
Modern metody analzy genomu - analza Mgr. Nikola Tom Brno, 2.4.2014.

BUDOWA GENOMU
1. Genom- zbiór wszystkich genów danego organizmu, czyli komplet jego informacji genetycznej; -termin ten oznacza również haploidalną liczbę chromosomów czyli „n”, -u eukariontów występuje w postaci molekuł DNA, -u roślin dzieli się na:
a) jądrowy: zawiera DNA skondensowane w postaci chromatyny, czyli łańcucha DNA o długości 146pz nawiniętego na zasadowe białka (zawierające głównie lizynę i argininę) histonowe złożone w oktamery z par histonów H2A, H2B, H3, H4, spiętych łącznikowym odcinkiem DNA o długości 30nm i histonem H1 który wiąże kolejne nukleosomy. -wielkość genomu jądrowego (wyrażona zawartością DNA w podstawowym kariotypie) jest zróżnicowana w zależności od gatunku rośliny, -zwykle im dłuższy cykl życiowy rośliny tym większy jej genom, oraz -zwykle więcej DNA mają rośliny nagonasienne niż drzewiaste rośliny okrytonasienne, choć są wyjątki.-wyróżniamy sekwencje kodujące (geny) i niekodujące, występujące w jednej lub wielu kopiach. -liczba genów u roślin jest zbliżona (20-30 tys.), a różnice w zawartości DNA wynikają głównie z różnej liczby powtórzeń sekwencji niekodujących. -genom jądrowy wpływa na fenotyp organizmu dwojako: przez ekspresję genów i przez fizyczną masę i objętość w jądrze i komórce.
b) roślinny mitochondrialny: genomy organellarne są zwykle koliste i superhelikalne, czasami współwystępują koliste i liniowe, najrzadziej wyłącznie liniowe, mogą być wieloczęściowe, -w liczbie kopii 1000 - 10 000 na komórkę, -są ściśle upakowane, pozbawione znacznych ilości sekwencji niekodujących, rzadko też pojawiają się introny genów -genomy mitochondrialne są zróżnicowane wielkościowo, zawierają od 6 do 2500 kpz i zwykle od ok. 5 do 100 genów kodujących mitochondrialne rRNA, tRNA i białka rybosomowe, -układ genów ulega częstym rearanżacjom w wyniku rekombinacji wewnątrzcząsteczkowych -wśród kopii DNA w mitochondrium wyróżnia się chromosom główny i cząsteczki subgenomowe.
c) plastydowy: wielkość genomów plastydów waha się w mniejszym zakresie niż mitochondrialnych, -układ genów jest zachowawczy, -geny kodują tRNA, rRNA, białka rybosomowe i translacyjne, polimerazy RNA oraz produkty zaangażowane w fotosyntezę.
2. Endosymbiotyczny transfer genów- w trakcie koewolucji plastydów i mitochondriów w komórce roślinnej wystąpił endosymbiotyczny transfer genów obu rodzajów organelli do genomu jądrowego, -którego najpowszechniej akceptowanym mechanizmem jest rekombinacja genomowego DNA organelli z DNA jądrowym, -współistnienie różnych genomów w różnych obszarach komórki wymaga ścisłej kontroli ich ekspresji, -międzyorganellarny transfer genów przebiega tylko od chloroplastów do mitochondriów.
3. Paradoks C (ang. constant) DNA- zjawisko: -nadmiaru zawartości DNA jądrowego w stosunku do podstawowego zespołu genów oraz -brak zależności między poziomem rozwoju ewolucyjnego organizmu a wielkością jego genomu; -C określa zawartość DNA w niezreplikowanym, podstawowym zespole chromosomów danego gatunku, gdzie: 1C występuje w gamecie, -zaś diploidalne komórki somatyczne przed replikacją mają 2C, -zaś po replikacji 4C.
4.Typy sekwencji DNA- można je podzielić wg liczby, długości, położenia, funkcji lub specyfiki, -długość sekwencji może wynosić od dwóch nukleotydów, przez sekwencje około długości DNA na jednym nukleosomie (180pz), do 10 000pz, -liczba kopii danej sekwencji może wynosić od 1 do kilku tysięcy w genomie haploidalnym.
DNA dzielimy na: I) geny z sekwencjami regulatorowymi: -pojedyncze, nazywane też unikatowymi, to większość genów strukturalnych, kodujących informacyjny RNA i -powtarzalne, którymi są geny kodujące transportujący i rybosomowy RNA oraz białka histonowe, występujące w powtórzeniach skupionych w rodziny genów; -ogółem geny stanowią od 0,02% do 30% zawartości genomu odwrotnie proporcjonalnie do jego wielkości (im większy genom tym procentowo mniej genów)
II)powtarzalne sekwencje pozagenowe:-wysoce powtarzalne szybko renaturujące – powyżej 105 kopii na genom haploidalny, -umiarkowanie powtarzalne wolniej renaturujące – 103 – 105 kopii/”n”,
1)tandemowe: występujące jedna za drugą w szeregu monomerów DNA niepoprzedzielanych innymi sekwencjami:a)telomery b)DNA satelitarny: najczęściej złożony z monomerów o długości 140-180pz lub 300-360pz, -o powtórzeniu do kilku milionów kopii, -charakteryzuje się odmiennym składem niż pozostały DNA jądrowy, np. wyjątkowym poziomem par A-T lub C-G,- występuje zwykle w odcinkach heterochromatyny subtelomerowej, przycentromerowej lub interstycjalnej większości lub wszystkich chromosomów danego gatunku – te charakterystyczne lokalizacje mogą być wykorzystywane jako marker do identyfikacji chromosomów,-nazwa pochodzi od powstawania odrębnego prążka dla tej frakcji DNA podczas wirowania w gradiencie gęstości chlorku cezu c)mikrosatelity to krótkie sekwencje o długości 1-6pz powtórzone tandemowo 10-12x. d)minisatelity to powtórzone odcinki satelitarnego DNA o długości 10-40pz
2)rozproszone: pojawiają się z różną częstotliwością w całym genomie, dzielą się na:

-retroelementy (transpozony klasy I) które przemieszczają się w genomie z udziałem RNA, na zasadzie amplifikacji i przeniesienia typu „kopiuj-wklej”, -ulegają standardowej transkrypcji na RNA, następnie odwrotna transkryptaza kodowana przez retrotranspozon przepisuje transkrypt na DNA, integrowany z genomem w tym samym lub innym miejscu/chromosomie co sekwencja wyjściowa, zwiększając liczbę kopii tej sekwencji:a)retrotranspozony LTR - z obecnością długich powtórzeń terminalnych, powszechne u wszystkich grup systematycznych roślin, od glonów i mszaków do nago- i okrytonasiennych, -o dł. sekw. od kilkuset do kilku tys. pz, a dł. całego retro elementu 3-20kpz,-sekwencja długiego powtórzenia terminalnego nie koduje białek,-sekwencja retrotranspozony zawiera geny:-gag kodujący białka do dojrzewania, upakowania i przygotowania RNA do integracji z genomem,-pol kodujący odwrotną transkryptazę, proteazę i RNazę H do replikacji i transpozycji,-int kodujący integrazę do katalizy insercji DNA retrotranspozonu w genomb)sekwencje SINE - krótkie rozproszone elementy jądrowe bez długich powtórzeń terminalnych, o dł. 100-500pz, nie mają genu pol ani intc)sekwencje LINE - długie rozproszone elementy jądrowe bez długich powtórzeń terminalnych, prostsze od elementów LTR, z genami gag i pol, o dł. 1-5kpz, są zakończone ogonem poliadenylowym
-transpozony DNA (klasy II), nie wymagające RNA do przemieszczenia się w genomie, są mniej powszechne u roślin, większość jest nieaktywna w wyniku metylacji, kodują m. in. transpozazę potrzebną do transpozycji tych sekwencji na zasadzie „wytnij-wklej”, powodują rearanżacje genomowe które mogą się stać przyczyną mutacji
5. Podział genów:a) kodujące białka zaangażowane w procesy: -przekształcania energii,-metabolizmu komórkowego,-wzrostu komórki,-proliferacji komórki,-syntezy DNA,-transportu,-transkrypcji,-translacji,-kierowania białek,-biogenezy komórki,-komunikacji komórkowej,-przenoszenia sygnału,-obronne,-starzenia,-śmierci,-zachowania homeostazy jonowej,-nieznane
-z analizy genomu rzodkiewnika wynika że procesy transkrypcji posiadają bardziej specyficzne mechanizmy u eukariontów, zaś procesy syntezy białka są już bardziej uwspólnione u eukariontów i są zachowawcze-w przeliczeniu globalnym na sekwencje egzonowe przypada 60% DNA, a 40% na sekwencje intronowe,-introny roślinne mają mniejszą średnią długość niż introny zwierzęce, mają więcej par A-T niż introny zwierzęce-introny roślinne dzielą się na 2 typy:a) U2 - introny o budowie klasycznej, o dominującym udziale w ogólnej liczbie intronów,b) U12 - introny o odmiennej budowie sekwencji na końcach, pozbawione traktu polipirymidowego między miejscem rozgałęzienia a końcem 3’ intronu, ich wycięcie wymaga specyficznych spliceosomów, zapewne pełnią ważne funkcje w procesach splicingu alternatywnego
b) kodujące różne RNA: -rRNA:występują tandemowo, należą do genów metabolizmu podstawowego, są eksprymowane w każdej komórce, należą do frakcji umiarkowanie powtarzalnej:- jedna jednostka strukturalna koduje 3 cząsteczki rRNA: 18S dla małej podjednostki, 5.8S, 25S dla dużej podjednostki rybosomy, transkrybowana z udziałem polimerazy I aby uzyskać prerybosomowy 45S RNA. -druga jednostka rDNA transkrybowana z udziałem polimerazy III koduje jedną 5S dla dużej podjednostki rybosomu,-liczba kopii 5S rDNA i 45S rDNA jest zróżnicowana gatunkowo, od kilkuset do kilku tysięcy w „n”, występują w różnych chromosomach i loci, różnią się nawet liczbą kopii w poszczególnych loci jednej komórki,-NTS- nietranskrybowany odcinek DNA o długości 1kpz, zawierającym sekwencję kontrolującą inicjację transkrypcji i promotor -ITS- wewnętrzna sekwencja transkrybowana znajdująca się między genami 18SrRNA (ITS 1) oraz między 5.8S i 25S (ITS 2) wycinana z dojrzewającego pre-rRNA-ETS- zewnętrzna sekwencja transkrybowana na końcu 5’, ulega transkrypcji do 45S pre-rRNA ale jest usuwana podczas dojrzewania rRNA-pojedyńcze powtórzenia kodujące rRNA mogą mieć długość 7-18kpz, ułożone są tandemowo w ściśle określonym obszarze chromosomu zwanym organizatorem jąderka w przewężeniu wtórnym chromosomu jąderko twórczego (NOR)
-tRNA:-kodowane przez 639 genów: 630 dla standardowych 20 aminokwasów, 8 psedugenów, 1 o nieznanym rodzaju kodowanego aminokwasu, żadnego dla inkorporowania selenocysteiny ani żadnego supresorowego transkrybowanych niezależnie polimerazą RNA III, nawet jeśli występują tandemowo-snRNA: małe jądrowe RNA, to tRNA występujące na terenie jądra, związane głównie ze splicingiem pre-mRNA, występuje 10 zespołów genów, z których największy tandem zawiera 7 kopii genu (5 na chromosomie 5 i 2 na chromosomie 1)-snoRNA: małe jąderkowe RNA, pełni funkcje ważne w dojrzewaniu prekursorów rRNA, metylacji 2’-O-rybozy i przekształcaniu urydyny w pseudourydynę, dzielą się ze względu na zawartość bloku C/D lub H/ACA

-mikroRNA: jednoniciowe RNA, 19-25 nukleotydów, ich funkcją jest naprowadzanie w procesie potranskrypcyjnego wyciszania genów przez blokadę transkrypcji lub rozcięcie mRNA dzięki sparowaniu z docelowym komplementarnym fragmentem mRNA
CYKL KOMÓRKOWY
Jest to szereg zmian biofizycznych i biochemicznych komórki zachodzących między końcem jednego i końcem następnego podziału. Składa się on z interfazy, czyli okresu między podziałami, oraz samego podziału, czyli mitozy lub mejozy. W interfazie zachodzi podwojenie materiału genetycznego, zaś w czasie mitozy podwojony materiał genetyczny jest rozdzielany w równych częściach do dwóch komórek potomnych. W interfazie cyklu komórkowego wyróżnia się fazę G1- między końcem mitozy a rozpoczęciem syntezy DNA, fazę S - syntezy DNA, oraz fazę G2- między końcem syntezy DNA a początkiem mitozy. W większości komórek roślin i zwierząt występuje pełny cykl tj. typu G1+S+G2+M, jednak u części komórek może on być skrócony, i tak w komórkach szybko proliferujących, rosnących w intrefazie np. nici spermatogeniczne w plemniach ramienic brak jest fazy G1, cykl typu S+G2+M. Zaś w kom. szybko proliferujących bez wzrostu kom. np. pierwsze podziały zygoty u ssaków, występuje cykl typu S+M.
FAZA G1- jest okresem życia komórki od końca mitozy do rozpoczęcia syntezy DNA. Komórki wchodzące w tą fazę są 2-krotnie mniejsze niż kom. matka. Czas trwania tej fazy jest najbardziej zmienny i wynosi od kilku do kilkunastu godzin. Faza ta charakteryzuje się intensywnymi procesami anabolicznymi, znacznym stopniem wymian chemicznych z otoczeniem oraz wzrostem innych przejawów aktywności jak ruchliwość, pinocytoza, transport przez błony itp., co prowadzi do wzrostu masy i objętości komórki. Ponadto zachodzą procesy związane z przygotowaniem do replikacji DNA tj. synteza prekursorów DNA oraz enzymów replikacyjnych.
We wczesnej fazie G1 komórka osiąga punkt restrykcyjny R i jeśli go przekroczy, wówczas podejmie syntezę DNA i zakończy cykl podziałem. Jeśli go nie przekroczy wchodzi w fazę spoczynkową G0. Mechanizm przechodzenia lub nie przez punkt R wiąże się z syntezą, nagromadzeniem i stopniem fosforylacji białek niestabilnych tzw. białek U, które są cyklinami.
FAZA S - przed każdym podziałem ilość DNA przypadająca na jądro podwaja się, dokonuje się to w ograniczonym czasie interfazy zwanym fazą syntezy (S). W fazie S ulega replikacji niemal cały jądrowy DNA -tzw. programowana synteza DNA - według sposobu semikonserwatywnego tj. podwójna spirala ulega rozdzieleniu a na każdej z jej obu nici syntetyzowana jest nowa. Istnieje też nieprogramowana synteza DNA dotycząca niewielkich jego fragmentów (synteza naprawcza). Jest ona następstwem uszkodzeń , mutacji nici DNA i nie jest związana z cyklem komórkowym.
FAZA G2 - obejmuje okres od zakończenia replikacji do rozpoczęcia mitozy i trwa kilka godzin. W tym czasie zachodzi synteza białek wrzeciona podziałowego gł. tubuliny oraz składników potrzebnych do odtwarzania błon otoczki jądrowej i plazmalemmy w telofazie i cytokinezie, jak również wyznaczenie płaszczyzny podziału (pierścień preprofazowy). Pod koniec fazy następuje uaktywnienie kinazy fazy M.(=MPF,=czynnik przyspieszający dojrzewanie) co prowadzi do rozpoczęcia i przeprowadzenia mitozy.
FAZA G0 - jest stanem spoczynkowym komórki - komórki funkcjonują lecz tracą zdolność odtwarzania materiału genetycznego i dzielenia się. Przejście w tą fazę może nastąpić u zwierząt z G1,u roślin z G1 lub G2. Komórki charakteryzują się obniżonym tempem metabolizmu, mniejszą aktywnością transkrypcyjną. Czas trwania tej fazy jest różny, od kilku dni do miesięcy i dłużej. Pod wpływem różnych bodźców komórki z fazy G0 mogą wchodzić w cykl komórkowy, zawsze do fazy w której nastąpiło jego przerwanie. Im dłużej komórki pozostają w fazie G0, tym więcej czasu zabiera im wejście w cykl po pobudzeniu.
MITOZA - dzieli się ją na kariokinezę i cytokinezę. W kariokinezie dzielonej na profazę, metafazę, anafazę i telofazę zachodzi kondensacja chromatyny, wytworzenie chromosomów, ich podział na chromatydy i przemieszczenie chromatyd do 2 potomnych komórek. Towarzyszy temu zanik jąderek, otoczki jądrowej oraz wytworzenie aparatu mitotycznego a następnie odbudowa jądra. W cytokinezie następuje podział cytoplazmy.
Regulacja cyklu komórkowego odbywa się przez uruchamianie kaskadowych reakcji fosforylacji i defosforylacji białek. Fosforylacja (przeniesienie grupy fosforanowej z ATP na odpowiednią resztę aminokwasową białka docelowego) jest katalizowana przez różnorodne kinazy białkowe, a defosforylacja przez fosfatazy. Substratami kinaz białkowych są różne białka jądra i cytoplazmy, a najczęściej fosforylowanymi aminokwasami tych białek są tyrozyna i treonina. Fosforylacja (i defosforylacja) jest jednym z najczęściej używanych przez komórkę sposobów zmiany aktywności białek.
Kinazy białkowe układu kontroli cyklu komórkowego są obecne w komórkach dzielących się podczas całego cyklu. Są jednak aktywowane tylko w odpowiednim okresie cyklu, po czym szybko tracą aktywność. Stąd aktywność każdej z tych kinaz cyklicznie zwiększa się i zmniejsza.
Aktywność kinaz białkowych zależy to od innego zestawu białek układu kontroli — od cyklin. Cykliny same nie mają aktywności enzymatycznej, ale muszą się przyłączyć do kinaz cyklu komórkowego, zanim kinazy te mogą zyskać aktywność enzymatyczną. Stąd kinazy układu kontroli cyklu komórkowego są nazywane kinazami białkowymi zależnymi od cyklin (Cdk - ang. cyclin-dpendent protein kinases). Nazwa cyklin pochodzi stąd, że przeciwnie niż poziom Cdk, ich stężenie zmienia się cyklicznie w cyklu komórkowym.
Cykliny występują w komórkach jako cykliny A i B oraz C, D i E. W czasie cyklu komórkowego cykliny A, C, D i E są syntetyzowane de novo i ich stężenie w komórce rośnie w miarę upływu cyklu, zaś cyklina B jest syntetyzowana w fazie G2. Maksymalne stężenie cyklin występuje w metafazie/ anafazie mitozy, po czym ulega ono obniżeniu na skutek trawienia ich przez proteazy.
Aktywacja kinaz zachodzi w dwóch krytycznych przedziałach czasowych (punktach kontrolnych) cyklu komórkowego: pod koniec fazy G2 (co prowadzi do przejścia G2 ® M , tj. zapoczątkowanie mitozy) oraz w fazie G1 (co prowadzi do przejścia G1 ® S , tj. zapoczątkowanie syntezy DNA). Każdy rodzaj kompleksu cyklina-Cdk działa na różny zestaw białek docelowych w komórce
Stężenie różnych typów cyklin zwiększa się, a potem gwałtownie maleje na skutek degradacji na drodze ubikwitynacji w określonym czasie cyklu komórkowego. Wzrost stężenia każdego typu cykliny wspomaga aktywację jej partnerskiej Cdk, a nagły jego spadek przywraca tę Cdk do stanu nieaktywnego. Powolne gromadzenie się cyklin, aż do krytycznego poziomu, jest jednym ze sposobów pomiaru odstępów czasu między jednym etapem cyklu a następnym w układzie kontroli cyklu komórkowego.
Przejście z późnej fazy G2 do M. dokonuje się przez aktywację kinazy fazy M, znanej jako czynnik wywołujący dojrzewanie (MPF - maturation promoting factor). Jest ona heterodimerem białkowym składającym się z białka o masie 34 kD i białka o masie 45 kD (cyklina). W kompleksie

tym białko p34 jest kinazą fosforylującą reszty seryny i treoniny wielu białek a cyklina (białko p34) nadaje aktywnemu kompleksowi powinowactwo do odpowiedniego substratu (białka, które ma być ufosforylowane).
Kinaza fazy M powstaje w fazie G2 w wyniku utworzenia kompleksu p34 z głównie z B. Kinaza MPF fosforyluje wiele kluczowych białek, zmieniając ich właściwości, np: rozpad otoczki jądrowej zachodzi przez w wyniku fosforylacji i demontażu biegnących pod otoczką jądrową filamentów laminy, podobnie fosforyluje białka towarzyszące mikrotubulom, co zmienia właściwości mikrotubul tak, że tworzą wrzeciono podziałowe, fosforyluje również histon H1 co powoduje kondensację chromosomów.
Regulacja fazy S odbywa się przez kontrolę przechodzenia komórki G1 ® S oraz przez kontrolę zakończenia syntezy DNA. Przypuszcza się, że białko p34 może łączyć się w fazie G1 głównie z cykliną A, D lub E, dając kompleks kinazy podobny do kinazy fazy M, nazywany kinazą fazy S. Aktywność takiej kinazy prowadzi komórki przez punkt startowy = restrykcyjny (w późnej fazie G1). Półokres trwania cyklin G1wynosi zaledwie ok. 15 min., co odpowiada klasie białek niestabilnych (białek U), które znane są od dawna i których nagromadzenie w komórce jest warunkiem przejścia G1 ® S.
Kinazy białkowe zależne od cyklin są regulowane nagromadzaniem i rozpadem cyklin
Regulacja stężenia cyklin ma ważny udział w synchronizacji zjawisk cyklu komórkowego. Na przykład, synteza składnika MPF — cykliny B, zaczyna się bezpośrednio po podziale i trwa stale podczas interfazy. Cyklina gromadzi się, stąd jej stężenie stopniowo zwiększa się i określa chwilę rozpoczęcia mitozy; jego późniejsze gwałtowne zmniejszenie się rozpoczyna wyjście z mitozy. Nagły spadek stężenia cykliny podczas mitozy jest spowodowany szybkim zniszczeniem cykliny w układzie proteolitycznym zależnym od ubikwityny. Wiele cząsteczek ubikwityny jest kowalencyjnie dołączonych do każdej cząsteczki cykliny, co kieruje ją do degradacji w proteosomach. Ta ubikwitynacja cykliny jest pośrednim wynikiem aktywacji kinazy MPF. Aktywacja MPF rozpoczyna proces prowadzący z opóźnieniem do ubikwitynacji i degradacji cyklin, co z kolei wyłącza kinazę.
Cykl komórkowy może zostać zatrzymany w G1 przez białkowe inhibitory Cdk
Układ kontroli cyklu komórkowego włącza zdarzenia cyklu w określonej kolejności. Na przykład, włącza mitozę tylko wtedy, gdy cały DNA został zreplikowany oraz pozwala komórce podzielić się na dwie dopiero po zakończeniu mitozy. Jeżeli jeden z etapów zostaje opóźniony, układ kontroli opóźnia aktywację następnych etapów tak, że ich sekwencja zostaje zachowana. Na przykład, ta właściwość samoregulacji układu kontroli zapewnia, że jeżeli synteza DNA zostaje zatrzymana z jakiegoś powodu w fazie S, to komórka nie wejdzie w fazę M z DNA zreplikowanym tylko w połowie.
Większość mechanizmów molekularnych odpowiedzialnych za zahamowanie biegu cyklu komórkowego w punktach kontrolnych jest słabo poznanych. W niektórych przypadkach są za to odpowiedzialne swoiste białkowe inhibitory Cdk; blokują one powstawanie bądź aktywność jednego albo kilku kompleksów cyklina-Cdk. Jeden z lepiej poznanych punktów kontrolnych zatrzymuje cykl komórkowy w G1 po uszkodzeniu DNA, co zapobiega replikacji przez komórkę uszkodzonego DNA. Uszkodzenie DNA powoduje nie poznanym dotąd mechanizmem zwiększenie stężenia i aktywności białka regulatorowego genów, nazwanego białkiem p53. Zaktywowane białko p53 zwiększa transkrypcję genu kodującego białkowy inhibitor Cdk, nazywanego p21. To zwiększa stężenie białka p21, które wiąże się z kompleksami cyklina-Cdk fazy S, odpowiedzialnymi za wprowadzenie komórki do fazy S i blokuje ich działanie. Zatrzymanie cyklu komórkowego w G1 daje komórce czas na reperację uszkodzonego DNA, zanim zostanie on zreplikowany. Gdy brak jest białka p53 albo jest ono nieaktywne, zachodzi nieograniczona replikacja uszkodzonego DNA, co zwiększa częstość mutacji i możliwości pojawienia się komórek nowotworowych. Mutacje genu p53, które pozwalają dzielić się komórkom z uszkodzonym DNA, stanowią ważny element w rozwoju większości nowotworów u człowieka.
Komórki mogą zdemontować swój układ kontroli i opuścić cykl komórkowy
Najbardziej radykalna dla układu kontroli cyklu komórkowego jest decyzja o zatrzymaniu podziałów w ogóle. Jest to inna sytuacja niż przerwa powodująca chwilowe opóźnienie w środku cyklu i ma specjalne znaczenie w organizmie wielokomórkowym. U człowieka np. komórki nerwowe i komórki mięśni szkieletowych powinny przetrwać przez całe życie organizmu bez podziałów; wchodzą one w zmodyfikowaną fazę G1 nazywaną G0. W G0 układ kontroli cyklu komórkowego jest częściowo zdemontowany, ponieważ brak w komórce wielu cyklin Cdk. Pewne typy komórek, np. komórki wątroby, prawidłowo dzielą się raz albo dwa razy w roku, natomiast pewne komórki nabłonkowe jelita dzielą się dwa bądź więcej razy dziennie, by stale odnawiać wyściółkę jelit. Większość naszych komórek mieści się między tymi skrajnościami: mogą się dzielić, gdy zajdzie taka potrzeba, ale zwykle dzielą się rzadko.
Wydaje się ogólną regułą, że komórki ssaków dzielą się tylko wtedy, gdy są pobudzane sygnałami dochodzącymi z innych komórek. Pozbawione tych sygnałów zatrzymują cykl komórkowy w punkcie kontrolnym fazy G1, i wchodzą w stan G0. Komórki mogą pozostawać w G0 przez dni, tygodnie, a nawet lata, zanim się ponownie podzielą. Stąd zmienność częstości podziałów komórek zależy od czasu, jaki komórki pozostają w G0 albo G1; gdy jednak komórka przejdzie punkt kontrolny G1, reszta cyklu komórkowego przebiega szybko, u ssaków typowo w ciągu 12-24 godzin.
Proliferacja komórek zależy od sygnałów z innych komórek
Organizmy jednokomórkowe takie jak bakterie i drożdże mają tendencję, by rosnąć i dzielić się tak szybko, jak to jest możliwe. Szybkość podziałów zależy głównie od dostępności substancji odżywczych w środowisku. Natomiast komórki organizmu wielokomórkowego są wyspecjalizowanymi członkami wysoce zorganizowanej społeczności, a ich proliferacja musi być kontrolowana. Pojedyncza komórka dzieli się tylko wtedy, gdy nowa komórka jest potrzebna organizmowi — ze względu na jego wzrost albo żeby zastąpić utraconą komórkę. Tak więc do proliferacji komórki zwierzęcej nie wystarczą substancje odżywcze. Musi ona jeszcze otrzymać pobudzający sygnał chemiczny od innych komórek, zwykle od sąsiadów. Takie działanie pozwala uniknąć użycia mechanizmów hamowania śródkomórkowego, które ograniczałoby wzrost komórki i blokowało przebieg cyklu komórkowego.
Ważnym przykładem hamowania podziałów komórki jest białko retinoblastoma (Rb), Wiąże się ono z określonymi białkami regulującymi geny, zapobiegając pobudzeniu transkrypcji genów koniecznych do podziałów. Na przykład zewnątrzkomórkowe sygnały, czynniki wzrostu (takie jak: płytkowy czynnik wzrostu, epidermalny czynnik wzrostu, czynnik wzrostu fibroblastów, czynnik wzrostu hepatocytów, erytropoetyna) pobudzają proliferację odpowiednich komórek. Między innymi powodują aktywację kompleksów cyklina-Cdk fazy G1, fosforylują one białko Rb zmieniając jego konformację tak, że uwalnia ono związane przez siebie czynniki transkrypcyjne - wtedy białka te mogą aktywować geny konieczne do przebiegu podziałów komórki.

Komórki zwierzęce mają zaprogramowane ograniczenie liczby podziałów
Nawet w obecności czynników wzrostu prawidłowe komórki zwierzęce w hodowli nie kontynuują wzrostu bez końca. Te typy komórek, które utrzymują zdolność do podziałów przez całe życie zwierzęcia, gdy pozostają w jego organizmie, zwykle przestają się dzielić po określonej liczbie podziałów w hodowli. Na przykład fibroblasty pobrane z płodu ludzkiego zanim przestaną się dzielić, przechodzą ok. 80 podziałów nawet wtedy, gdy mają wystarczająco dużo pożywienia, czynników wzrostu i miejsca do podziałów. Te zdolności komórek są jednak różne w zależności od wieku osoby, od której pobrano komórki. Fibroblasty pobrane od dorosłej 40-letniej osoby zatrzymują podziały już po ok. 40 cyklach.
Zjawisko to nazywamy starzeniem się komórki odpowiednio do starzenia się całego organizmu. Jednak ta analogia nie jest pewna. Ponieważ w hodowli fibroblasty zarodka myszy przestają się namnażać po 30 podziałach, jest możliwe, że starzenie się komórki może pomagać w wyznaczeniu wielkości ciała. Przypuszcza się, że mysz jest dlatego mniejsza niż my, gdyż jej komórki stają się niewrażliwe na pobudzenie czynnikami wzrostu już po mniejszej liczbie cykli podziałowych.
Komórki zwierzęce potrzebują sygnałów od innych komórek, by uniknąć programowanej śmierci komórki
Komórki zwierzęce potrzebują sygnałów od innych komórek nie tylko do proliferacji, lecz też do przeżycia. Pozbawione takich czynników przeżycia komórki aktywują śródkomórkowy program samobójczy i giną w procesie nazywanym programowaną śmiercią komórki. Ta konieczność otrzymywania od innych komórek sygnałów do przeżycia pomaga w utrzymaniu komórek tylko wtedy, gdy są one potrzebne i tam, gdzie są potrzebne. W tkankach rozwijających się i dojrzałych częstość programowanej śmierci komórek jest bardzo wysoka. Na przykład w rozwijającym się układzie nerwowym kręgowców ponad połowa komórek nerwowych zwykle obumiera wkrótce po ukształtowaniu się. U zdrowego człowieka w każdej godzinie miliony komórek giną w szpiku kostnym i jelicie.
Programowana śmierć komórek może służyć różnym celom. Na przykład przez programowaną śmierć komórek nasze ręce i stopy są rzeźbione podczas rozwoju zarodkowego- początkowo poszczególne palce rąk i nóg są słabo wyodrębnione a dopiero później są oddzielane, w miarę jak między nimi giną komórki. W innych przypadkach komórki giną, gdy struktury przez nie tworzone nie są potrzebne. Gdy kijanka przekształca się w żabę (metamorfoza), komórki ogona giną, a ogon niepotrzebny już żabie zanika. W jeszcze innych przypadkach śmierć komórek pomaga w regulacji liczby komórek. Na przykład w rozwijającym się układzie nerwowym śmierć komórek dostosowuje liczbę komórek nerwowych do liczby komórek docelowych wymagających unerwienia. Komórki nerwowe są w zarodku wytwarzane w nadmiarze i potem konkurują o ograniczone ilości czynników przeżycia wydzielanych przez komórki docelowe, z którymi się kontaktują. Komórki nerwowe otrzymujące wystarczającą ilość czynników przeżycia żyją, a inne giną.
W dojrzałych tkankach śmierć komórek równoważy proliferację, by zapobiec przerostowi narządów bądź ich kurczeniu się.
Programowana śmierć komórki zachodzi z udziałem śródkomórkowej kaskady proteaz
Komórki, które giną w wyniku ostrego urazu, obrzękają i pękają, uwalniając swoją zawartość do otoczenia komórek sąsiednich. Proces nazywany nekrozą (martwicą) komórki, co powoduje potencjalnie uszkadzającą odpowiedź zapalną. Natomiast komórka podlegająca śmierci programowanej umiera niezauważalnie, bez uszczerbku dla sąsiadów.
Typowy obraz śmierci programowanej w komórkach zwierzęcych to apoptoza. W trakcie apoptozy komórka kurczy się oddzielając się od sąsiednich komórek w tkance, cytoszkielet podlega zniszczeniu, otoczka jądrowa rozpada się, a jądrowy DNA jest cięty na fragmenty. Powierzchnia obumierającej komórki zmienia się - błona komórkowa pukla się do wewnątrz, odcinając kuliste fragmenty cytoplazmy (ciałka apoptotyczne) zawierające organelle i pocięty, skondensowany DNA jądrowy. Ciałka są szybko fagocytowane przez sąsiednie komórki albo przez makrofagi (wyspecjalizowane komórki fagocytujące), przez co nie następuje uwolnienie zawartości obumierającej komórki do otoczenia.
We wszystkich komórkach zwierzęcych funkcjonuje podobny układ odpowiedzialny za ten rodzaj kontrolowanej śmierci samobójczej. Składa on się z rodziny proteaz (enzymów rozcinających inne białka), które same są aktywowane proteolitycznym rozcięciem będącym odpowiedzią na sygnały indukujące programowaną śmierć komórki. Zaktywowane proteazy rozcinają i tym sposobem aktywują inne proteazy należące do tej samej rodziny białek, co razem stanowi kaskadę proteaz wzmacniającą efekt początkowy. Proteazy rozcinają następnie inne kluczowe białka w komórce, zabijając ją szybko i sprawnie. Na przykład jedna z proteaz rozcina jądrowe białka laminy powodując nieodwracalny rozpad blaszki jądrowej.
Układ śmierci samobójczej jest regulowany sygnałami z innych komórek. Niektóre działają jak sygnały zabijania, aktywując mechanizm samobójstwa komórki. W ten sposób działa hormon tarczycy w ogonie kijanki podczas metamorfozy. Inne działają jako sygnały przeżycia, hamując samobójstwo, by utrzymać komórkę przy życiu.
W organizmie wielokomórkowym programowana śmierć komórki jest zdarzeniem zwyczajnym, normalnym i ogólnie łagodnym. Natomiast niewłaściwa proliferacja i przeżywanie zbędnych komórek stanowią rzeczywiste niebezpieczeństwo – prowadzą do powstania nowotworów.
Mitoza zachodzi w komórkach somatycznych. Podział mitotyczny jest procesem ciągłym, w którym jeden etap przechodzi niepostrzeżenie w następny. Jednakże dla ułatwienia opisu przebiegu całego procesu podzielono go na cztery stadia (fazy):
1. Profazę2. Metafazę3. Anafazę4. Telofazę.
W telofazie zazwyczaj rozpoczyna się proces zwany cytokinezą. Profaza, rozpoczyna się z chwilą, gdy długie nici chromatynowe zaczynają ulegać kondensacji i uwidaczniają się chromosomy. Proces kondensacji

polega głównie na spiralizacji nici chromatynowych, powodującej skracanie i grubienie chromosomów. Zapobiega to splątywaniu się nici podczas przemieszczania się chromosomów do komórek potomnych. Każdy chromosom uległ podwojeniu podczas poprzedniej fazy S i składa się teraz z dwóch identycznych jednostek, zwanych chromatydami siostrzanymi. Każda chromatyda zawiera nie barwiące się przewężenie zwane centromerem. W rejonie centromeru siostrzane chromatydy są ze sobą ściśle połączone. Chemiczny charakter tego połączenia nie jest dotychczas całkowicie wyjaśniony, pewne dane wskazują jednak, że uczestniczy w nim specjalny rodzaj DNA i związane z nim specyficzne białka. W skład centromeru wchodzi struktura zwana kinetochorem, która służy jako miejsce przyczepu mikrotubul. Dzielącą się komórkę, niezależnie od jej aktualnego kształtu, opisuje się zwykle jako kule z równikiem, oznaczającym płaszczyznę środkową (równikową), i dwoma biegunami. Pomiędzy biegunami wykształca się system włókienek białkowych - mikrotubul tworzących wrzeciono podziałowe, zwane też mitotycznym lub kariokinetycznym. Wrzeciono podziałowe uczestniczy w rozdzielaniu chromosomów podczas anafazy. Powstawanie wrzeciona podziałowego przebiega odmiennie w komórkach roślinnych i zwierzęcych. W komórkach zwierzęcych każda z dwóch centrioli podwaja się w interfazie. Mikrotubule rozchodzą się promieniście w rejonach otaczających pary centrioli, które przemieszczają się do przeciwległych biegunów. Na biegunach tworzą się jeszcze dodatkowe, promieniście ułożone wiązki krótkich mikrotubul; wraz z zajmowanym obszarem tworzą one rejon astrosfery. Centriole pełnią najprawdopodobniej rolę organizatora wrzeciona podziałowego w komórkach zwierzęcych. Sądzi się, że ich udział jest niezbędny w procesie tworzenia się ciałka podstawowego rzęsek i wici. Dzięki wspomnianemu wyżej mechanizmowi każda z komórek potomnych otrzymuje parę centrioli. W profazie jąderko zmniejsza się stopniowo i w końcu zwykle zanika. Pod koniec profazy otoczka jądrowa pęka i do kinetochoru każdej z chromatyd przyłącza się kilka mikrotubul wrzeciona podziałowego. Podwojone chromosomy przemieszczają się pomiędzy biegunami i ostatecznie lokują w płaszczyźnie równikowej komórki, w połowie drogi między biegunami. Okres, w którym chromosomy lokują się w płaszczyźnie równikowej komórki (płytce metafazowej), nosi nazwę metafazy. Wrzeciono podziałowe jest już wtedy całkowicie uformowane. Składa się ono z licznych mikrotubul: jedne rozciągają się od każdego z biegunów do równika, gdzie zwykle zachodzą na siebie (mikrotubule biegunowe), drugie biegną od biegunów do kinetochorów (mikrotubule kinetochorowe). W metafazie podziału mitotycznego kinetochory chromatyd siostrzanych przymocowane są za pomocą mikrotubul do przeciwległych biegunów komórki. W komórkach zwierzęcych mikrotubule wrzeciona kończą się w rejonie otaczającym centriole, wypełnionym materiałem pericentriolarnym, lecz nie stykają się z centriolami. Podczas metafazy chromatydy są całkowicie skondensowane, wskutek czego są grube i łatwo rozróżnialne. Są one wtedy wyraźnie widoczne, dlatego właśnie w tym stadium robi się ich zdjęcia w celu wykrycia pewnych anomalii chromosomowych. Okres w którym siły utrzymujące łączność pomiędzy chromatydami siostrzanymi w rejonie centromeru zanikają nazywa się Anafazą. W tym momencie każda chromatyda staje się odrębnym, niezależnym chromosomem. Rozdzielone chromosomy wędrują teraz do przeciwnych biegunów. Kinetochory chromosomów, ciągle jeszcze połączone z mikrotubulami wrzeciona, posuwają się pierwsze, ciągnąc za sobą ramiona. Anafaza kończy się, gdy wszystkie chromosomy dotrą do biegunów. Pod wpływem jakich sił chromosomy się rozchodzą? Nie zostało to jeszcze w pełni poznane. Wnikliwa analiza zdjęć wykonanych w mikroskopie elektronowym pozwoliła na dwa ważne ustalenia:
1. Podczas anafazy mikrotubule ulegają skróceniu. Jedna z hipotez zakłada, że chromosomy przemieszczają się w miarę usuwania podjednostek tubuliny z końców mikrotubul, głównie w miejscu zetknięcia z kinetochorem. Jeśli kinetochor pozostaje mocno związany z mikrotubulami pomimo skracania się ich końców, to w efekcie następuje ruch chromosomów ku biegunom. 2. Podczas anafazy wrzeciono wydłuża się jako całość, co może powodować odpychanie chromosomów od siebie. Wrzeciono może wydłużać się, gdy mikrotubule wychodzące z przeciwległych biegunów ślizgają się względem siebie w rejonie równika. W ostatnim stadium - fazie mitozy, telofazie, następuje powrót do stanu właściwego interfazie. Chromosomy ulegają dekondensacji wskutek rozkręcania się. Wokół każdego zestawu chromosomów tworzy się otoczka jądrowa, w której budowie przynajmniej częściowo uczestniczą małe pęcherzyki stanowiące pozostałość po starej otoczce. Zanika wrzeciono podziałowe i uwidocznia się jąderko. Cytokineza, czyli podział cytoplazmy, którego efektem jest powstanie dwóch komórek potomnych, rozpoczyna się zwykle w telofazie, zatem mitoza i cytokineza zazębiają się w czasie. Cytokineza w komórkach zwierzęcych rozpoczyna się od utworzenia bruzdy, która obiega komórkę w rejonie równika. Bruzda, utworzona przez pierścień mikrofilamentów, stopniowo pogłębia się, aż w końcu dzieli cytoplazmę całkowicie, tak że powstają dwie komórki potomne - każda z odrębnym jądrem komórkowym. Zdumiewająca precyzja podziału komórkowego powoduje, że każde jądro otrzymuje dokładnie takie same chromosomy, jakie występowały w komórce rodzicielskiej, w dokładnie takiej samej liczbie. Zatem każda komórka organizmu wielokomórkowego (pominąwszy szczególne przypadki) ma taki sam zestaw chromosomów i zawiera identyczny zestaw genów. Jeżeli komórka na skutek nieprawidłowego przebiegu podziału komórkowego otrzyma więcej lub mniej chromosomów, niż powinna (tj. mniej lub więcej niż stanowi charakterystyczna liczba chromosomów dla danego gatunku), to może ona wykazywać liczne zaburzenia, a nawet może się okazać niezdolna do życia.
Centromer = przewężenie pierwotne w chromosomie metafazowym; średnica chromatydy w centromerze =250 nm ; średnica w ramionach = 750 nm.
Skład centromeru w chromosomie metafazowym:
DNA centromerowe, 2 kinetochory, kohezyna centromerowa Centromer jest częścią integralną - kinetochor przejściową
DNA centromerowe
heterochromatyna konstytucyjna o wysoce repetytywnych sekwencjach DNA bogatych w pary AT KINETOCHOR- jest strukturą białkową występującą po stronie zewnętrznej heterochromatyny centromerowej obu chromatyd; jest miejscem
kotwiczenia mikrotubul na centromerze Białka kinetochorowe konstytutywne (stałe) obecne podczas całego cyklu mitotycznego biorą udział w montowaniu kinetochoru Białka kinetochorowe fakultatywne (przejściowe)- są rekrutowane na DNA centromerowym dopiero przed mitozą (późne g2 do prometafazy) -
uczestnicą w funkcjonowaniu wrzeciona
Funkcje białek kinetochorowych
strukturalna punktu kontrolnego wrzeciona motoryczna
kohezyna mitotyczna- łączy chromatydy- dołączona w fazie S podjednostki SMC i Scc
SMC- białka podtrzymujące strukturę chromosomu

Scc- regulatorowe; kohezja chromatyd >>> podjednostki tworzą dimer; mogą się składać lub rozwierać
2 typy chromosomów
holocentryczne (orzęski, nicienie)- ma chromatynę centromerową w postaci wysepek wzdłuż chromosomu monocentryczne - centromerowe; chromatyna tylko w jednym miejscu (przewężenie pierwotne) >>> W centromerowych H3 zastąpiony przez CENP- A - zmodyfikowany histon> białka kinetochorowe mają powinowactwo do tego histonu
degradacja części kohezyny przez kinazę POLO >> w centromerze SEKURYNA chroni przed kinazą polo kinezyny -+- - zakotwiczenie MT do kinetochorów kinezyny --- - stabilizują wiązkowy układ MT dyneina ---- stabilizuje bieguny wrzeciona astralnego; generuje ruch chromosomów kinezyna bipolarna -+-- generuję ruch podczas anafazy B kinezyna katastroficzna - generuje ruch podczas anafazy A
zabezpieczenia między złymi połączeniami
1. punkt kontrolny wrzeciona 2. złe połączenia są destabilizowane przez AURORA B >>> fosforyluje kinazę katastroficzną- niszczy złe połączenia
IM= indeks mitotyczny= % komórek dzielących się w populacji wszystkich komórek - minimum 200 kom
IF= indeks fazowy= % udział każdej z faz w populacji komórek dzielących się
indeks mikrojąder= % komórek z mikrojądrami w 1000 komórek danej populacji
MEJOZA
Część I
Mejoza jest podziałem redukcyjnym (R!). Z jednej komórki diploidalnej (2n) powstają 4 haploidalne (1n).
Składa się z 2 rund:
1. podział redukcyjny2. podział wyrównawczy (ostateczna redukcja DNA 2c → 1c)
Pomiędzy podziałami mejotycznymi zachodzi interkineza, podczas której nie może zajść dodatkowa replikacja.
Mejoza różni się od mitozy w szczególności dwiema fazami:
Premejotyczna faza So Jest nieco dłuższa, częściowo zachodzi na profazę Io Zreplikowane w niej zostaje nie całe DNA, a tylko ok. 99.97% (ze względu na zaangażowanie mniejszej liczby replikonów). Wyjątek
stanowi Tetrachymena sp. Profaza I (profaza I-ego podziału mejotycznego)
o Jest wyjatkowo długa, np. u myszy trwa 12, u lilii – 6 dnio Ze względu na morfologię i aktywność chromosomów wyróżnia się w niej 5 kolejnych faz: leptoten, zygoten, pachyten, diploten,
diakineza(mnemonik: LeZyPaDiDia albo lepka zygota pod pachą diplodoka w diakinezie!)
1. PROFAZA I skracanie się chromosomów konfiguracja chromosomów homologicznych rekombinacja genetyczna w wyniku crossing-over synteza większości lub całej puli RNA, białek, lipidów i węglowodanów niezbędnych do utworzenia gamet i przebiegu wczesnych stadiów rozwoju
zarodkowego1.1. LEPTOTEN (gr. leptos = delikatny, cienki)
Początek kondensacji, pojawienie się chromosomów w postaci rozluźnionych nici chromatynowych Przyłączanie się telomerów do otoczki jądrowej i układanie się ich parami
1.2. ZYGOTEN Sparowane chromosomy homologiczne tworzą w wyniku koniugacji tzw. biwalenty (2 chromosomy homologiczne, 4 chromatydy). Proces
ten warunkuje kompleks synaptemalny, inaczej synaptynemalny lub synaptonemalny (SC)1.3. PACHYTEN (gr. pachus = gruby)
Kondensacja i skrócenie chromosomów homologicznych. Rekombinacja materiału genetycznego – crossing-over
1.4. DIPLOTEN (gr. diplos = podwójny)

Wyraźne podwojenie chromosomów homologicznych Zanikanie kompleksu synaptonemalnego, odpychanie się chromosomów homologicznych Ich uwolnienie z połączenia z otoczką jądrową Utrzymanie ich zasocjowania w miejscach chiazm (miejsc, w których zaszedł crossing-over)
1.5. DIAKINEZA2. METAFAZA I3. ANAFAZA I4. TELOFAZA
U myszy pachyten jest dużo dłuższy od pozostałych faz, u lilii podział czasowy jest równomierny.
Wyodrębniają się chromosomy, zaczyna się proces parowania. Chromosomy siostrzane (homologiczne) tworzą trwałą strukturę – biwalenty (tetrady – jeśli bierze się pod uwagę chromatydy). Chromosomy muszą się rozpoznać i odpowiednio ustawić (jest to skomplikowany i zagadkowy proces)
Tworzenie tzw. stadium bukietu:- Rozpoczyna się w leptotenie, istnieje w zygotenie (telomery ściśle skupione), zanika w pachytenie. /L-Z-P/- Chromosomy przyczepiają się końcami (telomerami) do otoczki jądrowej i dzięki MT* wędrują (ślizgają się) po niej.
* Jeżeli zdepolimeryzujemy MT, proces ten nie zachodzi.
Prowadzi to do skupienia się chromosomów przyczepionych do otoczki w jednym miejscu i powstania charakterystycznego ich pęczka przypominającego wyglądem bukiet.
- Dzięki temu sekwencje subtelomerowe znajdują się blisko siebie i mogą się łatwo odnaleźć. W ten sposób następuje parowanie się chromosomów homologicznych.
Parowanie chromosomów homologicznych:
- Chromosomy homologiczne zbliżają się na odl. ok. 100 nm. Zbliżanie może następować od strony telomerów, wzdłuż całych chromosomów lub tylko fragmentami (side-by-side).
- Pomiędzy nimi tworzy się struktura białkowa (rdzeń białkowy i poprzeczne białkowe włókna spajające).Synapsowanie:
- Tworzy się kompleks synaptonemalny (Synaptonemal Complex, SC).- Synapsa jest bardzo ścisłym połączeniem chromosomów.- Ma strukturę drabinkową (rys.).- Wystające z niej pętle DNA mogą mieć różną długość; ich wielkość zależy od: 1) rodzaju komórki, 2) gatunku, 3) miejsca na chromosomie (przy
końcach są mniejsze) – sprawdzono to, przenosząc daną sekwencję z jednego miejsca na drugie.Parowanie i synapsowanie to dwa odrębne procesy! Np. u pewnych mutantów drożdży występuje parowanie bez synapsowania, z kolei ... (drugi przykład?)
U Cenorabditis odkryto jeszcze jeden mechanizm rozpoznawania się chromosomów homologicznych:
HRR (Homology Recognising Region)
- po jednym HRR na każdym chromosomie- Wysunięto hipotezę, że do tego miejsca dołączana jest jakaś proteina, która przesuwa się wzdłuż chromosomów.- Być może ten mechanizm działa tylko u Cenorabditis, a przynajmniej nie występuje zawsze.
Procesy rozpoznawania się, parowania i synapsowania chromosomów homologicznych zachodzą w leptotenie i zygotenie; w pachyten wchodzą już całkowicie połączone chromosomy (biwalenty).
Wczesne węzły rekombinacyjne- pojawiają się w leptotenie i istnieją w zygotenie- występują wzdłuż SC- są stosunkowo małe- nie odpowiadają ściśle miejscom replikacji; może być ich nieco mniej, przemieszczają się po nici (?)- główne białka: Rad51, Dmc1
Późne węzły rekombinacyjne- wystepują w pachytenie (choć pojawiają się już w zygotenie), powstają jednocześnie z zanikaniem wczesnych węzłów- są to duże kompleksy- ich położenie ściśle odpowiada miejscom rekombinacji- główne białka: Msh4, Msh5, MLh1
Aby zaszła rekombinacja musi zajść przerwanie ciągłości dwuniciowego DNA.2-niciowe przerwy w DNA (Double-Strand Break, DSB)
- powstają w leptotenie

- ich tworzenie jest katalizowane przez białko Spo11 połączone kowalencyjnie z ...; na drodze trans-estryfikacji tworzy przerwy (lezje) w DNA; wykazuje powinowactwo (wg innej wersji – homologię???) do białek z grupy 2. topoizomeraz; wyizolowane z drożdży
Następnym etapem jest powiększanie tej przerwy (w kierunku 5’®3’) Działa tu białko Rad50 i Mre11.
Powstały wolny koniec „włazi” w nieprzerwane nici sąsiedniej chromatydy i tworzy D-pętlę /Rad51, Rad52, Dmc1/
W zygotenie – minimalna synteza DNA (tzw. zygptenowe DNA). Następuje też reperacja DNA względem podstawowej nici (DNA reperacyjne).W pachytenie zachodzi konwersja genów i synteza reperującego DNA na matrycy zrekombinowanej. Dlatego mówimy, że faza S w mejozie zachodzi na profazę I.W pachytenie tworzy się (i rozwiązuje się też w pachytenie) tzw. Holliday Junction (skrzyżowanie). /Zip1, Msh4, Msh5, MLh1 czyli białka zawarte w późnym węźle rekombinacyjnym – odpowiedzialne za rekombinację/
W wyniku przecięcia Holliday Junction przez ligazy powstają heterodupleksy z przemieszanymi odcinkami. (Należy zauważyć, że wychodziliśmy z dwóch homodupleksów homogonalnych, teraz powstały 2 heterodupleksy.)
W diplotenie:
- zanika otoczka jądrowa- chromosomy nieco się kondensują- zanika S.C., ale chromosomy nadal połączone w miejscach, gdzie zaszła rekombinacja genetyczna, tzw. chiazmach; chromatydy siostrzane
zespolone w centromerzeW zależności od tego, czy jest to oo- czy spermatogeneza, diploten i diakineza przebiegają nieco inaczej:
Oogenezao diploten
– trwa b. długo– komórka rośnie– chromosomy dekondensują i są b. aktywne transkrypcyjnie
o diakineza– przejście do metafazy– chromosomy rekondensują– ustaje transkrypcja
Spermatogenezao diploten i diakineza – b. krótkie, trudno wyróżnić
Metafaza
- biwalenty ustawione w powierzchni ekwatorialnej (płytka metafazalna); w przeciwieństwie do mitozy, nie kinetochory, a chiazmy- 1 miejsce kinetochorowe na 2 chromatydy (potem: 1 chromosom potomny)
Znowu istnieje różnica między oo- i spermatogenezą:
Centriole
Oogeneza – są lub nie ma (brak np. u myszy, szczura, małpy) – wrzeciono może być acentriolarne Spermatogeneza – obecne – wrzeciono zawsze centriolarle
Interkineza = „interfaza” pomiędzy 2 podziałami mejotycznymi
II podział mejotyczny – b. przypomina mitozę. U kręgowców rozpoczyna się dopiero po wniknięciu plemnika, w spermatogenezie nie ma przerwy.
Zmiany liczby chromosomów i zawartości DNA w czasie mejozy:
przed podziałem – 2n, 2c po fazie S – 2n, 4c po I podziale mejotycznym – 1n, 2c (oocyt, spermatocyt II rzędu) po II podziale mejotycznym – 1n, 1c
Powtórzyć oo- i spermatogenezę (pojęcia: oocyt I, II rzędu, ootyda itp.)!!!
U kręgowców oocyty wstrzymują rozwój w metafazie II podziału mejotycznego.
Mejoza u facetów trwa 24 h.

Pachyten i zygoten – synteza DNA. Dlatego mówi się, że w mejozie faza S zachodzi na profazę.
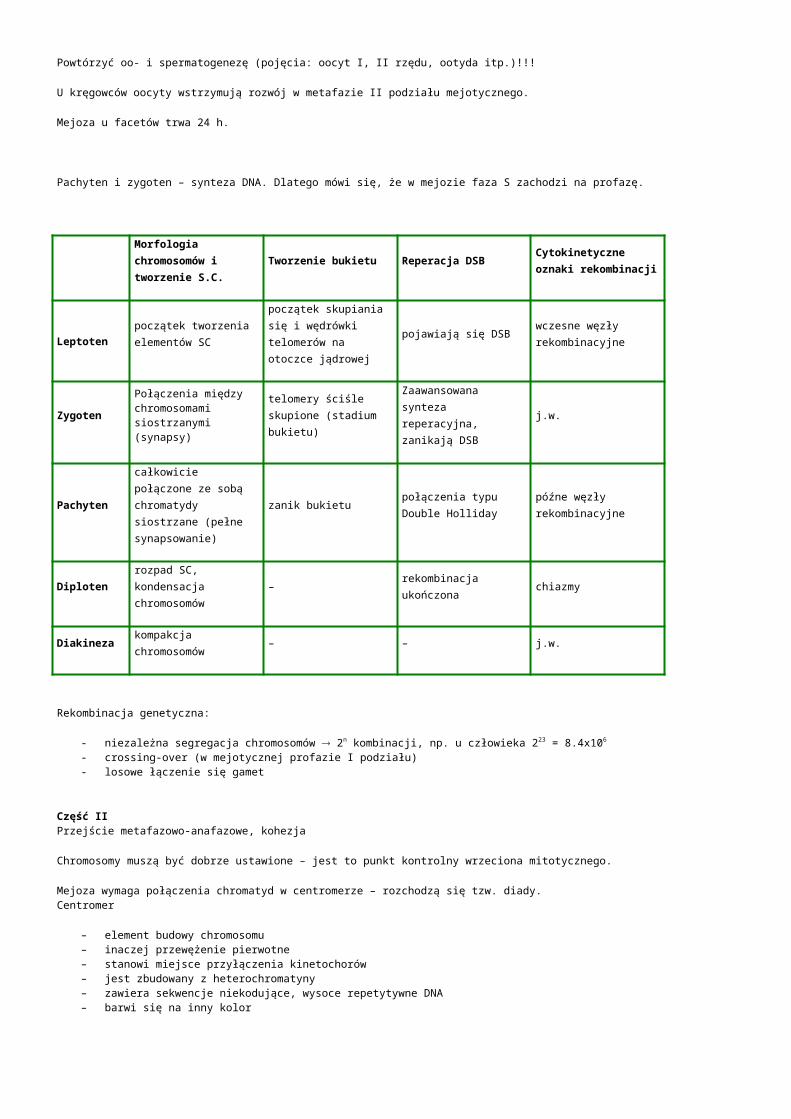
Morfologia chromosomów i tworzenie S.C.
Tworzenie bukietu Reperacja DSB Cytokinetyczne oznaki rekombinacji
Leptotenpoczątek tworzenia elementów SC
początek skupiania się i wędrówki telomerów na otoczce jądrowej
pojawiają się DSBwczesne węzły rekombinacyjne
ZygotenPołączenia między chromosomami siostrzanymi(synapsy)
telomery ściśle skupione (stadium bukietu)
Zaawansowana synteza reperacyjna, zanikają DSB
j.w.
Pachytencałkowicie połączone ze sobą chromatydy siostrzane (pełne synapsowanie)
zanik bukietupołączenia typu Double Holliday
późne węzły rekombinacyjne
Diplotenrozpad SC, kondensacja chromosomów
– rekombinacja ukończona chiazmy
Diakineza kompakcja chromosomów – – j.w.
Rekombinacja genetyczna:
- niezależna segregacja chromosomów ® 2n kombinacji, np. u człowieka 223 = 8.4x106
- crossing-over (w mejotycznej profazie I podziału)- losowe łączenie się gamet
Część IIPrzejście metafazowo-anafazowe, kohezja
Chromosomy muszą być dobrze ustawione – jest to punkt kontrolny wrzeciona mitotycznego.
Mejoza wymaga połączenia chromatyd w centromerze – rozchodzą się tzw. diady.Centromer
– element budowy chromosomu – inaczej przewężenie pierwotne– stanowi miejsce przyłączenia kinetochorów– jest zbudowany z heterochromatyny– zawiera sekwencje niekodujące, wysoce repetytywne DNA– barwi się na inny kolor
Kohezja (połączenie chromatyd siostrzanych) powstaje w fazie S cyklu komórkowego (w czasie replikacji DNA):
kohezja mitotyczna – w premitotycznej fazie S, kohezja mejotyczna – w premejotycznej fazie S.
® Chromosomal cohesin forms a ring. Gruber Haering & Nasmyth, 2003 (Cell 112: 765-777)
Such a process would be analogous to the entry of a climbing rope into carabiner, which is a ring with a gate.Rys.1
Główne białka tworzące obrączkę (kohezyny):
Mitoza:o Smc (Structural Mainterance of Chromosomes) Smc1 i Smc2o Scc (Sister Chromatid Cohesin): Scc1 i Scc3
Mejoza:o Smc1 i Smc2 – tak samo jak w mitozieo Rec8 (główna kohezyna mejotyczna) i Rec11 (od Recombination) – zamiast Scc
Rys. 2

Rozdzielenie biwalentów wymaga usunięcia kohezji między siostrzanymi chromatydami. Terminalizacja chemiczna zachodzi od centromeru w kierunku dystalnym.
® Disjunction of homologous chromosomes in meiosis I depends upon proteolytic cleavage of meiotic cohesin Rec8 by separin. Buonomo, Cyne, Fucus, Uhlmann & Nasmyth, 2000. (Cell 103: 387-398)
Przejście metafazowo-anafazowe wymaga uruchomienia separazy, która początkowo jest blokowana przez sekurynę. Proteoliza sekuryny prowadzi do aktywacji separazy.
Rys 3
Proteolizie ulega taż cyklina B kompleksu MPF (kinaza cdc2 – cyklina B).
W interkinezie zachowuje się stosunkowo wysoki poziom cykliny B i MAPK.
Zmiany zawartości DNA podczas mitozy i mejozy – przypomnienie.
U roślin nie ma wyraźnego punktu kontrolnego wrzeciona. Dlatego możliwe jest zapłodnienie ziarnem pyłku obcego gatunku (po obejściu barier związanych z zapyleniem i zapłodnieniem), a także powstawanie poliploidów. Czasem niewłaściwy pyłek padnie na niewłaściwy słupek – może powstać bezpłodna roślina, chyba, że nastąpi poliploidyzacja... Wtedy zostanie przywrócony stan diploidalności, ale nie powstaną tetrawalenty.
Roślina hybrydowa – nie rozmnaża się, brak chromosomów homologicznych, chromosomy homeologiczne rozchodzą się przypadkowo
Zablokowanie wrzeciona podziałowego (np. alkaloidem kolchicyną) -> poliploidyzacja
ENDOREPLIKACJA
endoreplikacja – zwielokrotnienie zawartości DNA poza typowym cyklem komórkowym
w ogóle nie następuje wejście w mitozę po fazach G1, S i G2 lub następuje przedwczesnezakończenie mitozy; poziom DNA po każdym cyklu endoreplikacyjnym się zwiększa;liczba chromosomów się zwiększa lub nie
gdy następuje powielenie całości DNA, mamy do czynienia z endomitozą (wtedy zwiększasię liczba chromosomów i następuje poliploidyzacja komórek) lub endoreduplikacją(wtedy liczba chromosomów się nie zwiększa, tylko liczba chromatyd chromosomów –powstają chromosmy politeniczne)
gdy następuje powielenie części DNA, mamy do czynienia z wybiórczą replikacją –amplifikacją (wtedy powielone zostają tylko pewne geny; odbywa się zazwyczaj pozachromosomami) lub niepełną replikacją (wtedy niewielkie fragmenty DNA, zazwyczajheterochromatynowe, nie zostają zreplikowane; powstają chromosomy politeniczne zfragmentami niedoreplikowanego DNA)
endoreplikacja u roślin: zjawisko powszechne do tego stopnia, że trudno by było wskazaćroślinę, u której jakieś komórki nie przeszłyby przez endoreplikację; endomitoza,endoreduplikacja i amplifikacja towarzyszą wczesnym stadiom rozwoju rośliny (lub jejorganów), np. w komórkach włosków liści, miękiszu liści (zwłaszcza u sukulentów),epidermy liści, miękiszu kory pierwotnej, włośników, prekursorów naczyń; niepełnareplikacja zachodzi w komórkach, których różnicowanie kończy się programowanąśmiercią komórkową, np. w komórkach wieszadełka, liścieni zarodka, bielma, ścian owocni,łożyska, zarodka w fazie kiełkowania
korzyści z endoreplikacji dla roślin: powielenie genomu wydaje się bardziejekonomicznym rozwiązaniem wzrostu rośliny (lub jej organu) niż intensyfikacja mitoz(endoreplikacja nie wymaga tak dużego zaangażowania związków wysokoenergetycznych,jakiego wymaga mitoza); do pewnego momentu powielenie DNA jest skorelowane zewzrostem komórek i ich ilością – komórek jest mniej i są większe (ale tylko do tetra-,oktoploidii)
endoreplikacja u zwierząt: wchodzą w nią nieliczne komórki, przede wszystkim organówzaangażowanych w sekrecję, związana z intensyfikacją metabolizmu (biologiczny sensendoreplikacji raczej nie polega na wzroście komórek, choć też jest z nim skorelowany);występuje np. u owadów w komórkach przetchlinek, cewek Malpighiego, ścian jelita,ślinianek (og. w gruczołach wydzielniczych), troficznych, follikularnych, oocytach; u

ssaków w megakariocytach i trofoblastach
endoreplikacja w cyklu komórkowym: jeżeli po replikacji do mitozy w ogóle niedochodzi, zachodzi endoreduplikacja; jeżeli przedwczesne zakończenie mitozynastępuje we wczesnej profazie, zachodzi endomitoza lub endoreduplikacja; jeżeliprzedwczesne zakończenie mitozy następuje we wczesnej anafazie, zachodziendomitoza; jeżeli zreplikowana zostaje niecała chromatyna (przede wszystkimeuchromatyna lub jej część), po czym nie dochodzi do mitozy, zachodzi amplifikacja lubniepełna replikacja
zajście endoreplikacji wymaga wyłączenia mechanizmów kontrolnych cyklukomórkowego*
ENDOMITOZA:
jej efektem jest poliploidyzacja komórek
megakariocyty: duże komórki wytwarzające wypustki (przypominające pseudopodia),osadzają się na ścianach naczyń krwionośnych i do światła naczyń odcinają obłonionefragmenty swojej cytoplazmy (zachodzi sekrecja) – prekursory płytek krwi; konieczny jestwięc intensywny wzrost tych komórek, który jest skorelowany z podwyższonym poziomemDNA; endomitoza inicjowana jest przez trombopoetynę – po zreplikowaniu DNA komórkawchodzi w mitozę: pojawia się aktywne MPF, degradowana jest otoczka jądrowa,chromatyna kondensuje, wyodrębniają się chromosomy, odbywa się normalna metafaza,zainicjowana zostaje anafaza, ale przebiega tylko anafaza A, po czym następujeprzedwczesne wyjście z anafazy (nie odbywa się anafaza B), przez co siostrzanechromosomy odsuwają się od siebie tylko na bardzo niewielki dystans, podczas telofazyzostaje odbudowana otoczka jądrowa i wszystkie chromosomy znajdują się w jednymjądrze – efektem jest powielenie liczby chromosomów; wyjście z mitozy następuje naskutek przedwczesnej lub zintensyfikowanej degradacji cykliny B
komórki miękiszowe (np. w liściach sukulentów): bardzo duże komórki; po zreplikowaniuDNA zahamowana zostaje degradacja otoczki jądrowej, nie powstaje wrzecionokariokinetyczne, kondensacja chromatyny jest niepełna, ale chromosomy dzielą się nachromatydy, dochodzi do degradacji kohezyny i liczba chromosomów ulega podwojeniuENDOREDUPLIKACJA:
jej efektem jest powstanie chromosomów politenicznych (złożonych z wielu chromatyd),o bardzo dobrze widocznym prążkowaniu (ze względu na ogromną ilość położonych oboksiebie chromatyd), posiadających puffy (pierścienie Balbianiego), które są miejscamigwałtownej dekondensacji, despiralizacji fragmentów chromatyny, będące przygotowaniemdo intensywnej transkrypcji na tych odcinkach, pojawiają się na chromosomach w różnychmiejscach, w zależności od tego, które fragmenty są aktualnie aktywne transkrypcyjnie;obecne także duże, rogalikowate struktury na chromosomach politenicznych – jąderka
aby zaszła endoreduplikacja konieczne: zablokowanie wejście w mitozę (nie może siępojawić aktywne MPF) oraz cykliczna replikacja DNA (replikacja --> faza G --> replikacja--> faza G..., faza G konieczna do tego, żeby chromatyna osiągnęła stan kompetencji doreplikacji – dołączone muszą zostać do chromatyny białka PRC (kompleksuprereplikacyjnego); w typowym cyklu komórkowym chromatyna jest kompetentna doreplikacji w fazie G1, aby ją ponownie uzyskać musi przejść mitozę – istnieje mechanizm,który chroni przed przedwczesnym zainicjowaniem replikacji DNA: aby zostałazainicjowana replikacja, z chromatyną musi zostać zasocjowany kompleksprereplikacyjny, składający się z wielu kompleksów białek, np. ORC – rozpoznającymiejsca inicjacji replikacji, kompleks MCM, cdc6; po zapoczątkowaniu replikacjiwszystkie białka (z wyjątkiem ORC) kompleksu prereplikacyjnego oddysocjowują odchromatyny i pozostają odłączone tak długo, jak długo działają aktywne kompleksycyklina/kinaza; ponowne zmontowanie kompleksu prereplikacyjnego na chromatynie jestmożliwe, kiedy spadek aktywności i poziomu cyklin będzie maksymalny – po degradacjicykliny A i B; cykliczność cyklów endoreduplikacyjnych zależy od zmian aktywnościkompleksu cyklina E/kinaza CDK (kom. zwierz.) oraz cyklina A/kinaza CDK i syntezybiałka cdk6 (kom. rośl.))
endoreduplikacja u roślin: w komórkach w nią wchodzących utrzymuje się wysoki poziombiałka ccs52 (cell cycle switch), które jest odpowiednikiem cdc20 u zwierząt (jestskładnikiem kompleksu ligazy ubikwitynowej/APC, decydującym o specyficznościsubstratowej ligazy ubikwitynowej), przez co nawet, gdy cyklina B się pojawi, jest od razudegradowana przez kompleks APC – brakuje jednego ze składników MPF, poza tymistnieje dodatkowa ścieżka zabezpieczająca przed wejściem w mitozę: utrzymuje się teżwysoki poziom białka CKI, które jest inhibitorem aktywności kinaz cyklinozależnych orazzahamowana jest aktywność fosfatazy, która defosforyluje w miejscach inhibitorowychkinazę – nie pojawia się więc aktywne MPF, przez co jest spełniony 1. warunekendoreduplikacji (zablokowanie wejścia w mitozę); 2. warunek (zapewnienie cyklicznościreplikacji DNA): utrzymuje się stały poziom aktywności kompleksu fazy G1 (cyklinaD/kinaza CDK), który fosforyluje białko FB, w wyniku czego uwolniony zostaje czynniktranskrypcyjny f2f, który promuje transkrypcję genów cykliny A i białka cdc6, powstajekompleks cyklina A/kinaza CDK, który inicjuje replikację DNA, po zainicjowaniu replikacji cyklina A jest ubikwitynowana i degradowana w proteasomach, następnie znów wfazie G musi dojść do fosforylacji FB... itd.; w fazie G zachodzi też reasocjacja białekkompleksu prereplikacyjnego na chromatynie
endoreduplikacja u zwierząt (owadów): zahamowanie wejścia w mitozę podobnie jak uroślin: w komórkach follikularnych, znajdujących się w jajnikach, zahamowana zostajeekspresja genu dla fosfatazy STRING cdc25, która może defosforylować kinazę cdk w jejmiejscach inhibitorowych (niski poziom STRING cdc25 = nieaktywna kinaza cdk1);poza tym promowana ekspresja genu białka Fzr (odpowiednika ccs52 u roślin),regulującego specyficzność substratową kompleksu APC/ligaza ubikwitynowa, któregowysoki poziom wywołuje degradację cykliny B – nie może więc powstać aktywne MPF;zapewnienie cyklicznej replikacji DNA: kluczową rolę w regulacji cyklu spełnia komplekscyklina E/cdk2; niski poziom białka Dacapo (CIP/KIP), inhibitora kinazy cdk2, pozwalana utworzenie aktywnego kompleksu cyklina E/cdk2, niezbędnego do inicjacji replikacji,po zainicjowaniu replikacji cyklina E ulega degradacji, następnie syntetyzowana jestponownie itd.; w trofoblastach ssaków poziom cykliny E się nie zmienia (nie ulegadegradacji), ale konieczna jest cykliczność aktywności kompleksu cdk2/cyklina E, którąwywołuje zmienny poziom białka p57, które jest inhibitorem kinaz cdk (CIP/KIP), p57akumulowane jest pod koniec S (wtedy zahamowana aktywność cdk2 i komplekscdk2/cyklina E przestaje być aktywny, co pozwala na oddysocjowanie od chromatyny białekkompleksu prereplikacyjnego i wejście w fazę G), pod koniec S p57 degradowane, coumożliwia reasocjację białek kompleksu prereplikacyjnego podczas fazy G, wtedy teżzaczyna być syntetyzowane białko p57 (ale maksimum akumulacji osiąga pod koniec S)(ścieżka Notch)
pętle inwersyjne są efektem zaistnienia w jednym ze skoniugowanych chromosomówpolitenicznych inwersji (chromosom, w którym nie doszło do inwersji, musi sięodpowiednio złożyć, aby dopasować się do chromosomu z inwersją)
niepełna endoreduplikacja: chromosomy mają powielone całe DNA, z wyjątkiem DNAcentromerowego – chromosomy połączone są okolicami centromerowymiAMPLIFIKACJA:
podlegają jej najczęściej geny, których ostatecznym produktem jest RNA
odbywa się najczęściej pozachromosomowo: odcinki mające być zamplifikowane sątranskrybowane – powstaje matryca rRNA, na której odwrotna transkryptaza syntetyzujerDNA; kopia rDNA ulega cyrkularyzacji i odbywa się replikacja: endonukleaza rozcinajedną z nici DNA, miejscem inicjacji replikacji jest koniec 3', asocjuje z nim polimeraza,która cyrkuluje po kolistej matrycy, katalizując syntezę komplementarnego DNA, przez coz każdym obrotem wydłuża się powielane DNA
Hybrydyzacja kwasów nukleinowych jest zjawiskiem związanym ze spontanicznym łączeniem się komplementarnych odcinków nici DNA ze sobą. Pod wpływem wysokiej temperatury lub substancji chemicznych tj. formamid, następuje rozdzielenie (denaturacja) podwójnej helisy DNA, lub RNA. Gdy wyeliminujemy czynnik denaturujący nastąpi odwrócenie procesu (renaturacja), czyli ponowne tworzenie podwójnej helisy. Wprowadzając do mieszaniny kwasów nukleinowych obce DNA lub RNA, w wyniku renaturacji, oprócz wyjściowych cząsteczek uzyskamy cząsteczki hybrydowe. Szybkość procesu zależy od kilku czynników, do których należą m.in.: długość hybrydyzujących fragmentów, komplementarność sekwencji oraz obecność substancji hamujących proces hybrydyzacji. Stosowanie sond jednoniciowych wiąże się z ryzykiem ich degradacji przez RNazy. Problem ten nie występuje w przypadku zastosowania sond dwuniciowych.
BŁONY KOMÓRKOWE
Błony występują we wszystkich znanych układach biologicznych zdolnych do samodzielnego życia. Oddzielają one komórkę od środowiska, a w komórkach Eukariota dzielą również wnętrze komórki na mniejsze obszary o zróżnicowanych funkcjach (budują struktury błoniaste: endoplazmatyczne retikulum, aparat Golgiego, pojedyncza błona otacza wakuolę, lizosomy, peroksysomy a podwójna jądro komórkowe, mitochondria i plastydy). Błony różnią się składem białek i fosfolipidów oraz nieznacznie właściwościami.
Błony biologiczne uczestniczą w:
· biernym lub czynnym, selektywnym transporcie jonów i substancji niejonowych,
· wydzielaniu produktów komórki do środowiska (egzocytoza) oraz pobieraniu makrocząsteczek do komórki (endocytoza),
· reakcjach na sygnały pochodzące ze środowiska (transdukcja sygnałów) poprzez receptory błonowe,
· przenoszeniu sygnałów do innych okolic komórki lub przekazywaniu ich do innych komórek,
· oddziaływaniu między komórką i podłożem oraz między komórkami.
Ich rolą jest też:
· oddzielenie wnętrza komórki od środowiska,

· oddzielanie w komórkach kompartymentów (przedziałów) o różnej koncentracji różnych substancji (enzymów, jonów, substratów),
· pośredniczenie w transporcie biernym i czynnym,
· wytwarzanie potencjału elektrochemicznego - różnej koncentracji jonów,
· miejsce przebiegu procesów (np. łańcuch transportu elektronów w mitochondriach i chloroplastach)
Teorie budowy błon
1. Model lipidowy - W roku 1895 Overton opierając się na fakcie, że substancje rozpuszczalne w tłuszczach wnikały do komórki bardziej efektywnie niż nierozpuszczalne - wydedukował, że lipidy muszą stanowić ważny składnik błony plazmatycznej.
2. Model dwuwarstwy lipidowej (1925) - Gortel i Grendel ekstrahując acetonem lipidy z błon erytrocytów ludzkich i obliczając powierzchnię błonki utworzonej przez ten ekstrakt, stwierdzili, że jest ona dwukrotnie większa od powierzchni wyjściowych krwinek. Sformułowali więc hipotezę, że błona komórkowa składa się z dwóch warstw lipidowych, sugerując uwodnienie obu ich stron tzn. polarne główki cząsteczek lipidów muszą być skierowane na zewnątrz, a niepolarne łańcuchy węglowodorowe ku sobie, do wnętrza podwójnej warstwy lipidowej.
3. Model trójwarstwowej błony (1935) - Dowson i Danielli korzystając z obserwacji Cole, że białka dodane do emulsji olejowo-wodnej w znacznym stopniu obniżają napięcie powierzchniowe pomiędzy wodą i kroplami oleju (napięcie takie jak w naturalnych błonach komórkowych) wysnuli hipotezę, że błony komórkowe zbudowane są symetrycznie z podwójnej warstwy lipidowej pokrytej po obu stronach warstwą białek.
4. Model płynnej mozaiki (1972) - Singer i Nicolson opublikowali teorię modelu płynnej mozaiki w której białka nie tworzą warstwy na powierzchni lipidów, lecz pływają w dwuwarstwie lipidowej zanurzone w różnym stopniu. Błona taka jest asymetryczna, płynna i dynamiczna.
Składniki błon biologicznych
Wszystkie błony w komórce zbudowane są z lipidów i białek, oraz mają wspólny plan budowy ogólnej.
Głównymi składnikami są lipidy i białka. Wzajemny stosunek tych składników może być różny w różnych błonach, a ich ułożenie też bywa zmienne.
Lipidy w błonach należą do trzech klas: fosfolipidów, glikolipidów i lipidów obojętnych (sterole). Podstawową strukturą błony jest dwuwarstwa lipidowa utworzona z fosfolipidów. Błona taka stanowi ośrodek, w którym lipidy i białka mogą przemieszczać się po powierzchni błony a także w poprzek błony.
Fosfolipidy zawierają dwie cząsteczki kwasów tłuszczowych połączone z dwoma spośród trzech atomów węgla glicerolu. Trzeci węgiel w glicerolu połączony jest z ujemnie naładowaną hydrofilową grupą fosforanową do której z kolei jest przyłączony mały związek hydrofilowy, taki jak cholina. Każda cząsteczka fosfolipidu zawiera więc hydrofobowy „ogon", złożony z dwóch łańcuchów kwasu tłuszczowego, oraz hydrofilową „głowę", gdzie znajduje się fosforan. Cząsteczki takie jak fosfolipidy, z regionami zarówno hydrofobowymi jak i hydrofilowymi, są nazywane cząsteczkami amfipatycznymi.
Zdolność fosfolipidów do tworzenia błon jest związana z ich amfipatycznym charakterem. Fosfolipidy rozprzestrzeniają się na powierzchni wody, tworząc pojedynczą warstwę cząsteczek fosfolipidowych, z hydrofobowymi „ogonami" skierowanymi ku górze, i hydrofilowymi „głowami" kontaktującymi się z wodą. Dwie takie jednocząsteczkowe warstwy mogą łączyć się na zasadzie „ogon z ogonem", tworząc dwuwarstwę fosfolipidową. Taka orientacja jest najbardziej korzystna pod względem energetycznym, gdyż pozwala na swobodny kontakt hydrofilowych głów z wodą, podczas gdy hydrofobowe łańcuchy kwasów tłuszczowych unikają kontaktu z wodą, gromadząc się w środku układu. Dodatkowo cząsteczki fosfolipidów mają w przybliżeniu jednakową szerokość, co również sprzyja układaniu się ich w podwójne warstwy cylindrycznych struktur.
Cząsteczka fosfolipidu w błonie nie jest sztywna. Oprócz ruchów obrotowych całej cząsteczki wokół swojej osi występuje rozchodzenie się i zginanie łańcuchów kwasów tłuszczowych. Mniej ruchliwa jest okolica polarna cząsteczki, natomiast schowane w głębi warstwy hydrofobowej końce łańcuchów węglowodorowych wykonują szybkie ruchy. Ruchliwość łańcucha węglowodorowego jest tym większa im jest on krótszy i ma liczniejsze wiązania nienasycone. Fosfolipidy łatwo przemieszczają się w obrębie jednej warstwy lipidowej błony (dyfuzja boczna) - zachodzi co około 10 -6 sekundy. Natomiast wymiana cząsteczek lipidów między jedną i drugą warstwą (tzw. ruchy flip-flop) może być bardzo wolna i zachodzić raz na kilkaset godzin.
W komórkach bakterii i drożdży, które muszą adaptować się do różnych temperatur, zarówno długość jak i stopień nienasycenia kwasów tłuszczowych są stale dopasowywane, tak aby utrzymać względnie stały poziom płynności błony: w wyższych temperaturach komórka wytwarza lipidy o łańcuchach dłuższych i zawierających mniej wiązań podwójnych, co sprzyja zachowaniu stabilności i płynności błony.
Płynność błon umożliwia fuzję błon ze sobą i mieszanie się ich składników, co przy podziale komórki zapewnia równomierne rozdzielenie budujących błonę cząsteczek pomiędzy komórki potomne.

Glikolipidy - są to cząsteczki lipidów połączone z łańcuchami polisacharydowymi. Zlokalizowane są w zewnętrznej warstwie błony. Domeny polarne glikolipidów wystają ponad powierzchnię błony komórkowej, prezentując swoje grupy polarne do środowiska. Jakkolwiek rola glikolipidów nie jest do końca poznana, to przypisuje się im rozmaite funkcje: 1) utrzymują asymetryczność błony komórkowej, 2) oddzielają komórki od środowiska i stabilizują błonę komórkową, 3) są receptorami dla niektórych hormonów peptydowych i toksyn bakteryjnych, 4) dzięki specyficznej kombinacji topograficznej reszt cukrowych w błonach erytrocytów określają grupy krwi (ABO). Glikolipidy są na tyle ważnymi składnikami błon, że w przypadku wad genetycznych związanych z ich metabolizmem występują duże zaburzenia rozwojowe, kończące się przedwczesną śmiercią noworodka. Warstwa glikolipidów pokrywa większość komórek zwierzęcych tworząc tzw. glikokaliks. Glikolipidy uzyskuja swoje grupy cukrowe w aparacie Golgiego.
Sterole - zbudowane są ze sztywnego poczwórnego pierścienia węglowego z bocznymi podstawnikami. W komórkach zwierzęcych głównym sterolem (steroidem) jest cholesterol, zaś u roślin występują fitosterole: sitosterol, kamposterol i stigmosterol. W błonie lokalizują się pomiędzy łańcuchami węglowodorowymi fosfolipidów. Cholesterol jest lipidem o słabych właściwośćiach amfipatycznych. Jego cząsteczka składa się z części hydrofobowej – steroidowej i łańcucha alifatycznego dołączonego do węgla 17 w pierścieniu D. Domena hydrofilowa reprezentowana jest przez grupę (OH -), związaną z 3. węglem w pierścieniu A. Cholesterol jest umiejscowiony w błonie komórkowej, podobnie jak glikolipidy, w jej zewnętrznej warstwie. W niej wiąże się swoją grupą hydroksylową z 1. węglem łańcucha alifatycznego kwasu tłuszczowego fosfolipidu. Cholesterol jest podstawowym czynnikiem regulującym przepuszczalność błon komórkowych. Położenie grupy hydrofobowej pomiędzy łańcuchami alifatycznymi fosfolipidów zapobiega przejściu fazowemu dużych obszarów błony ( zapobiega zbytniemu zbliżaniu się łańcuchów i uniemożliwia powstawanie pomiędzy nimi oddziaływań van der Waalsa, co prowadziło by do ich unieruchomienia i przejście w stan stały), utrzymuje wewnętrzną, hydrofobową część dwuwarstwy lipidowej w stanie płynnym. Natomiast grupy polarne cholesterolu uszczelniają oraz usztywniają i stabilizują zewnętrzne krawędzie dwuwarstwy lipidowej, zapobiegając niekontrolowanej migracji małych cząstek rozpuszczalnych w wodzie pomiędzy cząsteczkami fosfolipidów.
Białka błonowe umownie dzieli się na dwie grupy:
1. Białka które dają się łatwo usunąć z błony wodą, roztworami soli lub czynników chelatujących nie niszcząc dwuwarstwy lipidowej - są to białka powierzchniowe (peryferyjne) błony. Są one luźno związane z powierzchniami błony i często połączone z łańcuchami sacharydowymi (glikoproteiny) oraz kwasami tłuszczowymi czy długołańcuchowymi alkoholami, poprzez które polipeptydy te zakotwiczają się w obrębie błony. Białka powierzchniowe są cząsteczkami hydrofilnymi i najczęściej występują w rejonach, w których sterczą z błon fragmenty białek integralnych i są z nimi powiązane oddziaływaniami niekowalencyjnymi. Mogą również wiązać się z polarnymi fragmentami fosfolipidów. Część białek może znajdować się całkowicie poza rejonem błony, a jedynie wiązać się z nią za pomocą kowalencyjnego wiązania z cząsteczką lipidową błony.
2. Te które można wyizolować z błony do roztworu wodnego jedynie w postaci kompleksów z detergentem (solubilizacja detergentem - przeprowadzenie do roztworu wodnego kompleksów detergentu i składników błony) niszczącym uporządkowanie dwuwarstwy lipidowej - są to białka integralne na trwałe wbudowane w dwuwarstwę
Białka integralne mogą być zbudowane z jednej lub kilku podjednostek. Fragmenty cząsteczek białkowych mogą wyłaniać się na jednej lub na obu powierzchniach błony, bądź są prawie całkowicie schowane w części hydrofobowej dwuwarstwy lipidowej. Białka integralne mają w łańcuchu polipeptydowym przynajmniej jedną sekwencję składającą się z co najmniej 22 aminokwasów hydrofobowych, które pozwalają na zakotwiczenie się w błonie. W niektórych białkach reszty aminokwasów hydrofobowych tworzą kilka skupień, co sprawia, że łańcuch polipeptydowy kilkakrotnie przemierza dwuwarstwę lipidową. Koniec karboksylowy [C] łańcuchów polipeptydowych czasem jest skierowany do cytoplazmy, a koniec aminowy [N] na powierzchnię zewnętrzną błony, może też być przeciwnie. Białka mogą również kotwiczyć się w błonie poprzez kowalencyjnie związane z nimi łańcuchy kwasów tłuszczowych lub cząsteczkę glikofosfolipidu. Białka błonowe rozmieszczone są w błonie asymetrycznie. Ich ułożenie nie jest przypadkowe ale wynika ze specyficznych oddziaływań łańcucha polipeptydowego z dwuwarstwą lipidową. Wszystkie te cechy białek integralnych przyczyniają się do asymetrii błony. Większość białek integralnych błon biologicznych jest glikoproteinami
Funkcje białek błonowych
Wyróżnia się kilka klas funkcjonalnych białek błonowych:
1. Białka transportujące – uczestniczą w transporcie przez błony małych cząsteczek, tworzą kanały i pompy prowadząc transport kontrolowany (np. pompa sodowa, aktywnie wypompowuje z komórki jony sodu i wprowadza do niej jony potasu).
2. Białka wiążące – są elementami wyspecjalizowanych struktur odpowiedzialnych za utrzymywanie łączności pomiędzy komórkami lub z cytoszkieletem (np. integryny wiążące elementy wewnątrzkomórkowe filamenty aktyny z białkami substancji zewnątrzkomórkowej).
3. Białka receptorowe – pośredniczą w przekazywaniu informacji ze środowiska zewnętrznego do komórki, związanie cząsteczki sygnałowej indukuje zmiany w aktywności komórkowej (np. receptor płytkopochodnego czynnika wzrostu, który wytwarza wewnątrzkomórkowe sygnały powodujące wzrost i podział komórki).
4. Białka enzymatyczne – enzymy, których miejsca katalityczne znajdują się po jednej ze stron błony bądź w jej wnętrzu (np. cyklaza adenylanowa, w odpowiedzi na sygnały zewnątrzkomórkowe katalizuje wytwarzanie wewnątrzkomórkowego cyklicznego AMP, będącego wewnątrzkomórkowym przekaźnikiem)

Uważa się, że białka integralne pełniące funkcje transportowe, których łańcuch polipeptydowy wielokrotnie przemierza dwuwarstwę lipidową - tworzy przez błonę kanały. Modele kanałów błonowych przyjmują, że 22-aminokwasowe hydrofobowe odcinki łańcucha polipeptydowego tworzą struktury a-helisy, a kilka takich a- helis obok siebie stanowi ścianę kanału. Oprócz tych zewnętrznych a-helis mocujących kanał w dwuwarstwie lipidowej, wewnątrz kanału mogą biec dodatkowe, wewnętrzne odcinki łańcucha, zbudowane z hydrofilnych aminokwasów. Pełnią one właściwe funkcje transportowe np. białko kanałów wapniowych.
Właściwości błon
Półpłynność: dwuwarstwa lipidowa błony biologicznej jest w stanie półpłynnym lub inaczej płynno-krystalicznym. Ze względu na wysoki stopień uporządkowania ma ona właściwości krystaliczne (fosfolipidy ułożone w szeregi, biegunem polarnym na zewnątrz, apolarnym do środka). Z drugiej zaś strony podwójna warstwa lipidowa wykazuje właściwości płynne, bowiem pomimo tego uporządkowania łańcuchy węglowodorowe pozostają w ciągłym ruchu, co oznacza, że cząsteczki fosfolipidów mają swobodę rotacji i mogą dyfundować w obrębie pojedynczej warstwy błony, w której występują. Nadaje to podwójnej warstwie fosfolipidowej charakter cieczy krystalicznej, który bywa też określany jako półpłynny. Niektóre błony biologiczne w temperaturze optymalnej dla wzrostu komórki zawierają jednak pewne lipidy w formie krystalicznej. Krystaliczna struktura jest stanem, w którym cząsteczki lipidów są względem siebie uporządkowane, co powoduje ich wzajemne powiązanie a tym samym unieruchomienie .
Półpłynny charakter dwuwarstwy lipidowej w błonie komórek ma ważne znaczenie biologiczne dla organizmów żywych. Zachowanie odpowiedniej płynności błon umożliwia dyfuzję białek błonowych w płaszczyźnie obu warstw fosfolipidów i ich wzajemne oddziaływanie (np. podczas procesów transdukcji sygnałów), wzajemne zlewanie się błon (np. w czasie egzo- i endocytozy) i mieszanie się jej składników (zachodzące podczas podziałów komórkowych). Organizmy poikilotermiczne (zmienno cieplne, żyjące w środowisku o zmiennej temperaturze) takie jak bakterie i drożdże dostosowują skład lipidowy błon do temperatury otoczenia w jakim żyją. Temperatura przejścia fazowego ich błon staje się wyższa wówczas, gdy organizm dostosuje się do wzrostu w podwyższonej temperaturze, a niższa, gdy rośnie w hodowli o obniżonej temperaturze. To dostosowanie ma istotne znaczenie dla funkcji błony związanej z oddziaływaniem różnych cząsteczek białkowych i lipidowych wymagających jej półpłynnego stanu.
Dynamiczność: jest wyrażona w ruchach budujących błonę lipidów i białek. Cząsteczki fosfolipidów w błonie nie są sztywne. Mniej ruchliwe są ich okolice polarne, natomiast zanurzone w głębi warstwy hydrofobowej końce łańcuchów węglowodorowych wykonują szybkie ruchy, tym szybsze im te łańcuchy są krótsze i zawierają liczniejsze wiązania podwójne. Białka błony mogą natomiast być w jej płaszczyźnie przemieszczane dyfuzyjnie, wykonywać ruchy obrotowe w osi prostopadłej do powierzchni błony oraz wynurzać się z dwuwarstwy lipidowej lub w niej zanurzać.
Ruchliwość składników błon powoduje zamykanie wszelkich wyrw i ubytków. Błony w żywych komórkach nigdy nie tworzą wolnych krawędzi. Dzięki temu wnętrze komórki i poszczególnych jej przedziałów jest zawsze otoczone selektywnie przepuszczającą barierą. Ponieważ błona jest dwuwymiarowym płynem, wiele jej białek, podobnie jak i lipidów, może swobodnie poruszać się w obrębie płaszczyzny dwu-warstwy lipidowej. Można to w sposób łatwy i oczywisty wykazać, doprowadzając do fuzji komórki myszy z komórką ludzką, tworząc podwójnej wielkości komórkę hybrydową, a następnie śledząc rozmieszczenie białek błony komórkowej zarówno myszy, jak i człowieka. Aczkolwiek na początku białka te pozostaną na powierzchni swych odpowiednich połówek nowo powstałej komórki hybrydowej, to już po niecałej godzinie dwa zestawy tych białek zostaną równomiernie wymieszane na całej powierzchni komórki.
Jednakże obraz morza lipidów, w którym wszystkie białka pływają swobodnie, jest zbyt uproszczony. Komórki mają swoje sposoby ograniczenia lokalizacji poszczególnych białek błony komórkowej do pewnych pól dwuwarstwy, co prowadzi do powstania na powierzchni komórki wyspecjalizowanych funkcjonalnie obszarów, czyli domen błonowych.
Białka mogą być złączone z trwałymi strukturami na zewnątrz komórki, na przykład z cząsteczkami substancji międzykomórkowej. Białka błonowe mogą być także zakotwiczone do względnie nieruchomych struktur wewnątrz komórki, zwłaszcza do rozwiniętej pod powierzchnią błony komórkowej części cytoszkieletu, czyli kory komórki. W końcu, komórki mogą wytwarzać bariery ograniczające obecność poszczególnych składników błony do jednej domeny błonowej. Na przykład w komórkach nabłonka wyścielającego jelito ważne jest, aby białka transportujące, działające przy pobieraniu substancji odżywczych z jelita, występowały tylko w szczytowej powierzchni komórek (powierzchni zwróconej do światła jelita) oraz aby obecność innych białek, wyprowadzających rozpuszczone substancje z komórki nabłonkowej do tkanek i krwiobiegu, była ograniczona do powierzchni podstawnej i bocznej (rys. 11-37). To asymetryczne rozmieszczenie białek błonowych jest zachowywane dzięki barierze utworzonej wzdłuż strefy, w której komórka jest zespolona z przyległymi komórkami nabłonkowymi przez tak zwane, połączenia zamykające. W miejscu tym wyspecjalizowane białka łączące formują wokół komórki ciągły pas, gdzie kontaktuje się ona ze swoimi sąsiadami, wytwarzając ścisłe zespolenie pomiędzy przylegającymi do siebie błonami komórkowymi. Białka błonowe nie mogą w drodze dyfuzji przekroczyć tego połączenia.
Asymetryczność: polega na różnicach w budowie obu powierzchni błony, skierowanych na zewnątrz i ku wnętrzu komórki lub organelli. Dwie warstwy dwuwarstwy często zawierają różny skład fosfolipidów i glikolipidów a białka są wtopione w dwuwarstwę ze specyficzną orientacją przestrzenną, konieczną dla ich funkcji. W błonie komórkowej (plazmolemie) wyróżnia się dwie warstwy:
· warstwę lipidową zewnętrzną E (ang. exoplasmic) od strony środowiska,
· warstwę lipidową cytoplazmatyczną P. (ang. protoplasmic) od strony protoplazmy
Pomiędzy warstwami istnieją uchwytne różnice. Na przykład w błonie erytrocytu człowieka warstwa E zbudowana jest głównie z fosfolipidów cholinowych (fosfatydylocholin = lecytyn i sfingomielin), natomiast warstwa P zbudowana jest z fosfolipidów aminowych tzw. kefalin: fosfatydyloseryny i fosfatydyloetanoloaminy. Fosfolipidy warstwy P mają łańcuchy węglowodorowe zawierające więcej nienasyconych wiązań , a fosfatydyloseryny ponadto noszą jeden ładunek dodatni i dwa ujemne, mają więc przewagę ładunków ujemnych (asymetria jonowa). Asymetria dwuwarstwy lipidowej błony

komórkowej jest utrzymywana głównie przez obecność glikolipidów i glikosacharydów, które wchodzą w skład zewnętrznej (E) warstwy błony, a ich reszty cukrowe są eksponowane na zewnątrz komórki. Przykładem glikolipidów mogą być cząsteczki noszące własności grupowe ABO erytrocytów człowieka.
Półprzepuszczalność (selektywność): przez błonę mogą swobodnie przenikać tylko nieliczne związki np. H2O, CO2, glicerol; natomiast większość substancji, aby mogła przeniknąć przez błonę wymaga obecności w błonie odpowiednich układów transportujących, którymi są odpowiednie białka błonowe. Przepuszczalność błony dla danej substancji zależy od rozmiaru i ładunku jej cząsteczki. Na przykład cząsteczki wody z dużą szybkością przedostają się przez szczelinę w podwójnej warstwie lipidowej, powstałą na skutek chwilowego odchylenia się łańcucha kwasu tłuszczowego. Bez trudu przez dwuwarstwę przenikają gazy, np. tlen, CO2 i N2, małe cząsteczki polarne, np. glicerol, i niektóre większe cząsteczki apolarne (hydrofobowe), np. węglowodory. Cząsteczki większe, np. glukoza i jony różnej wielkości nie przedostają się z powodu zbyt dużych rozmiarów lub na skutek odpychania przez ujemnie naładowaną powierzchnię błony. Przepuszczalność dla tych związków wiąże się z występowaniem w błonie specyficznych białek transportujących. Wszystkie błony plazmatyczne są selektywnie przepuszczalne dla różnych rodzajów cząsteczek a wynika to z występowania w poszczególnych typach błon różnych zestawów białek transportujących. W odpowiedzi na zmianę warunków środowiska lub na aktualne zapotrzebowanie komórki błona może czasami stawać się barierą dla danej substancji, w innych natomiast okolicznościach może je aktywnie transportować. Kierując ruchem cząsteczek, komórka jest w stanie zapewnić stałość składu jonowego i cząsteczkowego swego wewnętrznego środowiska.
Zdolność do fuzji: ważną cechą podwójnych warstw lipidowych jest unikanie tworzenia układów z wolnymi końcami, czego wyrazem jest spontaniczne zamykanie się błon w struktury pęcherzykowate, oraz zdolność w określonych warunkach do łączenia się z innymi, podobnymi strukturami błonowymi. Fuzja (łączenie się, zlewanie) błon jest powszechnie występującym procesem i ma istotne znaczenie dla funkcjonowania komórki, np. podczas endocytozy, wydzielania i krążenia składników błon. Zachodzi też w wyspecjalizowanych komórkach, np. podczas egzocytozy (wydzielania) enzymów, neurohormonów, podczas łączenia się komórki jajowej z plemnikiem, łączenia się mioblastów. Fuzja zachodzi też w procesach patologicznych, np. w odpowiedzi zapalnej podczas tworzenia się komórek olbrzymich, podczas wnikania do komórki wirusów z otoczką.
Pochodzenie lipidów i białek błonowych
Fosfolipidy syntetyzowane są z CDP-glicerydów i L-seryny. Powstałe fosfatydyloseryny po dekarboksylacji przekształcają się w fosfatydyloetanoloaminy, a te z kolei podlegają metylacji do fosfatydylocholin. Fosfolipidy mogą być też pobierane ze środowiska otaczającego. Półokres trwania fosfolipidów błon in vivo może być stosunkowo długi (kilka tygodni -erytrocyt). Tam gdzie błony podlegają szybkiej wymianie, fosfolipidy maja szybszy obrót.
Pochodzenie glikolipidów błony komórkowej może być różne. Są one syntetyzowane w błonach śródplazmatycznych (ER) przez dołączanie cukrów. Mogą też być pobierane z zewnątrz.
Większość białek integralnych błon biologicznych jest glikoproteinami i jest syntetyzowana na rybosomach związanych z błonami siateczki śródplazamatycznej ziarnistej. Początkowy odcinek końca N łańcucha polipeptydowego syntetyzowany na rybosomie zbudowany z około 20 reszt aminokwasów ma charakter hydrofobowy (jest to odcinek sygnałowy) i dlatego może wnikać do dwuwarstwy lipidowej błony. Tam zostaje on otoczony przez białka tworzące kanał w dwuwarstwie lipidowej, przez który mogą przesuwać się dalsze, hydrofilne części łańcucha pilipeptydowego. Następnie syntetyzowany jest drugi odcinek sygnałowy polipeptydu złożony z reszt hydrofobowych aminokwasów. Podczas wnikania do błony przesuwa on białko w płaszczyźnie poza błonowy kanał białkowy. Ten hydrofobowy odcinek polipeptydu zakotwicza go w apolarnej dwuwarstwie lipidowej. Hydrofilny koniec C odcinka polipeptydu jest syntetyzowany najpóźniej, a jego odłączenie od rybosomu kończy syntezę łańcucha.
Ten opis wbudowywania białek w błonę odpowiada hipotezie odcinków sygnałowych.
Druga hipoteza wbudowywania białek integralnych błony opiera się na stwierdzeniu, że wspólną cechą integralnych białek bonowych jest ich nierozpuszczalność w roztworach wodnych. Ta cecha białek integralnych w większym stopniu zależy od ich konformacji, niż składu aminokwasowego. Sekwencje około 20 reszt aminokwasów hydrofobowych obecne w wielu białkach błonowych równie często znajdują się w białkach rozpuszczalnych. Białka przeznaczone do wbudowania w błonę w kontakcie z nią przyjmują konformację przestrzenną, która powoduje ich przemieszczenie się z fazy wodnej środowiska cytoplazmatycznego do fazy hydrofobowej dwuwarstwy lipidowej. Przykładem takich białek mogą być oksydaza cytochromowa, dehydrogenaza 3-fosforanu glicerolu, cytochrom b5, transferaza galaktozylowa. Taki sposób wbudowywania białek do błony określa się terminem fałdowania się białek zależnym od błony.
Glikokaliks
Powierzchnia komórek prokariotycznych i eukariotycznych pokryta jest różnej grubości otoczką zbudowaną z cukrów o różnym stopniu polimeryzacji. Błona komórkowa bakterii jest otoczona grubą ścianą komórkową i otoczką śluzową zbudowaną z wielocukrów. Ściany komórek roślinnych zbudowane są z wielocukru celulozy, który jest zasadniczym elementem szkieletowym tych komórek, określającym ich kształt i przeciwstawiającym się dużemu ciśnieniu osmotycznemu wnętrza komórki. Warstwa cukrowców pokrywająca powierzchnię komórek zwierzęcych nosi nazwę glikokaliks. Wszystkie cukrowce wchodzące w skład glikoprotein, proteoglikanów i glikolipidów występują tylko na powierzchni zewnętrznej błony (na powierzchni komórki) tworząc cukrowcowy „płaszcz”. Jest on ważnym elementem ochrony powierzchni komórki przed uszkodzeniem chemicznym i mechanicznym. Ponieważ oligosacharydy i polisacharydy wchłaniają wodę, powodują śliskość powierzchni komórki. Pozwala to komórkom ruchliwym, takim jak krwinki białe, przeciskać się przez wąskie przestrzenie i zapobiega przylepianiu się krwinek do siebie lub do ścian naczyń krwionośnych. Pokrywa węglowodanowa różnych typów komórek różni się istotnie zarówno składem reszt cukrowych jak i grubością. Składa się ona głównie z cukrów prostych, które występują zazwyczaj jako boczne łańcuchy związane kowalencyjnie z białkami błonowymi i lipidami. Ponadto obecne są w niej proteoglikany, związki białkowo-węglowodanowe syntetyzowane w komórce, wydzielane na zewnątrz i adsorbowane do powierzchni błony komórkowej. Mukopolisacharydowa otoczka komórki jest bardzo

wrażliwa na każdą fizjologiczną zmianę komórki. Przypuszcza się, że spełnia ona kilka funkcji: 1) kotwiczenie białek transbłonowych w dwuwarstwie lipidowej zapobiegające ich wypadnięciu do cytoplazmy, 2) utrzymywanie prawidłowego sfałdowania łańcucha polipeptydowego przez dołączone reszty cukrowe, 3) pełnią funkcję sygnałów kierujących białka transbłonowe do miejsca przeznaczenia w błonie, 4) charakterystyczny dla każdej komórki skład i konfiguracja reszt cukrowych są odpowiedzialne za wzajemne rozpoznawanie się komórek w procesie rozwoju organizmu i w czasie całego życia.
Kora komórki
Błona otaczająca komórkę (podobnie jak i błony wewnątrzkomórkowe) jest bardzo cienka i delikatna, dlatego też wzmocniona jest od strony wnętrza komórki „rusztowaniem” białek, tzw. szkieletowych, podczepionych do błony poprzez specyficzne białka transbłonowe. Białka szkieletowe kotwiczą się do niektórych białek transbłonowych, np. glikoforyny lub białka trzeciego szczytu elektroforetycznego, i są również powiązane wzajemnie, utrzymując kształt komórki i zapewniając błonie elastyczność i wytrzymałość. Rusztowanie to, zbudowane z sieci włóknistych białek zwane jest korą komórki albo membranoszkieletem. Głównym składnikiem kory jest białko spektryna. Występuje ono zawsze jako dimer dwóch wzajemnie helikalnie splecionych monomerów a i b. Dimery spektryny wiążą się ze sobą tworząc filamenty o długości ok. 100nm. Białko to buduje tuż pod plazmolemą sieć stanowiącą podporę dla błony komórkowej i utrzymującą kształt komórki. Sieć spektrynowa połączona jest z błoną przez białko łącznikowe ankirynę (łączy ją z białkem integralnym, białkiem trzeciego szczytu elektroforetycznego) oraz z cytoszkieletem komórki, głównie filamentami aktynowymi. Obok spektryny i ankiryny, kora komórki zawiera gęstą sieć filamentów aktynowych, które biegną do cytoplazmy, gdzie zostają poprzecznie powiązane w trójwymiarową sieć. Ta aktynowa sieć kory decyduje o kształcie i właściwościach mechanicznych błony komórkowej i powierzchni komórki. Przetasowania aktyny w obrębie kory stanowią molekularną podstawę zmian kształtu komórki i jej ruchów.
Substancja międzykomórkowa
Przestrzeń między komórkami w tkankach i narządach zwierzęcych wypełniona jest substancją międzykomórkową. Poprzez oddziaływanie na swoiste receptory błon, białka substancji międzykomórkowej mogą wpływać na morfologię, aktywność metaboliczną, wzrost i różnicowanie komórek. Jest ona zbudowana z włókien i substancji podstawowej. Wśród włókien substancji międzykomórkowej dominują włókna kolagenowe i elastynowe. Substancja podstawowa zawiera rozpuszczone prekursory białek tworzących włókna, proteoglikany, glikoproteiny i inne cząsteczki produkowane przez komórki. Należą do nich niektóre glikoproteiny występujące w otoczeniu komórek, wpływające na ich wzajemne oddziaływania oraz oddziaływania ze składnikami osocza. Substancja międzykomórkowa jest ściśle powiązana z błoną komórkową za pośrednictwem białek łącznikowych, takich jak np. fibronektyna czy laminina. Białka te uczestniczą w oddziaływaniach komórek z substancją międzykomórkową. Fibronektyna jest białkiem syntetyzowanym przez wiele komórek, w tym fibroblasty, komórki nabłonkowe, komórki śródbłonka. Białko to występuje w osoczu krwi i na powierzchni komórek. Wraz z proteoglikanami pełni ważne funkcję pomostu łączącego powierzchnię komórki z otaczającymi ją składnikami substancji międzykomórkowej. Jednym końcem cząsteczka fibronektyny wiąże się z włóknami kolagenowymi zaś drugim końcem z białkiem transbłonowym – integryną, która z kolei jest powiązana z cytoszkieletem komórki (filamentami aktynowymi). Innym poznanym dobrze białkiem międzykomórkowym wpływającym na funkcje komórek jest laminina. Jest główną niekolagenową glikoproteiną błon podstawnych, które spajają komórki nabłonka z tkanką łączną. Białko to warunkuje oddziaływanie między komórkami a błoną podstawną w procesach adhezji, migracji, proliferacji i różnicowania komórek.
Transport przez błony biologiczne
TRANSPORT
PRZEZ BŁONĘ PĘCHERZYKOWY
(z fragmentami błon)bez udziału nośników z udziałem nośników egzocytoza endocytoza:
· dyfuzja prosta
· dyfuzja złożona
dyfuzja ułatwiona
transport aktywny
· pinocytoza
· fagocytoza
· pierwotny
· wtórny
· translokacja grupowa
· endocytoza receptorowa

Transport bez udziału nośników:
Dyfuzja prosta - wypadkowe przemieszczanie się cząsteczek z obszarów o wyższym stężeniu do obszarów o niższym stężeniu, tak że ostatecznie rozkład cząstek staje się równomierny (dyfuzja jest zatem ruchem cząsteczek zgodnym ze spadkiem gradientu stężenia). Szybkość dyfuzji zależy od wielkości i kształtu cząsteczek, ich ładunku elektryczne go i temperatury otoczenia.
Dyfuzja złożona – przenikanie substancji zachodzi nie tylko pod wpływem gradient stężenia, ale i innych bodźców, jak np. gradientu potencjału elektrochemicznego czy gradientu ciśnienia
Osmoza - przemieszczanie się (dyfundowanie) wody z obszarów o wyższym jej stężeniu do obszarów o stężeniu niższym.
Transport z udziałem nośników - transport przez błony z uczestnictwem w przenoszeniu różnych, zlokalizowanych w błonie białek. W ten rodzaj transportu mogą być zaangażowane dwa mechanizmy: dyfuzji ułatwionej (wspomaganej) oraz aktywnego transportu.
W dyfuzji ułatwionej ruch cząsteczek odbywa się tylko w kierunku zgodnym ze spadkiem gradientu stężenia (od wyższego do niższego) - błona jest przepuszczalna dla przemieszczanej substancji, lecz obecność w błonie specyficznego nośnika, wiążącego czasowo transportowaną cząstkę przyspiesza jej przemieszczanie się przez błonę. Białko przenośnikowe nie ulega w tym procesie żadnym zmianom; po odłączeniu jednej cząsteczki może natychmiast wiązać się z drugą. Przykładem takiego nośnika jest białko transportujące glukozę przez błonę komórkową erytrocytów.
Transport aktywny - transport cząsteczek wbrew gradientowi stężeń, odbywający się kosztem energii metabolicznej. Energia do tego transportu pochodzi najczęściej z ATP, np. pompa sodowo-potasowa - zlokalizowana w błonach plazmatycznych grupa specyficznych białek, które wykorzystują energię pochodzącą z rozkładu ATP do wymiany jonów sodowych z wnętrza komórki na jony potasowe wnikające z zewnątrz. W tym wypadku wytwarzany gradient stężenia dotyczy cząstek obdarzonych ładunkiem, zatem w poprzek błony tworzy się nie tylko gradient stężenia, lecz i także gradient potencjału elektrycznego. Można wyróżnić trzy różne mechanizmy transportu aktywnego:
translokacja grupowa – gdy energia do transportu danej cząsteczki równa jest energii potrzebnej do wytworzenia nowych wiązań kowalencyjnych w transportowanej cząsteczce
transport aktywny pierwotny – gdy energia do transportu danej cząsteczki równa jest energii potrzebnej do wytworzenia nowych wiązań kowalencyjnych w nośniku
transport aktywny wtórny – gdzie aktywnie transportowana pierwsza substancja (np. Na+) tworzy gradient potencjału elektrochemicznego, który warunkuje transport innej substancji, np. cukru, aminokwasu, zgodnie z tym gradientem.
Transport przez błonę można podzielić inaczej na bierny, czyli bez nakładu energii ze strony komórki ( osmoza, dyfuzja prosta, złożona i dyfuzja ułatwiona) oraz na transport aktywny, wymagający dostarczenia energii metabolicznej
Przepuszczalność błony komórkowej dla danej substancji zależy od rozmiaru i ładunku jej cząsteczek. Błona jest przepuszczalna, gdy cząsteczki swobodnie przez nią przenikają, i odwrotnie - jest nieprzepuszczalna, gdy cząsteczki nie są w stanie się przez nią przedostać. Błona selektywnie przepuszczalna (półprzepuszczalna) przepuszcza tylko niektóre rodzaje cząsteczek, podczas gdy inne zatrzymuje.
Niektóre cząsteczki przenikają przez podwójną warstwę lipidową dość łatwo (szczeliną powstałą na skutek odchylenia się łańcucha kwasu tłuszczowego) np. cząsteczki wody, tlen, dwutlenek węgla, azot, małe cząsteczki polarne (np.glicerol) i niektóre większe cząsteczki niepolarne (np. węglowodory). Cząsteczki większe, np. glukoza i jony różnej wielkości nie przedostają się przez podwójną warstwę lipidową z powodu zbyt dużych rozmiarów lub na skutek odpychania przez naładowaną powierzchniową warstwę błony.
Półprzepuszczalność błony wiąże się z występowaniem w błonach specyficznych białek transportujących zwanych nośnikami (dotyczy to wszystkich błon plazmatycznych - otaczających i budujących różne struktury). Błony są selektywnie przepuszczalne dla różnych rodzajów cząsteczek. Zestaw białek transportujących zawarty w błonie komórkowej czy w błonie organelli wewnątrzkomórkowych ściśle określa, jakie substancje mogą wejść do komórki lub organelli oraz z nich wyjść. Aby nadać impuls i zapewnić poprawny, złożony ruch drobnych cząsteczek, zarówno wchodzących do komórki, jak i z niej wychodzących oraz przemieszczanych pomiędzy cytozolem a różnymi organellami komórki, każda błona w komórce zawiera charakterystyczny dla siebie zestaw przenośników. Tak więc w błonie komórkowej znajdują się przenośniki importujące substancje odżywcze, takie jak cukry, aminokwasy i nukleotydy; w wewnętrznej błonie mitochondrialnej znajdują się przenośniki do importu pirogronianu (w komórkach roślinnych także: jabłczanu i szczawiooctanu) i ADP oraz eksportu ATP itd. W odpowiedzi na zmianę warunków środowiska lub na aktualne zapotrzebowanie komórki błona komórkowa może stawać się barierą nie do przebycia dla cząstek danej substancji, w innych natomiast okolicznościach może je aktywnie transportować.
Nośniki są białkami błonowymi niezbędnymi do przenoszenia poprzez błony jonów oraz prawie wszystkich małych cząsteczek organicznych z wyjątkiem cząsteczek rozpuszczalnych w rozpuszczalnikach organicznych oraz małych cząsteczek nienaładowanych, które mogą przechodzić przez błonę w drodze dyfuzji prostej. Każdy nośnik jest wysoce selektywny i często transportuje tylko jeden typ cząsteczek. Wyróżnia się dwa rodzaje nośników: ruchome (przenośniki, permeazy) i nieruchome czyli kanały. Przenośniki są białkami integralnymi, które wiążą rozpuszczoną substancję po jednej stronie błony i przenoszą ją na drugą stronę poprzez zmianę konformacji przenośnika. Tą drogą mogą być transportowane zarówno małe cząsteczki organiczne, jak i nieorganiczne jony. Natomiast kanały tworzą w błonie małe hydrofilowe pory, przez które substancje mogą przechodzić w drodze dyfuzji. Większość kanałów białkowych przepuszcza tylko jony nieorganiczne i dlatego określa się je jako kanały jonowe. Komórki mogą wprawdzie przenosić selektywnie przez swe błony także makrocząsteczki, takie jak białka, ale wymaga to znacznie bardziej skomplikowanego mechanizmu.

Łańcuchy polipeptydowe przebadanych szczegółowo białek prowadzących transport przez błonę — zarówno przenośników, jak i kanałów — wielokrotnie przechodzą przez dwuwarstwę lipidową. Uważa się, że przechodząc wielokrotnie tam i z powrotem przez dwuwarstwę, łańcuch polipeptydowy tworzy wyścielone białkiem ciągłe przejście, pozwalające wybranym małym cząsteczkom hydrofilowym na przechodzenie poprzez błonę bez wejścia w bezpośredni kontakt z hydrofobowym wnętrzem dwuwarstwy lipidowej.
Zasadniczą różnicą między przenośnikiem a kanałem jest sposób, w jaki rozróżniają one rozpuszczone cząsteczki, transportując tylko pewne z nich, a inne nie. Kanały prowadzą to rozróżnienie na zasadzie ich wielkości i ładunku elektrycznego: gdy kanał jest otwarty, cząsteczki dosta tecznie małe i niosące odpowiedni ładunek mogą się prześlizgnąć jak przez wąskie, otwarte drzwi zapadkowe. Przenośnik działa bardziej jak jednokierunkowe drzwi obrotowe: pozwala wejść tylko tej cząsteczce, która pasuje do miejsca wiążącego na białku przenośnika i przenosi te cząsteczki poprzez błonę tylko pojedynczo, za każdym razem zmieniając swą konformację. Przenośnik specyficznie wiąże przenoszoną cząsteczkę w ten sam sposób, w jaki enzym wiąże swój substrat i to właśnie wymóg specyficznego wiązania nadaje transportowi selektywność. Aby całkowicie zrozumieć sposób, w jaki przenośnik przeprowadza cząsteczkę poprzez błonę, musielibyśmy znać szczegóły jego trójwymiarowej struktury, ale taka informacja istnieje na razie tylko w stosunku do nielicznych białek czynnych w transporcie przez błony. Jednym z nich jest bakteriorodopsyna, która działa jak aktywowana światłem pompa protonowa
W zasadzie najprostszą drogą umożliwiającą małym, rozpuszczalnym w wodzie cząsteczkom przejście z jednej strony błony na drugą jest stworzenie hydrofilowego kanału. Funkcję tę pełnią w błonach komórkowych białka kanałowe, tworzące wodne pory transbłonowe, umożliwiające bierny ruch małych, rozpuszczalnych w wodzie cząsteczek, zarówno między cytozolem i otoczeniem komórki, jak i między cytozolem i wnętrzem organelli.
Tylko nieliczne białka kanałowe tworzą względnie duże pory; przykładem są białka, które tworzą poleczenia komunikacyjne pomiędzy dwoma przylegającymi komórkami oraz poryny tworzące kanały w zewnętrznej błonie mitochondriów i pewnych bakterii. Jednak takie duże, działające bez ograniczeń kanały powodowałyby katastrofalne przecieki, gdyby bezpośrednio łączyły cytozol komórki z przestrzenią zewnątrzkomórkową. Dlatego też większość białek kanałowych w błonie komórkowej komórek zwierząt i roślin jest całkowicie odmienna i ma pory wąskie, o dużej selektywności. Prawie wszystkie te białka są kanałami jonowymi, prowadzącymi wyłącznie transport jonów nieorganicznych, głównie Na+, K+, Cl~, Ca2+.
Dwie ważne właściwości odróżniają kanały jonowe od prostych porów wodnych. Po pierwsze wykazują one selektywność jonową pozwalającą na przejście tylko niektórych jonów nieorganicznych. Selektywność jonowa zależy od średnicy i kształtu kanału jonowego oraz od rozmieszczenia w wyściółce kanału naładowanych reszt aminokwasowych. Kanał jest w pewnych miejscach dostatecznie wąski, aby zmusić jony do kontaktu ze ścianą kanału, przez co przechodzić mogą tylko te jony, które mają odpowiednią wielkość i ładunek. Na przykład wąskie kanały nie przepuszczą dużych jonów, a kanały wyścielone ładunkami ujemnymi uniemożliwią wejście jonów ujemnych ze względu na elektrostatyczne odpychanie ładunków jednoimiennych. Na tej zasadzie powstały kanały selektywne dla jednego tylko typu jonu, np. Na+ lub Cl-. Każdy jon w roztworze wodnym jest otoczony cienkim płaszczem cząsteczek wody; uważa się, że dopiero zdjęcie większości towarzyszących cząsteczek wody umożliwia przejście jonów jednego po drugim przez najwęższą część kanału. Ten etap transportu jonu ogranicza maksymalną szybkość przewodzenia jonów przez kanał. Tak więc w miarę wzrostu stężenia jonów ich przepływ przez kanał początkowo wzrasta proporcjonalnie do stężenia, ale następnie ulegnie wysyceniu przy maksymalnej szybkości.
Drugą ważną cechą odróżniającą kanały jonowe od prostych porów wodnych jest to, że kanały jonowe nie są ustawiczne otwarte. Transport jonów nie miałby dla komórki żadnej wartości, gdyby nie było sposobu kontrolowania ich przepływu i gdyby wiele tysięcy kanałów jonowych w błonie komórkowej było przez cały czas otwarte. Jak omówimy to później, większość kanałów jonowych jest bramkowana; mogą one przełączać się ze stanu otwartego w zamknięty przez zmianę konformacji, a przejście takie jest regulowane warunkami panującymi w środku i na zewnątrz komórki.
Kanały jonowe mają znaczną przewagę nad przenośnikami pod względem ich maksymalnej szybkości transportu. Przez jeden kanał może w ciągu każdej sekundy przejść ponad milion jonów, co jest szybkością 1000 razy większą niż największa znana szybkość transportu dokonywanego przez jakikolwiek przenośnik. Z drugiej strony, kanały nie mogą sprzęgnąć przepływu jonów z żadnym źródłem energii, co umożliwiłoby im prowadzenie transportu aktywnego. Tak więc funkcją większości kanałów jonowych jest uczynienie błony przejściowo przepuszczalną dla wybranych jonów nieorganicznych, głównie Na+, K+, Ca2+
i Cl~, pozwalając — w czasie otwarcia bramek kanałów — na szybkie dyfuzyjne przejście tych jonów poprzez błonę zgodnie z ich gradientami elektrochemicznymi.
W wyniku aktywnego transportu prowadzonego przez pompy i inne białka transportujące, większość stężeń jonowych po obu stronach błony jest daleko odsunięta od równowagi. Dlatego też po otwarciu kanału jony szybko przez niego przepływają. Takie szybkie wpływanie jonów wytwarza puls ładunku elektrycznego albo doprowadzonego do komórki (gdy jony wpływają), albo wyprowadzonego z komórki (gdy jony wypływają). Przepływ jonów zmienia napięcie istniejące w poprzek błony —potencjał błonowy — co zmienia siły elektrochemiczne stanowiące napęd do przemieszczania wszystkich innych jonów poprzez błonę. Zarazem, zmusza to inne kanały jonowe, specyficznie wrażliwe na zmiany potencjału błonowego, do otwarcia się lub zamknięcia w ciągu milisekund. Wynikająca stąd eksplozja aktywności elektrycznej może szybko przemieszczać się z jednego obszaru błony komórkowej do drugiego, przewodząc sygnały elektryczne. Ten typ sygnalizacji elektrycznej nie jest ograniczony do zwierząt, ale występuje też u pierwotniaków i roślin; np. mięsożerna roślina, rosiczka, używa sygnalizacji elektrycznej do wyczuwania obecności i złapania owadów.
Potencjał błonowy stanowi podstawę każdej aktywności elektrycznej w komórce, zarówno roślin, zwierząt, jak i pierwotniaków.
Główną metodą stosowaną do badania ruchu jonów i zachowania się kanałów jonowych w żywych komórkach są pomiary elektryczne. Techniki zapisu elektrycznego zostały tak wspaniale udoskonalone, że można obecnie wykrywać i mierzyć prąd elektryczny płynący przez pojedynczy kanał. Procedura znana jako zapis metodą patch-clamp pozwoliła odtworzyć zdumiewający obraz pracy indywidualnych kanałów jonowych.
W metodzie tej bardzo cienka rureczka szklana jest używana jako mikroelektroda do wytworzenia elektrycznego kontaktu z powierzchnią komórki. Mikroelektrodę uzyskuje się przez rozgrzewanie rurki szklanej i jej rozciągnięcie, co pozwala otrzymać niezwykle delikatną końcówkę o średnicy rzędu kilku

mikrometrów. Rurkę napełnia się wodnym roztworem przewodzącym prąd, a końcówką naciska się powierzchnię komórki. Przez delikatne zassanie wytwarza się szczelne złącze elektryczne pomiędzy błoną komórkową a ujściem mikroelektrody. Jeśli chcemy odsłonić cytozolową stronę błony, łatkę błony przychwyconą mikro-elektrodą delikatnie oddzielamy od komórki. W drugi, otwarty koniec mikroelektrody wprowadza się cienki metalowy przewód. Prąd wchodzący do mikroelektrody przez kanały jonowe w małej łatce błony zakrywającej końcówkę elektrody przechodzi poprzez przewód do aparatów pomiarowych, a stąd do łaźni z płynem, w której jest umieszczona komórka lub oderwana z niej łatka. Pomiary metodą patch-clamp umożliwiają uzyskanie zapisu działania kanałów jonowych we wszystkich typach komórek — nie tylko w dużych komórkach nerwowych, znanych ze swej aktywności elektrycznej, ale również w komórkach takich jak drożdże, zbyt małych, aby zachodzące w nich zjawiska elektryczne wykryć jakąkolwiek inną metodą.
Zmieniając stężenie jonów środowiska po którejkolwiek stronie łatki błony można sprawdzić, jaki jon będzie przechodził przez kanał. Przy odpowiednim obwodzie elektronicznym można ustalić napięcie istniejące w poprzek łatki błony, czyli potencjał błonowy, i utrzymywać go na stałym, dowolnie wybranym poziomie. W ten sposób można sprawdzić, jak zmiany potencjału błonowego wpływają na otwieranie się i zamykanie kanałów w błonach.
Gdy obszar błony zamkniętej końcówką elektrody jest dostatecznie mały, można natrafić na sytuację, w której będzie obecny tylko pojedynczy kanał jonowy. Nowoczesna aparatura elektryczna jest dostatecznie czuła, aby wykryć przepływ jonów poprzez pojedynczy kanał wyrażony niezwykle małym prądem (rzędu 10~12A). Zachowanie się takich prądów jest zazwyczaj zaskakujące; nawet przy utrzymaniu stałych warunków prądy nagle pojawiają się i znikają, tak jakby ktoś przypadkowo bawił się wyłącznikiem. Takie zachowanie sugeruje, że kanał ma ruchome części i przełącza się tam i z powrotem od jednej do drugiej konformacji. Ponieważ takie zachowanie pojawia się nawet przy doświadczalnym utrzymaniu stałości warunków, wskazuje, że prawdopodobnie białko kanału jest wybijane z jednej konformacji w drugą termicznymi ruchami cząsteczek w jego otoczeniu. Jest to jeden z nielicznych przypadków, w których można śledzić zmiany konformacyjne pojedynczej cząsteczki białka. Wyłaniający się obraz drgającej maszyny poddanej stałym poszturchiwaniom mógłby być z pewnością zastosowany do innych białek mających ruchome części.
Jeśli kanały w przypadkowy sposób przechodzą z konformacji otwartej do zamkniętej nawet wtedy, gdy warunki po każdej stronie błony są stałe, zastanawiające jest, w jaki sposób ich stan może być regulowany warunkami panującymi na zewnątrz komórki i w jej wnętrzu? Odpowiedź jest taka, że przy zmianie odpowiednich warunków przypadkowość zachowania zostaje zachowana, ale znacznie zmienia się prawdopodobieństwo. Jeśli na przykład zmienione warunki wykazują tendencję do otwierania kanału, to kanał będzie występował w konformacji otwartej znacznie częściej, aczkolwiek nie pozostanie otwarty w sposób ciągły. Gdy kanał jonowy jest otwarty, to jest otwarty całkowicie, a kiedy jest zamknięty, to też całkowicie.
Odkryto dotąd ponad sto typów kanałów jonowych i ciągle znajduje się nowe. Różnią się one między sobą głównie pod względem 1) selektywności jonów — a więc typem jonów, których przepływ umożliwiają i 2) bramkowania — a więc warunków wpływających na ich otwieranie i zamykanie.
W przypadku kanału bramkowanego napięciem prawdopodobieństwo otwarcia jest kontrolowane przez potencjał błonowy. W przypadku kanału bramkowanego ligandem, np. receptora acetylocholiny stan otwarcia jest kontrolowany związaniem określonej cząsteczki (liganda) z białkiem kanału. Otwarcie kanału aktywowanego przez stres jest kontrolowane siłą mechaniczną przyłożoną do kanału. Rzęsate komórki słuchowe w uchu są ważnym przykładem komórek, których działanie zależy od tego typu kanału. Drgania akustyczne otwierają kanały aktywowane przez stres powodując wpłynięcie jonów do komórek rzęsatych; powoduje to powstanie sygnału elektrycznego, który jest przenoszony z komórek włosowych do nerwu słuchowego przewodzącego sygnał do mózgu .
Kanały bramkowane napięciem odgrywają główną rolę w przewodzeniu sygnałów elektrycznych przez komórki nerwowe. Są one również obecne w wielu innych komórkach, takich jak komórki mięśniowe i jajowe, pierwotniaki, a nawet komórki roślin, gdzie umożliwiają przenoszenie sygnałów elektrycznych z jednej części rośliny do drugiej, na przykład podczas reakcji zamykania liści u mimozy. Kanały jonowe bramkowane napięciem mają wyspecjalizowane naładowane elektrycznie domeny białkowe nazywane czujnikami napięcia, które są niezwykle wrażliwe na zmiany potencjału błonowego: zmiany przekraczające określoną wartość progową wywierają na te domeny dostateczną siłę elektryczną, aby spowodować przełączenie się kanału z konformacji zamkniętej w otwartą lub odwrotnie.
Stężenie jonów wewnątrz komórki jest bardzo różne od ich stężenia na zewnątrz
Transport jonów poprzez błony komórkowe ma w biologii zasadnicze znaczenie. Komórki utrzymują wewnętrzny skład jonowy bardzo odmienny od tego, jaki istnieje w płynie otaczającym je, przy czym różnice te są kluczowe dla przeżycia i funkcjonowania komórek. W otoczeniu komórki substancjami rozpuszczonymi występującymi w największych ilościach są jony Na+, K+, Ca2+, Cl- i H+ (protony), a ich przechodzenie przez błonę komórkową stanowi istotną część wielu procesów komórkowych. Na przykład komórki zwierzęce pompują Na+ na zewnątrz, aby utrzymać małe stężenie Na+ w cytoplazmie. Pompowanie to pomaga w utrzymaniu równowagi ciśnień osmotycznych po obu stronach błony: jeśli ono zawiedzie, woda wpływa w drodze osmozy do komórki powodując jej pęcznienie i pęknięcie. Przemieszczanie jonów poprzez błony komórki pełni też zasadniczą rolę w działaniu komórki nerwowej.
Na+ jest najliczniejszym dodatnio naładowanym jonem (kationem) obecnym na zewnątrz komórki, natomiast K+ jest jonem najliczniej występującym w jej wnętrzu. Jeśli komórka ma nie być rozerwana przez siły elektryczne, ilość ładunków dodatnich wewnątrz komórki musi być zrównoważona przez prawie równą ilość ładunków ujemnych, przy czym to samo dotyczy ładunków w płynie otaczającym komórkę. Mały nadmiar dodatnich lub ujemnych ładunków, zagęszczonych w sąsiedztwie błony komórkowej jest dopuszczalny i pełni ważne funkcje elektryczne.
Duże stężenie Na+ na zewnątrz komórki jest zrównoważone głównie przez zewnątrzkomórkowe jony Cl-. Duże stężenie K+ wewnątrz komórki jest zrównoważone przez cały zestaw ujemnie naładowanych jonów (anionów) wewnątrzkomórkowych. Istotnie, większość związków wewnątrzkomórkowych ma ładunek ujemny: poza Cl-, komórki zawierają jony nieorganiczne, takie jak kwaśne węglany (HCO3
-), fosforany (PO4 3-), metabolity organiczne — zawierające ujemnie naładowane grupy fosforanowe i karboksylowe (COO-) — oraz makrocząsteczki, takie jak białka i kwasy nukleinowe, również zawierające

liczne grupy fosforanowe i karboksylowe. Organiczne cząsteczki naładowane ujemnie są czasem nazywane „utrwalonymi anionami" („fixed anions"), ponieważ nie mogą uciec z komórki przekraczając błonę komórkową.
Cząsteczki i jony przechodzą poprzez błonę w drodze transportu biernego lub aktywnego
Przy rozważaniu transportu ważnym pytaniem jest powód, dla którego zachodzi on w danym, a nie w innym kierunku. Jeśli tylko istnieje odpowiednia droga, przechodzenie cząsteczek z rejonów o ich dużym stężeniu do rejonów o małym stężeniu jest korzystne energetycznie, a więc przebiega spontanicznie. Taki ruch określamy jako bierny, ponieważ nie wymaga żadnej innej siły napędowej. Jeśli na przykład rozpuszczona substancja występuje poza komórką w stężeniu większym niż w komórce i gdy w błonie komórkowej jest obecny odpowiedni kanał lub przenośnik, substancja ta będzie spontanicznie przechodzić przez błonę do komórki w drodze transportu biernego (nazywanego też dyfuzją ułatwioną), bez wydatku energii ze strony transportującego białka.
Jednakże, aby przesunąć cząsteczkę lub jon wbrew gradientowi stężeń, transportujące białko musi wykonać pracę. Musi ono pokonać różnicę stężeń przez sprzężenie przenoszenia danych cząsteczek lub jonów z innymi procesami, które dostarczą energii. Prowadzone tą drogą przemieszczanie poprzez błonę określa się jako transport aktywny; jest on dokonywany tylko przez specjalne typy przenośników, które mogą do procesu transportu zaprząc określone źródła energii.
Napędem transportu biernego mogą być zarówno siły elektryczne, jak i gradienty stężeń
Prostym przykładem przenośnika pośredniczącego w transporcie biernym jest przenośnik glukozy obecny w błonie komórkowej komórek wątroby ssaków, a także wielu innych typów komórek. Buduje go łańcuch białkowy o 12 helisach transbłonowych. Uważa się, że białko to może przyjmować przynajmniej dwie konformacje, miedzy którymi oscyluje odwracalnie i przypadkowo. W jednej konformacji przenośnik eksponuje miejsca wiążące glukozę na zewnątrz komórki, a w drugiej eksponuje je do wnętrza komórki.
Gdy na zewnątrz komórki wątroby jest dużo glukozy (np. po posiłku), jej cząsteczki wiążą się do wystawionych na zewnątrz miejsc wiążących; gdy białko zmienia swą konformację, wprowadza te cząsteczki do wnętrza i uwalania je do cytozolu, gdzie stężenie glukozy jest małe. Odwrotnie, gdy poziom cukru we krwi jest niski (gdy jest się głodnym), hormon glukagon stymuluje komórkę wątroby do wytwarzania dużej ilości glukozy w drodze rozkładu glikogenu. W konsekwencji stężenie glukozy w komórce staje się większe niż na zewnątrz, a glukoza wiąże się do tych miejsc na przenośniku, które są eksponowane do wnętrza komórki; gdy białko zmieni swą konformację na przeciwną, glukoza zostaje wyprowadzona z komórki. Przepływ glukozy może następować w którymkolwiek kierunku, ale zgodnie z gradientem stężenia glukozy istniejącym poprzez błonę — do środka, jeśli więcej glukozy jest na zewnątrz komórki i na zewnątrz, jeśli sytuacja jest odwrotna. To właśnie tego typu białka transportujące, które umożliwiają przepływ substancji, ale nie biorą udziału w określeniu jego kierunku, prowadzą transport bierny. Chociaż bierny, transport ten jest jednak bardzo selektywny. Miejsca wiążące na przenośniku glukozy wiążą tylko D-glukozę, lecz nie, na przykład, jej zwierciadlane odbicie - L-glukozę, której komórki nie mogą używać do glikolizy. O kierunku biernego transportu glukozy, która jest cząsteczką nie naładowaną, decyduje po prostu gradient jej stężenia. W przypadku cząsteczek naładowanych elektrycznie, zarówno małych jonów organicznych, jak i nieorganicznych, w grę wchodzi dodatkowa siła. W poprzek większości błon komórkowych występuje różnica potencjałów, określana jako potencjał transblonowy wynikający z różnej koncentracji ładunków elektrycznych po dwóch stronach błony . Działa on z określoną siłą na każdą cząsteczkę, która niesie ładunek elektryczny. Cytoplazmatyczna powierzchnia błony komórkowej ma zazwyczaj potencjał ujemny ( więcej ładunków ujemnych) względem otoczenia komórki, a to powoduje tendencję do wprowadzania dodatnio naładowanych jonów lub cząsteczek do komórki, a wyprowadzania z niej jonów lub cząsteczek naładowanych ujemnie. Równocześnie jednak cząsteczki te będą miały tendencję do przemieszczania się w dół ich gradientu stężenia. Taki gradient jest też formą magazynowania energii, podobnie jak zgromadzona przed zaporą woda, która może zostać wykorzystana do napędzania innego układu transportującego.
Wypadkowa siła kierująca poprzez błonę jony lub naładowane cząsteczki składa się więc z dwóch sił składowych, z których jedna wynika z gradientu stężenia, a druga z napięcia istniejącego poprzez błonę. Tę wypadkową siłę określa się jako gradient elektrochemiczny dla danej przenoszonej jednostki. Ten właśnie gradient determinuje kierunek biernego transportu przez błonę. Dla pewnych jonów napięcie i gradient stężenia działają w tym samym kierunku, tworząc względnie stromy gradient elektrochemiczny. Tak jest na przykład w przypadku Na+, który jest naładowany dodatnio i którego stężenie jest większe na zewnątrz komórki niż w jej wnętrzu. Dlatego też, gdy tylko Na+ ma takie możliwości, będzie dążył do wejścia do komórki. Gdy napięcie i gradienty stężeń mają efekt przeciwstawny, wypadkowy gradient elektrochemiczny może być mały. Przykładem jest tu K+, jon naładowany dodatnio, którego stężenie wewnątrz komórki jest znacznie większe niż na zewnątrz. Właśnie z powodu przeciwstawnych efektów K+ ma mały gradient elektrochemiczny poprzez błonę, mimo jego dużego gradientu stężenia i dlatego wypadkowe przemieszczanie K+ przez błonę jest niewielkie.
Transport aktywny przemieszcza jony i cząsteczki wbrew ich gradientom elektrochemicznym
Komórki nie mogą polegać jedynie na transporcie biernym. Do zachowania wewnątrzkomórkowego składu jonowego komórek i do wprowadzania cząsteczek, których stężenie na zewnątrz jest mniejsze niż w komórce, niezbędny jest aktywny transport cząsteczek i jonów wbrew ich gradiento wi elektrochemicznemu. Istnieją trzy główne drogi, którymi komórki prowadzą transport aktywny: 1) przenośniki sprzężone sprzęgają transport przez błonę

jednej cząsteczki, zachodzący wbrew gradientowi, z transportem innej, zgodnym z gradientem; 2) pompy napędzane przez ATP sprzęgają transport wbrew gradientowi z hydrolizą ATP; 3) pompy napędzane światłem, znajdowane głównie w komórkach bakteryjnych (bakteriorodopsyna), sprzęgają transport wbrew gradientowi z wprowadzeniem energii ze światła.
Ponieważ substancja, która ma się przemieszczać zgodnie z gradientem, musi być uprzednio przetransportowana wbrew gradientowi, niezbędne jest powiązanie różnych form aktywnego transportu. Tak więc, w błonie komórkowej komórek zwierząt pompy napędzane przez ATP wyprowadzają z komórki Na+
wbrew jego gradientowi elektrochemicznemu, a następnie Na+ wpływa do komórek z powrotem już zgodnie z tym gradientem. Ponieważ Na+ wpływa do cytozolu poprzez przenośniki sprzężone z Na+, jego napływ stanowi napęd do aktywnego przemieszczenia wielu innych substancji do komórki wbrew ich gradientom elektrochemicznym. Gdyby pompa Na+ przestała działać, gradient Na+ prędko by się wyrównał, a transport poprzez przenośniki sprzężone z Na+
uległby zatrzymaniu. Dlatego też napędzana przez ATP pompa Na+ odgrywa centralną rolę w transporcie poprzez błony w komórkach zwierząt. W komórkach roślin, grzybów i wielu bakterii podobną rolę odgrywają napędzane przez ATP pompy, które wytwarzają protonowy gradient elektrochemiczny przez wy-pompowywanie H+ z komórki.
W procesie egzocytozy komórka pozbywa się produktów odpadowych lub też wytworzonych przez siebie specyficznych wydzielin w wyniku zlania się pęcherzyka z wydzieliną (lub wydaliną) z błoną komórkową. Egzocytoza polega zatem na wbudowaniu błony tworzącej pęcherzyk wydzielniczy w błonę komórkową. Jest to również podstawowy mechanizm powiększania się błon.
W procesie endocytozy komórka pochłania materiał pochodzący z zewnątrz. W wyniku fagocytozy komórka pochłania cząstki pożywienia lub bakterie. Proces ten polega na otoczeniu pochłanianych cząsteczek przez mikrofałdy błony komórkowej i utworzeniu wokół nich wakuoli. Gdy cząstki są już całkowicie otoczone, dochodzi do fuzji z lizosomami, w których następuje rozkład pochłoniętego materiału.
Inną formą endocytozy jest pinocytoza, w wyniku której komórka pobiera z zewnątrz materiał w postaci rozpuszczonej. Małe kropelki płynu zostają uwięzione w mikrofałdach błony komórkowej, z której odrywają się po stronie cytoplazmy drobne pęcherzyki. Płynna zawartość pęcherzyków przenika powoli do cytoplazmy, podczas gdy pęcherzyki powoli zmniejszają się stopniowo, aż w końcu znikają .
W endocytozie receptorowej specyficzne białka lub cząstki łączą się z receptorami białkowymi zlokalizowanymi w błonie komórkowej. Kompleksy cząstek z receptorami przesuwają się wzdłuż płaszczyzny błony do zagłębień opłaszczonych (po stronie cytoplazmatycznej) grzybkowatymi strukturami. W wyniku fuzji (zlania się) zagłębienia te przekształcają się w opłaszczone pęcherzyki. Struktury opłaszczające są białkami (klatrynami), które formują wokół pęcherzyka sieć. W kilka sekund po oderwaniu się pęcherzyka od błony do cytoplazmy białkowa sieć oddysocjowuje. Uwolnione z sieci pęcherzyki zlewają się z innymi podobnymi pęcherzykami, tworząc endosom - czyli większy pęcherzyk w którym transportowane cząstki nie są już związane z receptorami błonowymi. Endosom rozpada się z kolei na pęcherzyki dwóch rodzajów: jedne, zawierające receptory , powracają do błony, drugie - zawierające wchłonięte cząstki, zlewają się z lizosomem, gdzie zostają przetworzone, co umożliwia ich wykorzystanie przez komórkę. W taki sposób wchłaniany jest cholesterol do komórek zwierzęcych.
POŁĄCZENIA MIĘDZYKOMÓRKOWE
Komórki współpracujące ze sobą w zespołach (tkanki i narządy ) z reguły ściśle do siebie przylegają. W rejonach szczególnie ścisłego styku tworzą się specjalne struktury łączące je ze sobą, nazywane połączeniami międzykomórkowymi. Połączenie takie zbudowane jest z dwóch symetrycznych części, z których każda należy do jednej z komórek tworzących styk. Połączenia spełniają trzy zasadnicze funkcje:
1. Połączenia mechaniczne -zapewniają mechaniczne powiązanie sąsiadujących komórek
· Desmosom - pełni funkcje połączenia mechanicznego, jest strukturą lokalną zespalając komórki jedynie w pewnych punktach na podobieństwo zatrzasków. Ten typ połączenia występuje w komórkach nabłonka pokrywającego skórę i innych nabłonkach wielowarstwowych, oraz pomiędzy komórkami mięśnia sercowego. Charakteryzuje się bardzo dużą wytrzymałością mechaniczną i łatwym przenoszeniem naprężeń na sąsiednie komórki.
Odległość pomiędzy błonami sąsiednich komórek w obrębie desmosomu wynosi ok. 30nm. Desmosom składa się z obszarów o dużej gęstości, przylegających do cytoplazmatycznej strony każdej z dwóch zespolonych błon komórkowych sąsiednich komórek (są to tzw. płytki adhezyjne czyli dyskowate struktury zbudowane głównie z polipeptydów desmoplakiny, plakoglobiny i desmokalminy, które są podobne u różnych organizmów oraz z białkowych filamentów przenikających przestrzeń między komórkami. W płytce adhezyjnej zakotwiczają się, tworząc układy pętlowe, dochodzące z głębi cytoplazmy filamenty pośrednie: keratynowe (tonofilamenty) w nabłonkach, desminowe w komórkach mięśniowych.
Przestrzeń międzybłonowa wypełniona jest układami włókien białkowych o średnicy 8-12nm, zazębiających się ze sobą na kształt zamka błyskawicznego i spajającego błony sąsiednich komórek

Elementy składowe desmosomów odznaczają się ogromną odpornością na czynniki denaturujące białka. Połączenia te są uważane za najsilniejsze mechaniczne połączenia międzykomórkowe.
Desmosomy łączą się z filamentami pośrednimi cytoszkieletu i z innymi desmosomami. Poprzez desmosomy sieci cytoszkieletów sąsiadujących komórek są połączone w taki sposób, że mechaniczne naprężenia są przenoszone po całej tkance. Dzięki desmosomom komórki tworzą mocno spojone układy przestrzenne, a wymiana substancji między komórkami może się swobodnie odbywać poprzez przestwory międzykomórkowe.
· Półdesmosom jest jak gdyby „połówkami” desmosomu, nie zawierają jednak typowych dla desmosomu białek, desmoplakin i desmoglein. Występują one na pograniczu komórek nabłonkowych i błony podstawnej,
· Strefa przylegania - jest to połączenie o charakterze pasa biegnącego wokół komórki i typowo występuje w nabłonkach jednowarstwowych walcowatych. Błony sąsiednich komórek znajduja się bardzo blisko siebie (15-20 nm), a po ich cytoplazmatycznej stronie widoczne jest skupienie gęstego materiału który są białka winkulina (odpowiedzialna za przyczep mikrofilamentów aktynowych do błon biologicznych), i plakoglobina. Mikrofilamenty aktynowe stanowią stały element składowy tego połączenia, tworząc wiązkę biegnącą równolegle do przebiegu strefy. Jedynie niewielka część mikrofilamentów aktynowych zakotwicza się w tym obszarze i biegnie prostopadle w głąb cytoplazmy. W przestrzeniach pomiędzy błonami tego połączenia występuje białko nazywane cząsteczką przylegania komórkowego i któremu przypisuje się pełnienie funkcji „receptora” odpowiedzialnego za przyleganie błon.
Uważa się, że strefy przylegania, dzięki udziałowi w nich kurczliwych mikrofilamentów aktynowych, są dynamiczną formą połączenia międzykomórkowego i odgrywają pierwszoplanową role w zmianach kształtu zespołów komórkowych podczas morfogenezy.
Szczególne odmiany tego połączenia to powięź przylegania i punkt przylegania. Powięź przylegania jest jednym ze składników wstawki, czyli specjalnego systemu połączeń komórek mięśnia sercowego. Połączenie to, o przestrzennej formie nieregularnych płaszczyzn, lokalizuje się głównie na pionowych powierzchniach wstawki i jest miejscem zakotwiczenia w błonie komórkowej cienkich (aktynowych) miofilamentów. Punkty przylegania, opisywane pomiędzy komórkami śródbłonków naczyniowych i pomiędzy komórkami Sertoliego w jądrze, różnią się od strefy jedynie ograniczoną przestrzennie formą. W obrębie obu połączeń stwierdzono - podobnie jak w strefie przylegania - obecność winkuliny i plakoglobiny.
2. Połączenia barierowe uszczelniają przestrzeń międzykomórkową, zapobiegając swobodnej dyfuzji substancji pomiędzy komórkami
· Strefa zamykająca (złącza ścisłe, styk zwarty) - to ścisłe połączenia pomiędzy całymi obszarami błon komórkowych sąsiednich komórek. Podobnie jak strefa przylegania strefa zamykająca otacza komórkę w formie ciągłego pasa. Na jej przebiegu obserwuje się leżące blisko siebie punkty ścisłego styku błon komórkowych, w których dochodzi do zespolenia zewnętrznych warstw sąsiadujących błon. Kontakt jest tak ścisły, że zanika przestrzeń międzykomórkowa. Niektóre substancje (jony i cząsteczki) nie przenikają przez tak zespolone warstwy komórek. Połączone w ten sposób warstwy komórek oddzielają często narządy od jam ciała. Na przykład strefy zamykające pomiędzy komórkami wyściełającymi jelito zapobiegają wnikaniu substancji zawartych w jelicie do komórek ciała lub krwiobiegu przez przestwory międzykomórkowe. Takie zespolone warstwy komórek pełnią rolę selektywnych barier; substancje niezbędne organizmowi muszą zatem być transportowane do krwi przez cytoplazmę komórek nabłonkowych jelita, które mogą proces ten kontrolować i decydować o jego selektywności. Bariery utworzone przez strefy zamykające zapobiegają również przedostawaniu się do krwi różnego rodzaju toksyn i zbędnych substancji.
2. Połączenia komunikacyjne - umożliwiają komunikowanie się komórek ze sobą, poprzez bezpośrednią wymianę jonów i niskocząsteczkowych substancji biologicznie czynnych
· Złącza szczelinowe (neksus) spinają komórki jak mostki, podobnie jak desmosomy lecz na mniejszym obszarze. Połączenie to różni się od desmosomu tym, że oprócz funkcji zespalającej błony pełnią funkcję kanału łączącego cytoplazmy sąsiadujących komórek. Złącze szczelinowe zbudowane jest z białek integralnych tworzących regularny układ sześciokątny (konekson), przez którego wnętrze przebiega kanał o średnicy 1-2 nm. Mogą przez ten kanał przenikać małe cząstki nieorganiczne, np. jony i niektóre cząsteczki ważne biologicznie np. pochodne ATP, natomiast większe cząsteczki nie są przepuszczane. Liczba koneksonów występujących w obrębie jednego neksusa jest różna.
Złącza szczelinowe zapewniają szybkie przekazywanie informacji pomiędzy komórkami na drodze chemicznej i elektrycznej. Tego typu połączenia występują w komórkach trzustki: jeśli jedna komórka zostanie pobudzona do wydzielania insuliny, sygnał przedostanie się przez złącza szczelinowe do innych komórek, zapewniając skoordynowana odpowiedź całego narządu. Komórki mięśnia sercowego są również zespolone złączami szczelinowymi. Zapewniają one taki stopień elektrycznego sprzężenia, że skurcz komórek odbywa się synchronicznie.
Neksusy mają istotny udział w takich procesach jak:
· przewodnictwo elektryczne - przewodzenie bodźców m.in. pomiędzy komórkami mięśnia gładkiego i mięśnia sercowego odbywa się na drodze przepływu jonów przez neksusy;
· rozwój i różnicowanie tkanek - komórki przekazują sobie poprzez neksusy substancje sygnałowe oraz niezbędne do prawidłowego przebiegu tych procesów czynniki biologiczne,
· przekazywanie bodźców hormonalnych - komórki mogą wymieniać między sobą niskocząsteczkowe hormony, bądź cykliczne nukleotydy, będące wewnątrzkomórkowymi przekaźnikami;

· kooperacja metaboliczna - komórki połączone neksusami mogą przekazywać sobie wzajemnie niskocząsteczkowe metabolity, w ten sposób substancje wytworzone przez niektóre tylko komórki mogą być wykorzystywane przez cały ich zespół, a procesy przemiany materii ulegają w takim zespole koordynacji;
· synchronizacja funkcji komórek w zespole i zespołów komórkowych w procesach rozwoju komórkowego.
Opisane połączenia występują tylko w komórkach zwierzęcych. U roślin występuje tylko jeden typ połączeń międzykomórkowych zwany plazmodesmami. Są to pasma cytoplazmy otoczone błoną komórkową, które łączą protoplasty sąsiadujących komórek. Plazmodesmy pełnią rolę połączeń komunikacyjnych, przez które zachodzi wymiana cząsteczek pomiędzy komórkami.
WAKUOLA w komórkach roślin
Wakuola jest przestrzenią w cytoplazmie komórki otoczoną pojedynczą błoną komórkową zwaną tonoplastem, zawierającą sok komórkowy. Podstawowym składnikiem soku komórkowego (wakuolarnego) jest woda w której rozpuszczone są składniki mineralne i organiczne, mogą się tam znajdować ciała stałe - kryształy szczawianu wapnia. Wyżej opisane wakuole występują jedynie w komórkach roślinnych.
Ontogeneza (powstawanie) i rozwój wakuoli
W procesie różnicowania komórek wakuole powstają z:
· z siateczki śródplazmatycznej (ER) poprzez wytwarzanie pęcherzyków przez siateczkę lub lokalne rozszerzenia kanałów siateczki powstają tzw prowakuole. Prowakuole tworzą się w komórkach merystematycznych, a w miarę dojrzewania komórki powiększają się i zlewają ze sobą aż w komórce dojrzałej powstaje jedna dużą centralnie leżąca wakuola Część siateczki która ulega rozszerzeniu w wakuolę, jest gładka lub zawiera nieliczne rybosomy, stąd wniosek, że tonoplast jest zróżnicowaną formą błon (ER). Dowodem na pochodzenie wakuoli od ER jest obecność w wakuoli taniny uprzednio stwierdzonej w cysternach siateczki, jak też obecność w małych i dużych wakuolach komórek merystematycznych korzeni enzymów typowych dla siateczki
· z pęcherzyków aparatu Golgiego, które mogą łączyć się z istniejącymi już wakuolami rozbudowując je.
· przez rozpad dużych wakuol, np. u drożdży podczas pączkowania, czy też w komórkach przyszparkowych.
Składniki soku komórkowego (wakuolarnego)
1. Składniki nieorganiczne: woda, jony: kationy K, Na, Ca, Mg, Cu, aniony chlorkowe, fosforanowe, siarczanowe, azotanowe.
2. Składniki organiczne
· metabolity pośrednie - nadwyżki związków z różnych szlaków metabolicznych komórki czasowo gromadzone w wakuoli np.:
- kwasy organiczne (nadwyżki z cyklu kwasów trójkarboksylowych)
- aminokwasy (z hydrolizy białek zapasowych)
- cukry (sacharoza, maltoza, glukoza, fruktoza)
· metabolity wtórne - produkty różnych procesów komórkowych składowane w wakuoli:
- glikozydy antocyjanowe - (antocyjany = połączenia cukrów np. glukozy, galaktozy z hydroksy-pochodnymi trójpierścieniowego układu tzw. antocyjanidyn-aglikonów takich jak pelargonidyna, cyjanidyna, delfinidyna). Należą do nich: pelargonina, cyjanina, idaina, violanina, są to związki nadające zabarwienie od czerwonego do niebieskiego a ich odcień zależy od struktury aglikonu i pH.
- glikozydy flawonowe - (połączenie cukrów z pochodną flawonu): chryzyna, luteina, kemferol, kwercetyna , nadające żółte zabarwienie.
- alkaloidy zasady organiczne zawierające azot w układzie pierścieniowym, występują często w postaci soli są związkami przeważnie bezbarwnymi, silnie toksycznymi, mają zastosowanie w lecznictwie np.: nikotyna, skopolamina, kokaina, chinina, papaweryna, morfina, narkotyna, kodeina, strychnina

-garbniki - pochodne wielofenoli i kwasów fenolokarboksylowych oraz cukrów (występ. w korze korzeniach, kłączach, owocach, nasionach, liściach), mają właściwości toksyczne
· składniki zapasowe: białka (ziarna aleuronowe), cukry (inulina), tłuszcze (jedynie w wakuolach grzybów)
Funkcje wakuoli
· utrzymanie turgoru (odpowiedni stan napięcia ścian komórki) dzięki zawartości substancji osmotycznie czynnych, wciągających wodę do wakuoli,
· magazynowanie czasowe nadwyżek metabolitów pośrednich oraz składników zapasowych, które mogą być wykorzystane w warunkach niedoboru składników w środowisku
· gromadzenie substancji toksycznych dla komórki, wydalin - funkcja detoksykacyjna
· funkcja obronna - magazynowane w wakuoli związki gł. metabolity wtórne chronią rośliny przed zjadaniem przez zwierzęta ( nadają nieprzyjemny smak lub są toksyczne), wnikaniem pasożytów czy mikroorganizmów
· trawienie substancji zapasowych i autofagia (udział w różnicowaniu komórek). Wakuole pełnią funkcję lizosomu w komórkach roślinnych. Stwierdzono, że wakuole roślin wyższych zawierają wszystkie klasy enzymów hydrolitycznych (proteinazy, peptydazy, esterazy, glukozydazy i inne), mogą więc tam zachodzić procesy wewnątrzkomórkowego trawienia np. materiałów zapasowych na prostsze związki, które mogą być włączone w metabolizm komórki czy nawet fragmentów komórki - zjawisko autofagii. Wyróżnia się dwa mechanizmy tego procesu:
- pierwszy polega na inwaginacji (wpuklaniu) tonoplastu, co przypomina fagocytozę. Wynikiem postępującej inwaginacji jest powstawanie wewnątrzwakuolarnych pęcherzyków w których znajdują się fragmenty cytoplazmy wraz z organellami.
- drugi mechanizm sprowadza się do pojawienia się w cytoplazmie koncentrycznego układu małych fragmentów siateczki śródplazmatycznej, następnie powiększają się one i łączą ze sobą tworząc wakuolę.
Autofagia odgrywa dużą rolę podczas różnicowania (przebudowa komórki) oraz starzenia się komórek i stanowi prawdopodobnie jedno z ogniw ogólnej przemiany materii w roślinie.
· dzięki zawartości barwników wakuole nadają zabarwienie komórkom i całym organom np. kwiatom, liściom, owocom, nasionom itp.
Cząsteczki RNA są polimerami zbudowanymi z liniowych, nierozgałęzionych łańcuchów podjednostek, zwanych nukleotydami. Nukleotyd budują trzy części: cukier, grupa fosforanowa i zasada azotowa. W RNA cukrem jest ryboza, zasadami zaś są: adenina, cytozyna, guanina i uracyl.
Nukleotydy biorące udział w syntezie RNA to:
- adenozyno-5’-trifosforan (ATP),
- cytydyno-5’-trifosforan (CTP),
- guanozyno-5’-trifosforan (GTP) i
- urydyno-5’-trifosforan (UTP).
Polinukleotyd RNA zawiera wiązanie 3’-5’-fosfodiestrowe, które jest mniej stabilne niż w DNA, dlatego polimery RNA rzadko składają się z więcej niż kilku tysięcy nukleotydów. W RNA mogą powstawać pary zasad: A łączy się z U podwójnym wiązaniem wodorowym, a G z C potrójnym wiązaniem wodorowym. Cząsteczki RNA występują w formie jednoniciowej i dwuniciowej. Cząsteczki dwuniciowego RNA (dsRNA) nie mogą tworzyć tak regularnej struktury jak w przypadku B-DNA, ze względu na występowanie grupy hydroksylowej przy 2 atomie węgla rybozy. Zazwyczaj jednak RNA występuje w postaci jednoniciowego (ssRNA), silnie pofałdowanego polinukleotydu zawierającego odcinki dwuniciowe i tworzącego skomplikowane struktury przestrzenne. Fragmenty dwuniciowe przybierają przeważnie strukturę helisy, której długość nie przekracza zazwyczaj kilkudziesięciu par zasad. W przeciwieństwie do DNA stosunek zasad azotowych purynowych do pirymidynowych, w cząsteczce jednoniciowego RNA, nie jest zachowany.
Rodzaje RNA - rodzaje kwasów rybonukleinowych RNA
Kwas rybonukleinowy RNA dzieli się na kilka rodzajów:

- informacyjny lub matrycowy RNA (Zobacz: mRNA),
- rybosomalny RNA (Zobacz: rRNA),
- transportowy (transferowy) RNA (Zobacz: tRNA),
- heterogenny jądrowy RNA (hnRNA lub pre-mRNA) – jest to głównie produkt transkrypcji DNA, który jest następnie przetwarzany do mRNA,
- antysensowny RNA lub interferencyjny RNA (Zobacz: siRNA i miRNA). Interferencyjne RNA reguluje ekspresję genów kodujących białka,
- mały jądrowy RNA (snRNA) - pełniący funkcje enzymatyczne przy obróbce potranskrypcyjnej , czyli bierze udział w wycinaniu intronów z transkryptów,
- mały jąderkowy RNA (snoRNA) – wpływa na modyfikację pre-mRNA,
- mały cytoplazmatyczny RNA (scRNA).
Postać dwuniciowa kwasu RNA, analogiczna do dwuniciowego DNA, występuje przede wszystkim jako materiał genetyczny niektórych wirusów oraz wiroidów.
Ekspresja genów jest złożonym procesem regulowanym m.in przez mechanizm określany jako interferencja RNA (RNAi). Kluczowym składnikiem tego układu regulacyjnego jest dwuniciowy RNA (dsRNA), powodujący degradację homologicznych cząsteczek RNA. Ogół procesów komórkowych, prowadzących ostatecznie do wspomnianej degradacji RNA, określa się jako interferencję RNA. RNAi jest składową ogółu procesów posttranskrypcyjnego wyciszania ekspresji genów, który pierwotnie odkryto u roślin. Wprowadzenie dodatkowych kopii genów syntazy chalkonowej w celu zwiększenia ilości pigmentu w kwiatach petunii wbrew oczekiwaniom spowodowało, iż kwiaty tej rośliny były częściowo lub całkowicie pozbawione zabarwienia. W istocie nie nastąpiła ekspresja dodatkowo wprowadzonego genu, a supresja endogennego genu wytwarzającego pigment. Było to jedno z pierwszych doświadczeń wskazujących na istnienie zjawiska wyciszania ekspresji genów.
Cząsteczki RNA indukujące mechanizmy RNAi charakteryzują się specyficzną strukturą oraz specyficznymi właściwościami chemicznymi. Natywne siRNA, będące produktami nukleolitycznej degradacji długiego dsRNA przez rybonukleazę DICER, mają charakterystyczną budowę. Obie nici RNA o długości 21-23 nukleotydów, tworzą dupleks na odcinku 19 nukleotydów, pozostawiając na każdym końcu 3’ po dwa lub więcej niesparowane nukleotydy. Antysensowna nić siRNA tworzy komplementarny dupleks z docelowym mRNA. Modyfikacje w obrębie tej nici siRNA powodują obniżenie wydajności procesu RNAi lub też całkowity zanik tego zjawiska. siRNA posiadają na końcach 3’ wolną grupę hydroksylową, a na końcach 5’ grupę fosforanową. Wprowadzenie modyfikacji końca 5’ przez przyłączenie grupy 3-aminopropylofosforanowej lub grupy O-metylowej nie ogranicza efektywności procesu RNAi, jeśli modyfikacja dotyczy nici sensownej, natomiast modyfikacja nici antysensownej prowadzi do zaniku procesu RNAi. Wskazuje to, że koniec 5’ nici antysensownej siRNA pełni istotną funkcję w procesie RNAi. Sugeruje się, że miejsce hydrolizy mRNA jest zależne od położenia końca 5’ nici antysensownej siRNA, a 5’-terminalna grupa fosforanowa jest „molekularnym punktem odniesienia”, od którego mierzone jest miejsce hydrolizy mRNA (Tuschl i wsp.). Podstawienie 3’-terminalnej grupy hydroksylowej nici sensownej i antysensownej siRNA resztą fluoresceiny, puromycyny czy biotyną, lub też wprowadzenie 3’-terminalnego 2’,3’-dideoksynukleotydu lub grupy 3-aminopropylofosforanowej nie powoduje zahamowania efektu RNAi. Natomiast modyfikacja nici sensownej za pomocą 2’-O,4-C-etyleno-tymidyny i 2-hydroksy-etylofosforanu tymidyny nie wpływa na jakość RNAi, zaś modyfikacja tymi samymi czynnikami nici antysensownej lub obydwu nici powoduje całkowite zahamowanie RNAi.
Zaproponowano co najmniej dwa mechanizmy, według których może zachodzić proces wyciszania ekspresji genów. W pierwszym z nich inicjacja RNAi następuje przez konwersję dwuniciowego RNA do 21-23-nukleotydowych fragmentów za pomocą wielodomenowej rybonukleazy DICER, wywodzącej się z rodziny RNazy III. Produkty hydrolizy – krótkie dupleksy RNA, zwane siRNA, włączone są w kompleks nukleazowy RISC i kierują go do docelowej sekwencji mRNA. Endorybonukleaza obecna w kompleksie RISC wykorzystuje antysensowną nić siRNA do odnalezienia i degradacji komplementarnej sekwencji mRNA, dlatego też siRNA nazywany jest dupleksem kierującym. Nukleolityczne właściwości kompleksu RISC są istotą procesu RNAi. Przyłączenie siRNA aktywuje RISC, który następnie rozpoznaje i degraduje komplementarny mRNA - blokowana jest w ten sposób ekspresja genu. Drugi mechanizm RNAi, zaproponowany przez Lipardi i wsp., Sijen i wsp., Nishikura i wsp., polega na tzw. „degradacyjnym PCR”. W mechanizmie tym próbuje się wytłumaczyć katalityczny charakter RNAi u nicienia Caenorhabditis elegans. Stwierdzono, że jedynie kilka cząsteczek dsRNA wystarcza do degradacji mRNA. Inaktywacja ekspresji danego genu u tego nicienia utrzymuje się podczas podziałów komórkowych, przenoszona jest do nietransfekowanych komórek i tkanek, jak również pojawia się w następnym pokoleniu. Polimeraza RNA zależna od RNA (RdRP) na matrycy mRNA syntetyzuje nić komplementarną, tworząc nowy dwuniciowy RNA, rozpoznawany i hydrolizowany przez rybonukleazę DICER. W ten sposób tworzona jest nowa generacja siRNA. Starterem w reakcji polimeryzacji jest antysensowna nić siRNA. W reakcji tej szczególne znaczenie ma grupa hydroksylowa obecna na końcu 3’ nici antysensownej siRNA.
Obniżenie poziomu białka wywołane przez egzogenne podanie siRNA jest przejściowe, trwa zwykle 5-7 dni po transfekcji. Podanie plazmidów i wywołanie ekspresji RNA wewnątrz komórki powoduje dłuższy efekt wyciszania. Możliwość stabilnej ekspresji siRNA otwiera drogę do zastosowania zjawiska RNAi w terapii genowej, na przykład w leczeniu infekcji wirusowych. Wysoki koszt syntezy chemicznej RNA bez gwarancji, że wybrane sekwencje wywołają wydajne wyciszenie ekspresji docelowego genu, powoduje, że poszukuje się alternatywnych sposobów uzyskiwania siRNA. Jedną z metod syntezy dużej liczby transkryptów krótkich interferujących RNA jest metoda z udziałem polimerazy RNA z faga T7 (Donze i wsp.). Otrzymywane w ten sposób siRNA wywołują efekt wyciszenia genów egzo- i endogennych w różnych komórkach ssaków. Nici RNA, sensowną i antysensowną, o długości 21 nukleotydów każda, generowano w osobnych reakcjach polimeryzacji na matrycy DNA kodującego wybrany fragment danego genu. Tą metodą otrzymano RNA o strukturze typu spinki (shRNA), o długościach dupleksu 22-29 nukleotydów, z czteronukleotydową pętlą UUAA. Uzyskane transkrypty shRNA wprowadzano do komórek owadów i ssaków. Wykazano, że shRNA w cytoplazmie hydrolizowane są przez enzym DICER do postaci 22-nukleotydowych siRNA, wywołując degradację docelowego mRNA (Paddison i wsp.). Inną metodą uzyskiwania siRNA w warunkach in vitro jest metoda z wykorzystaniem RNazy III z E.coli. Powstałe w wyniku jej działania krótkie fragmenty (e-siRNA) mają taką samą strukturę jak natywne siRNA. Kontrolowana reakcja enzymatycznej hydrolizy prowadzi do wydajnego generowania siRNA o długości 20-25 nukleotydów. e-siRNA wytwarzane w ten sposób wydajnie obniżają ekspresję egzogennych i endogennych genów w różnych liniach komórkowych.
Planowanie sekwencji siRNA, które mogą interferować z ekspresją wybranego genu wymaga wiedzy na temat sekwencji kodującego mRNA. RNAi jest

procesem cytoplazmatycznym, dlatego też siRNA nie może być skierowane na sekwencje intronowe pre-mRNA, którego dojrzewanie zachodzi w jądrze. Musi być skierowane na sekwencje kodujące genu. siRNA o sekwencjach homologicznych do różnych miejsc mRNA tego samego genu, mogą indukować zróżnicowany poziom ekspresji genu. Selekcja sekwencji docelowej dokonywana jest doświadczalnie metodą prób i błędów. Najlepiej wybierać sekwencję odległą o 50-100 nukleotydów od miejsca inicjacji translacji. Najlepiej jest wybierać sekwencje siRNA posiadające na końcach 3’ reszty urydylowe. Zawartość G-C w projektowanych sekwencjach nie powinna przekraczać 30-40%. Należy unikać sekwencji bogatych w G zdolnych do tworzenia struktur tetrapleksowych. Zjawisko RNAi odgrywa bardzo ważną rolę w prawidłowym funkcjonowaniu komórki i całego organizmu. Zaburzenia tego procesu powodują szereg niekorzystnych zmian w organizmie. Indukowany RNAi stanowi obronę przed wirusami, ochronę genomu przed transpozonami oraz współuczestniczy w regulacji ontogenezy. RNAi wpływa na stabilność genomu poprzez ograniczenie integracji transpozonów, które wbudowując się mogą przerwać ciągłość genów. Inhibicja ekspresji genów poprzez RNAi może przebiegać na różnych poziomach. dsRNA u roślin indukuję metylację fragmentów genomu, a w przypadku modyfikacji sekwencji promotora następuje zablokowanie transkrypcji genu. RNAi może także modulować strukturę chromatyny. Opisano pierwsze udane eksperymenty wykorzystujące siRNA skierowane przeciwko genom białek rev i tat w liniach komórkowych transfekowanych prowirusem HIV-1, jak również genom receptora komórkowego cd4, białka strukturalnego gag czy białka regulatorowego nef. Działania anty-HIV mają także na celu hamowanie replikacji wirusa w komórkach gospodarza. Po zastosowaniu siRNA w limfocytach zaobserwowano obniżenie poziomu wirusa HIV w komórkach do 30-50 razy. Inhibicja infekcji wirusowej przez RNAi dotyczy także innych wirusów jak: RSV, który wywołuje szereg chorób układu oddechowego oraz wirusa polio. Ekspresja siRNA w wektorach retrowirusowych pozwala na kierowanie ich do komórek pierwotnych, co do tej pory było nieosiągalne. Wysoka specyficzność siRNA stwarza nadzieję ich zastosowania do wyciszania onkogenów. RNAi wykorzystuje się również jako narzędzie do badania własności genów u różnych organizmów poprzez „knock-out” mRNA i obserwacje funkcjonalnego efektu inhibicji ekspresji całych grup genów na poziomie komórki czy całego organizmu.
Kwas rybonukleinowy mRNA jest pojedynczą cząsteczką RNA (ssRNA), stanowiącą nośnik informacji genetycznej, zawartej w sekwencji zasad azotowych. Na jej podstawie polimeryzowane są aminokwasy według określonej kolejności. Dzięki temu procesowi powstaje produkt końcowy ekspresji informacji genetycznej, czyli białko. W nukleotydach mRNA zapisana jest, kodem genetycznym, sekwencja aminokwasów polipeptydów. Jeden kodon składa się z trzech nukleotydów.
Kodony tworzące kod genetyczny64 kodony tworzące kod genetyczny można podzielić na grupy. Kodony wchodzące w skład jednej grupy kodują jeden aminokwas. Kod genetyczny ma 4 kodony przestankowe: kodon inicjacyjny (AUG) oraz kodony terminacyjne (UAG, UAA, UGA). Procesy syntezy i dojrzewania mRNA u organizmów prokariotycznych i eukariotycznych znacznie różnią się od siebie.
Podstawową różnicą pomiędzy mRNA bakteryjnym a eukariotycznym, jest to, że mRNA bakterii w zasadzie nie podlega dojrzewaniu – nie zawiera intronów. Pierwotny transkrypt syntetyzowany przez polimerazę RNA jest od razu dojrzałym mRNA i jeszcze przed zakończeniem transkrypcji rozpoczyna się translacja. mRNA jest syntetyzowany na matrycy DNA. Rybonukleotydy są dodawane do końca 3’ transkryptu RNA na zasadzie komplementarnego łączenia zasad. W czasie procesu elongacji łańcucha mRNA tworzy się pęcherzyk transkrypcyjny, wewnątrz którego znajduje się hybryda DNA-RNA. Pęcherzyk przesuwa się po matrycy i uwalniany jest transkrypt. Zakończenie transkrypcji może przebiegać z udziałem samodzielnych terminatorów lub białka Rho. mRNA bakteryjny jest policistronowy, co oznacza że zawiera informacje dotyczące budowy wielu różnych białek.
mRNA eukariotyczne
W odróżnieniu od organizmów prokariotycznych wszystkie eukariotyczne mRNA mają czapeczkę przyłączoną na końcu 5’, większość z nich jest poliadenylowana przez dodanie serii adenozyn do końca 3’, wiele transkryptów zawiera introny i podlega procesowi składania, a niektóre są poddawane redagowaniu. Tworzenie się czapeczki to wieloetapowy proces, który rozpoczyna się w niedługim czasie po zajściu efektywnej inicjacji i oddaleniu się polimerazy od promotora. Pierwszym etapem tego procesu jest dodanie guanozyny do końca 5’ RNA. W drugim etapie następuje przekształcenie końcowej guanozyny w 7-metyloguanozynę. Czapeczka może być istotna dla eksportu mRNA z jądra, ale jej podstawową rolą jest udział w procesie inicjacji translacji. Proces elongacji łańcucha u eukariotów jest znacznie dłuższy, co jest spowodowane obecnością intronów oraz występowaniem nukleosomów. Praktycznie wszystkie eukariotyczne mRNA mają serię do 250 adenozyn na końcach 3’. Nukleotydy te nie są zakodowane w DNA, ale dodawane do transkryptu przez polimerazę RNA niezależną od matrycy zwaną polimerazą poli(A). Najczęściej poliadenylacja stanowi nieodłączną część procesu terminacji transkrypcji. Rola „ogona” poli(A) nie jest do końca znana. Zakłada się, że może mieć on wpływ na stabilność RNA oraz pełnić funkcje w inicjacji translacji. Powstały pre-mRNA u eukariotów podlega procesowi dojrzewania, który polega wycinaniu intronów. Eukariotyczny pre-mRNA może zawierać ponad 100 intronów, stanowiących znaczną część transkryptu. Muszą one zostać wycięte, a eksony połączone w odpowiedni sposób, aby transkrypt mógł funkcjonować jako dojrzały mRNA. Dwa podstawowe typy intronów to: GU-AG i AU-AC, które znajdują się w genach eukariotycznych kodujących białka.
Obróbka kwasu mRNA
Szlak wycinania intronów można podzielić na dwa etapy:
- cięcie po stronie 5’ intronu,
- cięcie po stronie 3’ intronu i połączenie eksonów.
Centralnymi składnikami aparatu do wycinania intronów typu GU-AG są snRNA zwane U1, U2, U4, U5 i U6. Są to krótkie cząsteczki u kręgowców mające długość od 106 do 185 nukleotydów. Wiążą się one z czynnikami białkowymi, tworząc małe jądrowe rybonukleoproteiny (snRNP), które łącznie z innymi czynnikami białkowymi tworzą serie kompleksów, które przeprowadzają dwa etapy reakcji składania, której dokładny mechanizm nie jest do końca znany.
Wycinanie intronów AU-AC przebiega identycznie jak GU-AG, ale bierze w nim udział odmienny zestaw czynników związanych z wycinaniem. Introny AU-AC zostały wykorzystane do badania modeli oddziaływań zachodzących w czasie wycinania intronów GU-AG. Badania te pomogły zdefiniować strukturę powstającą w wyniku łączenia się w pary komplementarnych zasad, wytworzoną pomiędzy U2 i U6 snRNA w kompleksie wycinającym introny GU-AG (Tarn i Steitz, 1996).

Redagowanie mRNA przebiegające na drodze modyfikacji chemicznych stwarza możliwość zmiany właściwości kodujących transkrypt, prowadząc do odpowiedniej zmiany sekwencji aminokwasowej zakodowanego białka.
Populacja cząsteczek mRNA obecnych w komórce w danym momencie jest wynikiem stanu równowagi między syntezą nowych mRNA w procesie transkrypcji i usuwaniem już istniejących mRNA w procesach degradacji. Bakteryjne mRNA są na ogół degradowane bardzo szybko z udziałem kompleksu multienzymatycznego zwanego degradosomem. Zawiera on endo- i egzonukleazy. Eukariotyczne mRNA są cząsteczkami żyjącymi dłużej od prokariotycznych. Szlaki degradacji u wyższych eukariotów są słabo poznane. Prawdopodobnie wstępem do degradacji mRNA jest utrata łańcucha poli(A) i usunięcie czapeczki.
Mitochondrialny mtRNA
W skład tej grupy RNA wchodzą cząsteczki RNA występujące w mitochondriach i stanowiące odpowiednik mRNA, rRNA i tRNA. Do mtRNA należą mt mRNA, kodujący 13 białek, mt rRNA oraz mt tRNA. W cząsteczkach mt tRNA kręgowców czasami brakuje niektórych elementów struktury. Na przykład ludzki mitochondrialny tRNASer nie ma ramienia D.
tRNA stanowi 10-12% ogólnej ilości kwasów rybonukleinowych w komórce. Jest on zbudowany z 70-90 nukleotydów. Charakteryzuje się wśród innych rodzajów RNA najmniejszą masą cząsteczkową, zawartą w granicach od 25 do 30 kDa. Bakterie zawierają 35-40, a eukarioty do 50 rodzajów cząsteczek tRNA. We wszystkich organizmach występuje przynajmniej kilka izoakceptorowych tRNA – są to różniące się od siebie cząsteczki tRNA, które są specyficzne w stosunku do tego samego aminokwasu. Transportujące RNA są najkrótszymi znanymi, funkcjonalnymi cząsteczkami RNA. Długość najkrótszych wynosi 74 nukleotydy, a najdłuższych rzadko przekracza 90 nukleotydów. Z powodu swoich małych rozmiarów oraz możliwości oczyszczenia poszczególnych rodzajów tRNA, cząsteczki te były jednymi z pierwszych zsekwencjonowanych kwasów nukleinowych. Sekwencje pierwszych cząsteczek tRNA poznano w 1965 roku dzięki doświadczeniom grupy Roberta Holleya z Cornell University w Nowym Jorku. Cząsteczki tRNA charakteryzują się specyficznym ułożeniem nukleotydów i określoną strukturą przestrzenną. Pomimo tego, że dana cząsteczka tRNA jest specyficzna wyłącznie dla określonego aminokwasu, struktura wszystkich tRNA jest zbliżona. W strukturze II rzędowej przyjmuje ona postać liścia koniczyny, w III rzędowej wszystkie cząsteczki tRNA przypominają kształtem literę L. Cząsteczki tRNA zawierają wiele nietypowych, modyfikowanych zasad, zazwyczaj 7-15 na molekułę, co stanowi około 10 do 20% wszystkich nukleotydów. Wśród nietypowych zasad charakterystyczne dla wszystkich cząsteczek tRNA są: 5,6-dihydrourydyna oraz pseudourydyna, mogą występować także pochodne metylowane, na przykład 2-metylo-guanozyna, O2,-metylo-cytydyna oraz 1-metylo-adenozyna.W strukturze II rzędowej tRNA można wyróżnić 5 zasadniczych regionów:
- Domena akceptorowa, składająca się z ramienia i pętli akceptorowej, z charakterystyczną sekwencją 5’-CCA-3’, do której przyłączany zostaje aminokwas.
- Domena antykodonowa (ramię i pętla antykodonowa, zawiera 3 nukleotydy zwane antykodonem, które w czasie translacji łączą się z mRNA.
- Domena dihydrourydyny (ramię i pętla DHU).
- Domena TΨC zbudowana z ramienia oraz pętli, w której występuje charakterystyczny układ zasad: rybotymidyna (T), pseudourydyna (Ψ) oraz cytozyna (C).
- Ramię zmienne, znajdujące się pomiędzy domeną antykodonową a domeną TΨC. Ramię to zawiera różną ilość nukleotydów.
Podobieństwa między cząsteczkami tRNA dotyczą nie tylko konserwatywnej struktury II-rzędowej. W niektórych pozycjach cząsteczek tRNA zawsze występują te same nukleotydy, a w innych zawsze występuje puryna lub zawsze pirymidyna. Zmodyfikowane nukleotydy prawie zawsze występują w tych samych pozycjach. Wiele konserwatywnych nukleotydów występujących zawsze w danej pozycji pełni ważną rolę w tworzeniu struktury III-rzędowej. W strukturze III-rzędowej tRNA wyróżniamy dwie główne domeny: I – domenę akceptorową (tzw. domena minihelisy, w skład której wchodzi ramię akceptorowe oraz pętla TΨC oraz II – domenę antykodonową, zbudowaną z ramienia i pętli antykodonowej oraz ramienia i pętli DHU. Wiele dowodów wskazuje na to, że te dwie domeny tRNA powstały niezależnie od siebie w procesie ewolucji. Każda z tych domen oddziałuje z innymi częściami rRNA rybosomu. Zasadniczym etapem w procesie translacji jest selekcja tRNA przez syntetazy aminoacylo-tRNA. Enzymy te determinują poprawność wyboru aminokwasu w biosyntezie białka. Ich podstawową funkcją jest swoiste rozpoznanie, a następnie kataliza dwuetapowej reakcji estryfikacji tRNA. Enzymy te podzielono na dwie klasy, zgodnie z obecnością w nich krótkich charakterystycznych sekwencji aminokwasowych. Małe aminokwasy są z reguły rozpoznawane przez syntetazy klasy II, większe i bardziej hydrofobowe – przez enzymy klasy I. Te dwie klasy różnią się strukturą domeny białkowej. W klasie I miejsce katalityczne enzymu tworzy pięć na przemian ułożonych helis α, tzw. struktura Rossmanna. Centrum aktywne syntetaz klasy II tworzy siedem antyrównolegle ułożonych struktur β-daszkowych oraz trzy helisy α. tRNA odgrywają zasadniczą rolę w translacji. Tworzą one połączenia między mRNA i syntetyzowanym polipeptydem. Jest to połączenie zarówno fizyczne, jak i informacyjne: tRNA wiąże się jednocześnie z mRNA i wydłużającym się polipeptydem oraz gwarantuje, że sekwencja aminokwasów syntetyzowanego polipeptydu jest zgodna z sekwencją zapisaną kodem genetycznym w nukleotydach mRNA.
Do lat 80-tych ubiegłego wieku sądzono, że jedynie białka wykazują właściwości katalityczne. Załamanie tej teorii nastąpiło podczas badań splicingu RNA mających na celu wykrycie enzymu katalizującego wycinanie intronów RNA u orzęska Tetrahymena thermophila. Thomas Cech zauważył wtedy, że proces ten zachodzi również, gdy białka są nieobecne, co stanowiło dowód na to, że samo RNA w specyficznych sytuacjach może wykazywać właściwości enzymatyczne. Podobne wnioski wysnuł podczas równoległych badań nad dojrzewaniem tRNA w rybosomach Sidney Altman. Za to odkrycie obaj naukowcy zostali uhonorowani Nagrodą Nobla w 1989 roku w dziedzinie chemii.
Budowa rybozymówStrukturę pierwszorzędową rybozymów stanowi jednoniciowe RNA, zbudowane z czterech rodzajów zasad nukleotydowych – adenina, guanina, cytozyna, uracyl. Mogą one łączyć się ze sobą za pomocą wiązań wodorowych, tworząc strukturę drugorzędową w postaci pętel, wybrzuszeń czy spinek. Te z kolei oddziałując między sobą tworzą strukturę trzeciorzędową. Do prawidłowego pofałdowania niezbędna jest obecność jonów dodatnich (kationów magnezowych lub sodowych), która będzie przeszkadzała elektrostatycznemu odpychaniu się ładunków ujemnych zgromadzonych na rybonukleotydach. Jedynie cząsteczki o odpowiedniej strukturze trzeciorzędowej wykazują właściwości katalityczne.
Rodzaje rybozymówZe względu na budowę oraz rodzaj katalizowanej reakcji rybozymy dzielimy na grupy przedstawione w Tab. 1.

Tab. 1. Rodzaje rybozymów
Rybozymy z pierwszej grupy katalizują reakcje wycinania z nici substratu produktu, który posiada na końcu 3’ grupę hydroksylową a na końcu 5’ - fosforan. Wyróżniamy tutaj dwa typy rybozymów: introny grupy I i II. Różnią się one rodzajem cząsteczki, która dokonuje ataku na miejsce splicingowe 5’. W intronach grupy I jest nią kofaktor guanozynowy, zaś grupy II – grupa 2’-hydroksylowa reszty adenylanowej intronu. Przykładem intronu grupy I jest rybozym Tetrahymena thermophila (intron genu 26S rRNA tego orzęska). Ze względu na budowę zaliczamy go do klasy rybozymów dużych, składa się bowiem z licznych części helikalnych oraz pętli wewnętrznych. Katalizuje reakcję wycinania intronów, czyli fragmentów niekodujących własnego łańcucha RNA. Należy do rybozymów katalizujących jeden obrót reakcji, co oznacza, że traci swoje właściwości po przeprowadzeniu reakcji, bowiem następuje w nim nieodwracalna zmiana strukturalna.
Rys. 1. Struktura drugorzędowa prekursora rRNA z Tetrahymena thermophila
Do kolejnej grupy rybozymów zaliczamy te cząstki, które katalizują reakcje prowadzące do powstania produktów o końcach 5’-hydroksylowym i cyklicznym – 2’, 3’-fosfodiestrowym. Rybozymy te różnią się między sobą strukturą centrum katalitycznego. Najczęściej spotykane to rybozymy typu „głowa młotka” (z ang. hammerhead) oraz typu „spinki” (z ang. hairpin). Rybozymy typu hairpin są rybozymami małymi i katalizują jeden obrót reakcji. Biorą udział m. in. w replikacji wirusa mozaiki tytoniowej. Podczas namnażania tworzy się łańcuch komplementarny do satelitarnego RNA, rybozym rozcina komplementarny łańcuch, liguje go tworząc matrycę do syntezy cząsteczek RNA. Rybozymy z tej grupy mogą różnić się między sobą budową. Część katalityczna jest niezmienna i zbudowana jest z dwóch heliakalnych ramion, na których znajdują się pętle wewnętrzne składające się zawsze z takich samych nukleozydów. Część nie katalityczna może zawierać różne struktury drugorzędowe.
Rys 2. Budowa rybozymu typu hairpin. Linia ciągła oznacza część aktywną katalitycznie, zaś przerywana- część nie katalityczną.
Rybozymy typu hammerhead to wiroidy ulegające autokatalitycznemu splicingowi. Zbudowane są z trzech ramion odchodzących promieniście od środka składającego się z niesparowanych nukleotydów.

Rys. 3. Schemat budowy rybozymu typu hammerhead
RNaza P bierze udział w dojrzewaniu końca 5’ tRNA u bakterii, archeowców czy Eukariota. In vivo występuje w postaci holoenzymu (katalizę wspomaga kofaktor). U bakterii kofaktor składa się z jednej cząsteczki białka, w archeowców z czterech, zaś u organizmów eukariotycznych z dziewięciu lub dziecięciu. In vitro prezentuje swoje właściwości nawet bez udziału części białkowej. Aktywność enzymatyczną warunkuje RNA M1, rozpoznając właściwe miejsca na nici pierwotnego transkryptu.
Zastosowanie rybozymówRybozymy jako cząstki zdolne do enzymatycznego cięcia cząsteczek mRNA znajdują szerokie zastosowanie w terapii genowej, specyficzne cięcie mRNA może bowiem osłabiać lub zapobiegać translacji odpowiednich białek. Wykorzystuje się je w genoterapii chorób wirusowych, blokuje się w ten sposób namnażanie wirusa brodawczaka ludzkiego (human papilloma virus – HPV) czy czynnika prowadzącego do transformacji nowotworowej, w wyniku któ®ej dochodzi do powstania raka skóry, sromu i ust.
Przedstawione informacje wskazują na to, że właściwości katalityczne rybozymów wynikają ze zmian konformacyjnymi w ich strukturze trzeciorzędowej. Rybozymy mogą przeprowadzać rekacje transestryfikacji (hairpin), transferu nukleotydu (introny), hydrolizy (RNA RNazy P) czy syntezę wiązania peptydowego (rybosom). Są ważnymi pod względem biologicznym cząsteczkami, wciąż prowadzone są badania nad ich aktywnością, ponieważ:1. rybozymy biorą udział w ważnych reakcjach, których zbadanie jest niezbędne do poznania metabolizmu komórki2. mają one prostszą budowę niż enzymy (cztery rodzaje monomerów) oraz wykazują komplementarność pomiędzy adeniną i uracylem oraz guaniną i cytozyną, przez co mechanizm ich działania jest prostszy3. selektywność ich działania może być wykorzystywana w terapii celowanej w RNA, choć do tego niezbędne jest poznanie stabilności chemicznej oligonukleotydów w środowisku komórkowym, transportu do wyznaczonych miejsc w komórce czy fałdowania do konformacji aktywnej4. możliwe jest poznanie fałdowania i aktywności pojedynczych cząstek, co dostarcza informacji, które są niedostępne z powodu uśredniania towarzyszącego badaniom zespołów cząsteczek w roztworze (np. wyznaczenie stałych kinetycznych)
Modyfikacja potranskrypcyjna
U organizmów eukariotycznych sekwencja nukleotydów pierwotnego transkryptu (pre-mRNA) ulega wieloetapowej obróbce zwanej procesem dojrzewania. Zachodzi ona głównie w jądrze komórkowy i tylko poddane niej cząsteczki mogą przemieszczać się z jądra do cytoplazmy i uczestniczyć w przebiegu translacji. Obróbka pierwotnych transkryptów może służyć do regulacji ekspresji informacji genetycznej. Obecność intronów umożliwia bowiem wykorzystywanie alternatywnych miejsc składania genów oraz taksowanie i duplikację eksonów, przyczyniając się do powiększenia zasobów informacji genetycznej.
Składanie (ang. Splicing), czyli precyzyjne wycinanie niekodujących intronów i łączenie pozostałych eksonów. Powstała sekwencja RNA zawiera ciągłą informację o strukturze budowanego białka i nazywana jest otwartą ramką odczytu (ang. open reading frame).

Rys. 1. Splicing
Introny mają różną długość i zaczynają się zwykle od sekwencji GU, a kończą na AG. Posiadają również ważny odcinek zwany miejscem rozgałęzienia (ang. branch site) biorący udział przy ich wycinaniu. Znajdujące się na stykach miejsca cięcia mają konserwatywną budowę, tzw. sekwencję konsensusu, która znacznie ułatwia ich identyfikację.
Rys. 2. Sekwencja konsensus
Istnieją różne mechanizmy składania transkryptu: • samowycinanie – zachodzi m.in. genach mitochondrialnych; intron jest wycinany w sposób autokatalityczny, tzn. bez udziału białek; • wycinanie z udziałem spliceosomów, czyli struktur złożonych z białek oraz czterech małocząsteczkowych RNA – snRNA (U1, U2, U5 i U4/U6). Przecięty przy końcu 5’ intron tworzy pętlę (produkt pośredni w kształcie lassa – ang. lariat intermediate). Dopiero potem przecinany jest koniec 3’, a całość uwalniana z pierwotnego transkryptu i trawiona enzymatycznie. Pozostałe eksony łączą się ze sobą.

Rys. 3. Sposób składania spliceosomu
Redagowanie (ang. Editing) Niektóre cząsteczki RNA podczas obróbki tracą lub zyskują nowe nukleotydy. Jest to jeden z mechanizmów regulacji ekspresji genów. Proces ten zachodzi w różnych częściach transkryptu, w kierunku od końca 3’ do 5’.W jego wyniku mogą powstawać nowe otwarte ramki odczytu, nowe miejsca startu translacji oraz sekwencje wyznaczające miejsce wycinania eksonów. U świdrowców bierze w nim udział korygujące RNA (ang. guide RNA, gRNA) spełniające rolę matrycy.
Modyfikacja cząsteczek mRNAZachodzi głównie w jądrze komórkowym i obejmuje oba końce nowopowstałego transkryptu: • do końca 5’ doczepiana zostaje czapeczka (7-metyloguanozyna), która ułatwia transport cząsteczki z jądra do cytoplazmy, zabezpiecza transkrypt przed działaniem enzymów nukleolitycznych oraz uczestniczy przy wycinaniu intronów i inicjowaniu translacji; • poliadenylacja końca 3’ , czyli przyłączenie ogona złożonego z poli(A) – nie zawierają go transkrypty genów kodujących białka histonowe. Rola tej modyfikacji nie jest do końca poznana, wiadomo jednak, że nie decyduje ona o pojawieniu się cząsteczki w cytoplazmie. Usunięcie czapeczki i ogona poli(A) obniża trwałość i stabilność transkryptu.
Po stronie 3’ i 5’ cytoplazmatycznego mRNA występują dodatkowe niekodujące białek regiony. Biorą one udział w przekształcaniu RNA, transporcie cząsteczki, jej degradacji oraz translacji.
Modyfikacja cząsteczek tRNATransportowy RNA przepisywany jest w postaci dużych prekursorowych cząsteczek, zawierających więcej niż 1 tRNA. Ulegają one przekształceniom nukleolitycznym katalizowanym przez rybonukleazy. Podczas dalszych etapów zachodzi wycinanie intronów ulokowanych w pobliżu pętli antykodonowej, alkilacja nukleotydów oraz dołączenie charakterystycznej końcówki CCA przy końcu 3’. Jest ona odpowiedzialna za swoiste łączenie się z transportowanym aminokwasem.
Modyfikacja cząsteczek rRNAU ssaków geny kodujące rRNA umiejscowione są na terenie jąderka. Pojedyncze cząsteczki silnie zmetylowanego, pierwotnego transkryptu 45S zawierają geny 18S, 5,8S i 28S rRNA i ulegają swoistym nukleolitycznym przekształceniom, podczas których degradowana jest ponad połowa oryginalnego pierwotnego transkryptu. Zachodzi również dalsza metyzacja oraz utworzenie podjednostek rybosomalnych, czyli połączenie rRNA z odpowiednimi białkami.
GENOM MITOCHONDRIALNY
Mitochondria są organellami komórkowymi przeprowadzającymi proces oddychania tlenowego. Przyjmuje się, że pochodzą one od bakterio-podobnych organizmów, które na drodze endosymbiozy zostały włączone do eukariotycznych komórek. Teorię tą potwierdza fakt, że mitochondria posiadają własny materiał genetyczny w postaci kilku (od 4 do 10) kopii kolistego nukleotydu o długość 16569 par zasad.
U ssaków, zawarta w nim informacja dziedziczona jest wyłącznie w linii żeńskiej. Dzieje się tak dlatego, że komórka jajowa posiada o wiele więcej cząsteczek mtDNA (ok. 100 000) niż plemniki (ok. 100), u których dodatkowo ulega on procesowi degradacji. Dzięki badaniom nad mitochondrialnym DNA pochodzącym od różnych grup etnicznych ustalono, że każdy współczesny Europejczyk jest potomkiem jednej z siedmiu kobiet żyjących w epoce kamiennej (od 10 do 50 tys. lat temu): Heleny, Veldy, Tary, Katriny, Ursuli, Yenii oraz Jasminy.
Sekwencję mtDNA poznano już w latach osiemdziesiątych. Wszystkie uzyskane dane dotyczące ludzkiego genomu mitochondrialnego zebrane są w bazie danych MITOMAP (http://www.mitomap.org/MITOMAP). Wiadomo, że zawiera on informację na temat 13 białek uczestniczących w procesie fosforylacji

oksydacyjnej, 22 typów tRNA oraz 2 rodzajów rRNA (12S w małej i 16S w dużej podjednostce rybosomowej). Pozostałe białka zaangażowane w proces oddychania są kodowane przez DNA jądrowe, syntetyzowane w cytoplazmie i jako gotowe cząsteczki wnikają do mitochondriów. Nici budujące podwójną helisę mtDNA określa się mianem ciężkiej H (zawiera informację na temat 12 białek i 14 tRNA) oraz lekkiej L (koduje 1 białko i 8 tRNA).
Kod genetyczny mitochondrialnego DNA różni się od uniwersalnego zapisu: UGA – kodon STOP w mitochondriach oznacza tryptofan, AUA – koduje metioninę zamiast izoleucyny, AGA, AGG - kodujące argininę w mitochondriach oznaczają sygnał STOP.
Cechą charakterystyczną mtDNA jest również duże zagęszczenie kodowanych odcinków. Tzw. pętla D (ang. D-loop) jest jedynym fragmentem niezawierającym genów. Zachodzi w niej inicjacja transkrypcji. Odcinek ten zawiera dwa hiperzmienne obszary, które różnią się między sobą u poszczególnych osobników. Właściwość ta wykorzystywana jest w genetyce populacyjnej i medycynie sądowej.
Rys. Ludzki mitochondrialny DNA
DNA mitochondrialny posiada mało sprawny system naprawczy i z tego powodu ewoluuje o wiele szybciej. Powstające zmiany mogą powodować zaburzenia funkcji fizjologicznych mitochondriów i prowadzić do występowania chorób, których objawy związane są przeważnie z tkankami o wysokim zapotrzebowaniu energetycznym - mięśniowej i nerwowej. Jednak ze względu na losową segregację mtDNA mogą dotyczyć właściwie wszystkich tkanek i narządów – szpiku kostnego, gruczołów i in.. Wielu chorobom towarzyszy zatem występowanie wielu różnych objawów klinicznych, które dodatkowo mogą się też różnić stopniem nasilenia u poszczególnych członków rodziny.
Przykładowe choroby: • encefalopatia z kwasicą mleczanową i napadami udaropodobnymi (ang. mitochondria encephalopathy with lactic acidosis and stroke-like episodes, MELAS), • zespół Lebera - dziedziczna neuropatia wzrokowa Lebera (ang. Leber’s hereditary optic neuropaty, LHON), • zespół Kearnsa-Sayre’a (ang. Kearns-Sayre syndrome, KSS),

• porażenie mięśni odwodzących oka (ang. chronic progressive external ophtalmoplegia, CPEO), • neuropatia obwodowa z ataksją i barwnikowym zwyrodnieniem siatkówki (ang. neurogenic muscle weaknes, atalia, retilis pigmentosa, NARP), • padaczka miokloniczna z nieprawidłowymi czerwonymi włóknami mięśniowymi (ang. myoclonic epilepsy with ragged red fibres, MERFF).
Efekty mutacji pojawiają się najczęściej w dorosłym wieku, a ich leczenie jest jedynie objawowe. Upośledzenie działania mitochondriów może być również wywołane zmianami w DNA jądrowym.
RECEPTORY
Receptory komórkoweDodano : 8.5.2011
Między różnymi organizmami występuje niewielkie zróżnicowanie receptorów. Powstały one przez zwielokrotnienie genu wspólnego przodka (teoria wspólnego przodka). Następowały kolejne modyfikacje.
Receptory dzielimy na 3 podstawowe nadrodziny:
1. jonotropowe – powiązane z kanałem jonowym (sygnałem jest przepływ jonów prowadzący do efektu elektrycznego) – ligand otwiera kanał jonowy2. metabotropowe – związane z układem białek G (wewnątrzkomórkowym sygnałem jest zaktywowana forma białka G związanego z błoną od strony
cytoplazmatycznej, dzięki nim działają hormony)3. katalityczne – związane z układem enzymatycznym (sygnałem jest wzbudzenie w cytoplazmatycznej domenie receptora aktywności
enzymatycznej, dzięki nim działają czynniki wzrostu, cytokiny) – receptory aktywujące kinaze tyrozynową i kinazę serynowo-treoninową
Receptory jonotropowe
służą szybkiemu przemieszczaniu sygnałów w synapsach, przekształcają sygnał chemiczny w elektryczny występuje w synapsach nerwowo-mięśniowych składają się z 5 podjednostek: 2 a, 2 b, ligand łączy się z podjednostkami a (domeną zewnątrzbłonową) i powoduje zmianę konformacji i w konsekwencji zmianę domeny
wewnątrzkomórkowej ligand powoduje otwarcie kanału – Na+ wpływa do komórki i powoduje depolaryzację a następnie skurcz
o RECEPTOR NIKOTYNOWY DLA ACETYLOCHOLINY jest to kanał dla Na+
nikotyna jest agonistą, substancja inna niż naturalny ligand łącząca się z receptorem i w małych ilościach pobudzająca receptor
o RECEPTOR GABAERGICZNY typu A (GABAA) występują w dużej ilości w centralnym układzie nerwowym – stanowią 1/3 wszystkich występujących tam receptorów ligandem jest kwas -aminomasłowy ma 5 podjednostek, a każda kodowana jest przez kilka różnych genów receptory tego typu różnią się między sobą nie jakościowo a ilościowo co jest związane z tym, że podjednostki receptorów
mogą być kodowane przez różne geny receptor obsługuje kanał dla jonów Cl – (które wchodzą do cytoplazmy) i wywołują hiperpolaryzację doprowadzając do
wygaszania potencjałów komórkowych za pomocą tych receptorów działa wiele leków – uspokajające, przeciwalergiczne, nasenne, anastetyki steroidowe,
(barbiturany – luminal, benzodiazepiny – relanium, tegretal) – działają depresyjnie na OUN (to agoniści niekompetycyjni – łączą się i pobudzają receptor ale w innych miejscach, a nie w tych w których łączy się naturalny ligand), takim agonistą jest także alkohol etylowy – działa w niewielkim stężeniu
o RECEPTOR GLICYNOWY WRAŻLIWY NA STRYCHNINĘ pentamer (5 podjednostek), 3a i 2b tworzy kanał dla jonów Cl-
występuje w rdzeniu kręgowym w neuronach pośredniczących – regulacja napięcia mięśni szkieletowych w trakcie życia człowieka jego budowa ulega zmianom. u dziecka i u płodu receptory wrażliwe na strychninę (domeny a2) są
zastępowane u dorosłego w a1
normalne działanie receptora zwiotcza mięśnie strychnina jest antagonistą – wywołuje zahamowanie receptorów – drgawki i wzrost napięcia mięśniowego, spazmy
RECEPTORY METABOTROPOWE
współpracują z białkami G pośredniczą w powstawaniu odpowiedzi na hormony, mediatory lokalne i przekaźniki nerwowe inna nazwa to serpentynowy – bo układa się jak serpentyna ich struktura oparta jest na siedmiokrotnym przejściu pojedynczego łańcucha polipeptydowego przez dwuwarstwę lipidową. białko G łączy się z receptorem na 3 pętli wewnętrznej i zmienia jej konformację
o białko G – heterotrimer, zbudowany z 3 podjednostek: a, b, ; w stanie niepobudzonym podjednostka a ma związany ze sobą GDP, a całe białko jest w stanie spoczynkowym, mogą być pobudzające bądź inhibitujące

gdy zewnątrzkomórkowy ligand łączy się z receptorem, receptor wiąże się z białkiem G i aktywuje je wywołując zmianę GDP na GTP
białko G rozpada się na dwa kompleksy aGTP i b kompleks aGTP ma aktywność GTP-azową (wywołuję ją cAMP) co prowadzi do ponownego powstania GDP i połączenia
się obu jednostek białka G aGTP działa na układ wzmacniacza enzymatycznego (najczęściej cyklaza adenylowa) – dochodzi do wzmożonej produkcji
substancji będącej informatorem II rzędu (najczęściej cAMP), która wywołuje różne efekty poprzez związanie się z kinazą zależną od cAMP-u i uwolnienie aktywnego enzymu
aktywność może wykazywać także kompleks b
Receptory metabotropowe: serotoninowy, dopaminoergiczny, histaminowy, GABAB , adrenergiczne (ligandem naturalnym jest adrenalina bądź noradrenalina – występuje w nich cyklaza adenylowa)
RECEPTORY Z BIAŁKIEM G INHIBITUJĄCEo badano endotoksyny białkowe działające na białka Go toksyna cholery – działa na układ białek G – blokuje aktywację przez układ wzmacniacza enzymatycznego działalność ATP-azy przez
co nie następuje ponowne połączenie podjednostek białka G, przez co ciągle są otwarte kanały Na w jelitach co prowadzi do ciągłego ich napływu i w konsekwencji do biegunek i odwodnienia
o toksyna krztuśca – działa na układ białek G, dochodzi do zablokowania wymiany GDP na GTP, co prowadzi do ciągłego wzrostu stężenie cAMP, jest to wyłączenie białka G hamującego czyli aktywacja białka G stymulującego czyli w konsekwencji wzrost cAMP
RECEPTORY KATALITYCZNE
są białkami transbłonowymi, których domeny wiążące ligandy znajdują się na zewnętrznej powierzchni błony, domena ta działa jak enzym związanie ligandu powoduje zbliżenie się w błonie dwóch cząsteczek receptora i wytworzenie ich dimeru co powoduje włączenie ich aktywności
kinazowejo O AKTYWNOŚCI KINAZY TYROZYNOWEJ
rodzaj I - kinaza tyrozynowa stanowi element budowy takiego receptora – jest domeną wiążącą, powoduje fosforylację reszt tyrozyny co prowadzi do aktywacji następnych receptorów
PGT – płytkowy czynnik wzrostu IGT – insulinopodobny czynnik wzrostu (insuliny) PDGF, EGF, IGF, UGF
rodzaj II - w swojej budowie nie mają domeny zbudowanej z kinazy tyrozynowej, ale aktywują kinazy tyrozynowe cytoplazmatyczne
cytokiny (układ immunologiczny) interleukiny I, II, VI interferony TNF – czynnik martwicy nowotworu
o Białko Ras monomer, należy do grupy białek o aktywności kinazy tyrozynowej łączy działalność białka G z receptorami o aktywności kinazy tyrozynowej białko to w stanie aktywnym (z GTP) inicjuje kaskadę kinaz, w której ostatnia kinaza działa na określone białka
regulatorowe genów jego mutacja jest odpowiedzialna za powstanie raka pęcherza moczowego u człowieka
o O AKTYWNOŚCI KINAZ układ receptorowy występuje rzadko mechanizm działania jest bardzo skomplikowany odpowiedzialny za działanie substancji regulujących powstawanie mezodermy, substancji aktywujących i hamujących
czynność przysadki oraz TGF - b
RECEPTORY WEWNĄTRZKOMÓRKOWE – cytoplazmatyczne
cząsteczka aktywująca receptor musi wniknąć do komórki substancje aktywujące są hydrofobowe – dzięki temu mogą przenikać przez błony : hormony steroidowe, hormony tarczycy, witamina D3, retinoidy
(pochodna witaminy A) działają bezpośrednio na układ genów wykazują wysoki stopień homologii (teoria wspólnego przodka) receptor łączy się z ligandem z cytoplazmy dostaje się do jądra komórkowego i tam dochodzi do transkrypcji układu genowego
Receptory sprzężone z białkami G (GPCRs – ang. G protein coupled receptors) stanowią najliczniejszą i bardzo zróżnicowaną grupę białek błonowych odpowiedzialnych za przekazywanie sygnałów przez dwuwarstwę lipidową do miejsc efektorowych znajdujących się we wnętrzu komórki [1]. Sekwencjonowanie ludzkiego genomu ujawniło występowanie ok. 800 różnych typów receptorów należących do rodziny GPCR (geny kodujące receptory GPCR stanowią powyżej 3% ludzkiego genomu), a ponad połowa z nich wykazuje potencjalne znaczenie dla przemysłu farmaceutycznego [2, 3].

Analiza porównawcza sekwencji receptorów GPCR oraz badania funkcji poszczególnych typów receptorów doprowadziły do podziału tej rodziny białek na klasy, które oznaczono literami od A do F [4, 5]. Pierwsza, najliczniejsza klasa A (nazywana również klasą podobnych do rodopsyny – ang. rhodopsin like), obejmująca ponad 80% wszystkich GPCR, to klasa receptorów rodopsyno-podobnych. Receptory wchodzące w skład tej grupy są jednymi z najlepiej zbadanych. Zaliczamy do nich, obok rodopsyny, między innymi: receptory adrenergiczne, opioidowe, adenozynowe, kanabinoidowe, receptory chemokin, dopaminowe i histaminowe. Klasę B stanowią receptory sekretyno-podobne. Do klasy C zaliczamy receptory glutaminergiczne i feromonowe. Kolejne klasy D i E tworzą odpowiednio receptory feromonów grzybów oraz receptory cAMP. Ostatnia klasa F to receptory frizzled/smoothened. Klasyfikacja ta pokrywa się w większości z nową klasyfikacją GRAFS [6], opartą na badaniach filogenetycznych. Nazwa GRAFS pochodzi od pierwszych liter wyodrębnionych rodzin receptorów, do których należą odpowiednio: receptory Glutaminergiczne, Rodopsyno-podobne, Adhezyjne, Frizzled i smakowe oraz Sekretyno-podobne.
Receptory sprzężone z białkami G uczestniczą w kaskadach sygnalizacyjnych pośrednicząc w przekazywaniu informacji przez liczne zewnątrzkomórkowe cząsteczki sygnałowe: hormony, przekaźniki neuronalne, małe białka, krótkie łańcuchy peptydowe, aminy, lipidy, nukleotydy czy pochodne aminokwasów i kwasów tłuszczowych [3, 7]. Dla każdej z wymienionych substancji istnieje receptor, bądź też grupa receptorów mogąca wiązać daną cząsteczkę, inicjującą jednocześnie przekazanie sygnału przez błonę komórkową. Receptory GPCR odgrywają kluczową rolę w wielu procesach fizjologicznych regulujących prace komórki, jej metabolizm, wzrost i obronę immunologiczną. Receptory sprzężone z białkami G pełnią ponadto bardzo różnorodne funkcje w organizmie człowieka. Zaliczamy do nich między innymi: rodopsynę [8, 9], białko fotoreceptorowe aktywowane przez światło, uczestniczące w procesie widzenia, receptory adrenergiczne [10-12] wywierające wpływ na ciśnienie tętnicze, receptory opioidowe [13, 14] modulujące poziom odczuwanego bólu, receptory muskarynowe kontrolujące pracę mięsni gładkich, a także receptory smakowe oraz węchowe.
Rys. 1. Schemat budowy receptora GPCR oraz jego lokalizacja w błonie komórkowej, IL:I do IL:III - pętle wewnątrzkomórkowe, EL:I do EL:III – pętle zewnątrzkomórkowe, helisy transbłonowe oznaczono cyframi od I do VII. N-koniec położony jest po stronie zewnątrzkomórkowej, C-koniec znajduje się po stronie cytoplazmatycznej błony. Rysunek wykonany na podstawie pracy [15]
Niezależnie od pełnionych funkcji oraz różnic w długości łańcucha białkowego receptory GPCR posiadają wspólne cechy strukturalne. Wszystkie zbudowane są z pojedynczego łańcucha polipeptydowego siedmiokrotnie przechodzącego przez dwuwarstwę lipidową (Rys. 1). Domenę transbłonową wszystkich receptorów GPCR tworzy siedem hydrofobowych α-helis (TMH:I do TMH:VII) połączonych pętlami, trzy pętle na zewnątrz komórki (EL:I do EL:III) oraz trzy wewnątrz komórki (IL:I do IL:III)). Koniec karboksylowy (C-koniec) znajduje się zawsze po stronie cytozolowej błony i zawiera miejsca fosforylacji przez odpowiednie kinazy GRK (ang. G protein coupled Receptor Kinase). Mający wolną grupę aminową koniec łańcucha białkowego (N-koniec) położony jest zawsze na zewnątrz komórki i zwykle zawiera miejsca glikozylacji. Na znajdującej się po obu stronach błony komórkowej powierzchni receptora, występują liczne aminokwasy o charakterze hydrofilowym, natomiast w obszarze błony przeważają aminokwasy o właściwościach hydrofobowych co powoduje silne zakotwiczenie białka w błonie lipidowej. Obecne pomiędzy helisami liczne wiązania wodorowe dodatkowo stabilizują strukturę receptorów GPCR.
Receptory sprzężone z białkami G wykazują duże podobieństwo sekwencyjne i strukturalne w rejonie transbłonowym [11, 16-20], natomiast długość łańcucha białkowego w obszarze N- jak i C-końca oraz pętli łączących helisy podlega dużemu zróżnicowaniu [21]. Znikome podobieństwo sekwencyjne tych fragmentów pozwala również przypuszczać, iż posiadają one odmienną strukturę przestrzenną, szczególnie w części zewnątrzkomórkowej (która wiąże odmienne ligandy), podczas gdy są one zadziwiająco podobne pod względem długości pętli w części cytoplazmatycznej (która służy do wiązania trimeru białka G).
Miejsce wiązania ligandów receptorów rodziny A zlokalizowane jest przeważnie we wnęce tworzonej przez siedem α-helis, jak ma to miejsce w przypadku receptora b2 adrenergicznego [10] czy rodopsyny [8]. Bezpośrednie otocznie miejsca aktywnego stanowią aminokwasy pochodzące z helis transmembranowych TMH:I, TMH:III, ,TMH:V, TMH:VI, oraz TMH:VII. W niektórych typach receptorów GPCR ligandy wiążą się również do dużej domeny tworzonej przez długi N-koniec receptora (receptory feromonów, receptory GABAB), [22, 23] jak i do aminokwasów zlokalizowanych w pętlach i w obszarze transmembranowym białka receptorowego (receptory neuropeptydowe) [1, 7, 24, 25].
Poznane dotychczas struktury krystaliczne receptorów GPCR (struktury rodopsyny nieaktywnej [8], struktura rodopsyny częściowo aktywnej [26], struktury opsyny [16], struktura receptora A2A adenozynowego [18], struktury receptora β1 adrenergicznego [11] oraz receptora β2 adrenergicznego [10]) stanowią receptory należące do klasy A, rodopsyno-podobnych. Pomimo faktu, iż podobieństwo sekwencyjne nie jest duże dla różnych typów receptorów klasy A (20-30% dla całej długości łańcucha białkowego), prezentowane struktury wykazują wysoką zbieżność w obszarze transbłonowym (po wzajemnym nałożeniu części łańcucha białkowego, wspólnego dla obszaru transmembranowego receptora β2 adrenergicznego oraz rodopsyny, średnie odchylenie kwadratowe

położenia współrzędnych atomów białka (RMSD) nie przekracza 3.2 Å). Wysokie podobieństwo strukturalne tego obszaru sugeruje, że proces przeniesienia sygnału przez błonę komórkową inicjowany aktywacją receptora może posiadać wspólny mechanizm dla większości przedstawicieli tej rodziny
FOSFORYLACJA HISTONÓW
Fosforylacji ulegają zarówno histony rdzeniowe, jak i łącznikowe. W przypadku histonów łącznikowych H1 i histonu H3 grupa fosforanowa zostaje przyłączona do reszt serynowych i treoninowych tych białek w sposób zależny od fazy cyklu komórkowego (patrz dalej), przy czym jej najwyższy poziom przypada na fazę mitozy. W histonie H1 fosforylacji ulegają reszty Ser/Thr położone w obrębie ogonów N- i C-końcowych (Rys. 7), podczas gdy fosforylacja tych reszt w histonie H3 ogranicza się do ogona N-końcowego (van Holde, 1989).
Co więcej, fosforylacja histonu H3 ogranicza się wyłącznie do stadium mitozy i jest niezbędnym warunkiem prawidłowej kondensacji i segregacji chromosomów w metafazie (Wei i wsp., 1999). Dowodu na taką rolę odwracalnej fosforylacji histonu H3 dostarczyły badania na makronukleusie Tetrahymena, w których doświadczalna zamiana seryny-10, podlegającej modyfikacji, na alaninę, prowadzi do zaburzeń kondensacji chromosomów mitotycznych i mejotycznych. Inne prace, przeprowadzone z wykorzystaniem modelu komórek mysich (Cobb i wsp., 1999) wskazują jednak, że fosforylacja histonu H3 nie może być wyłącznym czynnikiem wywołującym kondensację chromosomów w metafazie.
Obecnie uważa się, że fosforylacja histonów, podobnie jak acetylacja, powoduje rozluźnienie struktury chromatyny: i tak na przykład w mysich fibroblastach poddanych stymulacji czynnikami wzrostowymi i estrami forbolu histon H3 podlega szybkiej fosforylacji jednocześnie z aktywacją transkrypcyjną genów wczesnej odpowiedzi c-fos i c-jun. Ustalono, że ufosforylowana forma histonu wiąże się z uaktywnionymi formami tych genów (Mahadevan i wsp., 1991; Chadee i wsp., 1999).
Związek fosforylacji histonu H1 z cyklem komórkowym i stanem struktury chromatyny był od wielu już lat przedmiotem intensywnych badań. Nasilona fosforylacja histonu H1 podczas mitozy skłoniła niektórych badaczy do postawienia tezy, że modyfikacja ta jest czynnikiem inicjującym proces kondensacji chromosomów (Bradbury i wsp., 1973), a tym samym impulsem wywołującym podziały mitotyczne (Bradbury i wsp., 1974). Wykorzystanie do badań zwierzęcych linii komórkowych poddających się procesowi synchronizacji pozwoliło na ustalenie, że liczba grup fosforanowych przyłączanych początkowo do reszt serynowych w C-końcowej, a następnie do reszt treoninowych w aminowej części histonu łącznikowego zmienia się wraz fazą cyklu w ten sposób, że osiąga maksimum podczas metafazy mitotycznej (Hohman i wsp., 1976): podczas gdy na cząsteczkę histonu H1 w interfazie przypadają 1 3 grupy fosforanowe, w czasie mitozy (począwszy od jej najwcześniejszego stadium profazy, aż do ostatniego telofazy) przypada ich już od 3 do 6, czemu odpowiada zmiana struktury (kondensacja) chromatyny widoczna pod mikroskopem elektronowym (Gurley i wsp., 1978). Ważną cechą modyfikacji histonu H1 przez fosforylację jest jej zróżnicowany zakres w stosunku do kolejnych wariantów sekwencyjnych (izotypów) histonów łącznikowych obecnych w danym rodzaju komórek lub organizmów. Na przykład ludzka linia komórkowe HeLa posiada dwie główne subfakcje histonów H1: H1A i H1B (Ajiro i wsp., 1981a). Występują one w podobnych ilościach, lecz ich poziom fosforylacji różni się znacznie w trakcie całego cyklu komórkowego, nie tylko zaś w trakcie mitozy. Fosforylacja histonu H1B jest niemal dwukrotnie mocniejsza niż wariantu H1A i zachodzi w części aminowej białka w trakcie całego cyklu komórkowego, podczas gdy w białku H1A region ten ulega
fosforylacji wyłącznie w czasie mitozy. Dokładniejsze badania tego zjawiska pozwoliły na ustalenie, że oba warianty histonu łącznikowego zajmują różne, nieprzypadkowe miejsca w chromatynie, i że różnią się także liczbą miejsc podatnych na fosforylację zależną od replikacji DNA (Ajiro i wsp., 1981b). Także sześć wariantów sekwencyjnych histonu H1 myszy podlega zróżnicowanej modyfikacji fosforylacyjnej w kolejnych fazach cyklu komórkowego. I tak,
podczas gdy wariant H1b przyłącza w późnej fazie S trzy grupy fosforanowe, a w fazie M pięć, do histonu H1 przyłącza się ich w fazie S tylko jedna, a w fazie M - trzy (Talasz i wsp., 1996). Inne prace wykorzystujące przeciwciała specyficzne względem ufosforylowanych i zdefosforylowanych izoform H1 dowodzą, że odmienne fosfoformy histonu H1 zajmują odrębne domeny chromatynowe. I tak w makronukleusie Tetrahymena ufosforylowany histon H1 występuje na skraju ciałek chromatynowych i w otaczającej je euchromatynie, zaś histon H1 pozbawiony grup fosforanowych jest obecny w elektronowo gęstej heterochromatynie (Lu i wsp., 1995).
Głównym akceptorem grup fosforanowych w cząsteczkach histonu H1 pochodzących ze zwierzęcych komórek interfazowych jest ogon aminowy, w którym lokuje się blisko 60% wbudowywanych grup fosforanowych (Hohman, 1983). Z inną sytuacją mamy do czynienia w przypadku niższych eukariontów, takich jak śluzowiec Physarum polycephalum, w którego histonie H1 główne miejsca fosforylacji znajdują się we większej, C-końcowej części cząsteczki (Jerzmanowski i Maleszewski, 1985). Co więcej, fosforylacja histonów łącznikowych w niższych eukariontów wydaje się być znacznie bardziej intensywna i trwała histon H1 Physarum jest zdolny do przyjęcia podczas mitozy aż 20 grup fosforanowych, z których od ośmiu do szesnastu pozostaje przyłączone do cząsteczki H1 również w fazie G2.
Interesujący wydaje się być przypadek histonu H1 u Drosophila: stwierdzono, że istnieje związek pomiędzy bardzo obniżoną fosforylacją histonów H1 i H2A z chromatyny jej chromosomów politenicznych (w porównaniu z diploidalną chromatyną zarodkową), a znacznym spadkiem zawartości gęstej, nieaktywnej traskrypcyjnie heterochromatyny (Holmgren i wsp., 1985). Ponadto przedstawiono dowody, że fosforylacja histonu H1 Drosophila jest stała i niezależna od replikacji DNA, cyklu komórkowego, ani przejściowej stymulacji hormonalnej (Talmange i Blumenfeld, 1987). Dodatkowo, modyfikacja ta zachodzi w obrębie globularnego fragmentu cząsteczki, chociaż dochodzi do niej również w trzech innych miejscach położonych na końcu aminowym.
Dążąc do ustalenia związku fosforylacji histonu H1 i mitotycznej kondensacji chromosomów poddawano komórki bodźcom termicznym i działaniu inhibitorów enzymatycznych. Podniesienie temperatury hodowli komórek FM3A ponad dopuszczalną dla nich granicę zmniejsza fosforylację histonu H1 i wywołuje nienormalną kondensację chromatyny; również w komórkach tsBN2 poddanych działaniu niefizjologicznej temperatury dochodzi na hiperfosforylacji histonu H1 i do przedwczesnej kondensacji chromatyny (ang. premature chromatin condensation, PCC) (Matsumoto i wsp., 1980; Ajiro i wsp., 1983). Także defosforylacja
histonu H1 przy pomocy inhibitora topoizomerazy II, VM26 (Roberge, 1990), lub inhibitora kinazy białkowej, staurosporyny (Thng i wsp., 1994) prowadzi do dekondensacji chromatyny, co mogłoby wskazywać na rolę pełnioną przez H1 w wywołaniu tego procesu. Jednakże wiadomo, że doświadczenia z użyciem podobnych inhibitorów o szerokim spektrum działania obciążone jest potencjalnym niebezpieczeństwem wystąpienia efektów plejotropowych (Dou i wsp., 1999, patrz dalszy ciąg tego podrozdziału).
Głównym enzymem odpowiedzialnym za fosforylację białek jądrowych i wpływających na cykl komórkowy u eukariontów jest kinaza histonu H1 o masie 34 kDa (Woodford i Pardee, 1986), zidentyfikowana początkowo jako produkt genu cdc2+ Schizosaccharomyces pombe (Beach i wsp., 1982; Lohka, 1989). Kinaza cdc2 jest strukturalnym i funkcjonalnym homologiem czynnika wywołującego dojrzewanie (ang. maturation promoting factor, MPF), zdolnego do bezpośredniego wywołania mitozy w oocytach z pominięciem wymogu przejścia fazy syntezy białek komórkowych (Dunphy i wsp., 1988). Do tej pory udało

się wykryć obecność homologów kinazy p34cdc2 we wszystkich badanych układach eukariotycznych, wliczając w to ludzkie linie komórkowe (Lee i Nurse., 1988; Lohka, 1989). Świadczy to o konserwowanym charakterze tej modyfikacji białek komórkowych. W wielu różnych typach komórek podlegającym podziałom, p34cdc2 ulega przejściowemu pobudzeniu w trakcie przejścia z fazy G2 cyklu komórkowego w stadium mitozy (Arion i wsp., 1988; Lamb i wsp., 1990). Poziom kinazy p34cdc2 pozostaje stały podczas cyklu komórkowego, lecz jego aktywność podlega wahaniom, wynikającym z oddziaływania MPF z cyklinami - grupą białek, które charakteryzuje synteza i degradacja zależna od fazy cyklu komórkowego (Evans i wsp., 1983; Solomon i wsp., 1988; Murray i wsp., 1989). Cykliny (a w szczególności cyklina B) wiążą się do p34cdc2 i są zdolne do jego pośredniej i bezpośredniej kontroli (Draetta i wsp., 1989; Evans i wsp., 1983; Murray i wsp., 1989; Freeman i Donoghue, 1991).
Zanim histon H1 może zostać zmodyfikowany przez kinazę p34cdc2 (MPF) konieczne jest jego częściowe odłączenie od chromatyny (Jerzmanowski i Cole, 1992). Za proces takiego odłączenia mogą w warunkach fizjologicznych odpowiadać inne białka jądrowe, takie jak HMG, nukloplazmina lub topoizomeraza II, uczestnicząca bezpośrednio w kondensacji chromosomów (Adachi i wsp., 1991; Roth i Allis, 1992).
Kinaza p34cdc2 rozpoznaje najczęściej w białkach sekwencję (S/T)-P-X-(R/K) (seryna lub treonina prolina - aminokwas polarny - (arginina-lizyna, lub inny aminokwas zasadowy), modyfikując ją podczas mitozy (Kennelly i Krebs, 1991). Stwierdzono, że modyfikacja taka zdolna jest do zmniejszenia powinowactwa do DNA peptydów zawierających motyw SPKK pochodzących z histonów H1 i H2B występujących u jeżowca Strongylocentrotus purpuratus specyficznie w jądrach, zaś ich defosforylacja to powinowactwo zwiększa. Tym samym, wiązanie zdefosforylowanych domen SPKK do DNA łącznikowego sprzyja ścisłemu pakowaniu chromatyny plemników poprzez zbliżenie do siebie sąsiadujących nukleosomów (Green i wsp., 1993). Aktywność tej samej kinazy w procesie fosforylacji histonu H1 wykryto również w amitotycznych makronukleusach Tetrahymena (Roth i wsp., 1991), chociaż dzielą się one bez mitotycznej kondensacji chromosomów. Obserwacja ta podważyła związek pomiędzy fosforylacją histonu H1, a tworzeniem w trakcie podziałów komórkowych skondensowanej struktury chromatyny. Co więcej, ustalono, że również u jeżowca histon H1 obecny jest w silnie skondensowanej chromatynie plemników w postaci zdefosforylowanej (Green i Poccia, 1985).
Ostatnio Dou i współpracownicy (1999) przeprowadzili doświadczenie na Tetrahymena, polegające na wymianie w cząsteczce H1 wszystkich pięciu aminokwasów ulegających fosforylacji (seryn i treonin) na alaniny (otrzymując histon H1 niezdolny do podlegania fosforylacji) albo na reszty kwasu glutaminowego (co odpowiada trwale fosforylowanemu histonowi H1). Nie stwierdzono istotnych różnic w ogólnym fenotypie komórek Tetrahymena zawierających zmutowane cząsteczki histonu H1, chociaż druga mutacja wywierała efekt przypominający ten, jaki daje usunięcie histonu H1 w stosunku do aktywacji genu ngoA, oraz do represji innego genu, (którego funkcja nie jest znana) - CyP1. Z drugiej strony brak fosforylacji H1 w pierwszym mutancie powodował skrócenie czasu niezbędnego do pobudzenia ekspresji genu CyP1. Co więcej, ekspresja CyP1 w dzikich komórkach zachodzi wyłącznie po defosforylacji histonu H1. Taka obserwacja zestawiona z omówionymi poprzednio sprzecznymi danymi na temat związku fosforylacji histonu H1 ze strukturą chromatyny sugeruje, że fosforylacja H1 może być raczej specyficznym regulatorem ekspresji genów in vivo, zaś jej mechanizm może polegać na wydajnym odłączaniu histonu H1.
METYLACJA HISTONÓW
Metylacji podlegają histony rdzeniowe H2B, H3 i H4. Modyfikacja ta związana jest z acetylacją (Hendzel i Davie, 1989), choć występowanie obu tych modyfikacji nie jest od siebie uzależnione. Metylacja histonów jest modyfikacją stosunkowo trwałą i podlegającą powolnym przemianom. Analiza nieodwracalnej metylacji i odwracalnej fosforylacji histonu H1 w cyklu komórkowym u śluzowca Physarum polycephalum pozwoliła na stwierdzenie, że metylacja zachodzi wcześniej niż fosforylacja (Jerzmanowski i Maleszewski, 1985). Istnieją przesłanki do przypuszczeń, że metylacja związana jest z aktywacją transkrypcyjną, a być może również z procesami nowotworowymi, przynajmniej w modelowej ludzkiej linii komórkowej HeLa (Annunziato i wsp., 1995).
ACETYLACJA HISTONÓW
Histony rdzeniowe ulegają odwracalnej acetylacji wybranych reszt lizynowych ogonów N-końcowych. W nukleosomie znajduje się w sumie 26 miejsc acetylacji (Spencer i Davie, 1999). Modyfikacja histonów rdzeniowych poprzez acetylację prowadzi do głębokich zmian na wszystkich szczeblach struktur chromatynowcyh. Zaburzeniu ulega zwijanie chromatyny w struktury wyższego rzędu (Garcia-Ramirez i wsp., 1995), zwiększa się rozpuszczalność chromatyny przy fizjologicznej sile jonowej, stabilizowana jest rozwinięta struktura nukleosomu sprzyjająca transkrypcji (Walia i wsp., 1998). Przypuszcza się, że acetylacja ogonów histonów rdzeniowych utrudnia zwijanie ogonu N-końcowego, zaburzając tym samym powstawanie
struktur chromatynowych wyższego rzędu, a także odsłaniając nukleosomowe DNA oraz ułatwiając wiązanie i działanie czynników transkrypcyjnych (Workman i Kingston, 1998). Ponadto acetylacja może wpływać na oddziaływania białek represyjnych kontaktujących się z ogonami N-końcowymi. Na przykład, skutkiem acetylacji są zaburzenia wiązania domeny końcowej z represorem Tup1 (Edmondson i wsp., 1996).
Enzymami katalizującymi odwracalną acetylację histonów są acetylotransferazy (HAT) i histonowe deacetylazy (HDAC). Badania ostatnich lat pozwoliły na ustalenie, że pierwsze z nich pełnią jednocześnie funkcję koaktywatorów transkrypcji, podczas gdy drugie są jej korepresorami. Tym samym udało się udowodnić związek modyfikacji kowalencyjnej białek chromosomowych (acetylacji histonów rdzeniowych) z ekspresją genów. Jedną z pierwszych odkrytych acetylotransferaz histonowych było białko p55 Tetrahymena, spokrewnione z drożdżowym białkiem Gcn5 (Brownell i wsp., 1996), znanym już uprzednio jako aktywator
transkrypcji. Możliwe okazało się zniesienie działania Gcn5 poprzez zaburzenie jego oddziaływania z białkiem Ada2, będącego składnikiem kompleksu adaptorowego, niezbędnego do zajścia transkrypcji (Candau i wsp., 1997). Ponadto udowodniono, że Gcn5 wchodzi w skład dużych kompleksów białkowych, zdolnych do acetylacji in vivo histonów: są to kompleksy Ada i SAGA (Grant i wsp., 1997). Pierwszy zawiera białka Ada3 i Ada2 i ma masę cząsteczkową równą 0,8 MDa, zaś drugi (Spt-Ada-Gcn5-acetylotransferaza) ma masę 1,8 MDa i zawiera białka Spt, czynniki związane z transkrypcją przez polimerazę RNA II (TAFII) i białko Tra-1, będące homologiem białka TRRAP (ludzkiego białka transformacyjnego związanego z domenami transkrypcyjnymi) (Grant i wsp., 1998). To ostatnie białko jest zaś związane z chorobą AT (ataxia
teleangiectasia). Inną acetylotransferazą histonów jest ludzkie białko PCAF. Jego domena C-końcowa przypomina drożdżowe GCN5; i również ono stanowi część dużego kompleksu zawierającego ponad 20 białek, w tym PAF400, białko o masie 400 kDa, będące ludzkim homologiem białka TRRAP (Vassilev i wsp., 1998). Białko to odgrywa rolę w modulacji aktywności czynnika p53 w odpowiedzi na uszkodzenia DNA przez promieniowanie ultrafioletowe: PCAF acetyluje koniec karboksylowy białka p53, powodując jego wiązanie do specyficznych sekwencji w DNA (Liu i wsp., 1999). PCAF zdolny jest również do acetylacji niehistonowych białek HMG-17 i HMG-14 związanych z nukleosomami (Herrera i wsp., 1999), powodując ich zmniejszone powinowactwo do nukleosomu. Inną acetylotransferazą histonową jest koaktywator transkrypcyjny CBP/p300. Białko to jest w stanie acetylować wszystkie cztery histony wewnątrz nukleosomu, jak również liczne czynniki transkrypcyjne, takie jak p53 i GATA-1 (Liu i wsp., 1999). CBP jest ponadto składnikiem holoenzymu polimerazy II RNA (Davie i Chadee, 1998). Do pozostałych białek o niedawno odkrytych funkcjach acetylotransferaz histonowych należą m.in. Esa1 - składnik wielobiałkowego drożdżowego kompleksu NuA4 o masie 1,3 MDa, zdolnego do acetylacji histonów H2A i H4 wyizolowanego ostatnio również z
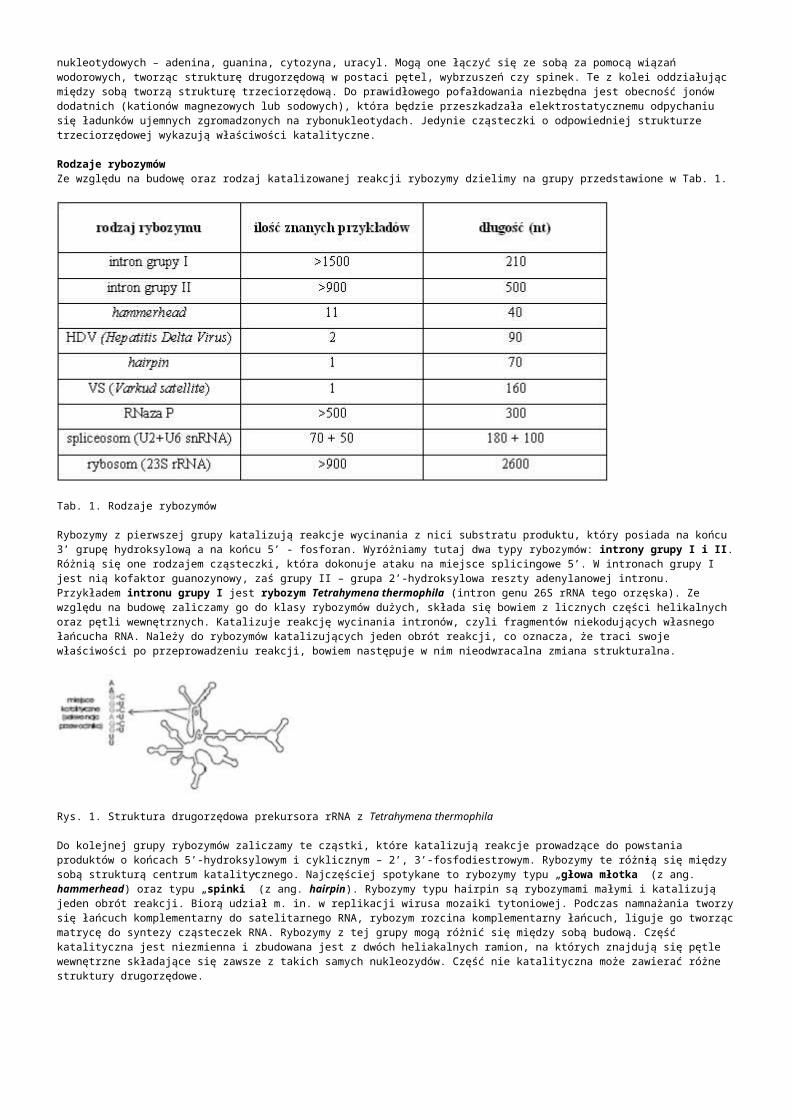
Tetrahymena (Ohba i wsp., 1999). Aktywność acetylotransferazy histonowej wykazują także duże kompleksy związane z rearanżacją (przebudową) struktur chromatynowych, a nie zawierające białka Gcn5: Swi/Snf i Srb/Mediator, konieczny do właściwej regulacji transkrypcji przez polimerazę II RNA (Carlson, 1997, patrz dalszy ciąg Wstępu).
Acetylacja histonów odgrywa ważną rolę w procesie transkrypcji. Histonowe acetylotransferazy drożdży (SAGA, NuA4, NuA3 i Ada) i Tetrahymena (NuA4) ułatwiają transkrypcję z matryc nukleosomowych (patrz dalej), lecz nie z nagiego DNA. Co istotne, enzymy wykazują działanie jedynie w obecności acetylokoenzymu A, źródła reszt acetylowych (Ohba i wsp., 1999; Steger i wsp., 1998). Acetylazy histonowe aktywują transkrypcję poprzez wzmocnienie etapów inicjacji i elongancji. Za proces wzmocnienia tego typu może odpowiadać złożona wielobiałkowa cząsteczka, zwana enhancosomem (cząstką wzmacniającą transkrypcję). Na przykład enhancosom IFN? złożony jest z białek NF-?B, IRF1, ATP2/c-Jun i HMG-I(Y), białka które jest zasadniczym elementem strukturalnym chromosomów. Po uformowaniu, kompleks taki przyłącza aktywnie białko CBP (poprzez podjednostkę p65 białka NF?B), które następnie przeprowadza acetylację histonów H3 i H4 w sąsiadujących ze sobą nukleosomach. Prowadzi to do przebudowy struktury chromatyny, czego wynikiem jest przyciągnięcie holoenzymu polimerazy RHA II i uruchomienie ekspresji genu IFN? (Merika i wsp., 1998).
Niedawno stwierdzono, że histony łącznikowe H1 i H5 mogą pełnić w chromatynie funkcję specyficznego represora acetylacji rdzeniowego histonu H3 (Herrera i wsp., 2000). Blokowanie acetylacji dotyczy wyłącznie histonu H3 związanego w mono- lub oligosomach (kilku nukleosomach ułożonych wzdłuż nici DNA), nie zaś wolnych histonów. Za inhibicję tego typu odpowiedzialne są ogony histonu łącznikowego wprowadzające zawadę steryczną dla aktywności acetylotransferazy histonowej. Mimo to, kompleks transacetylazy zawierający czynnik PCAF jest w stanie przełamać blokadę spowodowaną przez ogony histonów łącznikowych.
W przeciwieństwie do acetylacji, deacetylacja histonów prowadzi do zablokowania (represji) struktur chromatynowych. Histony ssaków są deacetylowane przez duże kompleksy zawierające wiele białek, takie jak kompleksy mSin3A i NuRD (Knoepfer i Eisenman, 1999; Kouzarides, 1999; Ayer, 1999). Pierwszy z nich zawiera korepresory mSin3, N-CoR, SMRT, RbAp48, RbAp4 i c-Ski (Nomura i wsp., 1999). W skład drugiego kompleksu, zdolnego do przebudowy chromatyny, wchodzą N-CoR, MTA2, Mi2 i RbAP46/48 (Grozinger i wsp., 1999). Kompleksy deacetylaz wykazują również aktywność wobec innych niż histony białek chromosomowych, takich jak acetylowane białka HMG i czynniki transkrypcyjne. Podobna przebudowa struktury chromatyny może wiązać się z jednoczesną aktywacją ekspresji genów przez indukcję deacetylaz histonowych w określonym punkcie różnicowania komórek. Na przykład ekspresja mysich deacetylaz mHDA1 i mHDA2, homologicznych z drożdżową deacetylazą RPD3, jest ściśle powiązana ze stanem zróżnicowania komórek, podobnie
jak ma to miejsce w przypadku jednego z wariantów rozwojowych histonu H1, H1 (patrz dalsza część Wstępu, Verdel i Khochbin, 1999).
Oprócz znaczenia w regulacji transkrypcyjnej, acetylacja i deacetylacja histonów pełni bardzo ważną rolę podczas rozwoju i różnicowania komórek. Białka p300 i PCAF stanowią ważny element procesu miogenezy (różnicowania komórek mięśniowych) (Puri i wsp., 1997), jak i szeregu innych szlaków rozwojowych, takich jak różnicowanie erytrocytów, limfocytów i melanocytów. Mutacje genów acetylaz wywołują poważne choroby genetyczne u ludzi, takie jak zespół Rubinsteina-Taybiego, objawiający się opóźnieniem umysłowym i charakterystycznymi deformacjami szkieletu (Giles i wsp., 1995). Znany jest także związek acetylaz i deacetylaz histonowych z innymi groźnymi stanami chorobowymi, w tym z procesami nowotworowymi (Archer i Hodin, 1999). Najsilniejszego na to dowodu dostarcza ostra białaczka mieloidalna (AML), w której odnajduje się przemieszczony gen CBP połączony albo do domniemanej acetylazy MOZ (Kamine i wsp., 1996) lub też do domniemanego regulatora homeotycznego białaczki mieszanego pochodzenia (MLL) (Sobulo i wsp., 1997). Z rakiem związane są także deacetylazy histonowe (HDAC). Na przykład mutacje w białku retinoblastoma (RB), będącym supresorem procesu nowotworzenia, które znoszą jego oddziaływania z HDAC, prowadzą do rozwoju raka, tak jak dzieje się to również w przypadku zablokowania tego oddziaływania przez onkoproteiny wirusowe (np. białko E7 wirusa HPV) (Kouzarides, 1999).
Metody hybrydyzacji molekularnej:
-Southern Blotting,
-DNA fingerprinting,
-Northern blotting,
-In situ hybridization (FISH).
FISH – istota procesu
FISH (ang. fluorescence in situ hybridization) jest techniką cytochemiczną polegającą na hybrydyzacji sekwencji DNA lub RNA ze specyficznymi sondami znakowanymi barwnikami fluorescencyjnymi. Dzięki reakcji sonda–nić DNA jesteśmy w stanie określić pozycję markera, czyli specyficznej, zazwyczaj oligonukleotydowej sekwencji, na badanej nici DNA. Stosując technikę FISH możemy również zidentyfikować interesujący nas chromosom (np. chromosom X lub Y - wykorzystanie FISH w badaniach przebiegu mejozy u zwierząt). Określenie in situ wskazuje na miejsce przeprowadzania reakcji hybrydyzacji, którym jest naturalne środowisko występowania DNA (chromosomy, komórki). Obserwacja zhybrydyzowanej sondy z komplementarną sekwencją DNA, jest możliwa dzięki wyznakowaniu sondy fluorochromem, który pod wpływem wzbudzenia światłem (UV-VIS) emituje promieniowanie (świeci). Do analizy badanego materiału wymagany jest mikroskop fluorescencyjny.

Mikroskop fluorescencyjny
Utrwalanie i hybrydyzacja
Przygotowanie komórek do doświadczenia polega na usunięciu cytoplazmy i utrwaleniu jąder komórkowych na szkiełku podstawowym (hybrydyzacja na poziomie DNA jądrowego) lub odpowiednie utrwalenie całych komórek (hybrydyzacja sond z mRNA występującym w cytoplazmie). Doświadczenie wykonuje się na skrawkach kriostatowych lub parafinowych oraz hodowlach komórek w monowarstwie. Prowadzone są także badania na skrawkach z materiału formalinowego. Przygotowane próby mogą być użyte bezpośrednio po utrwaleniu, lub przechowywane w roztworze soli fizjologicznej (opcjonalnie etanolu), w temperaturze 4 stopni Celsjusza.
- hybrydyzacja z RNA
Dzięki odpowiedniemu przygotowaniu komórek możemy uniknąć niespecyficznych absorpcji sondy do elementów komórkowych, oraz łatwiejszą penetrację cytoplazmy komórek. Skrawki poddajemy działaniu bezwodnika octowego, detergentu oraz 0,01 do 0,1 M HCL, których zadaniem jest zobojętnienie białek zasadowych, nieswoiście wiążących kwasy nukleinowe. W przypadku plemników czy błony śluzowej żołądka, zabiegi te są niewystarczające, toteż dodatkowo traktujemy je proteinazami (np. proteinazą K). Pracując nad wykrywaniem RNA sondami jednoniciowymi przykładamy dużą uwagę do czystości preparatów, gdyż niekontrolowane pojawienie się RNaz może zniszczyć cały wkład naszej pracy (RNazy występują np. w pocie). Problem ten nie istnieje, gdy wykrywamy DNA na poziomie jądra komórkowego lub chromosomów używając do tego celu sond dwuniciowych (dsDNA).
W celu uniknięcia degradacji przez RNazy sond jednoniciowych, hybrydyzujących z RNA, stosuje się sondy dwuniciowe.
- hybrydyzacja z DNA
Całość zabiegów niewiele różni się od tych opisanych powyżej. W sytuacji, kiedy planujemy otrzymać hybrydy na poziomie jądra, musimy pozbyć się środowiska, które go otacza. Jest to istotne szczególnie wtedy, gdy naszym celem jest wybarwienie chromosomów, oraz uzyskanie wysokiej rozdzielczości obrazu. W innym przypadku, gdy chcemy wybarwić określone chromosomy aby zidentyfikować ich położenia w komórce, postępujemy jak w przypadku hybrydyzacji RNA (nie usuwamy struktur komórkowych).Niewątpliwą różnicą pomiędzy hybrydyzacją DNA i RNA jest to, że w przypadku pierwszej, konieczna jest denaturacja nici tuż przed połączeniem z sondą. Również zastosowane sondy dwuniciowe wymagają rozbicia na dwie oddzielne nici. Brak denaturacji uniemożliwia połączenie sondy z komplementarnym odcinkiem kwasu nukleinowego.Aby denaturować bez konieczności niszczenia morfologii chromosomów suszymy je na szkiełku podstawowym, a następnie poddajemy działaniu formamidem. Inne utrwalacze stosowane w hybrydyzacji in situ także pozwalają uzyskać pozytywne efekty. Mogą to być utrwalacze precypitacyjne oparte na alkoholu, czy utrwalacze wiążące krzyżowo takie jak: formalina, paraformaldehyd czy aldehyd glutarowy. Ten ostatni może utrudniać penetrację sondy poprzez silne usieciowanie białek cytoplazmy – mówimy tu o sytuacji, gdy nie wyodrębniamy jądra ze struktur komórkowych. Wymaga się, aby stosowane odczynniki z jednej strony zachowywały badane kwasy nukleinowe w komórce, z drugiej zaś nie utrudniały penetracji przez sondy wewnątrz cytoplazmy. Dokonując wyboru utrwalacza zwracamy uwagę na rodzaj kwasów nukleinowych oraz sondy z uwzględnieniem jej masy cząsteczkowej.
Po wykonaniu czynności wstępnych nanosimy na preparat sondę wyznakowaną fluorescencyjnie i poddajemy kilkugodzinnej hybrydyzacji (RNA, DNA), pamiętając o konieczności wcześniejszej denaturacji (DNA, sondy dwuniciowe, dsRNA). Dla uniknięcia szybkiej renaturacji, zdenaturowaną nić DNA przenosimy do strefy niskiej temperatury (np. łaźnia lodowa). Zabieg ten nie jest konieczny, gdy inkubacja DNA z sondą w ośrodku denaturującym przebiega równocześnie (krótkotrwała denaturacja 2-3 min, w temp. 95 stopni Celsjusza w obecności formamidu).
- usuwanie niezwiązanych sond
Po etapie hybrydyzacji usuwamy nadmiar niezwiązanych sond kilkukrotnie opłukując preparat w odpowiednim buforze (wg określonej metodyki). Zaleca się

płukanie w roztworze o tej samej temperaturze w jakiej zachodziła hybrydyzacja. Płukanie można przeprowadzać w obecności skrawków nitrocelulozowych, które będą absorbować niezwiązane sondy. Uwidocznienie jąder komórkowych następuje po wybarwieniu barwnikiem specyficznym dla DNA (np. DAPI - 4,6-diamidyno-2-phenylindol). Obserwacji preparatu dokonujemy pod mikroskopem fluorescencyjnym.
- prehybrydyzcja
Właściwa hybrydyzacja powinna być poprzedzona prehybrydyzacją. Polega to na inkubacji sondy w mieszaninie zawierającej nieoznaczony DNA. Pozwala to zredukować niespecyficzność hybrydyzacji będącej wynikiem istnienia na sondzie sekwencji powtórzonych. Sytuacja ta może zaistnieć gdy zastosujemy długie sondy, szczególnie u organizmów eukariotycznych. Istnieje wówczas prawdopodobieństwo wystąpienia hybrydyzacji niespecyficznych w miejscach występowania powtarzalnych sekwencji.
Modyfikacje techniki FISH
Modyfikacją techniki FISH jest M-FISH (ang. multiplex fluorescence in situ hybridization). W przypadku pierwszej stosujemy jedną sondę znakowaną fluorescencyjnie, z kolei w drugiej używamy kilku sond znakowanych różnymi fluorochromami, które po wzbudzeniu dają różnobarwny obraz wyznakowanych chromosomów.Odmianą M-FISH jest technika molekularnego kariotypowania – analiza widmowa kariotypu SKY (ang. spectral karyotyping). Polega na wybarwianiu określonych regionów chromosomu przy użyciu komplementarnych sond molekularnych, wyznakowanych różnymi fluorochromami. W rezultacie otrzymujemy wielobarwny obraz kariotypu, gdzie każda para chromosomów ma inny kolor.
Fluorescencyjne sondy wykorzystywane w FISH
Dylematy związane ze stosowaniem sond znakowanych izotopami (patrz: znakowanie sond izotopami) doprowadziło do wytworzenia szeregu znaczników fluorescencyjnych, które charakteryzują się wysoką czułością oraz rozdzielczością (stąd nazwa techniki FISH).
Sondy stosowane w technice FISH dzielimy na:
- malujące (wcp – ang. whole chromosome paint)
- pokrywające chromosom lub poszczególne ramiona
- alfa-satelitarne (centromerowe, telomerowe)
- specyficzne dla poszczególnych sekwencji lub określonego locus
Wśród znaczników fluorescencyjnych, używanych do znakowania sond wymienić należy: rodaminę, fluoresceinę oraz kumarynę.
Znakowanie sond izotopami
Początkowe znakowanie sond izotopami promieniotwórczymi okazało się niezbyt dobrym rozwiązaniem. Wymagania jakie stawia technika hybrydyzacji in situ, czyli uzyskanie wysokiej czułości i rozdzielczości obrazu, w przypadku izotopów, były trudne do spełnienia. Uzyskanie wysokiej czułości przy znakowaniu promieniotwórczym wymaga zastosowania znacznika emitującego dużą energię, która z kolei jest przyczyną rozpraszania sygnału. W efekcie uzyskuje się obraz o słabej rozdzielczości. Izotopy a niskiej energii emisji pozwalają uzyskiwać wysokie rozdzielczości, jednak charakteryzują się stosunkowo małą czułością. Przedłużanie ekspozycji, w celu poprawy czułości jest przyczyną powstawania silnego tła. Do innych wad sond znakowanych radioaktywnie zaliczyć należy względy zdrowotne i środowiskowe, stąd też znaczniki te coraz bardziej tracą na popularności.
Radioizotopy:Tryt, Czas półtrwania: 12,35 lat, Energia emitowanych cząstek (MeV): 0,0186;Węgiel-14, Czas półtrwania: 5730 lat, Energia emitowanych cząstek (MeV): 0,156;Siarka-35, Czas półtrwania: 87,5 dnia, Energia emitowanych cząstek (MeV): 0,167;Fosfor-33, Czas półtrwania: 25,5 dnia, Energia emitowanych cząstek (MeV): 0,248;Fosfor-32, Czas półtrwania: 14,3, Energia emitowanych cząstek (MeV): 1,709;
P32 – fosfor radioaktywny, stosowany do znakowania DNA i RNAS35 – izotop siarki, głównie DNAH3 – tryt – znakowanie DNA (głownie)
FISH – zastosowanie w diagnostyce
Technika FISH znalazła szerokie zastosowanie w badaniach cytogenetycznych. Z najważniejszych wymienić należy: diagnostyka submikroskopowych aberracji chromosomowych, identyfikacja złożonych aberracji struktury chromosomów, identyfikacja dodatkowego materiału chromosomowego,

chromosomów markerowych czy diagnostyka aneuploidii chromosomowych.Najczęściej stosowane badania zespołów lub zaburzeń o podłożu genetycznym takich jak: zespoły Williamsa, Rubinstein-Taybiego, Wolfa-Hirshhorna, Prader-Willego, badanie przyczyn niepełnosprawności umysłowej powodowej w 50 procentach wadami genetycznymi, ponadto zaburzenia różnicowania płciowego (obojnactwo). Technika FISH umożliwia wykrywanie chorób nowotworowych, np. poprzez ocenę liczby kopii danego genu w komórkach nowotworowych. Dzięki niej możemy także identyfikować obecność określonych drobnoustrojów w materiale biologicznym, lub jako metoda skreeningowa zakażenia, służąca szybkiej identyfikacji w materiale biologicznym np. bakterii Achromobacter xylosoxidans czy Alcaligenes faecalis.FISH obok techniki PCR, jest nieocenioną pomocą w diagnostyce preimplantacyjnej pojedynczej komórki polocytu czy blastomeru, dzięki czemu pozwala uniknąć implantacji in vitro zarodka obarczonego wadami genetycznymi.
Enzymy restrykcyjne (restryktazy, endonukleazy restrykcyjne, enzymy trawienne) są białkami o aktywności enzymatycznej, które wyizolowano z bakterii. Endonukleazy restrykcyjne rozpoznają i przecinają specyficzne sekwencje w dwuniciowej cząsteczce DNA, w rezultacie czego otrzymuje się tzw. tępe i lepkie końce.
Enzymy restrykcyjne - izoschizomery
Izoschizomery są enzymami restrykcyjnymi, rozpoznającymi tę samą sekwencję DNA. Przyjmuje się, że pierwszy zidentyfikowany enzym restrykcyjny jest prototypem rodziny, a każdy kolejny jest jego izoschizomerem. Do izoschizomerów zaliczyć można wiele przykładów enzymów. Izoschizomerem enzymu AanI jest enzym restrykcyjny PsiI. Oba rozpoznają i tną sekwencję 5'-TTA/TAA-3'. Izoschizomery izolowane są zazwyczaj z różnych szczepów bakterii, w związku z czym mogą wymagać różnych warunków reakcji, różnych temperatur, stężenia soli czy czasu reakcji.
Enzymy restrykcyjne - neoschizomery
Poza izoschizomerami w przyrodzie występują także neoschizomery. O ile izoschizomery rozpoznają i tną daną sekwencję w identycznym miejscu, to w przypadku neoschizomerów jest trochę inaczej. Neoschizomer rozpoznaje tę sama sekwencję co tzw. prototyp (enzym restrykcyjny o określonych parametrach cięcia, wyizolowany jako pierwszy), jednak tnie ją w różnych miejscach. Neoschizomery zaliczane są do specyficznego podtypu izoschizomerów.Na przykład enzym AatII rozpoznaje i przecina sekwencje w następującym miejscu GACGT/C), przy czym jego neoschizomer ZraI rozpoznaje również tę samą sekwencję, jednak miejsce cięcia jest inne – GAC/GTC).
Badania DNA z wykorzystaniem enzymów restrykcyjnych
DNA lub produkt PCR trawimy enzymami, zwanymi endonukleazami restrykcyjnymi. Większość reakcji trawienia restryktazami przebiega nie dłużej niż 16 godzin. Obecnie, firmy oferują coraz szerszą gamę enzymów z tzw. grupy FastDigest. Trawienie zmodyfikowanymi enzymami FastDigest trwa zaledwie kilka, do kilkunastu minut (5-20 minut). Temperatura reakcji zależy od wybranego enzymu, a jej zakres może być bardzo szeroki, tj. od 37 stopni Celsjusza (np. AlwNI), do nawet 60-65 st. Celsjusza (np. BsiHKAI).
Trawienie restryktazami - zasady
- trawienie restryktazami przeprowadza się zwykle przy stężeniu 0.05-0.5µg DNA /µl;
- duża grupa enzymów restrykcyjnych, stosowanych w laboratorium, działa w jednym z trzech buforów, różniących się stężeniem soli (Tabela 1). Dane odnośnie wymaganych warunków reakcji, w tym stężenia soli, zawierają katalogi firm produkujących enzymy. Wskazane jest stosowanie buforów dostarczanych i zalecanych przez producenta danego enzymu;
- używane do trawienia bufory przygotowuje się w postaci stężonych roztworów (zazwyczaj x10);
- najczęściej używane bufory do enzymów restrykcyjnych to:

- jeśli zamierzamy trawić DNA (lub produkt reakcji PCR) dwoma enzymami, wymagającymi różnych buforów, należy najpierw przeprowadzić trawienie w buforze o niższym stężeniu soli, a następnie w buforze o wyższym stężeniu soli;
- ilość enzymu, jaką należy użyć w reakcji, oblicza się w oparciu o następującą definicję: 1 jednostka enzymu trawi kompletnie 1 µg DNA faga λ (ok. 50 kb), w czasie 1 godziny;
- ze względu na obecny w preparatach enzymatycznych glicerol, nie należy dodawać więcej enzymu niż 1/10 objętości mieszaniny reakcyjnej. Glicerol może hamować aktywność enzymu lub wpływać na jego specyficzność;
- niektóre enzymy, w pewnych warunkach reakcji trawienia, posiadają aktywność określaną jako aktywność "star". Jest to własność enzymów restrykcyjnych, polegająca na obniżeniu lub zaniku specyficzności ich działania. W wyniku zaniku specyficzności, cięcie DNA następuje w miejscach innych niż restrykcyjne. Wraz ze spadkiem specyficzności, cięciu podlegają miejsca coraz bardziej różne od oryginalnej sekwencji restrykcyjnej. Informacje o aktywności "star" podawane są przez producentów enzymów restrykcyjnych;
- przy sporządzaniu mieszaniny reakcyjnej enzym dodaje się na końcu. Ponadto, należy pamiętać, że enzym restrykcyjny musi być przetrzymywany w temperaturze -20 stopni Celsjusza, stąd też pobyt ER poza zamrażarką powinien trwać możliwie jak najkrócej;
- trawienie enzymatyczne prowadzi się w warunkach temperaturowych podanych przez producenta enzymu;
- przerwanie trawienia (zahamowanie aktywności enzymu) można wywołać na kilka sposobów. Najczęściej mieszaninę reakcyjną podgrzewa się do temperatury 65-80°C i utrzymuje przez 10-15 minut. Czasami producent zaleca dodanie do mieszaniny reakcyjnej EDTA, lub ekstrakcję fenolem. Związane jest to z właściwością enzymów - niektóre enzymy restrykcyjne są termostabilne i trudno ulegają termicznej inaktywacji.
Aktywność "star" enzymów restrykcyjnych
Pojawienie się aktywności "star", czyli niespecyficznego wykrywania i cięcia nici DNA w reakcjach z udziałem enzymów restrykcyjnych, zależy od wielu czynników. Aktywność „star” jest zwykle niepożądana, gdyż obniża specyfikę reakcji, wprowadza zmienność do przeprowadzanego eksperymentu, przez co utrudnia interpretację otrzymywanych wyników. Aktywność „star” pojawia się najczęściej w sytuacjach, kiedy reakcje prowadzi się używając kilku enzymów restrykcyjnych naraz (cięcie wielokrotne). Wówczas trudne jest zapewnienie warunków optymalnych dla każdego użytego enzymu.
Co stymuluje aktywność „star”?
- zbyt wysokie stężenie glicerolu, będącego składnikiem buforów do przechowywania enzymów oraz innych odczynników organicznych (DMSO, etanol, glikol i inne);
- zbyt wysokie stężenie enzymu względem DNA użytego do trawienia (po izolacji lub reakcji PCR);
- zbyt niska siła jonowa (<25mM);
- wysokie pH (>8,0)[2];
- zastąpienie jonów Mg2+ innymi jonami metali dwuwartościowych: (np.: jonami manganu, miedzi lub cynku).
Jak zapobiegać aktywności „star” enzymów restrykcyjnych?
Aby zapobiegać pojawianiu się niespecyficznych aktywności należy:- używać enzymów w minimalnej ilości, niezbędnej do pocięcia danej ilości DNA. Przykładowo, im mniejsza objętość enzymu, tym niższa stężenie glicerolu w próbce - należy przy tym pamiętać, że czas reakcji trawienia powinien zostać wydłużony;
- w miarę możliwości należy usunąć inne zanieczyszczenia organiczne, tj. np.: etanol używany do izolacji DNA;
- zapewnić odpowiednią siłę jonową oraz pH mieszaniny reakcyjnej (aktywność niektórych enzymów może być hamowana przez wysoką siłę jonową, z reguły wzrost siły jonowej powoduje spadek aktywności jonów, z kolei wyższe pH roztworu może być efektem używania niewystarczająco czystego DNA izolowanego metodą lizy alkalicznej);
- używanie jonów magnezu, jako czynnika wspomagającego pracę enzymu, jednocześnie należy unikać zanieczyszczeń innymi, dwuwartościowymi jonami.
TRANSKRYPCJA
Transkrypcja to proces przepisywania informacji genetycznej zawartej w DNA na cząsteczkę RNA. Jej podstawą jest reguła komplementarności (łac. cemplementum - dopełnienie, uzupełnienie) obowiązująca podczas parowania się zasad azotowych. Dzięki niej sekwencja nukleotydów w nowo-syntetyzowanym RNA jest jednoznacznie określona przez kolejność zasad na matrycowej nici kwasu dezoksyrybonukleinowego.
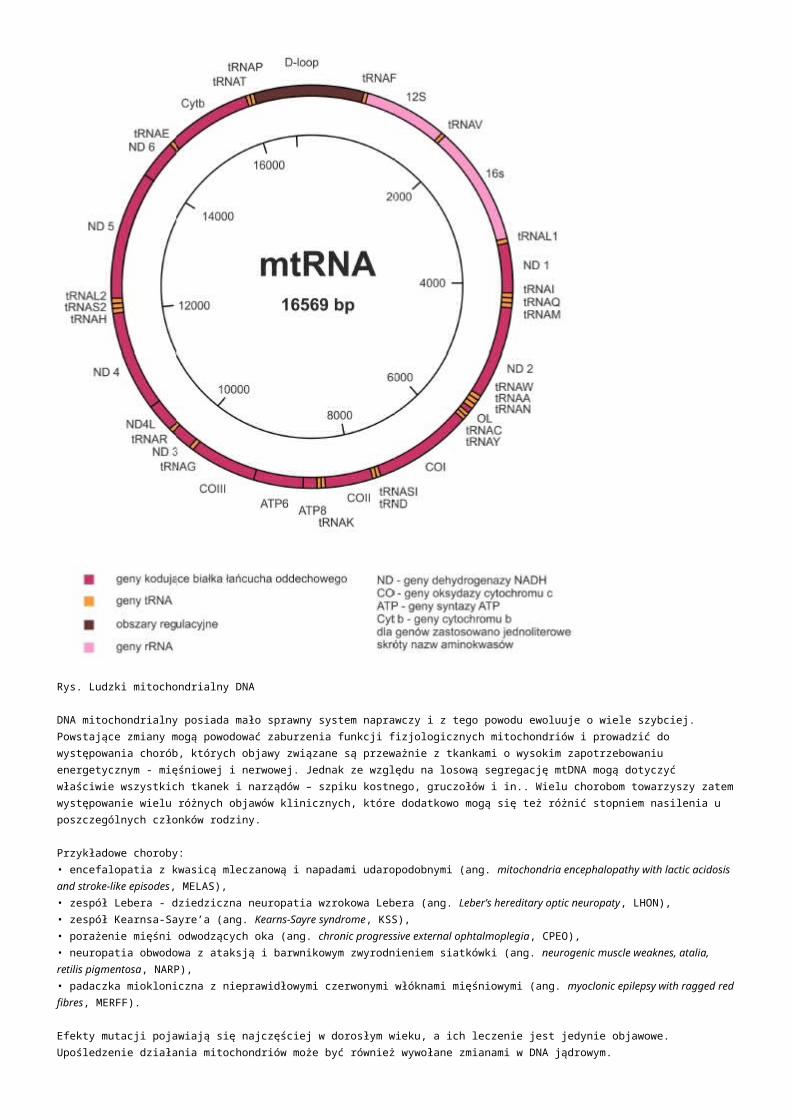
Rys. 1. Zasada komplementarności podczas transkrypcji
Transkrypcji podlega odcinek DNA znajdujący się między promotorem a terminatorem i nazywany jednostką traskrypcji. Nić DNA odczytywana jest w kierunku 3' → 5', natomiast cząsteczka RNA powstaje w kierunku 5' → 3'. Proces katalizowany jest przez polimerazę RNA.
Etapy transkrypcji: • Inicjacja – przyłączenie się polimerazy do promotora; • Elongacja - wydłużenie nici RNA. Zachodzi w bąblach transkrypcyjnych, które poruszają się wzdłuż matrycy DNA. Substratami są trifosfonukleotydy, od których odłączane są dwie grupy fosforanowe dostarczając w ten sposób energii potrzebnej do wytworzenia połączenia między nukleotydami; • Terminacja – zachodzi, gdy polimeraza RNA dotrze do sekwencji kończącej zwanej terminatorem.
Rys. 2. Wydłużanie (elongacja) transkryptu RNA
Transkrypcja u Eucaryota jest o wiele bardziej złożona niż u Procaryota. Obecność otoczonego błoną jądra komórkowego warunkuje rozdzielenie transkrypcji i translacji w czasie i przestrzeni, co umożliwia wielostopniową regulację ekspresji genów. Powstały łańcuch jest poddawany dalszym obróbkom – dodanie kapu na końcu 5’, ogona Poli(A) na końcu 3’ oraz splicing. U Procaryota oba procesy są ze sobą powiązane – translacja rozpoczyna się, gdy transkrypt jest

jeszcze syntetyzowany.
U bakterii występuje również tylko jeden rodzaj polimerazy RNA. Jest to kompleksowy enzym zbudowany jest z pięciu podjednostek. Dwie z nich - α wiążą białka regulatorowe, podjednostka β przyłącza substraty reakcji, natomiast β’ jest odpowiedzialna za wiązanie matrycy DNA. Razem tworzą one rdzeń enzymu, który przed syntezą łączy się z podjednostką σ odszukującą miejsce promotorowe na matrycy. U Procaryota promotor położony jest w sąsiedztwie części kodującej genu, a liczba powstających transkryptów zależy od ilości cząsteczek polimerazy, które biorą w niej udział.
U Eucaryota promotor zbudowany jest z kilku części regulatorowych. Promotor rdzeniowy poprzedza miejsce startu transkrypcji i pełni rolę zbliżoną do swojego bakteryjnego odpowiednika. W jego obrębie znajdują się sekwencje TATA-box oraz CAAT-box. Wzmacniacze (ang. enhancers) i wyciszacze (ang. silencers) umiejscowione są poza obszarem rdzenia i często występują w znacznej odległości od odcinka kodującego dane białko. Polimerazy u Eucaryota nie mogą same przyłączać się do promotora. Na ich powinowactwo wpływają niehistonowe białka pomocnicze, czyli tzw. czynniki transkrypcyjne (ang. transcription factors). Tworzą one kompleks transkrypcyjny, będący odpowiednikiem podjednostki σ u Procaryota. Ten system regulacji transkrypcji nie dotyczy genów metabolizmu podstawowego (ang. housekeping genes), kodujących białka niezbędne do przebiegu procesów życiowych komórki.
Czynniki transkrypcyjne dzieli się na trzy typy: • ogólne (ang. general factors) - inicjują syntezę RNA na wszystkich typach promotorów; • wiodące (ang. upstream factors) - łączą się z sekwencjami położonymi przed miejscem startu transkrypcji; • indukowalne (ang. inducible factors) - podobne do czynników wiodących, lecz syntetyzowane jedynie o określonym czasie i w określonych tkankach.
Białka regulatorowe posiadają charakterystyczne motywy sekwencyjne, które wyróżniają je spośród innych białek. Są to: • palec cynkowy (ang. zinc finger domain) – posiada w swojej strukturze jeden lub więcej jonów Zn2+ stabilizujących domenę. Mogą być one otoczone przez dwie reszty cysteinowe i dwie reszty histydynowe (klasa C2H2) lub przez cztery reszty cysteinowe (klasa C4); • zamek leucynowy (ang. leucine zipper) – to bogata w leucynę domena dimeryzacyjna, warunkująca hydrofobowe połączenie dwóch zwojów α-helisy; • helisa-skręt-helisa (ang. helix-turn-helix; HTH) – wiąże się do głównego rowka DNA; • helisa- pętla-helisa (ang. helix-loop-helix; HLH) – jest odpowiedzialna za dimeryzację.
U Eucaryota występują trzy rodzaje polimerazy RNA. Wszystkie są dużymi kompleksami białkowymi składającymi się z 8-12 podjednostek. • polimeraza I RNA – niewrażliwa na α-amanitynę, umiejscowiona w jąderku. Prowadzi transkrypcję wszystkich rodzajów genów rRNA z wyjątkiem 5S rRNA. Do swojej aktywności wymaga obecności jonów Mg2+ oraz czynników transkrypcyjnych: UBF1 (ang. upstream binding factor) i UCF. Rozpoznaje promotory leżące w górę od miejsca startu transkrypcji; • polimeraza II RNA – bardzo wrażliwa na α-amanitynę. Znajduje się w nukleoplazmie i jest odpowiedzialna za syntezę mRNA oraz większości snRNA. Wymaga obecności jonów Mg2+. Jej promotory zbudowane są z rdzenia (blok TATA – miejsce wiązania ogólnego czynnika TFIID) i dodatkowych sekwencji regulatorowych (ang. cis-acting elements) np. sekwencja CAAT, wzmacniacze (ang. enhancers), elementy RE (ang. response elements); • polimeraza III RNA – wrażliwa na wysokie stężenia α-amanityny. Usytuowana w nukleoplazmie, przeprowadza transkrypcję genów 5S rRNA, tRNA oraz małych jądrowych RNA. Wymaga obecności jonów Mn2+ oraz ogólnych czynników transkrypcyjnych: TFIIIA, TFIIIB i TFIIIC tworzących kompleks pre-inicjacyjny. Wykorzystuje trzy typy promotorów, z których dwa są za miejscem startu transkrypcji (ang. downstream), antomiast trzeci posiada bliski i daleki promotor.
W regulacji ekspresji genów ważną rolę odgrywa również struktura chromatyny. Mocno upakowana heterochromatyna, w okresach wzmożonej transkrypcji może ulegać dekondensacji. W proces ten zaangażowany jest także histon H1, który w odmienny sposób wiąże się do obszarów aktywnych transkrypcyjnie i rejonów wyciszonych.
TRANSLACJA
jest procesem tłumaczenia (ang. translation) informacji z „języka” kwasów nukleinowych na „język” aminokwasów budujących cząsteczkę peptydu lub białka. Literami tworzącymi zakodowaną informację są cztery zasady azotowe (Adenina - A, Cytozyna - C, Guanina - G i Uracyl – U), w łańcuchu polipeptydowym występuje natomiast 20 różnych aminokwasów. Z tego powodu informacja genetyczna zapisana jest za pomocą uniwersalnego, trzyliterowego kodu, dającego 64 różne kombinacje. Oznacza to, że choć każda trójka nukleotydów (triplet lub inaczej kodon) wyznacza ściśle określony aminokwas (jednoznaczność, zdeterminowanie kodu), to może być on zapisany na kilka różnych sposobów (degeneracja kodu).

Rys. 1. Kod genetycznyLewy margines - pierwsza litera kodu genetycznego; góra – druga; prawy margines – trzecia. Pod każdą trójką nukleotydów podano nazwę kodowanego aminokwasu.
Zapis pozbawiony jest również znaków przystankowych, tzn. jest bezprzecinkowy i stanowi ciąg niezachodzących na siebie tripletów, których kolejność ustala sekwencję aminokwasów w budowanym białku. Tą cechę kodu nazywamy kolinearnością.
Translacja jest procesem bardzo skomplikowanym i wymaga skoordynowanego współdziałania ponad stu różnych rodzajów makrocząsteczek: rybosomów, tRNA, mRNA oraz wielu białek pomocniczych.
Rybosomy
Synteza białka przebiega na rybosomach, czyli dużych, składających się z dwóch podjednostek zespołach rybonukleoproteinowych. Mogą one jednocześnie odczytywać informację zakodowaną w dwóch kodonach matrycowego mRNA. Przez wiele lat uważano, że główną funkcję podczas syntezy polipeptydów pełnią białka rybosomalne, natomiast rybosomalne RNA (rRNA) stanowi jedynie szkielet strukturalny. Obecnie wiadomo, że to nie prawda. Pozbawione białek rybosomy nadal mogą pełnić funkcje katalityczne podczas tworzenia się wiązań peptydowych w nowo syntetyzowanym łańcuchu aminokwasów. Ze względu na wielkość rybosomy dzielimy na małe - o stałej sedymentacji (stałej Svedberga) – 70S (mniejsza podjednostka 30S, większa 50S). Nie są one związane z błonami komórkowymi, występują u Procaryota oraz u Eucaryota w plastydach i mitochondriach. Rybosomy duże, zwane także eukariotycznymi charakteryzują się stałą sedymentacji 80S (mniejsza podjednostka 40S, większa 60S) i zwykle związane są z błonami szorstkiego retikulum endoplazmatycznego. Rzadko występują w cytoplazmie jako wolne organelle. Liczba znajdujących się w komórce rybosomów zależy od jej aktywności metabolicznej.
mRNA - matrycowy, informacyjny lub inaczej przekaźnikowy RNA (z ang. messenger RNA)
Tworzony jest w procesie transkrypcji, jako cząsteczka komplementarna do nici wiodącej DNA. Jego funkcją jest przenoszenie do aparatu translacyjnego zakodowanej informacji genetycznej. Po połączeniu się z rybosomem pełni on zatem funkcję matrycy wyznaczającej kolejność aminokwasów w budowanym białku. Translacja zachodzi w kierunku od końca 5’ do 3’.

tRNA - transportujący, transferowy RNA (ang. transfer RNA)
Cząsteczka ta zbudowana jest z 76- nukleotydowego, pojedynczego łańcucha przybierającego charakterystyczny kształt liścia koniczyny. Występuje w różnych postaciach, które różnią się między sobą sekwencją nukleotydów. Mogą w nim występować również inne zasady azotowe, tzn. metylocytozyna, hydroksymetylocytozyna, pseudouracyl, dihydrouracyl. Koniec 5’ łańcucha jest fosforylowany, natomiast 3’ zawiera wolną grupę hydroksylową łączącą się z grupą karboksylową odpowiedniego aminokwasu. Reakcja ta wymaga nakładu energii dostarczanej przez ATP i katalizowana jest przez enzym syntetezę aminoacylo-tRNA. Pośrodku każdej cząsteczki tRNA znajduje się tzw. pętla antykodonowa, determinująca rodzaj transportowanego aminokwasu. To właśnie zawarta w niej sekwencja (5’ – pirymidyna – pirymidyna – X –Y – Z – zmodyfikowana puryna – zmienna zasada – 3’) umożliwia odnalezienie właściwej cząsteczki przez specyficzną syntetazę. Jest ona również odpowiedzialna za rozpoznawanie właściwego tripletu na łańcuchu mRNA - determinuje zatem przyłączanie właściwego aminokwasu do powstającego polipeptydu. Niektóre tRNA rozpoznają więcej niż jeden kodon. Dzieje się tak, ponieważ parowanie ostatniej zasady jest mniej specyficzne niż dwóch pozostałych nukleotydów. Regułę tę nazywamy zasadą tolerancji.
Rys. 2. Ogólny schemat budowy cząsteczki tRNA
Proces syntezy białka zachodzi w cytoplazmie. Można podzielić go na trzy etapy, których przebieg zasadniczo nie różni się u organizmów eukariotycznych i prokoriotycznych, jednak u tych ostatnich translacja jest sprzężona z zachodzącą w jądrze transkrypcją i rozpoczyna się jeszcze przed ukończeniem syntezy mRNA. Kilka rybosomów może wtedy „tłumaczyć” zapisaną na mRNA informację.
Inicjacja translacji – polega na połączeniu się nici mRNA z podjednostkami rybosomy. Razem tworzą one tzw. kompleks translacyjny, zwany również „maszyną translacyjną”. Kodonem inicjującym translację na mRNA jest sekwencja AUG (rzadziej GUG). Startowa trójka nukleotydów ustawiona jest w miejscu P (peptydylowym, czyli zajmowanym przez tRNA połączony z polipeptydem) na rybosomie. Do niej przyłącza się inicjatorowy aminoacylo – tRNA. Pierwszym aminokwasem nowo zsyntetyzowanego łańcucha jest zawsze metionina (u Eucaryota) lub jej pochodna - formylometionina (u Procaryota).
Elongacja translacji – etap ten rozpoczyna się, gdy pierwszy aminoacylo-tRNA zajmie miejsce A (aminoacylowe, czyli zajmowany przez tRNA połączony z aminokwasem) na rybosomie. Budowa przestrzenna kompleksu translacyjnego umożliwia zbliżenie się grupy karboksylowej pierwszego aminokwasu do grupy aminowej drugiego aminokwasu. Proces tworzenia wiązań peptydowych między nimi wymaga nakładu energii oraz obecności enzymów rybosomowych. Uwolniona od polipeptydu cząsteczka tRNA opuszcza miejsce P i wraca do cytoplazmy, gdzie ponownie połączy się z właściwym sobie aminokwasem. Rybosom natomiast przesuwa się wzdłuż matrycy mRNA i w ten sposób w miejscu A pojawia się kolejna wolna trójka nukleotydów.
Cząsteczki aminokwasów przyłączane są do końcowej grupy karboksylowej tworzonego polipeptydu - białko syntetyzowane jest zatem w kierunku od końca N do końca C. Dalsze cykle elongacyjne zachodzą według tego samego schematu.
Terminacja translacji – zakończenie syntezy łańcucha polipeptydowego wyznaczają tzw. kodony stop – (UAA, UAG, UGA). Nie są one rozpoznawane przez cząsteczki tRNA i dlatego zajmując miejsce A na rybosomie wstrzymują wydłużanie łańcucha. Powstałe białko uwalniane jest z kompleksu dzięki tzw. czynnikom uwalniającym (RF – ang. release factor) i ulega dalszej obróbce, podczas której przybiera właściwą sobie formę przestrzenną.

Rys. 3. Przebieg wydłużania łańcucha polipeptydowego
BIAŁKA REKOMBINOWANE
Białka rekombinowane są to białka uzyskiwane z rekombinowanych genów, czyli cząsteczek DNA złożonych z różnych fragmentów kwasów nukleinowych. Ich zastosowanie pozwala na wydajną produkcję tych peptydów, które w naturalnych warunkach występują w niewystarczających ilościach, np. insuliny stosowanej w farmaceutyce i podpuszczki niezbędnej do produkcji serów.
Produkcja białek rekombinowanych
Białka są związkami wielkocząsteczkowymi zbudowanymi z aminokwasów. Stanowią one największa grupą związków organicznych i pełnią wiele ważnych funkcji życiowych w wytwarzających je organizmach. Między innymi wchodzą w skład struktur komórkowych i płynów ustrojowych (kolagen, keratyna, albuminy), transportują i magazynują różne substancje (hemoglobina), warunkują odpowiedź immunologiczną organizmu (przeciwciała), regulują procesy metaboliczne (hormony, enzymy) oraz procesy krzepnięcia krwi (fibrynogen). Niedobór lub niewłaściwe funkcjonowanie białek stanowi przyczynę wielu groźnych chorób. Jedynym możliwym sposobem ich leczenia jest regularne uzupełnianie powstałych braków, np. podawanie insuliny osobom cierpiącym na cukrzycę.
Wiele białek, głównie enzymów jest również powszechnie wykorzystywanych w przemyśle chemicznym (np. przy produkcji proszków do prania) i spożywczym (np. renina niezbędna do produkcji serów podpuszczkowych).
Dawniej wszystkie białka pozyskiwano z tkanek żywych organizmów. Jednak ze względu na nieustannie rosnące zapotrzebowanie, tego typu metody okazały się mało wydajne. Problem niedoboru rozwiązano dzięki rozwojowi biotechnologii. Zastosowanie technik rekombinacji DNA umożliwiło masową produkcję

prowadzoną w specjalnych systemach komórkowych, to znaczy w bakteriach, drożdżach, roślinach i zwierzętach, do których za pomocą odpowiednich wektorów wprowadzono namnożony transgen - fragment genu kodującego pożądane białko.
Aby możliwa była ekspresja zrekombinowanych genów, konieczne jest zastosowanie specjalnych wektorów ekspresyjnych. Nośniki te, oprócz elementów typowych dla wektora klonującego, zawierają również silny promotor, który umożliwia produkcję dużej ilości białka w komórce biorcy.
Wektory mogą być ponadto zaopatrzone w sekwencje regulujące stabilność syntetyzowanych peptydów i warunkujące sekrecję produktu docelowego, czyli jego wydzielanie poza komórkę gospodarza. Ich obecność znacznie ułatwia izolację i oczyszczanie pożądanej substancji.
Rys. 1. Plazmidowy wektor ekspresyjny
Wydajana ekspresja białek wymaga zastosowania odpowiednich warunków. Ich rodzaj zależy od typu syntetyzowanego produktu. Zachodzi zatem konieczność indywidualnej optymalizacji całego procesu.
Problem nadprodukcji
Głównym celem podczas wytwarzania białek rekombinowanych jest ich nadprodukcja. To właśnie ona możliwia korzystną ekonomicznie realizację celów naukowych (badań nad ich funkcją i strukturą), terapeutycznych (terapia i profilaktyka chorób zarówno organicznych, jak i zakaźnych) oraz przemysłowych (np. enzymy wykorzystywane w przemyśle).
Aby uzyskać ten efekt często stosuje się wektory plazmidowe, to znaczy takie, które mogą występować w komórce w dużej liczbie kopii. Ich nadmiar niesie jednak za sobą ryzyko destabilizacji całego systemu. Niektóre komórki tracą pobrane plazmidy i w efekcie rosną szybciej niż te, które utrzymują dodatkowe geny. Dominując w prowadzonej kulturze zmniejszają ogólną wydajność ekspresji. Aby rozwiązać ten problem zaczęto stosować wektory wbudowujące się w informację genetyczną gospodarza. Choć obniżają one ogólny poziom ekspresji pożądanych białek, to jednak gwarantują stabilność ich produkcji.
W niektórych wypadkach nadprodukcja może stanowić nawet 30% wszystkich białek komórkowych. Zbyt intensywna synteza prowadzi często do odkłania się białek i formowania tak zwanych nierozpuszczalnych ciał inkluzyjnych.
Białka pochodzące z nadekspresji określonych genów w bakteriach mogą być także niestabilne ze względu na aktywność enzymów proteolitycznych gospodarza. Produkty docelowe chroni się przed degradacją zmieniając sekwencję nukleotydową wektora. Do N końca kodowanego polipeptydu dodaje się jeden lub kilka aminokwasów, których sekwencja może być później wykorzystana również do identyfikacji transkryptów (ang. tag sequence). Na samym końcu dodatkowe aminokwasy są usuwane enzymatycznie lub chemicznie.
Stała ekspresja wyczerpuje możliwości energetyczne komórki. Oprócz pełnienia funkcji biofabryki, musi ona nadal pełnić wszystkie niezbędne do przetrwania funkcje życiowe. Stosowane wektory powinny zatem umożliwiać włączanie i wyłączanie syntezy białka tzn. indukcję ekspresji klonowanej sekwencji, np. za

pomocą temperatury lub dodawanych do pożywki metabolitów.
Bakteryjne systemy eskpresyjne
Pierwsze próby pozyskiwania białek rekombinowanych oparte były na bakteryjnych systemach ekspresyjnych. Tego typu metody nadal są stosowane do komercyjnej produkcji niektórych substancji.
Systemy bakteryjne najczęściej wykorzystują komórki pałeczki okrężnicy (Escherichia coli). Jest to organizm modelowy stosowany w eksperymentach inżynierii genetycznej i w związku z tym jego genom oraz warunki hodowli zostały bardzo dobrze poznane. Wiąże się to z szeroką dostępnością zarówno różnych szczepów, jak i wektorów, które mogą być użyte do badań. Inną zaletą E. coli jest stosunkowo niski koszt produkcji, gdyż bakteria ta rośnie dość szybko na tanich pożywkach.
Niepatogenne szczepy tej bakterii nie posiadają jednak efektywnych mechanizmów sekrecji białek do podłoża. W ich komórkach nie można również pozyskiwać niektórych typów cząsteczek, np. białek błonowych. W przypadku peptydów wytwarzanych do celów terapeutycznych, dużą niedogodnością jest także obecność lipopolisacharydu wchodzącego w skład ściany komórkowej wszystkich bakterii gram-ujemnych. Podczas izolacji może się on przedostawać do produkowanego białka i wywoływać gorączkę (to znaczy wykazywać właściwości pirogenne) w organizmie pacjenta.
Alternatywą jest stosowanie innych gatunków mikroorganizmów, np. gram-dodatnich bakterii Bacillus. Nie wytwarzają one lipopolisacharydu, co umożliwia późniejsze terapeutyczne zastosowanie uzyskiwanych w nich preparatów. Bakterie te posiadają również zdolność wydzielania dużych ilości białek do podłoża, czyli cechę znacznie ułatwiającą oczyszczanie pożądanych produktów.
Innym przykładem bakterii stosowanych do produkcji białek rekombinowanych są Lactococcus lactis, które stosuje się np. podczas otrzymywania peptydów błonowych.
Tabela 1. Szczepy E. coli najczęściej stosowane do produkcji rekombinowanych białek (według Staroń A. i in.)
System ten ma jednak swoje słabe strony i to niezależnie od rodzaju zastosowanych bakterii.
Żadne organizmy prokariotyczne nie wycinają niekodujących fragmentów DNA. Z tego względu należy w nich używać klonów komplementarnego DNA (ang. complementary DNA; cDNA) połączonych z prokariotycznymi sygnałami transkrypcji i translacji. cDNA jest uzyskiwany dzięki odwrotnej transkrypcji przeprowadzanej na matrycy mRNA i w związku z tym nie zawiera intronów.
Ważna jest również wielkość transgenu. Białka o masie cząsteczkowej większej niż 30 kDa mogą ulegać nieprawidłowemu fałdowaniu w komórkach bakteryjnych.
W niektórych przypadkach produkty wprowadzonych genów są uznawane za obce. Dzieje się tak nawet wtedy, gdy wytwarzają je geny zintegrowane z chromosomem gospodarza. Bakteria może wówczas odrzucać i niszczyć nieznane sobie peptydy. Problem ten pokonano przygotowując tak zwane białka fuzyjne, czyli inaczej białka chimeryczne. Składają się one z połączonych ze sobą dwóch lub większej ilości genów, które pierwotnie nie były ze sobą związane i odpowiadały za produkcję osobnych peptydów.
Geny kodujące pożądane białko łączy się z genami kodującymi białka typowo bakteryjne, a powstały w ten sposób produkt fuzyjny jest rozpoznawany przez gospodarza jako jego własny i nie ulega degradacji. Wydzielone z hodowli białko należy pod koniec rozszczepić enzymatycznie i dopiero w ten sposób uzyskuje się pożądany produkt.

Jednym z największych ograniczeń jest jednak fakt, że żadne bakterie nie są zdolne do przeprowadzania mechanizmów modyfikacji posttranslacyjnej, takich jak np. cięcia enzymatyczne, glikozylacja. W większości przypadków są one kluczowe dla prawidłowej aktywności białek eukariotycznych. Produkty wytwarzane w systemach bakteryjnych mogą być zatem niestabilne lub nieaktywne biologicznie.
Tabela. 2. Najczęściej spotykane problemy i ich możliwe rozwiązania w eksperymentach nadekspresji białek w komórkach E.coli (według Staroń A. i in.)
Eukariotyczne systemy ekspresyjne
Aby rozwiązać problem braku obróbki posttranslacyjnej opracowano systemy ekspresyjne opierające się na komórkach eukariotycznych. Uzyskiwane dzięki nim białka w większym stopniu przypominają formy natywne i mogą być wprowadzane do organizmu człowieka, a zatem służyć jako leki. W ten sposób otrzymuje się np. hormon wzrostu, β-interferon, białko krzepliwości krwi (czynnik VIII i IX), fibrynogen, erytropoetynę.
Ssacze wektory są wektorami wahadłowymi, inaczej bifunkcjonalnymi lub czółenkowymi (ang. shuttle). Oznacza to, że mają one dodatkową sekwencję charakterystyczną dla komórek prokariotycznych i dzięki temu mogą być namnażane w bakteriach. Dopiero potem DNA wprowadza się do komórek eukariotycznego gospodarza, to znaczy do miejsca, w których geny ulegną ekspresji. Najczęściej stosuje się elektroporację. Można też dodawać do zawiesiny komórek DNA wytrącony fosforanem wapnia.
Oprócz sekwencji promotorowych i sekwencji sygnałowych transkrypcji i translacji, eukariotyczne wektory ekspresyjne powinny posiadać również między innymi sekwencje sygnałowe poliadenylacji mRNA. Wektory te mogą występować w formie episomalnej (to znaczy pozostającej bez związku z genomem gospodarza) jako plazmidy lub też ulegać integracji z jednym z chromosomów komórki gospodarza.
W produkcji białek rekombinowanych szeroko stosowane są drożdże Saccharomyces cerevisiae, których genetyka i fizjologia zostały już dobrze poznane. Ich hodowla nie jest trudna, a wytworzone dzięki nim białka można łatwo oczyszczać wykorzystując do tego celu zjawisko sekrecji. Drożdżowe wektory ekspresyjne integrują się z genomem gospodarza lub też mają postać plazmidów zbudowanych na bazie endogennego plazmidu drożdżowego 2μ.
DNA wprowadza się do komórki na kilka sposobów, np. poprzez elektroporację lub usunięcie ściany komórkowej, to znaczy otrzymywanie protoplastów. Przykładowym produktem otrzymywanym w drożdżach jest insulina oraz, stosowany jako szczepionka, powierzchniowy antygen wirusowego zapalenia wątroby typu B.
Można także posługiwać się komórkami owadzimi, które wykorzystuje się do hodowli bakulowirusów. Jako wirusy infekujące bezkręgowce, są one naturalnie przystosowane do funkcji eukariotycznego wektora ekspresyjnego. Znajdujący się w ich genomie gen białka poliedryny pozostaje pod wpływem bardzo silnego promotora. Można go zastąpić genem docelowym, który w ten sposób łatwo zostaje przeniesiony do komórek gospodarza i ulega ekspresji na późnym etapie infekcji. Tak zwany system bakulowirusowy, jest bardzo efektywny pod względem sekrecji białek do podłoża. Kolejną jego zaletą jest to, że uzyskiwane w nim białka mają wzór glikozylacji zbliżony do wzoru swoich ssaczych odpowiedników. Komórki owadzie służą między innymi do produkcji antywirusowego białka - β-interferonu oraz erytropoetyny stosowanej u pacjentów cierpiących na anemię.

Powszechnie wykorzystuje się także mysie lub ludzkie linie komórek ssaczych. Przykładowo białko ludzkiego czynnika VIII (podawane osobom chorym na hemofilię) wytwarza się w liniach komórkowych chomika poddanych wcześniej transfekcji 186kpz fragmentem ludzkiego genomowego DNA. W porównaniu z komórkami mikroorganizmów komórki ssaków cechują się wolniejszym tempem translacji i fałdowania. W związku z tym są one lepszymi gospodarzami w przypadku produkcji białek błonowych. Ich wadą jest jednak stosunkowo niska wydajność idąca w parze z wysokimi kosztami całego procesu.
Obecnie produkcja białek rekombinowanych jest możliwa również z wykorzystaniem całych genetycznie zmodyfikowanych organizmów (ang. genetically modified organisms, GMO). Dzięki zastosowaniu metod inżynierii genetycznej zdołano skonstruować między innymi owce i kozy wydzielające do mleka odpowiedniki ludzkich białek. Wymienione czynniki najłatwiej byłoby uzyskać w surowicy krwi. Organizm zwierzęcia transgenicznego toleruje jednak tylko ograniczone ilości obcego produktu w swoich tkankach. Wydzielane na zewnątrz mleko nie tylko rozwiązuje ten problem, ale również jest produkowane w dużych ilościach i może być zbierane bez szkody dla wytwarzającego je zwierzęcia. Ponadto zawiera ono niewiele rodzajów białek, co znacznie ułatwia proces oczyszczania otrzymywanych w nim produktów. Doustne podawanie leku jest dogodne również z punktu widzenia pacjenta. W ten sposób produkuje się m.in. czynnik IX (krzepnięcie krwi) i białko osocza α1-antytrypsynę. Wykorzystanie zwierząt transgenicznych jest jednak bardzo kosztowne i budzi wiele kontrowersji na tle etycznym.
Produkcja białek w roślinach
Pierwsze eksperymenty dotyczące produkcji białek rekombinowanych w roślinach opisano około 25 lat temu. Wprowadzane geny ulegają ekspresji w określonych tkankach, np. w liściach, z których następnie oczyszcza się pożądany produkt.
Organizmy stosowane jako biofabryki nie są przeznaczone do celów spożywczych i nie mają nic wspólnego z genetycznie zmodyfikowaną żywnością. Pozostałe po nich odpadki poddaje się degradacji.
Produkcję białek rekombinowanych z użyciem roślin określa się mianem rolnictwa molekularnego (ang. molecular farming, plant biopharming, plant molecular farming (PMF)).
Najczęściej stosowanymi gatunkami są kukurydza, tytoń, ziemniaki i pomidory. Są to organizmy dobrze poznane zarówno pod względem genomu jak i najlepszych technik uprawy. Lista wytwarzanych w ten sposób białek (znajdujących się na różnych etapach badań) obejmuje między innymi przeciwciała i ich fragmenty, antygeny do produkcji szczepionek, enzymy i cytokiny. Otrzymywane produkty wykorzystuje się najczęściej w przemyśle farmaceutycznym, chemicznym i spożywczym.
Hodowla roślin jest bardzo wydajna i może być prowadzona na polu oraz w zamkniętych szklarniach. Zastosowanie tego typu biofabryk mogłoby zatem znacznie obniżyć koszty produkcji białek i w ten sposób wpłynąć na cenę i dostępność niektórych biofarmaceutyków. Zniwelowałoby to również ryzyko występowania zakażeń odzwierzęcych, gdyż powodujące je bakterie oraz priony nie mogą się rozwijać wewnątrz roślin. Ponadto, produkowane w ten sposób szczepionki mogłyby być podawane w postaci pokarmu, to znaczy bez konieczności iniekcji, która stanowi mniej przyjemną i bardziej inwazyjną metodę wprowadzania leków do organizmu.
Duże obawy wśród społeczeństwa wzbudza jednak ryzyko przypadkowego spożycia zmodyfikowanych roślin. Ich części, np. pyłki mogłyby zanieczyść maszyny rolnicze i przedostać się do upraw przeznaczonych na cele spożywcze. Wątpliwości budzi również wpływ roślin GM na mikroorganizmy glebowe.
Białka rekombinowane w medycynie
Pierwotnie potrzebne białka izolowano i oczyszczano z organizmów zwierzęcych lub uzyskiwano je od ludzkich dawców. Metoda ta była jednak mało wydajna. Ponadto, białka pobrane od innych gatunków wykazywały różnice funkcjonalne, a ich stosowanie niosło ze sobą ryzyko wstąpienia groźnych zakażeń, np. wirusami HIV, prionami CJD.
Zastosowanie białek rekombinowanych zwiększa wydajność produkcji leków i ich dostępność Niweluje problem niezgodności i ogranicza do minimum zagrożenie związane z przenoszeniem czynników chorobotwórczych.
Tabela. 3. Niektóre leki o charakterze rekombinowanych białek (według Ratledge C. i in.)
Grupa leków
cytokiny i antagoniści
Przykłady białek
Interferon alfa-2aInterferon alfa-2bInterferon alfacon-1Peginterferon alfa-2aPeginterferon alfa-2bInterferon beta-1aInterferon beta-1bInterferon gamma-1bAldesleukin (IL-2)Filgrastim (G-CSF)Pegfilgrastim Lenograstim (G-CSF)Molgramostim (GM-CSF)Sargramostin (GM-CSF)

Tasonermin (TNF-α)Becaplermin (PDGF-BB)Oprevelkin (IL-II)Anakinra (IL-IRA)
hormony i peptydy InsulinaInsulina lisproInsulina aspartInsulina glargineEpoetyna alfa (erytropoetyna)Epoetyna beta (erytropoetyna)Epoetyna delta (erytropoetyna)Darbepoetyna-alfaFolitropina alfaFolitropina betaGlukagonSomatotropinaLutropina-alfaTeriparatide (PTH I-34)Kalcytonina z łososiaTyrotropina-alfaChoriogonadotropina A2Osteogeniczne białko 1Dibotermina alfa (BMP-2)Pegvisomat (antagonista hGH)Nesirtide (peptyd natriuretyczny)
czynniki krzepliwości i inhibitory Eptacog alfaCzynnik antyhemofilowyMoroktokog alfa (muteina FVIII)Nonacog alfaDesirudynaLepirudynaDrotrekogina alfa (aktywowana białkiem C)Inhibitor α1-proteinazy
enzymy Alteplaza (t-PA)Reteplaza (muteina t-PA)Tenekteplaza (muteina t-PA)Monteplaza (muteina t-PA)Dornaza-alfa (RNaza)ImiglucerazaAgalzydaza alfaAgalzydaza betaResburykazaLaronidaza
szczepionki Szczepionka przeciw zapaleniu wątrobySzczepionka przeciw boreliozieSzczepionka di-per-te (przeciw błonicy, teżcowi I krztuścowi)Szczepionka przeciw rotawirusom
białka fuzyjne Denileukin diftitoksEtanerceptAlefacept
Tłustym drukiem zaznaczono białka stosowane w zmodyfikowanej formie (w porównaniu z oryginalnym białkiem ludzkim). Niektóre z przedstawionych białek zostały wprowadzone do handlu pod różnymi nazwami przez różne przedsiębiorstwa farmaceutyczne. Dokładniejsze informacje na temat poszczególnych leków białkowych znajdują się na stronach internetowych producentów.

Białka rekombinowane w przemyśle
Wykorzystanie technologii rekombinacji DNA pozwala na modyfikację uzyskiwanych białek. Jest to tak zwana inżynieria białek. Dzięki temu można polepszyć właściwości uzyskiwanych produktów, np. enzymy odporne na wysoka temperaturę lub o zmienionej specyficzności względem substratu.
Dużą popularnością cieszy się potencjalne zastosowanie roślin do uzyskiwania biodegradowalnych plastików (Metabolix company). Innym użyciem mogłaby być produkcja olejów przemysłowych, takich jak oleje hydrauliczne lub wysokowydajne biodiesle, nowe biopaliwa stałe, włókna i papiery.
Tabela. 4. Przykładowe białka rekombinowane stosowane w przemyśle (według Winter P.C. i in.)
Green fluorescent protein – czyli GFP, jest białkiem pochodzącym z meduzy Aquorea victoria. Za jego odkrycie Chalfie, Shimomura i Tsien otrzymali nagrodę Nobla w 2008 roku. GFP świeci na zielono w ultrafiolecie. Złożone jest z 238 aminokwasów a jego masa wynosi 26,9 kDa. To białko ma strukturę ß – baryłki, składającej się z jednej ß – kartki z α – helisami zawartymi w chromoforze, biegnącymi przez centrum. Łańcuchy boczne białka tej baryłki indukują specyficzną cyklizację reakcji w tripeptydzie Ser65 -Tyr66 – gly67, która prowadzi do powstania chromoforu. Ten proces posttranslacyjnej modyfikacji jest opisywany jako dojrzewanie.
FP mogą występować w różnych wariantach barwnych, np. żółtym – YFP (ang. yellow fluorescent protein), tj. Citrine, Venus i YPet, niebieskim – BFP (ang. blue fluorescent protein), tj. Azurite, mKalama 1, EBFP (ang. enhanced blue fluorescent protein – białko o wzmocnionej fluorescencji) i EBFP 2 a także RFP (ang. red fluorescent protein). Nomenklatura modyfikowanych GFP jest często zbieżna z powodu mapowania kilku wersji GFP do pojedynczej nazwy. Wersja GFP wrażliwa na redox (roGFP) była zmieniona poprzez wprowadzenie cysteiny do struktury ß – baryłki. Stan redox cystein determinuje fluorescencyjne właściwości roGTP. Mutanty GFP cechują się szybszym czasem dojrzewania fluoroforu, większą stabilnością i emisją silniejszego sygnału świetlnego niż formy dzikie. Te warianty GFP, które służyły do badania aktywności i funkcjonalności promotorów, charakteryzowały się krótszym okresem półtrwania.
Ze względu na niską toksyczność GFP jest może być szeroko stosowane. Pozwala to na detekcję odbywającą się bez niszczenia struktur mikroorganizmów oraz wykrywanie znaczonych komórek bez ich wcześniejszego wybarwienia. W odróżnieniu od innych białek wykazujących fluorescencję nie wymaga ono kofaktorów (jako aktywatorów) ani towarzyszących białek oraz ATP (jak lucyferaza). Do wzbudzenia jego fluorescencji wymagane jest jedynie światło mieszczące się w zakresie absorbancji, a do wzbudzania wystarczą dwa kwanty światła.
Produkt genu GFP jest włączany do genomu organizmu w rejon DNA, który koduje docelowe białko i jest kontrolowane przez tę samą regulatorową sekwencję. W komórkach, gdzie następuje ekspresja genów i są produkowane białka, GFP jest wytwarzany w tym samym czasie. Analiza czasu wygaśnięcia tego białka jest używana do zrozumienia wielu z biologicznych procesów, do których zalicza się fałdowanie białek, transport białek oraz dynamikę RNA. GFP umożliwia optyczną detekcję specyficznego typu komórek in vitro lub nawet in vivo (w żywych organizmach). Kilka spektralnych wariantów GFP modyfikowanych genetycznie jest przydatnych w analizowaniu położenia neuronów (mapowania mózgu) w Brainbow. Innym ciekawym zastosowaniem fluoryzujących białek jest użycie FP jako sensora neuronalnego potencjału błonowego, śledzenie receptorów AMPA w błonie komórkowej oraz badanie infekcji poszczególnymi wirusami grypy. Stworzono także nowe transgeniczne GFP, które mogą mieć istotne znaczenie dla terapii genowej. W biologii molekularnej GFP jest często wykorzystywane do wizualizacji np. lokalizacji białek w komórce przez utworzenie fuzji danego białka z GFP. Doskonale sprawdza się też jako cząsteczka reporterowa, służąca do badania m. in. aktywności promotorów lub wydajności transfekcji komórek. Ponadto plusem jest stabilność sygnału – GFP jest odporne na denaturację termiczną i utrzymuje fluorescencję do temperatury ok. 65ºC. Odporne jest również na denaturację chemiczną, gdyż wykazuje fluorescencję w zakresie pH od 6 do 12. Ze względu na zwartą strukturę nie ulega działaniu wielu enzymów proteolitycznych tj. trypsyny i papainy, aż do stężenia 1 mg/ml. To białko ma minimalny wpływ na dynamikę procesów zachodzących na powierzchni komórek ze względu na ekspresję zachodzącą w cytoplazmie. Jest ono ciągle syntetyzowane w komórkach, jedynie podczas replikacji u bakterii następuje minimalne obniżenie sygnału fluorescencji. Brak jest również tła emitowanego przez autochtoniczne populacje bakteryjne, które zakłócałoby detekcję.
Jedynym ograniczeniem w aplikacji genu gfp jest dostępność tlenu, ponieważ w warunkach beztlenowych nie dochodzi do prawidłowego formowania się odpowiedzialnego za świecenie fluoroforu. W konsekwencji następuje zmiana długości fali maksymalnego wzbudzenia oraz emisji. Do wad GFP należy również m. in. niestabilność plazmidów zawierających geny kodujące GFP – bardziej stabilne są inserty wprowadzone w chromosom bakteryjny, co pozwala na obniżenie ryzyka transferu genu kodującego GFP do genomów bakterii autochtonicznych. Ponadto w warunkach beztlenowych GFP nie emituje sygnału. Niezbadana jest zmienność ekspresji GFP w komórkach różnych gatunków oraz wpływ czynników środowiskowych na ekspresję GFP.
Reasumując, GFP jest jednym z najbardziej popularnych genów reporterowych w biologii molekularnej, dzięki możliwości przyżyciowego monitorowania in situ horyzontalnego transferu genów między mikroorganizmami, ich aktywności w różnych środowiskach oraz monitorowaniu poziomu ekspresji genów. Ekspresję genu gfp wykazano z użyciem wektorów plazmidowych jak i transpozonów. Fuzje badanych białek z GFP pozwalają na monitorowanie w czasie rzeczywistym migracji białek fuzyjnych w różnych układach i kompleksach wielobiałkowych. Właściwości fizykochemiczne tego białka wynikają bezpośrednio z kolejności i typu aminokwasów. Ewolucja faworyzowała trwałość GFP i ochronę centralnie położonego fluoroforu przed wygaszeniem.
1. MutarotacjaCukry występują głównie w formie pierścieniowej – zawartość formy łańcuchowej w stanie równowagi jest znikoma i w przypadku glukozy wynosi jedynie ok. 0,02%. Utworzenie formy pierścieniowej powoduje powstanie nowego centrum chiralnego i co za tym idzie dwóch diastereoizomerycznych odmian formy

pierścieniowej – anomerów α i β. W roztworze cukru ustala się stan równowagi pomiędzy formami α, β i łańcuchową, będącą stanem pośrednim w procesie przemiany jednego anomeru w drugi:
Rozpuszczając zatem kryształy cukru uzyskanego w czystej formie α lub β (jest to możliwe poprzez krystalizację w odpowiednich warunkach), po pewnym czasie uzyska się roztwór, w którym obecne będą wszystkie formy w stanie równowagi, w odpowiednich proporcjach zależnych od budowy chemicznej danego cukru. Przemianom tym towarzyszy zmiana kąta skręcania płaszczyzny światła spolaryzowanego. Natychmiast po sporządzeniu roztworu wartość skręcalności właściwej jest charakterystyczna dla czystego anomeru. Z czasem skręcalność zmienia się aż do momentu osiągnięcia stanu równowagi, kiedy odpowiada wypadkowej skręcalności obu form anomerycznych znajdujących się w stanie równowagi. Zmiana skręcalności właściwej roztworu cukru spowodowana ustalaniem się równowagi pomiędzy formami α i β w roztworze w wyniku zmiany konfiguracji przy anomerycznym atomie węgla nosi nazwę mutarotacji.2. Wpływ kwasów i zasad na węglowodany2.1. Reakcje przebiegające pod wpływem kwasów.Cukry są dosyć stabilne w słabo kwaśnym środowisku. Pod wpływem mocnych mineralnych kwasów o średnim stężeniu ulegają jednak różnym przemianom:2.1.1. Hydroliza polisacharydów.Oligo- i polisacharydy pod wpływem kwasów mineralnych (w praktyce najczęściej jest stosowany kilku- kilkunastoprocentowy roztwór HCl) ulegają hydrolizie, która w przypadku polisacharydów biegnie przez stadium oligosacharydów (hydrolityczny podział na mniejsze fragmenty) i w rezultacie prowadzi do otrzymania monosacharydów.2.1.2. Dehydratacja monosacharydówMonosacharydy pod wpływem średnio stężonych kwasów mineralnych ulegają procesowi dehydratacji, którego wynik zależny jest od struktury wyjściowego cukru: Pentozy ulegają dehydratacji z wytworzeniem aldehydu 2-furylowego (furano- 2-karbaldehydu):
Reakcja ta jest wykorzystywana jako przemysłowy proces otrzymywania furfuralu (inna nazwa zwyczajowa furano-2-karbaldehydu).Heksozy w wyniku reakcji dehydratacji dają 5-hydroksymetylofurano-2- karbaldehyd (5-hydroksymetylofurfural), który w środowisku kwaśnym może podlegać dalszej reakcji degradacji do kwasu lewulinowego:
2.2. W środowisku zasadowym, nawet w łagodnych warunkach monosacharydy ulegają rozlicznym przemianom tautomerycznym w reakcjach równowagowych, prowadzących do otrzymania mieszaniny związków. Procesy te są wynikiem ruchliwości atomów wodoru w pozycji α względem grupy karbonylowej i biegną poprzez stadium endiolu:Wyjściowy związek, jakim w powyższym schemacie jest glukoza, pod wpływem jonów OH- ulega przemianie tautomerycznej. W jej wyniku powstaje związek określany mianem endiolu, ponieważ przy wiązaniu podwójnym występują w nim dwie grupy OH. Związek ten może w wyniku kolejnej przemiany tautomerycznej odtworzyć grupę karbonylową, lecz teraz jest to możliwe na trzech drogach:

1. odtwarzana jest grupa aldehydowa, proton znajdujący się przy pierwszej grupie OH endiolu wraca na poprzednią pozycję w łańcuchu cukru i w wyniku otrzymuje się niezmienioną cząsteczkę glukozy;2. odtwarzana jest grupa aldehydowa, proton znajdujący się przy pierwszej grupie OH endiolu wraca na pozycję C-2 łańcucha cukru, lecz z drugiej strony łańcucha co powoduje otrzymanie diastereoizomeru glukozy – mannozy różniącej się od glukozy konfiguracją przy węglu C-2;3. utworzona zostaje grupa karbonylowa przy węglu C-2, na skutek przemiany tautomerycznej polegającej na przeniesieniu protonu z grupy hydroksylowej w pozycji 2 do węgla C-1, w wyniku czego powstaje cząsteczka fruktozy. Ogół powyżej przedstawionych przemian nosi nazwę reakcji epimeryzacji, jednakże zaznaczyć należy, że nie wszystkie występujące w nim związki są dla siebie epimerami. Zgodnie z definicją epimery to takie diastereoizomery, które różnią się konfiguracją wyłącznie przy jednym centrum chiralnym. Porównując związki występujące na schemacie powyżej, epimerami są dla siebie wyłącznie glukoza i mannoza – pozostałe związki nie są izomerami optycznymi, lecz strukturalnymi.Proces epimeryzacji cukrów wyjaśnia również, dlaczego ketozy (np. fruktoza) należą do cukrów redukujących. Analityczne próby na odczyn redukujący cukrów prowadzi się bowiem w warunkach zasadowych, w których w wyniku epimeryzacji pojawiają się w roztworze również odpowiednie aldozy, mogące bezpośrednio ulegać reakcji utlenienia.2.3. W silniej zasadowym środowisku następuje rozpad cząsteczek cukrów. Jest to proces równowagowy będący odwróceniem reakcji kondensacji aldolowej. Zachodzi on zatem zawsze w pozycji β względem grupy karbonylowej cukru:
W komórkach, w trakcie biologicznej degradacji oraz biosyntezy cukrów, pod wpływem odpowiednich enzymów zachodzą procesy analogiczne do wyżej przedstawionego procesu rozpadu cukru w reakcji odwrotnej do kondensacji aldolowej, jak i reakcje łączenia fragmentów w reakcji kondensacji aldolowej.3. Tworzenie osazonów – reakcja z fenylohydrazyną.Związki karbonylowe chętnie reagują ze związkami posiadającymi wolną grupę NH2. Jednym z często stosowanych w analizie jest fenylohydrazyna. Cukry, jako polihydroksylowe aldehydy i ketony, również łatwo reagują z tym odczynnikiem, dając łatwe do wydzielenia, krystaliczne pochodne zwane osazonami. Pierwszy etap reakcji monosacharydów z fenylohydrazyną biegnie analogicznie do reakcji prostych związków karbonylowych z tym odczynnikiem, dając prosty fenylohydrazon:

W przypadku cukrów reakcja nie zatrzymuje się na tym etapie, lecz powstający fenylohydrazon reaguje z kolejną cząsteczką fenylohydrazy w reakcji redoks, w której fenylohydrazyna utlenia grupę hydroksylową w pozycji α względem grupy karbonylowej wyjściowego cukru:
Uzyskany w ten sposób produkt pośredni, zawierający nową grupę karbonylową, kondensuje z trzecią cząsteczką fenylohydrazyny, dając końcowy produkt – osazon:
Przedstawiona powyżej reakcja jest ogólną reakcją charakterystyczną dla wszystkich monosacharydów (zarówno aldoz, jak i ketoz) jak i wszystkich aldehydów i ketonów zawierających grupę hydroksylową w pozycji α względem grupy karbonylowej. W analizie chemicznej cukrów osazony odgrywają bardzo ważną rolę jako pochodne umożliwiające identyfikację cukru – określa się temperaturę topnienia otrzymanego osazonu, jak i czas jego powstawania. Należy podkreślić, iż cukry, które mogą przechodzić w siebie w procesie epimeryzacji (a więc różniące się strukturą wyłącznie w pozycjach 1 i(lub) 2) dają ten sam osazon, natomiast czas jego powstawania będzie inny:

4. Utlenianie aldoz4.1. Kwasy aldonoweUtlenianie słabymi utleniaczami (odczynnik Tollensa, odczynnik Fehlinga, woda bromowa) prowadzi do utlenienia grupy aldehydowej. Do celów preparatywnych stosuje się wodę bromową, ponieważ pozostałe środki utleniające stosuje się w środowisku zasadowym, w którym zachodzą, omówione powyżej, procesy izomeryzacji.
4.2. Kwasy aldaroweUtlenianie aldoz silniejszymi utleniaczami (najczęściej stosowany jest kwas azotowy(V)) prowadzi do powstania kwasów aldarowych, posiadających dwie grupy karboksylowe – jedna powstaje w wyniku utlenienia grupy aldehydowej, druga – w wyniku utlenienia końcowej grupy CH2OH łańcucha cukru:
4.3. Kwasy uronowe.Bardzo ważna z biochemicznego punktu widzenia jest reakcja prowadząca do utlenienia ostatniej grupy CH2OH cukru do grupy karboksylowej z jednoczesnym zachowaniem nienaruszonej grupy aldehydowej. Utlenianie takie jest możliwe do przeprowadzenia wyłącznie w warunkach reakcji enzymatycznej. Na drodze syntezy chemicznej reakcji takiej w procesie jednoetapowym nie można zrealizować. Powstałe produkty utleniania noszą nazwę kwasów uronowych (kwas glukuronowy, galakturonowy itd.):

Związki te są powszechnie występującymi składnikami polisacharydów roślinnych i zwierzęcych, z których składają się substancje śluzowate i galaretowate takie jak np. pektyny, kwasy alginowe itp. W postaci monomerycznej, w organizmach zwierzęcych ich glikozydy stanowią formę wydalania toksycznych substancji z organizmu, które jako dobrze rozpuszczalne w wodzie mogą zostać łatwo usunięte wraz z moczem:
5. Redukcja monosacharydówGrupa karbonylowa cukrów łatwo ulega redukcji do grupy OH. Najczęściej w tym celu stosowany jest NaBH4. W wyniku otrzymuje się wielowodorotlenowy alkohol – alditol (np. glucitol czy mannitol):
Należy zwrócić uwagę, że w wyniku redukcji ketoz otrzymuje się dwa diastereoizomeryczne produkty – w miejsce grupy karbonylowej ketozy powstaje nowe centrum chiralne (oznaczone na poniższym schemacie gwiazdką):
6. GlikozydyCukry łatwo reagują z alkoholami w kierunku tworzenia acetalowych połączeń (forma pierścieniowa cukru jest hemiacetalem) zwanych glikozydami:
Z reguły otrzymuje się mieszaninę anomerycznych glikozydów, ponieważ w środowisku kwaśnym szybko następuje ustalenie równowagi pomiędzy formami α i β cukru. Możliwe jest eteryfikowanie wszystkich grup OH cukru, jednakże wymaga to silniejszych środków alkilujących. Często wykonywane dla potrzeb analizy cukrów metylowanie przeprowadza się np. przy użyciu jodku metylu:

Wiązanie glikozydowe przy anomerycznym atomie tlenu (tak jak wiązanie acetalowe) jest odporne na działanie zasad i odczynników utleniających, działających w środowisku zasadowym, natomiast łatwo ulega hydrolizie w środowisku kwaśnym. Wiązania eterowe, powstałe w wyniku alkilowania dalszych grup OH są bierne w sposób charakterystyczny dla eterów:
7. Tworzenie estrówCukry traktowane bezwodnikami kwasowymi ulegają łatwo reakcji estryfikacji – acylowaniu ulegają wszystkie grupy OH cukru:
Można również otrzymywać estrowe pochodne oligo i polisacharydów, które to pochodne są wykorzystywane jako cenne tworzywa sztuczne (np. wiskoza – octan celulozy).
Glikoliza (gr. „glik” - słodki i „lysis” - rozpuszczanie) jest łańcuchem reakcji prowadzącym do przekształcenia glukozy w pirogronian. Zachodzi ona w cytoplazmie prawie wszystkich organizmów żywych i służy do wytwarzania energii zgromadzonej w ATP oraz do dostarczania elementów budujących składniki komórki. U tlenowców stanowi zaledwie wstępny etap oddychania tlenowego.
Glikoliza składa się z dziesięciu reakcji i można ją podzielić na dwie fazy. Pierwsza z nich przekształca glukozę w fruktozo-1,6-bisfosforan i wymaga zużycia dwóch cząsteczek ATP na każdą cząsteczkę cukru. W drugim etapie zachodzi rozszczepienie fruktozo-1,6-bisfosforanu na dwa związki, ulegające wzajemnym przekształceniom. Jeden z nich, aldehyd 3-fosfoglicerynowy, ulega dalszym przemianom obejmującym utlenianie i fosforylację, a reakcjom tym towarzyszy utworzenie ATP.

Rys. 1. Glikoliza
Reakcja sumaryczna glikolizy jest następująca: Glukoza + 2Pi + 2ADP + NAD+ -> 2 cząsteczki pirogronianu + 2 ATP + 2NADH + 2H+ 2H2OPodczas przekształcania glukozy w dwie cząsteczki pirogronianu powstają zatem dwie cząsteczki ATP.
Tabela. 1. Zużycie i synteza ATP podczas glikolizy
Akceptorem elektronów podczas procesu utleniania aldehydu 3-fosfoglicerynowego jest NAD+, który musi być regenerowany, aby umożliwić stałe zachodzenie przemian glikolitycznych. Organizmy tlenowe wykorzystują do tego łańcuch oddechowy, podczas którego NADH przekazuje elektrony na tlen. W warunkach beztlenowych NADH redukuje pirogronian do mleczanu lub etanolu w procesach tak zwanej fermentacji mlekowej lub alkoholowej.

W warunkach fizjologicznych rekcje glikolizy są odwracalne. Wyjątek stanowią trzy procesy katalizowane przez heksokinazę, fosfofruktokinazę i kinazę pirogronianową. Najważniejszym punktem kontrolnym glikolizy jest reakcja regulowana przez fosfofruktokinazę, aktywną w sytuacji dużego zapotrzebowania na energię lub składniki budulcowe, tzn. w obecności AMP i fruktozo-2,6-bisfosforanu, sygnalizującyego duże stężenie glukozy w wątrobie. Hamują ją natomiast duże stężenia ATP i cytrynianu. Brak aktywności tego enzymu przyczynia się do akumulowania glukozo-6-fosforanu inaktywującego heksakinazę.
Ostatni etap kontrolny to reakcja kinazy pirogronianowej allosterycznie hamowanej przez ATP i alaninę, a aktywowanej przez fruktozo-1,6-bisfosforan. Małe stężenie glukozy we krwi pobudza fosforylację tego enzymu w wątrobie i hamuje tym samym jego aktywność oraz zużycie glukozy.
Do szlaku glikolitycznego mogą wejść również inne, powszechnie występujące monosacharydy, np. fruktoza, która powstaje w wyniku rozpadu sacharozy, czyli cukru spożywczego. Jej przemiany zachodzące w wątrobie określa się mianem szlaku przemian fruktozo-1-fosforanu.
Rys. 2. Szlak przemian fruktozo-1-fosforanu
Glikolizie może ulegać również galaktoza, będąca jednym ze składników laktozy - cukru występującego w mleku. Podczas czterech kolejnych etapów ulega ona przekształceniu w glukozo-6-fosforan. Sumaryczną reakcję katalizowaną przez galaktokinazę można zapisać równaniem:
Galaktoza + ATP -> glukozo-1-fosforan + ADP + H+
Glukozo-1- fosforan ulega następnie izomeryzacji do glukozo-6-fosforanu, a przemiana ta jest katalizowana przez enzym fosfoglukomutazę.
Znana jest niewielka liczb chorób, w których stwierdzono brak aktywności enzymów glikolitycznych. Ich zasadniczym objawem jest niedokrwistość hemolityczna. W intensywnie rosnących komórkach nowotworowych glikoliza przebiega znacznie szybciej, niż tego wymaga cykl Krebsa. Produkcja pirogronianu przewyższa zdolności jego metabolizowania, co prowadzi do nadmiernego wytwarzania mleczanu i zakwaszania środowiska w guzie.
Fermentacja mlekowa to enzymatyczny rozkład bogatszych w energię substancji organicznych do uboższych związków prostych, przebiegający w warunkach beztlenowych. Proces ten przeprowadzają różne gatunki bakterii, metabolizując cukry proste i dwucukry do kwasu mlekowego i innych związków tj. kwasu octowego czy dwutlenku węgla, z zastosowaniem enzymów.
Bakterie właściwej fermentacji mlekowej, ze względu na metabolizm dzielimy na: • homofermentatywne (fermentują cukrowce wytwarzając kwas mlekowy) • fakultatywnie heterofermentatywne (podczas fermentacji produkują tylko kwas mlekowy lub dodatkowo kwas octowy, etanol i dwutlenek węgla) • heterofermentatywne (fermentują cukrowce wytwarzając obok kwasu mlekowego produkty uboczne).
Bakterie te zaliczane są do rodzajów: • Lactococcus- paciorkowce homofermentatywne (Lactococcus lactis- paciorkowiec mlekowy, Lactococcus cremoris- paciorkowiec śmietanowy),• Leuconostoc- paciorkowce heterofermentatywne (Leuconostoc citrovorum- używany jako dodatek do zakwasów przy wyrobie masła),• Lactobacillus- pałeczki zarówno homo-, jak i heterofermentatywne (Lactobacillus bulgaricus- pałeczka używana do wyrobu jogurtu, Lactobacillus viridescens- wywołuje zielenienie mięsa peklowanego oraz surowych kiełbas).
Przebieg właściwej fermentacji mlekowej można przedstawić za pomocą następującego równania sumarycznego: C6H12O6 -> 2CH3 • CHOH • COOH + 94 kJ (22,5 kcal)
Fermentacji ulegają głownie heksozy, zaś pentozy i cukry złożone nie są metabolizowane w tym procesie. Oprócz bakterii właściwej fermentacji mlekowej wyróżniamy też tzw. bakterie fermentacji pseudomlekowej (rzekomej), tj. Escherichia i Micrococus. Cechą tego rodzaju fermentacji jest to, że oprócz kwasu mlekowego powstają produkty uboczne w postaci dwutlenku węgla, kwasu octowego czy alkoholu etylowego. Zachodzi w mleku zakażonym bakteriami pseudomlekowymi obok właściwej fermentacji mlekowej, a także podczas fermentacji podłoża roślinnego (np. kwaszenie ogórków czy kapusty), powodując jednocześnie obniżenie wartości końcowej produktu. W mleku powoduje gazowanie oraz

rozrywanie skrzepu.
Znaczenie procesu fermentacjiProces fermentacji mlekowej znalazł szerokie zastosowanie w przemyśle spożywczym, szczególnie w mleczarstwie: do produkcji fermentowanych napojów mlecznych, ukwaszania śmietanki spożywczej i mleka, do dojrzewania serów podpuszczkowych. Bakterie mlekowe biorą także udział przy kwaszeniu kapusty, ogórków czy innych warzyw. Powstający tu kwas mlekowy jest czynnikiem konserwującym te produkty na długi czas. Jako pierwsze rozwijają się bakterie niewłaściwej fermentacji mlekowej, które zakwaszając środowisko stwarzają doskonałe warunki dla rozwoju szczepów właściwych. Najpierw więc pojawiają się paciorkowce, a następnie pałeczki. W przemyśle mięsnym biorą udział w przemianach mikrobiologicznych, jakie zachodzą podczas produkcji i przechowywania wędlin surowych tj. metka czy salami. Pośrednio kształtują także zapach, barwę, konsystencję czy smak tych wędlin. W przemyśle piekarskim są składnikiem zakwasów chlebowych. Zakwaszają ciasto żytnie, nadając mu charakterystyczny smak i zapach, a jednocześnie chronią przed rozwojem szkodliwych bakterii gnilnych. Bakterie fermentacji mlekowej są tez składnikiem probiotyków, czyli preparatów i produktów żywnościowych zawierających żywe kultury bakterii, które wpływają korzystnie na organizm gospodarza, poprawiając równowagę flory jelitowej. Bakterie mlekowe mogą również wywoływać negatywne skutki swojego działania, m.in. w przemyśle mleczarskim. Pewna odmiana paciorkowca mlekowego wytwarza dużo śluzu i powoduje ciągliwość mleka. Pałeczki okrężnicy mogą powodować różne wady mleka (oborowy smak i zapach, porozrywany skrzep), wczesne wzdęcia serów czy wady masła. Także w gorzelnictwie, piwowarstwie i winiarstwie mogą wywoływać niepożądane skutki. Produkty uboczne powstające w procesie hetereofermentatywnym wpływają hamująco na drożdże. Bakterie te wpływają też na kwaśnienie czy zmętnienie piwa. Mogą też rozwijać się w leżakujących winach. Heterofermentatywne paciorkowce (Leuconostoc mesenteroides) są przyczyną śluzowacenia soków dyfuzyjnych w cukrownictwie. Również w przemyśle drożdżowym mogą stać się czynnikiem niepożądanym- jeśli rozwiną się w dużej ilości podczas ich produkcji, powodują obniżenie siły pędnej i zahamowanie ich rozwoju. Proces fermentacji mlekowej uruchamiany jest także w mięśniach szkieletowych ssaków w warunkach beztlenowych. Pozwala on produkcję ATP, poprzez redukcję pirogronianu (produktu ostatniego etapu glikolizy) do mleczanu przy jednoczesnym utlenieniu NADH do NAD przy udziale dehydrogenazy mleczanowej. Kwas mlekowy jest związkiem wysokoenergetycznym, jednak fermentacja mlekowa nie może zachodzić bez końca, gdyż kwas ten jest związkiem toksycznym dla komórek mięśni. Dlatego musi on być stale z nich usuwany- jest przenoszony do wątroby, przetwarzany na glukozę, która utlenia się lub staje substratem dla oddychania tlenowego.
Fermentacja alkoholowa
Fermentacja alkoholowa to beztlenowy rozkład cukrów prowadzony głównie przez drożdże z gatunku Saccaromyces cerevisiae, według ogólnego równania ustalonego już ponad 150 lat temu przez Gay Lussaca:
C6H12O6 = 2C2H5OH + 2CO2
W wyniku tego procesu powstaje również szereg produktów ubocznych, między innymi: gliceryna, kwas bursztynowy i kwas octowy. Produktami ubocznymi fermentacji są również wyższe alkohole i estry, które mają decydujący wpływ na bukiet smakowo-zapachowy produktu. Fermentacja jest od tysięcy lat głównym sposobem zabezpieczania żywności przed psuciem. Mikroorganizmy biorące udział w fermentacji powodują zmiany chemiczne i fizyczne, w wyniku których powstający produkt może być znacznie dłużej przechowywany oraz uzyskuje nowy smak i zapach. Fermentacja alkoholowa jest wynikiem działania dużej liczby enzymów, przy czym rezultatem jej jest wydzielenie się około 50 kcal/mol rozłożonej heksozy. Szereg reakcji omawianego procesu zachodzi także w komórkach drożdży, w mięsistych dużych owocach, w nasionach okrytych twardą łupiną i w korzeniach, jeśli jest dużo wody w glebie, zachodzi w korzeniach roślin bagiennych np. ryż.
Fermentacja etanolowa, jako proces 3 etapowyW przebiegu fermentacji alkoholowej można dostrzec trzy etapy, z których dwa pierwsze zdają się być identyczne z przebiegiem fermentacji mlekowej lub z procesem glikolizy zachodzącym w mięśniach (beztlenową przemianą cukru w kwas mlekowy).
a) pierwszy etap polega na tworzeniu się z glukozy (pod wpływem ATP, odpowiednich kinaz i izomeraz) – ostatecznie 1,6-dwufosforanu fruktozyb) drugi etap polega na rozpadzie dwufosforanu fruktozy na aldehyd fosfoglicerynowy i fosfodwuhydroksyaceton a następnie wytworzenie kwasu pirogronowego

c) trzeci etap fermentacji alkoholowej to dekarboksylacja kwasu pirogronowego a następnie zredukowanie, aldehydu octowego do alkoholu etanolowego pod wpływem zredukowanego NAD (dinukleityd nikotynoamidoadeninowy)
Pirogronian +H+ => dekarboksylaza pirogronianowa=> aldehyd octowy + CO2
aldehyd octowy +NADH +H+ => dehydrogenaza alkoholowa => etanol +NAD+
Ostatecznym rezultatem tych przemian jest powstanie dwóch cząsteczek etanolu i dwóch cząsteczek CO2 z cząsteczki rozłożonej heksozy.
Organizmy fermentacji alkoholowejWiele grzybów strzępkowych (Mucom, Rhizopus) oraz bakterii (Sarcina ventriculi) wytwarza etanol, jednakże praktyczne zastosowanie mają, tylko te u których etanol jest produktem głównym.W przemyśle stosowane są różne gatunki i rasy drożdży należących do rodzaju Saccharomyces. Fermentują one cukry proste tj.: glukoza, fruktoza, mannoza także maltoza i sacharoza. Podczas intensywnego przebiegu fermentacji, komórki drożdży są rozmieszczone w całej objętości płynu.Drożdże fermentacji górnej (Saccharomyces cerevisiae) prowadzą proces w wyższej temperaturze niż drożdże fermentacji dolnej (Saccharomyces carlsbergensis), sam proces przebiega bardziej intensywnie z wydzieleniem dużej ilości CO2.
Zastosowanie fermentacji alkoholowej
Fermentacja alkoholowa cukrów w obecności drożdży należy do szybko rozwijającej się gałęzi przemysłu i jest wykorzystywana w piwowarstwie, winiarstwie, gorzelnictwie, kandyzowaniu owoców, produkcji drożdży, pieczywa, a także w ochronie środowiska. Proces ten polega na utlenianiu węglowodanów przez odpowiednie szczepy drożdży, w wyniku czego powstaje alkohol etylowy, wykorzystywany do celów spożywczych oraz przemysłowych. Surowce stosowane w przemyśle fermentacyjnym dzielą się na trzy grupy. Do pierwszej zalicza się produkty cukrowe: melasę, soki owocowe, trzcinę cukrową, do drugiej - materiały zawierające skrobię (żyto, jęczmień i pszenicę), a do trzeciej produkty pocelulozowe (drewno i ługi posiarczynowe). Drożdże zawierają enzymy, które hydrolizują disacharydy na cukry proste. Jedynie cztery heksozy ulegają fermentacji pod wpływem drożdży: D-glukoza, D-mannoza, D-fruktoza i D-galaktoza. Ostatnia ulega fermentacji z trudnością i nie pod wpływem wszystkich gatunków drożdży. Końcowy produkt fermentacji, alkohol, wydziela się przez destylację. Wydajność obliczona w stosunku fermentujących cukrów wynosi około 90%.W wyniku hydrolizy celulozy zawartej w trocinach drzewnych powstaje glukoza. Tą właśnie metodą w niektórych państwach, jak np. Szwecja, Finlandia, Kanada, czy Brazylia, z odpadów drewna produkuje się glukozę, a z niej drożdże i etanol (fermentacja alkoholowa). Obecnie technologia produkcji bioetanolu celulozowego jest stale unowocześniana. Każdy surowiec, jak np. drewno, słoma, rdzenie kolby kukurydzy, śmieci organiczne, składa się z około 60% celulozy, którą można rozłożyć do glukozy wykorzystywanej do fermentacji i produkcji etanolu. Bioetanol celulozowy znajduje zastosowanie w motoryzacji jako paliwo samodzielne E100 (100% EtOH) lub jako komponent w paliwie (E5 - 5% obj. etanolu w benzynie oraz odpowiednio E10, E22, E85 i E95). Czysty etanol oraz paliwa E95 i E85 mogą być wykorzystywane w specjalnych lub przystosowanych silnikach spalinowych. Paliwo E5 stosuje się do wszystkich

samochodów z silnikami na benzynę w niektórych krajach Europy (np. w Szwecji), E10 wykorzystuje się do silników benzynowych w USA, natomiast E22 do silników niskoprężnych w Brazylii. Mieszając benzynę z bioetanolem celulozowym, otrzymuje się mieszaninę wzbogaconą w tlen, co polepsza parametry spalania, a tym samym obniża emisję tlenku węgla do atmosfery.Głównym problemem jest duży koszt technologii przerobu celulozy, co powoduje, że koszt produkcji etanolu celulozowego jest większy o 50% niż w przypadku tradycyjnych surowców. Jednak postęp w technologii przerobu etanolu jest tak szybki, że w ciągu najbliższych kilku lat należy oczekiwać znaczącego obniżenia kosztów produkcji etanolu z celulozy.
Fermentacja masłowa
Fermentacja masłowa to beztlenowy proces enzymatycznego rozkładu sacharydów na kwas masłowy, dwutlenek węgla i wodór. Produktami są również: kwas octowy, kwas bursztynowy i etanol. Zachodzi ona w komórkach bakterii Clostridium. Fermentacja masłowa jest rozpowszechniona w przyrodzie, to m.in. rozkład resztek roślinnych np. w zbiornikach wodnych czy bagnach. Fermentacja masłowa jest podstawą przemysłu produkcji kwasu masłowego oraz bierze udział w procesie roszenia łodyg roślin włóknodajnych jak len, konopie.Charakterystyczną cechą fermentacji masłowej jest wytwarzanie dużej ilości wodoru, oraz liczne reakcje kondensacji, uwodnienia, wtórnego uwodorowania itd.
Przebieg fermentacji masłowejFermentacja masłowa ma różny przebieg w zależności od gatunku bakterii, które ją przeprowadzają jak i od odczynu środowiska. Kwas masłowy jest głównym produktem fermentacji w środowisku obojętnym, zaś w środowisku kwaśnym te same bakterie przeprowadzają fermentację acetobutanolową. Bakterie masłowe odgrywają również istotną rolę w procesie moczenia łodyg lnu i konopi, ponieważ umożliwiają, na skutek fermentacji błonnika, oddzielenie włókien przędnych od tkanki korowej i zdrewniałej.
C6H12O6 + bakterie masłowe → CH3CH2CH2COOH + 2CO2 + 2H2 + ok. 15 kcal/mol
Bakterie fermentacji masłowejFermentację masłową prowadzą bakterie charakteryzujące się następującymi cechami:
• należą do rodzaju Clostridium,• są beztlenowcami,• mają kształt laseczek,• wytwarzają przetrwalniki (endospory) nadające komórce kształt buławki lub wrzeciona,• mają zdolność do rozkładu wielocukrów na cukry proste; rozkładają m.in. skrobię, dekstryny, błonnik, pektyny,• materiały zapasowe gromadzą w postaci wielocukrów podobnych do skrobi,• naturalnym środowiskiem ich występowania jest gleba.
Bakterie fermentacji masłowej:
• Clostridium butyricum• Clostridium pasteurianum• Clostridium tyrobutyricum• Clostridium pectinovorum• Butyrivibrio fibrisolvens• Eubacterium limosum• Bacteroides melaninogenicus• Treponema phagedenis• Sarcina• Butyribacterium• Fusobacterium• Megasphera
Rola kwasu masłowegoKwas masłowy (nazwa systematyczna: kwas butanowy) jest organicznym związkiem chemicznym z grupy kwasów karboksylowych. Występuje w zjełczałym maśle, nadaje lekko gorzki posmak wielu serom. Tworzy się głównie w jelicie grubym człowieka, gdzie jego głównym źródłem są niestrawne węglowodany i oligomery heksozy o rozmaitym stopniu polimeryzacji. Do węglowodanów pochodzenia roślinnego odpornych na działanie enzymów trawiennych należą: oporna skrobia, nieskrobiowe polisacharydy, oligosacharydy (prebiotyki – inulina, oligofruktoza, dwusacharydy (laktoza, stachioza, rafinoza) i alkohole (sorbitol, mannitol)). Do puli endogennych nie trawionych węglowodanów zalicza się: mucyny, oligosacharydy mleka (istotne źródło kwasu masłowego u noworodków i niemowląt, ponieważ mleko kobiece nie zawiera kwasu masłowego), siarczan chondroityny i inne. W wyniku fermentacji bakteryjnej powstają z nich kwasy octowy, propio¬nowy i masłowy oraz w znacznie mniejszych ilościach kwasy mlekowy, sukcynylowy i kapronowy, a także dwutlenek węgla i wodór.Kwas masłowy w temperaturze pokojowej jest oleistą, bezbarwną cieczą, która jest łatwo rozpuszczalna w wodzie i większości organicznych rozpuszczalników polarnych, takich jak aceton, etanol, eter dietylowy itp.W mniejszych stężeniach, tj. od 10 do 100 ppm nadaje potrawom lekko gorzki smak, z zauważalną nutą słodką. W nieco większych stężeniach posiada ostry, intensywny, trudny do zniesienia zapach zjełczałego tłuszczu.Jego produkcja opiera się najczęściej na tzw. "masłowej" fermentacji skrobi, w której do wodnego roztworu skrobi dodaje się bakterie normalnie stosowane do fermentacji serów (Bacillus subtilis). W laboratorium można go też otrzymać poprzez przepuszczanie tlenku węgla (CO) przez mieszaninę octanu sodu (CH3COONa) i etanolanu sodu (CH3CH2ONa) w temperaturze 205°C. W wielu procesach przemysłowych kwas masłowy powstaje jako produkt uboczny,

który najczęściej wyodrębnia się poprzez strącanie go wodorotlenkiem wapnia (Ca(OH)2), gdyż sól wapniowa kwasu masłowego jest nierozpuszczalna w wodzie.Kwas masłowy stosowany jest w przemyśle od początków XX w. W dzisiejszych czasach używany jest powszechnie w przemysłach takich jak:• spożywczy,• alkoholowy,• chemiczny,• farmaceutyczny,• perfumeryjny,• tworzyw sztucznych, zwłaszcza włókien syntetycznych.
Fermentacja masłowa mimo swojego dość szerokiego zastosowania w różnych gałęziach przemysłu, wywiera również negatywny wpływ na niektóre technologie produkcyjne. Ze względu na fakt, iż bakterie z rodzaju Clostridium tworzą spory trudne do zabicia w procesach technologicznych, mogą mieć negatywny wpływ na produkcję żywności. Przetrwalniki mogą występować w przetworach mięsnych, warzywnych czy serach, powodując psucie się żywności, gnicie ziemniaków, wzdęcia niesterylnych konserw mięsnych, warzywnych, czy mleka skondensowanego a także serów.
Fermentacja propionowa to proces biochemiczny prowadzony przez liczne gatunki bakterii propionowych (rodzajPropionibacterium), charakterystyczny dla oddychania beztlenowego. Polega na przemianie cukrów, mleczanów i innych związków na kwas propionowy, kwas octowy i dwutlenek węgla. Przebieg reakcji przedstawić można za pomocą schematu:
3CH3CHOHCOOH => 2CH3CH2COOH + CH3COOH + CO2 +H20
Przebieg fermentacji propionowejRedukcja mleczanu lub pirogronianu do propionianu zachodzi w szlaku metylomalonylo-CoA. Pirogronian ulega dekarboksylacji do szczawiooctanu przy współudziale karboksylotransferazy metylomalonylo-CoA i kompleksu biotyny z dwutlenkiem węgla. Szczawiooctan z kolei jest redukowany poprzez jabłczan i fumaran do bursztynianu. Ostatni etap jest sprzężony z fosforylacją. Bursztynian najpierw ulega acetylacji przez transferazę CoA (transferaza CoA bursztybylo-CoA: propionian) do bursztybylo-CoA, który przy współudziale koenzymu B12 (cyjanokobalamina) i mutazy metylomalonylo-CoA jest przekształcany do metylomalonylo-CoA.Oderwanie dwutlenku węgla od tego związku przez karboksylotransferazę metylomalonylo-CoA prowadzi do powstania propionylo-CoA. Transferaza CoA odrywa CoA od propionylo-CoA, przekształcając go w propionian, a koenzym jest przekazywany do bursztynianu. Należy zauważyć, że w procesie wytwarzania propianianu dwie grupy (CO2 i CoA) są przekazywane z produktu do prekursora nie występując w formie wolnej.Inną ważną cechą tego szlaku jest obecność w nim trzech kofaktorów (biotyna, koenzym A i koenzym B12). Większość bakterii propionowych wytwarza kwas propionowy właśnie w tym szlaku, który występuje również u Veilonella alcalescens i Selenomonas ruminantium.
Szlak akryloilo-CoA
Clostridium propionicum, Bacteroides ruminicola i Megasphaera elsdenii wytwarzają kwas propionowy w znacznie prostszym szlaku, w którym produktem pośrednim jest pochodna kwasu akrylowego, tj. akrylo-CoA.

Kwas propionowyKwas propionowy jest naturalnym produktem przemian metabolicznych. W żwaczu krowy dziennie powstaje około 1,5 litra kwasu propionowego na skutek działalności mikroorganizmów. Kwas ten jest także produktem metabolizmu zwierzęcego, który jest szybko wykorzystywany przez zwierzęta monogastryczne. W małych stężeniach występuje w produktach mlecznych, a także w moczu i pocie.Wyraźne działanie antymikrobiologiczne kwasu propionowego powoduje, iż ma on działanie przeciw grzybom, drożdżom i bakteriom. W stosunku do bakterii Gram dodatnich jego oddziaływanie jest słabsze niż do Gram ujemnych.Zastosowanie kwasu propionowego jest dość rozpowszechnione w przemyśle konserwującym. Jego sole nadają się doskonale do konserwacji mieszanek paszowych i ich komponentów. Ilości potrzebne do tego celu zależą od wielu czynników, które mogą być uwzględnione przy ocenie stosowania środka konserwującego. Należą tu m.in. wilgotność i wielkość skażenia mikrobiologicznego konserwowanego materiału, długość okresu przechowywania i warunki magazynowania (wahania temperatur w nocy i w dzień, wilgotność względna).Do korzyści wynikających z konserwacji kwasem propionowym należą m.in.: - ograniczenie strat składników pokarmowych, - zabezpieczenie przed powstawaniem mikotoksyn, - zmniejszenie obciążenia organizmu zwierzęcia dużą ilością mikroorganizmów i związane z tym działanie przeciw biegunkom powodowanym przez mikroorganizmy, - poprawa dziennych przyrostów, - zmniejszone pobranie paszy na kg przyrostu, - dobra smakowitość paszy, - dobra sypkość przygotowanej paszy.
Kolejnym zastosowaniem jest zakiszanie surowca co na dłuższy czas poprawia tlenową trwałość kiszonki. Z tego powodu dodatek kwasu propionowego zmniejsza ryzyko wystąpienia „wtórnej fermentacji" i strat powierzchniowych, co dotyczy także powierzchni zewnętrznych kiszonek, powstających podczas wyjmowania zakiszonego surowca. Dodatek kwasu propionowego może zmniejszyć aktywność enzymatyczną zakiszanego materiału i redukuje skażenie paszy przez owady, przez co ogranicza się wystąpienie ewentualnych strat.
Bakterie fermentacji propionowejBakterie propionowe występują w żwaczu i jelicie przeżuwaczy (bydło, owce), gdzie odgrywają rolę w tworzeniu kwasów tłuszczowych, przede wszystkim kwasu propionowego i octowego w żwaczu. Ich działanie polega na przekształceniu mleczanu, powstającego podczas różnorodnych fermentacji w żwaczu, do kwasu propionowego. Rzadko występują w mleku, nie są izolowane z gleby, ani wody, natomiast istnieje możliwość ich wyizolowania z hodowli wzbogaconej, stosując podłoże zawierające kwas mlekowy i ekstrakt drożdżowy.
Do najbardziej znanych gatunków bakterii należą:• Propionibacterium freudenreichii i jego podgatunek P. shermanii,• Propionibacterium acidi-propionici,• Propionibacterium agnes,• Veilonella alcalescens (Micrococcus lactilyticus),• Clostridium propionicum,• Selenomonas,• Micromonospora.
Bakterie z rodzaju Propionibacterium to gram-dodatnie, nieruchliwe laseczki, nie tworzące endospor. Bakterie propionowe nie rosną na podłożach stałych na powietrzu z powodu braku tolerancji na tlen atmosferyczny oraz zdolności do wzrostu i tworzenia ATP w wyniku beztlenowej fermentacji. Przez długi czas uważano bakterie propionowe za obligatoryjnie fermentujące. Stwierdzono jednak, że zawierają one enzymy takie jak katalazy, a także cytochromy. Wszyscy dotychczas zbadani przedstawiciele rodzaju Propionibacterium rosną zarówno w warunkach tlenowych, jak i beztlenowych.W warunkach tlenowych Propionibacterium przeprowadzają fermentację glukozy, sacharozy, laktozy, jak również mleczanu, jabłczanu, glicerolu i wielu innych związków do kwasu propionowego.
Zastosowanie bakterii propionowychBakterie propionowe są zaliczane do grupy przemysłowych, gdyż wytwarzają cenny produkt, jakim jest kwas propionowy. Propionian wapnia i sodu, otrzymywane w procesie fermentacyjnym, są wykorzystywane do utrwalania żywności.Bakterie propionowe znajdują zastosowanie w formie zakwasów w produkcji dojrzewających serów podpuszczkowych. Wydzielający się podczas fermentacji propionowej dwutlenek węgla powoduje powstawanie prawidłowych oczek w serach, np. typu edamskiego. Bakterie Propionibacterium i fermentacja propionowa są też wykorzystywane do produkcji witaminy B12 na skalę przemysłową.
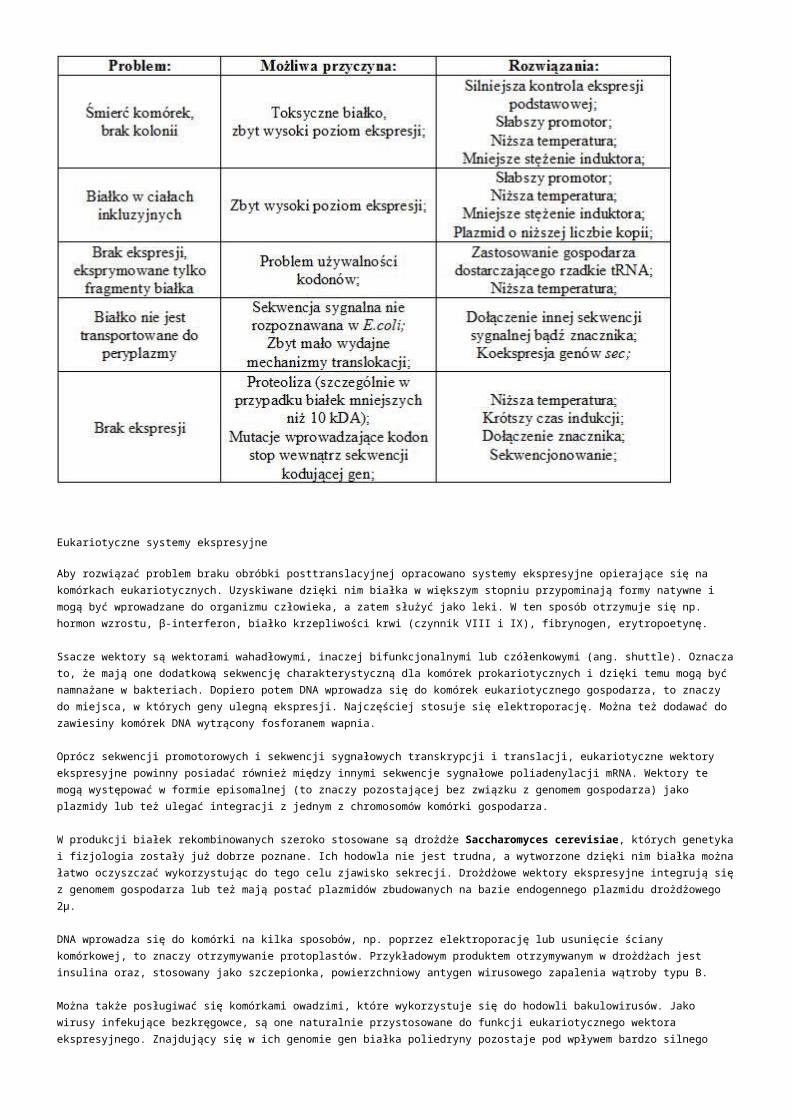
Fermentacja octowa
Fermentacja octowa to enzymatyczny proces utleniania alkoholu etylowego do kwasu octowego, katalizowana przez enzymy wytwarzane głównie przez bakterie octowe. Zachodzi także jako proces uboczny podczas innych przemian fermentacyjnych. Fermentacja octowa stanowi fundamentalny proces w przemysłowym otrzymywaniu octu (roztworu kwasu octowego). Kwas octowy stosowany jest również przy produkcji barwników, różnorodnych tworzyw sztucznych w tym m.in. sztucznego jedwabiu, środków zapachowych, rozpuszczalników i leków w tym aspiryny.
Przebieg procesu fermentacji octowejFermentacja octowa jest procesem biochemicznym w wyniku którego powstaje kwas octowy. Zdolnością wytwarzania tego kwasu, w wyniku niecałkowitego spalania alkoholu i zdobywaniem w ten sposób energii, odznaczają się przede wszystkim bakterie octowe, należące do rodzaju Acetobacter.Pod wpływem enzymów wytwarzanych przez bakterie octowe etanol utlenia się z wykorzystaniem tlenu z powietrza do kwasu octowego z wydzieleniem wody.
Fermentacja octowa przebiega w dwóch etapach:
1. CH3CH2OH + H2O → CH3COOH + 2H2 - 29,29 kJ
2. 2H2 + O2 → 2H2O +477,09Kj
Pierwsza reakcja jest endotermiczna i wymaga wydatkowania energii, wynoszącej 29,29 kJ/mol alkoholu. Druga zaś jest egzotermiczna i dostarcza 477,09 kJ/,mol. W sumie obie reakcje dostarczają 447,80kJ/mol:
CH3CH2OH + O2 → CH3COOH + H2O + 447,80kJ
U niektórych gatunków bakterii octowych występuje zjawisko nadmiernego utlenienia, czyli tzw. nadoksydacji. Mogą one np. dalej utleniać kwas octowy, zgodnie z zapisem reakcji:
CH3COOH +2 O2 → 2CO2 + 2H2O + 857,92kJ
Bakterie fermentacji octowejBakterie octowe należą do rodzaju Acetobacter. Są to pałeczki występujące pojedynczo, po dwie lub w łańcuszkach Mają skłonność do przechodzenia w postacie nieprawidłowe, czyli inwolucyjne, pod wpływem silnego zakwaszenia środowiska Wiele gatunków wytwarza otoczki śluzowe. Nie tworzą przetrwalników. Są Gram – ujemne. Są typowymi tlenowcami, mezofilami, najkorzystniejsze pH dla ich rozwoju wynosi od 4 do 6,5. Nie mają wysokich wymagań pokarmowych. Mogą utleniać alkohol etylowy do kwasu octowego.Kwas octowy, oprócz bakterii octowych, produkowany jest przez inne drobnoustroje, np. przy fermentacji alkoholowej, mlekowej (przez bakterie heterofermentatywne) i masłowej, ale powstaje on wtedy w małych ilościach, jako produkt uboczny. Istnieją również niektóre bakterie z rodzaju Clostridium (np. C. aceticum), które przenoszą wodór, odczepiony w reakcjach oksydacyjnych na dwutlenek węgla i wytwarzają w ten sposób również kwas octowy:
8H+ + 2CO2 → CHCOOH + 2H2O
OcetWe właściwej fermentacji octowej stosowanej w przemyśle w celu otrzymania metodą mikrobiologiczną octu, wykorzystywane są szybko i intensywnie kwaszące gatunki bakterii octowych, jak np. Acetobacter schutzenbachii, A. alei, A. orleanense i wiele innych. Bakterie te są Gram-dodatnie, występują w postaci krótkich, słabo ruchliwych pałeczek, skłonnych do tworzenia form „ stretto”.Ocet jest to 3,5 -15,5 % roztwór kwasu octowego z towarzyszącymi mu pozostałościami składników pochodzących z zacieru octowniczego lub wina ( przy produkcji octu winnego). Zacier octowni czy jest wodnym roztworem etanolu ( 6-12% obj.) z dodatkiem pożywki mineralnej ( i ewentualnie organicznej).Ocet posiada właściwości konserwujące żywność, działa bowiem bakteriostatycznie - hamuje rozwój drobnoustrojów niszczących żywność (drożdże, bakterie, pleśnie). Poza tym nadaje produktom spożywczym nowy bukiet smakowo-zapachowy. Jednak nadmiar octu w żywieniu jest szkodliwy. Każdy ocet niezależnie od tego, z jakiego surowca został zrobiony, zawsze pozostaje kwasem organicznym, substancją kwaśną o bardzo niskim pH. Zbyt duża ilość octu w diecie jest szkodliwa. Może dojść do zachwiania równowagi kwasowo - zasadowej co w rezultacie prowadzi do zakwaszenia organizmu. Zmniejsza się wówczas wchłanianie witamin i związków mineralnych, zwiększa natomiast ryzyko kamicy nerkowej. Duże ilości pokarmów bardzo kwaśnych stanowią też obciążenie dla nerek i wątroby. Mocny ocet stosowany bez umiaru może uszkodzić ponadto śluzówkę jamy ustnej i żołądka. Marynat z octem bezwzględnie powinny unikać małe dzieci, kobiety w ciąży, matki karmiące, oraz osoby cierpiące na wrzody żołądka, kamicę nerkową lub też uskarżające się na bóle wątroby.Głównym problemem w produkcji przemysłowej kwasu octowego jest stworzenie właściwych warunków tlenowych dla hodowli bakterii. Wyróżnia się trzy główne metody produkcyjne:
• Powierzchniową, w której bakterie (np. Acetobacter xylinum) rozwijają się w postaci kożuszka na powierzchni wina, rozlanego do płaskich naczyń lub kadzi fermentacyjnych.• Wiązaną, w której bakterie są „ związane” z materiałem nośnym, np. rozwijają się na wiórkach z drewna bukowego, wypełniających wysokie kadzie octownicze i omywanych przez cyrkulujący zacier.• Wgłębną, w której, dzięki wysoce wyspecjalizowanemu systemowi napowietrzania zacieru, rozmnażanie bakterii zachodzi nie tylko na powierzchni, ale i w

całej masie zacieru, np. w specjalnym fermentatorze, tzw. Acetatorze Fringsa.
Szkodliwe działanie bakterii octowych Bakterie octowe mogą powodować wiele wad w gotowym piwie lub winie, wywołując zmętnienie, kwaśnienie, zapach octu, kożuch na powierzchni. Mogą one także powodować psucie się marynat owocowych i warzywnych, tworząc kożuch na powierzchni i obniżając kwasowość środowiska. Bakterie octowe powodują również zakłócenia w produkcji drożdży piekarskich, ponieważ brzeczka melasowa jest dobrym środowiskiem do ich rozwoju.
Fermentacja cytrynowa
Fermentacja cytrynowa polega na wytwarzaniu kwasu cytrynowego z glukozy przy wykorzystaniu odpowiednich pleśni. Sumarycznie równanie reakcji prezentujące przebieg fermentacji przedstawić można za pomocą schematu:
3C6H12O6 + 9O2 + Aspergillus niger => 2C6H8O7 + 6CO2 + 10H2O + kcal
Kwas cytrynowyKwas cytrynowy (2-hydroksy-1,2,3-propanotrikarboksylowy) jest jednym z najważniejszych produktów otrzymywany na drodze biotechnologicznej. W formie krystalicznej kwas ten występuje w postaci bezwodnej lub jednowodnej. Jednowodna forma powstaje na drodze krystalizacji w temperaturze poniżej 36,6ºC, powyżej tej temperatury powstaje forma bezwodna. Kwas cytrynowy występuje w przyrodzie w owocach cytrusowych i ananasach. Owoce te do niedawna były głównym źródłem tego kwasu. Światowa produkcja kwasu cytrynowego przekracza 300 000 ton rocznie. Jego wykorzystanie wynika m.in. z działania konserwującego na skutek obniżania wartości pH środowiska. Jednocześnie jest związkiem nietoksycznym, o dobrych walorach smakowych i zapachowych.
Metody produkcji kwasu cytrynowegoProdukcję kwasu cytrynowego prowadzić można kilkoma metodami:
• powierzchniowa (statyczna), • wgłębna (dynamiczna), • na pożywkach stałych.
Metoda powierzchniowa (wydajność procesu 70– 80 %)W metodzie powierzchniowej w warunkach statycznych grzybnia Aspergillus niger rozwija się na powierzchni pożywki, tworząc gęstą plechę złożoną z mocno rozgałęzionych strzępek. W metodzie tej najczęściej stosowanym surowcem jest melasa buraczana. Sterylną pożywkę melasową rozlewa się do płytkich tac fermentacyjnych sporządzonych ze stali kwasoodpornej i umieszcza się w komorach z regulacją temperatury i napowietrzania. Kiedy temperatura pożywki obniży się do wartości 40°C, szczepi się ją zarodnikami pleśni zmieszanymi z węglem aktywnym jako nośnikiem. Po 24 godzinnej inkubacji pojawia się widoczna cienka grzybnia, której intensywny wzrost trwa do 48-72 godz., po czym słabnie na korzyść produkcji kwasu cytrynowego. Dojrzała grzybnia osiąga grubość 2-3 cm. Jej warstwa dolna stykająca się z podłożem jest odpowiedzialna za syntezę kwasu cytrynowego. Czas trwania procesu fermentacji wynosi 7-9 dni w temp. 30°C-34°C, wydajność procesu 70-80%. Podczas fermentacji obok kwasu cytrynowego (80%), do podłoża wydzielany jest kwas szczawiowy i kwas glukonowy oraz w śladowych ilościach kwasy z cyklu Krebsa. Po zlaniu spod grzybni przefermentowanego podłoża można ponownie wprowadzić świeżą pożywkę i wykorzystywać grzybnię kilkukrotnie. Materiał odpadowy jakim jest grzybnia pleśni (ok. 10 kg/1 m2 tac) może być wykorzystana jako źródło białka (ok. 20%).
Metoda wgłębna (wydajność procesu 80-90%)Wgłębna metoda biosyntezy kwasu cytrynowego w skali przemysłowej prowadzona jest w bioreaktorach o poj. 50-100 m3 z użyciem surowców o wyższym stopniu czystości niż w metodzie powierzchniowej, tj. cukru handlowego (sacharoza), soków cukrowniczych, mączek cukrowych, hydrolizatów skrobiowych. Rzadziej stosuje się melasę, ponieważ proces wgłębny z jej użyciem nie zapewnia odpowiedniej wydajności kwasu cytrynowego. W metodzie wgłębnej, w wyniku ciągłego mieszania, grzybnia rozwija się w całej objętości podłoża. Po 48 godzinach rozpoczyna się faza produkcji kwasu cytrynowego. Czas trwania procesu biosyntezy, do momentu wyczerpania źródła węgla, wynosi od 5 do 7 dni w temperaturze 30-32°C.
Zalety metody wgłębnej w porównaniu do powierzchniowej: • krótszy czas trwania procesu, • mniejsze zagrożenie rozwojem obcej mikroflory z uwagi na niskie pH i zamknięte bioreaktory, • możliwość wydzielania kwasu cytrynowego z pożywki na drodze krystalizacji, • mniejsze obciążenie ściekami.
Różnice miedzy metodą powierzchniową a wgłębną: W hodowli powierzchniowej obydwie fazy przebiegają w systemie jednostopniowym. W procesie wgłębnym pierwsza faza odbywa się w podłożu inokulacyjnym, o składzie różniącym się od fermentacyjnego, głównie ze względu na zmniejszoną ilość źródła węgla.
Na pożywkach stałych (wydajność procesu 20%)Metoda ta najpowszechniej rozwinęła się w Japonii. Surowcem w tej metodzie mogą być odpady przemysłu ziemniaczanego i młynarskiego np. otręby, a także wytłoki z trzciny cukrowej, wadliwa melasa buraczana czy trzcinowa, która z uwagi na niewłaściwy skład, nie mogła być wykorzystana w pozostałych metodach. Proces fermentacji prowadzi się w fermentorach tacowych (wilgotność podłoża 65-70%) w napowietrzonych komorach przez ok. 90 godz. Kwas wydziela się z podłoża poprzez ekstrakcję wodą. Metoda ta ma znaczenie marginalne, chociaż budzi też duże nadzieje ze względu na znacznie niższe obciążenie produkcji ściekami oraz wyższą tolerancję A. niger na obecność jonów metali i mikroelementów.

Mikroorganizmy fermentacji cytrynowej• szczepy pleśni Aspergillus niger, Aspergillus wentii, Pencillium luteum, Trichoderma viridae,• drożdze z rodzaju Candida (Candida lipolytica, Candida tropicalis, Candida intermedia), które syntetyzują kwas cytrynowy w obecności n-alkanów,• bakterie z rodzaju Arthrobacter.
Surowce do produkcji kwasu cytrynowegoPodłoże hodowlane odgrywa niezwykle istotną rolę w produkcji kwasu cytrynowego:
• źródłem węgla i energii są cukry (stężenie zwykle 10-22%, tj. ~ 10% dla hodowli wgłębnej, 16% dla hodowli powierzchniowej – melasa buraczana, syropy glukozowy lub fruktozowy), • kwasowość pożywki musi być wysoka (pH 2-3) – w tych warunkach kwas cytrynowy jest głównym produktem fermentacji; przy wyższych wartościach pH wytwarzane są znaczne ilości kwasu szczawiowego i glukonowego, • istotne jest wyeliminowanie soli metali (żelaza, manganu, cynku, etc.) – w tym celu dodaje się do podłoża żelazocyjanek potasu, który wytrąca jony metali, natomiast w nadmiarze działa jako inhibitor wzrostu i promotor wytwarzania kwasu, • niskie stężenie soli fosforowych i amonowych (zwykle siarczan amonu – źródło azotu) co limituje wzrost grzybni, • podłoże musi być intensywnie napowietrzane.
Zastosowanie kwasu cytrynowegoPrzemysł spożywczy: • przemysł owocowo-warzywnym, kwas cytrynowy służy jako inaktywator enzymów (oksydaz), które odpowiedzialne są za utlenianie związków fenolowych, powodujących ciemnienie owoców i warzyw, • w produkcji win, jako preparat zakwaszający i stabilizujący, • antyoksydant olejów, wiążąc jony metali (Fe, Cu, Zn) katalizujące proces jełczenia tłuszczów staje się stabilizatorem, • stabilizacji barwy, smaku i zapachu ryb, krewetek, owoców i warzyw składowanych w chłodniach.
Przemysł farmaceutyczny: • jako dodatek do tabletek, z kwaśnym węglowodanem sodu powoduje musujący efekt podczas rozpuszczania w wodzie, • jako preparat zapobiegający krzepnięciu krwi, używany w Stacjach Krwiodawstwa, • przy zatruciach ołowiem, kwas cytrynowy reagując z tym pierwiastkiem przekształca go w połączenia trudno przyswajalne przez organizm.
Przemysł kosmetyczny: • wchodzi w skład kosmetyków np. szamponów, toników, kremów, utrwalaczy do włosów.
Przemysł tekstylny: • stosowany przy wyrobie tkanin – nadaje im trwałość i niepodatność na gniecenie.
Chromatografia
Wprowadzenie do chromatografiiChromatografia jest techniką analityczną i preparatywną wykorzystującą rozdzielanie mieszanin substancji na poszczególne składniki, bądź ich grupy (frakcje), dzięki różnicom w zachowaniu się tych składników w układzie dwóch faz, w których jedna nie zmienia swego położenia (faza nieruchoma, stacjonarna), druga zaś porusza się względem pierwszej w określonym kierunku (faza ruchoma, roztwór rozwijający).
Ze względu na rodzaj dominującego mechanizmu procesu rozdziału chromatografie możemy podzielić na:a) adsorpcyjną - gdy fazą ruchomą jest ciecz lub gaz, a nieruchomą ciało stałe o właściwościach adsorbujących i odpowiednim rozdrobnieniu (np. żel krzemionkowy, tlenek glinowy, węgiel aktywny itp.). Rozdział jest powodowany różnicami współczynników adsorpcji.b) podziałową - gdy fazą ruchomą jest ciecz lub gaz, a stacjonarną ciecz zatrzymana na odpowiednim nośniku (bibuła, ziemia okrzemkowa, kulki szklane itp.). Rozdział jest wynikiem różnic współczynników podziału.c) jonowymienną - gdy fazą ruchomą jest ciecz, a stacjonarną wymieniacz jonowy. Rozdział zależy od różnic w sile wiązania składników przez wymieniacz.d) żelową - gdy fazą ruchomą jest ciecz, a nieruchomą granulowany, jednorodny spęczniały żel. Rozdział jest powodowany różnicami w zdolnościach dyfundowania do cząsteczek żelu, a więc różnicami w wielkości cząsteczek składników.
W zależności od rodzaju fazy rozdzielczej i natury eluentu czyli substancji w której rozpuszcza się badaną mieszaninę rozróżnia się następujące techniki chromatograficzne:a) chromatografia gazowa – to analityczna technika chromatograficzna, w której fazą nośną jest gaz (zazwyczaj hel, argon lub wodór). Chromatografia gazowa jest najczęściej stosowaną metodą do szybkiej analizy złożonych mieszanin związków chemicznych oraz oceny czystości tych związków.b) chromatografia cieczowa –chromatografia, w której eluentem jest ciecz. Istotą każdej chromatografii cieczowej jest rozdział analizowanej mieszaniny na poszczególne związki chemiczne poprzez przepuszczanie roztworu tej mieszaniny przez stałe lub żelowe złoża. Na skutek oddziaływań międzycząsteczkowych między związkami tworzącymi mieszaninę a złożem jedne z nich przechodzą przez złoże szybciej a inne wolniej.c) chromatografia kolumnowa - w której faza rozdzielcza jest umieszczona w specjalnej kolumnie, przez którą przepuszcza się następnie roztwór badanej mieszaniny. Przepływ roztworu przez kolumnę można wymuszać grawitacyjnie lub stosując różnicę ciśnień na wlocie i wylocie kolumny.d) chromatografia planarna - rozdział chromatograficzny jest prowadzony, gdy faza stacjonarna jest w postaci cienkiej warstwy.
Chromatografię planarną można jeszcze podzielić na:- chromatografię bibułową (PC) której podstawą działania jest podział substancji rozdzielanych miedzy fazę nieruchomą, którą stanowi woda unieruchomiona przez celulozę dzięki jej higroskopijności, oraz fazę ruchomą rozpuszczalnik organiczny lub mieszaninę rozpuszczalników, często nawet z wodą, wędrujące po bibule dzięki siłom kapilarnym. Celuloza stanowiąca nośnik dla fazy stacjonarnej (wody) ma niewielkie właściwości adsorpcyjne i prawie nie zniekształca przebiegu rozdzielania opartego na podziale substancji między wodę a wędrujący rozpuszczalnik. Jeżeli współczynniki podziału substancji naniesionych na bibułę są różne, to w trakcie wędrówki po bibule następuje ich rozdzielenie- chromatografię cienkowarstwową (TLC) - w której fazę nieruchomą nanosi się w postaci cienkiej równomiernej warstewki na płytkę szklaną, arkusz folii aluminiowej czy tworzywa sztucznego.

Chromatografia cienkowarstwowa
Chromatografia cienkowarstwowa (TLC – z ang. Thin Layer Chromatography) jest najpopularniejszą metodą szybkiego sprawdzania postępu reakcji chemicznej i czystości produktu. Może być także wykorzystywana do identyfikacji związków zarówno organicznych jak i nieorganicznych. W chromatografii cienkowarstwowej mieszanina substancji jest rozdzielana na poszczególne składniki ze względu na różnice w szybkości ich przemieszczania się na podłożu wykonanym z aktywnego adsorbenta stanowiącego fazę nieruchomą. Poszczególne składniki mieszaniny z różną siła wiążą się z polarnym adsorbentem, a następnie wymywane są z niego rozpuszczalnikiem stanowiącym fazę ruchomą. Im bardziej polarna jest badana substancja, tym silniej wiąże się z fazą stałą i tym trudniej jest wymywana przez eluent. Im bardziej polarny jest eluent, tym więcej polarnych centrów aktywnych fazy stałej jest przez niego blokowane, co zmniejsza „opory” przesuwania się eluowanych substancji.W chromatografii cienkowarstwowej faza stacjonarna jest naniesiona w postaci cienkiej warstwy o grubości od 0,1 do 0,25 mm na płytkę szklaną lub metalową lub z tworzywa sztucznego o wymiarach 200x200mm, 200x50 mm lub100x50 mm. Naniesiona warstwa musi być mechanicznie trwała i odporna na ocieranie Najczęściej do fazy stacjonarnej dodaje się 0.1 - 10 % tzw. lepiszcza, którym może być gips, skrobia, sole kwasów poliakrylowych i inne. Ze względu na detekcję substancji warstwy chromatograficzne zawierają także często trwale zaadsorbowany wskaźnik fluorescencyjny.
W chromatografii cienkowarstwowej, podobnie jak w kolumnowej, do rozdzielania są wykorzystywane te same mechanizmy oddziaływań międzycząsteczkowych, a więc adsorpcja, wymiana jonowa, podział, czy warunki układu faz odwróconych. Jednakże ciągle dominującą rolę (w przeciwieństwie do chromatografii kolumnowej) odgrywa adsorpcja, a najczęściej stosowanymi adsorbentami są:
A) Żel krzemionkowy - jest jednym z najbardziej popularnych adsorbentów stosowanych w chromatografii cienkowarstwowej. Jest on otrzymywany przez hydrolizę krzemianu sodu, jego kondensację i polimeryzację w trakcie prażenia. Jego struktura wygląda w przybliżeniu następująco:
Aktywność żelu krzemionkowego zapewniają grupy Si-OH (silanolowe) obecne na jego powierzchni. Wytwórcy kontrolują aktywność na etapie prażenia. Żel stosowany w TLC ma średnicę ziaren w zakresię 5-10 mm. Typy żelu krzemionkowego oznaczane są często w sposób następujący:- Żel krzemionkowy G – zawiera 13% gipsu jako czynnika wiążącego- Żel krzemionkowy H bez środka wiążącego- Żel krzemionkowy F254 z fluorescentem- Żel krzemionkowy UV254 z fluorescentemJako fluorescent zazwyczaj stosowany jest siarczek cynku. Żel krzemionkowy ma odczyn lekko kwaśny i może być stosowany do rozdziału sterydów, aminokwasów, węglowodorów, tłuszczów, alkaloidów itp.
B) Tlenek glinu - aktywność tlenku glinu związana jest z obecnością zarówno atomów tlenu jak i glinu. Metoda produkcji polega na kondensacji uwodnionego wodorotlenku glinu. Może on być produkowany w trzech odmianach kwasowości powierzchni: formie kwasowej, zasadowej i obojętnej. Zasadowy tlenek glinu z w/w jest najbardziej popularny.
Tlenek glinu jest używany do rozdziału sterydów, barwników, witamin i alkaloidów.
C) Celuloza jest naturalnym polisacharydem zbudowanym z cząsteczek glukozy połączonych wiązaniami beta (1,4) glikozydowymi. Płytki TLC pokryte są cząsteczkami celulozy o podobnej wielkości ziaren, co powoduje regularny przepływ rozpuszczalnika. Celuloza jest stosowana do rozdziału związków hydrofilowych, rozpuszczalnych jonów nieorganicznych i kwasów nukleinowych, które mogą zbyt silnie oddziaływać z tlenkiem glinu lub krzemionką. Oprócz tych typowych sorbentów w chromatografii cienkowarstwowej stosuje się także proszek poliamidowy, modyfikowane celulozy o właściwościach jonowymiennych oraz żele organiczne o właściwościach sit molekularnych np. Sephadex, BioGel P i inne.
D) Z nieorganicznych adsorbentów stosowanych w chromatografii cienkowarstwowej można wymienić szkło sproszkowane, krzemian wapnia, hydroksyapatyt, siarczan wapnia, tlenek cyrkonu, tlenek tytanu a nawet tlenek żelaza.
Natomiast fazę ruchomą stanowią zazwyczaj rozpuszczalniki organiczne. Powinny one być neutralne w odniesieniu do rozdzielanych substancji, wykazywać znaczną lotność par, co znacznie ułatwia osiągnięcie równowagi ciecz-para, i niską gęstość, co przyspiesza usunięcie rozpuszczalnika z powierzchni płytki. W celu określenia siły oddziaływań pomiędzy fazą ruchomą a powierzchnią nośnika wprowadzono parametr siły rozpuszczalnika e0. Jest to energia adsorpcji na jednostkę powierzchni standardowego adsorbenta (e0 jest równe zero na tlenku glinu eluowanym pentanem). Generalnie, im większe wartości e0 tym silniej rozpuszczalnik oddziałuje z powierzchnią adsorbenta i tym łatwiej cząsteczki związku będą się przemieszczały co prowadzi do większej wartości Rf (Warto zauważyć, iż im większa polarność eluenta, tym większe e0). Wartości Rf substancji w każdej fazie ruchomej będą zależne od różnicy w wartości siły rozpuszczalnika. Parametr mocy rozpuszczalnika, który może być zmierzony daje szereg elucyjny rozpuszczalników zamieszczony w tabeli poniżej.


Dobór odpowiedniego układu rozpuszczalników opiera się na zasadzie: podobny rozpuszcza się w podobnym. Zazwyczaj wykonuje się kilka prób (zmieniając polarność eluentu), dążąc do osiągnięcia wartości Rf pomiędzy sąsiednimi plamkami przynajmniej 0.1 mm. Przy czym najniższa z plamek powinna przesunąć się z linii startowej przynajmniej o kilka milimetrów. Często także korzysta się z danych literaturowych. W wielkim uproszczeniu zjawiska zachodzące w komorze chromatograficznej można przedstawić następująco: faza ruchoma dzięki siłom kapilarnym migruje wzdłuż warstwy sorbentu (fazy stacjonarnej) i w zależności od energii oddziaływań substancji z fazami wykazują one różny stopień retencji, tzn. mają różną drogę migracji i znajdują się w różnych miejscach warstwy.
Rys.1. Schematyczny obraz procesów zachodzących w czasie rozwijania chromatogramu na płytce chromatograficznej
Najbardziej powszechnym sposobem rozwijania płytek chromatograficznych jest sposób liniowy, który w najprostszej postaci wymaga tylko kontaktu jednego końca płytki (warstwy chromatograficznej) z rozpuszczalnikiem. Płytkę chromatograficzną ustawia się w pozycji pionowej bądź pod kątem w kilku mililitrach rozpuszczalnika w odpowiednim pojemniku zwanym komorą chromatograficzną Rozwijanie liniowe może być jednokierunkowe lub dwukierunkowe. Płytkę można rozwijać pojedyncze lub wielokrotne. W rozwijaniu jednokierunkowym faza ruchoma przesuwa się w jednym kierunku zaś rozwijanie dwukierunkowe polega na tym, że po pierwszym rozwinięciu płytkę suszy się w celu usunięcia rozpuszczalnika a następnie zanurza w tym samym rozpuszczalniku lub innym, ale obróconą o 90 stopni. Przy wielokrotnym rozwijaniu płytkę po każdej analizie suszy się i ponownie rozwija stosując taką samą fazę ruchomą lub inną. Efektem tego sposobu rozwijania jest zwężenie pasma stężeniowego substancji i związane z tym zwiększenie sprawności układu jak również zwiększenie czułości metody.Po rozwinięciu płytki chromatograficznej i wysuszeniu wizualizacje „plamek” na warstwie chromatograficznej można dokonać m.in. metodami fizycznymi, chemicznymi lub biologicznymi.Do metod fizycznych zaliczamy fotometrie absorpcyjną, fluorescencje, fosforescencje (w przypadku substancji znakowanych izotopami promieniotwórczymi) oraz metody radiometryczne. Metody te nie powodują uszkodzenia próbki co ma istotne znaczenie jeśli stosuje się chromatografie cienkowarstwową do celów preparatywnych. Do wizualizacji plamek można także wykorzystać metody chemiczne. Wówczas rozdzielane substancje przeprowadza się w substancje barwne stosując do tego celu reagenty chemiczne, które ulegają reakcji z wybranymi grupami funkcyjnymi. Stosunkowo często stosowanym wywoływaczem jest stężony kwas siarkowy. Proces wywoływania polega na spalaniu substancji organicznych i pojawianiu się ciemnych plam na warstwie chromatograficznej. Inne popularne wywoływacze zostały zamieszczone w tabeli 4.Natomiast biologiczne metody wywoływania chromatogramów mają zastosowanie w badaniach substancji czynnych, występujących w żywych ustrojach, np. enzymów, koenzymów, aktywatorów i inhibitorów enzymatycznych. Na przykład w badaniach przemian enzymatycznych nanosi się badany materiał na bibułę i rozpyla roztwór preparatu enzymatycznego. Inkubację przeprowadza się w termostacie w temp. 37-38°C, a po jej zakończeniu, rozwinięciu chromatogramu i wywołaniu właściwym testem, identyfikuje się produkty reakcji enzymatycznej.
Tabela 4. Roztwory stosowane do wizualizacji substancji na płytkach TLC

Wykonanie analizy TLC
Badane substancje (10-20 mg) rozpuszcza się w niewielkiej ilości rozpuszczalnika ok. 0.5 ml. Substancja powinna być dobrze rozpuszczalna w zastosowanym rozpuszczalniku, najczęściej stosuje się roztwory w metanolu, acetonie, chloroformie. W przypadku substancji bardzo polarnych np. aminokwasy do roztworu można dodać 1 kroplę wody. Przygotowuje się płytkę chromatograficzną o wymiarach zależnych od ilości substancji jakie zamierza się nałożyć na płytkę. Na przygotowanej płytce zaznacza się przy użyciu zwykłego ołówka punkty, na które następnie nanosi się roztwory substancji badanych, przy pomocy szklanej kapilary, w odległości ok. 0.5 cm od bocznej krawędzi płytki oraz ok. 0.5 cm od dolnej krawędzi. Roztwór nanosi się lekko dotykając powierzchni płytki końcem kapilary(czynność można powtórzyć kilkakrotnie). Dobrze naniesiona substancja tworzy na płytce tzw. „plamkę” o średnicy ok. 2 mm. Po nałożeniu wszystkich substancji płytkę suszy się, a następnie ostrożnie zanurza się dolną krawędzią w roztworze rozwijającym, znajdującym się w komorze chromatograficznej, tak by nie zanurzyć plamek. Komora powinna być umieszczona pod sprawnie działającym wyciągiem. Rolę komory chromatograficznej z powodzeniem spełnia zwykły słoik przykryty szkiełkiem zegarkowym. Dla lepszego nasycenia komory parami rozpuszczalnika, ścianę boczną komory wyściela się paskiem z bibuły. Czoło rozpuszczalnika powoli przemieszcza się po płytce, aż do osiągnięcia linii końcowej tj. ok. 5 mm od górnej krawędzi płytki. Wówczas płytkę ostrożnie wyjmujemy z komory. Rozpuszczalnik pozostały na płytce usuwa się za pomocą zwykłej suszarki do włosów. W przypadku związków absorbujących w UV, wizualizację plamek dokonuje się przez włożenie płytki do komory lampy UV, pod warunkiem, że adsorbent znajdujący się na płytce zawiera dodatek fluorescenta. Wtedy na żółto zabarwionej w świetle UV płytce substancje widać jako ciemne plamki. Inna metoda wizualizacji plamek polega na umieszczeniu płytki w komorze jodowej lub krótkotrwałym zanurzeniu w naczyniu z odpowiednim roztworem wywołującym a następnie wygrzaniu płytki w podwyższonej temperaturze (zazwyczaj nie wyższej niż 120 oC). Wraz z przesuwaniem się rozpuszczalnika przemieściły się nałożone substancje. Szybkość ich przemieszczania jest ich cechą indywidualną i zależy od ich powinowactwa do podłoża płytki (adsorbcja) i wymywania (desorpcja). W wyniku różnej szybkości migracji poszczególnych substancji w danych warunkach, na płytce chromatograficznej powstaje seria plamek w różnej odległości od linii startowej. Położenie plamek zaznacza się przez lekkie obrysowanie ich krawędzi ołówkiem bezpośrednio po wywołaniu. Następnie obliczamy tzw. współczynnik Rf (współczynnik retencji). Mierzy się odległość środka plamki od linii startowej (w mm) i dzieli przez dystans pomiędzy linią startową i końcową też w mm. Pomiaru i obliczenia Rf dokonuje się dla każdej plamki. Na rys.2 przedstawiono sposób pomiaru Rf.
Rys. 2. Sposób pomiaru i obliczania Rf.
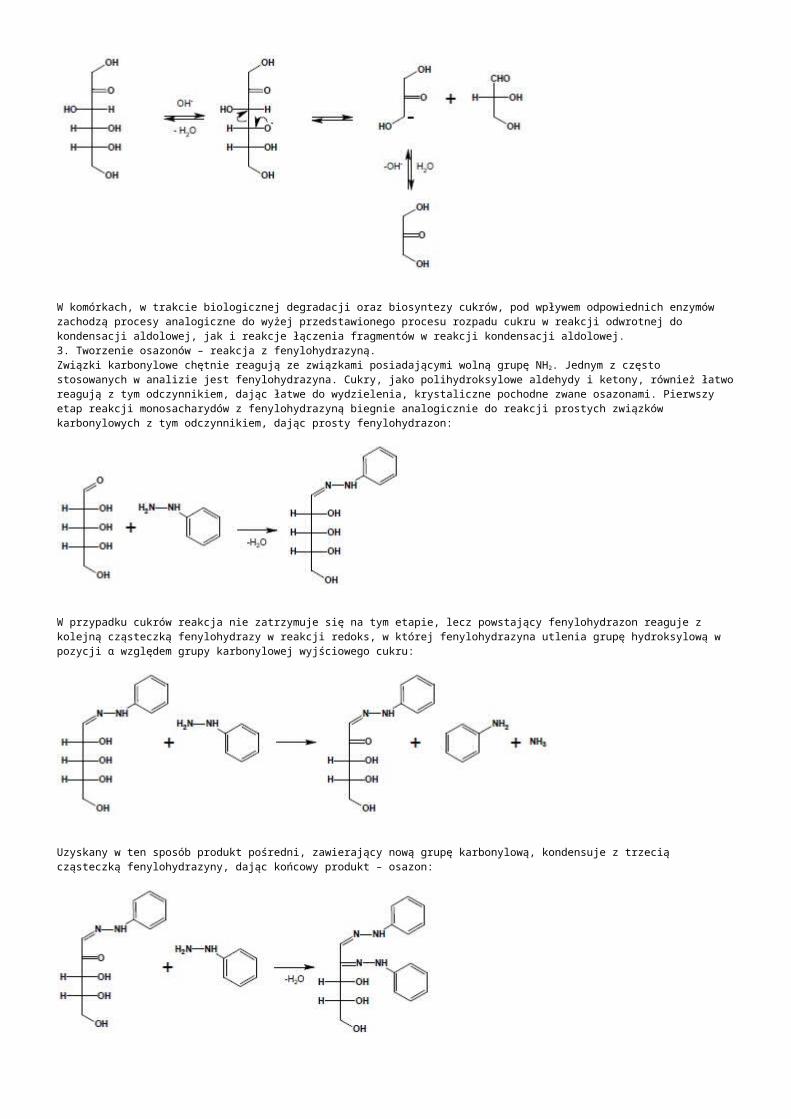
Odmianą chromatografii cienkowarstwowej (TLC) jest preparatywna chromatografia cienkowarstwowa (PTLC) którą stosuje się w celu izolacji znacznie większych ilości substancji, które następnie mogłyby być stosowane dla celów preparatywnych. Jest to możliwe gdyż stosuje się płytki o znacznie większych wymiarach niż analityczne. Najczęściej stosowane są płytki wymiarach 20x20 cm i grubości warstwy 1,0-2,0 mm. Postępowanie w przypadku stosowania preparatywnej chromatografii cienkowarstwowej jest podobne jak w przypadku stosowania konwencjonalnej czy wysokosprawnej warstwy. Na warstwę chromatograficzną nakłada się rozdzielaną mieszaninę substancji w formie paska lub plamek obok siebie. Ilość nakładanej substancji zależy od grubości warstwy żelu oraz od trudności rozdzielczych w danym układzie. Po rozwinięciu płytki w komorze zawierającej układ rozwijający i po wysuszeniu płytkę wywołuje się stosując metody nieinwazyjne jak np. wizualizację za pomocą lampy UV.
Chromatografia jako metoda oczyszczania białekPreparacja białek jest niezwykle skrupulatnym i wieloetapowym procesem. Wymaga zabiegów, w których wykorzystuje się zarówno fizyczne jak i chemiczne właściwości danej molekuły. Wydzielanie białek w stanie natywnym, o możliwie najwyższym stopniu oczyszczenia, wymagaodpowiedniego dobrania technik i warunków preparacji. Pozwala to w późniejszych etapach na dokładne określenie struktury przestrzennej oraz molekularnych mechanizmów działania pozyskiwanego białka. Już ponad kilka tysięcy białek udało się oczyścić w stanie aktywnym w oparciu o takie ich cechy jak: wielkość, rozpuszczalność, ładunek i specyficzne powinowactwo wiązania.
Na odpowiednim kroku preparacji małe cząsteczki można oddzielić od białka metodą dializy przez błonę półprzepuszczalną. Generalnie metodę tą stosuje się do odsalania roztworu białek, do wymiany w roztworze białka jednego buforu na inny, bądź do oddzielenia związków drobnocząsteczkowych od makrocząsteczek. Jednakże jeśli chcemy rozdzielić mieszaninę białek różniących się wielkością, dogodnym krokiem będzie zastosowanie filtracji żelowej (sączenia molekularnego). Rozdział następuje podczas przepływu mieszaniny białek (lub białka i soli) przez kolumny napełnione porowatym nośnikiem, zbudowanym z silnie uwodnionego polimeru typu dekstranu lub agarowy ( powszechnie stosowane żele to: Sephadex, Sepharose, Bio-gel). Podczas rozdziału małe cząsteczki wnikają do wnętrza owych ziaren żelu, podczas gdy duże nie. W rezultacie małe cząsteczki pozostają rozproszone w roztworze wodnym, znajdującym się w ziarnach żelu oraz pomiędzy nimi, natomiast duże znajdują się jedynie w roztworze wodnym wypełniającym przestrzenie pomiędzy ziarnami żelu. Objętość kolumny, która jest dostępna dla dużych cząstek jest mniejsza niż dla cząstek mały, w związku z czym większe białka przepływają przez kolumnę szybciej i wypływają jako pierwsze.
W artykule tym przyjrzymy się bliżej chromatografii jonowymiennej, jako jednej z najbardziej popularnych metod rozdziału mieszanin wieloskładnikowych. Separacja makromolekuł w chromatografii jonowymiennej polega na odwracalnej adsorpcji obdarzonych ładunkiem elektrycznym makromolekuł na złożu jonowymiennym (jonowymieniaczu). Wymieniacz jonowy jest substancją, która w zetknięciu z roztworem wymienia część swoich jonów na jony z roztworu. W jego strukturze można wyróżnić nierozpuszczalną matrycę połączoną wiązaniami kowalencyjnymi z określonymi grupami funkcyjnymi, które ulegają jonizacji i mogą w sposób odwracalny asocjować z jonami przeciwnego znaku, tzw. przeciwjonami. Ze względu na ładunek jonów które podlegają wymianie wyróżniamy dwa rodzaje jonowymieniaczy:
* Kationity (polianiony) wymieniają kationy z roztworem* Anionit (polikation) wymieniają aniony z roztworem
Kationy i aniony będące jonami wymiany, czyli tzw. przeciwjonami pochodzą z roztworu buforowego używanego do równoważenia złoża. Podczas rozdziału chromatograficznego są one zastępowane przez obdarzone ładunkiem cząsteczki białka.
Tab.1. Grupy funkcyjne jonowynieniaczy. /* Dla amin czwartorzędowych nie podano wartości pK, ponieważ niezależnie od wartości pH otoczenia grupy te zawsze zachowują swój ładunek elektryczny.
W obrębie obu grup wymieniaczy spotkać można różne grupy funkcyjne odpowiedzialne za ekspozycję odpowiedniego ładunku elektrycznego. W zależności od wartości pK, charakteryzującej grupę funkcyjną, wymieniacz zaliczany jest do jonowymieniaczy silnych bądźsłabych. I tak np. DEAE- słabo zasadowy anionit, QAE- silnie zasadowy anionit, CM- słabo kwaśny kationit, SP-; SE- słabo kwaśne kationity.
Jeśli chodzi o nośniki stosowane w tego typu chromatografii jest to głównie celuloza, Sepharose, Sephadex oraz Bio-Gel. Inną bardzo ważna cechą charakteryzującą złoże jest pojemność jonowymieniacza, czyli liczba równoważników wymienianych jonów (w miliwatach) przypadająca na 1 g złoża.
Możemy wyróżnić kilka etapów rozdziału białek w chromatografii jonowymiennej:
* Naniesienie mieszaniny białek na kolumnę chromatograficzną* Adsorpcja białek o odpowiednim ładunku* Desorpcja, czyli elucja białek związanych do złoża* Regeneracja złoża
Rys.1 Etapy chromatografii jonowymiennej. (Zmodyfikowano na podstawie: http://courses.cm.utexas.edu/jrobertus/ch339k/overheads-1/ch5_ion-exchange.jpg )
Wybór warunków chromatografii jonowymiennej:1. Jeśli znamy pI białka:• Białka zasadowe - pI tych białek przypada w zakresie pH zasadowego: stosujemy kationit (polianion). Nanosimy białko w pH niższym niż pI. Np. białko o pI 8 będzie kationem w pH np. 6,5 i zwiąże się do kationitu. Elucja: bufor o wyższym pH niż pH nanoszonego roztworu białka• Białka kwaśne – o pI przypadającym w pH kwaśnym: stosujemy anionit (polikation). Nanosimy próby w buforze o pH wyższym niż pI białka. Np. białko o pI 2,5 będzie anionem e pH 3,7 i zwiąże się do anionitu. Elucja: bufor o pH niższym niż to w którym nanoszono białko.
2. Elucje białka z kolumny można prowadzić dwoma sposobami:• Zmiana pH buforu, którym przemywamy złoże
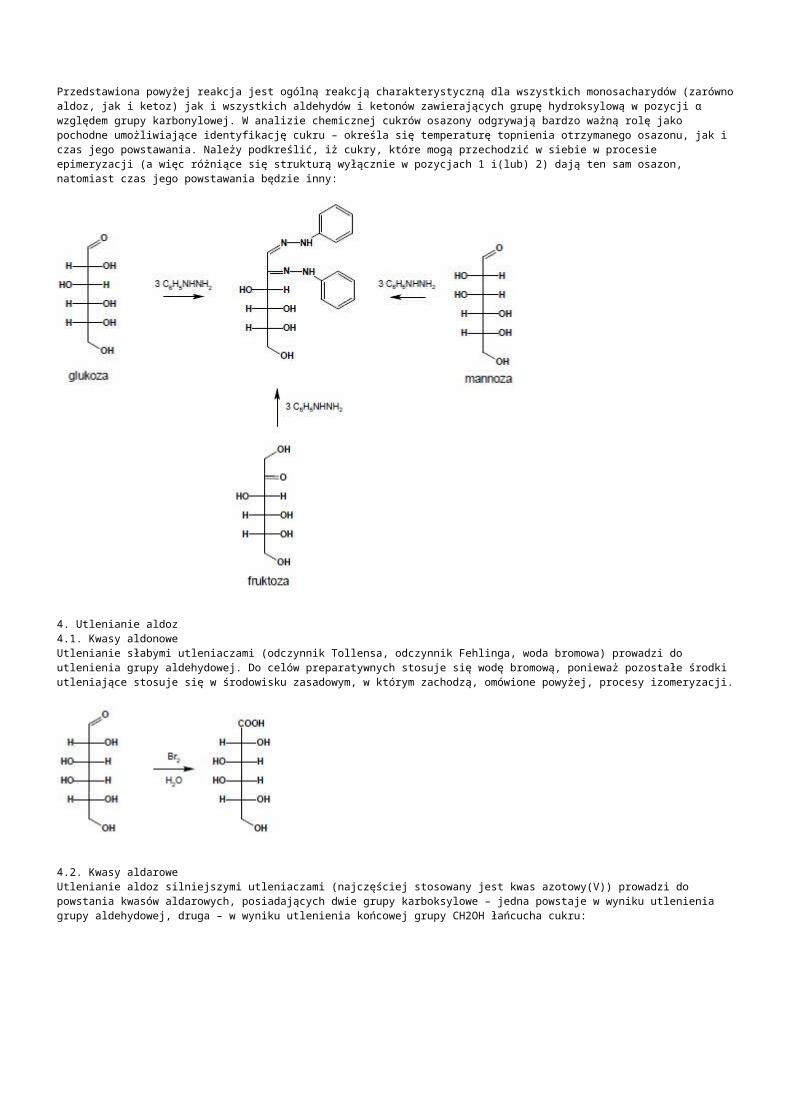
• Zwiększenie siły jonowej eluentu
Bufory jakich używamy w chromatografii jonowymiennej:1. Dla kationitów: fosforanowy, octanowy, cytrynianowi, maleinowy, weronalowi, HEPES2. Dla anionitów: Tris, bis-Tris, etanoloamina, imidazolWracając do drugiego sposobu elucji, możemy tego dokonać zwiększając stężenie wcześniej używanego buforu, bądź dodając do buforu soli obojętnej (KCl, NaCl). Mając na względzie jakoś i dokładność prowadzonego rozdziału, należy odpowiednio dobrać szybkość przepływu oraz gradient użytej soli (liniowy, schodkowy). Im wolniejszy będzie przepływ i mniej gwałtowny przyrost stężenia np. NaCl tym rozdział będzie dokładniejszy.
Podsumowanie:
Wady i zalety chromatografii jonowymiennej
Zalety:- wysoka rozdzielczość metody oraz znaczna jej selektywność,- możliwość stosowania próbek o objętościach znacznie przekraczających objętość kolumny i o bardzo niskiej koncentracji separowanych makromolekuł,- możliwość zatężania rozdzielanych makromolekuł przy pracy w technice gradientowej,- duża pojemność kolumny (wysokość co najmniej 15 cm).
Wady:- konieczność stosowania próbek w roztworze o niskiej sile jonowej i dokładnie określonej wartości pH,- rozdzielone molekuły wymywane są zwykle w eluencie o wysokiej zawartości soli, którą trzeba później usunąć innymi technikami.
Chromatografia jest techniką analityczną i preparatywną, która umożliwia rozdział mieszaniny na poszczególne składniki lub frakcje. Wykorzystuje ona różnice w zachowaniu się poszczególnych związków w układzie dwufazowym, w którym jedna z faz nie zmienia swojego położenia (faza stacjonarna), druga natomiast porusza się względem pierwszej w określonym kierunku (roztwór rozwijający). W przypadku chromatografii cieczowej (ang. Liquid Chromatography) faza ruchoma jest cieczą.
Układ chromatograficzny
Układ chromatograficzny składa się z trzech składników:
• mieszaniny poddawanej rozdzieleniu;
• fazy stacjonarnej, sorbentu, czyli stałego lub żelowego złoża, które może oddziaływać międzycząsteczkowo ze związkami tworzącymi analizowaną mieszaninę. Istotna dla rozdziału jest zarówno wielkość powierzchni sorpcyjnej, jak i stopień jej obsadzenia grupami funkcyjnymi;
• fazy ruchomej, czyli eluentu lub inaczej czynnika wymywającego. W przypadku chromatografii cieczowej jest nim ciekły rozpuszczalnik, który przenosi analizowaną substancję przez złoże. Dobry eluent powinien zapewniać wystarczającą selektywność układu chromatograficznego, a przy tym nie reagować ani z fazą stacjonarną, ani z substancjami rozdzielanymi (wyłączając zamierzone działania). Powinien on również charakteryzować się możliwie niską lepkością, niskim ciepłem parowania i nie absorbować światła w tych zakresach fali, które będą wykorzystywane przy detekcji.
Proces chromatograficzny to wielokrotna sorpcja i desorpcja substancji z fazy stacjonarnej do fazy ruchomej i odwrotnie, tzn. z fazy ruchomej do stacjonarnej. Różne związki oddziałują z każdą z faz w charakterystyczny dla siebie sposób i z tego względu przemieszczają się z różną szybkością, co umożliwia ich rozdział. Czas ich wędrówki określa się mianem retencji.
Elucja
Proces wymywania składników nazywa się elucją. Istnieją dwa sposoby jej prowadzenia:
• izokratyczna - podczas której skład eluentu jest stały;
• gradientowa - w której dzięki zmianie składu fazy ruchomej następuje wzrost siły elucyjnej (np. skład eluentu w trakcie rozdzielania zmienia się od 10% do 90% metanolu w wodzie). Faza ruchoma o odpowiedniej mocy do rozdzielenia składników o dużej retencji nie rozdziela składników wymywanych na początkowym etapie. Faza ruchoma odpowiednia dla składników o słabej retencji nie wymywa składników silnie zatrzymywanych lub też powoduje ich oddzielenie dopiero po bardzo długim czasie. Elucja gradientowa rozwiązuje ten problem dzięki systematycznemu zwiększaniu siły elucyjnej i stanowi najskuteczniejszy sposób rozwiązania tzw. ogólnego problemu elucji. Stosuje się ją w przypadkach, kiedy analizowana mieszanina składa się z substancji znacznie różniących się od siebie właściwościami sorpcyjnymi.
Wyniki analizy
Wyniki przeprowadzonej analizy przedstawia się zazwyczaj w postaci chromatogramu, czyli wykresu pików odpowiadających poszczególnym składnikom. Obraz ten uzyskuje się dzięki zastosowaniu odpowiedniego detektora.

Rys. 1. Chromatogarm elucyjny; tm - zerowy czas retencji lub inaczej czas retencji niezatrzymywanej, wyznaczany dzięki przepuszczeniu przez kolumnę substancji, która nie oddziałuje z fazą stacjonarną; tR -zredukowany czas retencji, czyli okres, jaki upływa między wprowadzeniem próbki, a pojawieniem się maksimum piku rozpatrywanego składnika; tR’ - zredukowany czas retencji, związany z przebywaniem substancji w kolumnie tylko w wyniku oddziaływania tej substancji z fazą stacjonarną (tR’ = tR - tm)
Chromatogram może dostarczać dwojakich informacji:
• jakościowych - liczba pików może oznaczać ilość substancji zawartych w badanej mieszaninie, natomiast ich lokalizacja na chromatogramie pozwala określić rodzaj, tzn. właściwości fizykochemiczne rozdzielanych substancji;
• ilościowych - wielkość rejestrowanego sygnału detektora, tzn. wysokość albo powierzchnia piku chromatograficznego jest funkcją stężenia lub też masy analitu w badanej próbce.
W przypadku chromatografii bibułowej obraz analizy chromatograficznej uzyskuje się bezpośrednio na fazie stacjonarnej, w postaci barwnych palm pochodzących od poszczególnych składników mieszaniny.
Chromatografia preparatywna i procesowa
Chromatografia cieczowa pozwala na identyfikację badanych związków. Jest ona zatem techniką analityczną o ogromnym zakresie zastosowań.
Stanowi ona również jedną z metod wyodrębniania czystych postaci poszczególnych składników mieszanin złożonych. Ze względu na ilość pozyskiwanych substancji wyróżnia się chromatografię preparatywną (dostarczającą niewielkich ilości substancji i jedynie sporadycznie) oraz chromatografię procesową lub inaczej produkcyjną (gdy ilość pozyskiwanego produktu jest znacznie większa, a proces prowadzony jest w sposób cykliczny lub ciągły). Obie znalazły zastosowanie m.in. w izolacji produktów biotechnologii (białek, polisacharydów, fosfolipidów, określonych fragmentów DNA lub RNA), produktów naturalnych pochodzenia roślinnego lub zwierzęcego, lantanowców i transuranowców, izomerów optycznych, produktów syntezy leków, składników kosmetyków, polimerów o niskim stopniu polidyspersyjności, a także użytkowych ilości substancji używanych do badań i mikrosyntez w zakresie chemii organicznej, biochemii, mikrobiologii.
W odróżnieniu od warunków analitycznych, w warunkach chromatografii preparatywnej dąży się do maksymalnego wykorzystania tzw. przeładowania kolumny, dzięki któremu proces rozdzielczy jest bardziej efektywny ekonomicznie.
Rys. 2. Rodzaje chromatografii cieczowej
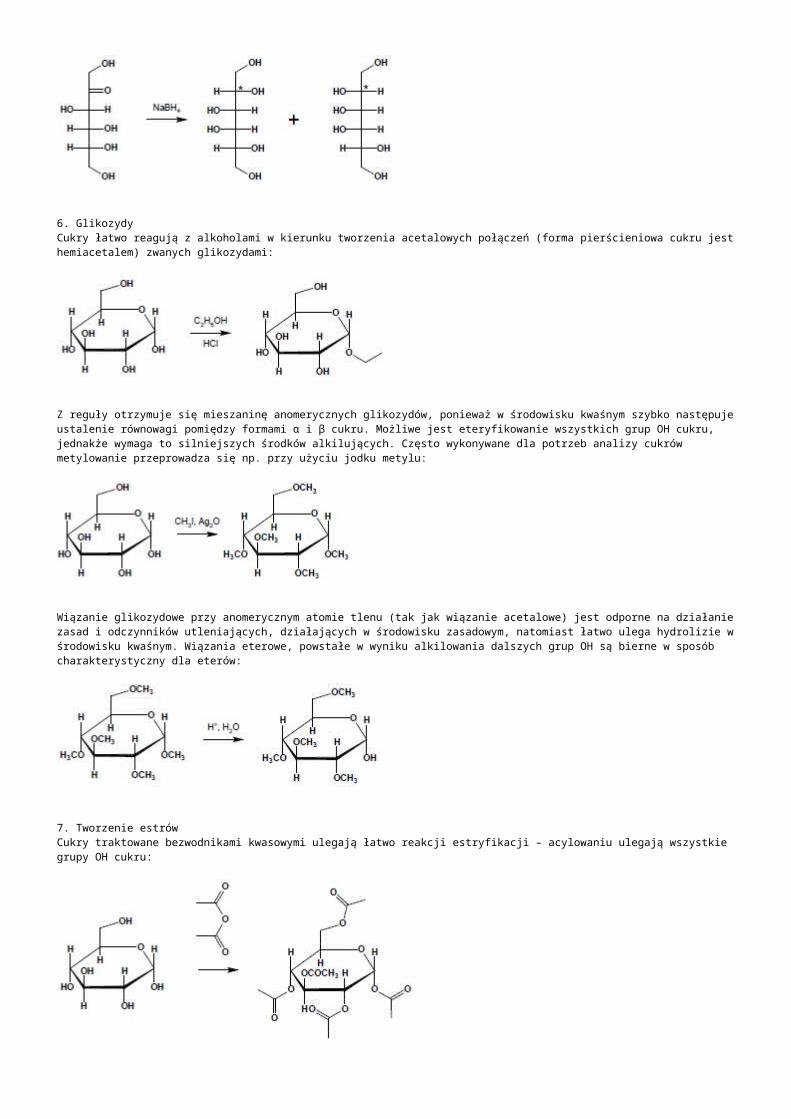
Klasyfikacja chromatografii cieczowej ze względu na różne mechanizmy rozdziału
• Układy chromatograficzne adsorpcyjne (ciecz - ciało stałe), w których wyróżnia się:
Układ faz normalnych (NP), wykorzystujący interakcje pomiędzy polarnymi grupami funkcyjnymi rozdzielanych składników, a polarnymi centrami aktywnymi znajdującymi się na powierzchni fazy stacjonarnej. Podstawowym zjawiskiem decydującym o rozdzielaniu jest adsorpcja na powierzchni bardziej polarnego sorbentu.
Od pierwszych lat stosowania chromatografii cieczowej, fazą stacjonarną były substancje nieorganiczne, takie jak krzemian magnezu, węglan magnezu, tlenek magnezu lub też węglan wapnia i inne. Fazę ruchomą stanowiły natomiast rozpuszczalniki organiczne. Rozwój wysokosprawnej chromatografii cieczowej (HPLC) doprowadził do stosowania organicznych faz stacjonarnych, których właściwości są bardzo zbliżone do adsorbentów nieorganicznych.
Chromatografię w układzie faz normalnych stosuje się jako alternatywę dla układów faz odwróconych podczas rozdzielania związków niejonowych i raczej nisko oraz średnio polarnych, które nie mogą ulec rozpuszczeniu w środowisku wodnym. Dodatkowo, układy faz normalnych posiadają zdolność do wysoce selektywnego rozdzielania izomerów strukturalnych, a w przypadku stosowania chiralnych faz stacjonarnych, również do rozdzielania izomerów optycznych.
Mimo tych zalet, w chromatografii cieczowej dominują obecnie tzw. układy faz odwróconych (RP), czyli takie, w których faza stacjonarna jest mniej polarna niż faza ruchoma. Nazwa ich wynika jedynie z historii chromatografii. Fazę ruchomą stanowi tu mieszanina wody i rozpuszczalnika organicznego. Retencja związku zależy od chemicznej natury cząsteczek substancji, eluentu i sorbentu. Substancje opuszczają kolumnę w kolejności od najbardziej polarnych do niepolarnych, a w szeregach homologicznych od nisko- do wysokocząsteczkowych. Miarą różnic retencji poszczególnych substancji jest ich selektywność hydrofobowa, chemiczna i steryczna.
• Podziałowe (ciecz-ciecz)
Analizowane substancje rozdzielane są pomiędzy dwie ciekłe, niemieszające się fazy: ruchomą i stacjonarną, osadzoną statycznie na nośniku (związaną z nim siłami przylegania), bądź też generowaną w sposób dynamiczny w czasie przepływu eluentu przez kolumnę.
Zgodnie z prawem podziału Nernsta, stosunek stężeń w obu fazach w stanie równowagi jest wielkością stałą i charakterystyczną w danych warunkach dla danej substancji. Oznacza to, że jeżeli stężenie badanego składnika w fazie ruchomej jest zbyt duże, substancja ta przechodzi do fazy ciekłej stacjonarnej i odwrotnie, gdy faza ruchoma ma mniejsze stężenie, składnik częściowo przechodzi z fazy stacjonarnej do fazy ruchomej. Proces ten trwa aż do momentu ustalenia się odpowiedniego stosunku stężeń w obu fazach.
• Chromatografię żelową, wykluczenia sterycznego, sitową
Ten typ chromatografii eliminuje, o ile to możliwe, zjawisko sorpcji. Składniki mieszaniny rozdzielane są na tzw. sitach molekularnych, czyli ziarnach tworzących pory międzyziarnowe przez które płynie ciecz. Zawierają one również mikropory wewnętrzne, w których przestrzeni eluent praktycznie nie płynie. W ten sposób związki rozdzielane są ze względu na swoją wielkość i masę molową.
• Chromatografię jonowymienną
Ten typ chromatografii korzysta z różnic w ładunku wypadkowym odmiennych cząstek, a rozdział mieszaniny następuje w wyniku odwracalnej wymiany jonów z fazą stacjonarną. Cząsteczki o małej gęstości wypadkowych ładunków dodatnich wypływają z kolumny jako pierwsze, a cząsteczki o większej gęstości ładunków - dopiero w następnej kolejności.
Białka naładowane ujemnie (anionowe) można rozdzielać chromatograficznie na kolumnach wypełnionych dodatnio naładowaną dietyloaminoetylocelulozą (DEAE-celulozą). Natomiast białka naładowane dodatnio można rozdzielać na kolumnie z ujemnie naładowaną karboksymetyocelulozą (CM – celulozą).
Do tej grupy chromatografii zalicza się również tzw. technikę wykluczania jonowego, w której mechanizm rozdzielania wykorzystuje zjawisko powstawania tzw. membrany Donnana.
Alternatywą dla chromatografii jonowymiennej jest chromatografia par jonowych, stosowana głównie w układach faz odwróconych, gdy jonowe, albo bardzo silnie polarne fragmenty cząsteczek substancji rozdzielanych są maskowane odpowiednim organicznym przeciwjonem. Powstający w ten sposób kompleks zachowuje się zatem jak substancja neutralna.
• Chromatografię powinowactwa
Technika ta wykorzystuje duże powinowactwo wielu białek do specyficznych grup chemicznych (np. oddziaływanie enzym - koenzym) albo innego typu oddziaływania zachodzące pomiędzy fazą stacjonarną, a rozdzielanymi składnikami (np. tzw. chromatografia “metalopowinowactwa” - oddziaływania Ni...S).
Można ją stosować bez kolumny, tzn. wykonywać rozdzielenie w naczyniu laboratoryjnym albo bezpośrednio w bioreaktorze.

Rys. 3. Chromatografia powinowactwa konkanawaliny A (kolor żółty) na stałym nośniku zawierającym kowalencyjnie przyłączone reszty glukozy. Związane białko można uwolnić dodając stężony roztwór glukozy
Podział chromatografii cieczowej ze względu na sposób prowadzenia rozdziału
Chromatografię cieczową można również podzielić na:
• kolumnową, w której adsorbent stanowi wypełnienie kolumny chromatograficznej.
Składniki mieszaniny przemieszczają się ku dołowi z szybkością zależną od siły oddziaływań ich cząsteczek z adsorbentem. Poszczególne związki wędrują z różną szybkością i dzięki temu można je kolejno odbierać na dole kolumny.
Przepływ fazy ruchomej może być grawitacyjny lub wymuszony za pomocą ciśnienia pod jakim eluent jest wprowadzany do kolumny (ang. High Performance Liquid Chromatography - HPLC, wysokosprawna chromatografia cieczowa).
Rys. 4. Kolumna chromatograficzna
• planarną, gdzie faza stacjonarna jest uformowana w postaci płytki lub płasko rozprowadzona na odpowiednim nośniku.
Zalicza się tutaj chromatografię bibułową, w której rozdział substancji wykonuje się na odpowiednio dobranej bibule oraz cienkowarstwową (z ang. Thin Layer Chromatgraphy, TLC), w której sorbent jest osadzony na podłożu, tzn. folii lub płytki wykonanej ze szkła, aluminium lub tworzywa sztucznego.
Chromatografię cienkowarstwową po raz pierwszy wykorzystano w 1938 roku do oznaczania zanieczyszczeń leków. Jej znaczny rozwój nastąpił po 1956 roku, dzięki odkryciu tzw. urządzenia Stahla, umożliwiającego w prosty sposób przygotowanie warstw chromatograficznych na płytkach szklanych. Jego odkrywca był również twórcą podstaw teoretycznych i metodycznych tego typu chromatografii.
Główną zaletą TLC jest możliwość przechowywania płytek z rozdzielonymi substancjami. Pozwala ona również określić stopień rozdzielenia substancji na każdym etapie rozwijania chromatografu, a co za tym idzie przerwać ten proces w dowolnym momencie. W przypadku kolumny chromatograficznej zarówno detekcja składników, jak i ocena stopnia ich rozdzielenia możliwe są dopiero, gdy związki te opuszczą kolumnę.
Znaczącym ulepszeniem okazało się zastosowanie tzw. densytometrów, czyli detektorów służących do automatycznego dozowania próbki oraz komputerowego opracowywania wyników. Możliwości rozdzielcze tej techniki zwiększyło dodatkowo wprowadzenie tzw. wysokosprawnej chromatografii cienkowarstwowej (ang. High Performance Thin Layer Chromatography - HPTLC).

Tab.1. Porównanie chromatografii cienkowarstwowej z wymuszonym przepływem fazy ruchomej (konwencjonalnej) oraz wysokosprawnej
Rys. 5. Schematyczny obraz procesów zachodzących w czasie rozwijania chromatogramu na płytce chromatograficznej w warunkach konwencjonalnej chromatografii cienkowarstwowej (A) oraz schemat chromatografu cienkowarstwowego mieszaniny substancji (B)
Postęp rozwoju chromatografii zarówno cienkowarstwowej jak i kolumnowej wiąże się głównie z zastosowaniem mniejszych ziaren fazy stałej, dostępnością gotowych warstw chromatograficznych oraz instrumentalizacją
Sensory chemiczne są to urządzenia przetwarzające informację chemiczną na parametry nadające się do obróbki. Jednym z ich rodzajów są tak zwane biosensory, w których część receptorową tworzy materiał biologiczny. Przedrostek „-bio” odnosi się zatem do rodzaju czujnika, nie zaś do typu oznaczanej substancji. Biosensory cechują się dużą selektywnością i powinowactwem względem badanego związku i przez to stanowią ważny krok w rozwoju metod analitycznych.
Tak się zaczęło
Początek badań nad biosensorami datuje się na rok 1962, w którym Clark i Lyons opisali tak zwaną elektrodę enzymatyczną. Ten pierwszy bioczujnik służył do wykrywania glukozy i wykorzystywał oksydazę glukozową immobilizowaną na platynowej elektrodzie.
Budowa sensorów
W skład każdego biosensora wchodzą dwa podstawowe elementy:
• warstwa analitycznie aktywna, czyli część receptorowa lub inaczej element czuły - tutaj następuje proces rozpoznania międzymolekularnego receptor-analit, w trakcie którego wytworzony zostaje sygnał chemiczny lub fizykochemiczny, np. dotyczący stężenia jakiegoś konkretnego składnika próbki. W

biosensorach warstwę analitycznie aktywną tworzy materiał biologiczny, najczęściej unieruchomiony na odpowiednim nośniku. Jego rodzaj wpływa na selektywność i czułość względem badanej substancji. Ważne jest, aby część receptorowa była stabilna w warunkach prowadzonego eksperymentu. Istotna jest także gęstość upakowania sond na powierzchni przetwornika, np. w przypadku biosensora opartego na reakcji hybrydyzacji, zbyt duża gęstość może utrudniać proces rozpoznania, natomiast zbyt luźna wzmaga ryzyko wystąpienia niespecyficznej adsorbcji na powierzchni elektrody;
• element przetwornikowy (transducer) - dzięki niemu sygnał, otrzymany w warstwie analitycznie aktywne,j zostaje zamieniony na parametr mierzalny, tzn. nadający się do obróbki. Wynik pomiaru może być przedstawiany w formie analogowej, odejmowany od sygnału odniesienia, a także przetworzony na sygnał cyfrowy i opracowywany statystycznie.
Rys.1. Ogólny schemat budowy biosensora.
Parametry użytkowe biosensorów
O praktycznej przydatności biosensorów decydują:
• dokładność, • powtarzalność wyników; • czułość, powinowactwo – wartość emitowanego sygnału na jednostkę stężenia; • zakres dynamiczny – zakres stężeń, w których czułość jest większa od zera; • selektywność, czyli specyficzność względem badanej substancji – zdolność sensora do pomiaru stężenia jednego chemicznego składnika w obecności innych; • czas odpowiedzi lub inaczej szybkość czasu detekcji; • czas życia, czyli trwałość sensora, uzależniona zarówno od jego rodzaju, jak i trybu stosowania i przechowywania.
Podział biosensorów ze względu na rodzaj przetwornika
Rodzaj zastosowanego przetwornika warunkuje rodzaj odbieranego sygnału i wpływa na czułość sensora. Wyróżnia się m.in. bioczujniki:
• optyczne, wykorzystujące pomiary świetlne dokonywane w aparatach optycznych, takich jak np. spektrofotometry, fluorometry, lumenometry oraz fotometry; • piezoelektryczne, które przetwarzają zmiany kształtu na napięcia elektryczne i wskazują najczęściej na zmiany masy, np. mikroorganizmów; • termometryczne; • magnetyczne; • elektrochemiczne - potencjometryczne lub amperometryczne.
Podział biosensorów ze względu na element czuły
Dobór części czułej bioreceptora warunkowany jest rodzajem identyfikowanego analitu. Ze względu na typ użytego materiału biologicznego wyróżnia się:
• biosensory enzymatyczne Na przykład biosensory bazujące na tyrozynazie lub peroksydazie i stosowane do monitoringu związków fenolowych, hydrolazy fosforoorganicznej oraz cholinoesterazy (fosfoorganiczne pestycydy są inhibitorami cholinoesterazy). Ich wadą jest słaba długoterminowa aktywność. Alternatywą mogą być biosensory jednorazowe.
• immunosensory

W tym przypadku warstwę czułą tworzą przeciwciała monoklonalne (tzn. pochodzące z jednego klonu limfocytu B i w związku z tym wykazujące w jednakową swoistość względem danego antygenu) lub poliklonalne (takie, które różnią się miedzy sobą składem łańcuchów ciężkich i lekkich, i przez to wykazują różne powinowactwo względem epitopów danego antygenu).
Można je wykorzystywać do bezpośredniej detekcji lub też w systemach, w których wiązany antygen konkuruje z analitem o miejsce na przeciwciele.
W produkcji biosensorów istotna jest stabilność użytych immunoglobulin, które powinny być odporne na zmiany środowiska, takie jak np. wysoka lub niska temperatura. Jest to niezbędne zwłaszcza w przypadku przeprowadzania analiz ciągłych.
Wadą tego typu biosensorów jest stosunkowo duża cena, wynikająca z wysokich kosztów produkcji odpowiednich przeciwciał. Dodatkowo nie mogą być one wykorzystywane w przypadku zbyt małych molekuł lub cząstek z niską immunogennością i wysoką toksycznością.
• biosensory wykorzystujące kwasy nukleinowe Jest to stosunkowo nowa grupa biosensorów, która szybko znalazła zastosowanie podczas wykrywania mutacji w sekwencjach genetycznych, zanieczyszczeń w przemyśle farmaceutycznym oraz w badaniach nad uwolnieniem genetycznie zmodyfikowanych mikroorganizmów do środowiska.
Tego typu biosensory mogą zawierać pojedyncze lub dwuniciowe łańcuchy DNA lub RNA, hybrydyzujące ze specyficznymi dla siebie sekwencjami komplementarnymi, niskocząsteczkowymi związkami chemicznymi (toksyny, leki) lub też białkami wykazującymi powinowactwo do kwasów nukleinowych.
Istnieją również typy biosensorów, w których elementem czułym jest kwas peptydonukleinowy (ang. Peptide Nucleic Acid, PNA) unieruchomiony na nośniku za pomocą wiązań peptydowych lub za pośrednictwem cząsteczek łącznikowych. W odróżnieniu od DNA i jego analogów, PNA nie zawiera pentoz ani grup fosforanowych, jednak mimo to jest w stanie naśladować DNA i w podobny sposób łączyć się z komplementarnymi sekwencjami. Może się on również łączyć się z odpowiednimi cząsteczkami RNA.
Rys. 2. Hybrydyzacja cząsteczki analitu z komplementarną sondą biosensora.
Najbardziej pożądane są kilkunastonukleotydowe sekwencje DNA i RNA, wykazujące wysoką specyficzność i powinowactwo względem wiązanego analitu. Nazywa się je aptamerami, od łacińskiego słowa aptus, to znaczy przyczepiony, dopasowany.
Ich zastosowanie pozwala otrzymać sondy wykazujące powinowactwo do praktycznie każdego badanego związku. Siła z jaką aptamery wiążą się z rozpoznawaną molekułą, jest porównywalna z oddziaływaniami zachodzącymi między przeciwciałem monoklonalnym i jego specyficznym antygenem. Tak wysoka stabilność kompleksu warunkowana jest niezwykłym dopasowaniem przestrzennej struktury aptameru i analitu.
Ze względu na małe rozmiary aptamery mogą być również gęsto upakowane na powierzchni biosensora. Ponadto, cechują się one dużą stabilnością, którą można dodatkowo zwiększyć stosując chemicznie modyfikowane nukleotydy. Dzięki temu, przez wiele cykli, łańcuchy nie tracą swoich właściwości, nawet wtedy, kiedy są poddawane termicznej denaturacji oraz renaturacji.
Aptamery nie wymagają stosowania kultur komórkowych ani organizmów zwierzęcych i w związku z tym można je uzyskiwać w dużych ilościach oraz stosunkowo małym kosztem. Sekwencje o pożądanych właściwościach fizycznych i chemicznych selekcjonuje w warunkach in vitro dzięki tzw. metodzie SELEX (ang. systematic evolution of ligands by exponential enrichment).

Rys. 3. Poszukiwanie aptameru RNA w bibliotece RNA za pomocą metody SELEX.
• translacyjne biosensory Łącząc aptamer RNA z domeną regulacyjną RNA można, za pomocą właściwego ligandu, regulować ekspresję danego genu. Są to tzw. biosensory translacyjne.
Połączenie aptameru z ligandem wywołuje zmiany konformacyjne czujnika RNA. Prowadzi to do uwolnienia domeny antysensowej (uaktywnienie sekwencji) lub jej związania w stabilną pętlę (unieczynnienie). Próg wykrywalności reguluje się za pomocą zmian w sekwencji RNA.
Do produkcji tego typu sensorów można stosować aptamery specyficzne dla różnych ligandów oraz sekwencje antysensowne dla różnych genów.
• biosensory receptoroweTen typ biosensorów składa się z błonowych białek receptorowych wyzwalających białkowe kaskady sygnałowe.
Stosowane receptory mogą być konstruowane de novo na poziomie molekularnym, tak, aby wiązały określony ligand, np. kancerogeny, trinitrotoluen TNT, medycznie istotne metabolity (L-lactate) i związki związane z kondycja psychiczną (serotonina).
• biosensory wykorzystujące wirusy Do konstruowania tego typu biosensorów najczęściej stosuje się bakteriofagi, czyli wirusy infekujące bakterie. Działają one z wysoką selektywnością i czułością, a ponadto cechują się dobrą chemiczną i termiczną stabilnością. Mogą być one również łączone z innymi biomolekułami i nanomateriałami oraz immobilizowane na powierzchni przetwornika urządzenia analitycznego.
Wirusy o powinowactwie do konkretnego ligandu odnajduje się dzięki tzw. fagowej ekspresji peptydów (ang. phage display). Specyficzność względem badanego analitu można również uzyskać dzięki odpowiednim modyfikacjom.
W produkcji biosensorów wykorzystuje się zarówno fagi nielityczne (takie jak M13 i fd), stosowane jako substytuty przeciwciał, jak i fagi lityczne (T4 lub T7). Te ostatnie niszczą komórki gospodarza u uwalniają zawarte w niej markerów, np. enzymy. Ich zastosowanie umożliwia wykrywanie specyficznych szczepów bakterii.
• biosensory wykorzystujące mikroorganizmyPierwsze biosensory tego typu wykorzystywano do detekcji BZT, czyli biochemicznego zapotrzebowania tlenu (ang. biochemical oxygen demand, B.O.D.). Obecnie stosuje się je również do pomiarów związków toksycznych zawartych w środowisku.
Ich główną zaletą jest niska cena i to, że są łatwe do skonstruowania.
Obecnie wykorzystuje się również mikroorganizmy modyfikowane genetycznie i posiadające na swojej powierzchni odpowiednie detektory, takie jak np. pojedyncze łańcuchy fragmentów przeciwciał (scFvs). Ich użycie pozwala ominąć kosztowny etap oczyszczania enzymów.
• biosensory transkrypcyjneTranskrypcja jest pierwszym etapem ekspresji genów, a jej regulacja stanowi jedną z metod odpowiedzi na zmieniające się warunki środowiska. Maszyneria

transkrypcyjna, tj. geny ulegające ekspresji, ich promotory, polimeraza RNA i czynniki transkrypcyjne, mogą być wykorzystywane do tworzenia odpowiednich biosensorów.
Ten typ czujników wykorzystuje m.in. promotory, których aktywność warunkowana jest zmianami środowiska, tzn. zmiany w ekspresji genów zachodzą w sposób zależny od obecności induktora, czyli odpowiedniego ligandu. Jest to tzw. system indukowanej ekspresji, którą można uwidaczniać za pomocą ekspresji fluorescencyjnego reportera lub też zachodzących zmian fenotypowych, np. formowanie biofilmu, apoptoza.
Niekiedy pojedyncze sygnały są niewystarczające. W takich wypadkach wykorzystuje się tzw. bramki AND, które przekształcają złożone sygnały i pozwalają na odpowiednie dobranie wrażliwości biosensora.
Rys.4. Przykład biosensora transkrypcyjnego. Na przedstawionym przykładzie transkrypcyjna bramka logiczna AND (układ realizujący fizycznie prostą funkcję logiczną, tutaj koniunkcję, iloczyn), została zaprojektowana w taki sposób, aby odbierać i przekazywać jedynie równoczesną obecność dwóch analitów: kwasu salicylowego i arabinozy. Na jednym wejściu do bramki znajduje się promotor, który w odpowiedzi na pojedynczy sygnał, tzn. obecność arabinozy, aktywuje transkrypcję genów T7 RNA polimeraz. Gen ten zawiera wewnętrznie kodowany kodon amber (stop), blokujący translację transkryptu. Kluczem do odblokowania translacji jest aktywacja drugiego wejścia. Innymi słowy, polimeraza T7RNA może ulegać dokładnej ekspresji tylko w obecności dwóch sygnałów środowiskowych.
Zastosowanie biosensorów
Biosensory pozwalają na identyfikację obcego DNA, białek, szeregu metabolitów i skażeń chemicznych (toksyny) oraz biologicznych (wirusy, bakterie).
Atrakcyjność stosowania bioczujników wynika głównie z ich wysokiej czułości i selektywności względem badanej substancji. Dzięki tej właściwości pozwalają one w sposób prosty i szybki oznaczyć interesujący nas składnik nawet w złożonej mieszaninie, np. w produkcie spożywczym.
Bioczujniki mogą być stosowane zwykle przez kilka tygodni, a nawet miesięcy i wykonywać w tym czasie setki pomiarów. Obniża to znacznie koszty analizy. Dokonywane pomiary nie wymagają również pracochłonnego przygotowywania badanej próby. Dzięki temu biosensory stanowią niezastąpione narzędzie wykorzystywane podczas monitoringu środowiska, tzn. obserwacji wybranych cech środowiska z wykorzystaniem ciągłych lub stale powtarzanych pomiarów.
Stosowane są one m.in. do określania ilości metali ciężkich, pestycydów, BZT, azotu, fosforu, lotnych substancji organicznych, patogenów, substancji rakotwórczych i toksycznych (pestycydów, mykotoksyn, antybiotyków) w powietrzu, wodzie i glebie. Ich użycie uzupełnia dane uzyskane z analiz laboratoryjnych, a analizy te są niezbędne do zapewnienia bezpieczeństwa i zdrowia oraz do kontrolowania skuteczności fizycznej, chemicznej i biologicznej kontroli zanieczyszczeń.
Biosensory wykorzystuje się również do kontroli żywności m.in. przy identyfikacji organizmów genetycznie zmodyfikowanych.

Ponadto umożliwiają one wykrywanie śladowych ilości materiałów wybuchowych używanych w bioterroryzmie.
Rys. 5. Przykładowe zastosowanie biosensorów.
Dzięki miniaturyzacji, biosensory umożliwiają uzyskanie dużej czułości przy minimalnej ingerencji w funkcjonowanie komórki. Zastosowanie w żywych komórkach biosensorów bazujących na fluorescencyjnych białkach pozwala na wizualizowanie procesów zachodzących w komórkach w czasie rzeczywistym (ang. real time).
Pomaga to zrozumieć, w jaki sposób komórka odpowiada na odbierane sygnały środowiskowe i jest bardzo pomocne m.in. przy poszukiwaniu nowych celów terapeutycznych w walce z niektórymi chorobami. Tego typu biosensory uwidaczniają aktywność kinaz i fosfataz, dynamikę przekaźników drugorzędowych i metabolitów takich jak glukoza, a także wizualizują sposoby łączenia się receptorów i efektorów. Dzięki nim poznano wiele mechanizmów czasoprzestrzennej regulacji zdarzeń zachodzących wewnątrz sieci sygnalnych.
Apoptoza to jeden z rodzajów śmierci komórkowej. Jej nazwa pochodzi z języka greckiego i oznacza opadanie płatków kwiatów, bądź liści. Pierwsza część wyrazu – „apo” nawiązuje do pozornego wyciekania komórek do przestrzeni międzykomórkowej, natomiast „ptosis” dotyczy usunięcia komórek z tkanki.

Rys. 1. Rodzaje śmierci komórkowej
Apoptoza jest niezbędna do zachowania homeostazy tkankowej. Jej biologiczna rola polega na eliminowaniu zużytych i uszkodzonych komórek, które mogłyby być szkodliwe dla organizmu. Do samobójczej śmierci zdolna jest większość komórek, jednak łatwość z jaką wchodzą one na drogę apoptozy zależy od ich typu i stopnia rozwoju. Proces ten zaobserwowano zarówno podczas embriogenezy (kształtowanie układu nerwowego ssaków, tworzenie komórek soczewki oka) jak i u organizmów dorosłych (np. segregacja dojrzewających limfocytów T w grasicy).
Zmiany zachodzące w komórce podczas apoptozy
Zmiany morfologiczne:- zmiana symetrii błony komórkowej przy zachowaniu jej integralności; - odwodnienie i zmiana kształtu komórki; - kondensacja chromatyny oraz jej agregacja przy nukleoplazmie; - tworzenie się pęcherzyków poprzez otoczanie błoną komórkową fragmentującej cytoplazmy (ang. blebbing); - odpączkowywanie ciałek apoptotycznych (odrywanych od komórki macierzystej pęcherzyków).
Zmiany biochemiczne: - translokacja fosfatydyloseryny PS do zewnętrznej warstwy błony komórkowej; - aktywacja fosfataz i kinaz białkowych; - wydzielenie cytochromu c i czynnika AIF (ang. apoptosis inducing factor) z mitochondriów; - aktywacja kaskady kaspaz; - cięcie DNA na mono- i oligonukleosomy (powstaje drabinka nukleosomowa) przez endonukleazę CAD (ang. caspase-activated deoxyribonuclease); - zużycie energii zgromadzonej w ATP.
Odpowiedź tkankowa indukowana jest przez: a) zmiany fizjologiczne (w dopływie hormonów do komórki, spadek poziomu czynników troficznych i wzrostowych); b) działanie toksyn (zewnętrznych czynników uszkadzających i toksyn komórkowych: wolnych rodników, ekscytotoksyn, jonów wapnia); - zachodzi w pojedynczych komórkach rozsianych w prawidłowej tkance; - fagocytoza przez rezydujące komórki żerne oraz wędrujące makrofagi; - nie wywołuje odpowiedzi zapalnej. Aktywacja fizjologicznej śmierci komórki może zachodzić pod wpływem wielu różnych czynników: • biologicznych, takich jak zakażenia (np. Helicobacter pylori, wirusami), niedobór czynników wzrostu, sygnały śmierci wysyłane przez inne komórki, produkty limfocytów Tc (np. cytokiny, perforyny); • chemicznych, czyli stresu oksydacyjnego wywołanego obecnością reaktywnych form tlenu, chemoterapeutyków stosowanych w terapii przeciwnowotworowej; • fizycznych, to jest promieniowania jonizującego, szoku termicznego.
Apoptoza to aktywny, zaprogramowanym przez komórkę proces. Związany jest on z aktywacją specyficznych genów, które pozwalają na zachowanie równowagi między procesami proliferacji i śmierci komórek. Ekspresję genów zaangażowanych w proces samobójczej śmierci reguluje m.in. czynnik transkrypcyjny NFкB. Przykładem genów specyficznych dla śmierci fizjologicznej są proapoptotyczne geny ced-3 i ced-4. Biorą one udział w procesie morfogenezy u nicienia Caenorhabditis elegant. Ssaczym homologiem ced-3 jest gen kodujący należące do kaspaz białko ICE (ang. interleukin-1β-converting enzyme). Negatywnym regulatorem aktywności tych genów jest antyapoptotyczny gen ced-9 działający podobnie jak ssaczy gen bcl-2. Jego ekspresja jest cechą charakterystyczną dla tych tkanek, u których apoptoza ma za zadanie regulowanie ich rozwoju oraz przyczynia się do wymiany komórek na nowe. Typowym przykładem genów aktywowanych nie tylko podczas procesu apoptozy są protoonkogeny będące czynnikami transkrypcyjnymi, np. c-myc, c-fos, c-jun. W odpowiedzi komórkowej uczestniczą również geny niektórych cyklin i kinaz cyklinozależnych oraz geny takie jak Rb oraz „strażnik genomu”, czyli gen p53. Wydłuża on fazę G1 cyklu komórkowego umożliwiając w ten sposób naprawę uszkodzonego DNA i dzięki czemu zapobiega przekazywaniu komórkom potomnym zaburzeń genetycznych. W przypadku zbyt dużych uszkodzeń DNA gen ten indukuje proces apoptozy.

Enzymy biorące udział w procesie apoptozy:
• Kaspazy (ang. cysteine-dependent aspartate specific protease) - proteinazy cysteinowe, enzymy trawiące białka jądrowe i cytoplazmatyczne. Ich charakterystyczną cechą jest to, że tną swoje substraty zawsze w miejscach zlokalizowanych obok reszty asparaginianowej. Kaspazy składają się z dwóch różniących się wielkością podjednostek. W mniejszej z nich zlokalizowane są reszty aminokwasowe tworzące centrum aktywne enzymu. W większej znajduje się reszta cysteinowa, której kaspazy zawdzięczają swoją nazwę. Proteinazy cysteinowe produkowane są przez komórkę w formie nieaktywnych zymogenów. Obie podjednostki rozdzielone są w nich domeną o charakterze regulatorowymi, a do N-końca dołączona jest dodatkowa prodomena. Ulegająca proteolitycznemu cięciu prokaspaza tworzy dwa fragmenty: duży (17-20 kDa) i mały (10-12 kDa), które następnie łączą się ze sobą w formę aktywną enzymu. Aktywacja kaspaz zachodzi w sposób schierarchizowany (tzw. „kaskada kaspaz”), na drodze proteolizy i oligomeryzacji. Jako pierwsze aktywowane zastają kaspazy inicjatorowe (-2, -8, -9, -10). Ich rola polega na aktywacji tzw. kaspaz efektorowych (-3, -6, -7), spośród których najważniejsza jest kaspaza 3, odpowiedzialna za hydrolizę białek cytoszkieletu (m. in. aktyny i spektryny) oraz błony otaczającej jądro komórkowe. Przeprowadza ona również aktywną proteolizę inhibitora DNazy DFF40/CAD (ang. DNA fragmentation factor 40kDa/caspase-activated deoxyribonuclease) - enzymu degradującego DNA. • enzymy endonukleolityczne odpowiedzialne za degradację kwasów nukleinowych komórki: nukleazy NUC-18, DNaza I oraz DNaza II. Są one zależne od obecności jonów wapnia i magnezu oraz hamowane działaniem jonów cynku. • transglutaminazy tworzące ciałka apoptotyczne, przeprowadzające polimeryzację białek cytoplazmatycznych oraz tworzące pomiędzy nimi wiązania krzyżowe.
Rys. 2. Aktywacja kaspazy
Istnieją dwie podstawowe drogi prowadzące do apoptozy komórki. Wywoływane są one różnymi czynnikami oraz cechują się całkowicie odmiennym przebiegiem w fazie początkowej.
Szlak zależny od mitochondriów, czyli inaczej szlak wewnętrzny
Na skutek uszkodzenia DNA, pozbawienia komórek czynników wzrostu, niedotlenienia lub za pośrednictwem onkogenów, w błonie mitochondriów utworzone zostają kanały przez które uwalniane są czynniki apoptotyczne. Zmiany przepuszczalności błon mitochondrialnych powodują spadek ich potencjału elektrochemicznego, zmianę wewnątrzkomórkowego pH oraz utratę zgromadzonego GSH, NAD(P)H i jonów wapnia. Powstawanie kanałów może również prowadzić do produkcji reaktywnych form tlenu (RFT) indukujących tzw. stres oksydacyjny. Proces apoptozy regulują białka z rodziny Bcl-2. Ich funkcja nie jest jednorodna, gdyż należą do niech zarówno czynniki proaoptotyczne jak i antyapoptotyczne. Ich wzajemne oddziaływania decydują o śmierci lub przeżyciu komórki.
Tab. 2. Białka z rodziny Bcl-2

Spośród czynników apoptotycznych uwalnianych z przestrzeni międzybłonowej mitochondriów na szczególną uwagę zasługują cytochrom c oraz białko Apaf 1 (ang. apoptotic protease activating factor-1). Razem tworzą one kompleks zwany apoptosomem, uczynniajacy kaspazę 9, czyli bezpośredni aktywator kaspazy 3. Innymi uwalnianymi czynnikami są endonukleaza G i AIF. Ostatni czynnik po uwolnieniu do cytoplazmy transportowany jest do jądra komórkowego, gdzie następnie aktywuje odpowiednie kaspazy przeprowadzające fragmentację chromatyny jądrowej.
Rys. 3. Wewnętrzny szlak apoptotyczny
Szlak zależny od receptorów śmierci, czyli szlak zewnętrzny
Rozpoczyna się on od pobudzenia błonowych receptorów śmierci DR (ang. Death Receptor) należących do nadrodziny TNF, charakteryzującej się posiadaniem wewnątrzkomórkowej domeny śmierci DD (ang. Death Domain). Wiąże ona zarówno ligand, jak i cytoplazmatyczne białka adaptorowe (np. FADD (ang. Fas-Associated Death Domain)) tworząc kompleks DISC (ang. Death Inducing Signaling Complex), który uczynnia kaspazę 8 - bezpośredni aktywator kaspazy 3. W ten sposób uruchomiona zostaje kaskada kaspaz, prowadząca do apoptozy komórki. Kaspazy mogą być również aktywowane przez ceramidy. Są one przekaźnikami sygnału apoptotycznego, łączącymi szlak zewnętrzny ze szlakiem mitochondrialnym.

Rys. 4. Zewnętrzny szlak indukcji apoptozy
MIKROROZMNAŻANIE
Mikrorozmnażanie, czyli rozmnażanie klonalne lub mikropropagacja, jest jednym z najważniejszych praktycznych zastosowań roślinnych czystych kultur. Termin ten oznacza wykorzystanie hodowli in vitro do masowego rozmnażania wegetatywnego roślin. Oczywiście cząstka "mikro-" w nazwie tej techniki odnosi się jedynie do ilości wyjściowego materiału biologicznego, natomiast liczba uzyskiwanych w ten sposób roślin potomnych może być bardzo wysoka. Poza wysokim współczynnikiem rozmnożenia (proporcją liczby uzyskanych roślin do wyjściowej liczby roślin użytych do rozmnażania) zaletą tej techniki jest bardzo duże wyrównanie genotypowe i fenotypowe produkowanych roślin, charakterystyczne zresztą dla wszystkich metod rozmnażania wegetatywnego. Wyrównaniu właściwości roślin sprzyja też fakt, że mikrorozmnażanie prowadzi się w dokładnie kontrolowanych warunkach laboratoryjnych. Ścisła izolacja hodowanego materiału od zewnętrznego środowiska, ułatwia otrzymanie roślin wolnych od patogenów i szkodników a także uniezależnia produkcję od miejscowych warunków geograficznych, klimatycznych itp. Z drugiej strony, praca w warunkach laboratoryjnych pociąga za sobą znaczne koszty produkcji, bo urządzenie laboratorium i jego eksploatacja są niewątpliwie wielokrotnie droższe w porównaniu z bardziej tradycyjnymi metodami produkcji roślin. W związku z tym technika ta wykorzystywana jest obecnie głównie w odniesieniu do roślin, których cena jednostkowa jest stosunkowo wysoka, na przykład roślin ozdobnych, drzew i krzewów owocowych lub leśnych, a także cennych materiałów hodowlanych i gatunków zagrożonych wyginięciem. Argumentem przemawiającym za wykorzystaniem tej techniki w produkcji sadzonek drzew leśnych jest fakt, że w odniesieniu do tych gatunków nie istnieją, albo mało skuteczne są prostsze metody rozmnażania wegetatywnego, natomiast kwitnienie i owocowanie tych roślin następuje dopiero gdy osiągną wiek kilkudziesięciu lat. Do mikrorozmnażania wystarczy pobrać niewielki fragment młodej siewki, gdy tylko stwierdzimy, że właśnie ta roślina wykazuje korzystne cechy fenotypowe. Ze względu na pracochłonnośc i wysokie koszty produkcji mikrorozmnażanie nie ma obecnie znaczenia w towarowym rolnictwie, bo przy masowości produkcji rolniczej cena pojedynczej rośliny musi być tu bardzo niska. Nawet jednak rośliny typowo rolnicze, np. ziemniak, bywają poddawane mikrorozmnażaniu, lecz nie przez rolników - producentów, ale hodowców. Dzięki temu z pojedynczych wyselekcjonowanych roślin można uzyskać zdrowy i wyrównany materiał mateczny, który potem dodatkowo powiela się metodami już bardziej tradycyjnymi i tańszymi.
Można wyróżnić następujące główne sposoby mikrorozmnażania:
1. hodowle wierzchołków pędów; jest to forma wzrostu uorganizowanego - wierzchołki pędów rosną, ukorzeniają się, po czym zostają adaptowane do warunków ex vitro i wykorzystane jako sadzonki); odcinek pędu stosowany w tej metodzie ma zwykle długość od kilku milimetrów do kilku centymetrów i zawiera zarówno merystem wierzchołkowy jak i towarzyszące mu zawiązki liści; możliwe jest też regenerowanie roślin ze znacznie mniejszych eksplantatów (długości poniżej 1 mm), zawierających niemal wyłącznie tkankę merystematyczną; jednakże izolowanie merystemów, a nie wierzchołków pędów, będąc techniką dość pracochłonną, stosuje się w nieco innych celach niż mikrorozmnażanie i omówione jest dokładniej w dalszej części tego skryptu
2. pobudzenie rozwoju pąków bocznych w hodowlach odcinków pędów to metoda bardziej uniwersalna i dużo częściej stosowana niż poprzednia; eksplantatami są węzły łodygi (odcinki z których wyrastają liście); u nasady liści występują uśpione pąki boczne (zwane też pachwinowymi), które w wyniku izolacji eksplantatu, a czasami także pod wpływem występujących w pożywce cytokinin, wznawiają wzrost i tworzą pędy, wykorzystywane następnie podobnie jak w poprzedniej metodzie; również ta metoda opiera się na zjawisku uorganizowanego wzrostu in vitro - komórki eksplantatu realizują program rozwojowy, który został im przypisany jeszcze w warunkach in vivo);

3. powstawanie pąków przybyszowych bezpośrednio w tkankach eksplantatu - o tej drodze regeneracji mówimy, gdy zawiązki pędów tworzą się w kulturze in vitro od nowa, w wyniku przeprogramowania komórek eksplantatu, tzn. eksplantat nie zawierał pąków; utworzyły się dopiero w kulturze in vitro; mamy tu więc do czynienia nie tylko ze wzrostem uorganizowanym, ale wcześniej jeszcze z organogenezą pędową (kaulogenezą); jeśli organogeneza nie jest poprzedzona powstaniem kalusa nazywamy ją bezpośrednią; poza kaulogenezą, drugą podstawową formą organogenezy jest ryzogeneza, czyli tworzenie się przybyszowych korzeni; o ile jednak z pędu zazwyczaj łatwo i szybko daje się odtworzyć pełną roślinę, to z korzenia udaje się to dosyć rzadko, dlatego ryzogeneza praktycznie nie bywa stosowana do mikrorozmnażania roślin
4. powstawanie pąków przybyszowych z komórek kalusa - organogeneza pędowa pośrednia; zjawisko podobne do opisanego w poprzednim punkcie, ale w tym przypadku komórki eksplantatu najpierw odróżnicowują i namnażają się, wytwarzając kalus, a dopiero niektóre komórki kalusa przystępują do odtwarzania rośliny;
5. powstawanie zarodków somatycznych bezpośrednio w tkankach eksplantatu - bezpośrednia somatyczna embriogeneza (to bardzo pożądany sposób rozmnażania roślin - odpada konieczność ukorzeniania pędów; zarodki tworzą się od razu, nie trzeba czekać na rozwój kalusa; zwykle jednak nie jest tak pięknie - częściej udaje się zastosować metodę poniższą)
6. powstawanie zarodków somatycznych z komórek kalusa - somatyczna embriogeneza pośrednia (wadą tej metody jest obserwowana często niestabilność genetyczna komórek kalusa; zachodzą w nich spontanicznie zmiany, które powodują, że regenerowane w ten sposób rośliny nie są tak wyrównane jak można by oczekiwać po metodzie wegetatywnego rozmnażania).
Przedstawiona tu terminologia nie jest całkiem powszechnie uznawana. Niektórzy badacze za mikrorozmnażanie uznają tylko techniki wymienione w punktach 1-2. W ujęciu nieco mniej rygorystycznym do mikrorozmnażania zalicza się wszystkie powyższe techniki, poza opartymi na somatycznej embriogenezie (p. 5, 6). Niemniej, na naszych zajęciach będziemy trzymać się powyższego schematu (podążając z grubsza za sugestią prof. Murashigego) i do mikrorozmnażania zaliczać będziemy wszystkie metody wymienione w punktach 1-6.
Wykorzystując wierzchołki pędów i pąki kątowe do mikrorozmnażania roślin nie oczekujemy od komórek eksplantatu jakiejś radykalnej zmiany zachowania, w stosunku do tego jakie wykazywały in planta, czyli w hodowanej ex vitro, nie uszkodzonej roślinie. Wprawdzie pąki kątowe w warunkach in vivo są zwykle uśpione, ale już sam fakt oddzielenia ich od wierzchołka łodygi wyzwala je spod wierzchołkowej dominacji (narzuconej przy pomocy auksyn transportowanych w dół łodygi) i pobudza ich rozwój. Dlatego do rozmnażania roślin przy pomocy wierzchołków pędów i pąków pachwinowych wystarczyć mogą pożywki nie zawierające żadnych fitohormonów. Z drugiej jednak strony, dodanie do pożywki fitohormonów, zwłaszcza cytokinin, przyspiesza przełamywanie dominacji wierzchołkowej, nasila rozgałęzianie się pędów i zwiększa współczynnik rozmnożenia. U niektórych gatunków od samej cytokininy korzystniej działa jej kombinacja z auksyną, pobudzającą wydłużanie się pędów. Również giberelina GA3 oraz kwas ferulowy mogą sprzyjać namnażaniu pędów i ich prawidłowej budowie (długości międzywęźli, wielkości liści).

Spośród auksyn, w mikrorozmnażaniu roślin opartym na hodowlach wierzchołków pędów i pąków kątowych, a także na organogenezie pędowej, najczęściej wykorzystuje się IAA, NAA lub IBA. 2,4-D natomiast okazuje się zwykle zbyt silną auksyną do tego celu i zamiast tworzenia się pędów pobudza wzrost kalusa (natomiast w metodach opartych na somatycznej embriogenezie, najczęściej wykorzystywaną auksyną jest właśnie 2,4-D, patrz niżej).
Dwie pierwsze z omawianych tu metod mikrorozmnażania nie różnią się istotnie od tradycyjnego rozmnażania roślin przy pomocy sadzonek wierzchołkowych i odkładów (jakkolwiek zadowalają się znacznie mniejszą ilością materiału wyjściowego). Wydaje się, że wszystkie gatunki rozmnażane tymi tradycyjnymi metodami nadają się też do rozmnażania in vitro za pomocą wierzchołków pędów lub pąków kątowych. Wśród roślin do rozmnażania których te metody rzeczywiście są wykorzystywane w produkcji na dużą skalę, można wymienić m. in. maliny, wiśnie, śliwy, jabłonie, winorośl, a także dalie, gerbery, storczyki, ziemniaki i wiele innych. U tych gatunków z jednego eksplantatu można w ciągu roku "wyprowadzić" tysiące roślin potomnych. Co kilka tygodni (zwykle co miesiąc) pędy odcinane są od tkanki macierzystej i pasażowane na świeżą pożywkę namnażającą.
Pędy ziemniaka, zwłaszcza gdy są hodowane na pożywce o niskim potencjale osmotycznym (zawartości sacharozy podwyższonej np. do 60 g/l), w niskiej temperaturze (ok. 10°C) i przy krótkim dniu (8 godzinnym) mogą tworzyć tzw. mikrobulwy, czyli bulwy o średnicy zaledwie kilku milimetrów. Tworzenie się mikrobulw nazywamy tuberyzacją in vitro (od ang. tuber - bulwa) i w pracach nad mikrorozmnażaniem roślin traktujemy zwykle jako proces pożądany. Dodatkowo można go pobudzić dodając do pożywki pochodną kwasu jasmonowego zwaną kwasem tuberonowym. Mikrobulwy, można wysiewać do gleby jak nasiona, a przy tym są znacznie wygodniejsze w manipulacji i odporniejsze na uszkodzenie niż młode sadzonki.
Organogeneza pędowa, bezpośrednia lub pośrednia, pozwala na uzyskanie jeszcze większego współczynnika rozmnożenia niż hodowle wierzchołków pędów czy pąków kątowych. Pod wpływem odpowiedniego zestawu fitohormonów, obejmującego w przypadku tej metody zazwyczaj zarówno cytokininę, jak i auksynę, niektóre komórki ekskplantatu lub namnożonego na nim kalusa wytwarzają zawiązki pędów - pąki. Czasami w obrębie jednego eksplantatu może się ich tworzyć kilkadziesiąt. Tak bywa na przykład u popularnych roślin ozdobnych z rodziny ostrojowatych, takich jak sępolia (Saintapaulia), syningia (Sinningia) czy skrętnik (Streptocarpus), u których łatwo jest pobudzić powstawanie pędów przybyszowych w niewielkich (kilkumilimetrowych) fragmentach blaszki liściowej i ogonków liściowych. Znamienne, że rośliny te łatwo, choć nie aż tak wydajnie, dają się rozmnażać z liści również w warunkach ex vitro. Rośliną bardzo wydajnie dającą się rozmnażać w ten sposób jest też tytoń, ale takich przykładów można znaleźć dużo więcej. Już pod koniec ubiegłego wieku szacowano, że lista roślin, dla których opublikowano metody mikrorozmnażania (którąkolwiek z powyższych metod) przekroczyła sześćset gatunków i obejmowała praktycznie wszystkie rośliny ważne gospodarczo. U roślin cebulowych, np. lilii, tulipanów, jako eksplantaty wykorzystuje się często kawałki liści spichrzowych, a wynikiem hodowli jest tworzenie się drobnych, kilkumilimetrowych cebulek - mikrocebul.
Wynik hodowli zależy od właściwości biologicznych rośliny, składu pożywki (a zwłaszcza rodzaju i stężeń fitohormonów) a także warunków hodowli. Za wzorcowe, "klasyczne", uważa się dziś badania wpływu auksyn i cytokinin na hodowle tkanek tytoniu, przeprowadzone przez Skooga. W doświadczeniach tych okazało się, że najszybsze tworzenie kalusa następowało przy umiarkowanych, niezbyt wysokich stężeniach auksyn i cytokinin. Zwiększenie zawartości auksyn sprzyjało tworzeniu się przybyszowych korzeni, a podwyższenie zawartości cytokinin pobudzało organogenezę pędową.
W procedurach mikrorozmnażania roślin w oparciu o wierzchołki pędów, pąki kątowe i organogenezę pędową wyróżnić można kilka typowych etapów (nie wszyscy autorzy wyodrębniają punkty pierwszy i ostatni; pozostałe natomiast etapy są na tyle jednoznacznie rozumiane, że producenci ogłaszają krótko np. "sprzedam rośliny po III etapie mikrorozmnażania"):
1. Dobór materiału roślinnego2. Założenie czystej (aksenicznej) kultury3. Namnażanie pędów4. Przygotowanie pędów do wykorzystania w warunkach ex vitro
a. wydłużanieb. ukorzenianiec. (ewentualne wstępne hartowanie)
5. Przesadzenie roślin do gleby, hartowanie
Na etapach oznaczonych tu numerami 0 - 1 uwzględnia się wszystkie kryteria, o których wspominaliśmy przy omawianiu ogólnych zasad doboru eksplantatu (por. rozdz. 1), a ponadto zwraca się uwagę na te cechy biologiczne i użytkowe roślin matecznych, które odziedziczone przez rośliny potomne, będą wyznaczać ich wartość handlową (np. barwa i wielkość kwiatów, pokrój rośliny). W celu sprawdzenia skuteczności odkażania można niektóre eksplantaty umieścić na typowych podłożach mikrobiologicznych, które lepiej niż pożywki dla roślin pobudzają wzrost drobnoustrojów i pozwalają na szybsze ich wykrycie. Badanie obecności charakterystycznych drobnoustrojów w eksplantatach, albo nawet w roślinach matecznych, z których eksplantaty dopiero będą izolowane, nazywa się czasem z angielska indeksacją (ang. indexing for pathogens), zwłaszcza jeśli wykorzystuje się w nich pożywki różnicujące, metody biochemiczne, albo rośliny wskaźnikowe, dające bardzo charakterystyczne, silne reakcje na obecność jakiegoś drobnoustroju i pozwalające na jego identyfikację.
Na etapie namnażania otrzymuje się bardzo dużą liczbę drobnych (kilkumilimetrowych) zawiązków pędów. Przenosząc je na pożywkę zawierającą znacznie mniejsze stężenia regulatorów wzrostu, lub wcale nie zawierającą fitohormonów, zatrzymuje się proces namnażania pędów, a pobudza ich wzrost. Czasami na etapie wydłużania pędów wykorzystuje się pożywki zawierające węgiel aktywny, który pochłania pozostałości fitohormonów przenoszone wraz z materiałem roślinnym z poprzedniego etapu hodowli. Ukorzenianie pędu i jego wzrost mogą zachodzić na tej samej pożywce. W razie potrzeby ukorzenianie może być pobudzane przez dodatek niewielkiej ilości auksyny takiej jak IAA, IBA lub NAA.
Konieczność hartowania regenerowanych in vitro roślin wynika z dużej różnicy pomiędzy warunkami środowiska in vitro i ex vitro. Przede wszystkim chodzi tu wilgotność powietrza (w kulturach in vitro często bliską 100%) oraz obecność cukru i innych związków organicznych w pożywce, przy ich niemal zupełnym braku w glebie. Zabieg hartowania, a nawet ukorzeniania, może być przeprowadzony (a zwłaszcza kontynuowany) już po przesadzeniu roślin do gleby. Przez okres 1 - 2 tygodni utrzymywana jest wysoka wilgotność powietrza (rośliny mogą być osłonięte tunelami foliowymi) i dość niskie natężenie światła (zacienienie). Później stopniowo usuwa się osłony zmuszając rośliny do adaptowania się do warunków właściwej uprawy ex vitro.
Metodą mikrorozmnażania, która powinna dać się stosunkowo łatwo automatyzować, a więc okazać się stosunkowo mało pracochłonną, tanią, jest wykorzystanie somatycznych zarodków. Skoro mówimy o zarodkach somatycznych, to dla jasności sprawy uzgodnijmy od razu jakie w ogóle rodzaje roślinnych zarodków będziemy wyróżniać. Ogólnie dzieli się je na zygotyczne i niezygotyczne czyli apomiktyczne. Somatyczne należą oczywiście do tej drugiej grupy. Do zarodków apomiktycznych poza somatycznymi, zalicza się jeszcze zarodki partenogenetyczne, powstające z żeńskiego gametofitu, a zwłaszcza należącej do niego komórki jajowej i androgeniczne, powstające z mikrospory (niedojrzałego ziarna pyłku) lub męskiego gametofitu (kiełkującego ziarna pyłku). Wszystkimi zarodkami innymi niż somatyczne będziemy się zajmować w dalszej części tego podręcznika. Dodajmy, że czasami obserwujemy zarodki somatyczne wyrastające na powierzchni innych, starszych zarodków, somatycznych lub zygotycznych. Taki szczególny rodzaj zarodków somatycznych nazywamy przybyszowymi lub dodatkowymi.

Zarodki somatyczne i zygotyczne są oczywiście podobne nie tylko z nazwy, ale wykazują podobną budowę i podobne przemiany rozwojowe - pojedyncza komórka, drobne agregaty komórkowe, zarodek kulisty (globularny), sercowaty, torpedowaty (hodowcy roślin czasem mówią "stadium torpedo" bo torpeda to po angielsku właśnie torpedo) i stadium liścieniowe. W wyniku wzrostu, zarodki przekształcają się ostatecznie w rośliny. Wydaje się, że somatyczną embriogenezę (SE) można wywołać u wszystkich gatunków roślin, ale widoczne są duże różnice gatunkowe i genotypowe w zdolności do SE. Stosunkowo łatwo uzyskuje się to zjawisko np. u marchwi, kalafiora selera, rzepaku, cytryny, kawy, a z gatunków rolniczych - lucerny.
Somatyczną embriogenezę pobudzają warunki hodowli, a mianowicie obecność auksyny (zwłaszcza silnych, syntetycznych auksyn jak 2,4-D i pikloram), odpowiedni (wysoki) stosunek azotu amonowego do azotanowego, dość wysokie pH (ok. 7). W niektórych hodowlach indukująco może działać obecność określonych aminokwasów (zwłaszcza kwasu glutaminowego i proliny) lub kwasów organicznych. Również obecność cytokininy sprzyja zapoczątkowaniu SE, a szczególnie skuteczna pod tym względem bywa zeatyna. Działanie egzogennej auksyny konieczne jest tylko we wstępnej fazie somatycznej embriogenezy. Później (po okresie kilku dni do miesiąca) hodowle przenoszone są na pożywkę, która albo auksyny nie zawiera, albo zawiera jej dużo mniej niż pożywka indukująca (tę drugą pożywkę nazywamy różnicującą). Na pożywce indukującej powstają masy komórek prazarodkowych, tzw. PEMy (od ang. proembryogenic masses), które niewiele różnią się wyglądem od zwykłego kalusa i tworzą tzw. kalus embriogenny czyli zarodkotwórczy (po angielsku zarodkotwórczy to embryogenic, stąd w polskich tekstach pojawia się też czasem słowo "embriogeniczny", znaczące dokładnie to samo co embriogenny). Gdyby te skupiska komórek nie zostały następnie przeniesione na pożywkę różnicującą, to pozostałyby w formie embriogennego kalusa, powiększającego rozmiary, lecz nie tworzącego uorganizowanych struktur.
Tylko niektóre komórki eksplantatu reagują na działanie czynników indukujących SE. Komórki te nazywamy kompetentnymi. Inne w tych samych warunkach wytwarzają tylko kalus, czy zawiesinę, albo w ogóle nie reagują. Kompetentymi okazują się na ogół komórki epidermalne lub położone tuż pod nimi subepidermalne, a także komórki powierzchniowej strefy kalusa, lub hodowane pojedynczo komórki parenchymatyczne. Komórki embriogenne mają charakterystyczną budowę - zwykle są stosunkowo drobne, izodiametryczne, zawierają dużo cytoplazmy i duże jądro komórkowe, a wakuole - drobne , silnie barwiące się błękitem toluidyny, a więc zawierające dużo białka. W chloroplastach tych komórek można dostrzec ziarna skrobi, natomiast zanika gradient protonowy, co sugeruje, że fotosynteza ulega w nich zahamowaniu.
Somatyczna embriogeneza jest procesem bardzo wszechstronnie badanym, co wynika zarówno z jej znaczenia praktycznego, jak i poznawczego. W tym procesie wyjątkowo wyraźnie przejawia się rozwojowa elastyczność i totipotencja komórek. Podczas SE zachodzą podobne przemiany jak w rozwoju zarodków zygotycznych, ale są łatwiej dostępne obserwacji, bo nieukryte pod tkankami nasienia i owocu. Nagromadzające się stopniowo obserwacje sugerują, że SE może być ściśle powiązana z reakcjami roślinnych tkanek na różnorodne czynniki stresowe.
Działanie egzogennej auksyny polega prawdopodobnie na zakłóceniu metabolizmu auksyny endogennej i nagromadzaniu się w komórkach dużych ilości IAA. Poza omówionymi wyżej, dokładnie udokumentowanymi induktorami SE, jako czynniki pobudzające ten proces opisywano sporadycznie metale ciężkie i wysoką temperaturę, a także egzogenny kwas abscysynowy i betainę - substancje, które rośliny same wytwarzają, gdy znajdą się w warunkach stresu. Wytwarzane w embriogennych komórkach cząsteczki RNA kodują tzw. białka związane z patogenezą i białka szoku termicznego. Wśród białek intensywnie wydzielanych przez embriogenne komórki wykrywa się chitynazy, glukanazy i białka osmotynopodobne. Bardzo charakterystyczne dla komórek embriogennych jest powlekanie ścian komórkowych kalozą i nagromadzanie w ścianie komórkowej białek arabinogalaktanowych (AGP). Białka te zawierają glukozaminę i ich obecność wyjaśnia zapewne uaktywnianie się chitynaz podczas embriogenezy. Do niedawna zjawisko to było zupełnie niezrozumiałe, gdyż wydawało się, że w roślinach nie ma żadnego substratu dla tych enzymów. Okazuje się jednak, że białka AGP "nadtrawione" chitynazami wykazują zwiększoną zdolność do nadawania komórkom stanu kompetencji i indukowania SE. Dodając AGP do pożywki z kulturami, które po dłuższym okresie hodowli utraciły zdolność reagowania na typowe induktory SE, przywrócono tym kulturom kompetencję rozwojową. Poszukując markerów molekularnych SE, wykryto gen SERK, kodujący kinazę receptorową somatycznej embriogenezy (Somatic Embryogenesis Receptor Kinase). ). Jego uaktywnienie się jest najwcześniejszym znanym obecnie objawem nabycia przez komórki somatyczne zdolności (kompetencji) do rozwoju zarodkowego.
W niektórych doświadczeniach obserwowano, że pierwszy podział komórki embriogennej zachodzi w szczególny sposób, przypominający podział zygoty, a mianowicie jest niesymetryczny - jego wynikiem są dwie komórki różniące się wielkością. W rozwoju zarodków zygotycznych komórka mniejsza, określana jako wierzchołkowa, po kilku kolejnych podziałach przekształca się w zarodek właściwy, a komórka większa, zwana podstawową przekształca się w wieszadełko, twór ułatwiający odżywianie zarodka właściwego. Wieszadełko wraz z drobnym, mało jeszcze zróżnicowanym zarodkiem właściwym, nazywane jest łącznie prazarodkiem. Podobnie w somatycznej embriogenezie obserwuje się, jak wspomniano wyżej, komórki zwane prazarodkowymi, i powstające z nich odpowiedniki zarodka właściwego. Typowego wieszadełka w somatycznej embriogenezie nie daje się zwykle zaobserwować.
Zarodki somatyczne można masowo wytwarzać w płynnej pożywce, w specjalnych bioreaktorach (fermenterach). Otrzymane jednak w warunkach in vitro zarodki są zbyt delikatne, aby je po prostu wysiać do gleby, tak jak się to robi ze zwykłymi nasionami. Dlatego najpierw zarodki te poddawane są zabiegom przygotowawczym, np. łagodnemu suszeniu albo otoczkowaniu - umieszcza się je więc jakby w kapsułkach z substancji ochronnej, takiej jak alginian wapnia lub potasu, czy syntetyczne żywice (np. glikol polioksyetylenowy). Tak zabezpieczone zarodki somatyczne nazywane są sztucznymi nasionami, albo nasionami syntetycznymi, albo somatycznymi. Układem nieco prostszym od kapsułek z pojedycznymi zarodkami jest tzw. wysiew cieczowy (ang. fluid drilling), przy którym zarodki są po prostu zawieszone w pewnej objętości gęstej ochronnej cieczy i wraz z nią wylewane są do gleby.
Wyróżnia się więc kilka rodzajów sztucznych nasion:
a. odwodnione nieotoczkowane b. odwodnione otoczkowanec. uwodnione otoczkowane (oczywiście ich znaczne uwodnienie nie jest zamierzonym wynikiem jakiegoś zabiegu technologicznego, ale efektem
hodowli na pożywce zoptymalizowanej do szybkiego wzrostu kultury)d. uwodnione nie otoczkowane indywidualnie, ale stosowane w układzie siewu cieczowego.
Nasiona typu (c) i (d) są metabolicznie aktywne, a więc oddychają, zużywają tlen i złożone metabolity, a nagromadzają produkty oddychania; z tego powodu są nietrwałe i wkrótce po wytworzeniu powinny być wysiane do gleby. Natomiast nasiona typu (a) i (b) dają się długo przechowywać, bo w wyniku odwodnienia komórek ich metabolizm jest zahamowany, ale sam proces osuszania powoduje spadek żywotności wielu zarodków. Tak więc otrzymanie wysokiej jakości sztucznych nasion z zarodkami odwodnionymi jest trudne, gdy jednak ta sztuka się uda, nasiona takie można przechowywać stosunkowo długo. Czasami podobnym zabiegom ochronnym poddaje się inne niż zarodki fragmenty roślin, które wysiane do gleby mogą przekształcić się w kompletne rośliny. Mogą to być np. mikrobulwy, mikrocebule, pąki. Takie laboratoryjnie wytworzone "rozmnóżki" roślin można również nazwać sztucznymi nasionami. Uogólniając więc nieco, możemy przyjąć, że sztuczne nasiona to zarodki somatyczne lub inne drobne eksplantaty (mikrobulwy, mikrocebule, pąki) otrzymane in vitro i przygotowane do wysiania do gleby (zabezpieczone przed uszkodzeniami). To przygotowanie polega najczęściej na otoczkowaniu substancją zwiększającą mechaniczną wytrzymałość eksplantatów. Funkcje ochronne otoczki można wspomagać wysycając ją fungicydami, fitoncydami lub odpowiednio wybiórczymi herbicydami. Czasami rozważa się też dodawanie substancji zapasowych do otoczki, dzięki czemu pełniłaby nie tylko rolę okrywy nasiennej, ale także sztucznego bielma, odżywiającego zarodek podczas wzrostu. Wzrost w pełni wykształconego zarodka, wznowiony po przerwaniu spoczynku, nazywa się oczywiście kiełkowaniem. Tego terminu unika się jednak gdy mowa o somatycznych zarodkach i sztucznych nasionach, głównie dlatego, że nasiona te z reguły nie przechodzą spoczynku więc faza powstawania zarodka (wzrostu zarodkowego) i jego przekształcania się w siewkę

(kiełkowania) nie dają się tu wyraźnie rozgraniczyć. Zamiast więc o kiełkowaniu sztucznych nasion mówi się zwykle o ich konwersji (ang. conversion) czyli przekształcaniu się w rośliny. Przekształcanie się zarodka somatycznego (lub sztucznego nasienia) w prawidłowo zbudowaną roślinę nazywamy konwersją. Częstość konwersji (w %) jest podstawowym wskaźnikiem jakości sztucznych nasion.
Próby przemysłowej produkcji sztucznych nasion zaczęto podejmować w 80-tych latach ubiegłego stulecia. Obecnie produkowane są m. in. sztuczne nasiona marchwi, selera, lucerny, papryki, winorośli, kalafiora, soi i ryżu. Mimo to do dziś wykorzystuje się je raczej sporadycznie. Jedną z najważniejszych przyczyn są wysokie koszty produkcji. Produkcja w warunkach sterylnych, na sztucznych pożywkach, nawet gdy prowadzona jest w wielkiej skali, w instalacjach przemysłowych, nie może konkurować pod względem ceny z pozyskiwaniem nasion zygotycznych metodami tradycyjnej uprawy polowej. Nie do końca zostały też rozwiązane problemy techniczne, dotyczące wydajności indukcji somatycznej embriogenezy i synchronizacji tego procesu, co jest warunkiem automatyzacji produkcji. Utrudnieniem jest nietrwałość sztucznych nasion (trudności w ich przechowywaniu) oraz niewystarczająca żywotność. Ważnym problemem jest też zróżnicowanie genetyczne roślin otrzymywanych ze sztucznych nasion. Właściwie należałoby oczekiwać, że zmienności takiej wcale nie będzie, ponieważ jak już wspominaliśmy, sztuczne nasiona można uznać za formę wegetatywnego rozmnażania roślin. Nie działają więc podczas ich powstawania takie mechanizmy odpowiedzialne za różnicowanie genetyczne jak segregacja genów (towarzysząca mejozie) i losowy dobór gamet (mający miejsce podczas zapłodnienia). Mimo to, rośliny otrzymywane z somatycznych zarodków czy sztucznych nasion, wykazują często wyraźne zróżnicowanie, i to również o podłożu genetycznym. Zjawisko to obserwuje się zresztą w mniejszym lub większym natężeniu także przy innych metodach mikrorozmnażania roślin.
Fitoestrogeny to grupa niesteroidowych związków pochodzenia roślinnego o budowie i funkcji podobnej do naturalnych estrogenów. Obecne są we wszystkich częściach roślin – kwiatach, owocach, liściach, nasionach oraz korzeniach. Wyróżniamy trzy klasy fitoestrogenów: flawonoidy, ligniny i stilbeny. Występują one zazwyczaj w postaci nieaktywnych glikozydów lub w formie prekursorowej. Ich formy aktywne powstają w przewodzie pokarmowym w wyniku złożonych przemian enzymatyczno-metabolicznych. Najbogatszym źródłem fitoestrogenów są: soja i jej przetwory, nasiona roślin strączkowych (soczewica, fasola, bób, groch), winogrona oraz liście zielonej herbaty.
Znaczenie fitoestrogenów u roślinFitoestrogeny u roślin pełnią funkcje grzybobójcze, antyutleniające i budulcowe. Mogą być też barwnikami kwiatów i chronić roślinę przed promieniami UV. Biorą również udział w kiełkowaniu pyłku i sygnalizowaniu stresu.
Klasyfikacja fitoestrogenów
Rys. 1. Podział fitoestrogenów. źródło: Kraszewska O., Nynca A., Kamińska B., Ciereszko R., 2007, Fitoestrogeny I. Występowanie, metabolizm i znaczenie biologiczne u samic. Postępy biologii komórki, 34(1): 189-205 (zmienione)
Ligniny to grupa obejmująca sekoizolaricirezinol oraz matairezinol. Występują one głównie w nasionach lnu i słonecznika a także w pełnoziarnistych produktach zbożowych, ziołach, warzywach (marchew, cebula), owocach (jabłka, wiśnie, jagody) i orzechach. Obecne są także w czarnej i zielonej herbacie
oraz kawie. Związki te po przedostaniu się do układu pokarmowego zwierząt ulegają przekształceniu w enterodiol i enterolakton. Różnica w budowie polega na obecności grupy hydroksylowej w pozycji meta pierścienia aromatycznego u lignanów zwierzęcych. Dzięki temu są one bardziej stabilne chemicznie, biochemicznie i biologicznie. Do stilbenów zaliczamy m.in. resweratrol występujący w skórce owoców tj. winogrona, morwy oraz w orzeszkach ziemnych. Wykazuje silne działanie przeciwutleniające oraz jest nietoksycznym fungicydem. Flawonoidy stanowią bardzo złożoną grupę (Rys. 1.). Wśród nich najpopularniejsze to izoflawony do których zaliczamy: genisteinę, daidzeinę, formononetynę, biochaninę A i glicyteinę. Ich najbogatszym źródłem są: warzywa strączkowe: soja, soczewica, fasola szparagowa czy groch oraz produkty sojowe: kasza i mąka, tofu i mleko sojowe. Kolejne grupy flawonoidów, czyli izoflawanony i izoflawany są metabolitami daidzeiny, obecnymi jedynie w organizmach zwierzęcych. Kumestany występują w lucernie, nasionach soi oraz kiełkach koniczyny. Kumestrol jako najlepiej poznany przedstawiciel tej klasy jest izoflawonoidem o największym potencjale estrogennym.
Budowa fitoestrogenówFitoestrogeny wykazują podobieństwa w budowie strukturalnej do naturalnych i syntetycznych estrogenów. U roślin są syntetyzowane z fenylopropanoidów i prostych fenoli. Flawonoidy to polifenole, których podstawowym elementem strukturalnym jest jednostka diarylpropanowa (C6-C3-C6). Zbudowane są najczęściej z dwóch pierścieni benzenowych (A i B) połączonych łańcuchem trójwęglowym, który u większości tworzy razem z atomem tlenu trzeci pierścień (C). Większość flawonoidów posiada pierścień fenylowy B w pozycji C-2. Izoflawonoidy powstają na skutek izomerycznego przeniesienia pierścienia B w pozycję C-3.
Rys. 2. Wzory strukturalne wybranych fitoestrogenów. źródło: Kraszewska O., Nynca A., Kamińska B., Ciereszko R., 2007, Fitoestrogeny I. Występowanie, metabolizm i znaczenie biologiczne u samic. Postępy biologii komórki, 34(1): 189-205
Biologiczne efekty fitoestrogenówIch działanie na komórki docelowe może być korzystne lub szkodliwe. Obserwuje się hamujący wpływ tych związków na rozwój niektórych typów nowotworów, osteoporozy czy chorób naczyniowych u ludzi i zwierząt. Mogą również wykazywać aktywność jako endokrynogenny czynnik zakłócający funkcjonowanie m.in. układu rozrodczego. Ze względu na strukturalne podobieństwo do 17-β-estradiolu (żeńskiego hormonu płciowego) wykazują zdolność do wiązania się z receptorami estrogenowymi α i β. Mogą przez to działać jako agoniści lub antagoniści tych receptorów, konkurując z estradiolem o miejsce wiązania.
CYTOKINY
Cytokiny to glikoproteidy o masie cząsteczkowej od kilku do kilkunastu kDa, uwalniane są przez aktywowane komórki różnych tkanek. Nazwa ich wywodzi się z greckich słów citos, czyli komórka oraz kinesis – ruch. Ich odkrycie przez R. Levi-Montalcini oraz S. Cohen zostało uhonorowane Nagrodą Nobla w dziedzinie fizjologii i medycyny w roku 1986 roku. Mają istotny wpływ na procesy zapalenia: regulują ekspresję cząstek adhezyjnych na komórkach śródbłonka, indukują syntezę prostaglandyn, wpływają na syntezę białek ostrej fazy, aktywują komórki uczestniczące w zapaleniu: neutrofile, makrofagi, komórki tuczne. Kontrolują wszystkie fazy odpowiedzi immunologicznej – indukcyjną, efektorową i wygaszającą. Do tej pory opisanych jest ponad sto kilkadziesiąt cytokin o działaniu pro- i przeciwzapalnym. Przewaga cytokin prozapalnych doprowadza do ogólnoustrojowej reakcji zapalnej, zaś przeciwzapalnych – do odpowiedzi przeciwzapalnej. W miejscu swojego działania wpływają na komórki docelowe za pośrednictwem receptorów.
Cechy cytokin: • Plejotropia – zdolność do wpływania na różne typy komórek i wywoływanie w nich odmiennych działań• Redundancja – zdolność różnych cytokin do wywoływania jednakowego efektu• Synergizm – współdziałanie cytokin, co zwiększa ich aktywność

• Antagonizm – wzajemne blokowanie efektów działania• Zdolność do indukcji kaskad dodatnich i ujemnych sprzężeń zwrotnych
Sposoby oddziaływnia cytokin na komórki: • Autokrynowe – oddziaływanie na komórkę, przez którą zostały wydzielone po połączeniu się z odpowiednim receptorem• Parakrynowe – oddziaływanie na sąsiadującą komórkę w stosunku do komórki wydzielniczej• Endokrynowe – oddziaływanie na komórki innych narządów
Podział cytokin ze względu na właściwości:
• Cytokin pozapalne: IL-1, IL-6, IL-8, IL-15, IL-17, IL-18, IL-23, mediator TNF
Interleukina 1β (IL-1 β) jest wydzielana przez komórki immunologiczne, glejowe, naskórka, śródbłonka i mięśni szkieletowych. Induktorami jej wytwarzania są: IL-2, IL-3, IL-12, TNFα oraz sama IL-1. Jest czynnikiem chemotaktycznym, przyciągającym i aktywującym neutrofile w miejscu reakcji zapalnej. Indukuje syntezę IL-6 i IL-8 przez makrofagi, fibroblasty, komórki śródbłonka i mięśni, IL-2 i receptora dla IL-2 przez limfocyty T, wytwarzanie przeciwciał przez limfocyty B, proliferację fibroblastów. Stymuluje wydzielanie prostaglandyn i czynnika aktywującego płytki, zwiększa przepuszczalność śródbłonka, indukuje aktywność prokoagulacyjną.
Interleukina 6 (IL-6) wytwarzana jest przez komórki immunologiczne śródbłonka, tkanki łącznej, tłuszczowej, naskórka i komórki mięśni szkieletowych. Jest aktywatorem limfocytów T, reguluje wzrost i różnicowanie limfocytów B, stymuluje uwalnianie białek opiekuńczych HSP (z ang. heat shock proteins) z wątroby i cytokin IL-ra i LI-10. Poprzez synergistyczne działanie z IL-3 i IL-4 pobudza erytropoezę i anigogenezę.
Interleukina 8 (IL-8) jest uwalniana z monocytów, komórek śródbłonka i mięśni szkieletowych w odpowiedzi na rosnące stężenie IL-1β, TNFα i reaktywne formy tlenu. Jest najlepiej poznanym czynnikiem chemotaktycznym przyciągającym i aktywującym neutrofile, połączone z wytwarzaniem reaktywnych form tlenu w tych komórkach.
Interleukina 15 (IL-15) jest wytwarzana przez makrofagi, komórki dendrytyczne, śródbłonka i mięśni szkieletowych. Induktorami jej wydzielania są IL-1β, TNFα, IL-4, IFNγ. Odgrywa ważna rolę w procesach rozwoju, proliferacji i przeżycia komórek NK (z ang. natural killer). Jest silnie działającym czynnikiem chemotaktycznym dla leukocytów oraz inhibitorem procesu apoptozy limfocytów.
Interleukina 18 (IL-18) wytwarzana jest przez makrofagi, komórki nabłonkowe, osteoblasty, adipocyty i komórki mięśniowe. Aktywacja jej następuje z udziałem kaspaz, czyli enzymów kontrolujących proces apoptozy. Uwalnianie tej cytokin może być sygnałem rozpoczynającej się samobójczej śmierci komórki.
TNFα (z ang. tumour necrosis factor), inaczej czynnik martwicy nowotworu należy do grupy cytokin obejmującej 20 cząsteczek. Wytwarzany jest przez monocyty, makrofagi, komórki tkanki łącznej, tłuszczowej, naskórka i mięśni szkieletowych. Stymuluje powstawanie i różnicowanie limfocytów B, T i komórek NK, wpływa na wzrost fibroblastów, syntezę kolagenu, fibronekryny i kwasu hialuronowego, działa chemotaktycznie na neutrofile.
• Cytokiny przeciwzapalne: IL-4, IL-10, IL-13
Interleukina 10 (IL-10) jest cytokiną uwalnianą przez komórki immunologiczne, naskórka i prawdopodobnie komórki mięśni szkieletowych. Minimalizuje skutki reakcji zapalnej na wysiłek fizyczny poprzez hamowanie wytwarzania pozapalnych cytokin TNFα i IL-8 oraz pobudza syntezę antagonisty receptora dla IL-1 w mięśniach. Zmniejsza też wytwarzanie reaktywnych form tlenu przez makrofagi i neutrofile.
Biotechnologiczne pozyskiwanie cytokin
Obecnie większość cytokin można pozyskać na szeroką skalę z zastosowaniem technologii rekombinacji genów. Umieszczenie takich genów w bakteriach (Escherichia. coli), drożdżach lub komórkach jajników chomika umożliwia produkcję pożądanych cytokin w wymaganych ilościach. Dzięki temu możliwe jest ich zastosowanie w terapii chorób nowotworowych i stanach niewydolności hematopoezy.
Obecnie dostępnych jest kilka preparatów rekombinowanego INF-α: Intron A (Schering Plough), Roferon A (Hoffmann La Roche) czy Wellferon (Glaxo Wellcome), będący naturalnym ludzkim INF-α. Są to jednorodne białka o stopniu czystości 95%. Zawierają one 166 aminokwasów i różnią się między sobą rodzajem aminokwasu w pozycji 24. Naturalny ludzki INF-α. otrzymywany jest z ludzkich komórek limfoblastoidalnych, stymulowanych wirusem Sendai i stanowi mieszaninę różnych podtypów INF-α. Wszystkie komercyjne preparaty mają podobne wskazania i znajdują zastosowanie w leczeniu nowotworów i przewlekłego WZW.
Receptor Tyrosine Kinases and Ras
The receptor tyrosine kinases (RTKs) are the second major type of cell-surface receptors that we discuss in detail in this chapter (see Figure 20-3d, right). The ligands for RTKs are soluble or membrane-bound peptide/protein hormones including nerve growth factor (NGF), platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), epidermal growth factor (EGF), and insulin. Binding of a ligand to this type of receptor stimulates the receptor’s intrinsic protein-tyrosine kinase activity, which subsequently stimulates a signal-transduction cascade leading to changes in cellular physiology and/or patterns of gene expression (see Figure 20-6). RTK signaling pathways have a wide spectrum of functions including regulation of cell proliferation and differentiation, promotion of cell survival (Section 23.8), and modulation of cellular metabolism.
Some RTKs have been identified in studies on human cancers associated with mutant forms of growth-factor receptors, which send a proliferative signal to cells even in the absence of growth factor. One such mutant receptor, encoded at the neu locus, contributes to the uncontrolled proliferation of certain human breast cancers (Section 24.3). Other RTKs have been uncovered during analysis of developmental mutations that lead to blocks in differentiation of certain cell types in C. elegans, Drosophila, and the mouse.

In this section we discuss activation of RTKs and how they transmit a hormone signal to Ras, the GTPase switch protein that functions in transducing signals from many different RTKs. The second part of RTK-Ras signaling pathways, the transduction of signals downstream from Ras to a common cascade of serine/threonine kinases, is covered in the next section.
Go to:
Ligand Binding Leads to Autophosphorylation of RTKs
All RTKs comprise an extracellular domain containing a ligand-binding site, a single hydrophobic transmembrane α helix, and a cytosolic domain that includes a region with protein-tyrosine kinase activity. Binding of ligand causes most RTKs to dimerize; the protein kinase of each receptor monomer then phosphorylates a distinct set of tyrosine residues in the cytosolic domain of its dimer partner, a process termed autophosphorylation (Figure 20-21). Autophosphorylation occurs in two stages. First, tyrosine residues in the phosphorylation lip near the catalytic site are phosphorylated. This leads to a conformational change that facilitates binding of ATP in some receptors (e.g., the insulin receptor) and binding of protein substrates in other receptors (e.g., FGF receptor). The receptor kinase activity then phosphorylates other sites in the cytosolic domain; the resulting phosphotyrosines serve as docking sites for other proteins involved in RTK-mediated signal transduction.
Figure 20-21
General structure and activation of receptor tyrosine kinases (RTKs). The ligands for some RTKs, such as the receptor for EGF depicted here, are monomeric; ligand binding induces a conformational (more...)
As described later, the subunits of some RTKs, including the insulin receptor, are covalently linked. Although these receptors exist as dimers or tetramers even in the absence of ligand, binding of ligand is required for autophosphorylation to occur. Presumably, ligand binding induces a conformational change that activates the kinase.
The phosphotyrosine residues in activated RTKs interact with adapter proteins containing SH2 or PTB domains. These proteins couple the activated receptors to other components of the signal-transduction pathway but have no intrinsic signaling properties. Before examining the structure and function of adapter proteins, we discuss the role of Ras, the other key signaling molecule in pathways triggered by activation of RTKs. As we will see later, several membrane-associated enzymes that function in signal transduction also bind to specific phosphotyrosines in activated RTKs.
Go to:
Ras and Gα Subunits Belong to the GTPase Superfamily of Intracellular Switch Proteins
Ras is a GTP-binding switch protein that, like the Gα subunits in different G proteins, alternates between an active on state with a bound GTP and an inactive off state with a bound GDP. As discussed in the previous section, Gα is directly coupled to GPCRs and transduces signals to various effectors such as adenylyl cyclase. In contrast, Ras is not directly linked to RTKs.
Activation of both Ras and Gα is triggered by hormone binding to an appropriate cell-surface receptor. Ras activation is accelerated by a protein called guanine nucleotide – exchange factor (GEF), which binds to the Ras · GDP complex, causing dissociation of the bound GDP (Figure 20-22). Because GTP is present in cells at a higher concentration than GDP, GTP binds spontaneously to “empty” Ras molecules, with release of GEF. In contrast, formation of an active Gα · GTP complex does not require an exchange factor.
Figure 20-22
Cycling of the Ras protein between the inactive form with bound GDP and the active form with bound GTP occurs in four steps. By mechanisms discussed later, binding of certain growth factors to (more...)
Hydrolysis of the bound GTP deactivates both Ras and Gα. The average lifetime of a GTP bound to Ras is about 1 minute, which is much longer thanthe lifetime of Gα · GTP. The reason for this difference is that the deactivation of Ras, unlike the deactivation of Gα, requires the assistance of another protein: a GTPase-activating protein (GAP), which binds to Ras · GTP and accelerates its intrinsic GTPase activity by a hundredfold (see Figure 20-22). Mammalian Ras proteins have been studied in great detail because mutant Ras proteins are associated with many types of human cancer. These mutant proteins, which bind but cannot hydrolyze GTP, are permanently in the “on” state and cause neoplastic transformation (Chapter 24).

The differences in the cycling mechanisms of Ras and Gα are reflected in their structures. Ras (≈170 amino acids) is smaller than Gα proteins (≈300 amino acids), but its three-dimensional structure is similar to that of the GTPase domain of Gα. Recent structural and biochemical studies show that Gα also contains another domain, a helical domain that apparently functions like GAP to increase the rate of GTP hydrolysis by Gα (see Figure 20-19). In addition, the direct interaction between an activated receptor and Gα · GDP promotes release of GDP and binding of GTP, so that a separate exchange factor is not required.
Both Gsα and Ras are members of a family of intracellular GTP-binding switch proteins collectively referred to as the GTPase superfamily. This family also includes other Gα subunits (e.g., Giα), the Rab proteins which regulate fusion of vesicles within cells (Section 17.10), and the Rho family proteins which regulate the actin cytoskeleton (Section 18.2). The many similarities between the structure and function of Ras and Gsα, and the identification of both proteins in all eukaryotic cells, indicate that a single type of signaltransducing GTPase originated very early in evolution. The gene encoding this protein subsequently duplicated and evolved to the extent that cells today contain a superfamily of such GTPases, comprising perhaps a hundred different intracellular switch proteins. These related proteins control many aspects of cellular growth and metabolism.
Go to:
An Adapter Protein and GEF Link Most Activated RTKs to Ras
The first indication that Ras functioned downstream from RTKs in a common signaling pathway came from experiments in which cultured fibroblast cells were induced to proliferate by treatment with a mixture of platelet-derived growth factor (PDGF) and epidermal growth factor (EGF). Microinjection of anti-Ras antibodies into these cells blocked cell proliferation. Conversely, injection of a constitutively active mutant Ras protein (i.e., RasD), which hydrolyzes GTP very inefficiently and thus persists in the active state, caused the cells to proliferate in the absence of the growth factors. These findings are consistent with studies showing that addition of fibroblast growth factor (FGF) to fibroblasts leads to a rapid increase in the proportion of Ras present in the GTP-bound active form.
But how does binding of a growth factor (e.g., EGF) to an RTK (e.g., the EGF receptor) lead to activation of Ras? Two cytosolic proteins — GRB2 and Sos — provide the key links (Figure 20-23). An SH2 domain in GRB2 binds to a specific phosphotyrosine residue in the activated receptor. GRB2 also contains two SH3 domains, which bind to and activate Sos. GRB2 thus functions as an adapter protein for the EGF receptor. Sos functions as a guanine nucleotide – exchange protein (GEF), which helps convert inactive GDP-bound Ras to the active GTP-bound form.
Figure 20-23
Activation of Ras following binding of a hormone (e.g., EGF) to an RTK. The adapter protein GRB2 binds to a specific phosphotyrosine on the activated RTK and to Sos, which in turn interacts (more...)
Genetic analysis of mutants blocked at particular stages of differentiation have provided considerable insight into RTK signaling pathways. Most of these genetic studies were done in the worm C. elegans and in the fly Drosophila. Mutants in these species in which development of specific cells is blocked were particularly useful in elucidating the pathway from an RTK to Ras shown in Figure 20-23. To illustrate the power of this experimental approach, we consider development of a particular type of cell in the compound eye of Drosophila.
The compound eye of the fly is composed of some 800 individual eyes called ommatidia (Figure 20-24a). Each ommatidium consists of 22 cells, eight of which are photosensitive neurons called retinula, or R cells, designated R1 – R8 (Figure 20-24b). An RTK called Sevenless (Sev) specifically regulates development of the R7 cell. In flies with a mutant sevenless (sev) gene, the R7 cell in each ommatidium does not form (Figure 20-24c). Since the R7 photoreceptor is necessary for flies to see in ultraviolet light, mutants that lack functional R7 cells are easily isolated.
Figure 20-24
The compound eye of Drosophila melanogaster. (a) Scanning electron micrograph showing individual ommatidia that compose the fruit fly eye. (b) Longitudinal and cut-away views of a (more...)

During development of each ommatidium, a protein called Boss (Bride of Sevenless) is expressed on the surface of the R8 cell. This membrane-bound protein binds to the Sev RTK on the surface of the neighboring R7 precursor cell, signaling it to develop into a photosensitive neuron (Figure 20-25a). In mutant flies that do not express a functional Sev RTK, interaction between the Boss and Sev proteins cannot occur, and no R7 cells develop (Figure 20-25b).
Figure 20-25
Genetic analysis of induction of the R7 photoreceptor in the Drosophila eye. (a) During larval development of wild-type flies, the R8 cell (tan) in each developing ommatidium expresses (more...)
To identify intracellular signal-transducing proteins in the Sev RTK pathway, investigators produced mutant flies expressing a temperature-sensitive Sev protein. When these flies were maintained at a permissive temperature, all their ommatidia contained R7 cells; when they were maintained at a nonpermissive temperature, no R7 cells developed. At a particular intermediate temperature, however, just enough of the Sev RTK was functional to mediate normal R7 development. The investigators reasoned that at this intermediate temperature, the signaling pathway would become defective, and thus no R7 cells would develop, if the level of another protein involved in the pathway was reduced. A recessive mutation affecting such a protein would have this effect because, in diploid organisms like Drosophila, a heterozygote containing one wild-type and one mutant allele of a gene will produce half the normal amount of the gene product; hence even if such a recessive mutation is in an essential gene, the organism will be viable. However, a fly carrying a temperature-sensitive mutation in the sev gene and a second mutation affecting another protein in the signaling pathway would be expected to lack R7 cells at the intermediate temperature.
By use of this screen, researchers identified genes encoding three important proteins in the Sev pathway (see Figure 20-23):
A Ras protein exhibiting 80 percent identity with its mammalian counterparts A GEF called Sos (Son of Sevenless) exhibiting 45 percent identity with its mouse counterpart An SH2-containing adapter protein exhibiting 64 percent identity to human GRB2
These three proteins have been found to function in other RTK signal-transduction pathways initiated by ligand binding to different receptors and used at different times and places in the developing fly. Recessive lethal mutations in these essential genes can be identified by the strategy described here much more easily than by the procedure described in Chapter 8 (see Figure 8-10).
In subsequent studies, researchers introduced a mutant rasD gene into fly embryos carrying the sevenless mutation. As noted earlier, the rasD gene encodes a Ras protein that has reduced GTPase activity and hence is present in the active GTP-bound form even in the absence of a hormone signal. Although no functional Sev RTK was expressed in these double-mutants (sev−; rasD), R7 cells formed normally, indicating that activation of Ras is sufficient for induction of R7-cell development (Figure 20-25c). This finding is consistent with the results with cultured fibroblasts described earlier.
Go to:
SH2 Domain in GRB2 Adapter Protein Binds to a Specific Phosphotyrosine in an Activated RTK
To identify proteins that associate with phosphotyrosine residues in the cytosolic domain of activated EGF receptors, scientists used an expression cloning strategy. cDNAs synthesized from mRNAs isolated from human brain-stem tissue were inserted into a λgt11 expression vector, which then was plated on a lawn of E. coli cells (see Figure 7-21). When the resulting cDNA library was screened using a fragment of phosphorylated human EGF receptor as the probe, two cDNA clones were identified. One encoded a subunit of PI-3 kinase that contains an SH2 domain and the other encoded GRB2, a homolog of the SH2-containing adapter protein identified in the Drosophila Sev pathway. Thus GRB2 and its Drosophila homolog are adapter proteins that function downstream from RTKs but upstream of Ras in both flies and mammalian cells.
GRB2 and similar adapter proteins bind to different phosphotyrosine residues on RTKs via the conserved SH2 domain. This domain derived its full name, the Src homology 2 domain, from its homology with a region in the prototypical cytosolic tyrosine kinase encoded by src. The three-dimensional structures of SH2 domains in different proteins are very similar. Each binds to a distinct sequence of amino acids surrounding a phosphotyrosine residue. The unique amino acid sequence of each SH2 domain determines the specific phosphotyrosine residues it binds. The SH2 domain of the Src tyrosine kinase, for example, binds strongly to any peptide containing the critical core sequence of phosphotyrosine – glutamic acid – glutamic acid – isoleucine (Figure 20-26a). These four amino acids make intimate contact with the peptide-binding site in the Src SH2 domain. Binding resembles the insertion of a two-pronged “plug” — the phosphotyrosine and isoleucine residues of the peptide — into a two-pronged “socket” in the SH2 domain. The two glutamic acids fit snugly onto the surface of the SH2 domain between the phosphotyrosine socket and the hydrophobic socket that accepts the isoleucine residue. Variations in the nature of the hydrophobic socket in different SH2 domains allow them to bind to phosphotyrosines adjacent to different sequences, accounting for differences in their binding specificity.
Figure 20-26
Models of SH2 and SH3 domains bound to short target peptides. The peptides, positioned above the SH2 and SH3 domains, are shown as space-filling models with the backbone in yellow and the (more...)
Activated RTKs also can recruit signaling molecules through a different domain called the phosphotyrosinebinding (PTB) domain. While SH2-binding specificity is largely determined by residues C-terminal to the phosphotyrosine, PTB-binding specificity is determined by specific residues five to eight residues N-terminal to the phosphotyrosine residue.
Go to:

Sos, a Guanine Nucleotide – Exchange Factor, Binds to the SH3 Domains in GRB2
In addition to one SH2 domain, which binds to phosphotyrosine residues in RTKs, GRB2 contains two SH3 domains, which bind to Sos, a guanine nucleotide – exchange factor. SH3 domains, which contain ≈55 – 70 residues, are present in a large number of proteins involved in intracellular signaling. Although the three-dimensional structures of various SH3 domains are similar, their specific amino acid sequences differ. SH3 domains selectively bind to proline-rich sequences in Sos and other proteins; different SH3 domains bind to different proline-rich sequences.
Proline residues play two roles in the interaction between an SH3 domain in an adapter protein (e.g., GRB2) and a proline-rich sequence in another protein (e.g., Sos). First, the proline-rich sequence assumes an extended conformation that permits extensive contacts with the SH3 domain, thereby facilitating interaction. Second, a subset of these prolines fit into binding “pockets” on the surface of the SH3 domain (Figure 20-26b). Several nonproline residues also interact with the SH3 domain and are responsible for determining the binding specificity. Hence the binding of peptides to SH2 and SH3 domains follows a similar strategy: certain residues provide the overall structural motif necessary for binding, and neighboring residues confer specificity to the binding.
Following hormone-induced activation of an RTK (e.g., the EGF receptor), a complex containing the activated RTK, GRB2, and Sos is formed on the cytosolic face of the plasma membrane (see Figure 20-23). Formation of this complex depends on the dual binding ability of GRB2. Receptor activation thus leads to relocalization of Sos from the cytosol to the membrane, bringing Sos near to its substrate, membrane-bound Ras · GDP. Biochemical and genetic studies indicate that the C-terminus of Sos inhibits its nucleotideexchange activity and that GRB2 binding relieves this inhibition. Binding of Sos to Ras · GDP leads to changes in the conformation of two regions of Ras, switch I and switch II, thereby opening the binding pocket for GDP so it can diffuse out (Figure 20-27). Because GTP is present in cells at a concentration some 10 times higher than GDP, GTP binding occurs preferentially, leading to activation of Ras. The activation of Ras and Gsα thus occur by similar mechanisms: a conformational change induced by binding of a protein — Sos and an activated GPCR, respectively — that opens the protein structure so bound GDP is released to be replaced by GTP. As we discuss in the next section, binding of GTP to Ras, in turn, induces a specific conformation of switch I and II that allow Ras · GTP to activate downstream effector molecules.
Figure 20-27
Structures of Ras · GDP-Sos complex and Ras · GTP determined by x-ray crystallography. (a) Sos (shown as a trace diagram) binds to two switch regions of Ras · GDP, (more...)
Several other proteins, including GAP, bind to specific phosphotyrosines in activated RTKs. This binding localizes GAP close to Ras · GTP, so it can promote the cycling of Ras (see Figure 20-22); exactly how GAP interacts with Ras and perhaps other components of the RTK-Ras pathway is unclear.
Go to:
SUMMARY
Receptor tyrosine kinases (RTKs), which bind to peptide/protein hormones, may exist as dimers or dimerize during binding to ligands. Ligand binding leads to activation of the kinase activity of the receptor and autophosphorylation of tyrosine residues in its cytosolic domain (see
Figure 20-31). The activated receptor also can phosphorylate other protein substrates. Ras is an intracellular GTPase switch protein that acts downstream from most RTKs. Like Gsα, Ras cycles between an inactive GDP-bound form
and active GTP-bound form. Ras cycling requires the assistance of two proteins, GEF and GAP, (see Figure 20-22), whereas Gsα cycling does not. Unlike GPCRs, which interact directly with an associated G protein, RTKs are linked indirectly to Ras via two proteins, GRB2 and Sos (see Figure
20-23). The SH2 domain in GRB2, an adapter protein, binds to specific phosphotyrosines in activated RTKs. The two SH3 domains in GRB2 then bind
Sos, a guaninenucleotide exchange factor, thereby bringing Sos close to membrane-bound Ras · GDP and activating its exchange function. Binding of Sos to inactive Ras causes a large conformational change that permits release of GDP and binding of GTP. Normally, Ras activation and the subsequent cellular response is induced by ligand binding to an RTK. However, in cells that contain a
constitutively active Ras, the cellular response occurs in the absence of ligand binding.


















