
本論文-製本版ousar.lib.okayama-u.ac.jp/files/public/5/54370/...2 近いin...
69
博士論文 経口製剤投与後の薬物血中濃度の 変動要因の解析に関する研究 平成 28 年 3 月 杉原 正久 岡山大学大学院 医歯薬学総合研究科 博士後期課程創薬生命科学専攻
Transcript of 本論文-製本版ousar.lib.okayama-u.ac.jp/files/public/5/54370/...2 近いin...

博士論文
経口製剤投与後の薬物血中濃度の
変動要因の解析に関する研究
平成 28 年 3 月
杉原 正久
岡山大学大学院
医歯薬学総合研究科
博士後期課程創薬生命科学専攻


目次
総論の部 ..................................................................................................... 1
緒言 ........................................................................................................... 1
第 1 章 経口投与製剤のヒト BE 試験における変動要因 ..................................... 4
1-1 データベースの作成 ......................................................................... 5
1-2 個体内変動と個体間変動の相関 .......................................................... 8
1-3 薬物の膜透過性、溶解性と変動の相関 ............................................... 13
1-4 薬物の膜透過性、溶解性データを用いた BE 試験の被験者数の推定と
その妥当性の検証 ....................................................................... 16
1-5 代謝酵素の影響 ............................................................................. 17
1-6 考察 ............................................................................................ 21
第 2 章 胃排出速度の変動に及ぼす製剤の影響 ............................................... 25
2-1 腸溶性顆粒の胃排出速度に及ぼす粒子径の影響の検討 .......................... 25
2-1-1 セチリジン塩酸塩腸溶性顆粒の調製 .............................................. 26
2-1-2 イヌ経口投与試験 ...................................................................... 29
2-2 2 種のランソプラゾール製剤のヒト BE 試験における吸収の変動に関する検
証 ............................................................................................... 33
2-3 考察 ............................................................................................ 39
結論 ......................................................................................................... 42
謝辞 ......................................................................................................... 44
実験の部 .................................................................................................. 45
第 1 章 実験の部 .............................................................................. 45
第 2 章 実験の部 .............................................................................. 54

引用文献 .................................................................................................. 61

1
総論の部
緒言
医薬品の開発過程では、薬物投与後の血中濃度の測定およびその解析が、様々なステ
ージで繰り返し実施される。開発の初期ステージでは、主にマウスやラットなどの小動
物における血中濃度を測定することによって、経口吸収性、体内動態、薬効薬理(PK/PD)
等の薬物の基本的な特性が評価される。開発の中後期のステージ、特に経口剤の製剤開
発の過程では、イヌなどの大動物を用いた試験により、ヒトにおける吸収性、体内動態
を推定するとともに、臨床製剤化のための製剤処方が決定される。さらに、臨床試験で
は、健常な被験者あるいは患者における血中濃度の測定が実施され、ヒトにおける体内
動態を確認するとともに、有効性、安全性が検証される。一方、臨床ステージの後期以
降では、プロトタイプ製剤から市販製剤への処方変更、剤形変更や、上市後の剤形追加、
またジェネリック医薬品の開発においては、先発医薬品との生物学的同等性
(Bioequivalence)を検証する目的で、ヒトにおける同等性試験(BE 試験)が実施さ
れる。
BE 試験は、同一有効成分を同量含有する異なる 2 つの製剤の治療学的同等
性を保証するために行う様々な試験の総称であり、一般的には被験者に投与し
た 後 の 血 中 の 有 効 成 分 濃 度 を 測 定 す る こ と に よ り 、 生 物 学 的 利 用 率
(Bioavailability)を比較する試験のことを指す。BE 試験は、通常、対照製
剤と試験製剤を交互に投与する 2 剤 2 期のクロスオーバーで実施され、血中
濃度-時間曲線下面積(AUC)と最高血中濃度(Cmax)を評価パラメータと
して、その対数値の平均値の差の 90%信頼区間が log(0.80)~ log(1.25)であれ
ば同等と判定される 1~3)。 123
本邦においては、経口剤の BE 試験の実施には、 in vitro での溶出試験によ
って試験製剤と対照製剤の溶出挙動を、数種類の試験液と試験条件下で確認す
ることが必須となっている 3)。しかし、これらの試験で溶出挙動に類似性が得
られたとしても、ヒト BE 試験において必ずしも同等となるとは限らない。そ
の理由として、次の二つのケースが考えられる。
一つは、製剤自体が非同等の場合である。これは、in vitro での薬物の溶出
性とヒト消化管内での in vivo での溶出性が異なるために、 in vitro 溶出試験
では二つの製剤の真の違いを検出できない場合であり、現在、より in vivo に

2
近い in vitro 試験系の構築を目指して、多くの研究者が in vitro-in vivo
correlation(IVIVC)の検証に取り組んでいる 4~6)。もう一つは、経口投与後
の薬物の血中濃度推移の個体内変動が大きく、製剤の同等性が検証できない場
合である。BE 試験においては、個体間変動と個体内変動の 2 つのばらつきが
観察される。クロスオーバー法を用いた臨床試験では、被験者間の変動(個体
間変動)はキャンセルアウトされるため、同等性の判定には影響を及ぼさない
のに対し、被験者内の変動(個体内変動)の大きい薬物では、AUC や Cmax
の信頼区間の幅が大きくなり BE の検証が難しくなる。 456
血中濃度推移に大きな個体内変動が認められる薬物の BE の検証において、
欧米では reference-scaled average BE (RSABE) アプローチ 7)が用いられて
いる。これは、対照製剤を繰り返し 2 回投与する 2 剤 3 期(partial replicate
design)や、2 剤 4 期( full replicate design)のプロトコールで試験を行う
ことにより、その個体内変動を求め、その大きさからクライテリアを広げる方
法である。一方、日本では信頼区間の幅が広い場合でも、平均値の差が
log(0.90)~ log(1.11)の範囲内であれば同等と判定することが可能であるもの
の、これを適用するためには数種の溶出試験条件において二つの製剤の溶出性
が類似していることを証明する必要があり、より緻密な製剤設計とその検証の
ための試験が要求される。
経口剤の BE 試験において、同じ被験者であっても、試験日や投与のタイミ
ングの違いによって薬物の血中濃度推移に変動が生じる理由として、消化管内
の pH、水分量、内容物あるいは消化管運動性など、被験者の消化管の生理的
状態の変化が、薬物の溶解や吸収に影響を及ぼすことが考えられる。したがっ
て、その様な生理的状態の影響を受けやすい薬物あるいは製剤ほど、大きな個
体内変動が生じることになる。
経口投与された薬物の吸収挙動を考える上で、それぞれの薬物の物理化学的
特性から、吸収における律速段階を把握しておくことは極めて重要である。
Amidon らは、医薬品の経口吸収性を簡単に整理・理解するための指標として
biopharmaceutics classification system(BCS)を提唱した(Figure 1)8)。BCS では
薬物を溶解性と膜透過性によって Class 1 から Class 4 の 4 種類に分類し、それぞれの
経口吸収における律速段階を明らかにしている。溶解性と膜透過性がともに高い Class
1 に分類される薬物は、経口吸収に問題はなく、投与後小腸からほぼ完全に吸収される。
したがって、Class 1 薬物の吸収における律速段階は胃から小腸への移行過程となる。

3
膜透過性が高いものの溶解性が低い Class 2 の薬物では、消化管内での溶解過程が吸収
の律速となり、小腸滞留時間内に溶解されなかった薬物は未吸収となる。一方、溶解性
は高いものの膜透過性の低い Class 3 の薬物の吸収は膜透過過程によって律速され、吸
収率は膜透過性に依存する。膜透過性と溶解性ともに低い Class 4 の薬物では、両過程
によって吸収が律速されるために、吸収率の改善は困難であり経口剤には不適な薬物と
判断される。
本研究では、BCS 理論に基づいて、大きな個体内変動を示す薬物の特性を明らかに
することを目的として、まず、実際のヒト BE 試験の結果から、薬物血中濃度推移にお
ける個体内および個体間変動と薬物の溶解性、膜透過性との関係を網羅的に解析した。
さらに、薬物の消化管からの吸収における個体内変動に及ぼす製剤的な要因として粒子
径の違いに着目し、粒子径の違いによって胃排出速度がどのように変動するのかに関す
る検討を行った。以下、得られた結果を 2 章にわたり論述する。
Figure 1. Biopharmaceutics classification system8)
Class 1
溶解性、膜透過性には問題なし⇒ 胃排出過程が律速
Class 2
溶解性に問題
⇒ 消化管内での溶解過程が律速
Class 3
膜透過性に問題
⇒ 消化管膜透過過程が律速
Class 4
溶解性、膜透過性ともに問題⇒ 吸収は低く改善は困難
膜透
過性
高
低
溶解性高 低

4
第1章 経口投与製剤のヒト BE 試験における変動要因
ジェネリック医薬品が認可されるためには、新たに製造した製剤(試験製剤)
が市販されている先発製剤(標準製剤)と生物学的に同等であることを、ヒト
を用いた臨床試験によって証明する必要がある。厚生労働省から公示された
「後発医薬品の生物学的同等性試験のガイドライン」 3)によれば、ヒト BE 試
験に用いる被験者の例数として「同等性を判定するのに十分な例数で試験を行
う.例数が不足したために同等性が示せない場合には,本試験と同じ方法によ
り例数追加試験を 1 回行うことができる.追加試験は本試験の例数の半分以
上の例数で行う」とされている。したがって、ヒト BE 試験の効率化を図り、
必要以上の被験者に対して試験を実施することを避けるために、試験計画を立
案する段階で、対象となる製剤の BE を証明するために必要な試験規模(被験
者の例数)を予め予測することは極めて重要である。
製剤間に差が無い場合、BE を証明するために必要な例数は、投与後の薬物
血中濃度の個体内変動の大きさに依存する。当然、対象となる薬物あるいは製
剤の生体内挙動の個体内変動が大きくなるほど、製剤間の BE の検証は困難と
なり、より多くの例数が必要となる。Yamashita と Tachiki 9)は、経口剤の
BE 試験で非同等と判定されるリスクについての要因解析を行い、BCS Class
1 と Class 3 に分類される薬物で、投与後の AUC/dose 比が 18 × 10-6 h/mL 以
上となる場合には、概ね 24 例以下の被験者による同等性検証が可能であり、
大きなリスクはないことを示した。逆に、AUC/dose 比が 18 × 10-6 h/mL 以下
となる薬物では、経口投与後の血中濃度の個体内変動が大きくなり必要例数が
増える、つまり、BE の検証が難しくなることを報告している。これは Class1
薬物の場合はクリアランスが大きくなることと、Class 3 薬物の場合は膜透過
性が悪くなることによる吸収率の低下が原因であると考察している。また、
Sakuma ら 10)は異なる企業間の製剤での臨床試験結果を比較し、Class 1 と
Class 3 の薬物では製剤処方の違いに関わらず同様な結果が得られることを報
告している。一方、BCS で Class 2 または Class 4 に分類される薬物について
は、AUC/dose 比と BE 試験の必要例数間には明確な関連は認められておらず、
溶解性の低い薬物では消化管内での製剤からの溶出が以後の吸収に大きく影
響するため、血中プロファイルのばらつきには薬物自体の特性だけでなく、製
剤処方も影響している可能性が指摘されている。

5
この様に、経口剤のヒト BE 試験における個体内変動の要因として、対象となる薬物
の物理化学的および動態的な特性と、製剤的な特性の二つのファクターが考えられるも
のの、BCS class 2 に分類される難溶解性薬物を含めて、個体内変動の原因と
なる個々の要因に関する定量的な評価は行われていない。本章においては、ま
ず、薬物自体の要因についての詳細な解析を行い、得られた結果に基づいて
種々薬物の個体内変動を予測することが可能であるかどうかを検証すること
を目的として、沢井製薬株式会社で行った BE 試験のデータを基に、以下の様
な検討を行った。
1-1 データベースの作成
沢井製薬株式会社で行った 113 の BE 試験(速放性製剤、74 化合物)について、薬
物の物理化学的性質、薬物動態特性およびヒトBE試験の結果から経口投与後のAUC、
Cmax、Tmax(最高血中濃度到達時間)、MRT(平均滞留時間)に関する情報を入力
し、データベースを作成した(実験の部 Tables 7, 8)。
剤形別の内訳は、素錠が 31 製剤、フィルムコーティング錠 67 製剤、口腔内崩壊錠 5
製剤、カプセル剤 8 製剤、顆粒剤とドライシロップがそれぞれ 1 製剤であった(Figure
2)。薬効別では、抗うつ剤などの精神神経用薬、血圧降下剤、高脂血症薬、
糖尿病薬などの生活習慣病薬、抗アレルギー薬、抗菌薬など広範囲の薬物が含
まれていた。

6
データベースにおける各薬物の BCS Class は、溶解性の指標として次式に
従って求めた Dose Number(Do)を、膜透過性の指標として Drug Delivery
Foundation で公開されている BCS データベース 11)に記載されている cLogP
を用いて各クラスに割付けた。
Do/
(Eq. 1)
Eq. 1 において、Dose は投与量(mg)、Cs は水に対する溶解度(mg/mL)、
Vo は製剤投与時の飲水量(mL)である。薬物の Dose として、アメリカ食品
医薬品局(Food and Drug Administration; FDA) 12 )および欧州医薬品庁
(European Medicines Agency; EMA)2)によって公示された BCS ガイドライ
ンでは、その薬物の最大投与量を使うこととされているが、今回は含量違い製
剤間の差も検証するため、実際に BE 試験を行った製剤に含有されている薬物
量を用いた。また、Vo としては日本のガイドラインで推奨されている飲水量
である 150mL を用いた(FDA, EMA のガイドラインでは 250mL)。水に対す
Figure 2. The breakdown of pharmaceutics of 113 oral drug products.
UCT : uncoated tablet, FCT : film coated tablet, ODT : oral
disintegrating tablet, CAP : capsule, GR : granule, DS : dry syrup
UCT, 31
FCT, 67
ODT, 5CAP, 8
GR, 1 DS, 1

7
る溶解度 Cs は、ebastine、 itraconazole 以外は沢井製薬で測定した値を用い
た 。 ebastine 、 itraconazole に つ い て は ADMET Predictor version 7.2
(Simulation Plus, Inc., Lancaster, CA, USA)で計算した値を用いた。なお、
溶解度が 1000 mg/mL 以上の場合は>1000 と表記し、1000 mg/mL として計
算した。
今回の解析に用いた 74 化合物 113 製剤について、Do と cLogP の関係を
Figure 2 に示した。Kasim らは、溶解性のクライテリアを Do=1、膜透過性の
クライテリアを cLogP=1.5 とすることによって、薬物の BCS クラスを簡便
に判別できることを示している 13)。Figure 3 においても、Do=1、cLogP=1.5
をクライテリアとして BCS クラス分類を行った結果、55%の製剤が BCS
Class 1 に分類され、24%が Class 2、17%が Class 3 に分類されたが、Class
4 に分類されたものは 4%と極めて少なかった。これは Class 4 薬物の経口剤
としての開発の難しさを表していると考えられる。また、本研究では、それぞ
れの製剤に含有される薬物量を用いて Do を算出したため、cefcapene pivoxil
と cefdinir については、低含量製剤が Class 3、高含量製剤が Class 4 に分類
され、製剤含量によってクラスが異なる結果となった。

8
1-2 個体内変動と個体間変動の相関
先に述べたように、BE 試験においては、個体内、個体間の 2 つの変動が観
察される。個体間変動は、性別、年齢、体格や遺伝的な違いで吸収、分布、代
謝、排泄の各過程に違いが生じ、結果として被験者間に薬物の血中濃度の違い
として表れる。一方、個体内変動は、同一の製剤を同一の被験者に投与したと
きに現れる結果の違いのことであり、被験者の体調などによる生理的条件の変
化や、日間変動等が要因と考えられる。データを解析するに当り、それぞれの
指標として、以下に示す 2 つのパラメータを用いた。
Figure 3. BCS Classification of 113 oral drug products according to dose number (Do)
and cLogP.
The point of axes intersection is Do = 1 and cLogP = 1.5.

9
個体内変動(Vintra)を表す指標として、BE 試験のパラメータの分散分析の
結果から得られる残差の変動 ANOVA CV(%)を用いた(Eq.2)。
ANOVACV % 100 exp 1 Eq. 2 2:分散分析における残差平均平方
個体間変動(Vinter)の指標としては、BE 試験における試験製剤のパラメー
タの相対標準偏差 RSD(%)用いた(Eq.3)。
相対標準偏差 RSD(%)標準偏差
平均値 Eq. 3
分散分析の残差は、総変動から薬剤と被験者の変動を差し引いたものであり、
薬剤間差については考慮されているが、厳密には同一の製剤で検討することが
必要である。本研究では、BE 試験の結果を用いたので、試験製剤と標準製剤
のばらつきの違いによる差も含まれることになる。しかし、いずれも生物学的
同等性が検証され、後発医薬品として承認されているものであるので、製剤に
違いはないと判断した。
通常、BE 試験はクロスオーバー法で行われており、個体間変動については
キャンセルアウトされるため、個体間変動の大きさは、BE 試験の検証には影
響を及ぼさない。一方、個体内変動が 25~30%以上の薬物では、90%信頼区
間で生物学的同等性を証明しようとすると、実現不可能なほどの例数で試験を
行わなければならなくなる 14)とされている。本研究では個体内変動の大きさの
クライテリアとしては 25%を用いた。
AUC、Cmax、Tmax、および MRT における個体内変動(Vintra)と個体間変動(Vinter)
の関係を示した(Figure 4A, 4B)。Vintraと Vinterの絶対値を直接比較することはでき
ないものの、AUC および Cmax に関して、Class 3 の薬物では、全薬物から計算した 0
を通る回帰直線より上側に多く分布しており、相対的に Vintraが大きくなる傾向が認め
られ、この傾向は特に AUC において顕著であった。一方、Class 1 と Class 2 薬物で
は Vintra が相対的に大きくなる薬物もあれば、Vinter が相対的に大きくなる薬物もあり、
一定の関係性は認められなかった。Class 4 薬物は、膜透過性が低く、溶解性も低いこ
とから、個体内、個体間変動とも大きくなることが予想されたが、本研究における結果

10
はいずれについても、大きな変動は観察されなかった。
Tmax および MRT は、ともに時間に関するパラメータである。Tmax では Vintraと
Vinter の広がりがその他のパラメータと異なり、両パラメータが相関する傾向が認めら
れた。BE 試験において、Tmax は薬物の血中濃度が最も高かったサンプリング時間を
表しており、その変動の幅はサンプリングポイントの幅に制限されることから、個体内
と個体間変動に大きな差が出なかったと考えられる。また、MRT にも Vintra と Vinter
の間に特徴的な関係は認められなかった。経口投与後の MRT は薬物の吸収と血中から
の消失にかかる平均時間の和として表されることから、吸収過程における変動を解析す
るためのパラメータとしては不適当と考えられた。
以上、AUC と Cmax において Vintraと Vinterの間に同様な関係が認められた
ものの、Cmax は薬物の吸収率および吸収速度に依存するパラメータであり、
その変動には BE 試験実施時の採血時間の設定の仕方などの人為的な要因も
影響を及ぼす可能性があることから、以下の検討では AUC を用いた解析を行
うこととした

11
Figure 4A. Correlation of intrasubject variability (Vintra) and intersubject variability
(Vinter) in AUC and Cmax.
The dotted line is a regression analysis of the 113 formulations.

12
Figure 4B. Correlation of intrasubject variability (Vintra) and intersubject variability
(Vinter) in Tmax and MRT.
The dotted line is a regression analysis of the 113 formulations.

13
1-3 薬物の膜透過性、溶解性と変動の相関
Figure 4 において Class 3 薬物では相対的に Vintra が大きくなる傾向が認め
られた。Class 3 薬物の消化管からの吸収は膜透過によって律速されることか
ら、ヒト消化管膜に対する透過性(Peff)と AUC の変動の関係を検討した。
今回の検討では Peff として、ADMET Predictor 7.2 (Simulations Plus Inc.)
によって各薬物の物理化学的性質に基づき算出した値を用いた。
Figure 5 に、すべての製剤に関して Peff と Vintra (a)および Vinter (b)の関係
を示した。全体としては Peff と AUC の変動の間に明確な相関は認められなか
ったものの、Class 3 薬物に限った場合には個体内変動で r=-0.775(p<0.001)、
個体間変動で r=-0.715(p<0.001)と有意な相関性が認められた。これは、
薬物の膜透過性が低くなるほど、経口投与後の吸収に大きな変動が観察される
リスクが高まることを示す結果であり、膜透過が著しく低い薬物では、BE の
検証により多くの被験者が必要になるものと考えられた。
Figure 5. Effect of drug permeability (Peff) on intra- and intersubject variability in
AUC.
Figure shows the relation of Peff to Vintra (a) or Vinter (b), respectively. The
green dotted line represents a regression line only for the Class 3 drugs.

14
次に、薬物の溶解性と吸収おける個体内・個体間変動の関係について解析を
試みた。Figure 6 に Do と Vintra (a)および Vinter (b)の関係を示した。この場
合も、全体としては Do と AUC の変動の間に相関は認められなかったものの、
Class 2 薬物の場合には、有意では無かったものの、Do が大きくなるほど個
体内変動が大きくなる傾向が示された。一方、個体間変動にはその様な傾向は
認められなかった。
BCS Class 2 に分類される難溶解性薬物では、固形製剤として経口投与した
後の吸収が、薬物の消化管内での溶解速度もしくは溶解度によって律速される
ことが知られている 15)。溶解速度律速とは、消化管内での薬物の溶解速度が膜
透過速度よりも低いために、消化管内滞留時間内に吸収が完了しないケースで、
この時、薬物の吸収率は投与量に依存せず一定となり、吸収に飽和は観察され
ない。また、微紛化などの製剤学的な工夫によって溶解速度を上昇させること
により、比較的容易に吸収率の改善が可能である 16)。一方、溶解度律速の場合
には、消化管内での薬物の溶解度が低いために投与された薬物が完全に溶解せ
ず、吸収率が低くなる。また、投与量を増加しても吸収量が一定以上には増加
しないため、吸収率に低下が観察される(吸収飽和) 17)。
吸収がいずれの過程によって律速されているかは、対象となる薬物の
Maximum absorbable dose(MAD, 最大吸収量) 18~20)を指標とすることで、
Figure 6. Effect of Do on intra- and intersubject variability in AUC. Fig shows the
relation of Do to Vintra (a) or Vinter (b), respectively.

15
判定可能である。MAD とは、薬物が飽和溶解度で消化管内に存在したときに
滞留時間内に吸収される得る最大量を表しており、次式によって算出される
(Eq.4)。 181920
MAD (Eq. 4)
ここで SA は小腸の表面積、SITT は薬物の小腸滞留時間である。MAD が薬
物の投与量(Dose)より小さい場合は、薬物の溶解度が低いために、投与さ
れたすべての薬物が一定時間(SITT)内で消化管から完全に吸収されない、
と考えられることから、吸収は溶解度律速となる。逆に、MAD が Dose より
大きいにもかかわらず吸収率が 100%とならない場合には溶解速度律速と考
えられる。
Eq.1 と Eq.4 から MAD / dose は次式のように表すことができる。
MADdose
(Eq. 5)
Eq. 5 において SA、SITT および Vo は定数であるので、MAD / Dose は Peff /
Do に比例することとなる。そこで、この Peff / Do を新しいパラメータとして
定義し、各薬物の AUC の変動との相関を検討した(Figure 7)。Figure 7 中
の縦の破線は、SA、SITT にそれぞれヒトにおける生理学的な値として用いら
れている 800cm2 20)、3.5 時間 21, 22)、Vo に BE 試験で用いた飲水量 150mL を
代入して求めた Peff / Do の値(0.149 × 10−4 cm/s)を示している。この値に
おいて MAD/Dose は 1 となる。
BCS Class 2 薬物では、個体内変動の値が 25%以上を示す薬物は、Peff / Do
が 0.149 × 10−4 cm/s より大きくなる、すなわち、MAD>Dose となる薬物で
は 12 個中 1 個であったのに対し、Peff / Do が 0.149 × 10−4 cm/s より小さくな
る、すなわち MAD<Dose となる薬物の場合、25 個中 5 個あり、吸収におけ
る個体内変動が大きくなる傾向が示された。一方、個体間変動にはその様な傾
向は認められなかった。Class 2 薬物で MAD が投与量より小さい場合、その
吸収は溶解度律速となるため、消化管内の溶液量や滞留時間などの変化に伴っ
て吸収可能な薬物量が変動した結果、経口投与後の AUC に大きな個体内変動

16
が生じたものと推察される。一方、Class 2 薬物でも、吸収が溶解速度律速で
ある場合には、MAD の変化は吸収量の変動には直接影響しないため、個体内
変動も比較的小さかったものと考えられる。
1-4 薬物の膜透過性、溶解性データを用いた BE 試験の被験者数の推定とその
妥当性の検証
これまでの解析結果を基に、薬物の膜透過性、溶解性のデータからヒト BE
試験における被験者数を推定し、沢井製薬において実施された実際の BE 試験
の情報と照らし合わせることによって、その妥当性の検証を試みた。
Dilleti らは、ANOVA CV が 25%のとき、検出力 80%を達成するのに必要な例数は、
製剤間に真の差がない場合、1 群 12 例と報告している23)。本研究において、Figure 5a
に示した Class 3 薬物の回帰直線で、個体内変動が 25%を上回ると予測される膜透過
性は 0.27 × 10−4 cm/s であった。膜透過性が 0.27 × 10−4 cm/s 程度の値を示す
Suplatast Tosilate 製剤に関する沢井製薬における実際の BE 試験では、1 群
10 例の予試験において同等性が検証できなかったため、さらに例数を増やし
て本試験を行い、同等性が検証されている。さらに、本研究で用いた薬物のう
ち、0.27 × 10−4 cm/s 以下の膜透過性を示す薬物は 4 例(5 製剤)であったが、
そのうち 3 試験は 1 群 12~14 例で試験が実施され、同等性が検証されている。
Figure 7. Effect of Peff / Do on intra- and intersubject variability in AUC. Fig shows
the relation of Peff / Do to Vintra (a) or Vinter (b), respectively.
The vertical dotted line marks a threshold of Peff / Do (0.149 × 10−4 cm/s).

17
以上の結果は、膜透過性が 0.27 × 10−4 cm/s を下回る Class 3 薬物を含む製剤
の BE 試験では、多くの場合 1 群 12 例以上の試験が必要であることを示して
いることから、本研究での解析結果の妥当性を示唆するものと考えられる。
一方、BCS Class 2 に分類される薬物で吸収が溶解度律速であった 13 化合
物、15 製剤のヒト BE 試験のうち、3 試験では 1 群 10 例の予試験では同等性
が検証されず、例数を増やした本試験によって同等性の検証が可能であった。
また、他の 6 試験においても 1 群 12~16 例で BE 試験を行うことによって、
同等性が検証されており、吸収が溶解度律速となる薬物を含む製剤では、1 群
12 例以上の試験を設定することが妥当であると考えられた。今回パラメータ
として用いた Peff / Do と Vintra の間に有意な直線関係が認められなかったため、
正確な例数設計を行うことは困難であったものの、以上の結果は、膜透過性か
らの推定と同様、本研究の解析結果の妥当性を示唆するものと考えられる。
薬物の膜透過性、溶解性に関する情報は、簡単な in vitro 試験での測定、あ
るいは in silico での推定が可能であることから、実際の BE 試験前に対象と
なる薬物の Peff および Peff / Do を算出することにより、BE 試験の効率化が可
能になるものと期待される。
1-5 代謝酵素の影響
これまでの検討において、BCS Class 2 および Class 3 の薬物について、経
口投与後の吸収に大きな個体内変動が生じる要因を明らかにしてきた。しかし、
Figure 3 では、Class 2、Class 3 の薬物だけではなく、いくつかの Class 1
薬物においても大きな個体内、個体間変動が認められている。Figure 5~7 で
明らかにした要因では、これら Class 1 薬物の変動の大きさを説明することは
できず、薬物の物理化学的性質とは直接関係のない他の要因が経口投与後の血
中濃度推移に影響している可能性が考えられる。
経口投与後の生物学的利用率(Bioavailability、BA)は消化管からの吸収
率(Fa)と、小腸での初回通過代謝の回避率(Fg)および肝臓での初回通過代
謝の回避率(Fh)の積として表わされる。Class 1 薬物は膜透過性、溶解性と
も高く、Fa はほぼ 100%となると考えられることから、経口投与後の血中濃度
推移に大きな個体内変動が見られる要因として、Fg・Fh が変動している可能
性が考えられる。そこで、その要因として薬物代謝酵素の影響について検討を
行った。

18
Table 1 に、CYP450 系の代謝酵素のうち、多くの薬物の代謝に関与するこ
とが報告されている CYP3A4 および CYP2D6 の基質となる薬物を抽出して示
した。さらに Figure 8 に、CYP3A4 および CYP2D6 の基質となる薬物の血中
AUC の Vintra と Vinter の関係を示した。CYP2D6 の基質となる薬物はほとん
どが回帰直線の下側に分布し、個体内変動に比べて、個体間変動が大きくなる
傾向を示したのに対し、CYP3A4 の基質薬物では上側に分布するものが多く、
個体内変動が大きくなる傾向が認められた。
Table 1 List of API’s related to CYP450
Compound CYP
isoform Km (µM) Ref
Alprazolam CYP3A4 575 [24] Amiodarone Hydrochloride CYP3A4 310 [24] Amlodipine Besilate CYP3A4 Atorvastatin Calcium CYP3A4 33 [24] Benidipine Hydrochloride CYP3A4 3.8 [25] Brotizolam CYP3A4 595 [26] Cabergoline CYP3A4 Cilnidipine CYP3A4 Clarithromycin CYP3A4 49 [24] Donepezil Hydrochloride CYP3A4 Doxazosin Mesilate CYP3A4 Ebastine CYP3A4 3.85 [27] Fluconazole CYP3A4 Fluvastatin Sodium CYP3A4 Fluvoxamine Maleate CYP2D6 Itraconazole CYP3A4 0.0444 [27] Lafutidine CYP3A4 &
2D6
Loratadine CYP3A4 & 2D6
Milnacipran Hydrochloride CYP3A4 Mosapride Citrate CYP3A4 Nilvadipine CYP3A4 Paroxetine Hydrochloride CYP2D6 Pranlukast CYP3A4 Pravastatin Sodium CYP3A4 Propiverine Hydrochloride CYP3A4 Quazepam CYP3A4 Risperidone CYP2D6 106 [24] Simvastatin CYP3A4 21 [24] Tamoxifen Citrate CYP3A4 &
2D6 98 [24]
Tandospirone Citrate CYP3A4 & 2D6 7.21 [28]
Terbinafine Hydrochloride CYP3A4 Ticlopidine Hydrochloride CYP3A4 Toremifene Citrate CYP3A4 124 [24] Zolpidem Tartrate CYP3A4 114 [24]

19
CYP2D6 には、種々の遺伝子多型(SNPs)が報告されており、日本人では
代謝活性が低い Poor Metabolizer の割合は 1%以下とされているものの、代謝
機能が Extensive Metabolizer の半分以下となる Intermediate Metabolizer
が 30~40%存在することが報告されており 29)、それら被験者の試験への組み
入れの分布により、個体間変動が大きくなったものと推察された。一方、
CYP3A4 については、代謝機能に大きな影響をもたらす遺伝子多型は知られ
ておらず、そのことが個体間変動が比較的小さかった要因と考えられる。
CYP3A4 の基質薬物のうち、酵素に対する親和性 Km 値が文献 24~28)で報告
されている薬物について Vintra との相関を調べたところ、有意な相関(r=-
0.717, p<0.001)が認められた(Figure 9)。この結果は、CYP3A4 に対する
高い親和性が、経口投与後の BA の個体内変動の要因であることを示す結果と
考えられる。CYP3A4 は、ヒトにおいて、肝臓 CYP 酵素の含有量の 40%、小
腸では 82%を占める最も含有量の高いアイソフォームであるため 30)、その活
性の変動は薬物の BA に大きな影響を与える。小腸の CYP3A4 は、食品中に
含まれるフラボノイド、カテキンなどで阻害されることが報告されていること
から 31~34)、その影響によって同一の被験者における代謝活性が変動し、大き
な個体内変動を生じた可能性が考えられる。 31323334
Figure 8. Intrasubject variability (Vintra) and intersubject variability (Vinter) in AUC
of CYP3A4 and CY2D6 substrate drugs.
The dotted line indicates a regression analysis of the 113 formulations.

20
Figure 9. Effect of Km value in CYP3A4 mediated metabolism on intrasubject
variability (Vintra) in AUC. The Km values were quoted from the literatures
(references 24~28) those obtained in the in vitro experiment using human
liver microsomes.

21
1-6 考察
本章では、沢井製薬で BE 試験を実施した経口速放性製剤 74(化合物 113)試験に
ついて、経口吸収における個体内変動の要因を明らかにする目的でヒト BE 試験のデー
タを解析した。その結果、経口投与製剤における血中濃度推移の高い個体内変動をまね
くリスクファクターを、以下のように同定することが出来た。
1)吸収が膜透過律速となる薬物(BCS Class 3)において、膜透過性が極めて低い
薬物
2)溶解度の低い薬物(BCS Class 2)において、吸収が溶解度によって律速される
薬物
3)すべての BCS Class の薬物で CYP3A4 代謝酵素に高い親和性を有する薬物
上記の 1)および 2)に関しては、いずれも消化管からの吸収における律速過程に起
因した要因であり、特に 1)の低膜透過性に関しては、Yamashita と Tachiki 9)の報告
を支持する結果であった。彼らの報告では、非同等となるリスクを AUC / Dose(= F /
CLtot)と言うパラメータを用いて評価しているため、膜透過性の程度とリスクとの関
係を定量的に評価することは困難であった。今回、Class 3 薬物の膜透過性と個体内変
動の間に有意な相関が認められたことから、個々の薬物の膜透過性データから、BE 試
験における血中 AUC の個体内変動の程度、さらには必要被験者数の大まかな推定が可
能となり、実際の医薬品開発への適用が期待される。
Tanaka ら35)は BCS Class 1 と Class 3 薬物の経口吸収率に及ぼす消化管管腔内水分
量の影響に関してラットを用いた検討を行い、消化管内水分量が多い場合には Class 3
薬物(atenolol)の吸収率が顕著に低下すること、および、Class 1 薬物(metoprolol)
では水分量の影響は小さいことを報告している。これは、水分量が多い場合には消化管
内の薬物濃度が低く保たれるために、膜透過性の低い Class 3 薬物は十分に吸収されな
かったためと推察されている。消化管内の水分量は、被験者の体調(下痢、便秘など)
や摂食状況など、生理的な要因で大きく変化するため 36~38)、同じ被験者でも試験日時
によって異なっている可能性がある。したがって、対象となる薬物のヒト小腸膜透過性
が低い場合には個体内変動のリスクを考慮した症例設定を行い、BE 試験の効率化を図
ることが重要と考えられる。363738
一般に、薬物の難溶解性は経口投与後の吸収におけるばらつきの原因の一つと考えら

22
れている。しかしながら、これまで、溶解度とばらつきの関係を定量的に評価した研究
結果は報告されておらず、本研究においても、Figure 6 では、溶解性(Do)は個体内
変動および個体間変動には、いずれも相関が認められなかった。同じような結果は、
Yamashita と Tachiki も報告しており 9)、AUC および Cmax の 90%信頼区間の幅と
Do の間に相関は認められてない。これらの理由の一つとして、多くの難溶解性薬物の
製剤では、製剤処方の最適化によって溶解性を改善し消化管内での溶解速度のばらつき
が抑えられていることが考えられる。
BCS Class 2 薬物の吸収は、溶解過程により律速されるが、その溶解過程には溶解度
律速と溶解速度律速の 2 種類がある。このうち、溶解速度律速の場合、微粒子化などの
比較的シンプルな製剤改良によって溶解速度を上昇させることが可能であるため、その
様な製剤として投与した場合には、吸収におけるばらつきは低減され、個体内変動も抑
えられる。したがって、Class 2 薬物であっても吸収が溶解速度律速であれば、通常、
個体内変動のリスクにはならないものと考えられる。
一方、吸収が溶解度によって律速される薬物の吸収改善を行うためには、その溶解度
自体を上昇させる必要があり、塩形成、固体分散体、共結晶、非晶質化などを利用した
過飽和製剤化39)が適用される。過飽和溶解とは、熱力学的に平衡な溶解度以上に溶質が
溶媒中に溶解する現象で、通常、溶解している溶質濃度は、一過性の過飽和溶解の後、
時間の経過とともに平衡溶解度まで低下する。一般に、過飽和状態は熱力学的に不安定
であり、経口投与された薬物の過飽和溶解は消化管内の pH、内容物、水分量などによ
る影響を受け、過飽和の程度およびその持続時間は容易に変動することが知られている。
したがって、過飽和製剤化された薬物の吸収は、これら要因によって大きな個体内変動
を示す可能性が考えられた。本研究では、MAD から導いた新たなパラメータ、Peff / Do
を導入することによって、難溶解性薬物の律速過程を簡便に判別し、溶解度律速となる
薬物では個体内変動が大きくなることを示した。さらに、これら薬物ヒト BE 試験の結
果より、溶解度律速となる薬物を含む臨床製剤の多くは BE を検証するために 12 例以
上の症例が必要であったことから、BE 試験の被験者数を決定する上で、本パラメータ
の有用性が示された。
Figure 7 において、溶解度律速薬物の Vintraと Peff / Do との間に有意な相関が得られ
なかった理由として、図中、Itraconazole と Simvastatin の 2 つの薬物が極めて大き
な Vintraを示したことが挙げられる。これら 2 種の薬物は、いずれも CYP3A4 の良好な
基質であることが報告されていることから、消化管内での溶解のばらつきに加えて、
CYP3A4 による初回通過代謝のばらつきも関与している可能性が考えられる。実際、

23
この二つの薬物を除いた残りの薬物では、両パラメータの間に有意な相関(R=0.659,
p<0.05)が得られていることから、今後、個体内変動に関する様々な要因を同定し、そ
れらを用いた重回帰分析等を行うことによって、より正確な被験者数の推定が可能にな
るものと期待される。
一方、個体間変動に関して Class 2 薬物の律速過程との関連性が認められなかった理
由として、吸収における個体間変動には、消化管の生理的条件の変化に加えて、胃酸や
胆汁酸などの分泌能、消化管(特に胃)の形状、消化管トランスポーターの発現量など、
各被験者の基本的な薬物吸収能に関する要因が複合的に関わって起きており、今回の様
に条件を絞った解析を行っても、特定の変動要因を抽出できなかったものと考えられる。
Davit らは 180 の薬物の 1010 試験に及ぶ BE 試験についてレビューし、初回通過代
謝が BA のばらつきの最も重要な要因の一つであることを明らかにしている40)。そこで
本研究においても、多くの薬物の代謝に関わっていることが知られている、CYP3A4 と
CYP2D6 の基質なる薬物について、個体内変動と個体間変動の関係を調べた結果、
CYP3A4 で代謝される薬物は Vintraが大きく、CYP2D6 で代謝される薬物は Vinterが大
きくなることを示した。さらに、CYP3A4 の基質薬物に関して、その Km値と Vintraの
間に有意な相関が認められたことから、CYP3A4 による初回通過代謝の変動が、BA に
おける大きな個体内変動の要因であることを明らかとした。CYP3A4 は、ヒト小腸
における全 CYP 酵素量の 82%を占める最も含有量の高いアイソフォームであ
る 30)ことから、特に小腸での初回通過代謝の変動が大きな要因と推察される。
通常、BE 試験を実施するにあたっては、CYP3A4 で代謝される薬物の場合、グレー
プフルーツジュース41~42
43)やセントジョーンズワート44)などの CYP3A4 を阻害あるいは
誘導することが知られている食品、薬物の使用は禁止している。しかし、被験者が休薬
期間中にこれらの食品・薬物を服用しているか否かは被験者の自己申告に頼っている。
また、緑茶などの一般食品などでも CYP3A4 の阻害が報告されている 34)ことから、
CYP3A4 に対する親和性の高い薬物の場合、これらの変動要因を排除するためには、
休薬期間も含めた入院措置など、プロトコールへの介入が今後の課題と考えられる。
今回、CYP3A4 による個体内変動の程度を推定する目的で、それぞれの薬物の Km
値を用いた。Km値は、ミクロソームなどを用いた in vitro 代謝実験で比較的簡便に測
定することが可能であるため、BE 試験前に必要な被験者数を推定する上で有用なパラ
メータと考えられる。しかしながら BA や血中濃度推移への CYP3A4 代謝の全体的な
影響を評価するためには、CYP3A4 による代謝固有クリアランス(Vmax/Km)あるいは
代謝占有率(fm)などの評価も必要と考えられる。これまで、これら値を実測した報告

24
例は少ないものの、平成 26 年に厚生労働省から公表された「医薬品開発と適正な情報
提供のための薬物相互作用ガイドライン」45)では、経口投与時のクリアランスに対する、
in vivo における代謝の寄与率(Contribution Ratio, CR)の重要性が説かれており、今
後、その様な情報がインタビューフォーム等で公開されれば、より詳細な解析が可能に
なるものと期待される。
以上、本章では、薬物自体の物理化学的および動態的特性の観点から、BE 試験にお
ける薬物血中濃度推移の個体内変動の要因を明らかとし、それら薬物特性から BE 試験
における必要例数の予測についても可能であることを示した。ただし、本研究では各要
因を分離して評価しているが、薬物によってはその個体内変動に様々な要因が関与して
いる可能性があるため、今後、複数の要因が関与する場合の評価方法の検討が必要であ
ると考えられる。次章においては、製剤的な観点から経口投与後の吸収の変動要因に関
して検討を行った結果について論述する。

25
第2章 胃排出速度の変動に及ぼす製剤の影響
第 1 章では、薬物の物理化学的および動態的特性に基づいて、消化管からの吸収に
おける変動要因に関する解析を行った。一方、固形製剤を経口投与した場合には、製剤
側の要因によって薬物の吸収に個体内変動が生じる可能性がある。通常、錠剤、カプセ
ル剤などの固形製剤は、投与後、まず消化管内で崩壊して二次粒子となる。製剤が胃内
で崩壊した場合、胃内での薬物の溶解速度が十分に速ければ、薬物は速やかに溶解し溶
液として小腸に移行する。しかしながら、薬物の胃内での溶解度が低い、あるいは二次
粒子が腸溶性である場合には、粒子のままで胃内から排出されるため、小腸への移行速
度は粒子の特性に影響される。
胃から小腸への製剤の排出は、MMC(Migrating Motor Complex)46, 47)と呼ばれる
消化管の運動サイクルに大きく依存する。MMC の生理学的意味は、その強収縮により、
胃や小腸内の食物残渣やバクテリアを洗浄し押し流すものと考えられている48)。MMC
は 4 つの Phase からなっており、Phase Ⅰと呼ばれる静止期の後、Phase Ⅱの間欠期
で不規則な弱い収縮が起きる。さらに Phase Ⅲの強収縮期において、housekeeping
wave とも呼ばれる強く頻繁な収縮によって胃内容物をすべて排出した後、Phase Ⅳの
移行期へとつながっていく。ジェネリック医薬品の開発では、ヒト BE 試験を実施する
前に in vitro において製剤の崩壊、薬物の溶出性を確認し、標準製剤と同等な製剤を調
製・選択する。しかし、これら in vitro 試験では、製剤およびその二次粒子の消化管内
移行性を評価することはできないため、胃排出速度に違いがあった場合には非同等の原
因となる。また、製剤あるいは二次粒子の粒子径や比重などによって胃での MMC の影
響の受け方が異なる 49~52)ことから、製剤によっては同じ被験者でも投与のタイミング
によって胃排出速度が大きく変化し、その後の吸収における個体内変動を引き起こす可
能性が考えられる。49505152
この様に粒子径の違いが、胃排出速度に影響を及ぼすことは知られているが、その違
いが実際の薬物の吸収および血中濃度推移の変動に及ぼす影響、特に個体内変動にどの
ような影響を与えるかは明らかではない。そこで本章では、粒子径の違いが個体内変動
に及ぼす影響を明らかにすることを目的として、種々の製剤を調製し検討を行った。
2-1 腸溶性顆粒の胃排出速度に及ぼす粒子径の影響の検討
これまで、製剤中の粒子の大きさや比重が胃排出挙動に及ぼす影響について、腸溶性
製剤などを用いて国内外で多くの研究が行われており、一般的には直径 2 mm 未満の粒

26
子については、比較的容易に胃幽門を通過し十二指腸・小腸へと排出されるため、食事
等の影響を受けにくく、吸収速度の変動も小さくなることが報告されている 49, 53, 54)。
本項では、製剤の粒子径の影響をより詳細に解析することを目的として、薬物、剤形、
添加剤等が同じで粒子径のみの違う腸溶性顆粒を調製し、ビーグル犬を用いた経口投与
試験を実施した。モデル化合物としては第 1 章で検討したデータベースから、それ自体
の吸収における個体内変動の小さい薬物として、小腸での膜透過性、溶解性が高く、か
つ代謝の影響の少ないセチリジン塩酸塩を用いた。
2-1-1 セチリジン塩酸塩腸溶性顆粒の調製
本項における腸溶性顆粒の調製の目的は、粒子径と胃排出時間の関係の評価である。
薬物自体は酸にも安定であるので、耐酸性はそれほど重要ではないため、胃から排出さ
れた腸溶性顆粒から薬物が速やかに溶出することを優先し、耐酸性の目標値は 120 分
で 10 %以下と設定した。中性状態における溶出は速い方が望ましいため、5 分で 30 %、
10 分で 80 %を目指して調製した。
Table 2. Formulations of enteric-coated granules
Component (g)enteric-coated
granules(200µm)
enteric-coatedgranules(600µm)
enteric-coatedgranules
(1200µm)
Core Particle Celphere®
CP-102 600.0 ― ―
Celphere®
CP-507 ― 600.0 ―
Ceolus®
PH-101 ― ― 590.0
L-HPC (LH-21) ― ― 6.0HPC-L ― ― 4.0Water ― ― 600.0
Cetirizine Dihydrochloride 1.0 1.0 1.0HPC-SSL 10.0 10.0 10.0Talc 1.0 1.0 1.0Water 190.0 190.0 190.0
Enteric Layer Eudragit®
L30D-551) 240.0 72.0 48.0
Macrogol 6000 24.0 7.2 4.8Talc 24.0 7.2 4.8Water 800.0 240.0 160.0
Total1) 900.0 698.4 669.6
ActiveCompoundLayer
1) The amount of solid

27
Table 2 に、今回調製した 3 種の腸溶性顆粒の処方を示す。製造方法は、核粒子にレ
イヤリングする方法を用いた。粒子径 200 µm および 600 µm の顆粒には、核粒子とし
て結晶セルロースの球形核粒子である粒子径 100~200 µm の Celphere® CP-102 と、
粒子径 500~700 µm の Celphere® CP-507 を用いた。粒子径 1200 µm の顆粒の核粒子
は、結晶セルロースを造粒し、分級したものを用いた。それぞれの核粒子に転動流動層
装置を用いてセチリジン塩酸塩をレイヤリングし、さらに腸溶性コーティング基剤であ
る Eudragit® L30D-55 でコーティングした後、分級し、粒度を調整した。また、Figure
10 には、今回調製した 3 種類の腸溶性顆粒の顕微鏡写真および粒子径分布の測定結果
を、Table 3 には粒子径分布より得られた D50値および粒子の密度を示した。3 種の腸
溶性顆粒はほぼ設計した値の粒子径、粒度分布を示し、密度についてもほぼ同様であっ
た。
Figure 10. Photographs and particle size distribution of three kinds of cetirizine
enteric-coated granules
enteric-coated granules
(200 µm)
enteric-coated granules
(600 µm)
enteric-coated granules
(1200 µm)

28
Table 3 Characteristics of three kinds of cetirizine enteric-coated granules
enteric-coated
granules (200 µm)
enteric-coated
granules (600 µm)
enteric-coated
granules (1200 µm)
Particle size (D50) 204 µm 662 µm 1203 µm
Density 1.43 g/cm3 1.46 g/cm3 1.43 g/cm3
各顆粒からのセチリジン塩酸塩の溶出試験(パドル法)の結果を Table 4 に示した。耐
酸性については目標通り 120 分で 10 %程度以下と十分な値が得られていた。また、中
性(pH 6.8)での溶出は 5 分で 30 %、10 分で 80 %以上を目標に調製したが、200 µm
の粒子で若干低めになった。これは溶出試験のベッセル底部へのマウント形成の影響に
よるもので、粒子が細かいため形成されたマウントの内部への水の浸透が弱まったため、
溶出が抑えられたと考えられた。そこでフロースルーセル溶出試験で粒子自体からの溶
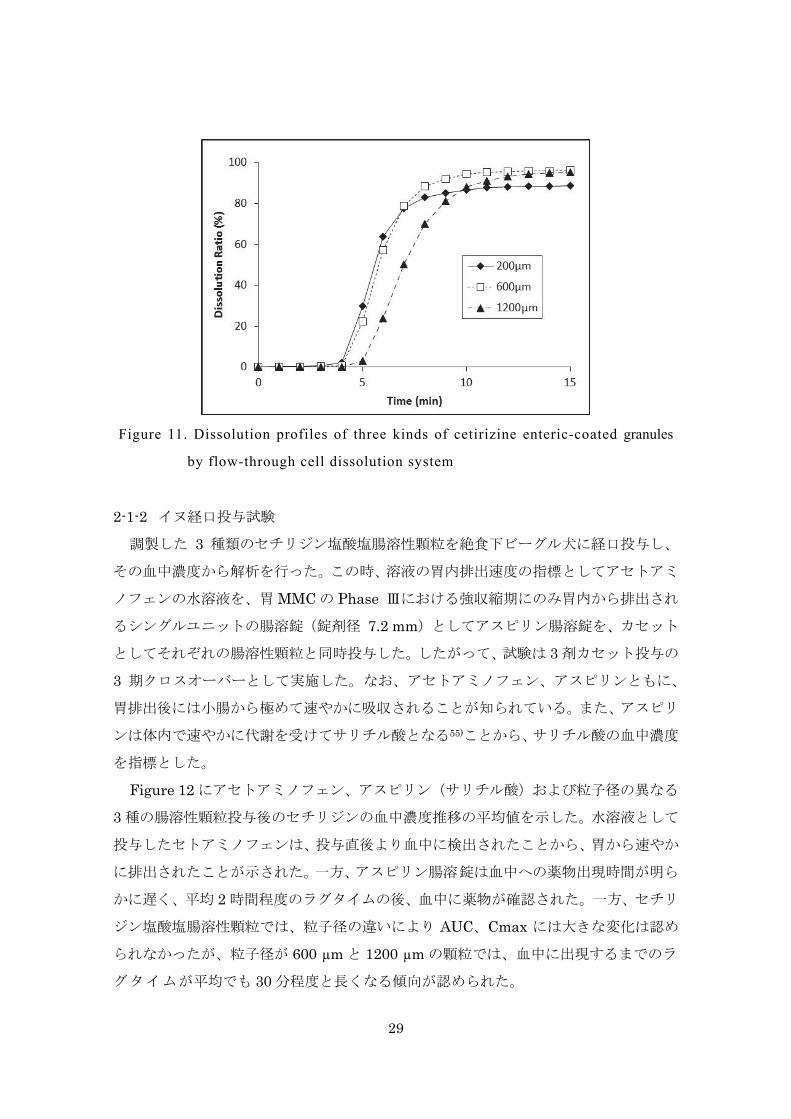
出性を確認したところ、3 種類の腸溶性顆粒からの溶出速度はほぼ同程度であった
(Figure 11)。3 種類の腸溶性顆粒は粒子径のみが違う同一処方、同一剤形の腸溶性
顆粒であり、これらを用いた経口投与試験により、胃排出速度に及ぼす粒子径の影響を
明らかにすることが可能と考えられた。
Table 4 Assay and characteristics of dissolution of three kinds of cetirizine
enteric-coated granules
enteric-coated
granules (200 µm)
enteric-coated
granules (600 µm)
enteric-coated
granules (1200 µm)
Assay 101.0 % 95.0 % 101.0 %
Dissolution
pH1.2, 120min 10.8 % 6.3 % 6.8 %
pH6.8, 5min 38.0 % 31.7 % 39.4 %
pH6.8, 10min 44.8 % 75.1 % 79.4 %
29
2-1-2 イヌ経口投与試験
調製した 3 種類のセチリジン塩酸塩腸溶性顆粒を絶食下ビーグル犬に経口投与し、
その血中濃度から解析を行った。この時、溶液の胃内排出速度の指標としてアセトアミ
ノフェンの水溶液を、胃 MMC の Phase Ⅲにおける強収縮期にのみ胃内から排出され
るシングルユニットの腸溶錠(錠剤径 7.2 mm)としてアスピリン腸溶錠を、カセット
としてそれぞれの腸溶性顆粒と同時投与した。したがって、試験は 3 剤カセット投与の
3 期クロスオーバーとして実施した。なお、アセトアミノフェン、アスピリンともに、
胃排出後には小腸から極めて速やかに吸収されることが知られている。また、アスピリ
ンは体内で速やかに代謝を受けてサリチル酸となる55)ことから、サリチル酸の血中濃度
を指標とした。
Figure 12 にアセトアミノフェン、アスピリン(サリチル酸)および粒子径の異なる
3 種の腸溶性顆粒投与後のセチリジンの血中濃度推移の平均値を示した。水溶液として
投与したセトアミノフェンは、投与直後より血中に検出されたことから、胃から速やか
に排出されたことが示された。一方、アスピリン腸溶錠は血中への薬物出現時間が明ら
かに遅く、平均 2 時間程度のラグタイムの後、血中に薬物が確認された。一方、セチリ
ジン塩酸塩腸溶性顆粒では、粒子径の違いにより AUC、Cmax には大きな変化は認め
られなかったが、粒子径が 600 µm と 1200 µm の顆粒では、血中に出現するまでのラ
グタイムが平均でも 30 分程度と長くなる傾向が認められた。
Figure 11. Dissolution profiles of three kinds of cetirizine enteric-coated granules
by flow-through cell dissolution system

30
Fig
ure
12
. C
on
cen
trat
ion
– t
ime
pro
file
s af
ter
ora
l ad
min
istr
atio
n a
s en
teri
c-co
ated
par
ticl
es,
ente
ric-
coat
ed t
able
t an
d
solu
tio
n.
Res
ult
s re
pre
sen
ted
mea
n (
n=
6) ±
S
D.

31
投与後、最初に薬物が血中に初めて検出された時間を、それぞれの薬物の吸収ラグタ
イムと考え、イヌごとの値を Figure 13 に示した。アセトアミノフェンでは個体、試験
時期にかかわらず常に最初のポイント(15 分)で血中に薬物が観察され、溶液は胃 MMC
の Phase に関わらず、幽門から速やかに排出され、小腸に移行することが確認された。
一方、アスピリン腸溶錠の場合、吸収ラグタイムは個体ごと、および試験時期ごとに大
きく変動しており、本腸溶錠のように大きな錠剤は、胃 MMC の Phase Ⅲの強収縮期
にのみ小腸へ排出されるものと考えられた。この強収縮はイヌやヒトでは 90~120 分
のサイクルで起きるとされており 46)、投与のタイミングによって胃排出時間が変動す
ることによって、経口投与後の吸収に個体間変動のみでなく、大きな個体内変動が生じ
たものと判断された。
Figure 14 に、各粒子径の腸溶性顆粒投与後のセチリジンおよびサリチル酸について
吸収ラグタイムの分布を示した。なお、図には示していないが、溶液として投与したア
セトアミノフェンは先に示したように全て 15 分で血中に検出されている。粒子径 200
µm の腸溶性顆粒の場合、0.5 ± 0.2 時間(平均 ± 標準偏差)と他の顆粒と比較してラ
Figure 13. Time for the first appearance of cetirizine and salicylic acid in plasma
after oral administration of acetaminophen solution and enteric-coated
aspirin tablet to each beagle dogs (n=6).

32
グタイムの個体間変動の分布は小さく、ラグタイムも短いことが示された。一方、粒子
径 600 µm の腸溶性顆粒ではラグタイムは 1.3 ± 0.7 時間、1200 µm の腸溶性顆粒では
1.1 ± 0.6 時間と長くなり、その分布も広がることが明らかとなった。統計的に有意な差
は示されなかったが、粒子径の違いによって胃 MMC の影響の受け方が異なっており、
その結果、吸収ラグタイムに変動が生じたものと推察された。
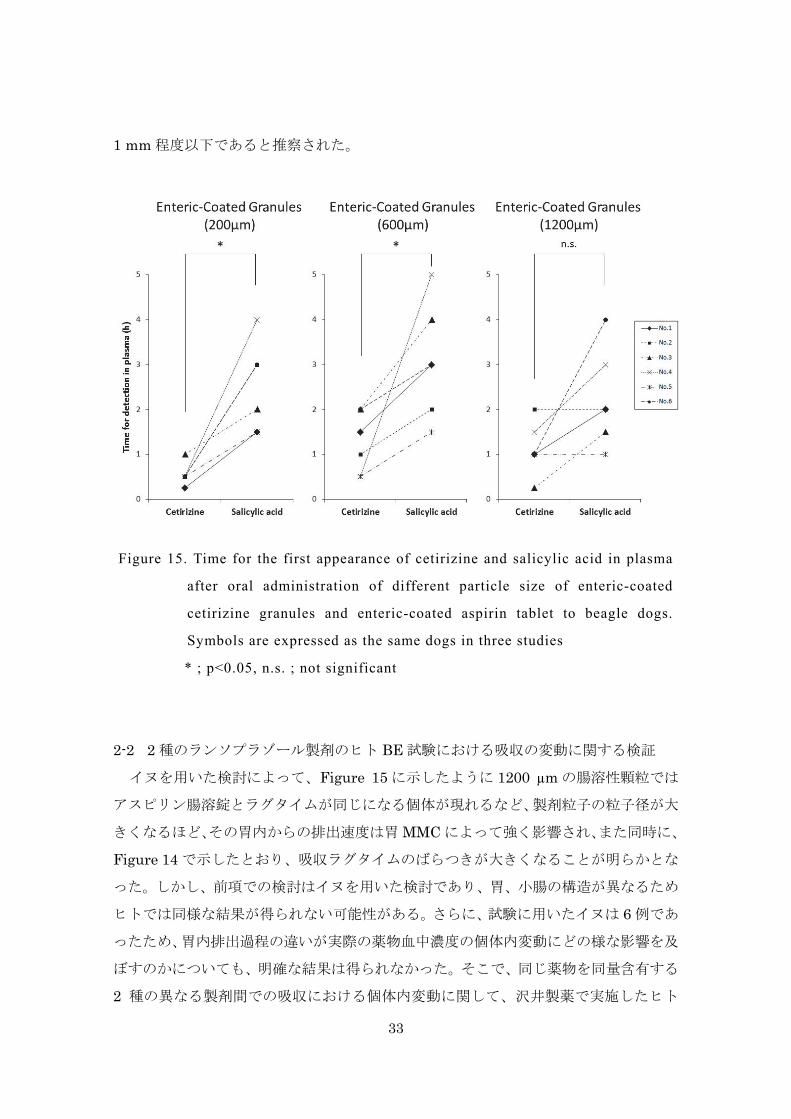
Figure 15 は同時投与したセチリジン塩酸塩腸溶性顆粒とアスピリン腸溶錠の吸収
ラグタイムをイヌ個体ごとに比較した結果を示している。粒子径 200 µm、600 µm の
腸溶性顆粒の場合、いずれの個体においても、アスピリン腸溶錠に比べてラグタイムは
短くなったのに対して、1200 µm の腸溶性顆粒の場合では、アスピリン腸溶錠との差
が小さく、同じラグタイムの個体も認められた。以上の結果は、胃内の一次粒子の粒子
径が大きくなるほど、その胃排出は胃 MMC の影響を強く受けること、また、特に粒子
径が 1000 µm を超える場合には、アスピリン腸溶錠と同様に Phase Ⅲにおいて排出さ
れる可能性が高くなることを示唆するものと考えられる。
一般に、胃幽門部には通常 2 mm 程度の隙間が開いており、溶液および 2 mm 以下
の小さな固体は、胃の収縮がない場合でも幽門を通過できると考えられている 49, 54)。
しかし、本研究で得られた結果より、胃排出における粒子径の閾値は、イヌにおいては
Figure 14. Distribution of time for the first appearance of cetirizine and salicylic
acid in plasma after oral administration of different particle size of
enteric-coated cetirizine granules and enteric-coated aspirin tablet to
beagle dogs
33
1 mm 程度以下であると推察された。
2-2 2 種のランソプラゾール製剤のヒト BE 試験における吸収の変動に関する検証
イヌを用いた検討によって、Figure 15 に示したように 1200 µm の腸溶性顆粒では
アスピリン腸溶錠とラグタイムが同じになる個体が現れるなど、製剤粒子の粒子径が大
きくなるほど、その胃内からの排出速度は胃 MMC によって強く影響され、また同時に、
Figure 14 で示したとおり、吸収ラグタイムのばらつきが大きくなることが明らかとな
った。しかし、前項での検討はイヌを用いた検討であり、胃、小腸の構造が異なるため
ヒトでは同様な結果が得られない可能性がある。さらに、試験に用いたイヌは 6 例であ
ったため、胃内排出過程の違いが実際の薬物血中濃度の個体内変動にどの様な影響を及
ぼすのかについても、明確な結果は得られなかった。そこで、同じ薬物を同量含有する
2 種の異なる製剤間での吸収における個体内変動に関して、沢井製薬で実施したヒト
Figure 15. Time for the first appearance of cetirizine and salicylic acid in plasma
after oral administration of different particle size of enteric-coated
cetirizine granules and enteric-coated aspirin tablet to beagle dogs.
Symbols are expressed as the same dogs in three studies
* ; p<0.05, n.s. ; not significant

34
BE 試験の結果に基づく調査を行った。
Figure 16に、主薬としてランソプラゾールを含有する腸溶性顆粒を含む 2種の製剤、
ランソプラゾール OD 錠 30 mg およびランソプラゾールカプセル 30 mg の BE 試験の
結果から、試験製剤の血中濃度プロファイルを示した。また、Table 5 にその薬物動態
パラメータの平均値を示した。ここに示すデータは、それぞれ独立した BE 試験におけ
る結果であり、被験者や試験時期が異なる上、例数も 22 例と 28 例と異なっているた
め、吸収率等についての直接的な比較は困難である。しかし、両製剤投与後のランソプ
ラゾール血中濃度推移を比較したところ、カプセル剤投与時のみ吸収に明らかなラグタ
イムが認められた。さらに、Table 5 に示したように、同じ薬物を同量含む製剤である
にもかかわらず、血中 AUC の個体内変動に大きな違いが認められた。すなわち、個体
間変動(Vinter)に関してはランソプラゾール OD 錠とカプセル剤で大きな差は検出さ
れなかったのに対し、個体内変動を表す Vintraの値はカプセル剤において 31 %と OD 錠
に比べて明らかに大きな値を示した。
Figure 16. Concentration – time profiles of two formulations of lansoprazole after
oral administration in each BE study. Results represented mean ± SD

35
Table 5. Pharmacokinetic parameters after administration of lansoprazole
formulations.
AUC
(ng/h/mL)
Cmax
(ng/mL) Tmax (h)
Vintra (%)
(AUC)
Vinter (%)
(AUC)
Lansoprazole OD Tablet 2721 ± 973 1108±276 1.7±0.9 8.9 35.8
Lansoprazole Capsule 2289 ± 1075 1034±438 2.8±1.4 31.2 47.0
先に述べたようにランソプラゾール OD 錠とランソプラゾールカプセルは、どちら
も腸溶性コーティングが施された二次粒子を含む製剤である。そこで、各製剤の二次粒
子の粒子径を測定したころ、OD 錠で 330 µm、カプセル剤で 1140 µm と明らかな違い
が認められた。また、粒子の密度は比較的類似した値であった(Table 6)。
Table 6. Characteristics of two kinds of enteric-coated fine particles in lansoprazole
OD Tablet and capsule
OD Tablet Capsule
Photographs
Particle size (D50) 327 µm 1143 µm
Density 1.35 g/cm3 1.48 g/cm3
さらに、in vitro におけるそれぞれの製剤からのランソプラゾールの溶出パターンを
Figure 17 に示した。日本薬局方溶出試験第 1 液(pH 1.2)では、両製剤とも 120 分間
での溶出率は 5 %以下であり、腸溶顆粒としての耐酸性は確保されていた。また、中性
付近の溶出試験液として日本薬局方溶出試験第 2 液(pH 6.8)で試験を行った結果、若
干のラグタイムを認めた後、ランソプラゾールが速やかに溶出するほぼ同様なパターン

36
が得られたことから、両製剤の溶出性には有意な差がないことが確認された。In vitro
試験の結果から in vivo 消化管内での溶出挙動を完全に把握することは困難であるが、
今回の結果から、製剤間の溶出性の違いが血中 AUC の個体内変動の違いの要因である
可能性は小さいものと判断された。
Figure 18 は、ランソプラゾール OD 錠、ランソプラゾールカプセルにおいて、被験
者ごとに、薬物が最初に血中に検出されるまでの時間の分布を示したものである。ラン
ソプラゾール OD 錠では、投与後 30 分以内に半数以上の被験者で、1時間以内には全
ての被験者で薬物が血中に検出された。それに対して、ランソプラゾールカプセルでは、
血中に薬物が検出されたのは、投与後1時間では約 40 %に過ぎず、2 時間以上経過し
てようやく薬物が血中に検出される被験者も散見された。ランソプラゾールは、セチリ
ジンと同様小腸膜透過性が高く、溶解した後、速やかに吸収されることから、Figure 18
Figure 17. Dissolution profiles of two formulations of lansoprazole OD
tablet and capsule
JP-I : Japanese Pharmacopeia, 1st fluid for dissolution test (pH
1.2) ; JP-II : Japanese Pharmacopeia, 2nd fluid for dissolution
test (pH 6.8)

37
に示された結果は、2 つの製剤の二次粒子の胃排出速度の違いに起因するものと考えら
れる。これまでの報告では、ヒトにおいて、直径 2 mm 未満の粒子は胃の MMC の影
響を受けることなく比較的容易に胃幽門を通過し、十二指腸・小腸へと排出されるとさ
れていた 49, 54)。しかしながら、今回用いた 2 種類の腸溶性コーティングの二次粒子は
いずれも 2 mm 以下であり、また密度も同様であったにもかかわらず、胃排出速度には
大きな違いが認められたことは、極めて興味深い知見と考えられる。
以上の解析結果より、ランソプラゾールカプセルにおいて、血中 AUC に大きな個体
内変動が生じた理由として、同じ被験者においても投与のタイミングによって胃 MMC
の Phase が異なっていた結果、胃排出速度に大きなばらつきが生じたためと考えられ
た。そこで、ランソプラゾールカプセル投与後、特徴的な血中濃度プロファイルを示し
た被験者を Figure 19 に示した。No.2, 25 の被験者では、吸収のラグタイムには大きな
差が認められているものの、AUC、Cmax に対する影響は比較的小さい。一方、No.13,
15 の被験者では、吸収ラグタイムとともに、AUC や Cmax にも大きな差が認められて
Figure 18. Time for the first appearance of lansoprazole in plasma after oral
administration of two formulations of lansoprazole OD tablet and
capsule

38
おり、この様なケースが BE 試験における大きな個体内変動を招いたとものと推察され
た。
Figure 19. Concentration–time profiles of characteristic subjects after oral
administration of lansoprazole capsule
open circle : reference product, closed circle : test product

39
2-3 考察
本章では、経口投与後の薬物血中濃度推移の個体内変動に関する製剤的な要因を明ら
かにすることを目的として、製剤粒子の粒子径と胃排出速度の関係に関してイヌを用い
た経口投与試験、およびヒト BE 試験データの解析による検証を行い、以下の様な知見
を得た。
1)製剤粒子が胃内で溶解しない場合、粒子径が大きくなるほど胃排出に遅延および
ばらつきが認められ、その結果、薬物の血中濃度推移に大きな個体内変動が生じ
ることが明らかとなった。
2)イヌ、ヒトとも小さな粒子は溶液と同様に速やかに胃内から排出され、その閾値
は 1 mm 程度であることが示唆された。
一般に、錠剤やカプセル剤を製造する際には、流動性の付与、結合性の付与、含量均
一性の確保などを目的として、粉体を造粒した顆粒を調製し、これを打錠あるいはカプ
セルに充填する56)。経口投与された製剤は、消化管内で、一旦この造粒物(二次粒子)
にまで崩壊した後、薬物が溶出(溶解)する。上記の 1)、2)の知見は、製剤自体ま
たはその二次粒子が胃内で溶解しないケースで、腸溶性製剤および BCS Class 2 に分
類される難溶解性薬物のうち、中性あるいは酸性薬物の製剤に認められる現象である。
Class 2 薬物でも、弱塩基性薬物は、通常、胃内の低 pH 条件下で速やかに溶解し、溶
液として胃から排出されると考えられるが、高齢者などの様に胃酸分泌が低下した患者
では、胃内での溶解が遅延した結果、吸収のばらつきが認められる可能性がある。
粒子径に依存した胃排出パターンの違いは、胃 MMC の影響の受け方の違いとして
説明される。すなわち、2)で示した様に、1 mm 以下の小さな粒子は、胃幽門部の隙
間を通過することが可能であるため、胃 MMC の Phase によらず溶液とともに速やか
に排出されるのに対し、1 mm 以上の粒子が排出されるためには、MMC Phase Ⅲの強
く頻繁な収縮(housekeeping wave)が必要とされる。MMC Phase Ⅲは 90~120 分
のサイクルで繰り返さることが知られており、同じ被験者であっても服薬のタイミング
によっては胃排出時間が大きく異なり、それが吸収ラグタイムのばらつきおよび血中濃
度推移の個体内変動につながると考えられる。
一般に、製剤試験に使われる崩壊試験器では、試験器の底部の網目の開きは 2 mm
程度であり、この網目を通り抜ければ崩壊したと見なされ、それ以上の検討は行われな

40
いことが多い。これまで、胃幽門部の隙間を通過可能な粒子径の閾値に関して、腸溶性
の粒子等を用いた多くの研究が行われており 49, 53, 54)、ヒトにおける閾値は 2 mm 程度
と報告されている。したがって、崩壊試験の結果が同等であれば、生じた二次粒子の胃
排出に大きな違いは生じないと考えられてきた。一方、今回の検討において、イヌでは
1200µm の腸溶性顆粒で、腸溶性錠剤と同じラグタイムを示す個体が現れる(Figure 15)
こと、ヒトでは 1100µm の腸溶性顆粒を含んだカプセル製剤で、半数以上が 1 時間を
超えるラグタイムを示す(Figure 18)ことから、ともに 1 mm 以上の粒子の胃排出パ
ターンは MMC の影響を強く受けることが示唆された。これは、製剤設計を行う上で重
要な知見であり、薬物によっては、崩壊によって生じる二次粒子の粒子径に関して、よ
り詳細な検証を行うことが推奨される。
医薬品開発の前臨床段階で製剤設計を行う場合、溶出試験などの in vitro 試験の後、
通常、イヌを用いた経口投与試験によって製剤の性能を評価するとともに、ヒトにおけ
る吸収速度や吸収率を推定する。イヌは臨床製剤と同じ大きさの製剤を投与可能である
ことが、経口剤の試験に用いられる主たる理由であるが、イヌとヒトでは胃や小腸の形
状、消化管内溶液組成、さらには代謝酵素やトランスポーター等の活性に種差が存在す
ることから、ヒトでの吸収を正確に予測することが困難なケースが報告されている 57~59)。
特に、イヌの胃は水平方向に前胃と後胃の二つに隔てられた形状であり、ヒトとは大き
く異なることが知られている。したがって、今回、薬物は異なるものの、製剤の粒子径
と胃排出パターンの関係、および胃幽門部を通過可能な粒子径の閾値が、イヌとヒトで
ほぼ同等であったことは極めて興味深い。少なくとも製剤の粒子設計に関しては、ヒト
予測のための in vivo 試験としてイヌを用いることの妥当性が示されたものと考えられ
る。575859
通常、胃内排出の遅れは、薬物血中濃度推移における Cmax の低下および Tmax の
延長をまねくと考えられる。ランソプラゾール製剤の BE 試験の解析において、Table 5
に示した様に粒子径の大きなカプセル剤投与後の血中 AUC にも大きな個体内変動が認
められた理由として、胃内に長時間滞留することにより、溶出した薬物が胃酸による分
解を受けた可能性や、吸収速度の低下によって小腸上皮あるいは肝臓中の薬物濃度が低
く維持されたため、初回通過代謝の影響を強く受けた可能性が考えられる(Figure 19
の被験者 No 13, 15 のケース)。
本研究において、製剤の粒子径を最適化することにより、個体内変動や個体間変動の
低減が可能であることが示された。ただし、ヒト BE 試験において、BE 判定に必要な
パラメータとして要求される薬物の血中 AUC や Cmax の個体内変動に関する製剤的な

41
リスクファクターとしては、今回検討を行った粒子径の他にも、製剤の崩壊や溶出にお
ける pH 依存性、製剤添加物と胆汁酸、食事成分などの消化管内容物との相互作用、な
どの要因が考えられる。したがって、今後これら要因に関しても本研究と同様な解析を
行うことにより、個体内変動の小さな製剤の開発が可能になるものと期待される。

42
結論
医薬品の開発において様々な形で血中薬物濃度を測定し評価しているが、正
確な評価のためには、その変動要因、特に個体内変動の要因について理解して
おくことは重要である。本研究では、固形製剤として経口投与された薬物の血
中濃度推移における個体内変動、個体間変動に及ぼす製剤および薬物の特性に
ついて検討した結果、以下のような結論を得た。
[1]経口投与製剤のヒト BE 試験における変動要因
沢井製薬で BE 試験を実施した 74 化合物、113 の速放性製剤について、薬
物血中濃度推移の個体内変動を、薬物の物理化学的・動態学的特性に基づいて
解析した。まず、膜透過性と変動の関係を調査し、吸収が膜透過律速となる薬物(BCS
Class 3)において、膜透過性と個体内変動に負の相関が認められ、膜透過性が極めて
低い薬物については、個体内変動が大きくなることを示した。一方、溶解度の低
い薬物(BCS Class 2)については、Peff / Do という新しいパラメータを用い、溶解律
速と溶解度律速に分離して評価することにより、溶解度律速の BCS Class 2 薬物は大
きな個体内変動を示す可能性が高いことを明らかにした。また、溶解や膜透過性
に問題が少ないと考えられる薬物が大きな個体内変動を示す要因として、代謝酵素によ
る影響を検討し、CYP3A4 に対する親和性(Km値)が個体内変動と負の相関が認めら
れることを示した。
これらの情報を in vitro 実験等によって事前に把握することで、個体内変動の大き
さを予測し、適切な評価をし得る実験計画の作成が可能になると考えられた。
[2]胃排出速度の変動に及ぼす製剤の影響
製剤粒子の粒子径と胃排出速度の関係に関して、イヌを用いた経口投与試験、および
ヒト BE 試験データの解析による検証を行い、経口投与後の薬物血中濃度推移の個体内
変動に関する製剤的な要因を検討した。まず、3 種類の粒子径のみが違う腸溶性顆粒を
調製し、ビーグル犬にマーカー薬物とカセット投与することにより、粒子径が胃排出に
与える影響を調査した。その結果、粒子径が大きくなるほど胃排出に遅延およびばらつ
きが認められることを示した。また、粒子径の違う腸溶性顆粒を含む 2 種類の製剤のヒ
ト BE 試験データを解析した結果、粒子径の違いが薬物の血中濃度推移に、大きな個体
内変動を生じさせることを示した。

43
上記の検討の結果、イヌ、ヒトとも小さな粒子は溶液と同様に速やかに胃内から排出
され、その閾値は 1 mm 程度であることを明らかにした。錠剤などの製剤を設計す
る際、特に胃で溶解しない酸性薬物やコーティング顆粒などでは二次粒子の崩
壊性や溶出性にも注意し、精査した上で製剤設計を行う必要性と、その際にイ
ヌを使うことの有用性を示すことができた。
以上、対象となる薬物および製剤について、経口投与後の吸収過程における
個体内変動のリスクを予め予測することで、より安定した血中濃度推移を示す
製剤の処方設計に役立つとともに、ヒト BE 試験の効率化を図る上で有益な情
報になるものと期待される。

44
謝辞
終わりに臨み、本研究の実施に当たり終始御懇篤なる御指導並びに御鞭撻を
賜りました岡山大学薬学部檜垣和孝教授、摂南大学薬学部山下伸二教授に衷心
より深甚なる謝意を表します。
また、本論文の作成に際して、有益なる御助言と御校閲を賜りました岡山大
学薬学部狩野光伸教授、合葉哲也准教授、四宮一昭准教授に謹んで深く感謝の
意を表します。
さらに、種々の有益なる御助言並びに御指導を賜りました岡山大学薬学部大
河原賢一准教授、摂南大学薬学部片岡誠講師に衷心より感謝いたします。
本研究に際し、実験に御協力いただきました沢井製薬(株)製剤研究部岡村
康史グループマネージャー、主任研究員保坂昌一博士、杉田優研究員、生物研
究部薬物動態グループ出口修平主幹研究員、竹内達主任研究員、小島由紀子研
究員をはじめグループ員に深く感謝いたします。
また、本研究の機会を与えてくださり、格別なる御配慮を賜りました沢井製
薬(株)特別顧問横浜重晴博士に深く感謝いたします。
最後にいつも支えてくれた家族に心から感謝いたします。

45
実験の部
第 1 章 実験の部
[1]データベースの作成
沢井製薬で 1997 年から 2012 年までに行った速放性製剤の BE 試験、74 化
合物、113 製剤についてデータベース化を行った(Tables 7, 8)。
全ての BE 試験は「後発医薬品の生物学的同等性試験ガイドライン」3)に従
って健康成人を用い、「医薬品の臨床試験の実施の基準」(GCP) 60)に則り、
治験審査委員会で承認を得た治験実施計画書を遵守して実施した。
cLogP は DDF 11)の BCS データベースに記載のあるものはここから、その
他は文献情報を基にした。
AUC、Cmax 等の薬物動態パラメータの算出と、信頼区間の算出、分散分
析については生物学的同等性試験システム BESTS((株)CAC エクシケア)
を用いて算出した。
[2]溶解度の測定
37℃における溶解度をフラスコ振とう法で測定した。薬物の溶解度が飽和
に達するまでじゅうぶんに振とうした後、速やかにフィルターろ過、または遠
心分離を行い、良溶媒で希釈して測定した。測定法はそれぞれの薬物の定量法
を準用した。

46
Tab
le 7
ph
ysic
och
emic
al p
rope
rtie
s of
113
ora
l dru
g pr
odu
cts
of 7
4 A
PIs
Com
pou
nd
dosa
gefo
rma
stre
ngt
hcL
ogP
Pef
f (×
10-4
cm/s
) S
olu
bili
ty(m
g/m
L)
Do
Pef
f/D
o (×
10-4
cm/s
)B
CS
clas
sA
lpra
zola
m
UC
T
0.8m
g2.
205
5.52
0.
086.
67E
-02
8.28
E+
011
Am
lodi
pin
e B
esil
ate
FC
T
2.5m
g3.
434
0.71
2.
227.
51E
-03
9.46
E+
011
FC
T
5mg
1.50
E-0
2 4.
73E
+01
1
Ato
rvas
tati
n C
alci
um
F
CT
5m
g 4.
457
1.73
0.
152.
22E
-01
7.79
E+
001
FC
T
10m
g 4.
44E
-01
3.89
E+
001
Ben
azep
ril H
ydro
chlo
ride
U
CT
5m
g 1.
824
0.53
12
82.
60E
-04
2.04
E+
031
Ber
apro
st S
odiu
m
FC
T
20μ
g 2.
044
2.05
10
02.
67E
-06
7.69
E+
051
Bet
axol
ol H
ydro
chlo
ride
F
CT
5m
g 2.
81.
96
>10
003.
33E
-05
5.88
E+
041
FC
T
10m
g 6.
67E
-05
2.94
E+
041
Bro
tizo
lam
O
DT
0.
25m
g2.
358
5.72
0.
028.
33E
-02
6.86
E+
011
Cab
ergo
lin
e U
CT
0.
5mg
4.17
11.
15
0.2
1.67
E-0
2 6.
90E
+01
1 U
CT
1m
g 3.
33E
-02
3.45
E+
011
Cet
iriz
ine
Hyd
roch
lori
de
OD
T
5mg
2.08
0.94
>
1000
3.33
E-0
5 2.
82E
+04
1 F
CT
10
mg
6.67
E-0
5 1.
41E
+04
1
Cib
enzo
lin
e S
ucc
inat
e F
CT
50
mg
3.07
3.6
23.7
51.
40E
-02
2.57
E+
021
FC
T
100m
g2.
81E
-02
1.28
E+
021
Clo
tiaz
epam
F
CT
5m
g 4.
117.
45
0.05
6.67
E-0
1 1.
12E
+01
1
Don
epez
il H
ydro
chlo
ride
F
CT
5m
g 4.
63.
42
125
2.67
E-0
4 1.
28E
+04
1 F
CT
10
mg
5.33
E-0
4 6.
41E
+03
1 D
oxaz
osin
Mes
ilat
e U
CT
1m
g 2.
481.
39
6.67
1.00
E-0
3 1.
39E
+03
1
UC
T
2mg
2.
00E
-03
6.95
E+
021
Epi
nas
tin
e H
ydro
chlo
ride
D
S
10m
g/g
3.50
62.
72
100
3.33
E-0
4 8.
16E
+03
1 F
CT
20
mg
1.33
E-0
3 2.
04E
+03
1 F
exof
enad
ine
Hyd
roch
lori
de
FC
T
60m
g 1.
960.
59
1.98
2.02
E-0
1 2.
92E
+00
1
Flu
vast
atin
Sod
ium
F
CT
20
mg
4.04
82.
51
47.3
2.82
E-0
3 8.
90E
+02
1 F
CT
30
mg
4.23
E-0
3 5.
94E
+02
1
Flu
voxa
min
e M
alea
te
FC
T
25m
g 3.
321
2.19
41
.74.
00E
-03
5.48
E+
021
FC
T
50m
g 7.
99E
-03
2.74
E+
021
FC
T
75m
g 1.
20E
-02
1.83
E+
021
Gli
mep
irid
e U
CT
1m
g 3.
960.
27
0.10
26.
54E
-02
4.13
E+
001
UC
T
3mg
1.96
E-0
1 1.
38E
+00
1

47
(Tab
le 7
) C
ontd
…
Com
pou
nd
dosa
gefo
rma
stre
ngt
hcL
ogP
Pef
f (×
10-4
cm/s
) S
olu
bili
ty(m
g/m
L)
Do
Pef
f/D
o (×
10-4
cm/s
)B
CS
clas
s
Imid
apri
l Hyd
roch
lori
de
UC
T
2.5m
g1.
531
0.35
91
1.83
E-0
4 1.
91E
+03
1 U
CT
5m
g 3.
66E
-04
9.56
E+
021
UC
T
10m
g 7.
33E
-04
4.78
E+
021
Itop
ride
Hyd
roch
lori
de
FC
T
50m
g 2.
651.
75
>10
003.
33E
-04
5.25
E+
031
Lim
apro
st A
lfad
ex
UC
T
5μg
3.44
1.38
50
06.
67E
-08
2.07
E+
071
Los
arta
n P
otas
siu
m
FC
T
50m
g 4.
112
0.87
33
69.
92E
-04
8.77
E+
021
FC
T
100m
g1.
98E
-03
4.38
E+
021
Mos
apri
de C
itra
te
FC
T
5mg
3.43
62.
73
0.05
6.67
E-0
1 4.
10E
+00
1
Nil
vadi
pin
e F
CT
2m
g 3.
038
1.84
0.
11.
33E
-01
1.38
E+
011
FC
T
4mg
2.67
E-0
1 6.
90E
+00
1
Par
oxet
ine
Hyd
roch
lori
de
FC
T
10m
g 4.
242.
43
8.64
7.72
E-0
3 3.
15E
+02
1 F
CT
20
mg
1.54
E-0
2 1.
57E
+02
1 P
ram
ipex
ole
Hyd
roch
lori
de
UC
T
0.12
5mg
1.76
1.35
>
1000
8.33
E-0
7 1.
62E
+06
1
Pra
vast
atin
Sod
ium
U
CT
5m
g 2.
048
1.07
>
1000
3.33
E-0
5 3.
21E
+04
1 U
CT
10
mg
6.67
E-0
5 1.
61E
+04
1
Pro
pive
rin
e H
ydro
chlo
ride
F
CT
10
mg
3.87
43.
27
167
3.99
E-0
4 8.
19E
+03
1 F
CT
20
mg
7.98
E-0
4 4.
10E
+03
1
Ris
peri
don
e
OD
T
0.5m
g
2.71
13.
35
0.09
3.70
E-0
2 9.
05E
+01
1 F
CT
1m
g 7.
41E
-02
4.52
E+
011
FC
T
2mg
1.48
E-0
1 2.
26E
+01
1 F
CT
3m
g 2.
22E
-01
1.51
E+
011
Tam
oxif
en C
itra
te
FC
T
20m
g 6.
818
5.62
0.
316
4.22
E-0
1 1.
33E
+01
1
Tan
dosp
iron
e C
itra
te
FC
T
5mg
1.5
2.98
25
1.33
E-0
3 2.
24E
+03
1 F
CT
10
mg
2.67
E-0
3 1.
12E
+03
1
Tem
ocap
ril H
ydro
chlo
ride
U
CT
2m
g 2.
102
0.48
0.
26.
67E
-02
7.20
E+
001
UC
T
4mg
1.33
E-0
1 3.
60E
+00
1 T
iclo
pidi
ne
Hyd
roch
lori
de
FC
T
100m
g4.
388
9.52
78
8.55
E-0
3 1.
11E
+03
1 T
orem
ifen
e C
itra
te
UC
T
40m
g 6.
535.
52
0.53
5.07
E-0
1 1.
09E
+01
1 T
ran
dola
pril
U
CT
1m
g 4
0.51
0.
778.
67E
-03
5.88
E+
011
Zol
pide
m T
artr
ate
FC
T
5mg
2.82
66.
94
8.9
3.75
E-0
3 1.
85E
+03
1 F
CT
10
mg
7.49
E-0
3 9.
26E
+02
1

48
(Tab
le 7
) C
ontd
…
Com
pou
nd
dosa
gefo
rma
stre
ngt
hcL
ogP
Pef
f (×
10-4
cm/s
) S
olu
bili
ty(m
g/m
L)
Do
Pef
f/D
o (×
10-4
cm/s
)B
CS
clas
sA
mio
daro
ne
Hyd
roch
lori
de
UC
T
100m
g9.
133.
12
0.43
1.54
E+
002.
03E
+00
2
Ben
idip
ine
Hyd
roch
lori
de
FC
T
2mg
5.70
91.
04
0.01
1.33
E+
007.
80E
-01
2 F
CT
4m
g 2.
67E
+00
3.90
E-0
1 2
FC
T
8mg
5.33
E+
001.
95E
-01
2 B
ical
uta
mid
e F
CT
80
mg
2.70
70.
99
0.00
441.
21E
+02
8.17
E-0
3 2
Car
vedi
lol
FC
T
2.5m
g4.
041
1.43
0.
013
1.27
E+
005.
64E
+00
2 C
efdi
tore
n P
ivox
il
FC
T
100m
g2.
711
0.81
0.
016.
67E
+01
1.22
E-0
2 2
Cil
nid
ipin
e F
CT
10
mg
5.54
1.1
0.00
0024
2.78
E+
033.
96E
-04
2
Cla
rith
rom
ycin
F
CT
50
mg
1.97
0.32
0.
013.
33E
+01
9.60
E-0
3 2
FC
T
200m
g1.
33E
+02
2.40
E-0
3 2
Eba
stin
e F
CT
5m
g 7.
062
2.72
0.
0021
1.58
E+
011.
72E
-01
2 F
CT
10
mg
3.16
E+
018.
61E
-02
2 E
cabe
t S
odiu
m
GR
0.
667g
/g4.
932.
23
0.01
6.67
E+
023.
35E
-03
2 E
todo
lac
FC
T
200m
g3.
523.
06
0.11
1.21
E+
012.
52E
-01
2 It
raco
naz
ole
CA
P
50m
g 6.
046
1.85
0.
0073
4.55
E+
014.
07E
-02
2 L
orat
adin
e U
CT
10
mg
5.05
17.
61
0.00
088
7.58
E+
011.
00E
-01
2
Mel
oxic
am
UC
T
5mg
2.29
22.
52
0.00
556.
07E
+00
4.15
E-0
1 2
UC
T
10m
g 1.
21E
+01
2.08
E-0
1 2
Pio
glit
azon
e H
ydro
chlo
ride
U
CT
15
mg
3.53
2.96
0.
027
3.70
E+
007.
99E
-01
2 U
CT
30
mg
7.41
E+
004.
00E
-01
2 P
ran
luka
st
CA
P
112.
5mg
5.06
50.
13
0.00
032.
50E
+03
5.20
E-0
5 2
Qu
azep
am
UC
T
15m
g 4.
8610
.29
0.00
058
1.72
E+
025.
97E
-02
2 U
CT
20
mg
2.30
E+
024.
48E
-02
2 R
ebam
ipid
e F
CT
10
0mg
1.96
51.
62
0.00
88.
33E
+01
1.94
E-0
2 2
Sim
vast
atin
U
CT
5m
g 4.
481
3.83
0.
0001
22.
78E
+02
1.38
E-0
2 2
Ter
bin
afin
e H
ydro
chlo
ride
U
CT
12
5mg
5.96
8.55
0.
145.
95E
+00
1.44
E+
002
Zal
topr
ofen
F
CT
80
mg
4.5
5.26
0.
0045
1.19
E+
024.
44E
-02
2

49
(Tab
le 7
) C
ontd
…
Com
pou
nd
dosa
gefo
rma
stre
ngt
hcL
ogP
Pef
f (×
10-4
cm/s
) S
olu
bili
ty(m
g/m
L)
Do
Pef
f/D
o (×
10-4
cm/s
)B
CS
clas
sA
ctar
it
FC
T
100m
g0.
473.
83
6.68
9.98
E-0
2 3.
84E
+01
3 A
len
dron
ate
Sod
ium
U
CT
35
mg
-5.6
420.
16
39.1
5.97
E-0
3 2.
68E
+01
3 A
nas
troz
ole
FC
T
1mg
1.28
4.49
0.
88.
33E
-03
5.39
E+
023
Cef
cape
ne
Piv
oxil
Hyd
roch
lori
de
FC
T
75m
g 0.
486
0.43
0.
68.
33E
-01
5.16
E-0
1 3
Cef
din
ir
CA
P
50m
g -0
.482
0.59
0.
369.
26E
-01
6.37
E-0
1 3
Fam
otid
ine
OD
T
10m
g -0
.611
0.35
1.
93.
51E
-02
9.98
E+
003
OD
T
20m
g 7.
02E
-02
4.99
E+
003
Flu
con
azol
e C
AP
10
0mg
-0.4
44.
06
8.33
8.00
E-0
2 5.
07E
+01
3 L
afu
tidi
ne
FC
T
10m
g 1.
220.
69
0.4
1.67
E-0
1 4.
14E
+00
3 M
eth
otre
xate
C
AP
2m
g -0
.53
0.24
0.
091
1.47
E-0
1 1.
63E
+00
3
Mil
nac
ipra
n H
ydro
chlo
ride
F
CT
15
mg
1.42
2.02
62
51.
60E
-04
1.26
E+
043
FC
T
25m
g 2.
67E
-04
7.58
E+
033
FC
T
50m
g 5.
33E
-04
3.79
E+
033
Niz
atid
ine
CA
P
75m
g -0
.202
1.02
40
1.25
E-0
2 8.
16E
+01
3 O
lopa
tadi
ne
Hyd
roch
lori
de
FC
T
5mg
1.09
1.03
29
.91.
11E
-03
9.24
E+
023
Sod
ium
Ris
edro
nat
e F
CT
2.
5mg
-2.6
220.
2 52
.73.
16E
-04
6.32
E+
023
FC
T
17.5
mg
2.21
E-0
3 9.
03E
+01
3 S
upl
atas
t T
osil
ate
CA
P
100m
g0.
490.
28
>10
006.
67E
-04
4.20
E+
023
talt
irel
in
UC
T
5mg
-1.4
20.
18
833
4.00
E-0
5 4.
50E
+03
3 C
efca
pen
e P
ivox
il H
ydro
chlo
ride
F
CT
10
0mg
0.48
60.
43
0.6
1.11
E+
003.
87E
-01
4 C
efdi
nir
C
AP
10
0mg
-0.4
820.
59
0.36
1.85
E+
003.
19E
-01
4 C
efpo
doxi
me
Pro
xeti
l F
CT
10
0mg
-0.4
10.
88
0.29
2.30
E+
003.
83E
-01
4 L
-Car
boci
stei
ne
FC
T
500m
g-2
.608
0.31
1.
71.
96E
+00
1.58
E-0
1 4
Tos
ufl
oxac
in T
osil
ate
FC
T
150m
g0.
190.
43
0.83
1.21
E+
003.
55E
-01
4 a
UC
T :
un
coat
ed t
able
t, F
CT
: fi
lm c
oate
d ta
blet
, OD
T :
oral
dis
inte
grat
ing
tabl
et, C
AP
: ca
psu
le, G
R :
gran
ule
,
DS
: dr
y sy
rup

50
Tab
le 8
BE
stu
dy p
aram
eter
s of
113
ora
l dru
g pr
odu
cts
of 7
4 A
PIs
Com
pou
nd
dosa
gefo
rm
BC
Scl
ass
actu
al
dose
(m
g)
AU
C
Vin
tra
AU
C
Vin
ter
Cm
ax
Vin
tra
Cm
ax
Vin
ter
Tm
ax
Vin
tra
Tm
ax
Vin
ter
MR
T
Vin
tra
MR
T
Vin
ter
Alp
razo
lam
U
CT
1
0.8
5.0
18.5
9.9
19.4
53.0
75.6
2.9
10.5
A
mlo
dipi
ne
Bes
ilat
e F
CT
1
2.5
10.7
23.2
13.3
25.2
15.4
12.2
1.8
3.9
F
CT
1
56.
019
.46.
119
.75.
88.
22.
03.
8 A
torv
asta
tin
Cal
ciu
m
FC
T
1 5
13.7
50.1
24.1
47.8
95.0
100.
310
.812
.8
F
CT
1
108.
944
.424
.739
.411
0.4
104.
83.
89.
5 B
enaz
epri
l Hyd
roch
lori
de
UC
T
1 5
16.2
27.3
36.7
27.9
42.2
31.4
25.6
24.1
B
erap
rost
Sod
ium
F
CT
1
0.04
35.6
41.7
45.2
48.6
57.9
44.4
21.7
24.8
B
etax
olol
Hyd
roch
lori
de
FC
T
1 5
5.3
15.9
5.1
15.8
20.3
15.7
3.6
5.4
F
CT
1
104.
023
.86.
018
.211
.324
.81.
710
.5
Bro
tizo
lam
O
DT
1
0.25
11.2
28.9
16.5
30.0
48.5
49.0
3.9
6.5
Cab
ergo
lin
e U
CT
1
0.5
25.6
36.1
29.0
42.1
72.6
63.5
4.4
8.8
U
CT
1
121
.535
.821
.628
.681
.479
.15.
19.
4 C
etir
izin
e H
ydro
chlo
ride
O
DT
1
56.
217
.99.
718
.944
.938
.34.
88.
8
FC
T
1 10
4.7
15.1
11.5
17.5
55.5
61.8
2.8
14.2
C
iben
zoli
ne
Su
ccin
ate
FC
T
1 50
4.7
21.7
11.5
21.2
24.4
39.1
3.3
13.4
FC
T
1 10
04.
217
.412
.522
.022
.530
.13.
88.
0 C
loti
azep
am
FC
T
1 5
7.3
35.8
24.2
35.2
47.5
61.5
5.2
33.7
D
onep
ezil
Hyd
roch
lori
de
FC
T
1 5
4.7
16.9
10.4
18.8
25.4
21.5
2.2
4.8
F
CT
1
104.
418
.010
.421
.728
.638
.71.
38.
3 D
oxaz
osin
Mes
ilat
e U
CT
1
117
.436
.220
.631
.340
.732
.113
.420
.6
U
CT
1
219
.836
.716
.520
.546
.743
.313
.514
.7
Epi
nas
tin
e H
ydro
chlo
ride
D
S
1 5
14.4
31.7
11.7
37.9
47.1
42.8
13.8
8.9
F
CT
1
2021
.623
.327
.037
.250
.493
.84.
942
.7
Fex
ofen
adin
e H
ydro
chlo
ride
F
CT
1
6032
.936
.738
.846
.835
.942
.38.
110
.9
Flu
vast
atin
Sod
ium
F
CT
1
2022
.638
.861
.852
.086
.484
.140
.038
.8
F
CT
1
3021
.932
.941
.246
.362
.869
.822
.232
.3
Flu
voxa
min
e M
alea
te
FC
T
1 25
9.5
52.8
14.5
52.4
17.3
18.4
4.6
7.7
F
CT
1
509.
894
.314
.061
.217
.022
.24.
210
.4
F
CT
1
7510
.447
.410
.331
.618
.124
.32.
310
.9
Gli
mep
irid
e U
CT
1
18.
226
.118
.426
.333
.740
.716
.023
.8
U
CT
1
36.
420
.412
.822
.528
.337
.412
.419
.7

51
(Tab
le 8
) C
ontd
…
Com
pou
nd
dosa
gefo
rm
BC
Scl
ass
actu
al
dose
(m
g)
AU
C
Vin
tra
AU
C
Vin
ter
Cm
ax
Vin
tra
Cm
ax
Vin
ter
Tm
ax
Vin
tra
Tm
ax
Vin
ter
MR
T
Vin
tra
MR
T
Vin
ter
Imid
apri
l Hyd
roch
lori
de
UC
T
1 2.
528
.536
.128
.341
.930
.723
.87.
58.
4
UC
T
1 5
33.1
49.0
37.8
54.7
21.2
23.4
7.1
10.4
UC
T
1 10
20.1
39.1
26.1
43.3
31.5
26.9
4.2
7.7
Itop
ride
Hyd
roch
lori
de
FC
T
1 50
5.4
25.1
12.0
27.3
28.2
32.9
4.7
12.0
L
imap
rost
Alf
adex
U
CT
1
0.00
525
.236
.839
.850
.834
.431
.414
.618
.5
Los
arta
n P
otas
siu
m
FC
T
1 50
11.6
36.6
41.4
54.4
70.7
62.6
31.6
30.2
FC
T
1 10
011
.436
.732
.643
.342
.658
.526
.035
.0
Mos
apri
de C
itra
te
FC
T
1 5
21.9
51.6
31.1
43.6
47.3
61.1
10.4
18.5
N
ilva
dipi
ne
FC
T
1 2
19.4
50.5
31.5
71.4
36.8
27.0
9.8
8.6
F
CT
1
419
.641
.525
.848
.228
.527
.58.
511
.6
Par
oxet
ine
Hyd
roch
lori
de
FC
T
1 10
17.9
155.
119
.312
0.2
49.1
85.9
9.9
30.9
FC
T
1 20
17.3
82.9
13.0
67.6
28.9
38.8
14.8
22.2
P
ram
ipex
ole
Hyd
roch
lori
de
UC
T
1 0.
125
7.8
23.1
8.9
22.0
24.8
44.4
3.4
8.1
Pra
vast
atin
Sod
ium
U
CT
1
560
.773
.065
.783
.229
.933
.98.
514
.2
U
CT
1
1029
.550
.933
.763
.528
.629
.611
.79.
4 P
ropi
veri
ne
Hyd
roch
lori
de
FC
T
1 10
20.5
50.9
19.4
53.5
35.4
41.6
5.1
22.5
FC
T
1 20
26.7
49.7
32.9
74.2
38.7
45.9
5.6
11.1
R
ispe
rido
ne
OD
T
1 0.
511
.573
.116
.254
.732
.040
.76.
617
.5
F
CT
1
122
.059
.229
.556
.242
.434
.210
.022
.0
F
CT
1
218
.366
.328
.442
.829
.125
.36.
430
.5
F
CT
1
315
.970
.725
.044
.230
.435
.57.
032
.6
Tam
oxif
en C
itra
te
FC
T
1 20
6.6
19.9
6.5
16.0
19.8
22.1
3.2
5.2
Tan
dosp
iron
e C
itra
te
FC
T
1 5
18.4
67.9
21.0
71.8
43.5
12.6
7.3
13.3
FC
T
1 10
15.3
75.2
17.1
73.8
35.5
45.9
10.6
13.2
T
emoc
apri
l Hyd
roch
lori
de
UC
T
1 2
9.0
19.2
16.9
21.5
29.1
27.4
5.3
6.9
U
CT
1
49.
617
.712
.519
.526
.719
.16.
18.
8 T
iclo
pidi
ne
Hyd
roch
lori
de
FC
T
1 10
020
.670
.122
.871
.024
.628
.010
.811
.8
Tor
emif
ene
Cit
rate
U
CT
1
408.
825
.311
.020
.324
.525
.84.
213
.3
Tra
ndo
lapr
il
UC
T
1 1
12.3
42.7
22.8
41.9
54.1
40.8
23.3
22.4
Z
olpi
dem
Tar
trat
e F
CT
1
519
.137
.829
.039
.469
.210
1.1
15.0
28.6
FC
T
1 10
11.3
35.1
27.0
32.5
94.1
93.8
12.6
19.1

52
(Tab
le 8
) C
ontd
…
Com
pou
nd
dosa
gefo
rm
BC
Scl
ass
actu
al
dose
(m
g)
AU
C
Vin
tra
AU
C
Vin
ter
Cm
ax
Vin
tra
Cm
ax
Vin
ter
Tm
ax
Vin
tra
Tm
ax
Vin
ter
MR
T
Vin
tra
MR
T
Vin
ter
Am
ioda
ron
e H
ydro
chlo
ride
U
CT
2
100
11.9
29.8
15.5
29.5
20.5
23.6
3.0
6.7
Ben
idip
ine
Hyd
roch
lori
de
FC
T
2 2
36.2
61.3
34.0
55.3
51.5
41.5
12.4
14.5
FC
T
2 4
18.2
57.6
23.6
58.4
46.3
42.5
10.6
15.5
FC
T
2 8
20.4
94.8
24.6
70.7
35.7
49.3
12.0
14.6
B
ical
uta
mid
e F
CT
2
8025
.433
.620
.423
.050
.658
.59.
117
.9
Car
vedi
lol
FC
T
2 2.
516
.038
.623
.451
.724
.344
.97.
68.
5 C
efdi
tore
n P
ivox
il
FC
T
2 10
030
.235
.035
.438
.630
.337
.19.
611
.7
Cil
nid
ipin
e F
CT
2
1023
.134
.227
.740
.538
.140
.012
.515
.5
Cla
rith
rom
ycin
F
CT
2
5020
.750
.526
.252
.454
.459
.18.
711
.4
F
CT
2
200
14.1
36.1
13.6
24.8
39.7
40.7
5.3
12.7
E
bast
ine
FC
T
2 5
13.6
26.0
15.0
29.4
25.0
17.9
4.6
9.4
F
CT
2
1016
.724
.122
.927
.616
.921
.44.
06.
8 E
cabe
t S
odiu
m
GR
2
1000
23.2
38.2
23.6
35.9
90.5
76.9
13.9
18.0
E
todo
lac
FC
T
2 20
09.
217
.725
.526
.373
.139
.411
.217
.0
Itra
con
azol
e C
AP
2
5044
.947
.535
.043
.317
.924
.233
.836
.4
Lor
atad
ine
UC
T
2 10
10.5
137.
217
.390
.819
.744
.815
.836
.3
Mel
oxic
am
UC
T
2 5
6.6
23.1
12.5
16.6
28.0
32.8
5.2
13.1
UC
T
2 10
7.0
30.7
14.7
22.3
44.9
21.9
6.6
16.3
P
iogl
itaz
one
Hyd
roch
lori
de
UC
T
2 15
15.6
27.3
20.7
27.4
39.5
52.6
12.4
18.3
UC
T
2 30
15.5
21.9
16.4
24.0
53.0
59.9
7.1
15.0
P
ran
luka
st
CA
P
2 11
2.5
33.7
53.3
30.2
63.0
28.9
32.9
9.9
13.4
Q
uaz
epam
U
CT
2
1512
.337
.827
.042
.745
.341
.49.
014
.1
U
CT
2
2019
.939
.629
.633
.735
.740
.59.
415
.8
Reb
amip
ide
FC
T
2 10
016
.941
.920
.941
.834
.945
.615
.018
.3
Sim
vast
atin
U
CT
2
538
.570
.941
.875
.746
.254
.320
.524
.4
Ter
bin
afin
e H
ydro
chlo
ride
U
CT
2
125
21.5
48.3
41.2
52.2
37.2
35.5
21.6
32.2
Z
alto
prof
en
FC
T
2 80
14.3
26.6
44.4
38.9
79.9
83.9
22.3
24.1

53
(Tab
le 8
) C
ontd
…
Com
pou
nd
dosa
gefo
rm
BC
Scl
ass
actu
al
dose
(m
g)
AU
C
Vin
tra
AU
C
Vin
ter
Cm
ax
Vin
tra
Cm
ax
Vin
ter
Tm
ax
Vin
tra
Tm
ax
Vin
ter
MR
T
Vin
tra
MR
T
Vin
ter
Act
arit
F
CT
3
100
7.4
13.5
20.5
30.8
31.7
40.8
9.6
20.6
A
len
dron
ate
Sod
ium
U
CT
3
3542
.755
.844
.353
.640
.947
.67.
711
.1
An
astr
ozol
e F
CT
3
14.
918
.35.
913
.935
.542
.33.
39.
3 C
efca
pen
e P
ivox
il H
ydro
chlo
ride
F
CT
3
7533
.637
.231
.435
.338
.038
.613
.317
.0
Cef
din
ir
CA
P
3 50
16.9
27.1
19.8
24.5
17.9
33.9
10.0
16.7
F
amot
idin
e O
DT
3
109.
220
.016
.029
.928
.344
.63.
97.
4
OD
T
3 20
17.5
20.5
17.0
23.2
20.2
42.1
5.2
9.9
Flu
con
azol
e C
AP
3
100
3.1
10.2
11.3
13.8
61.8
60.4
1.6
3.8
Laf
uti
din
e F
CT
3
109.
323
.211
.622
.427
.540
.64.
211
.3
Met
hot
rexa
te
CA
P
3 2
8.6
13.6
23.1
24.1
43.5
39.2
8.1
9.4
Mil
nac
ipra
n H
ydro
chlo
ride
F
CT
3
155.
315
.59.
820
.626
.629
.84.
58.
2
FC
T
3 25
3.9
14.4
10.7
15.6
38.4
43.8
4.1
5.8
F
CT
3
504.
314
.413
.715
.625
.148
.03.
69.
1 N
izat
idin
e C
AP
3
754.
715
.019
.028
.422
.734
.56.
416
.7
Olo
pata
din
e H
ydro
chlo
ride
F
CT
3
54.
516
.217
.522
.547
.055
.39.
413
.5
Sod
ium
Ris
edro
nat
e F
CT
3
2.5
43.2
68.8
40.7
77.6
41.7
60.8
8.7
14.1
FC
T
3 17
.527
.267
.836
.059
.553
.864
.614
.325
.3
Su
plat
ast
Tos
ilat
e C
AP
3
100
26.5
41.7
27.8
51.3
21.8
35.0
7.6
11.4
ta
ltir
elin
U
CT
3
534
.688
.338
.310
2.9
28.9
33.5
10.0
11.6
C
efca
pen
e P
ivox
il H
ydro
chlo
ride
F
CT
4
100
18.9
29.5
18.6
32.2
38.9
33.2
14.5
16.6
C
efdi
nir
C
AP
4
100
13.1
27.1
15.3
28.2
23.5
37.1
12.0
17.0
C
efpo
doxi
me
Pro
xeti
l F
CT
4
100
13.2
16.6
12.4
16.8
10.0
8.7
3.3
10.1
L
-Car
boci
stei
ne
FC
T
4 50
011
.618
.721
.127
.633
.330
.413
.718
.9
Tos
ufl
oxac
in T
osil
ate
FC
T
4 15
024
.930
.523
.129
.362
.063
.37.
78.
1

54
第 2 章 実験の部
[1]実験材料
アスピリン腸溶錠はゼンアスピリン錠 100(沢井製薬(株))を使用した。
アセトアミノフェンは和光純薬工業(株)、セチリジン塩酸塩は相模化成(株)
から購入した。結晶セルロース(粒)Celphere® CP-102 および CP-507、結
晶セルロース Ceolus® PH-101 は旭化成ケミカルズ(株)、低置換度ヒドロキ
シプロピルセルロース(LH-21)は信越化学工業(株)、ヒドロキシプロピル
セルロース(HPC-L および HPC-SSL)は日本曹達(株)、タルクは富士タル
ク工業(株)、メタクリル酸コポリマーLD(Eudragit® L30D-55)は EVONIK
Industries、マクロゴール 6000 は日油(株)のものを使用した。サリチル酸
はナカライテスク(株)、サリチル酸 -d4 体は Toronto Research Chemicals、
オロパタジン塩酸塩は住友化学(株)のものを用いた。その他の試薬は和光純
薬工業(株)の試薬特級グレードのものを用いた。
[2]セチリジン塩酸塩腸溶性顆粒の製造
200 µm と 600 µm の腸溶性顆粒には核粒子として結晶セルロースの球形核
粒子である Celphere® CP-102 と Celphere® CP-507 を用いた。1200 µm の核
粒子は結晶セルロース Ceolus® PH-101 と低置換度ヒドロキシプロピルセルロ
ース(LH-21)、ヒドロキシプロピルセルロース(HPC-L)を混合し、高速撹
拌造粒機(FS-GS-5、(株)アーステクニカ)を用いて、水で造粒し、60℃に
て乾燥後、得られた顆粒を 12 号篩および 18 号篩を用いて分級した。
それぞれの核粒子に、セチリジン塩酸塩、ヒドロキシプロピルセルロース
( HPC-SSL)、タルクを水に分散した液を転動型流動層装置( Multiplex
MP-01、(株)パウレック)を用いて、給気温度:55~60℃、転動盤回転数:
300 rpm、噴霧速度:4 g/min、噴霧圧:1.8 kg/cm2 にてコーティングした。
得られた素顆粒に、メタクリル酸コポリマーLD(Eudragit® L30D-55)、マ
クロゴール 6000、タルクを水に分散した液を転動型流動層装置(MP-01)を
用いて、給気温度:57~60℃、転動盤回転数:300 rpm、噴霧速度:4 g/min、
噴霧圧:1.8 kg/cm2 にてコーティングした。
200 µm と 600 µm の腸溶性顆粒についてはそれぞれ 50 号篩、18 号篩を用
いて分級した。

55
[3]セチリジン塩酸塩腸溶性顆粒の物性評価
それぞれの製剤の含まれる腸溶性顆粒の物性評価については、粒子径はレー
ザー回折型粒度分布測定装置(LS13 320, BECKMAN COULTER)、真密度は
乾式型真密度測定計(AccuPyc1340, Micromeritics)、表面観察はデジタルマ
イクロスコープ(VHX-1000, KEYENCE)を用いた。
含量試験は以下に示す条件で、HPLC 法にて実施した。
・HPLC:L-2200((株)日立ハイテクサイエンス)
・抽出溶媒:メタノール
・移動相:リン酸水溶液 /アセトニトリル=3/2
・カラム:Inertsil ODS-3,4 mmφ × 150 mm(GL サイエンス(株))
・カラム温度:40℃
・流速:0.8 mL/min
溶出試験はパドル法には NTR-6100A(富山産業(株))を用いた。各製剤
セチリジン塩酸塩として 1 mg 相当量を取り、試験液には日局溶出試験第Ⅰ液
(pH 1.2)および溶出試験第Ⅱ液(pH 6.8)を用い、900mL、50 回転 /分で行
った。
フロースルーセル溶出試験は CE-7smart(SOTAX AG)を用いた。各製剤
セチリジン塩酸塩として 1 mg 相当量を取り、セルには直径 22.6 mm のもの
を用い、流速 30 mL/min で日局溶出試験第Ⅰ液(pH 1.2)を 2 分間(60 mL)
送液した後、溶出試験第Ⅱ液(pH 6.8)を 13 分間(390mL)送液した。
測定は HPLC 法(HPLC 条件については含量試験と同様)にて行った。

56
[4]セチリジン塩酸塩腸溶性顆粒のイヌ投与試験
イヌへの投与試験は投薬から採血まで株式会社トランスジェニックに委託
した。
(1)使用動物
前日より絶食したビーグル犬(雄、15 か月齢、体重 12~14kg)6 頭にセ
チリジン塩酸塩腸溶性顆粒製剤、アセトアミノフェン溶液、アスピリン腸
溶錠をカセット投与した。
(2)投与薬剤
・セチリジン塩酸塩腸溶性顆粒
200 µm 製剤(Lot. 0807):製剤 891 mg 中にセチリジン塩酸塩として 1
mg 含有
600 µm製剤(Lot. 0806):製剤 735 mg 中にセチリジン塩酸塩として 1mg
含有
1200 µm 製剤(Lot. 0704):製剤 663 mg 中にセチリジン塩酸塩として
1mg 含有
投与量はセチリジン塩酸塩として 0.5 mg/body とし、単回強制経口投与
する。
・アセトアミノフェン水溶液
アセトアミノフェン(Lot. PKH0042):蒸留水に HCl を添加して pH を
2.0 に調整した媒体を用い、0.2 mg/mL の濃度になるように溶解する(用
時調製)。
投与量は 10 mg/body(50 mL)を単回強制経口投与する。
・アスピリン腸溶錠
ゼンアスピリン錠 100(製造番号:11Z01):1 錠中アスピリンを 100 mg
含有
投与量は 1 回 1 錠(アスピリン 100 mg)を単回強制経口投与する。
(3)試験群構成および投与方法
塩酸セチリジン塩酸塩腸溶性顆粒製剤の 3 剤を下表に従いクロスオーバ
ーで投与する。
投与方法は、最初にアスピリン腸溶錠を動物の咽喉深部に置き口を閉じ
嚥下させる。錠剤の飲込みを確認後,カテーテルを胃に入れカテーテルチ
ューブ内にセチリジン塩酸塩腸溶性顆粒製剤を落し入れ、シリンジを接合

57
してアセトアミノフェン水溶液(50 mL)を注入する。投与後 pH2.0 水溶
液 5 mL/body でカテーテル内をフラッシングする。
【セチリジン塩酸塩腸溶性顆粒製剤の投与スケジュール】
動物番号 セチリジン塩酸塩腸溶性顆粒製剤
1 回目 2 回目 3 回目
1 200 µm 600 µm 1200 µm
2 200 µm 600 µm 1200 µm
3 600 µm 1200 µm 200 µm
4 600 µm 1200 µm 200 µm
5 1200 µm 200 µm 600 µm
6 1200 µm 200 µm 600 µm
(4)採血および血液の処理方法
採血ポイント:投与後 0.25, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8 および 24 時間
(11point)
(ただし 1 回目実施前については,投与前採血も実施)
採血部位:橈側皮静脈
血液処理:ヘパリン処理
遠心分離:6000×g,3 min,4℃
採血量:1 mL/1 時点
保存: -20℃保存
[5]血漿中薬物濃度測定
(1)アセトアミノフェン、セチリジン
試料血漿 0.2 mL に内部標準(オロパタジン)溶液および McIlvaine 緩
衝液 (pH5)を加えて希釈し、平衡化した固相カラム (Oasis® WAX,30 μm(30
mg) Extraction Plate, Waters)にロードする。McIlvaine 緩衝液 (pH 5)、水
で洗浄した後、メタノールで溶出させる。溶出液を窒素気流下乾固し、残
渣に水/メタノール (9/1)0.4 mL を加えて再溶解し、この液 10 µL を高速液
体クロマトグラフ質量分析法により分析を行う。

58
HPLC:LC-10Avp((株)島津製作所)
・分析カラム:Scherzo SM-C18(2 × 50 mm,3 µm)(インタクト(株))
・移動相 A:2 mmol/L ギ酸アンモニウム溶液
・移動相 B:アセトニトリル /40 mmol/L ギ酸アンモニウム溶液 (95/5)
時間 (min) 移動相 B(%) 流量 (mL/min)
Initial 0 0.4
3.00 95 -
5.20 95 -
5.25 0 -
6.80 Stop
質量分析計:API4000(AB Sciex)
・Ionization:ESI
・Ionspray Voltage:4500 V
・Heated Gas Temperature:700℃(設定値)
・Detection Mode:MRM(Positive Ion Detection Mode)
・Monitored Ion, Collision Energy and Scan Time
セチリジン:m/z 389 → m/z 201 CE:25 eV 100 msec
アセトアミノフェン:m/z 152 → m/z 110 CE:23 eV 100 msec
内部標準物質:m/z 338 → m/z 247 CE:33 eV 100 msec

59
(2)サリチル酸
試料血漿 0.1mL に内部標準(サリチル酸 -d4 体)溶液およびアセトニト
リル 0.9mL を加えて除タンパクを行う。遠心分離後、アセトニトリル層
0.6mL をとり、1mol/L 水酸化ナトリウム 0.02mL を加えて加温 (40 度 , 30
分間 )し、アセチルサリチル酸をサリチル酸に加水分解した後、アセトニト
リル層 0.1mL を水/ギ酸混液 0.9mL を加えて希釈する。
この液 5µL を LC-MS/MS により分析を行う。
HPLC:LC-10Avp((株)島津製作所)
・分析カラム:Inertsil ODS-3(2.1 × 50 mm, 3 µm)
(GL サイエンス(株))
・移動相 A:2 mmol/L ギ酸アンモニウム溶液
・移動相 B:アセトニトリル /40 mmol/L ギ酸アンモニウム溶液 (95/5)
時間 (min) 移動相 B(%) 流量 (mL/min)
Initial 20 0.5
1.50 60 -
2.00 100 -
3.00 100 -
3.10 20 -
4.50 Stop
質量分析計:API4000(AB Sciex)
Ionization:ESI
Ionspray Voltage: -4000 V
Heated Gas Temperature:600℃(設定値)
Detection Mode:MRM(Negative Ion Detection Mode)
Monitored Ion , Collision Energy and Scan Time
サリチル酸:m/z 137 → m/z 93 CE: -22 eV 150 msec
内部標準物質:m/z 141 → m/z 97 CE: -20 eV 150 msec
[6]統計処理
統計解析は繰り返しのある Student’s t-test で行い、p<0.05 で有意差あり
とした。

60
[7]ランソプラゾール製剤の BE 試験
それぞれの BE 試験は「後発医薬品の生物学的同等性試験ガイドライン」 3)
に従って健康成人を用い、「医薬品の臨床試験の実施の基準」(GCP) 60)に則
り、治験審査委員会で承認を得た治験実施計画書を遵守して実施した。
[8]ランソプラゾール製剤の物性評価
ランソプラゾール製剤の物性評価には[3]と同様の装置を用いた。
溶出試験は RT-3std((株)大日本精機)を用いた。試験液には日局溶出試
験第Ⅰ液(pH 1.2)および溶出試験第Ⅱ液(pH 6.8)を用い、パドル法、900mL、
50 回転 /分で行った。測定は紫外可視吸光度測定法(UV-2200A、(株)島津製
作所)で行った。

61
引用文献 1 Guidance for Industry, Bioavailability and Bioequivalence Studies
Submitted in NDAs or INDs —General Considerations. US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research 2014. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM389370.pdf.
2 Guideline, the investigation of bioequivalence. Committee for Medicinal Products for Human Use (CHMP), European Medicines Agency (EMA) 2010. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf.
3 後発医薬品の生物学的同等性試験ガイドライン , 厚生労働省 , 2012. http://www.nihs.go.jp/drug/be-guide/GL061124_BE.pdf.
4 Takeuchi, S.; Tsume, Y.; Amidon, G. E.; Amidon, G. L., Evaluation of a three compartment in vitro gastrointestinal simulator dissolution apparatus to predict in vivo dissolution. J Pharm Sci 2014, 103 (11), 3416-22.
5 Tsume, Y.; Mudie, D. M.; Langguth, P.; Amidon, G. E.; Amidon, G. L., The Biopharmaceutics Classification System: subclasses for in vivo predictive dissolution (IPD) methodology and IVIVC. Eur J Pharm Sci 2014, 57, 152-63.
6 McAllister, M., Dynamic dissolution: a step closer to predictive dissolution testing? Mol Pharm 2010, 7 (5), 1374-87.
7 Davit, B. M.; Conner, D. P.; Fabian-Fritsch, B.; Haidar, S. H.; Jiang, X.; Patel, D. T.; Seo, P. R.; Suh, K.; Thompson, C. L.; Yu, L. X., Highly variable drugs: observations from bioequivalence data submitted to the FDA for new generic drug applications. AAPS J 2008, 10 (1), 148-56.
8 Amidon, G. L.; Lennernäs, H.; Shah, V. P.; Crison, J. R., A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res 1995, 12 (3), 413-20.
9 Yamashita, S.; Tachiki, H., Analysis of risk factors in human bioequivalence study that incur bioinequivalence of oral drug products. Mol Pharm 2009, 6 (1), 48-59.
10 Sakuma, S.; Tachiki, H.; Uchiyama, H.; Fukui, Y.; Takeuchi, N.; Kumamoto, K.; Satoh, T.; Yamamoto, Y.; Ishii, E.; Sakai, Y.; Takeuchi, S.; Sugita, M.; Yamashita, S., A perspective for biowaivers of human bioequivalence studies on the basis of the combination of the ratio of AUC to the dose and the biopharmaceutics classification system. Mol Pharm 2011, 8 (4), 1113-9.
11 Access BCS Database, Drug Delivery Foundation, http://www.ddfint.org/bcs-database/.
12 Guidance for Industry, Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System. US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research, 2015.

62
http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070246.pdf
13 Kasim, N. A.; Whitehouse, M.; Ramachandran, C.; Bermejo, M.; Lennernäs, H.; Hussain, A. S.; Junginger, H. E.; Stavchansky, S. A.; Midha, K. K.; Shah, V. P.; Amidon, G. L., Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol Pharm 2004, 1 (1), 85-96.
14 後発医薬品の生物学的同等性試験ガイドライン Q&A, 厚生労働省 , 2012. http://www.nihs.go.jp/drug/be-guide/QA120229_BE.pdf.
15 Sugano, K.; Okazaki, A.; Sugimoto, S.; Tavornvipas, S.; Omura, A.; Mano, T., Solubility and dissolution profile assessment in drug discovery. Drug Metab Pharmacokinet 2007, 22 (4), 225-54.
16 Takano, R.; Takata, N.; Shiraki, K.; Higo, S.; Hayashi, Y.; Yamashita, S., A theoretical-empirical analysis on the initial dissolution rate of drugs from polydispersed particles. Biol Pharm Bull 2009, 32 (11), 1885-91.
17 Takano, R.; Furumoto, K.; Shiraki, K.; Takata, N.; Hayashi, Y.; Aso, Y.; Yamashita, S., Rate-limiting steps of oral absorption for poorly water-soluble drugs in dogs; prediction from a miniscale dissolution test and a physiologically-based computer simulation. Pharm Res 2008, 25 (10), 2334-44.
18 Johnson, K. C.; Swindell, A. C., Guidance in the setting of drug particle size specifications to minimize variability in absorption. Pharm Res 1996, 13 (12), 1795-8.
19 Curatolo, W., Physical chemical properties of oral drug candidates in the discovery and exploratory development settings. Pharmaceutical Science & Technology Today 1998, 1 (9), 387-393.
20 Yu, L. X., An integrated model for determining causes of poor oral drug absorption. Pharm Res 1999, 16 (12), 1883-7.
21 Dressman, J. B.; Amidon, G. L.; Reppas, C.; Shah, V. P., Dissolution testing as a prognostic tool for oral drug absorption: immediate release dosage forms. Pharm Res 1998, 15 (1), 11-22.
22 Hirtz, J., The gastrointestinal absorption of drugs in man: a review of current concepts and methods of investigation. Br J Clin Pharmacol 1985, 19 Suppl 2, 77S-83S.
23 Diletti, E.; Hauschke, D.; Steinijans, V. W., Sample size determination for bioequivalence assessment by means of confidence intervals. Int J Clin Pharmacol Ther Toxicol 1992, 30 Suppl 1, S51-8.
24 Bu, H. Z., A literature review of enzyme kinetic parameters for CYP3A4-mediated metabolic reactions of 113 drugs in human liver microsomes: structure-kinetics relationship assessment. Curr Drug Metab 2006, 7 (3), 231-49.
25 Yoon, Y. J.; Kim, K. B.; Kim, H.; Seo, K. A.; Kim, H. S.; Cha, I. J.; Kim, E. Y.; Liu, K. H.; Shin, J. G., Characterization of benidipine and its enantiomers' metabolism by human liver cytochrome P450 enzymes. Drug Metab Dispos 2007, 35 (9), 1518-24.
26 Senda, C.; Kishimoto, W.; Sakai, K.; Nagakura, A.; Igarashi, T., Identification of human cytochrome P450 isoforms involved in the metabolism of brotizolam. Xenobiotica 1997, 27 (9), 913-22.
27 Niwa, T.; Murayama, N.; Emoto, C.; Yamazaki, H., Comparison of kinetic

63
parameters for drug oxidation rates and substrate inhibition potential mediated by cytochrome P450 3A4 and 3A5. Curr Drug Metab 2008, 9 (1), 20-33.
28 Niwa, T.; Shiraga, T.; Ishii, I.; Kagayama, A.; Takagi, A., Contribution of human hepatic cytochrome p450 isoforms to the metabolism of psychotropic drugs. Biol Pharm Bull 2005, 28 (9), 1711-6.
29 杉山雄一 ; 楠原洋之編 , 分子薬物動態学 , 南山堂 , 2008 30 Paine, M. F.; Hart, H. L.; Ludington, S. S.; Haining, R. L.; Rettie, A. E.;
Zeldin, D. C., The human intestinal cytochrome P450 "pie". Drug Metab Dispos 2006, 34 (5), 880-6.
g metabolism by induction of cytochrome P450 3A4 enzyme. JAMA 2003, 290 (11), 1500-4.
31 「医薬品開発と適正な情報提供のための薬物相互作用ガイドライン (最終
案 ) の公表について」 , 厚 生労働省医薬食品局審査管理課事務連絡 , 32 Code, C. F.; Marlett, J. A., The interdigestive myo-electric c omplex of the stomach and small bowel of dogs. J Physiol 1975, 246 (2),
289-309. 33 Itoh, Z.; Honda, R.; Takeuchi, S.; Aizawa, I.; Takayanagi, R., An
extraluminal force t ransducer for recording contractile activity of the gastrointestinal smooth
muscle in the conscious dogs: its construction and implantation. Gastroenterol Jpn 1977, 12 (4), 275-83.
34 Vantrappen, G.; Janssens, J.; Hellemans, J.; Ghoos, Y., The interdigestive motor complex of normal subjects and pat
35 Tanaka, Y.; Goto, T.; Kataoka, M.; Sakuma, S.; Yamashita, S., Impact of Luminal Fluid Volume on the Drug Absorption After Oral Administration: Analysis Based on In Vivo Drug Concentration-Time Profile in the Gastrointestinal Tract. J Pharm Sci 2015.
36 Schiller, C.; Fröhlich, C. P.; Giessmann, T.; Siegmund, W.; Mönnikes, H.; Hosten, N.; Weitschies, W., Intestinal fluid volumes and transit of dosage forms as assessed by magnetic resonance imaging. Aliment Pharmacol Ther 2005, 22 (10), 971-9.
37 Sjögren, E.; Abrahamsson, B.; Augustijns, P.; Becker, D.; Bolger, M. B.; Brewster, M.; Brouwers, J.; Flanagan, T.; Harwood, M.; Heinen, C.; Holm, R.; Juretschke, H. P.; Kubbinga, M.; Lindahl, A.; Lukacova, V.; Münster, U.; Neuhoff, S.; Nguyen, M. A.; Peer, A.; Reppas, C.; Hodjegan, A. R.; Tannergren, C.; Weitschies, W.; Wilson, C.; Zane, P.; Lennernäs, H.; Langguth, P., In vivo methods for drug absorption - comparative physiologies, model selection, correlations with in vitro methods (IVIVC), and applications for formulation/API/excipient characterization including food effects. Eur J Pharm Sci 2014, 57, 99-151.
38 Chen, M. L.; Straughn, A. B.; Sadrieh, N.; Meyer, M.; Faustino, P. J.; Ciavarella, A. B.; Meibohm, B.; Yates, C. R.; Hussain, A. S., A modern view of excipient effects on bioequivalence: case study of sorbitol. Pharm Res 2007, 24 (1), 73-80.
39 Brouwers, J.; Brewster, M. E.; Augustijns, P., Supersaturating drug delivery systems: the answer to solubility-limited oral bioavailability? J Pharm Sci 2009, 98 (8), 2549-72.

64
40 Davit, B. M.; Conner, D. P.; Fabian-Fritsch, B.; Haidar, S. H.; Jiang, X.;
Patel, D. T.; Seo, P. R.; Suh, K.; Thompson, C. L.; Yu, L. X., Highly variable drugs: observations from bioequivalence data submitted to the FDA for new generic drug applications. AAPS J 2008, 10 (1), 148-56.
41 Bailey, D. G.; Spence, J. D.; Munoz, C.; Arnold, J. M., Interaction of citrus juices with felodipine and nifedipine. Lancet 1991, 337 (8736), 268-9.
42 Markowitz, J. S.; Donovan, J. L.; DeVane, C. L.; Taylor, R. M.; Ruan, Y.; Wang, J. S.; Chavin, K. D., Effect of St John's wort on dru
43 Malhotra, S.; Bailey, D. G.; Paine, M. F.; Watkins, P. B., Seville orange juice-felodipine interaction: comparison with dilute grapefruit juice and involvement of furocoumarins. Clin Pharmacol Ther 2001, 69 (1), 14-23.
44 Markowitz, J. S.; Donovan, J. L.; DeVane, C. L.; Taylor, R. M.; Ruan, Y.; Wang, J. S.; Chavin, K. D., Effect of St John's wort on drug metabolism by induction of cytochrome P450 3A4 enzyme. JAMA 2003, 290 (11), 1500-4.
45 「医薬品開発と適正な情報提供のための薬物相互作用ガイドライン (最終
案 ) の公表について」 , 厚生労働省医薬食品局審査管理課事務連絡 , http://www.nihs.go.jp/mss/T140710-jimu.pdf
46 Code, C. F.; Marlett, J. A., The interdigestive myo-electric complex of the stomach and small bowel of dogs. J Physiol 1975, 246 (2), 289-309.
47 Itoh, Z.; Honda, R.; Takeuchi, S.; Aizawa, I.; Takayanagi, R., An extraluminal force transducer for recording contractile activity of the gastrointestinal smooth muscle in the conscious dogs: its construction and implantation. Gastroenterol Jpn 1977, 12 (4), 275-83.
48 Vantrappen, G.; Janssens, J.; Hellemans, J.; Ghoos, Y., The interdigestive motor complex of normal subjects and patients with bacterial overgrowth of the small intestine. J Clin Invest 1977, 59 (6), 1158-66.
49 Davis, S. S.; Hardy, J. G.; Fara, J. W., Transit of pharmaceutical dosage forms through the small intestine. Gut 1986, 27 (8), 886-92.
50 Gruber, P.; Rubinstein, A.; Li, V. H.; Bass, P.; Robinson, J. R., Gastric emptying of nondigestible solids in the fasted dog. J Pharm Sci 1987, 76 (2), 117-22.
51 Rhie, J. K.; Hayashi, Y.; Welage, L. S.; Frens, J.; Wald, R. J.; Barnett, J. L.; Amidon, G. E.; Putcha, L.; Amidon, G. L., Drug marker absorption in relation to pellet size, gastric motility and viscous meals in humans. Pharm Res 1998, 15 (2), 233-8.
52 Clarke, G. M.; Newton, J. M.; Short, M. D., Gastrointestinal transit of pellets of differing size and density. International Journal of Pharmaceutics 1993, 100 (1-3), 81-92.
53 Meyer, J. H.; Elashoff, J.; Porter-Fink, V.; Dressman, J.; Amidon, G. L., Human postprandial gastric emptying of 1-3-millimeter spheres. Gastroenterology 1988, 94 (6), 1315-25.
54 Itoh, T.; Higuchi, T.; Gardner, C. R.; Caldwell, L., Effect of particle size and food on gastric residence time of non-disintegrating solids in beagle dogs. J Pharm Pharmacol 1986, 38 (11), 801-6.

65
55 Levy, G., Clinical pharmacokinetics of salicylates: a re-assessment. Br
J Clin Pharmacol 1980, 10 Suppl 2, 285S-290S. 56 Makino T, New challenges for granulation technology : desigin and super
scale-up of fluidized-bed granulation for tablet manufacturing. J. Jpn. Soc. Pharm. Mach. & Eng. 2005,14(1):5-15
57 緒方宏泰 , 鮫島政義編 , 医薬品のバイオアベイラビリティと生物学的同等性
試験 , 薬業時報社 , 1992 58 Chiou, W. L.; Jeong, H. Y.; Chung, S. M.; Wu, T. C., Evaluation of using dog
as an animal model to study the fraction of oral dose absorbed of 43 drugs in humans. Pharm Res 2000, 17 (2), 135-40.
59 Kalantzi, L.; Persson, E.; Polentarutti, B.; Abrahamsson, B.; Goumas, K.; Dressman, J. B.; Reppas, C., Canine intestinal contents vs. simulated media for the assessment of solubility of two weak bases in the human small intestinal contents. Pharm Res 2006, 23 (6), 1373-81.
60 医薬品の臨床試験の実施の基準に関する省令 , 厚生労働省 , http://law.e-gov.go.jp/htmldata/H09/H09F03601000028.html


![BIOFARMACJA - kongresfarmaceutyczny.plkongresfarmaceutyczny.pl/uploads/article/files/93dd0c3f226370e... · IVIVC @ 0 20 40 60 80 100 0 3 6 9 [h] 12 @ A B C. Prosty test in vitro nie](https://static.fdocuments.pl/doc/165x107/5b78d59a7f8b9a331e8c6b0b/biofarmacja-k-ivivc-0-20-40-60-80-100-0-3-6-9-h-12-a-b-c-prosty-test.jpg)















