
Miniaturyzacja w technikach separacyjnych. Polimerowe … · 2008-05-19 · 4.6 mm ID), D) kolumna...
41
Miniaturyzacja w technikach separacyjnych Polimerowe monolityczne fazy stacjonarne – wytwarzanie i charakterystyka Michał Szumski, Bogusław Buszewski Uniwersytet Mikołaja Kopernika, Wydział Chemii Katedra Chemii Środowiska i Bioanalityki ul. Gagarina 7, 87-100 Toruń e-mail: [email protected]
Transcript of Miniaturyzacja w technikach separacyjnych. Polimerowe … · 2008-05-19 · 4.6 mm ID), D) kolumna...

Miniaturyzacja w technikach separacyjnych Polimerowe monolityczne fazy stacjonarne
– wytwarzanie i charakterystyka
Michał Szumski, Bogusław Buszewski
Uniwersytet Mikołaja Kopernika, Wydział ChemiiKatedra Chemii Środowiska i Bioanalityki
ul. Gagarina 7, 87-100 Toruńe-mail: [email protected]

Rys historyczny
1903 – odkrycie chromatografii przez Michaiła Cwieta; 21 marca – wykład na Uniwersytecie w Warszawie.
1952 - Martin i Synge – Nagroda Nobla za wprowadzenie chromatografii podziałowej.
1957 – Martin Golay wprowadza kolumny kapilarne do chromatografii gazowej.
1964 – John C. Giddings proponuje zastosowanie takich kolumn w chromatografii cieczowej.
1967 – Csaba Horvath i współ. stosują kolumny o średnicy wewnętrznej 1mm do separacji rybonukleotydów.
1974 – Victor Pretorius (University of Pretoria) wskazuje na obiecujące możliwości zastosowania przepływu elektroosmotycznego zamiast pompowania cieczy przez kolumnę pakowaną. Pierwsze eksperymenty są nieudane, gdyż stosuje się szklane kolumny o zbyt dużej średnicy wewnętrznej co w przypadku stosowania wysokich napięć powoduje trudności z odprowadzaniem ciepła z kolumny.
1977 – Daidô Ishii i współpracownicy preparują kolumny teflonowe o średnicy wewnętrznej 0.5 mm i długości 5-30 cm, jako pierwsi tworzą celę przepływową o pojemności 0.1-0.3 μl z odcinka kapilary kwarcowej. Pierwsze próby analizy gradientowej w skali mikro.
1978 – Takao Tsuda i Miloš Novotny preparują kolumny przez wyciąganie kolumny szklanej o 0.25 mm I.D. i 5.5 mm O.D. napakowanej 30 μm ziarnem. Możliwe było otrzymanie kolumn o dc/dp 2-10, np. dp = 30 μm, dc = 70 μm, L = 12 m.
1985 – Frank Yang – koncepcja chromatografii zunifikowanej GC-SFC-HPLC.
1990 – Andreas Manz i współ. – chromatograf cieczowy w formie chip-u.
1991 – Chipy do CE, CEC, CE z derywatyzacją przed- i pokolumnową.
Lata 80 i 90 – rozwój mikro-HPLC i technik elektromigracyjnych (CE, MEKC, CEC). Unifikacja HPLC - CEC

C D E
Ewolucja kolumn do chromatografii cieczowej A) Kolumny Cwieta z lat 1903-1908, B) kolumna Sthala, C) klasyczna kolumna analityczna (250 x 4.6 mm ID), D) kolumna o zmniejszonej średnicy „microbore” (250 x 1.0 mm ID), E) kolumna kapilarna ze złączkami (250 mm x 100 μm ID), F) kolumna preparatywna (250 x 45 mm ID), G) czip COMOSS, collocatedmonolithic support structure
A B
FG
Ewolucja kolumn

Miniaturyzacja w chromatografii oznacza stosowanie kolumn o coraz mniejszych średnicach.
Początki miniaturyzacji w chromatografii cieczowej sięgają końca lat ’70. Gwałtowny rozwój rozpoczął się jednak w latach ’90 i związany był min. z postępami w technologii wytwarzania kapilar kwarcowych, najważniejszego (i jak dotąd najlepszego) materiału na mikrokolumny.
dc = 4.6 mmkolumny
konwencjonalne
dc = 1.0 mmkolumny o
zmniejszonej średnicy
kolumny kapilarne
dc = 320 μm dc = 100 μm

• Znaczne zmniejszenie zużycia faz ruchomych i stacjonarnych. Można (i należy) dozować niewielkie ilości próbek;
• Stosując mikrokolumny uzyskuje sięwiększą czułość masową;
• Możliwe jest wytwarzanie i stosowanie długich kolumn co znacznie zwiększa rozdzielczość układu separacyjnego
• Możliwość programowania temperatury;
• Unifikacja – stosowanie jednej kolumny (kapilary) w różnych technikach chromatograficznych, np. HPLC, GC, CE, CEC
• Problemy z preparatykąkolumn – napełnianie kolumn oraz ich stabilność, szczególnie w warunkach elektrochromatografii
• Problemy z detekcją, szczególnie spektrofotometryczną
Zalety i wady miniaturyzacji

Unifikacja
Koncepcja Franka Yanga (1985)
F.J. Yang Microbore column chromatography. A unified approachto chromatography. Marcel Dekker Inc. New York-Basel 1985
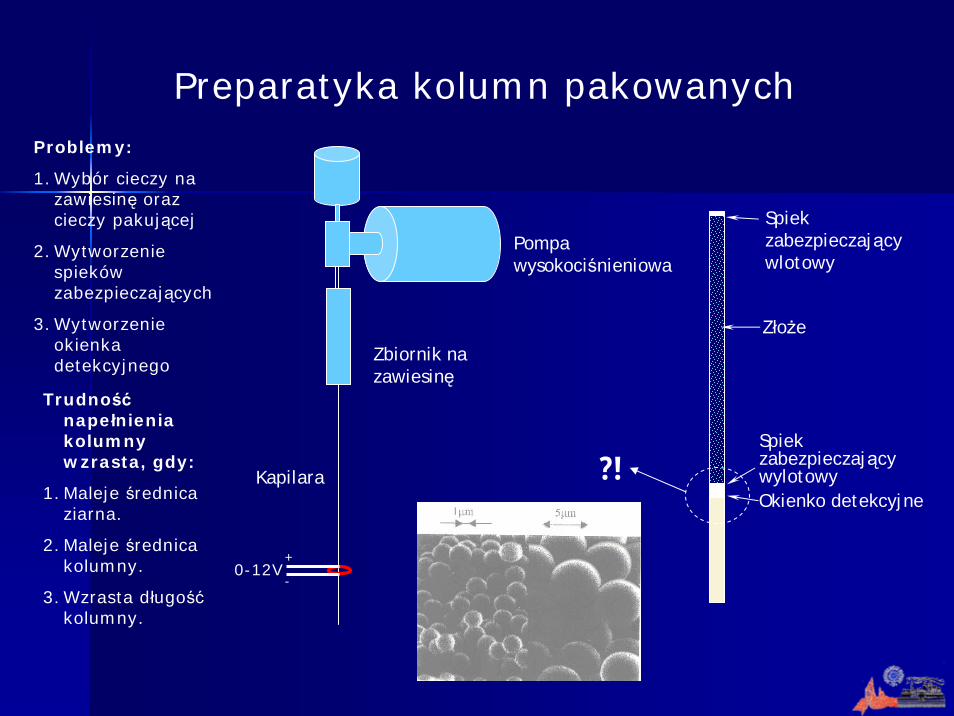
Preparatyka kolumn pakowanych
Pompa wysokociśnieniowa
Zbiornik na zawiesinę
KapilaraOkienko detekcyjne
Spiek zabezpieczający wlotowy
Złoże
Spiek zabezpieczający wylotowy?!
Problemy:
1. Wybór cieczy na zawiesinę oraz cieczy pakującej
2. Wytworzenie spieków zabezpieczających
3. Wytworzenie okienka detekcyjnego
Trudnośćnapełnienia kolumny wzrasta, gdy:
1. Maleje średnica ziarna.
2. Maleje średnica kolumny.
3. Wzrasta długośćkolumny.
0-12V+
-

Złoża monolityczne
złoże zbudowane z ziaren złoże monolityczne (monolith, rod, continuous bed)
Złoże monolityczne: ciągła, jednolita struktura porowata, w postaci szkieletu połączeń, sporządzona poprzez polimeryzacjęlub zespolenie w inny sposób (spieczenie, osadzenie, skompresowanie) materiału wewnątrz kapilary posiadająca własności chromatograficzne.
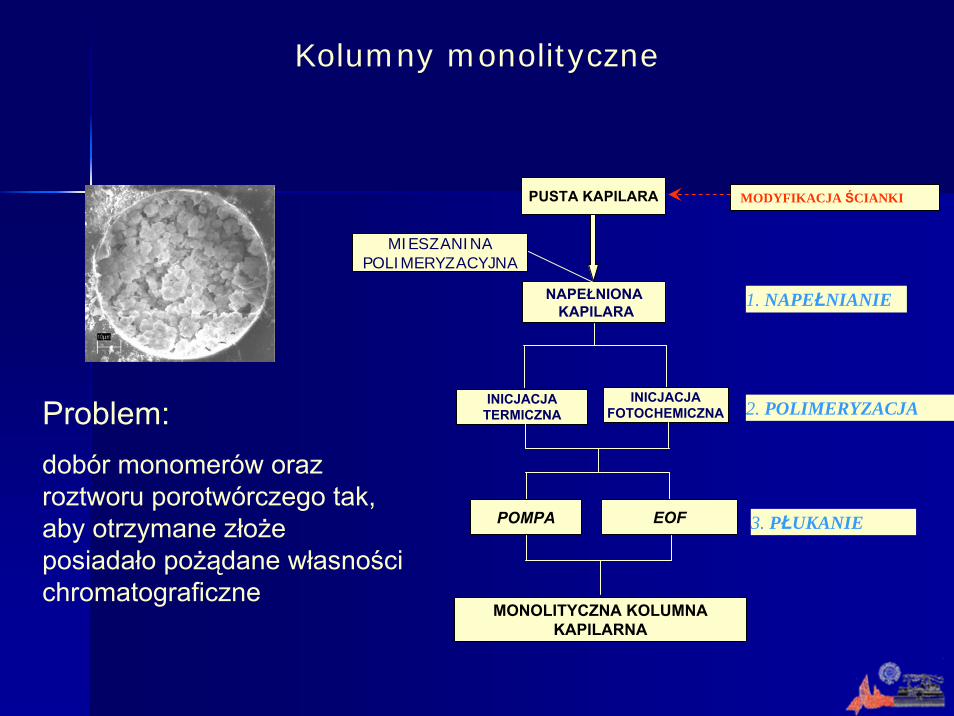
PUSTA KAPILARA
NAPEŁNIONAKAPILARA
MIESZANINA POLIMERYZACYJNA
INICJACJA TERMICZNA
INICJACJA FOTOCHEMICZNA
POMPA EOF
MONOLITYCZNA KOLUMNA KAPILARNA
1. NAPEŁNIANIE
2. POLIMERYZACJA
3. PŁUKANIE
MODYFIKACJA ŚCIANKI
Problem:dobór monomerów oraz roztworu porotwórczego tak, aby otrzymane złoże posiadało pożądane własności chromatograficzne
Kolumny monolityczne
O
OH
O
OH
O
NH2
O
NHSO3H
O
O
O
O
O
OO
O
O
OO
OO
OO CNCH3
CH3
N NCN
CH3CH3
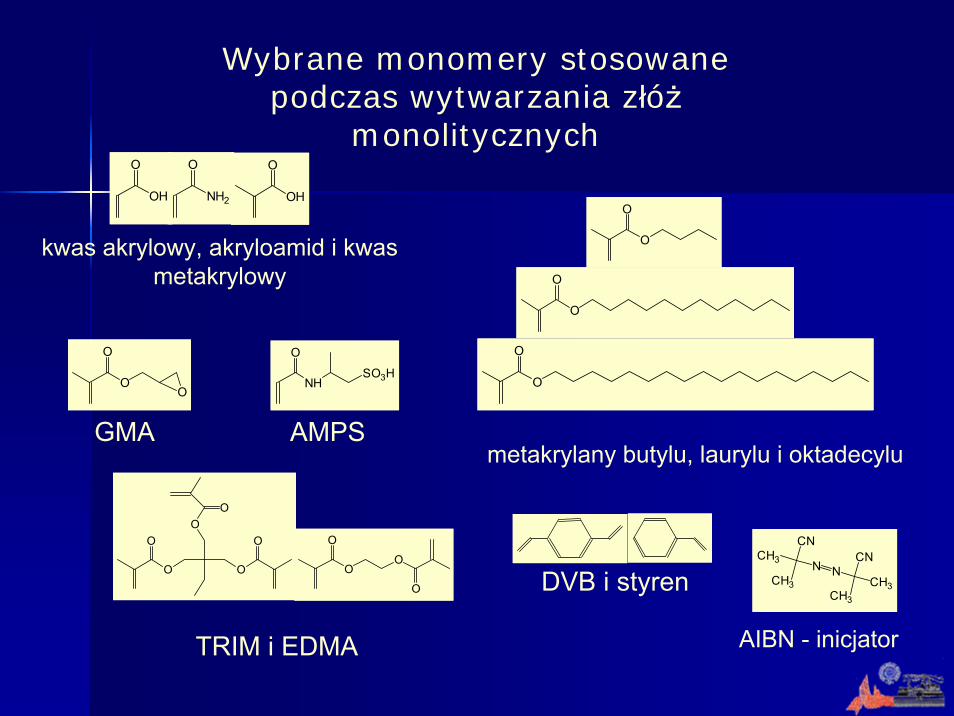
Wybrane monomery stosowane podczas wytwarzania złóż
monolitycznych
O
OO
O DVB i styren
AIBN - inicjatorTRIM i EDMA
GMA AMPS
kwas akrylowy, akryloamid i kwas metakrylowy
metakrylany butylu, laurylu i oktadecylu
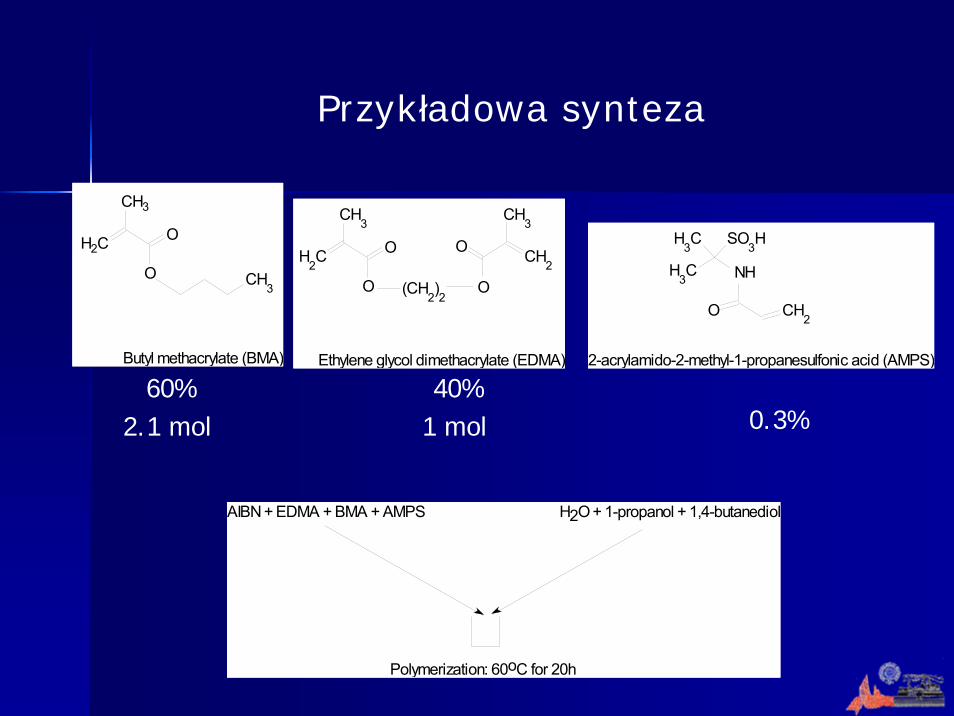
Przykładowa synteza
CH3
OH2C
O CH3
Butyl methacrylate (BMA)
CH3
O
H2C O O CH
2
CH3
O(CH2)2
Ethylene glycol dimethacrylate (EDMA)
NH
CH2
O
SO3HH
3C
H3C
2-acrylamido-2-methyl-1-propanesulfonic acid (AMPS)
AIBN + EDMA + BMA + AMPS H2O + 1-propanol + 1,4-butanediol
Polymerization: 60oC for 20h
40/60
60%2.1 mol
40%1 mol 0.3%
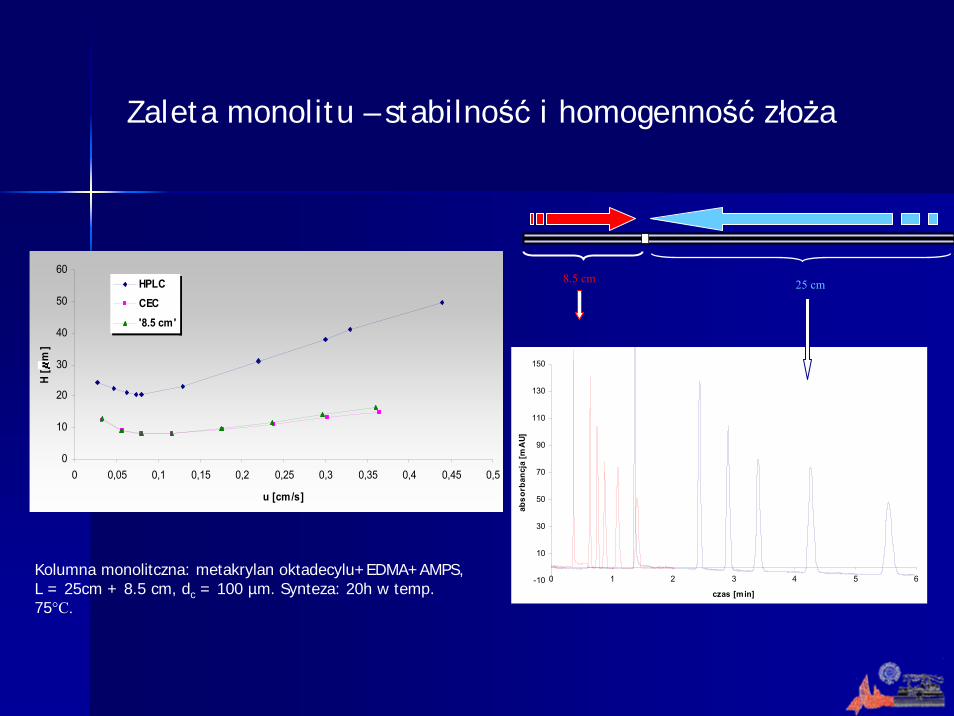
Zaleta monolitu – stabilność i homogenność złoża
0
10
20
30
40
50
60
0 0,05 0,1 0,15 0,2 0,25 0,3 0,35 0,4 0,45 0,5
u [cm/s]
H [
m]
HPLCCEC'8.5 cm'
-10
10
30
50
70
90
110
130
150
0 1 2 3 4 5 6
czas [min]
abso
rban
cja
[mA
U]
8.5 cm 25 cm
Kolumna monolitczna: metakrylan oktadecylu+EDMA+AMPS, L = 25cm + 8.5 cm, dc = 100 µm. Synteza: 20h w temp. 75°C.
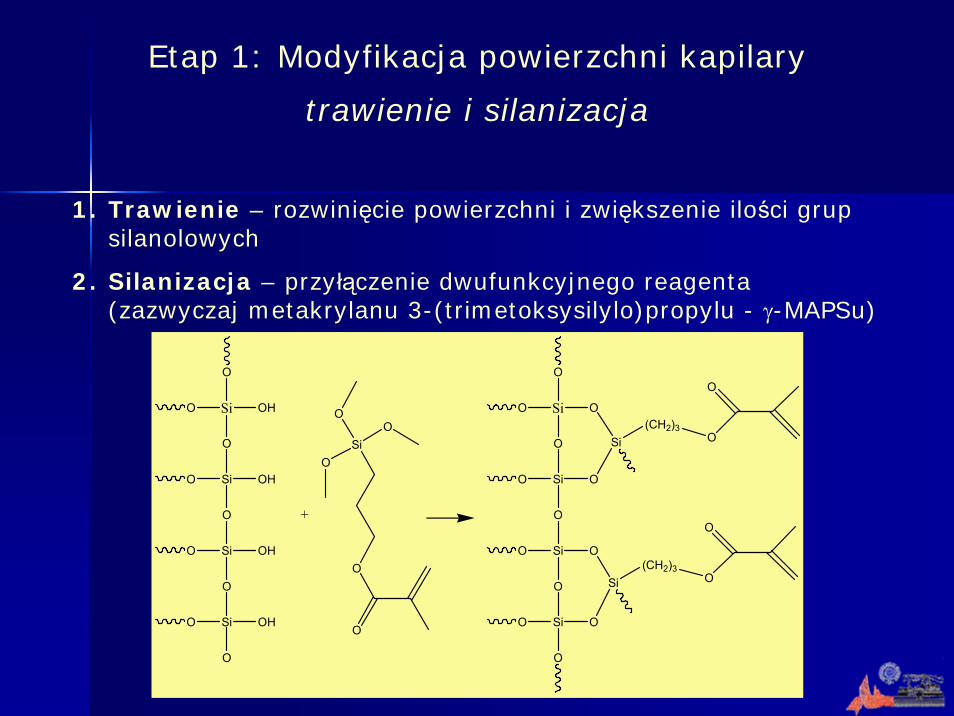
Etap 1: Modyfikacja powierzchni kapilary
trawienie i silanizacja
SiO
O
O
O
O
Si
O
O OH
O
Si OHO
O
Si
O
OHO
Si OHO
O
+
Si
O
O O
O
Si OO
O
Si
O
OO
Si OO
O
Si
Si
(CH2)3
(CH2)3
O
O
O
O
1. Trawienie – rozwinięcie powierzchni i zwiększenie ilości grup silanolowych
2. Silanizacja – przyłączenie dwufunkcyjnego reagenta (zazwyczaj metakrylanu 3-(trimetoksysilylo)propylu - γ-MAPSu)

Modyfikacja powierzchni kapilary
dlaczego ważna?
γ-MAPS spolimeryzowany w kapilarze
Monolit nie związany ze ściankąkapilary
1. Stabilność kolumny – monolit nie może być wypchnięty z kapilary
2. Sprawność kolumny – przepływ fazy ruchomej przez przestrzenie martwe między złożem a ścianką pogarsza sprawność kolumny.
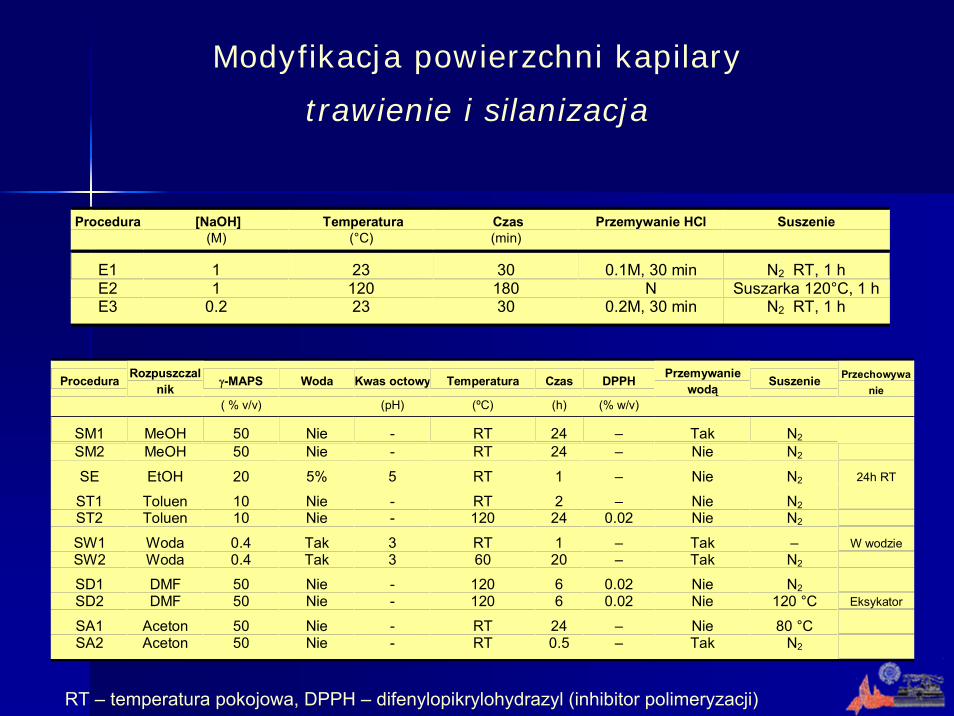
Modyfikacja powierzchni kapilary
trawienie i silanizacja
Procedura Rozpuszczalnik γ-MAPS Woda Kwas octowy Temperatura Czas DPPH Przemywanie
wodą Suszenie Przechowywanie
( % v/v) (pH) (ºC) (h) (% w/v)
SM1 MeOH 50 Nie - RT 24 – Tak N2 SM2 MeOH 50 Nie - RT 24 – Nie N2
SE EtOH 20 5% 5 RT 1 – Nie N2 24h RT
ST1 Toluen 10 Nie - RT 2 – Nie N2 ST2 Toluen 10 Nie - 120 24 0.02 Nie N2
SW1 Woda 0.4 Tak 3 RT 1 – Tak – W wodzie SW2 Woda 0.4 Tak 3 60 20 – Tak N2
SD1 DMF 50 Nie - 120 6 0.02 Nie N2 SD2 DMF 50 Nie - 120 6 0.02 Nie 120 °C Eksykator
SA1 Aceton 50 Nie - RT 24 – Nie 80 °C SA2 Aceton 50 Nie - RT 0.5 – Tak N2
Procedura [NaOH] Temperatura Czas Przemywanie HCl Suszenie (M) (°C) (min)
E1 1 23 30 0.1M, 30 min N2 RT, 1 h E2 1 120 180 N Suszarka 120°C, 1 h E3 0.2 23 30 0.2M, 30 min N2 RT, 1 h
RT – temperatura pokojowa, DPPH – difenylopikrylohydrazyl (inhibitor polimeryzacji)
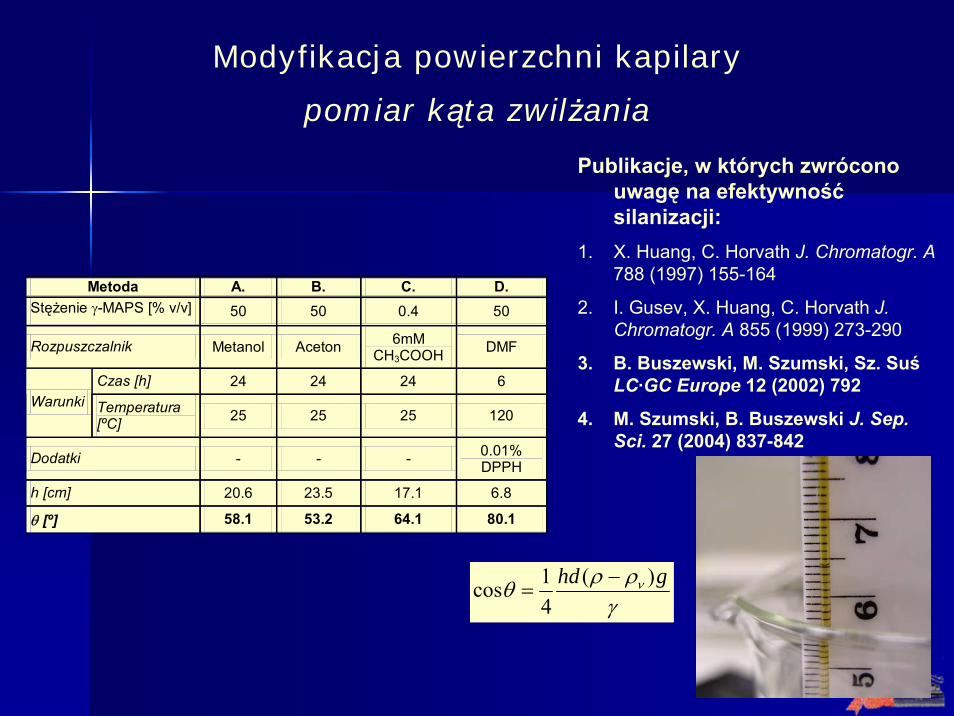
Modyfikacja powierzchni kapilary
pomiar kąta zwilżania
Metoda A. B. C. D. Stężenie γ-MAPS [% v/v] 50 50 0.4 50
Rozpuszczalnik Metanol Aceton 6mM CH3COOH DMF
Czas [h] 24 24 24 6 Warunki Temperatura
[ºC] 25 25 25 120
Dodatki - - - 0.01% DPPH
h [cm] 20.6 23.5 17.1 6.8
θ [º] 58.1 53.2 64.1 80.1
Publikacje, w których zwrócono uwagę na efektywnośćsilanizacji:
1. X. Huang, C. Horvath J. Chromatogr. A788 (1997) 155-164
2. I. Gusev, X. Huang, C. Horvath J. Chromatogr. A 855 (1999) 273-290
3. B. Buszewski, M. Szumski, Sz. SuśLC·GC Europe 12 (2002) 792
4. M. Szumski, B. Buszewski J. Sep. Sci. 27 (2004) 837-842
γρρθ ν ghd )(
41cos −
=
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
01002003004005006007008009001000
BE [eV]
inte
nsity
[cou
nts/
s]
before silanizationafter silanization
C 1s lines
0
500
1000
1500
2000
2500
3000
3500
4000
275,0280,0285,0290,0295,0300,0305,0
BE [eV]
inte
nsity
[cou
nts/
s]
O 1s
C 1s
Si 2p
C, C-H
C-OH-COO
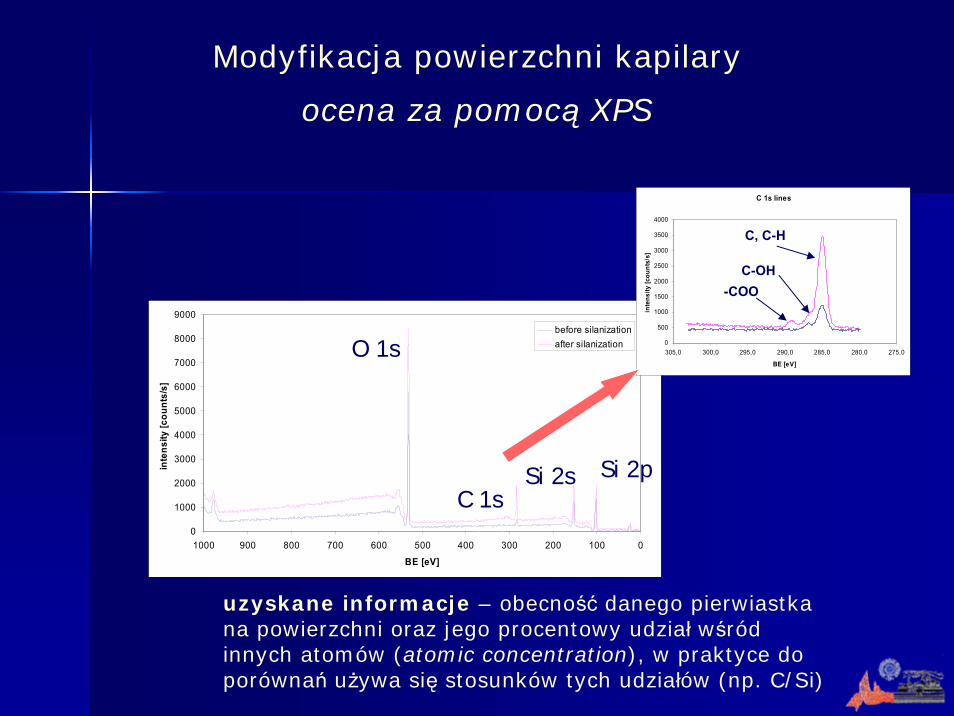
uzyskane informacje – obecność danego pierwiastka na powierzchni oraz jego procentowy udział wśród innych atomów (atomic concentration), w praktyce do porównań używa się stosunków tych udziałów (np. C/Si)
Modyfikacja powierzchni kapilary
ocena za pomocą XPS
O 1s
C 1sSi 2s Si 2p

1. Usunąć ca. 7 mm warstwy poliimidowej w środku ~8 cm odcinka kapilary o dc=1mm.
2. Naciągnąć dimetakrylan 1,6-hexanodiolu zawierający 1% AIBN.
3. Umieścić kapilarę w masce (szczelina 2mm) i zatkać końce.
4. Przeprowadzić fotopolimeryzację.
5. Napełnić kapilarę acetonitrylem i przyłączyć do pompy HPLC.Obserwować narost ciśnienia i zanotować przy jakim ciśnieniu polimer zostanie wypchnięty z kapilary.
Uzyskana informacja:
• stosując jednostki ciśnienia (MPa) możemy pokazać jak mocno polimer jest związany ze ścianką kapilary lub przylega do niej
Modyfikacja powierzchni kapilary
test adhezji

Modyfikacja powierzchni kapilary
chropowatość powierzchni
0
5
10
0
5
10
0
5
10
15
20
25
0
5
10
15
20
25
2 4
21
21
21
Kapilara trawiona w 120°CKapilara nietrawiona
Profile uzyskane podczas testu adhezji (badanie wytrzymałości polimeru na wypchnięcie z kapilary)….
…i obrazy uzyskane za pomocą mikroskopu siłatomowych (AFM)
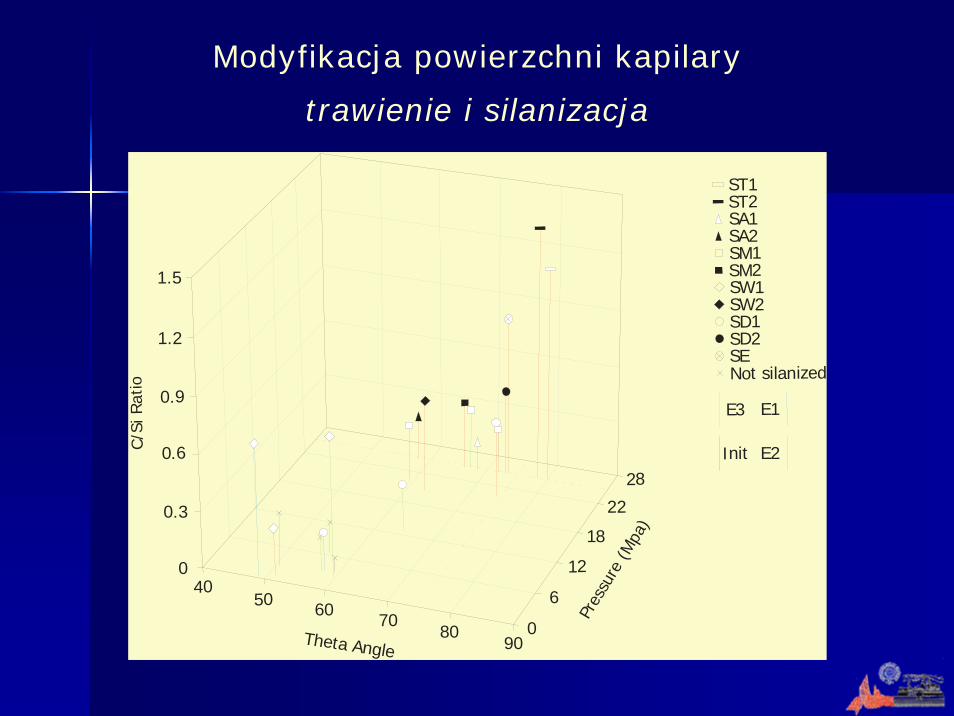
Modyfikacja powierzchni kapilary
trawienie i silanizacja
Pres
sure
(Mpa
)Theta Angle
1.5
1.2
0.9
0.6
0.3
040
50 6080
7090
0
6
12
18
22
28
C/S
i Rat
io Not silanizedSESD2SD1SW2SW1SM2SM1SA2SA1ST2ST1
Init E2
E3 E1

Czynniki wpływające na strukturę monolitycznych faz stacjonarnych
modyfikacja ścianki
stosunek monomer/monomer sieciujący
stosunek monomery/roztwór porotwórczy
skład roztworu porotwórczego
zawartość inicjatora
Dla polimeryzacji termicznej:
temperatura procesu
Dla fotopolimeryzacji:
ilość energii doprowadzonej do układu (natężenie promieniowania,czas naświetlania)
rodzaj źródła promieniowania
temperatura?

Fotopolimeryzacja - wady i zalety
☺ Proces jest bardzo szybki (zazwyczaj 0.5 ÷ 2 h) w porównaniu do polimeryzacji termicznej (16 - 24 h).
☺ Polimeryzację można prowadzić w wybranych miejscach kapilary lub kanału chipu – inne miejsca mogą być zasłonięte.
☺ Proces można prowadzić w różnych temperaturach co ma znaczenie przy wytwarzaniu polimerów z odciskiem molekularnym.
☺ Ze względu na szybkość procesu unika się efektów związanych z grawitacją.
Możliwa jest fotodegradacja polimeru.
Z uwagi na ograniczoną zdolność penetracji promieniowania UV mogąpojawić się różnice w strukturze monolitu. Z tego względu można wytwarzać polimery o relatywnie małych średnicach.
Należy stosować kapilary przejrzyste dla promieniowania UV (są dużo droższe i charakteryzują się mniejszą wytrzymałością mechaniczną).
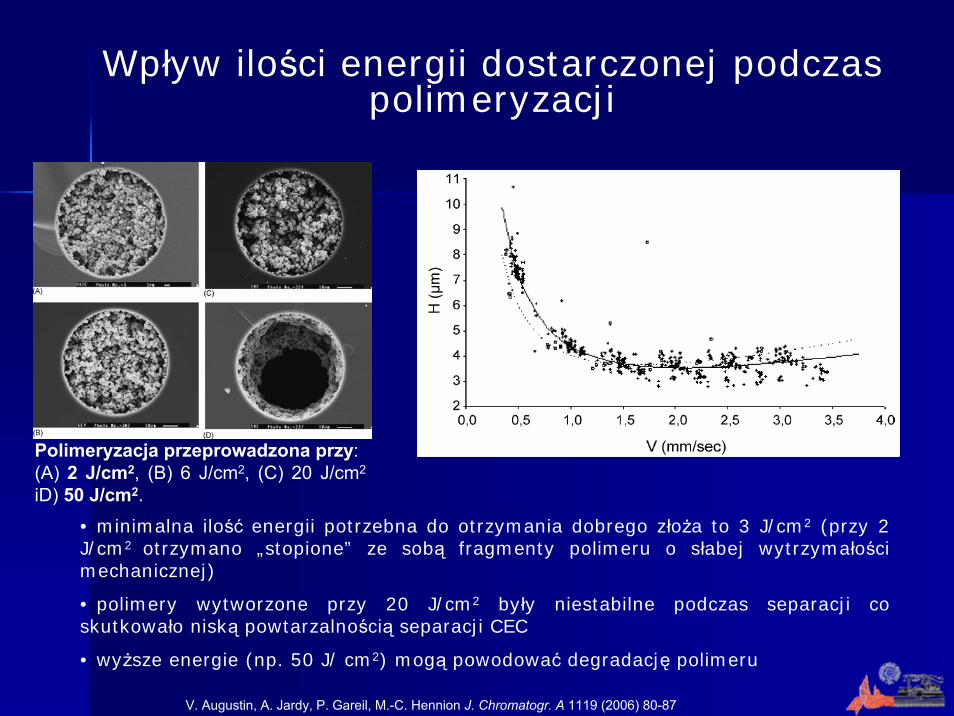
Wpływ ilości energii dostarczonej podczas polimeryzacji
V. Augustin, A. Jardy, P. Gareil, M.-C. Hennion J. Chromatogr. A 1119 (2006) 80-87
Polimeryzacja przeprowadzona przy:(A) 2 J/cm2, (B) 6 J/cm2, (C) 20 J/cm2
iD) 50 J/cm2.
20 J/cm2
4-6-8-12 J/cm2
• minimalna ilość energii potrzebna do otrzymania dobrego złoża to 3 J/cm2 (przy 2 J/cm2 otrzymano „stopione” ze sobą fragmenty polimeru o słabej wytrzymałości mechanicznej)
• polimery wytworzone przy 20 J/cm2 były niestabilne podczas separacji co skutkowało niską powtarzalnością separacji CEC
• wyższe energie (np. 50 J/ cm2) mogą powodować degradację polimeru

Wpływ źródła promieniowania
sześć 15W świetlówek UV, ciągłe naświetlanie
1500 W lampa Xe, światło pulsujące
J. Courtois, E. Byström, K. Irgum, Polymer 47 (2006) 2603

Wpływ temperatury
większość złóż monolitycznych otrzymanych w procesie fotopolimeryzacji syntetyzowano w „temperaturze pokojowej”. Niektórzy badacze starali sięutrzymywać temperaturę poniżej 30°C, z uwagi na nagrzewanie się komory reakcyjnej od włączonych lamp UV.
Schweitz i współ. prowadzili syntezę MIP w -20 ÷ -27°C (e.g. L. Schweitz, et al. Anal. Chem. 69 (1997) 1179)
Courtois et al. stosowali PEG jako składnik roztworu porotwórczego. Aby uniknąć wytrącania się PEG z mieszaniny była ona podgrzewana do ~45°C przed wprowadzeniem do kapilary. Z tego samego powodu temperatura podczas fotopolimeryzacji wynosiła ok. 35°C (nadmuch azotu). (J. Courtois, E. Byström, K. Irgum, Polymer 47 (2006) 2603)

A B
C
3
2
4
1
1 – termostat (-20°C ÷ +100°C)
2 – izolowany wymiennik ciepła
3 – płytka szklana
4 – czujnik temperatury
Procedura:
1. przygotowanie ścianki kapilary;
2. przygotowanie mieszaniny polimeryzacyjnej składającej się z 40% monomerów (60% BMA + 40% EDMA + 0.3% AMPS), 60% roztworu porotwórczego (59.55% 1-propanol + 30.45% 1,4-butanodiol + 10 % woda) i 2% BME, przedmuchiwanie azotem przez 10 min.;
3. wprowadzenie mieszaniny do kapilary;
4. polimeryzacja przez 2h po czym przepłukanie kapilary metanolem;
Zestaw do fotopolimeryzacji
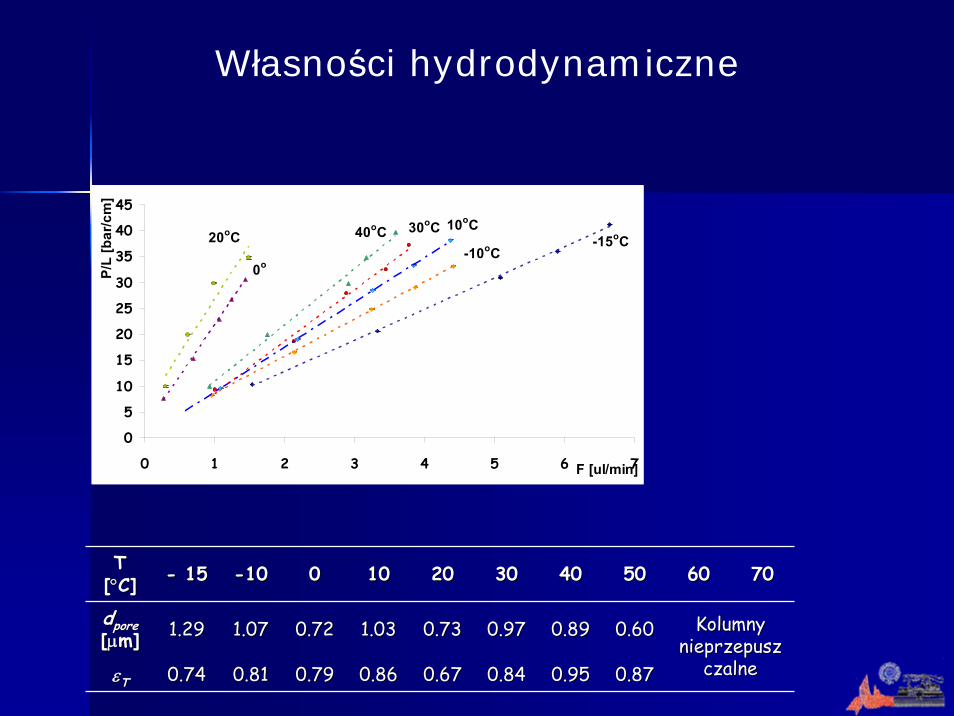
Własności hydrodynamiczne
0
5
10
15
20
25
30
35
40
45
0 1 2 3 4 5 6 7F [ul/min]
P/L
[bar
/cm
]
-15oC-10oC
10oC
0o
20oC30oC40oC
T T [[°°C]C] -- 1515 --1010 00 1010 2020 3030 4040 5050 6060 7070
ddporepore[[μμm]m] 11..2929 11..0707 00..7272 1.031.03
0.860.86
00..6060
00..8787
00..7373 00..9797 00..8989 Kolumny Kolumny nieprzepusznieprzepusz
czalneczalneεεTT 00..7474 00..8181 00..7979 00..6767 00..8484 00..9595
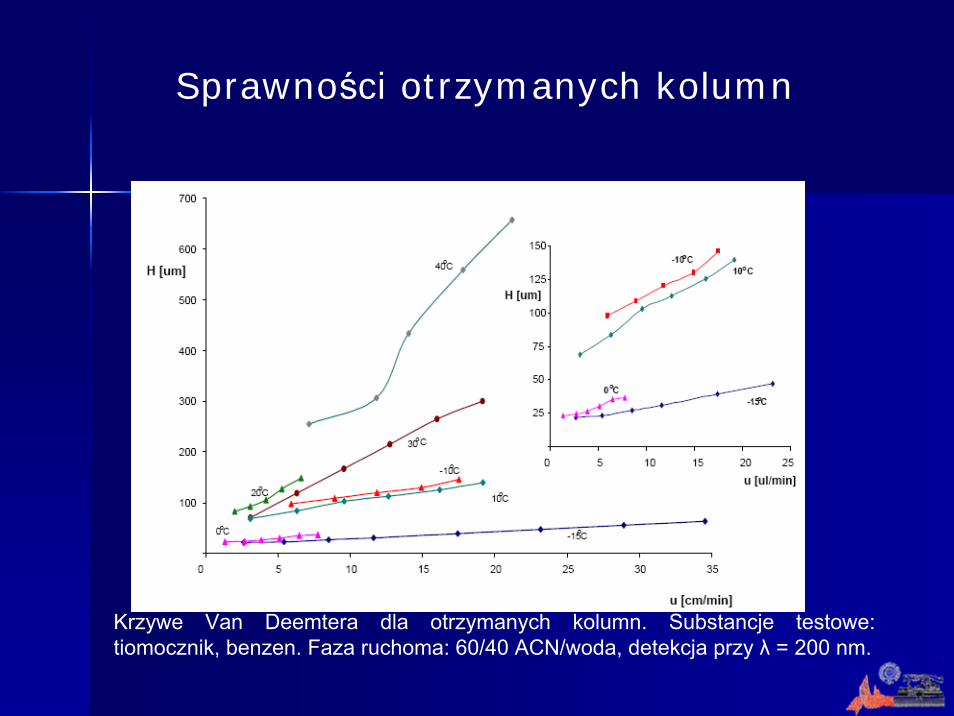
Krzywe Van Deemtera dla otrzymanych kolumn. Substancje testowe: tiomocznik, benzen. Faza ruchoma: 60/40 ACN/woda, detekcja przy λ = 200 nm.
Sprawności otrzymanych kolumn
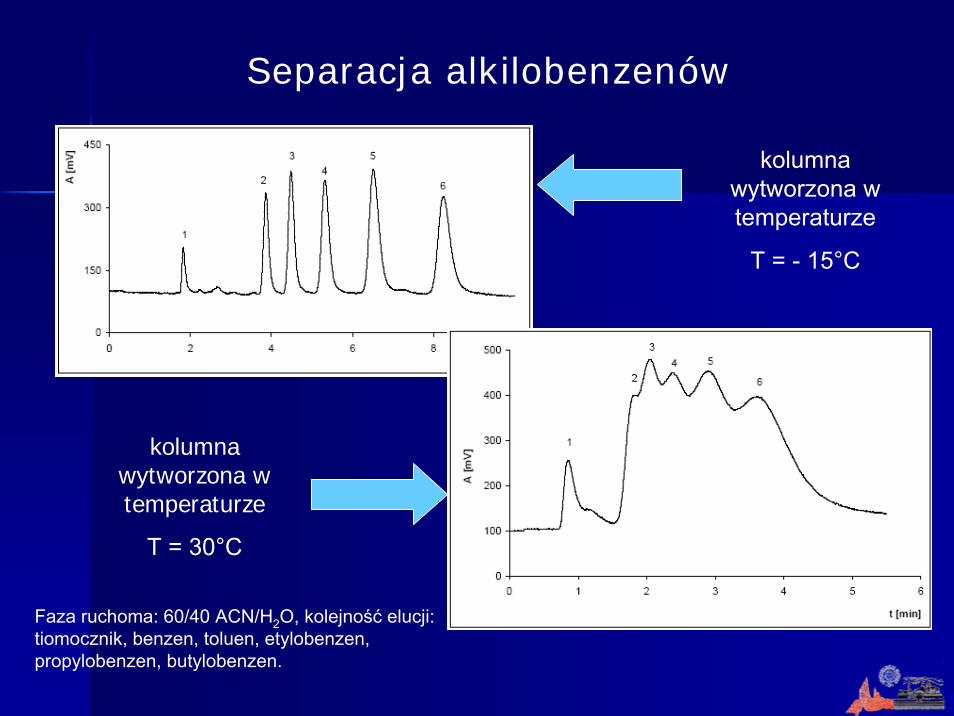
Separacja alkilobenzenów
kolumna wytworzona w temperaturze
T = - 15°C
kolumna wytworzona w temperaturze
T = 30°C
Faza ruchoma: 60/40 ACN/H2O, kolejność elucji: tiomocznik, benzen, toluen, etylobenzen, propylobenzen, butylobenzen.

Ocena porowatości złóż monolitycznych
W ocenie porowatości złóż monolitycznych stosuje się:
• pomiary powierzchni właściwej (SBET) – wymagana jest próbka makroskopowa
• rozkład porów metodą intruzji rtęci (MIP)….. – wymagana jest próbka makroskopowa
• ….lub za pomocą chromatografii wykluczania (SEC) – wykonuje się w kolumnie, stosuje się THF jako fazę ruchomą (pęcznienie niektórych polimerów)
• skaningową mikroskopię elektronową (SEM) – można stosować próbkęmakroskopową i monolit w kapilarze
czy rozkład porów w kapilarze odpowiada makroskopowej próbce dodatkowej? czy intruzja rtęci nie powoduje destrukcji polimeru?

1. Rozkład złoża monolitycznego (struktury globularne, cylindryczne, itp.) w przekroju poprzecznym kolumny.
2. Próba oceny porowatości złoża monolitycznego – porównanie z porozymetrią rtęciową.
Ocena porowatości złóż monolitycznych
Transmisyjna mikroskopia elektronowa- TEM

Przygotowanie próbki:
• zatopienie w żywicy (Spurr resin)
• usunięcie kapilary kwarcowej za pomocą np. HF
• wykonanie ultracienkich (<100nm) skrawków za pomocąmikrotomu
• utrwalanie, wybarwianie w celu uzyskania lepszego kontrastu
Ocena porowatości złóż monolitycznychTEM polimerów

Metakrylan ołowiu – dodany do mieszaniny polimeryzacyjnej (5.5 mmol GMA + 0.5 mmol Pb(MA)2)
Czerwień rutenowa – [(NH3)5RuORu(NH3)4ORu(NH3)5]Cl6 dodana do mieszaniny polimeryzacyjnej
OsO4 4 % roztwór wodny, reakcja ze złożem monolitycznym w kolumnie przez 5 h
Kwas fosfowolframowy (PTA) 5% roztwory wodne i etanolowe, 24 h
Ocena porowatości złóż monolitycznychTEM monolitów – zwiększanie kontrastu

Ocena porowatości złóż monolitycznychTEM monolitów
Pb(MA)2Ru red

OsO4 PTA w H2O PTA w EtOH
Ocena porowatości złóż monolitycznychTEM monolitów – zwiększanie kontrastu

Ocena porowatości złóż monolitycznychTEM monolitów – zwiększanie kontrastu
Podejście 2: Dodatek metakrylanu ołowiu do żywicy (1.8%).
10µm
A B
C D
Obrazy TEM: A. Chromolith (skala 10 µm),B. TEM1 (skala 5 µm), C. TEM6 (skala 10 µm) D. TEM4 (skala 5 µm)
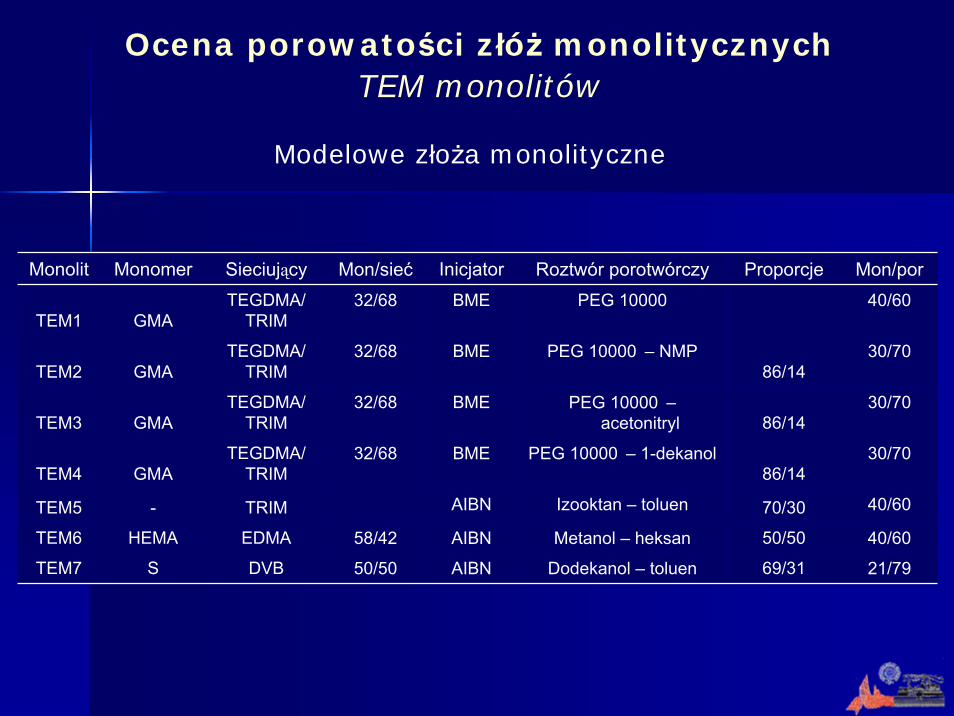
Monolit Monomer Sieciujący Mon/sieć Inicjator Roztwór porotwórczy Proporcje Mon/por
TEM1 GMATEGDMA/
TRIM32/68 BME PEG 10000 40/60
TEM2 GMATEGDMA/
TRIM 32/68 BME PEG 10000 – NMP
86/1430/70
TEM3 GMATEGDMA/
TRIM32/68 BME PEG 10000 –
acetonitryl 86/1430/70
TEM4 GMATEGDMA/
TRIM 32/68 BME PEG 10000 – 1-dekanol
86/1430/70
TEM5 - TRIM AIBN Izooktan – toluen 70/30 40/60
TEM6 HEMA EDMA 58/42 AIBN Metanol – heksan 50/50 40/60
TEM7 S DVB 50/50 AIBN Dodekanol – toluen 69/31 21/79
Modelowe złoża monolityczne
Ocena porowatości złóż monolitycznychTEM monolitów

Obróbka obrazów i pomiaryCh
rom
olith
TEM
3
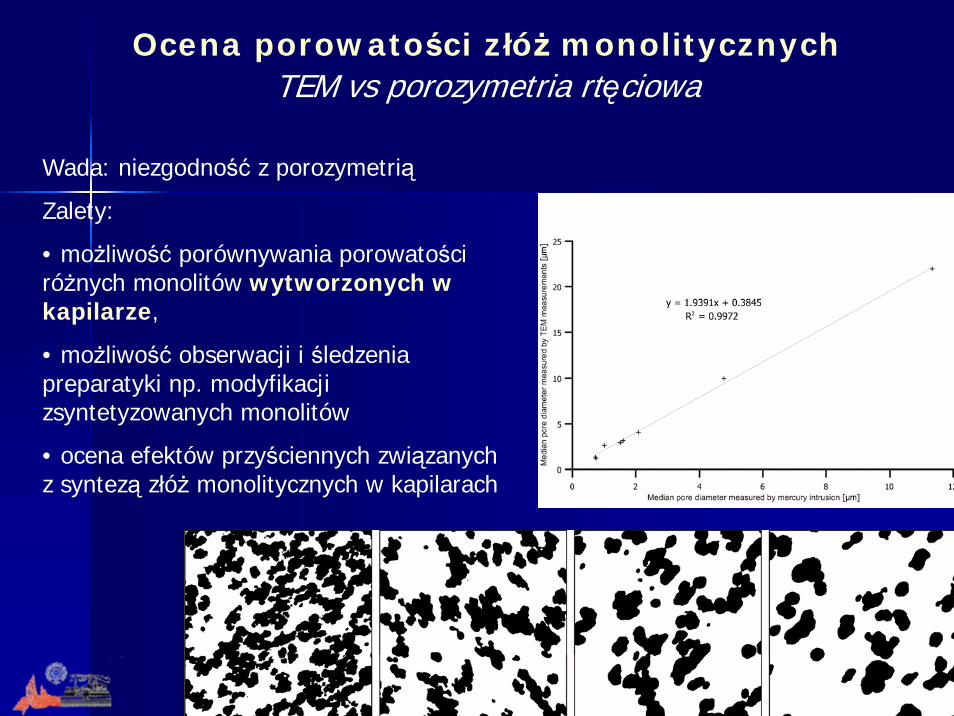
Wada: niezgodność z porozymetrią
Zalety:
• możliwość porównywania porowatości różnych monolitów wytworzonych w kapilarze,
• możliwość obserwacji i śledzenia preparatyki np. modyfikacji zsyntetyzowanych monolitów
• ocena efektów przyściennych związanych z syntezą złóż monolitycznych w kapilarach
Ocena porowatości złóż monolitycznychTEM vs porozymetria rtęciowa

Podsumowanie
• złoża monolityczne mogą stanowić doskonalą alternatywę dla kolumn pakowanych, szczególnie kapilarnych (trudnośćnapełniania kolumn kapilarnych fazą stacjonarną)
• otrzymanie „dobrej” kapilarnej kolumny monolitycznej związane jest z modyfikacją ścianki kapilary (trawienie i silanizacja)
• podczas fotopolimeryzacji należy kontrolować temperaturę
• oceniając wyniki badań porozymetrycznych należy pamiętać, że złoże w kapilarze może nie odpowiadać makroskopowej próbce monolitu
• transmisyjna mikroskopia elektronowa może dostarczyćdodatkowych informacji o strukturze złoża zsyntetyzowanego w kapilarze

Acknowledgements:
prof. Bogusław Buszewski
mgr Paulina Nadolna
Coworkers from Umeå University in Sweden:
prof. Knut Irgum, head of the RPAC (Reactive Polymers in AnalyticalChemistry) group at the Department of Analytical Chemistry
Dr. Julien Courtois
Emil Byström
Dr. Petrus Hemström
Dr. Andrei Shchukarev from Department of Inorganic Chemistry (XPS guy)
Lenore Johannsson from Swedish Defence Research Agency (FOI) (microtomation and TEM)
Per Hörsted from Department of Medical Biosciences, Pathology (SEM guy)










![QUANTITATIVE ANALYSIS OF CAST IRON … · column) on the example of interdendritic eutectics in a commercial Al alloy ... flake graphite in cast iron [20] (1 column - LM, planar metallographic](https://static.fdocuments.pl/doc/165x107/5b3327227f8b9aae458c7ee5/quantitative-analysis-of-cast-iron-column-on-the-example-of-interdendritic.jpg)







