
Sekwencjonowanie wczoraj i dziś - pg.gda.pl · • Zgodnie z przewidywaniami genomy ł ik i d l k...
25
Sekwencjonowanie Sekwencjonowanie Sekwencjonowanie wczoraj i dziś Sekwencjonowanie wczoraj i dziś dr hab. Beata Krawczyk Katedra Mikrobiologii PG Publikacja współfinansowana ze środków Unii Europejskiej w ramach Europejskiego Funduszu Społecznego
Transcript of Sekwencjonowanie wczoraj i dziś - pg.gda.pl · • Zgodnie z przewidywaniami genomy ł ik i d l k...

SekwencjonowanieSekwencjonowanieSekwencjonowaniewczoraj i dziś
Sekwencjonowaniewczoraj i dziśjj
dr hab. Beata Krawczyk
Katedra Mikrobiologii PG
Publikacja współfinansowana ze środków Unii Europejskiej w ramach Europejskiego Funduszu Społecznego

CelCel sekwencjonowaniasekwencjonowaniaCel Cel sekwencjonowaniasekwencjonowania
dostarcza wielu cennych informacji o strukturze i funkcji genów,
znajomość pełnej sekwencji DNA badanych organizmów umożliwia zrozumienie molekularnych mechanizmów ich f k j i i l jifunkcjonowania i ewolucji
umożliwia szukanie mutacji

• Naukowcy zakończyli sekwencjonowanie pełnego genomu neandertalczyka, wymarłego około 30 tys. lat temu bliskiego y , y g y g
krewniaka człowieka
• Zgodnie z przewidywaniami genomy ł i k i d l k
Archeologia molekularna -Historia człowieka
człowieka i neandertalczyka są w 99,5% identyczne (człowieka i
szympansa - w 98%).
http://migg.wordpress.com/2006/11/16/neandertalczyk-powraca/

Pochodzenie człowieka
Analiza fragmentów jądrowego i mitochondrialnego DNA ze
szczątków paleontologicznych i ą p g yarcheologicznych pozwala badać
pochodzenie człowieka
http://migg.files.wordpress.com/2007/03/101371_journalpbio0020340g005-m.jpg

Historia rozwoju metod sekwencjonowania DNA
19531953 ‐ opracowanie modelu podwójnej helisy DNA przez Jamesa Watsona i19531953 opracowanie modelu podwójnej helisy DNA przez Jamesa Watsona i Francisa Cricka19721972 ‐ opracowanie sposobów izolacji materiału do dalszych badań. Rozwój technologii rekombinacji umożliwiający izolację dowolnych fragmentów DNA i ich g j ją y ję y greplikację. 19771977 ‐ Allan Maxam i Walter Gilbert ogłaszają publikację pt.: Sekwencjonowanie DNA przez chemiczną degradację. W tym samym czasie Sanger opublikował też metodę sekwencjonowania DNA przez syntezę katalizowaną enzymatycznie. 19801980 ‐ Fred Sanger i Walter Gilbert otrzymali Nagrodę Nobla. 1982 ‐ utworzono Gen bank ‐ publicznie dostępną bazę danych sekwencji DNA Andre Marion i Samuel Eletr utworzyli firmę Applied Biosystems, która w tym okresie zdominowała automatyczne sekwencjonowanie DNA 19821982 ‐ Akiyoshi Wada zbudował roboty sekwencyjne dla firmy Hitachi. 19841984 ‐ kompletna sekwencja wirusa Epstein‐Barr, 170 kb. 1997 1997 ‐ zsekwencjonowano 5Mb genom bakterii. 20012001, 15 lutego ‐ Konsorcjum HUGO opublikowało sekwencję genomu ludzkiego w N tNature. 2001,2001, 16 lutego ‐ firma Celera Genomics opublikowała sekwencję genomu ludzkiego w Science.

MetodaMetoda MaxamaMaxama GilbertaGilberta (1977)(1977)Metoda Metoda MaxamaMaxama‐‐GilbertaGilberta (1977)(1977)
• Polega na użyciu związków chemicznych do specyficznegoPolega na użyciu związków chemicznych do specyficznego rozczepienia DNA
• Przeprowadza się 4 niezależne reakcje, w których wykorzystuje się 4 różne odczynniki specyficzne dla poszczególnych zasad:
1. reakcja „G” – DMS piperydyna
2 reakcja G+C” kwas mrówkowy piperydyna2. reakcja „G+C ‐ kwas mrówkowy, piperydyna
3. reakcja „T+C” – hydrazyna, piperydyna
4. reakcja „C” – hydrazyna + NaCl, piperydyna j „ y y , p p y y

Metoda Sangerametoda terminacji wydłużania łańcuchametoda terminacji wydłużania łańcucha
(tzw. metoda dideoksy)
Wykorzystuje polimerazę DNA, która ma zdolność do syntezy wiernej, komplementarnej kopii jednoniciowego DNA matrycowego
oraz
używa 2’3’dideoksynukleotydów jako substratówużywa 2 3 dideoksynukleotydów jako substratów

Enzymy używane w Enzymy używane w sekwencjonowaniusekwencjonowaniupowinny charakteryzować się: powinny charakteryzować się:
1. wysoką procesywnością
2. brakiem aktywności egzonukleazy 5’→3’2. brakiem aktywności egzonukleazy 5 →3
3. Brakiem aktywności egzonukleazy 3’→5’

Enzymem wykorzystywanym do reakcji wydłużania startera początkowo był fragment polimerazy DNA Klenowa (brak aktywności egzonukleolitycznej5’ 3’)Fragment Klenowa polimerazy z E.coli zastąpiono naturalną lub modyfikowaną polimerazą DNA z fagaT7T7wprowadzenie dideoksynukleotydów jest mniej zakłócone przez lokalne sekwencje nukleotydów,zakłócone przez lokalne sekwencje nukleotydów, prążki o wyrównanej intensywności

Z t i li t fil j b kt ii• Zastosowanie polimerazy z termofilnej bakterii Thermus aquaticus (Taq DNA polimeraza)
• ‐ pozwala na prowadzenie reakcji terminacji• ‐ pozwala na prowadzenie reakcji terminacjiłańcucha w temperaturze 65‐70°C, co minimalizuje artefakty spowodowane II‐go rzędową strukturą DNA
• Tabor i Richardson w 1995r zamienili resztę f l l i T DNA li tfenyloalaniny Taq DNA polimerazy na resztę tyrozyny (termostabilny enzym sekwencyjny nie rozróżnia dideoksynukleotydów odrozróżnia dideoksynukleotydów od deoksynukleotydów).

MatrycaMatryca w postaciw postaci ssss DNADNAMatryca Matryca w postaci w postaci ssss DNADNA
• DNA sklonowany na wektorze plazmidowym
• DNA sklonowany na wektorze M13wektorze M13DNA sklonowany na wektorze M13wektorze M13
• DNA sklonowany na fagmidzie
• Zastosowanie PCRPCR do otrzymania jednoniciowego DNAj g

produkty PCR do produkty PCR do sekwencjonowaniasekwencjonowania
Matryca DNA
B
DNAPCR z jednym biotynylowanymstarterem
BBiotynylowany produkt PCR
B
Wychwycenie przez streptawidynę sprzężoną z
streptawidyna
B P M Pp y ę p ę ą
kuleczkami magnetycznymiParamagnetic
particle
B P M PS k j i
Denaturacja alkaliczna
Sekwencjonowanieobu nici DNA

Starter wyznacza Starter wyznacza sekwencjonowanysekwencjonowany region na matrycowym DNAregion na matrycowym DNA
1.1. Starter Starter uniwersalny uniwersalny (do amplifikacji fr. o dł. 750 pz)
DNA przeznaczone do sekwencjonowania może być wklonowany w jedno ze zgrupowanych miejsc klonowania w
i i l kt ó M13 ii
DNA wektora
regionie lac wektorów M13 serii mpco pozwala na używanie ciągle tego samego startera, ułatwia również izolację pojedynczych nici DNA do sekwencjonowania
3’ 5’DNA wektora DNA wstawki
sekwencjonowania
2 Startery wewnętrzne2 Startery wewnętrzne2. Startery wewnętrzne 2. Startery wewnętrzne
3’ 5’

DetekcjaDetekcjaDetekcja Detekcja Ż• Żel sekwencyjny 6‐20%;
• 7 molarny mocznik (zapobiega tworzeniu się struktur II d h) d ż d i l ść d i ł j d i i hrzędowych): duża rozdzielczość, rozdział jednoniciowych
(zdenaturowanych) fragmentów DNA
• elektroforeza przebiega przy natężeniu prądu wystarczającym• elektroforeza przebiega przy natężeniu prądu wystarczającym do podgrzania żelu do ok.70°C, co przeciwdziała powstawaniu II rz. struktur.
• Autoradiografia
• Nieizotopowe metody identyfikacji: chemiluminescencyjne,Nieizotopowe metody identyfikacji: chemiluminescencyjne, chromogeniczne, fluorogeniczne
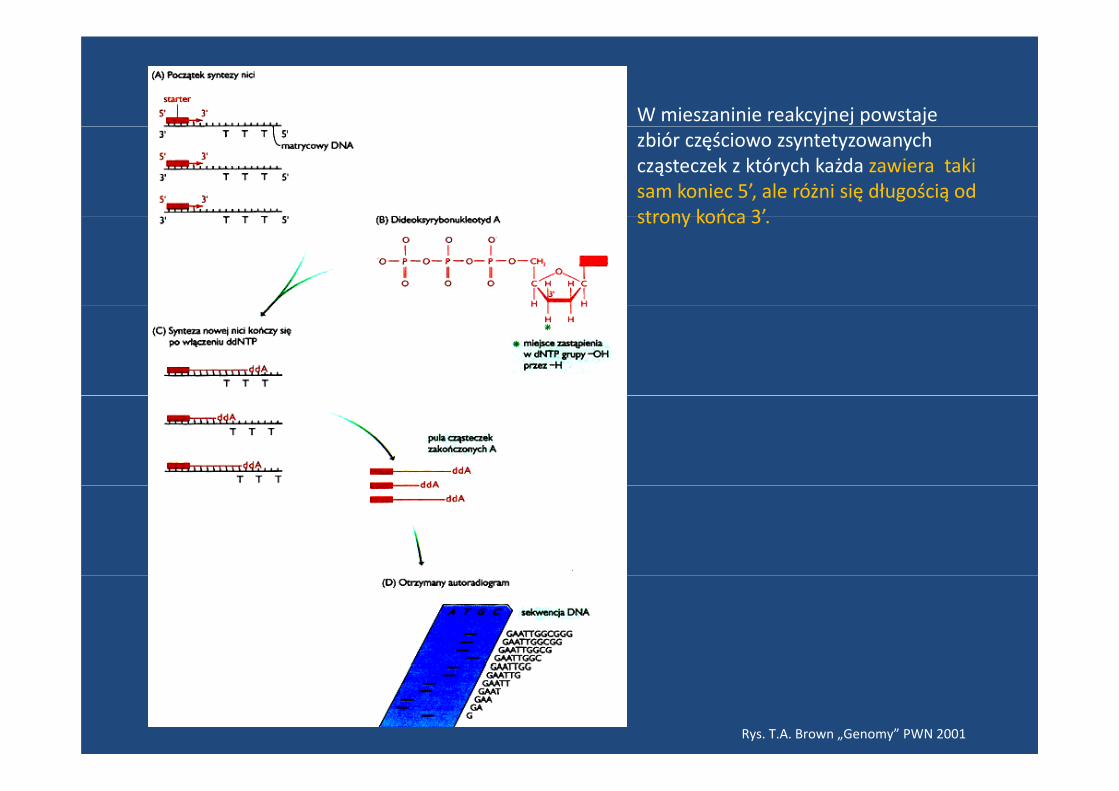
W mieszaninie reakcyjnej powstaje zbiór częściowo zsyntetyzowanych cząsteczek z których każda zawiera taki sam koniec 5’, ale różni się długością od strony końca 3’strony końca 3 .
Rys. T.A. Brown „Genomy” PWN 2001

Automatyczne Automatyczne sekwencjonowaniesekwencjonowanie z wykorzystaniem z wykorzystaniem dideoksyrybonukleotydówdideoksyrybonukleotydów znakowanych fluorescencyjnieznakowanych fluorescencyjniedideoksyrybonukleotydówdideoksyrybonukleotydów znakowanych fluorescencyjnie znakowanych fluorescencyjnie
(1987 (1987 rr ProberProber i i wspwsp.).)
Wydruk z sekwenatora, sekwencja w postaci serii szczytów, z których każdy p y , y yodpowiada innemu nukleotydowi
Rys. T.A. Brown „Genomy” PWN 2001

NoweNowe sekwenatorysekwenatoryNowe Nowe sekwenatorysekwenatory
• Rozdział w kapilarach, a nie w żelu poliakryloamidowym
• 96 kanałów – 96 sekwencji (2 godziny); 1000 sekwencjowań w i d bciągu doby
S k j iS k j i kli S ikli S i 19921992•• SekwencjonowanieSekwencjonowanie cykliczne Sears i cykliczne Sears i wspwsp. 1992. 1992
• Wyróżnia się dwiema cechami, których nie ma metoda tradycyjna:tradycyjna:
materiałem wyjściowym jest dsDNA (np. produkty PCR)
i k i ś i kl i d k j inie ma konieczności klonowania przed sekwencjonowaniem
wystarcza niewielka ilość DNA

Produkt PCR + wyznakowany starter +dNTP + polimeraza TaqPreparatywny PCR –namnożenie wybranego f ż i fi h
Mieszanina reakcyjna
fragmentu genu przy użyciu pary specyficznych starterów
d DNAyj
Zamiast startera barwnikami fl j i ż k ćddATP1 starter
dsDNA
ddATPddATP ddTTPddTTP ddCTPddCTP ddGTPddGTP
asymetryczny PCR (liniowy)
fluorescencyjnymi można wyznakować dideoksynukleotydy
ddATP1 starter
ddA
Sekwencjonowanie cykliczne‐denaturacja
ł i 20 45
asymetryczny PCR (liniowy)ddA
ddA
ddA‐przyłączanie startera‐wydłużanie startera‐terminacja
20‐45 cykli
Denaturacja produktów
Elektroforeza i odczyt sekwencjiPorównywanie z sekwencjąz dostępnych baz danych takich jak GenBankGenBank i EMBLi EMBL.

PirosekwencjonowaniePirosekwencjonowaniePirosekwencjonowaniePirosekwencjonowanie
ATACCTATAAC------
GG A ------
1 ATACCTATAAC------
GG A ------
1
Polimeraza DNA
+ dNTP
PPi +APS 2Polimeraza DNA
+ dNTP
PPi +APS 2+ dNTP sulfurylaza
ATP
+ 34 apyraza
+ dNTP sulfurylaza
ATP
+ 34 apyraza +lucyferaza
34
Lucyferyna oxylucyferyna
apyraza +lucyferaza
34
Lucyferyna oxylucyferyna
apyraza
dNDPs + dNMPs + fosforan
i
ADP + AMP + fosforan
dNDPs + dNMPs + fosforan
i
ADP + AMP + fosforanEtap 1: w obecności enzymów (polimerazy DNA, apyrazy, lucyferazy, sulfurylazy ATP) i substratów (adenozyno 5’-fosfosiarczanu APS i D-lucyferyny), sekwencyjny starter jest przyłączany do matrycy DNA uzyskanej w wyniku PCR i wydłużany przez polimerazę DNA. Podczas inkorporacji każdego z nukleotydów, nieorganiczny pirofosforan PPi jest usuwany w równomolarnych ilościach. Etap 2: w obecność APS, sulfurylaza ATP odwraca PPi do ATP. Etap 3: wytworzone ATP na etapie 2 jest wykorzystywane przez lucyferazę do katalizy konwersji lucyferyny do oxylucyferyny, która emituje światło w ilości proporcjonalnej do ilości zużytego ATP. Etap 4. Podczas reakcji polimeryzacji, apyraza degraduje niewintegrowane dNTP. Apyraza również degraduje nadmiar ATP wytworzonych z wyniku działania sulfurylazy ATP.

Pi k j iPi k j iPirosekwencjonowaniePirosekwencjonowaniestarter
Matrycowy DNA
starter
dATP degradacja
dTTP degradacja
dGTP Chemiluminescencja = G
dCTP degradacja
dATPChemiluminescencja =
GA
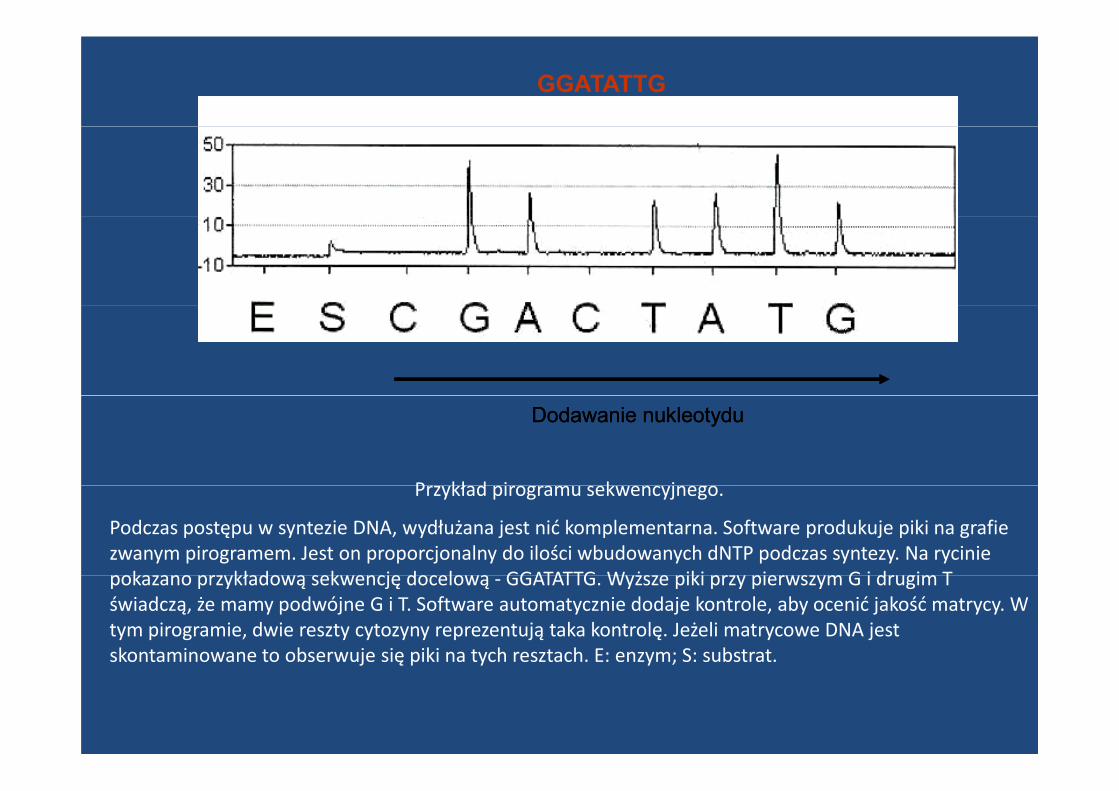
GGATATTG
Dodawanie nukleotyduDodawanie nukleotydu
P kł d i k jPrzykład pirogramu sekwencyjnego.
Podczas postępu w syntezie DNA, wydłużana jest nić komplementarna. Software produkuje piki na grafie zwanym pirogramem. Jest on proporcjonalny do ilości wbudowanych dNTP podczas syntezy. Na rycinie poka ano pr kłado ą sek encję docelo ą GGATATTG W żs e piki pr pier s m G i dr gim Tpokazano przykładową sekwencję docelową ‐ GGATATTG. Wyższe piki przy pierwszym G i drugim T świadczą, że mamy podwójne G i T. Software automatycznie dodaje kontrole, aby ocenić jakość matrycy. W tym pirogramie, dwie reszty cytozyny reprezentują taka kontrolę. Jeżeli matrycowe DNA jest skontaminowane to obserwuje się piki na tych resztach. E: enzym; S: substrat. j ę p y y

Kolekcja izolatów
Izolacja DNA
Amplifikacja PCR fragmentów genów 400‐500 pz
Sekwencjonowanie (dideoxy)
Kolekcja danych
Określenie sekwencji nukleotydowej
Zgromadzenie danych sekwencji nukleotydowychAnaliza
Profile alleliczne i identyfikacja typu sekwencji (ST)
Przydział ST do kompleksu klonalnego
Nowe sekwencjealleli
i weryfikacja ST
danych
Analiza S k jiBadanie populacji Badania epidemiologiczne Sekwencji
ML
Diagram badawczy MLSTDiagram badawczy MLST

• Sekwencje nukleotydowe dla każdego z fragmentów genu są traktowane
jako allele, tworząc profil alleliczny, czy typ sekwencji ST (ang. Sequence
Type) dla badanego szczepu.
• Izolaty o tym samym profilu allelicznym są zaliczane do tej samej grupy y y y p y ą j j g py
klonalnej.
• Wariantem metody MLST jest sekwencjonowanie wielu regionów między• Wariantem metody MLST jest sekwencjonowanie wielu regionów między‐
genowych. Pozwala to na osiągnięcie wyższego stopnia zróżnicowania niż
dk k ó d b lw przypadku sekwencjonowania genów podstawowego metabolizmu
komórkowego.
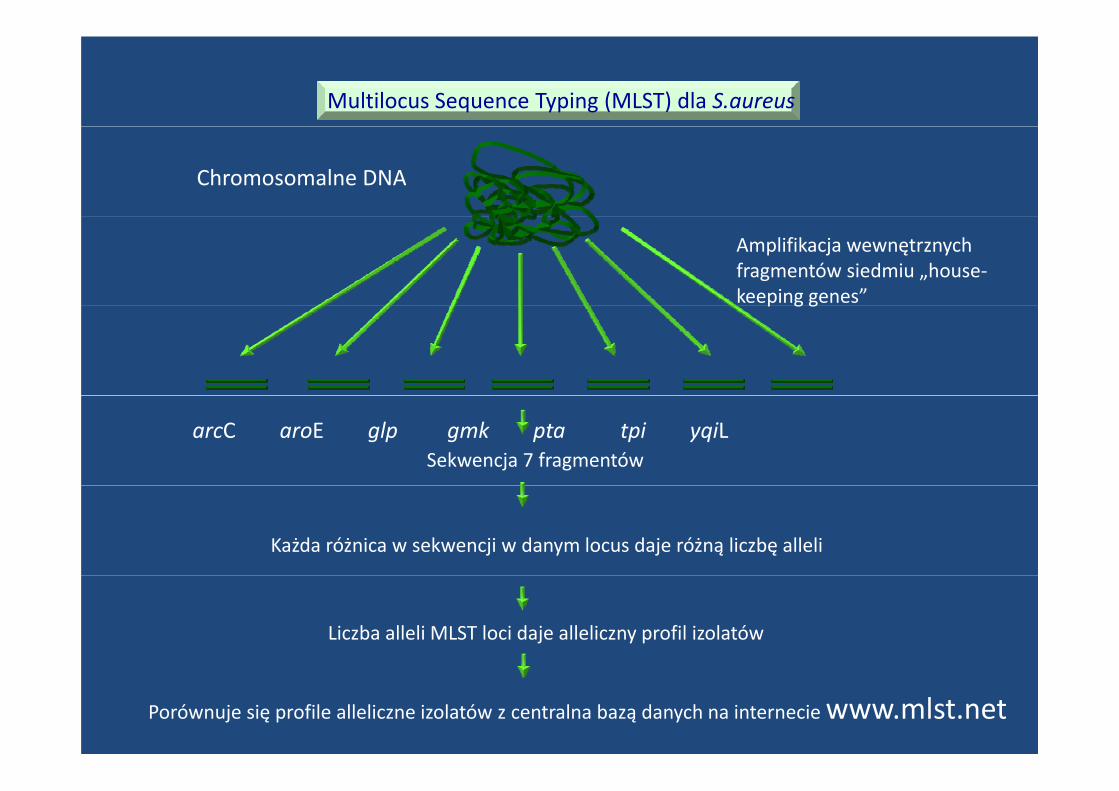
Multilocus Sequence Typing (MLST) dla S.aureus
Chromosomalne DNA
Amplifikacja wewnętrznychfragmentów siedmiu „house‐keeping genes”p g g
Sekwencja 7 fragmentówarcC aroE glp gmk pta tpi yqiL
Każda różnica w sekwencji w danym locus daje różną liczbę alleli
Liczba alleli MLST loci daje alleliczny profil izolatów
Porównuje się profile alleliczne izolatów z centralna bazą danych na internecie www.mlst.net

MLSTMLSTMLSTMLSTMaiden M C J, Bygraves J A, Feil E, Morelli G, Russell J E, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant D A, Feavers I M, Achtman M and Spratt B G (1998) Multilocus sequence typing: A portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. USA, 95: 3140‐3145
Urwin R, Maiden MC. (2003) Multi‐locus sequence typing: a tool for global id i l T d Mi bi l O t 11(10) 479 87epidemiology. Trends Microbiol. Oct;11(10):479‐87.
David M. Aanensen* and Brian G. Spratt (2005) The multilocus sequence typing network: mlst net Nucleic Acids Research Vol 33typing network: mlst.net Nucleic Acids Research, Vol. 33,
Maiden MC. Multilocus sequence typing of bacteria.Annu Rev Microbiol. 2006;60:561‐882006;60:561‐88


















