alkoholowymicukrów. W takim tetranukleotydzie zasady purynowe i pirymidynowe przeplatają się wzajemnie w myślprzedstawionego schematu.
Koncepcja Levene’a wydawała się tym bardziej prawdopodobna, Ŝe w jego pracowni stwierdzonopotencjometrycznym miareczkowaniem, iŜ w RNA z droŜdŜy na kaŜde cztery atomy fosforu występują czterygrupy hydroksylowe dysocjujące jako pierwszorzędowe i jedna grupa drugorzędowa. Tam teŜ z hydrolizatuDNA otrzymano dwufosforanowe estry tymidyny i dezoksycytydyny i wysunięto słuszny pogląd, Ŝe w DNAkwas fosforowy wiąŜe C-3’ jednego nukleozydu z C-5’ drugiego. Późniejsze prace podwaŜyły częściowokoncepcje Levene’a. Potencjometryczne miareczkowania dawały róŜne wyniki dla róŜnych kwasównukleinowych. Okazało się teŜ, Ŝe są to związki wielkocząsteczkowe o bardzo duŜych nieraz masachcząsteczkowych. Izolowano cząsteczki RNA o cięŜarach cząsteczkowych w granicach 104 – 106 i większe, acięŜary cząsteczkowe DNA dochodzą nawet do setek milionów. Oznaczanie cięŜaru cząsteczkowegoprzeprowadza się głównie metodą sedymentacji, lecz stosowane są równieŜ metody oparte o zjawisko dyfuzji,rozpraszania światła i inne. RównieŜ dane analityczne, uzyskiwane w większości analizą chromatograficznąwskazywały na brak tych prawidłowości w rozmieszczeniu zasad, jakich naleŜałoby spodziewać się, gdybyłańcuchy DNA zbudowane były z fragmentów tetranukleotydowych.
Słuszna natomiast okazała się koncepcja dotycząca rodzaju wiązań pomiędzy sąsiadującymi nukleotydami.Przeprowadzając hydrolizę łańcucha RNA swoistymi esterazami (enzymami rozszczepiającymi wiązaniaestrowe) otrzymano zarówno nukleozydo-3’-fosforany, jak i nukleozydo-5’-fosforany. Głównym więcwiązaniem między nukleotydami jest wiązanie estrowe kwasu ortofosforowego z grupą wodorotlenową C-5’jednego nukleozydu oraz z grupą wodorotlenową C-3’ drugiego nukleozydu w myśl przedstawionego schematu.
Schemat powyŜszy upraszamy bardzo często w sposób podanyna rysunku obok. Pionowe kreski oznaczają pentozę, a litera P –reszty fosforanowe. Skośne wiązanie symbolizuje wiązaniedwuestrowe pomiędzy C-3’ i C-5’.
Początkowo przypuszczano, Ŝe przy C-2’ mogą powstawaćrozgałęzienia łańcucha RNA, nie stwierdzono jednak występowaniatakich rozgałęzień, gdyŜ wyizolowane nukleotydy, będące mieszaniną nukleozydo-2’-fosforanów z nukleozydo-3’-fosforanami, tworzą się z cyklicznych nukleotydów, powstałych w czasie hydrolitycznego działania
Created by Neevia Document Converter trial version http://www.neevia.com

122
trzustkowej rybonukleazy (RN-aza). Jest to enzym o dobrze poznanej budowie. RN-aza trzustkowa swoiściekatalizuje hydrolizę łańcucha RNA w sąsiedztwie zasad pirymidynowych. Jako produkt pośredni powstajecykliczny nukleotyd pirymidynowy, z którego, działaniem tejŜe rybonukleazy, powstaje 3’- lub 2’-nukleotydpirymidynowy.
Opierając się na podobnych spostrzeŜeniach Todd zaproponował schemat modelu budowy łańcucha DNA.PoniewaŜ w dezoksyrybotydach brak tlenu przy C-2’, więc – analogicznie jak w RNA – głównym, jeŜeli niejedynym wiązaniem między nukleotydami w łańcuchu DNA, jest wiązanie typu dwuestrowego pomiędzy C-3’jednego nukleotydu a C-5’ drugiego.
Ze schematu widać, Ŝe nukleotydy DNA nie mogą tworzyć odmian cyklicznych i tym między innymitłumaczyć moŜna odmienne właściwości łańcucha DNA, np. odporność na alkaliczną hydrolizę.
Budowa przestrzenna DNA. Badania przestrzennej budowyłańcuchów kwasów nukleinowych dotyczyły głównie DNA. Pierwszekoncepcje na ten temat wysunął w 1938r. W. T. Astbury. W oparciu ozałoŜenia teoretyczne, uwzględniające duŜą gęstość DNA i wymiaryatomowe oraz na podstawie zdjęć rentgenowskich Astbury wyraziłpogląd, Ŝe cząsteczki DNA tworzą sztywne kolumny ściśle nałoŜonychna siebie nukleotydów, podobnie do monet ułoŜonych wrulon jedna na drugiej. Jednak późniejsze prace S. Furbergaz Londynu oraz L. Paulinga i R. B. Coreya z Kaliforniiwskazywały na śrubowy kształt łańcuchów DNA. Dwajostatni wysunęli w 1953r. koncepcję, Ŝe cząsteczka DNAzbudowana jest z trzech łańcuchów polinukleotydowychsplecionych wzajemnie na kształt liny. Koncepcja ta zostaław tymŜe roku podwaŜona przez J. D. Watsona i F. K. C.Cricka. Opierając się na badaniach rentgenowskich,szczególnie na zdjęciach wykonanych przez M. H. F.Wilkinsa, doszli oni do wniosku, Ŝe cząsteczka DNAzbudowana jest z dwóch łańcuchów wijących się liniąśrubową i splecionych wzajemnie. KaŜdy z tych łańcuchówzbudowany jest z reszt fosforanowych połączonychwiązaniem estrowym z cząsteczkami dezoksyryboz, poprzezC-3’ jednego nukleotydu i C-5’ następnego. PoniewaŜ obałańcuchy wiją się prawoskrętnie, toteŜ sekwencje atomów igrup układają się w kaŜdym ze splecionych łańcuchów wkierunku przeciwnym. Oznacza to, Ŝe wiązanie międzynukleotydami w jednym łańcuchu jest 3’→5’, a w drugim5’→3’, co Watson i Crick zaznaczyli na swoim modelu.Dwie splatające się wstąŜki symbolizują łańcuchypowstające z powiązania się fosforanów z cukrami. Poziomepręty obrazują pary zasad, wiąŜących oba łańcuchy. Liniapionowa symbolizuje oś włókna (Watson i Crick, 1953).
Warto zauwaŜyć, Ŝe powszechnie uŜywane terminy „spiralny” lub „spirala” niezbyt ściśle oddająkonfigurację splatających się łańcuchów. Łańcuchy zwijają się nie w kształcie spirali, która jest linią płaską, leczjak gdyby okręcają się wokół walca, tak jak kręcone schody, których poręcze zbudowane są z cukrów i kwasówfosforowych, a stopnie z powiązanych par zasad. Dlatego termin „śrubowy” właściwiej oddaje kształtsplecionych łańcuchów DNA.Jak wykazano metodami rentgenowskimi, grubość takiego łańcucha, splecionego w kształt helisy, (od C-1 jednejzasady do C-1 zasady komplementarnej) wynosi około 1,1 nm. Jeden skok utworzonej linii śrubowej wynosi 3,4nm, a poniewaŜ jest tam rozmieszczone 10 par powiązanych zasad, więc odległość pomiędzy sąsiadującymiparami wynosi 0,34 nm.W modelu zaproponowany przez Watsona i Cricka zewnętrzną część cząsteczki splecionej z dwóch łańcuchówstanowią fosforany powiązane z cukrami, zasady zaś znajdują się wewnątrz splecionego łańcucha. Oryginalnymrysem tej struktury jest sposób wiązania obu łańcuchów poprzez zasady purynowe i pirymidynowe. Zasadajednego łańcucha połączona jest wiązaniami wodorowymi z zasadą drugiego łańcucha. Watson i Crick w swoim
Created by Neevia Document Converter trial version http://www.neevia.com

123
załoŜeni przyjęli, Ŝe odległości pomiędzy łańcuchami są stałe. PoniewaŜ wymiary zasad purynowych ipirymidynowych są róŜne, wobec tego łączyć się moŜe tylko zasada purynowa jednego łańcucha z zasadąpirymidynową drugiego. Wiązania wodorowe mogą występować między atomem N-1 puryny i atomem N-1pirymidyny oraz między podstawnikami atomów C-6 puryny i C-6 pirymidyny. Ze stereochemicznego punktuwidzenia najlepsze warunki wzajemnego wiązania ma para adenina – tymina. Kierunek wiązania wodorowegomiędzy aminowym atomem azotu adeniny a grupą ketonową tyminy jest równoległy do wiązania biegnącego odatomu N-1 puryny do atomu N-1 pirymidyny.
Drugą parą zasad, która równieŜ ma korzystne warunki wiązania jest guanina i cytozyna. Stwierdzono, Ŝewiązanie między guaniną i cytozyną jest bardziej trwałe niŜ wiązanie w poprzedniej parze zasad. Jak widać zewzoru, między tymi zasadami istnieje moŜliwość powstania trzeciego wiązania wodorowego i tym naleŜytłumaczyć stwierdzone róŜnice w trwałości połączenia obu par omawianych zasad. Zasady układające się w parywedług podanych reguł (adenina – tymina, guanina – cytozyna) nazywamy zasadami komplementarnymi(uzupełniającymi).
Koncepcja Watsona i Cricka, za którą wspólnie z Wilkinsem uzyskali w 1962r. nagrodę Nobla, znalazłapotwierdzenie ze strony chemii analitycznej. W wielu pracowniach stwierdzono, Ŝe stosunki molarne puryn dopirymidyn, a takŜe stosunki zasad uzupełniających (A do T i G do C) są bliskie jedności.
Skład nukleotydowy RNAMateriał AMP GMP CMP UMP
Trzustka szczura 20,5 31,3 26,9 21,3Wątroba szczura 20,2 31,1 27,1 21,6Wątroba cielęca 23,1 28,9 28,1 20,0Grasica cielęca 19,2 33,2 25,9 21,9Azotabacter vinelandii 24,2 30,6 25,7 19,5Escherichia coli 26,0 30,7 24,1 19,2Wirus mozaiki tytoniowej 30,0 25,8 18,5 25,8Wirus grypy 24,8 17,9 24,5 32,7Wirus polio 29,0 24,0 22,0 25,0Wirus bakterii F2 (MS2) 22,0 26,0 27,0 25,0
Jak widać z zestawienia podanego w powyŜszej tabeli, skład zasad jest charakterystyczny dla danegogatunku, przewaŜnie taki sam we wszystkich jego narządach. U przebadanych zwierząt wyŜszych ten skład jestbardzo podobny, z duŜą przewagą par zasad AT. Natomiast u niektórych krabów morskich spotyka się odmianyDNA, w którym przewaga AT jest bardzo duŜa, a zawartość zasad GC jest wynosi zaledwie parę procent, np. uCancer borealis – około 3 %.
Znacznie większa rozmaitość składu występuje w świecie drobnoustrojów, wśród roślinjednokomórkowych, bakterii, wirusów i bakteriofagów. W niektórych z tych organizmów stwierdzono wyŜszązawartość GC niŜ AT, w innych łańcuchach DNA jest jednopasmowy, co odbija się oczywiście na składziezasad. Brak jest wówczas omawianych regularności. Przykładem jest bakteriofag ØX174, którego skład zasadwskazuje, Ŝe nie są one połączone w komplementarne pary (A:T=0,75, G:C=1,30). Stwierdzono natomiast, Ŝe powprowadzeniu faga ØX174 do komórki Escherichia coli, gdzie następuje jego replikacja i namnaŜanie się,występuje on w postaci dwupasmowej, a stosunki A:T i G:C są wówczas bliskie jedności.
Budowa przestrzenna RNA. W niektórych odmianach RNA, szczególnie pochodzenia zwierzęcego, np.rozpuszczalny RNA szczurzej wątroby, stosunki puryn do pirymidyn, a takŜe stosunki zasad komplementarnychsą bliskie jedności, z tym zastrzeŜeniem, Ŝe w RNA miejsce tyminy zastępuje uracyl. W oparciu o takie wynikipoczątkowo przypisywano cząsteczce RNA budowę dwułańcuchową, w myśl załoŜeń teorii Watsona i Crickadla DNA. Obecnie przyjmuje się, iŜ pojedyncza cząsteczka RNA zbudowana jest z jednego łańcuchapolinukleotydowego. Wyjątkiem jest RNA transportujący (tRNA), gdzie parowanie się niektórych odcinków nicipozwala tworzyć dwuniciowe fragmenty struktury II-rzędowej, przedstawianej modelowo jako liść koniczyny.
Created by Neevia Document Converter trial version http://www.neevia.com

124
WITAMINYWitaminy są związkami organicznymi pobieranymi z poŜywieniem, które nie stanowią plastycznego
materiału strukturalnego ani źródeł energii, ale są niezbędne jako katalizatory, bez których niemoŜliwy jestprawidłowy przebieg reakcji metabolicznych.
Jedne z nich znajdują się we wszystkich komórkach zwierzęcych, gdyŜ po wchłonięciu łączą się jako grupyprostetyczne z odpowiednimi białkami, aby tworzyć czynne enzymy. Z tego powodu nazywa się je witaminamiprostetycznymi. Do grupy tej naleŜą rozpuszczalne w wodzie witaminy grupy B oraz witamina K. Witamina B1jest koenzymem w dekarboksylazie kwasu pirogronowego i α-ketoglutarowego, B2 w Ŝółtym enzymieoddechowym, B6 w dekarboksylazach i transaminazach, PP w dehydrogenazach, a kwas pantotenowy wkoenzymie A. Do drugiej grupy naleŜą witaminy nie tworzące koenzymów, mniej równomiernie rozmieszczonew róŜnych komórkach, nie uczestniczące w całej przemianie materii, ale przeznaczone do specjalnych funkcjiczynnościowych, które spełniają jak gdyby indukcyjnie na drodze wytwarzania w organizmie innych ciałczynnych. Do witamin indukcyjnych naleŜą: A, C, D i E.
Witaminy dzieli się równieŜ zgodnie z kryterium rozpuszczalności. Witaminy A, D, E i K są rozpuszczalnew tłuszczach, zaś B, C, H i P – w wodzie.
ChociaŜ witaminy pobiera się w zasadzie z poŜywienia, to jednak wiele z nich pochodzi z drobnoustrojówŜyjących w przewodzie pokarmowym zwierząt i człowieka, a niektóre powstają nawet w organizmachzwierzęcych z wchłanianych substancji wyjściowych, zwanych prowitaminami. Niedostateczna ilość witamin wwewnętrznym środowisku moŜe być skutkiem zbyt małej podaŜy w poŜywieniu, upośledzonego wchłaniania,zwiększonego zapotrzebowania bądź teŜ działania specjalnych ciała antagonistycznych, zwanychantywitaminami. Występują wówczas mniej lub bardziej typowe zespoły objawów, które składają się na obrazchoroby z niedoboru, zwany hipowitaminozą lub awitaminozą. Niektóre z witamin podawane sztucznie wnadmiernej ilości wywołują odwrotne zaburzenia czynności i zmiany chorobowe, zwane hiperwitaminozą.Antywitaminy znane są w stosunku do wszystkich witamin prostetycznych, które odznaczają się ponadtopowstawaniem w mikroflorze przewodu pokarmowego i brakiem hiperwitaminoz. Odwrotnie jest z witaminamiindukcyjnymi, które nie posiadają antywitamin, nie powstają w przewodzie pokarmowym, a ich nadmiarprowadzi do hiperwitaminoz.
Antywitaminami są najczęściej związki bardzo podobne do odpowiedniej witaminy. Przez to komórkimogą być w pewnym stopniu oszukane – antywitamina bywa bowiem przyjęta jako witamina, zajmuje jejmiejsce, ale nie spełnia jej funkcji. Na tej zasadzie działają niektóre leki przeciwbakteryjne i nie tylko. Dlaprzykładu moŜna podać w parach witaminę i jedną z jej antywitamin: B1 – oksytiamina, PP – hydrazyd kwasuizonikotynowego, kwas para-aminobenzoesowy – sulfonamidy, K – dikumarol. Niektóre ciała działają znówjako antywitaminy przez to, Ŝe odpowiednią witaminę rozkładają lub wiąŜą chemicznie, np. B1 i tiaminaza czybiotyna i awidyna.
Witamina A (akseroftol, retinol)Witamina A, rozpuszczalna w tłuszczach została odkryta i rozszyfrowano jej strukturę chemiczna w roku
1931 a od 1947 wytwarza się ją syntetycznie. Witamina A i jej róŜne chemiczne formy (aldehydowe, estrowe,kwasowe) występuje tylko w produktach zwierzęcych (A1 – retinol C20H29OH, A2 – 3-dehydroretinolC20H27OH – rysunki).
Z kolei prowitamina A, którą tworzy cała grupa związków (około 600) określanych mianem karotenoidówwystępuje tylko w świecie roślinnym. Wśród karotenoidów za najwaŜniejszy uwaŜa się β-karoten (C40H56,rysunek poniŜej), gdyŜ z niego organizm najłatwiej tworzy retinol, zgodnie z równaniem reakcji:C40H56 + 2 H2O ---> 2 C20H29OH.
Witamina A bierze udział w przemianie ogólnoustrojowej, a zwłaszcza w metabolizmie białek, głównieaminokwasów siarkowych, warunkując prawidłowe rogowacenie nabłonka. Jest niezbędna do utrzymania wdobrym stanie rogówki oka, wszystkich błon śluzowych przewodu pokarmowego, przewodu moczowego,
Created by Neevia Document Converter trial version http://www.neevia.com

125
układu rozrodczego, skóry i płuc. Jest niezbędna dla normalnego widzenia gdyŜ odgrywa rolę w procesietworzenia rodopsyny, decydującej o moŜliwości widzenia o zmierzchu przy słabym oświetleniu. Witamina Awywiera wpływ na czynniki odpornościowe ustroju dlatego teŜ często określana jest jako witamina anty-infekcyjna. Bierze udział w budowie i wspomaganiu naturalnych barier chroniących organizm przedzakaŜeniem, wspomaga naturalny system samoobrony organizmu przed zakaŜeniem i chorobami. W okresiewzrostu organizmu bierze udział w tworzeniu szkliwa nazębnego i właściwego rozstawienia uzębienia. Bierzeudział w metabolizmie i hormonów steroidowych. Stymuluje wydzielanie hormonu tyreotropowego,jednocześnie częściowo unieczynniając tyroksynę w tkankach. Aktywizuje układy enzymatyczne wątroby.UwaŜa się, Ŝe przyjmowanie odpowiednich ilości witaminy A moŜe złagodzić skutki działania na wątrobętoksycznych substancji chemicznych.
W niedoborze witaminy A występują:- kruche, wolno rosnące paznokcie z podłuŜnym bruzdowaniem,- suche, łamliwe włosy,- szorstka, sucha skóra,- suche spojówki oraz zmiany chorobowe na rogówce,- osłabione, wraŜliwe dziąsła,- wysypka skórna,- brak apetytu,- zmniejszona odporność na zakaŜenia,- kurza ślepota czyli niewidzenie o zmierzchu,- zaburzenia widzenia,- zahamowanie wzrostu,- uczucie zmęczenia.
Podawanie duŜych dawek witaminy A przez dłuŜszy okres moŜe spowodować hiperwitaminozę witaminyA z takimi objawami jak:- uczucie zmęczenia i dyskomfortu,- podenerwowanie, draŜliwość,- nudności i wymioty,- zmniejszony apetyt,- bóle głowy,- krwawienie z dziąseł,- zaŜółcenie skóry,- powiększenie wątroby i śledziony,- wypadanie włosów,- świąd skory,- suchość skóry,- depresja.
Źródłem tej witaminy są produkty pochodzenia zwierzęcego: mleko pełne, śmietana, masło, produktymleczarskie, jaja, niektóre tłuste ryby, wątroba, tran Ŝółtka jaj.
Zawartość witaminy A w 100g jadalnych wybranych produktów:- wątroba wołowa - 8866 µg,- wątroba wieprzowa - 3883 µg,- masło śmietankowe - 762 µg,- ser podpuszczkowy 40% tłuszczu w.s.m. - 400 µg,- jaja kurze świeŜe - 375 µg.
Prowitamina A, zwana teŜ β-karotenem znajduje się głównie w produktach roślinnych takich jak: marchew,dynia, szpinak, boćwina, sałata, zielony groszek, szczypior, koper, pietruszka, jarmuŜ, sałata, fasolkaszparagowa pomidory, morele, wiśnie, śliwki i pomarańcze.
Dzienne zapotrzebowanie na witaminę A jako równowaŜniki retinolu [µg] (wg zaleceń Iśiś w W-wie)(równowaŜnik retinolu określa ile jest w nim wykorzystanej witaminy A, która moŜe pochodzić równolegle zretinolu i beta-karotenu): dzieci: 1-3: 400, 4-6: 500, 7-9: 700, młodzieŜ: c10-20: 1000, d10-12: 800, d13-15: 600,d16-20: 800, męŜczyźni: 1000, kobiety: 800, c2: 1000, k: 1200.
Witamina B1 (tiamina, czynnik przeciw beri-beri, czynnik antyneurotyczny, aneuryna)Witamina B1, rozpuszczalna w wodzie,
odgrywa zasadniczą rolę w procesach oddychaniatkankowego, głównie w przemianiewęglowodanów, jest częścią składową koenzymukarboksylazy (pirofosforanu tiaminy). Wzmagaczynność acetylocholiny, hamuje esterazę
Created by Neevia Document Converter trial version http://www.neevia.com

126
cholinową, działa synergicznie z tyroksyną i insuliną, pobudza wydzielanie hormonów gonadotropowych.Tiamina przyspiesza gojenie się ran i wykazuje działanie uśmierzające ból.
Przyjęty pokarm, naduŜywanie alkoholu powodują opóźnianie wchłaniania witaminy natomiast obecnośćdwutlenku siarki w winach lub atmosferze, czarne porzeczki powodują rozkład witaminy.
Stany niedoboru witaminy B1 prowadzą do zaburzeń oddychania tkankowego, czynności układunerwowego, wątroby i mięśnia sercowego. Do najczęstszych objawów niedoborowych tiaminy, które najczęściejspotyka się wśród osób naduŜywających alkohol (alkohol rozkłada w znacznym stopniu tiaminę) naleŜy:- zaburzenia czynności centralnego układu nerwowego: uczucie osłabienia i zmęczenie, oczopląs, zaburzenia
pamięci, koncentracji, zaburzenia pamięci i depresja ,- niewydolność krąŜenia: przyspieszona akcja serca, powiększenie wymiarów serca, obrzęki kończyn górnych
i dolnych ,- zaburzenia ze strony przewodu pokarmowego: utrata łaknienia, nudności, wymioty, biegunki, bóle brzucha.
brak apetytu, spadek wagi.Ostrą postać awitaminozy B1, zwaną beri-beri, spotyka się w stanach głodowych oraz u dzieci
odŜywianych np. samym ryŜem. Charakteryzuje się zaburzeniami czucia, osłabieniem mięśni kończyn dolnych,uogólnionymi obrzękami do jam ciała.
W przypadku przedawkowania witaminy B1 mogą wystąpić:- osłabienie, zmęczenie,- zawroty głowy,- obrzęki,- drŜenie mięśni,- zaburzenia rytmu serca,- reakcje alergiczne,- poty,- nudności,- duszność.
Źródłem Witaminy B1 są produkty zboŜowe grubego przemiału, mięso, wędliny (szczególniewieprzowina), rośliny strączkowe - groch, fasola oraz , droŜdŜe, orzechy, nasiona strączkowe, ryby, owoce iwarzywa. Tiamina jest zgromadzona głównie w warstwie zewnętrznej ziaren zbóŜ dlatego teŜ procesytechnologiczne polegające na łuskaniu i polerowaniu zubaŜają mąkę w tę witaminę.
Dzienne zapotrzebowanie na witaminę B1 [mg] (wg zaleceń Iśiś w W-wie): niemowlęta: 0-0,5: 0,7, 0,5-1:0,8, dzieci: 1-3: 0,9, 4-6: 1,1, 7-9: 1,2, młodzieŜ: c10-12: 1,5, c13-18: 1,7, d10-12: 1,3, d13-15:1,5, d16-18: 1,6,męŜczyźni: m1,8/ud2,0, kobiety: 19-25: m1,7/u1,9/d2,0, 26-60: m1,7/u1,9/d2,0, c2: 1,9, k: 2,2, >60: 1,4.
Witamina B2 (G, ryboflawina, laktoflawina, hepatoflawina, owoflawina)Witamina B2 rozpuszczalna w wodzie, jest koenzymem wielu
enzymów flawonowych – dehydrogenaz, odgrywających waŜną rolę wprocesach oksydoredukcyjnych, głównie w przemianiewęglowodanów. Wpływa na syntezę kwasów tłuszczowych z białek iwęglowodanów. Odgrywa równieŜ waŜną rolę w biochemicznychprzemianach w siatkówce oka przez co warunkuje prawidłowefunkcjonowanie wzroku. Witamina B2 eliminując z organizmuczłowieka trujące substancje zawarte w alkoholu i tytoniu, zapobieganowotworom przełyku. Ryboflawina współuczestniczy z witaminą A wprawidłowym funkcjonowaniu błon śluzowych, dróg oddechowych,śluzówki przewodu pokarmowego, nabłonka naczyń krwionośnych iskóry. Nie rozkłada się w czasie gotowania jednak traci aktywność podwpływem światła.
Objawy hipowitaminozy (niedoboru) witaminy B2:- opóźnianie wzrostu,- uszkodzenie gałek ocznych i rogówki,- pogorszenie ostrości wzroku, światłowstręt, łzawienie, łatwe męczenie się oczu i okołorogówkowe
wrastanie naczyń,- swędzenie w okolicy ujścia pochwy,- wypadanie włosów,- brak koncentracji,- zawroty głowy,- bezsenność,- zaburzenia oddechowe,- obrzmienie lub pękanie błony śluzowej jamy ustnej,
Created by Neevia Document Converter trial version http://www.neevia.com

127
- zapalenie czerwieni warg, języka lub błon śluzowych,- pleśniawki,- zajady jamy ustnej,- łojotok,- pelagra,- choroby układu nerwowego,- dystrofie mięsni,- łuszczące się okolice nosa i ust, czoło i uszy.
Przy duŜym przedawkowaniu mogą wystąpić nudności i wymioty.Witamina B2 znajduje się w droŜdŜach, wątrobie, mleku i jego przetwory, jajach, jarzynach, mięsie.
Znaczna ilość ryboflawiny występuje w ciemnym pieczywie, mięsie, podrobach, rybach, w grochu, fasoli i wdroŜdŜach .
Dzienne zapotrzebowanie na witaminę B2 [mg] (wg zaleceń Iśiś w W-wie): niemowlęta: 0-0,5: 0,8, 0,5-1:0,9, dzieci: 1-3: 1,0, 4-6: 1,3, 7-9: 1,4, młodzieŜ: c10-12: 1,9, c13-15: 2,0, c16-18: 2,2, d10-12: 1,6, d13-18: 2,0,męŜczyźni: m2,4/2,6/d2,8, >60: 2,2, kobiety: 19-60: m1,6/u1,8/d2,2, c2: 2,4, k: 2,6, >60: 2,0.
Witamina B3 (PP, niacyna, nikotynamid, niacynamid, pelagranina, kwas nikotynowy)Niacyna współdziała z ryboflawiną i tiaminą w procesach metabolicznych. Jako składnik koenzymów
niacyna uczestniczy w procesach utleniania i redukcji w organizmie.Uczestniczy w procesach regulacji poziomu cukru we krwi, w regulacjipoziomu cholesterolu w organizmie, w regulacji przepływu krwi wnaczyniach czy teŜ w utrzymaniu odpowiedniego stanu skóry.Współdziała w syntezie hormonów płciowych (estrogeny, progesteron).Niacyna nie jest magazynowana w organizmie ludzkim a jej nadwyŜkizostają wydalone z moczem, dlatego teŜ w bieŜącej diecie muszą znajdować się odpowiednie ilości witaminyB3.
Długotrwały niedobór prowadzi do:- wystąpienia pelagry tzw. rumień lombradzki - szorstkość i zaczerwienienie skóry,- zaburzenia w metabolizmie cukrów,- zaburzenia procesu oddychania komórkowego,- dysfunkcji układu trawiennego (biegunki, spadek masy ciała, osłabienie),- zakłócenia w funkcjonowaniu układu nerwowego (bezsenność, zawroty głowy, bóle głowy, zapalenie
nerwów, zaburzenia pamięci ).Nadmierne spoŜywanie cukru, słodyczy lub napojów słodzonych prowadzi do utraty niacyny.Stosowanie przez dłuŜszy czas duŜych dawek moŜe powodować:
- uszkodzenie wątroby,- niemiarowość pracy serca,- dolegliwości skórne (pieczenie i swędzenie),- podniesienie poziomu glukozy we krwi.
Witamina B2 naleŜy do witamin powszechnie występujących w świecie roślin i zwierząt. Najwięcej jejznajduje się w wątrobie, mięsie, rybach, orzechach oraz ziarnach zbóŜ. Kwas nikotynowy znajduje się wwiększej ilości produktów pochodzenia roślinnego. Niacyna powstaje takŜe z zawartego w produktachtryptofanu (z 60 mg tryptofanu powstaje 1mg niacyny).
Dzienne zapotrzebowanie na witaminę B3 [mg] (wg zaleceń Iśiś w W-wie): niemowlęta: 0-0,5: 8, 0,5-1:9, dzieci: 1-3: 11, 4-6: 14, 7-9: 17, młodzieŜ: c10-12: 20, c13-15: 22, c16-20: 24, d10-12: 18, d13-18: 20,męŜczyźni: m21/u23/d25, >60: 20, kobiety: m19/u21/d22, c2: 21, k: 23, >60: 18.
Witamina B5 (kwas pantotenowy, Bios II, czynnik przesączalny II)Kwas pantotenowy naleŜy do najmniej trwałych witamin z grupy B. Jego cząsteczka jest estrem kwasu 2,4-
dihydroksy-3,3-dimetylomasłowy oraz β-alaniny.Jest jednym z elementów składowych koenzymu A,którego synteza jest uzaleŜniona od podaŜy kwasupantotenowego w diecie. Kwas pantotenowy jakoskładnik koenzymu A wpływa na przemianytłuszczowo – białkowe obniŜając znacznie poziomcholesterolu, a takŜe wspomagając proces pigmentacji włosów zapobiega ich siwieniu. Bierze udział w synteziehormonów sterydowych i innych związków chemicznych. Uczestniczy w syntezie hemu do hemoglobiny icytochromów, w wytwarzaniu przeciwciał a takŜe bierze udział w regeneracji komórek skóry i błon śluzowych.
Objawami niedoboru witaminy B5 jest:- zespół piekących stóp,
Created by Neevia Document Converter trial version http://www.neevia.com

128
- zmęczenie, osłabienie,- bóle głowy i brzucha,- nudności i wymioty,- zmniejszenie odporności immunologicznej,- zmiany w skórze,- zaburzenia pigmentacji włosów.
Objawy nadmiaru witaminy B5 obserwuje się rzadko. U niektórych osób stwierdzono objawy uczulenia.Największe ilości kwasu pantotenowego zawierają: droŜdŜe, wątroba wieprzowa, wątroba cielęca, jaja,
niełuskane ziarno pszenicy, płatki owsiane, pełnoziarnisty chleb, mięso wieprzowe, mięso wołowe, chleb biały,mleko, ziemniaki, szpinak, marchew, kapusta.
Zapotrzebowanie na ten składnik nie zostało jeszcze jednoznacznie określone.
Witamina B6 (pirydoksyna – pirydoksol, pirydoksal, pirydoksamina, adermina)Witamina B6, rozpuszczalna w wodzie, obejmuje grupę trzech naturalnych związków pirydynowych
ulegających w organizmiewzajemnym przekształceniom iwykazujących podobne działaniebiologiczne. Pirydoksyna,pirydoksal i pirydoksamina sąkoenzymem dla ponad 50 róŜnychenzymów. Witamina B6 bierzeudział w przemianie białek jako koenzym dekarboksylazy i transaminaz aminokwasów. Szczególnie waŜny jestjej udział w przemianie tryptofanu (dającej kwas nikotynowy i serotoninę). Jako koenzym fosforylazy wpływana glikogenezę i glikogenolizę w mięśniach. Zmniejsza wydzielanie prolaktyny. Pirydoksyna bierze równieŜudział w syntezie białek i kwasów nukleinowych. Jest niezbędna do produkcji hemoglobiny. Podnosi odpornośćimmunologiczną organizmu i uczestniczy w tworzeniu przeciwciał.
Niedobór witaminy B6 występuje tylko wyjątkowo, np. u małych dzieci karmionych sztucznymiodŜywkami oraz u alkoholików. Do najczęściej spotykanych objawów naleŜą:- stany zapalne skóry - dermatitis (łojotokowe zmiany na twarzy, podraŜnienie języka i błon śluzowych jamy
ustnej,- zapalenie błony śluzowej jamy ustnej (języka, kącików warg),- zmiany w ośrodkowym układzie nerwowym (apatia, bezsenność, nadwraŜliwość, napady drgawek),- zwiększona podatność na infekcje,- niedokrwistość makrocytarna.
Organizm dobrze toleruje duŜe dawki witaminy B6. Przy dłuŜszym stosowaniu pirydoksalu w ilości 2gdziennie mogą wystąpić zaburzenia neurologiczne i uczucie zmęczenia.
Głównym źródłem witaminy B6 są produkty zboŜowe i ziemniaki (pirydoksyna) oraz mięso i wędliny(pirydoksal i pirydoksamina). Szczególnie bogate w pirodyksynę są: wątróbka, ziarna soi, kiełki pszeniczne,orzechy włoskie, ryby, banany, szpinak, awokado, mąka pełnoziarnista, drób
Dzienne zapotrzebowanie na witaminę B6 [mg] (wg zaleceń Iśiś w W-wie): niemowlęta: 0-0,5: 0,8, 0,5-1:1, dzieci: 1-3: 1,2, 4-6: 1,4, 7-9: 1,6, młodzieŜ: c10-12: 1,8, c13-15: 2,0, c16-18: 2,4, d10-12: 1,6, d13-15: 1,7,d16-18: 1,8, męŜczyźni: m2,2/u2,4/d2,6, >60: 2,4, kobiety: 19-25: m1,8/u2,0/d2,2, c2: 3,0, k: 2,9, >60: 2,2.Czynniki zwiększające zapotrzebowanie na pirydoksynę to:- stres,- miesiączka,- ciąŜa,- niewydolność serca,- podeszły wiek,- zbyt niski poziom cukru we krwi,- zaŜywanie pigułek antykoncepcyjnych.
Witamina B7 (H, biotyna, koenzym R)Witamina H naleŜy do witamin z grupy witamin B, a więc rozpuszczalna w wodzie. Witamina ta pełni rolę
przenośnika dwutlenku węgla w róŜnych procesach przemianymaterii. Wytwarzana jest przez bakterie Ŝyjące w przewodziepokarmowym. Bierze udział w metabolizmie białek i tłuszczów,uczestniczy w syntezie kwasów tłuszczowych, jak teŜ przywchłanianiu witaminy C. Współdziała w przemianie aminokwasówi cukrów jak równieŜ uczestniczy z witaminą K w syntezieprotrombiny białka odpowiedzialnego za prawidłowe krzepnięcie
Created by Neevia Document Converter trial version http://www.neevia.com

129
krwi. Wpływa na właściwe funkcjonowanie skóry oraz włosów, zapobiega siwieniu włosów oraz łysieniu.Biotyna jest bardzo odporna na ogrzewanie oraz na działanie kwasów i zasad, stąd obróbka kulinarna manieznaczny wpływ na zawartość tej witaminy. Związki utleniające inaktywują biotynę.
Niedobór witaminy H moŜe powodować:- bóle mięśniowe,- osłabienie i apatię,- łysienie,- łuszczycowe zmiany skóry na dłoniach, nogach i ramionach,- wysuszenie i przebarwienia skóry oraz błon śluzowych,- wypryski skórne,- pogorszenie metabolizmu tłuszczów,- podwyŜszenie poziomu cholesterolu i barwników Ŝółciowych we krwi,- depresję.
Do tej pory nie określono toksycznego wpływu biotyny na organizm ludzki.Biotynę znaleziono prawie we wszystkich tkankach roślin i zwierząt, przy czym w większych ilościach w
młodszych lub szybko rosnących niŜ w dojrzałych. Biotyna znajduje się głównie w mleku, w warzywach, wmięsie, Ŝółtkach jaj, mące sojowej, nie łuskanym ryŜu. Źródłem biotyny są owoce, orzechy, kalafior. Dość duŜobiotyny znajduje się w droŜdŜach, wątrobie, nerkach, trzustce i nadnerczach. W surowym jajku znajduje sięawidyna, która wiąŜe trwale biotynę i uniemoŜliwia jej wykorzystanie, jednak podczas gotowania jaja następujedenaturacja awidyny dzięki czemu biotyna staje się w pełni przyswajalna przez nasz organizm.
Witamina H naleŜy do witamin, które, podobnie jak kwas pantotenowy, nie są umieszczane w normachŜywieniowych. Dziennie potrzebujemy jej zaledwie 0,3 mg.
Witamina B8 (cholina)Cholina jest zasadą azotową występującą w tkankach roślinnych i zwierzęcych. Powstaje w organizmie z
kolaminy (etanoloaminy, 2-aminoetanolu: HO-CH2-CH2-NH2 – jest to najprostszy aminoalkohol). Wchodzi wskład fosfolipidów zwanych lecytynami (patrz rozdział poświęcony estrom),przez co wspomaga metabolizm tłuszczów oraz neurohormonu acetylocholiny(estryfikacja kwasem octowym), dlatego jest waŜna dla przewodzenianerwowego. W organizmie jest źródłem grup metylowych w niektórychprocesach biochemicznych. Sole choliny są stosowane w leczeniu schorzeńwątroby i naczyń krwionośnych. Jej niedobór powoduje wzrost ciśnienia krwi, krwotoki oraz deformacje kości.Jej źródłem są Ŝółtka jaj i warzywa liściaste. Dzienne zapotrzebowanie wynosi 500 – 900 mg.
Witamina B10 (kwas para-aminobeznoesowy, PABA)Witamina ta słuŜy bakteriom przewodu pokarmowego do syntezy kwasu foliowego (jest
jego składnikiem), działa jako koenzym w metabolizmie białek, wspomaga tworzenie sięczerwonych ciałek krwi. Objawami niedoboru są: zmęczenie, depresja, nerwowość,niebezpieczeństwo ataku serca, obstrukcja oraz zaburzenia przemiany materii. Występuje wdroŜdŜach i w wątrobie. Antymetabolitami są sulfonamidy.
Większość ochronnych kremów do opalania zawiera PABA na olejowym podłoŜu. Filtry tezatrzymują nie tylko część szkodliwych promieni, ale takŜe długość fal, które niosą ze sobąkorzystny wpływ słońca. Niektóre badania wskazują, Ŝe PABA moŜe bezpośrednio niszczyć komórki naskórka.
Witamina B11 (B4, B9, M, folan, folacyna, kwas listny, kwas pteroiloglutaminowy)Kwas foliowy to witamina z grupy B. Jest głównym czynnikiem biorącym udział w procesie podziału
komórek organizmu. Kwas foliowy reguluje róŜne procesy metaboliczne w organizmie, uczestniczy w synteziepuryn, pirymidyn i niektórych aminokwasów. Współuczestniczy z witaminą B12 w regulacji tworzenia idojrzewania czerwonych krwinek.
Objawy hipowitaminozy (niedoboru):- zaburzenia rozwojowe u płodu (wady cewy nerwowej),- niedokrwistość megaloblastyczna,- nadpobudliwość,- trudności w zasypianiu,- słaby wzrost,- problemy z trawieniem,- niedoŜywienie,- biegunka,- utrata apetytu,- osłabienie,
Created by Neevia Document Converter trial version http://www.neevia.com

130
- rozdraŜnienie,- bóle głowy,- zaburzenia zachowania.
U niektórych osób mogą tworzyć się szkodliwe kryształy folacyny w moczu, mogą równieŜ wystąpićalergiczne odczyny skórne. SpoŜycie dziennie ponad 15mg kwasu foliowego moŜe powodować zaburzeniaukładu nerwowego i pokarmowego.
Kwas foliowy jest bardzo szeroko rozpowszechnione w naturze. Występują on w tkankach wielu roślin izwierząt. DuŜe jego ilości występują w szpinaku, brokułach, orzechach, słoneczniku, wątróbce, kiełkachpszenicy i owocach, na przykład w bananach i pomarańczach oraz w droŜdŜach.
Dzienne zapotrzebowanie na kwas foliowy [mg] (wg zaleceń Iśiś w W-wie): niemowlęta: 0-1: 35, dzieci:1-3: 70, 4-6: 90, 7-9: 105, młodzieŜ: c10-15: 200, c16-18: 240, d10-12: 190, d13-15: 200, d16-18: 220,męŜczyźni: m280/ud300, >60: 340, kobiety: m270/u290/d310, c2: 450, k: 530, >60: 320.
Witamina B12 (kobalamina, cyjanokobalamina,czynnik przeciwanemiczny, czynnik wątrobowy,
faktor Castle'a)Witamina B12, rozpuszczalna w wodzie,
jest ogólną nazwą związków tzw. korynoidów ozbliŜonej budowie chemicznej i podobnej funkcjifizjologicznej. W cząsteczce kobalaminy,składającej się z rdzenia porfirynowego, znajdujesię atom kobaltu, z którym moŜe być związanyanion, np. cyjankowy CN- (cyjanokobalaminaC62H88-92O13N4PCo – najwaŜniejsza z grupywitamin B12), hydroksylowy OH-
(hydroksykobalamina), azotynowy NO2-
(nitrokobalamina), chlorkowy Cl- lubsiarczanowy SO4
2-.Witamina B12 jest krwiotwórczym
czynnikiem wątroby, którego czynnośćhemopoetyczna polega na udziale w syntezieprotoporfiryn i przekształceniu kwasu foliowegow folinowy. Witamina B12 łącznie z kwasemfoliowym jest niezbędna w syntezie rybozydówpurynowych szpiku i tworzeniu krwinekczerwonych i białych. Ponadto uczestniczy wdojrzewaniu komórek nabłonkowych, wykazujewłaściwości neurotropowe i ochraniające miąŜszwątroby. Bierze udział w syntezie aminokwasówi kwasów nukleinowych. Pełni ponadto funkcjekoenzymu w transmetylazach oraz w przemianach kwasów organicznych, np. bursztynowego imetylojabłkowego. Witamina B12 uczestniczy w syntezie metioniny. Aby prawidłowo wchłaniać w jelitach,niezbędny jest tzw. czynnik Castle'a, produkowany w Ŝołądku. Dlatego część chorób Ŝołądka powoduje niedobórwitaminy B12.
Zapasy witaminy B12 zgromadzone w wątrobie zdrowego człowieka wystarczają do pokryciazapotrzebowania człowieka na około 3 lata. Do niedoboru dochodzi zazwyczaj u jaroszy, u osób ze schorzeniamizwiązanymi z nieprawidłową produkcją czynnika Castle'a w Ŝołądku lub zaburzeniami wchłaniania w jelitach.Wśród objawów dominują:- zaburzenia powstawania ciałek krwi, zwłaszcza czerwonych -niedokrwistość złośliwa, magaloblastyczna
(choroba Addisona - Biermera),- zmiany zwyrodnieniowe błony śluzowej Ŝołądka,- zaburzenia Ŝołądkowo jelitowe i brak apetytu,- stany zapalne ust,- zaburzenia w układzie nerwowym (zaburzenia czucia, niezborność ruchów, zmęczenie, drętwienie rąk i nóg,
trudności w chodzeniu),- zaburzenia wzrostu u dzieci,- niemiły zapach ciała,- hiperhomocysteinemia,- jąkanie się,- depresja,
Created by Neevia Document Converter trial version http://www.neevia.com

131
- dolegliwości miesiączkowe.Nie są znane objawy przedawkowania witaminy B12. Przy stosowaniu przez dłuŜszy czas megadawek tej
witaminy zaobserwowano u niektórych ludzi objawy uczuleniowe.Witamina B12 nie jest syntezowana ani przez zwierzęta, ani przez rośliny wyŜsze, ale tylko przez bakterie.
Głównym jej źródłem są produkty zwierzęce, zwłaszcza wątroba i mleko, ryby. W niewielkim stopniu jesttworzona przez bakterie w przewodzie pokarmowym, skąd jest wchłaniana tylko w obecności tzw. czynnikakrwiotwórczego wewnętrznego (białko apoerytreina), wydzielanego przez śluzówkę Ŝołądka. Bogatym źródłemkobalaminy (powyŜej 20 µg w 100g) są ryby (szczupaki), wątroby i nerki: wieprzowe, wołowe, cielęce idrobiowe.
Dzienne zapotrzebowanie na witaminę B12 [µg] (wg zaleceń Iśiś w W-wie): niemowlęta: 0-0,5: 0,5, 0,5-1: 1,5, dzieci: 1-3: 2,0, 4-6: 2,5, 7-9: 3,0, młodzieŜ: 3,0, męŜczyźni: 3,0, kobiety: 3,0, c2: 4,0, k: 4,9, >60: 2,5.
Witamina B13 (kwas orotowy)Związek ten posiada zdolność przetwarzania stosunkowo mało wartościowych
substancji w bardzo wartościowy budulec. Witamina B13 bierze udział w metabolizmiekwasu foliowego i witaminy B12. Prawdopodobnie ułatwia funkcjonowanie wątroby.Witaminy tej bardzo potrzebują ludzie starsi, których organizm gorzej przyswaja spoŜytepokarmy. Ponadto zapobiega przedwczesnemu starzeniu się. Występuje m.in. w:warzywach korzeniowych, serwatka i kwaśnym mleku.
Witamina B14 (kwas folinowy, tetrahydrofoliowy, tetrahydropteroiloglutaminowy)Jest to uwodorniona pochodna kwasu foliowego – witaminy B11. Znalazła zastosowanie w medycynie przy
leczeniu anemii.
Witamina B15 (kwas pangamowy, calgam)Jej cząsteczka to sól wapniowa estru kwasu glukonowego i
dwumetyloglicyny. Witamina ta zwiększa przyswajanie tlenu przez tkanki,usuwając objawy nieznacznego ich niedotlenienia (hipoksji). Ma takŜe pewnewłaściwości psychotropowe – zwiększa koncentracje umysłu i zdolnośćskupienia się, wzmaga aktywność Ŝyciową i polepsza samopoczucie.Wspomaga ponadto metabolizm białek oraz stymuluje układ gruczołówdokrewnych, uczestniczy w procesach metylacji związków chemicznych i wbiosyntezie kreatyny, zwiększa aktywność enzymu dehydrogenazybursztynianowej. Stosowana w miaŜdŜycy (zwłaszcza naczyń mózgowych,wieńcowych, kończyn dolnych), w rozedmie płuc, w przewlekłym zapaleniumiąŜszu wątroby, w początkowym okresie marskości wątroby, w przewlekłym alkoholizmie, w chorobachskóry. MoŜna ją podawać przed długotrwałymi zawodami i w okresie odnowy, wpływa bowiem na wzrostzapasów glikogenu w mięśniach i w wątrobie, uodparnia na hipoksję i ma właściwości lipotropowe. Niedobórpowoduje zaburzenia w oddychaniu tkankowym, zapalenie skóry ze świądem oraz zapalenie wątroby. Jejźródłem są: rośliny, mięso i droŜdŜe.
Witamina B17 (letril, amigdalina)W przyrodzie amigdalina pełni rolę inhibitora wzrostu. Wydzielana jest do gleby przez drzewo brzoskwini
i orzecha, obecna jest w nasionach śliwki, wiśni, brzoskwini, moreli i czeremchy. Uwolniona do gleby ulegarozkładowi na glukozę, kwas cyjanowodorowy oraz aldehyd benzoesowy (patrz poniŜszy rysunek). Wywołuje toefekt allelopatyczny – wokół wydzielającej amigdalinę rośliny nie ma Ŝadnych konkurentów.
Interesujące jest zastosowanie amigdaliny do leczenia wszelkiego rodzaju nowotworów. Bazą preparatówjest wyciąg z pestek moreli. Powstające cyjanki silnie niszczą tkankę nowotworową, a powstała glukozazwiększa ilość energii w organizmie, wzmacniając w ten sposób komórki zdrowe.
Created by Neevia Document Converter trial version http://www.neevia.com

132
Inozytol (z grupy B)Inozytol jest cyklicznym alkoholem zawierającym sześć grup hydroksylowych. Jest składnikiem niektórych
fosfolipidów, np. fosfotydyloinozytolu, potrzebnego do tworzenia lecytyny. Syntetyzowany z glukozy przezdrobnoustroje i rośliny. Zwierzętom musi być dostarczany z pokarmem. W duŜych ilościach występuje wwątrobie, mięśniu sercowym, w mózgu, nerkach, krwi. Dzienne zapotrzebowanie wynosi przeciętnie 1 g. Jegoniedobór powoduje podwyŜszenie poziomu cholesterolu we krwi, stłuszczenie wątroby, obstrukcję, egzemę iwypadanie włosów. Ester heksafosforanowy i. (kwas fitynowy) w poŜywieniu (ziarna zbóŜ) moŜe być przyczynąniedoboru wapnia, cynku i Ŝelaza. W lecznictwie heksaoctan i. jest stosowany w schorzeniach wątroby i wmiaŜdŜycy. Przemiany fosforanów i. mają znaczenie w przekazywaniu informacji ze środowiska do komórki.
Witamina C (kwas askorbinowy, dehydroaskorbinowy)Witamina C naleŜy do grupy witamin rozpuszczalnych w wodzie. Pełni zasadnicze
funkcje w procesach oksydoredukcyjnych ustroju. Jest aktywatorem białkowej przemianyaminokwasów aromatycznych a przez fosforylację glukozy wpływa na syntezę hormonówsteroidowych kory nadnerczy i syntezę insuliny. Witamina C jest nieodzowna w prawidłowymfunkcjonowaniu tkanki łącznej, gdzie uczestniczy w syntezie kwasu hialuronowegoniezbędnego do syntezy kolagenu oraz w procesach kostnienia. Bierze takŜe udziałhematopoezie, w przyswajaniu Ŝelaza i syntezie hemoglobiny. Odgrywa waŜną rolę wprocesach odpornościowych, działa odtruwająco. Witamina C wpływa teŜ na wzrost i rozwójkomórek organizmu, utrudnia przenikanie do nich wirusów. Ma właściwościbakteriostatyczne, a nawet bakteriobójcze w stosunku do niektórych drobnoustrojówchorobotwórczych. Przyspiesza procesy gojenia. Uczestniczy w regeneracji witaminy E.
Neutralizowanie przez witaminę C szkodliwego wpływu wolnych rodników chroni organizm przedzmianami nowotworowymi. Przypuszcza się, Ŝe witamina ta ogranicza produkcję działających nowotworowonitrozamin, czyli substancji powstałych z połączenia azotynów (dodawanych do konserwowanych produktówŜywnościowych, m.in. do mięsa) z substancjami znajdującymi się w Ŝołądku. Ponadto łagodzi ona nieprzyjemneobjawy uboczne radioterapii stosowanej w leczeniu nowotworów.
Suszenie, solenie, promienie ultrafioletowe, stosowanie benzoesanu sodu czy kwasu salicylowego dokonserwowania przetworów warzywnych i owocowych oraz niektóre leki np. sulfonamidy, pochodnebarbiturowe niszczą witaminę C. Długotrwałe przyjmowanie aspiryny trzykrotnie zwiększa wydalanie witaminyC, dlatego teŜ naleŜy wtedy zwiększyć dawki tej witaminy.
Zbyt mała jej ilość w diecie manifestuje się:- osłabieniem, bólami mięśniowymi, apatią, brakiem apetytu,- bladością skóry i błon śluzowych,- pękaniem drobnych naczynek krwionośnych,- bólem głowy,- krwawieniem z dziąseł,- duŜą podatnością na choroby zakaźne,- zwiększoną łamliwością kości, wolniejszym gojeniem się ran,- wstępowaniem szkorbutu (gnilca) objawiającego się obrzękami i krwawieniem z dziąseł oraz wypadaniem
zębów.Brak witaminy C w poŜywieniu cięŜarnej stwarza zagroŜenie teratogenne dla płodu. Zbyt mała ilość
witaminy C powoduje zwiększone wydalanie witaminy B6.Stosowanie wysokich dawek powoduje zakwaszenie moczu, upośledzając w ten sposób wydalanie stałych
kwasów i zasad. Kwaśny odczyn moczu moŜe powodować wytrącanie się moczanów i cystynianów oraztworzenie się kamieni w drogach moczowych, nie naleŜy więc podawać duŜych dawek witaminy C chorym zeskłonnością do dny, cystynurii lub tworzenia się kamieni moczanowych.
Głównym źródłem witaminy C są kalafiory, kapusta – zwłaszcza kiszona, kiszone ogórki, nać pietruszki,chrzan, pomidory, jagody, owoce dzikiej róŜy, jabłka, w końcu cytrusy.
Dzienne zapotrzebowanie na witaminę C [mg] (wg zaleceń Iśiś w W-wie): dzieci: 1-3: 45, 4-6: 50, 7-9:65, młodzieŜ: 70, męŜczyźni: 70, kobiety: 70, c2: 80, k: 100, >60: 60. Palacze tytoniu powinni dostarczać swemuzatrutemu organizmowi nie mniej niŜ 200 mg kwasu askorbinowego na dobę, chyba Ŝe rzucą palenie.
Witamina DWitamina D obejmuje witaminę D1 (kalcyferol), D2 (ergokalcyferol C28H43OH – rysunek lewy) oraz D3
(cholekalcyferol C27H43OH – rysunek prawy). Witamina D1 znajduje się w tranie, D2 jest wytwarzana wroślinach wystawionych na działanie promieni ultrafioletowych, natomiast witamina D3 powstaje w skórze ludzii zwierząt i jako jedną z niewielu witamin organizm moŜe wyprodukować sam pod wpływem promieni
Created by Neevia Document Converter trial version http://www.neevia.com

133
słonecznych, które przemieniają zawarty w skórze człowieka 7-dehydrocholesterol (tzw. prowitamina D3)przemienia się w cholekalcyferol.
UwaŜa się, Ŝe juŜ dziesięć minut słonecznej kąpieli codziennie w czasie letnich miesięcy zapewniaodpowiednią dawkę tej witaminy na cały rok. Jednak naleŜy brać pod uwagę indywidualne zapotrzebowania, np.to, Ŝe dzieci potrzebują więcej witaminy niŜ dorośli, a ponadto Ŝe wraz z wiekiem zmniejsza się zdolnośćorganizmu do wytwarzania tej witaminy pod wpływem promieni ultrafioletowych. RównieŜ osoby znajdującesię w zanieczyszczonym środowisku mają mniejsze szanse na odpowiednią ilość witaminy D w organizmie.
Witamina D pobudza wchłanianie wapnia i fosforu, a takŜe zapobiega nadmiernemu wydalaniu tychpierwiastków z moczem przez co ma wpływ nie tylko na prawidłowe kształtowanie się kości u dzieci idorosłych, na ich odpowiednią gęstość i na stan naszego uzębienia. W przypadku gdy brakuje wapnia wpoŜywieniu pobierany jest on z kości. Obecność witaminy we krwi wpływa korzystnie na system nerwowy i naskurcze mięśni w tym serca. Odpowiednia ilość wapnia umoŜliwia sprawne przewodzenie impulsównerwowych. Witamina D zapobiega i łagodzi stany zapalne skóry, reguluje wydzielanie insuliny, a tym samymwpływa na odpowiedni poziom cukru w organizmie. Korzystnie wpływa na słuch, gdyŜ decyduje o dobrymstanie kostek ucha wewnętrznego. Oddziałuje na komórki szpiku kostnego produkujące komórki obronne(monocyty).
Niedobór witaminy D jest przyczyną występowania krzywicy u dzieci i młodzieŜy (krzywica dziecięca), atakŜe u dorosłych (krzywica późna). W krzywicy kości ulegają zniekształceniu i osłabieniu, wykrzywiają się podwpływem cięŜaru ciała szybko rosnącego dziecka. Kości i nadgarstki stają się powiększone, klatka piersiowazaczyna przypominać pierś gołębia, u dzieci powstają opóźnienia we wzroście zębów. Dziecko częściej poci się istaje się nadmiernie aktywne. U osób dorosłych, którzy mają utrudniony dostęp do słońca lub spoŜywająprodukty pozbawione witaminy D, moŜe pojawić się rozmiękczenie kości, zwane osteomalacją, które prowadzido częstych złamań i skrzywień układu kostnego. Niedobór witaminy D u dorosłych sprzyja powstawaniuosteoporozy polegającej na zmniejszeniu się masy i gęstości kości, które stają się porowate, kruche i łamliwe.
Zbyt mała ilość witaminy D moŜe powodować zapalenie spojówek, a takŜe stany zapalne skóry. Częstodochodzi do osłabienia organizmu co prowadzi do zmniejszonej odporności na zaziębienia. Skutkiem niedoboruwitaminy D bywa takŜe pogorszenie słuchu. Następstwem braku wapnia i fosforu jest osłabienie i wypadaniezębów.
Witamina D w duŜych ilościach, większych niŜ zalecane, jest toksyczna. Wynikiem nadmiaru tej witaminymoŜe być:- nudności,- biegunka,- spadek masy ciała,- łatwe męczenie się,- nadmierne pocenie się,- brak apetytu, utrata łaknienia,- senność,- opóźnienie w rozwoju dziecka,- zaburzenia rytmu pracy serca,- wzmoŜone oddawanie moczu,- ból oczu,- bóle szczęk, stawów i mięśni,- bóle głowy,- świąd skóry,- zwiększenie ryzyko powstania miaŜdŜycy,- zwiększenie ryzyko powstania kamicy nerkowej.
Doskonałym źródłem witaminy D jest tran i oleje rybne, które znajdują się w rybach, takich jak łosoś,tuńczyk, śledź, makrela czy sardynki. Witaminę tę znajdziemy równieŜ w mleku (najlepiej wzbogaconymdodatkowo przez dodanie tej witaminy), a takŜe w wątrobie, białku jaj oraz przetworach mlecznych, takich jakser, masło czy śmietana. Jednak ilość witaminy D w mleku zaleŜy od sposobu Ŝywienia krów i ich przebywaniana słońcu. Profilaktycznie wzbogacane są o tę witaminę nowoczesne margaryny i mleko w proszku.
Created by Neevia Document Converter trial version http://www.neevia.com

134
Dzienne zapotrzebowanie na witaminę D [µg] (wg zaleceń Iśiś w W-wie): dzieci: 10, młodzieŜ: c10-12:10, c13-20: 7,5, d10-12: 10, d13-20: 7,5, męŜczyźni: 5, kobiety: 5, c2: 10, k: 10, >60: 5.
Witamina E (tokoferole, tokotrienole)Istnieje co najmniej osiem postaci witaminy E zwanych tokoferolami, które są rozpuszczalne w tłuszczach.
Witamina E związana jest z przemianą azotową i oddychaniem. Witamina E bierze udział w przemianie materii,jako związek antyoksydacyjny i zwiększającypodaŜ tlenu, zapobiega utlenianiu witaminy A ,nienasyconych kwasów tłuszczowych i innychlipidów, uniemoŜliwiając tym samy tworzeniu siętoksycznych produktów i zapobiega rozwojowimiaŜdŜycy naczyń krwionośnych. Witamina EobniŜa podwyŜszony poziom lipidów w surowicykrwi. Aktywując układy enzymatyczne oddychaniatkankowego, uczestniczy w procesach odtruwania,chroni komórki przed stłuszczeniem, podtrzymuje czynność tkanki mięśniowej, ułatwia przyswajanie tlenu przezerytrocyty. Witamina E pobudza produkcję substancji przeciwzakrzepowych, zmniejszając ryzyko rozwojuschorzeń naczyń krwionośnych. Witamina E jest niezbędna u męŜczyzn do prawidłowej produkcji spermy.Dlatego niedobór witaminy E moŜe prowadzić do bezpłodności. Witamina E współdziała z witaminami A, C ikarotenoidami, zmniejszając ryzyko rozwoju chorób nowotworowych.
Na szczęście witamina E występuje powszechnie, praktycznie nie spotykamy się z jej niedoborem.JednakŜe hipowitaminoza moŜe powodować:- rozdraŜnienie,- osłabienie zdolności koncentracji,- zaburzenia funkcjonowania i osłabienie mięśni szkieletowych,- rogowacenie i wczesne starzenie się skóry,- gorsze gojenie się ran,- pogorszenie wzroku,- niedokrwistość,- bezpłodność,- zwiększone ryzyko chorób sercowo naczyniowych.
Witamina spoŜywana przez dłuŜszy czas w dawkach większych niŜ 1000 mg octanu α-tokoferolu na dobę uosób dorosłych, moŜe powodować:- zmęczenie,- bóle głowy,- osłabienie mięśni,- zaburzenia widzenia.
Witamina E powstaje tylko w roślinach. Jej naturalnym dostawcą są kiełki pszenicy, całe zboŜa, zielonewarzywa liściaste jak sałata, szpinak oraz kapusta, czosnek, orzechy, awokado oraz oleje roślinne. NaleŜypamiętać o tym, Ŝe oleje tłoczone na zimno zawierają znacznie więcej witaminy E niŜ te produkowaneprzemysłowo gdyŜ proces uszlachetniania niszczy aŜ 75% naturalnej witaminy.
Stopień wchłaniania witaminy E jest odwrotnie proporcjonalny do wielkości jednorazowej dawki i np. zdawki jednorazowej 100 mg ulega resorpcji 80%, z dawki jednorazowej 500 mg - tylko 68%. Obecnie dlaporównania aktywności róŜnych związków witaminy E stosowany jest równowaŜnik α-tokoferolu (RT).Największą aktywność spośród róŜnych form witaminy E wykazuje α-tokoferol = 100% bioaktywności.
- 1 mg RT = 1 mg α-tokoferolu- 1 mg RT = 2 mg β-tokoferolu- 1 mg RT = 4 mg γ-tokoferolu- 1 mg RT = 5 mg α-tokotrienolu- 1 mg RT = 25 mg β-tokotrienolu- 1 mg RT = 25 mg γ-tokotrienoluDzienne zapotrzebowanie na witaminę E (RT [mg]) (wg zaleceń Iśiś w W-wie): dzieci: 0-1: 4, 1-3: 6, 4-6:
7, 7-9: 10, młodzieŜ: 10, męŜczyźni: 10, kobiety: 10, c2: 12, k: 14, >60: 10.
Witamina FW skład tej witaminy wchodzą trzy wielonienasycone kwasy tłuszczowe:
linolowy, 9,12-oktedecenodienowy: CH3-(CH2)4-CH=CH-CH2-CH=CH-(CH2)7-COOH,linolenowy, 9,12,15-oktedecenotrienowy: CH3-CH2-CH=CH-CH2-CH=CH-CH2-CH=CH-(CH2)7-COOH,arachidonowy, 5,8,11,14-ejkozanotetraenowy: C19H31COOH CH3-(CH2)4-(CH=CH-CH2)4-(CH2)2-COOH.
Created by Neevia Document Converter trial version http://www.neevia.com

135
Substancje te nazwano witaminą, chociaŜ nią nie są, gdyŜ zapotrzebowanie dzienne na nie wynosi od 5 do 10 g –stanowią zatem składnik plastyczny i energetyczny.
Witamina K (filochinon, menachinon, farnochinony, witamina przeciwkrwotocza)Mianem witaminy K określamy grupę związków nierozpuszczalnych w wodzie, ale
rozpuszczalnych w tłuszczach. Do grupy tej naleŜą: witamina K1 (fitomenadion – nadole po lewej), witamina K2 (menachinon-30 – na dole po prawej) oraz witamina K3(menadion, 2-metylo-1,4-naftochinon – obok). Witaminę K1 pozyskuje się z poŜywienia,K2 jest produkowana przez bakterie jelitowe, natomiast witamina K3 jest syntetyzowana,tzn. produkowana sztucznie.
Witamina K jest koenzymem syntezy protombiny w wątrobie czyli białka osocza krwi, które bierze udziałw procesie krzepnięcia krwi. Witamina K uszczelnia sródbłonki naczyń krwionośnych, zapobiega krwawieniomwewnętrznym oraz krwotokom, jest czynnikiem zmniejszającym nadmierne, obfite krwawienia miesiączkowe.Uczestniczy w formowaniu tkanki kostnej, wpływa na przemiany metaboliczne kwasów nukleinowych.Witamina K posiada właściwości przeciwbakteryjne oraz przeciwgrzybicze, wykazuje równieŜ działanieprzeciwzapalne i przeciwbólowe.
Z niedoborem witaminy K spotykamy się rzadko, gdyŜ organizm moŜe ją wytwarzać sam za pomocąmikroorganizmów, które znajdują się w jelicie cienkim. Brak witaminy K mogą odczuwać ci, którzy jedzą zamało zielonolistnych warzyw. Poza tym niedobór tej cennej substancji występuje szczególnie u dorosłychpodczas kuracji antybiotykowej, jeśli pacjent naduŜywa leków, osłabia to bowiem bakterie przewodupokarmowego produkujące tę witaminę. Awitaminozę mogą spowodować teŜ zaburzenia we wchłanianiupokarmów ograniczające trawienie tłuszczów, jak równieŜ uszkodzenia wątroby. U osób z niedoborem witaminyK stwierdza się obniŜony poziom protrombiny we krwi oraz krwotoki z nosa, układu pokarmowego imoczowego. Niedobór witaminy K występuje czasem u niemowląt, gdyŜ ich organizm nie moŜe od razuwytworzyć w przewodzie pokarmowym odpowiedniej ilości bakterii potrzebnych do jej powstania. Abyzapobiec takiej sytuacji, kobietom cięŜarnym podaje się uzupełniające dawki witaminy K lub wstrzykuje się jądziecku po porodzie.
Witamina K nie jest toksyczna, ale nadmierne dawki mogą wpłynąć niekorzystnie na pracę wątrobywywołując Ŝółtaczkę i uszkodzenia tkanki mózgowej u niemowląt. Zbyt duŜe dawki witaminy K mogąpowodować poty oraz uczucie gorąca.
DuŜe ilości witaminy K występują w zielonych warzywach, kalarepie, kalafiorze, pomidorach,truskawkach oraz wątróbce. Witaminę K moŜna znaleźć w ciemnozielonych i liściastych warzywach, takich jak:brokuły, rzepa, szpinak, sałata, kapusta. Oprócz tego zawierają ją owoce awokado, brzoskwinie, ziemniaki,białko jaj, ser i wątroba. Źródłem tej witaminy są takŜe Ŝyjące w przewodzie pokarmowym człowieka bakterie.
NaleŜy podkreślić, Ŝe po antybiotykoterapii powinno się pamiętać o zwiększonej dostawie tej witaminy wpokarmach co jest związane ze zmniejszeniem jej produkcji przez bakterie znajdujące się w przewodziepokarmowym. RównieŜ zjełczałe tłuszcze, naduŜywanie środków przeciwbólowych, salicylanów niszczywitaminę K utrudniając jej wchłanianie lub prowadząc do przedwczesnego jej wydalania z organizmu.
Dzienne zapotrzebowanie wynosi:- u dzieci wynosi 1 mg,- u dorosłych 4 mg.
Witamina P (bioflawonoid rutyna, eskulina)Rutyna jest glikozydem zbudowanym z dwóch cząsteczek
cukrów prostych – glukozy i ramnozy, osadzonych naszkielecie flawonu kwercetyny (3,5,7,3’,4’-pentahydroksyflawon, rysunek). Rutyna działa ochronnie nakolagen i elastynę, będące podstawowymi elementamiwchodzącymi w skład ścian naczyń krwionośnych, dziękiczemu uszczelnia, wzmacnia i zwiększa elastyczność naczyńwłosowatych. Jest silnym przeciwutleniaczem – chroni kwasaskorbinowy przed utlenieniem, a przez to wzmaga jego
Created by Neevia Document Converter trial version http://www.neevia.com

136
działanie. Występuje m. in. w rucie, gryce, bratkach, czarnym bzie, rdeście i pączkach brzozy. Stanowi składnikleków, np. rutinoscorbinu.
Witamina P4 (trokserutyna)Witamina ta jest flawonową pochodną rutyny (O-β-hydroksyetylorutozyd). Jako lek w postaci handlowej
występuje pod nazwami: Troxerutin i Venotrex. Trokserutyna zwiększa napięcie i elastyczność naczyń Ŝylnych,zmniejsza przepuszczalność, kruchość i łamliwość naczyń oraz usprawnia przepływ krwi w naczyniach Ŝylnychw obrębie kończyn dolnych. Zapobiega obrzękom i wykazuje działanie przeciwzapalne. Po doustnym podaniuwchłania się w ok. 15-20%. Wydalana jest głównie z kałem i Ŝółcią.
Witamina T (termityna)Związek ten cechuje złoŜona budowa, bliska prawdopodobnie witaminie B12. Określa się ja mianem
pseudo-witaminy, gdyŜ nie jest czynnikiem niezbędnym do Ŝycia. W świecie owadów stanowi bodziecwzrostowy, powodujący wyrastanie olbrzymów i Ŝołnierzy u mrówek i termitów.
Witamina UJest to jeden z najsłabiej poznanych związków o charakterze witaminy. Przypisuje się jej prostą budowę
chemiczną. Ponadto z pewnością zawiera w cząsteczce co najmniej jeden atom azotu. Źródła określają ją jakos-metylometioninę bądź diazofenylomocznik. Wymieniana jest w składzie niektórych kremów poprawiającychwygląd skóry.
Created by Neevia Document Converter trial version http://www.neevia.com






![Skrypt [2008]](https://static.fdocuments.pl/doc/165x107/5571fda24979599169998e67/skrypt-2008.jpg)










