







Języki
Strony
Prawny


Teorie klasyfikacji

klasyfikacja fenetyczna (numeryczna)
• opiera się na filozofii nominalistycznej
• taksony tworzone są wyłącznie na podstawie
zaobserwowanego podobieństwa
• systematyka powinna być wolna od wszelkiej teorii
(a zwłaszcza od teorii ewolucji)
• filogeneza jako ciąg zdarzeń jest niepoznawalna
• analizowane jest ogólne podobieństwo, maksimum
dostępnych cech, niezależnie od ich natury
• wszystkie cechy mają tą samą wagę

klasyfikacja fenetyczna (numeryczna)
• fenogram nie ma wymiaru historycznego
• obliczana jest „odległość” między gatunkami w dowolnej
liczbie wymiarów
• rezultatem jest dendrogram ilustrujący stosunki podobieństwa
fenetycznego - fenogram
• końcowe taksony to tzw. OTU (operational taxonomic units)

nowa systematyka
Huxley J.S. 1940. The New Systematics. London
• gatunek jest złożony z populacji
• zmienność populacji jest kluczowa dla procesu
ewolucyjnego
• zmienność populacji jest oparta o zmienność genetyczną
testowanie wyników morfologicznych w oparciu
o dane ogólnobiologiczne

nowa nowa systematyka
• podstawową jednostką systematyki jest populacja
• zmienność jest zasadniczym elementem istoty
i definicji populacji
• populacje są systemami dynamicznymi, które ewoluują
Gatunek można definiować wyłącznie w kategoriach dynamiki,
ewolucji, genetyki, zależności wewnątrzpopulacyjnych, a nie
w kategoriach stałych struktur morfologicznych

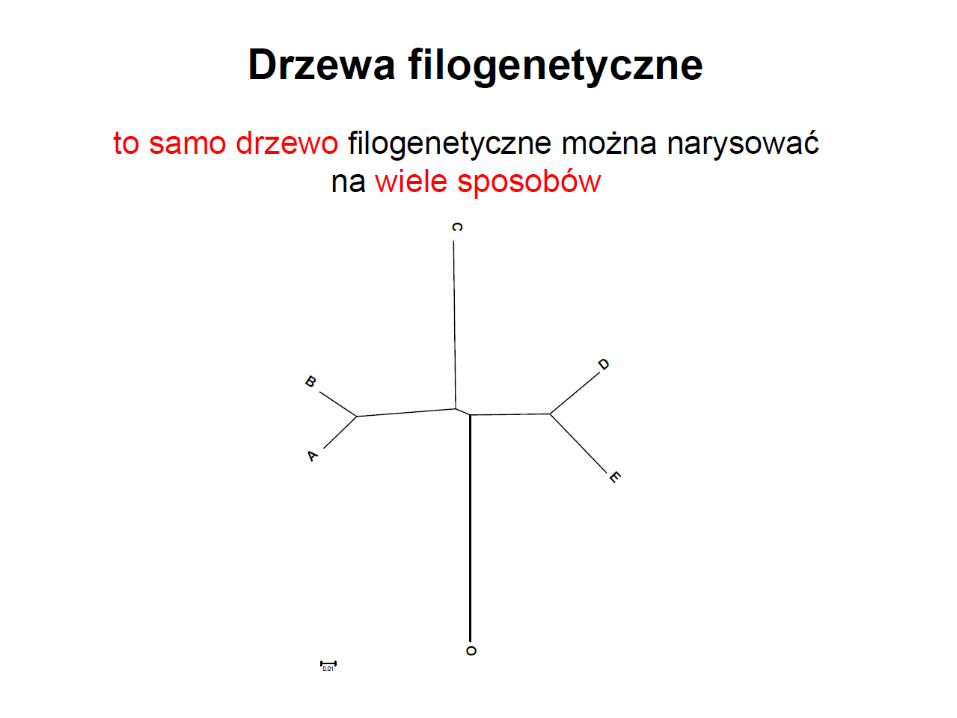
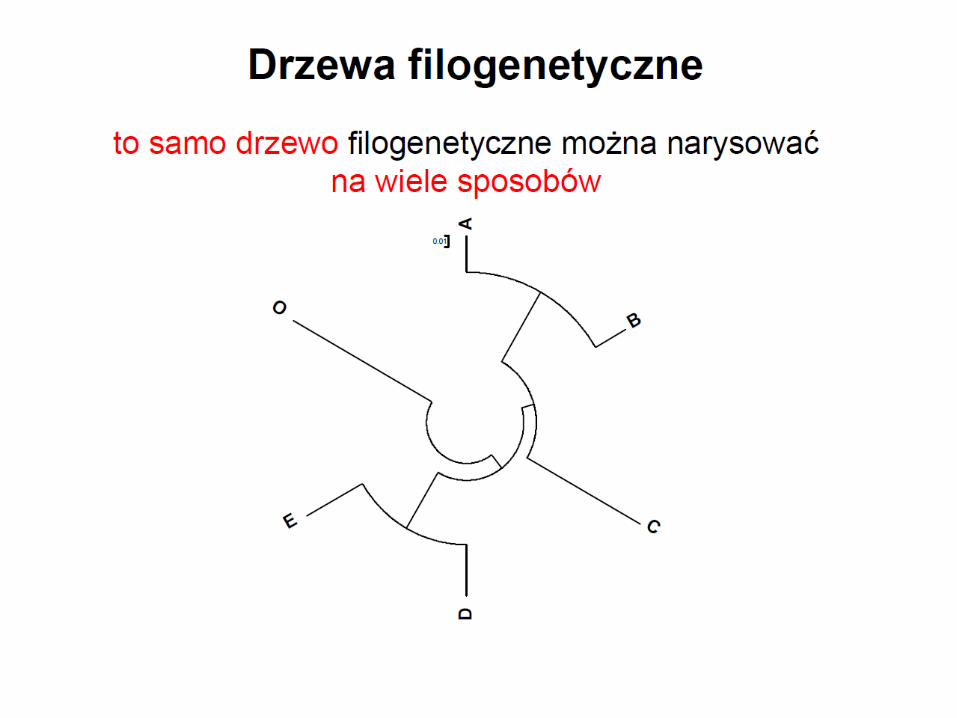
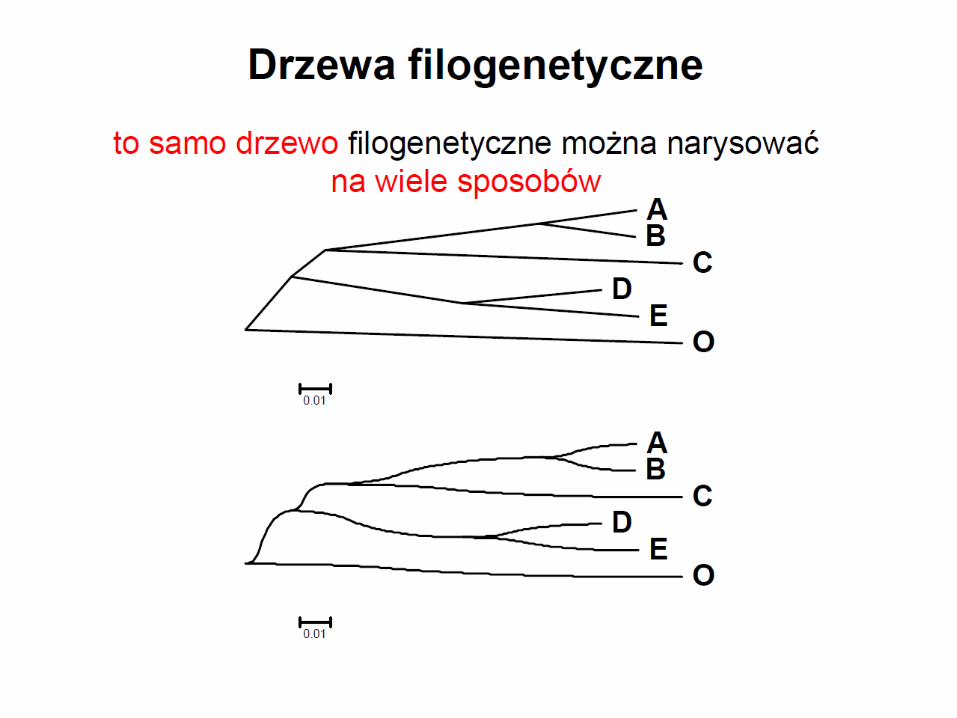



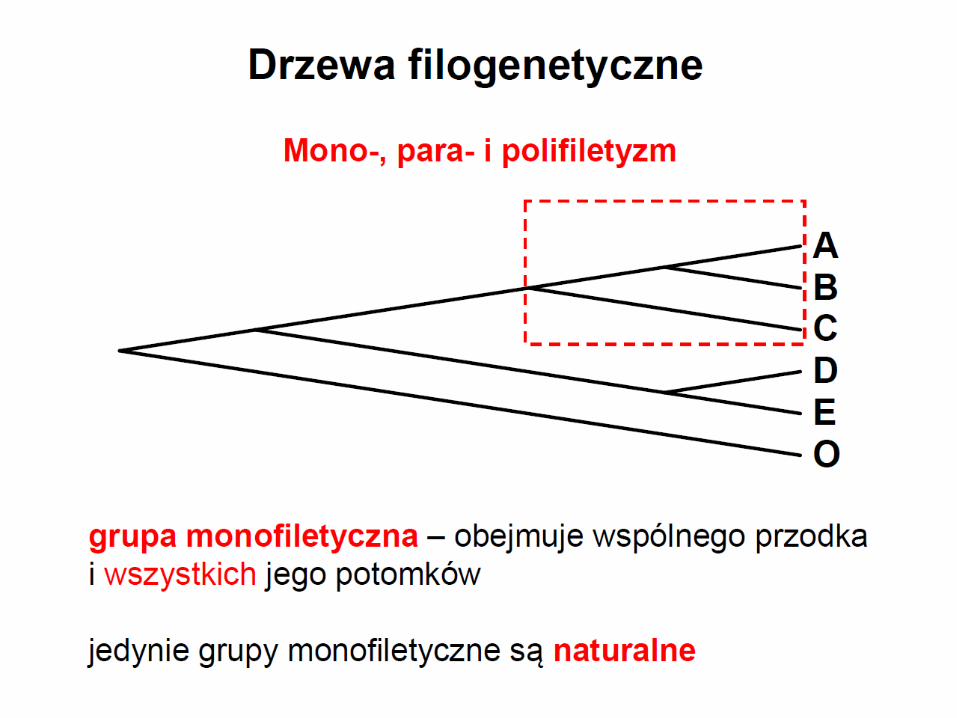
Monofiletyzm
grupa monofiletyczna zawiera swój takson macierzysty
i wszystkich jego potomków
wszyscy członkowie grupy musza posiadać co najmniej
jedną cechę apomorficzną

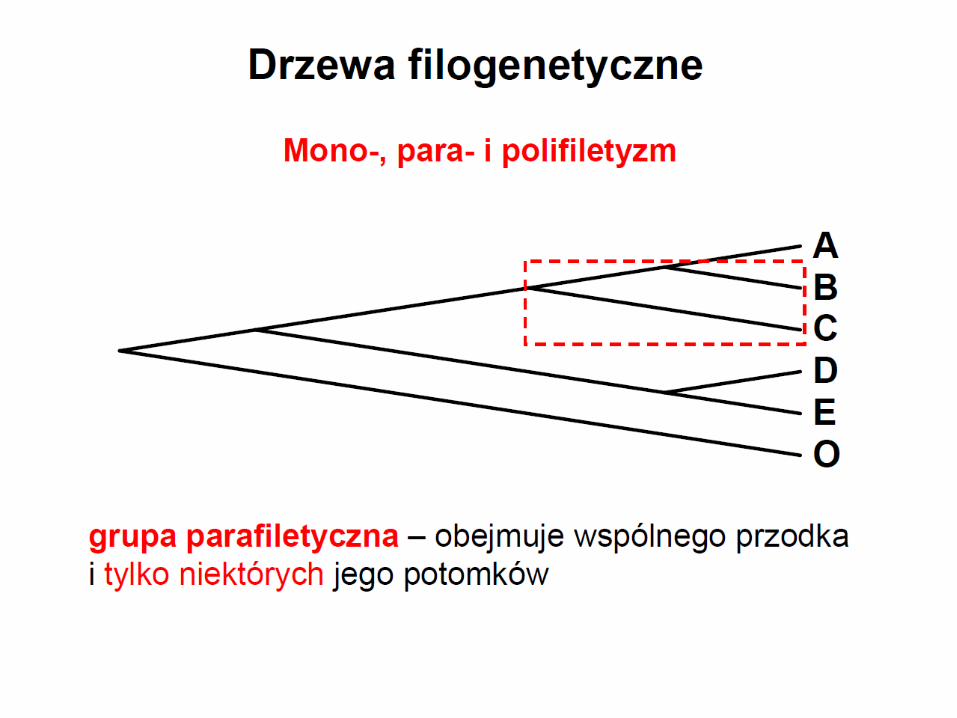
Parafiletyzm
grupa parafiletyczna zawiera swój takson macierzysty
i niektóych jego potomków

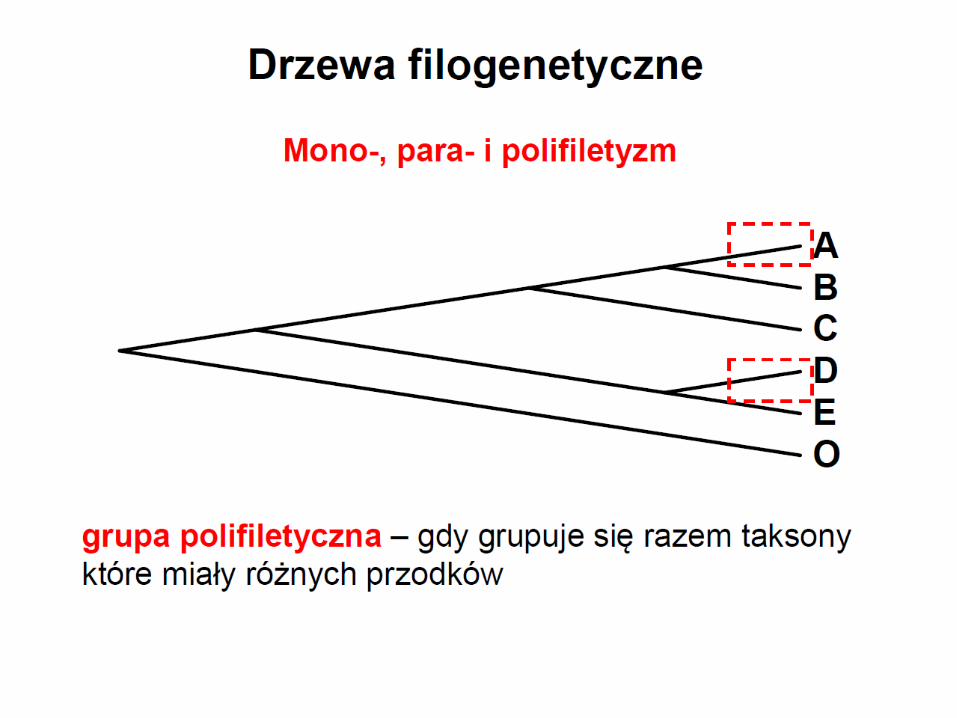
Polifiletyzm
grupa polifiletyczna zawiera potomków różnych przodków
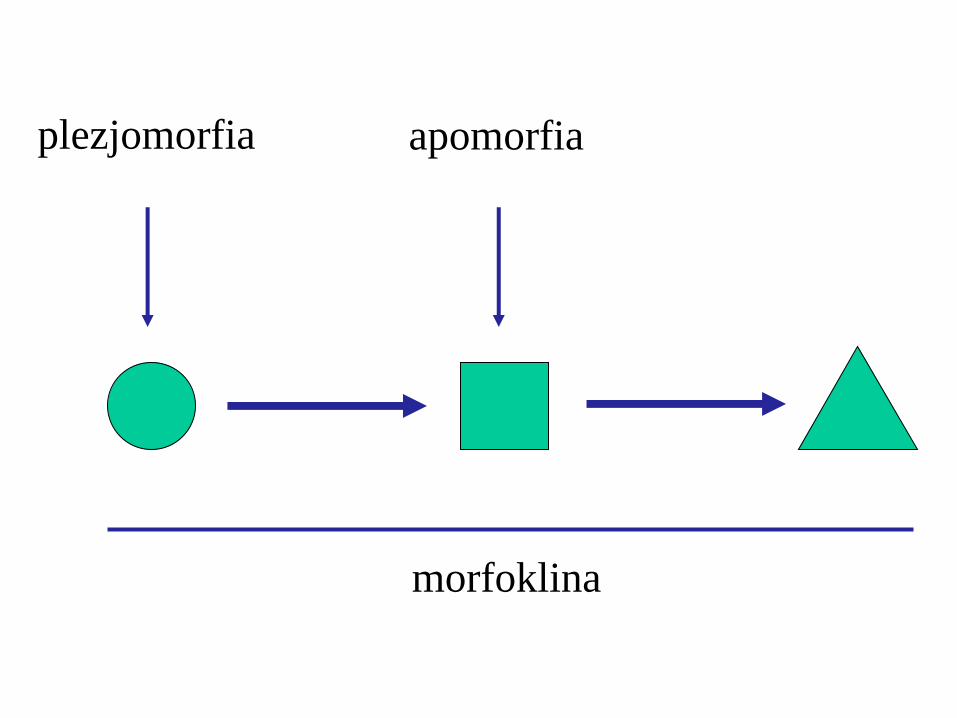
morfoklina
apomorfia plezjomorfia
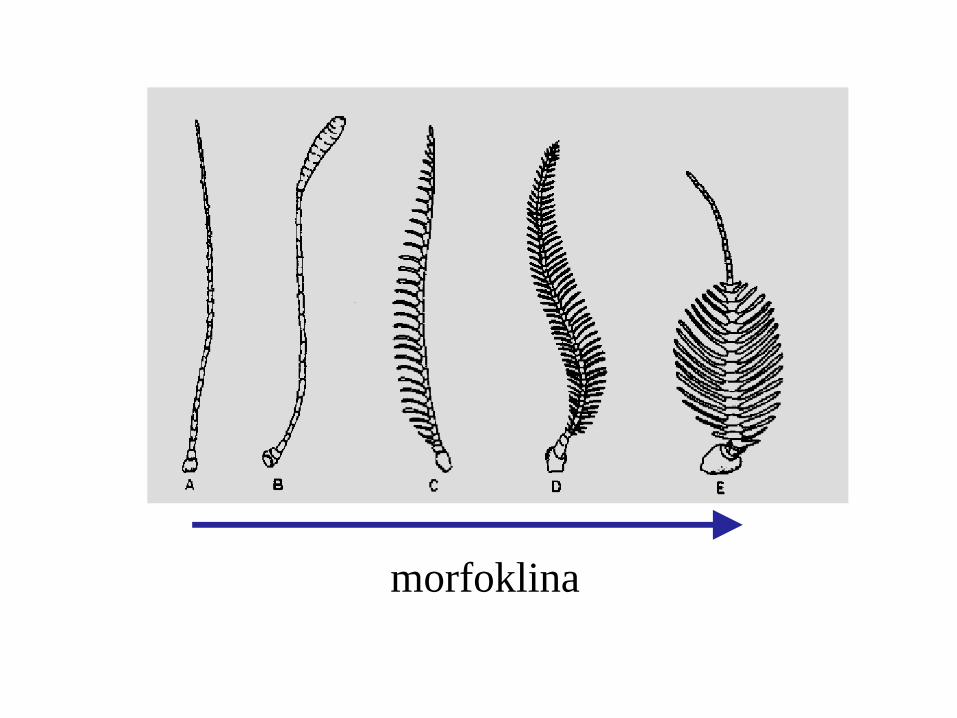
morfoklina
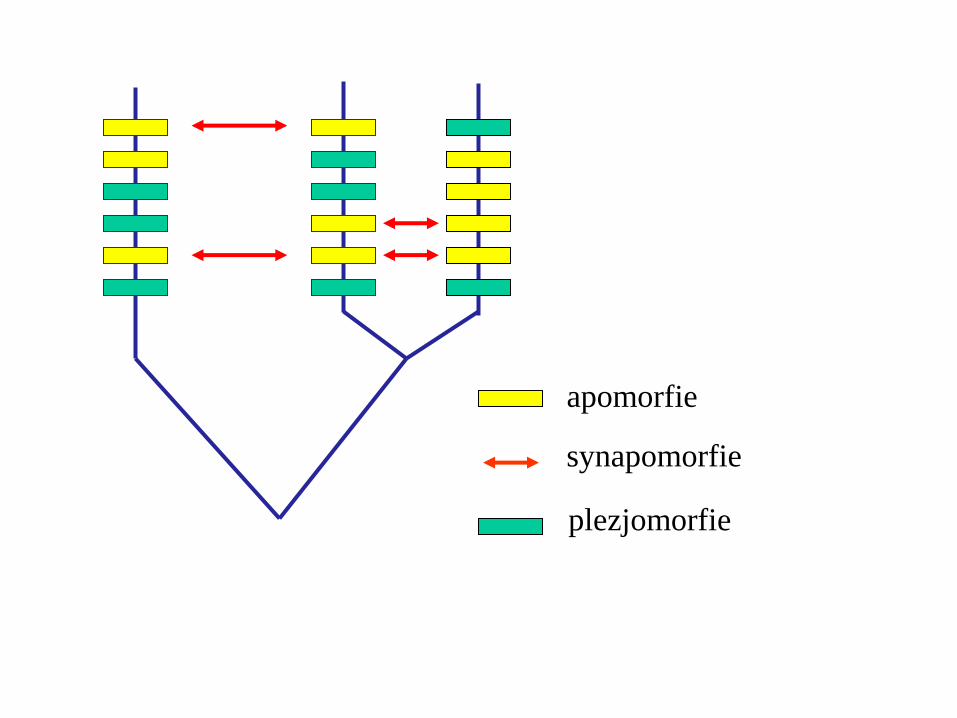
apomorfie
plezjomorfie
synapomorfie
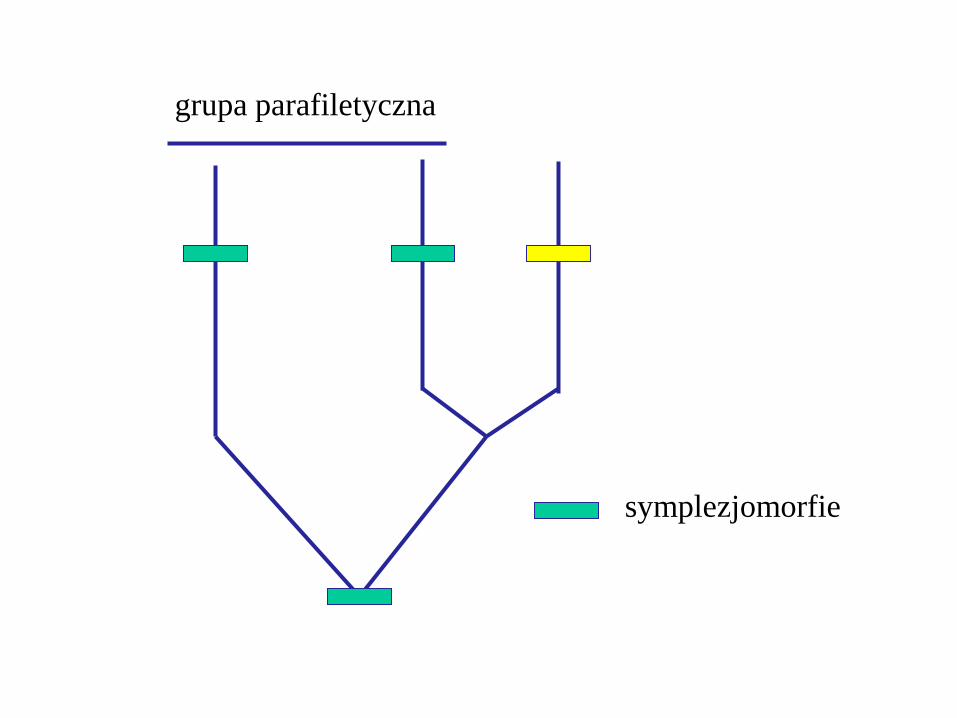
symplezjomorfie
grupa parafiletyczna

grupy siostrzane
dwie monofiletyczne grupy, pochodzące od
jednego gatunku macierzystego
razem tworzą grupę monofiletyczną
wyższej rangi
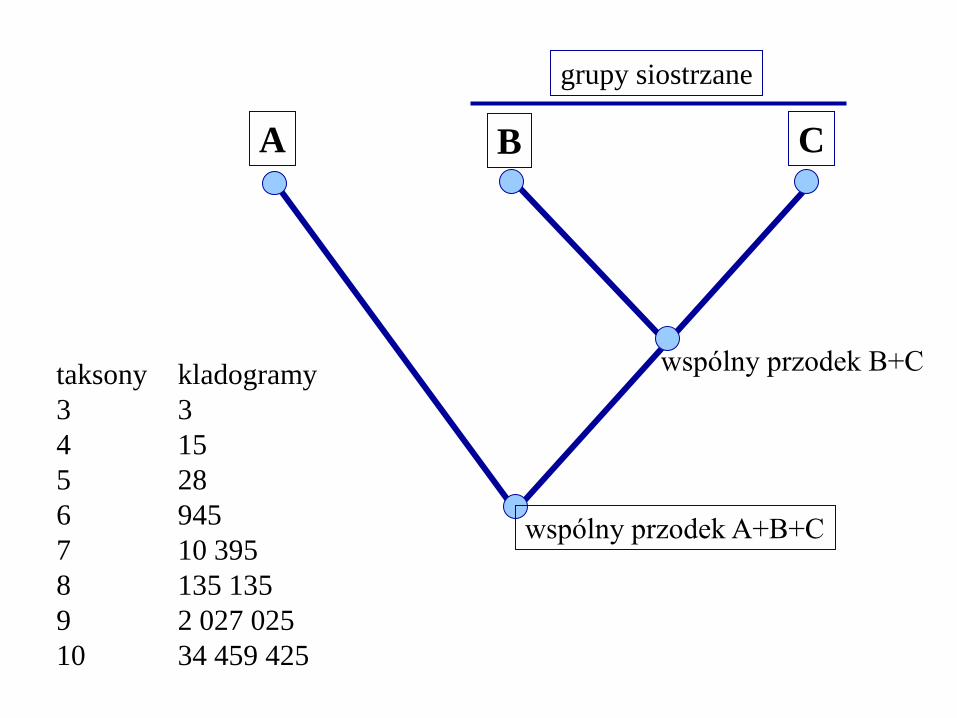
A B C
wspólny przodek B+C
wspólny przodek A+B+C
grupy siostrzane
taksony
3
4
5
6
7
8
9
10
kladogramy
3
15
28
945
10 395
135 135
2 027 025
34 459 425
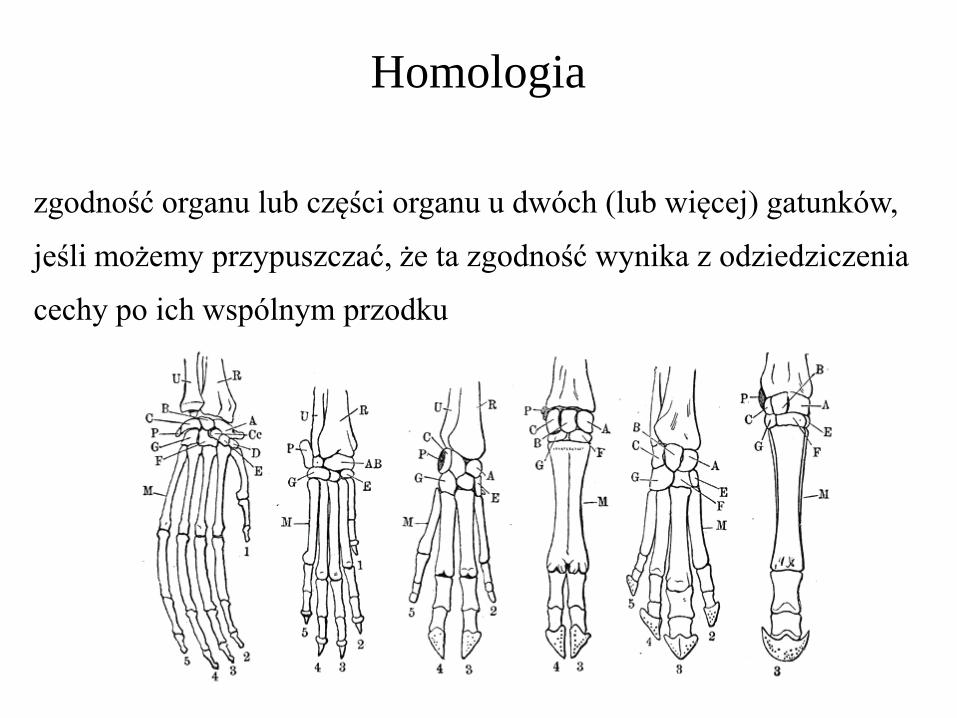
Homologia
zgodność organu lub części organu u dwóch (lub więcej) gatunków,
jeśli możemy przypuszczać, że ta zgodność wynika z odziedziczenia
cechy po ich wspólnym przodku
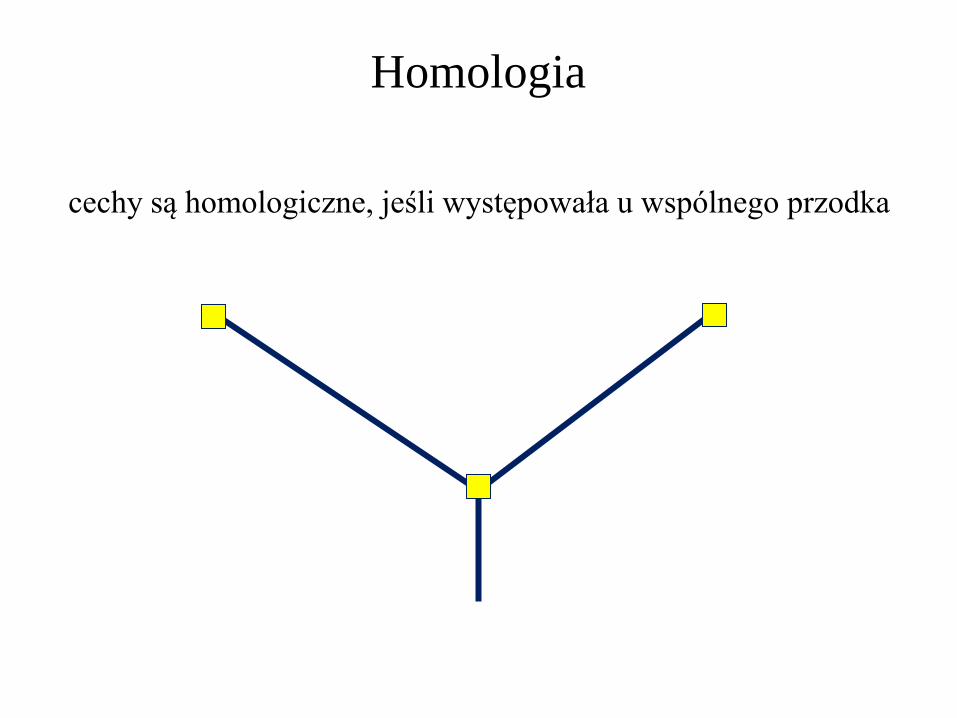
Homologia
cechy są homologiczne, jeśli występowała u wspólnego przodka
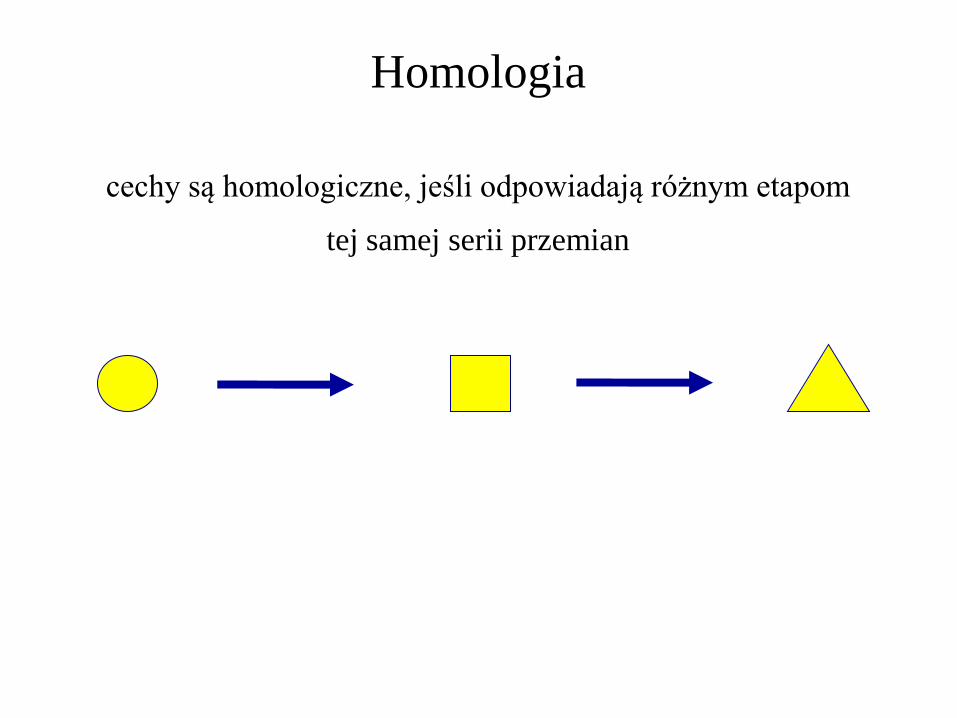
Homologia
cechy są homologiczne, jeśli odpowiadają różnym etapom
tej samej serii przemian

Homoplazja
występowanie u różnych taksonów podobnych cech
nie będących homologiami
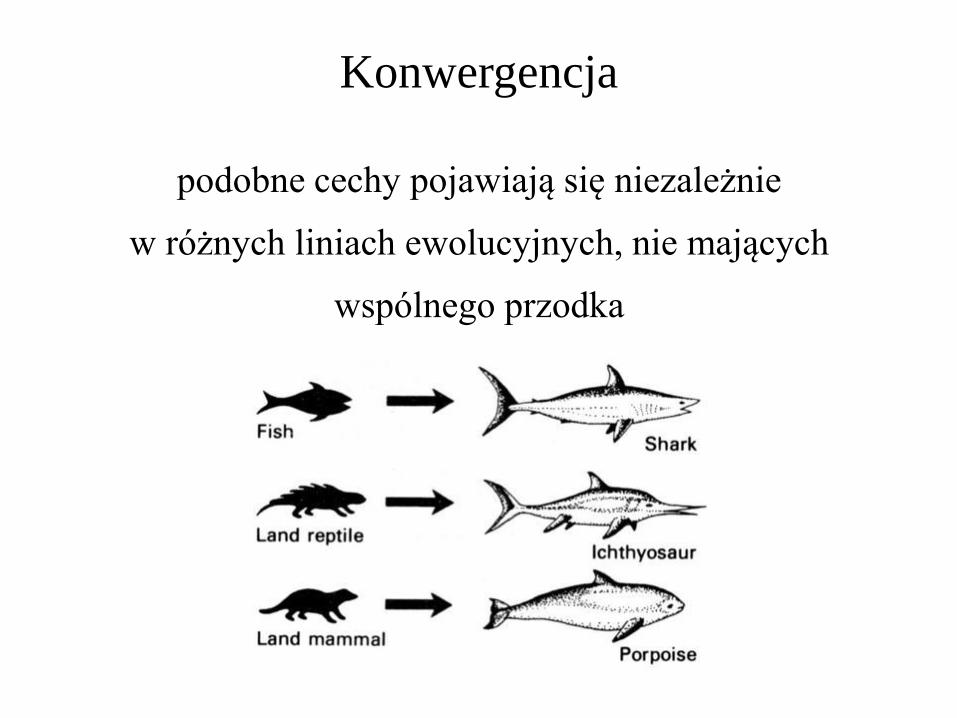
Konwergencja
podobne cechy pojawiają się niezależnie
w różnych liniach ewolucyjnych, nie mających
wspólnego przodka

Paralelizm
ta sama cecha pojawia się niezależnie w dwóch (lub więcej)
liniach mających bliskiego wspólnego przodka
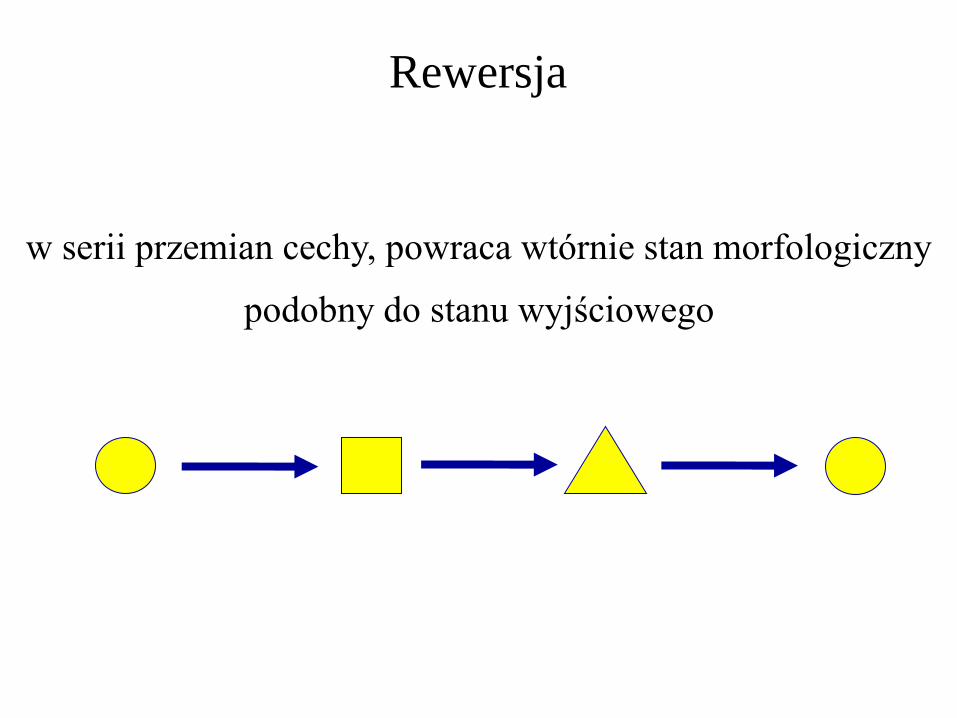
Rewersja
w serii przemian cechy, powraca wtórnie stan morfologiczny
podobny do stanu wyjściowego

Analogia
cechy konwergentne spełniają podobne funkcje

Analiza cech
polega na odtworzeniu serii przemian cech homologicznych
(morfoklin)

Kryterium embriologiczne
rozkład cech pojawiających się w ontogenezie wcześnie
jest bardziej ogólny (plezjomorfia)
niż cech pojawiających się późno (apomorfia)

Kryterium porównania pozagrupowego
jeśli istnieje cecha homologiczna spotykana wewnątrz
grupy monofiletycznej i również poza tą grupą,
to jest ona cechą plezjomorficzną
jeśli cecha homologiczna jest spotykana
wyłącznie wewnątrz badanej grupy monofiletycznej
to jest ona cechą apomorficzną

Kryterium porównania wewnątrzgrupowego
kryterium złożoności
jeśli różne stany cechy mogą być ułożone w serię
o wzrastającej złożoności, to jest prawdopodobne,
że stan najprostszy jest stanem plezjomorficznym

Kryterium porównania wewnątrzgrupowego
kryterium korelacji
niektóre cechy są ze sobą ściśle powiązane,
znajomość polaryzacji jednej morfokliny
może ułatwić określenie polaryzacji dla drugiej

Kryterium porównania wewnątrzgrupowego
kryterium powszechności
jeśli pewien stan cechy jest bardzo rozpowszechniony
w badanej grupie, a inny jest dużo rzadszy,
to stan częstszy jest prawdopodobnie
plezjomorficzny

Kryterium paleontologiczne
stan cechy pojawiający się w serii skamielin najwcześniej
jest najbardziej plezjomorficzny

Analiza parsymoniczna
zgodna z zasadą tzw. brzytwy Ockhama
najlepszy jest model filogenezy, który zakłada
najmniejszą liczbę zmian ewolucyjnych

kladyzm
• organizmy są szeregowane i klasyfikowane wyłącznie
w zależności od współczesności ich wspólnego pochodzenia
• status kategorii zależy od położenia punktów odgałęzienia
na drzewie filogenetycznym
• klasyfikacja musi być odbiciem genealogii
• taksony muszą być monofiletyczne

kladyzm
1. wszystkie organizmy rozmnażające się wyłącznie płciowo
mają tylko jedną historię genealogiczną (filogenezę)
2. cechy są przekazywane dziedzicznie z gatunku macierzystego
na gatunki potomne, a więc przynajmniej niektóre z cech
noszonych przez te ostatnie stanowią dowód pochodzenia
3. zmiany niektórych cech przodków nadają poszczególnym
liniom ich specyficzne właściwości

klasyfikacja ewolucyjna
• klasyfikacja ewolucyjna oparta jest na prostym fakcie,
że w przyrodzie występują łatwe do rozgraniczenia grupy
• taksonomia ewolucyjna wyjaśnia istnienie takich grup
gady ptaki ssaki
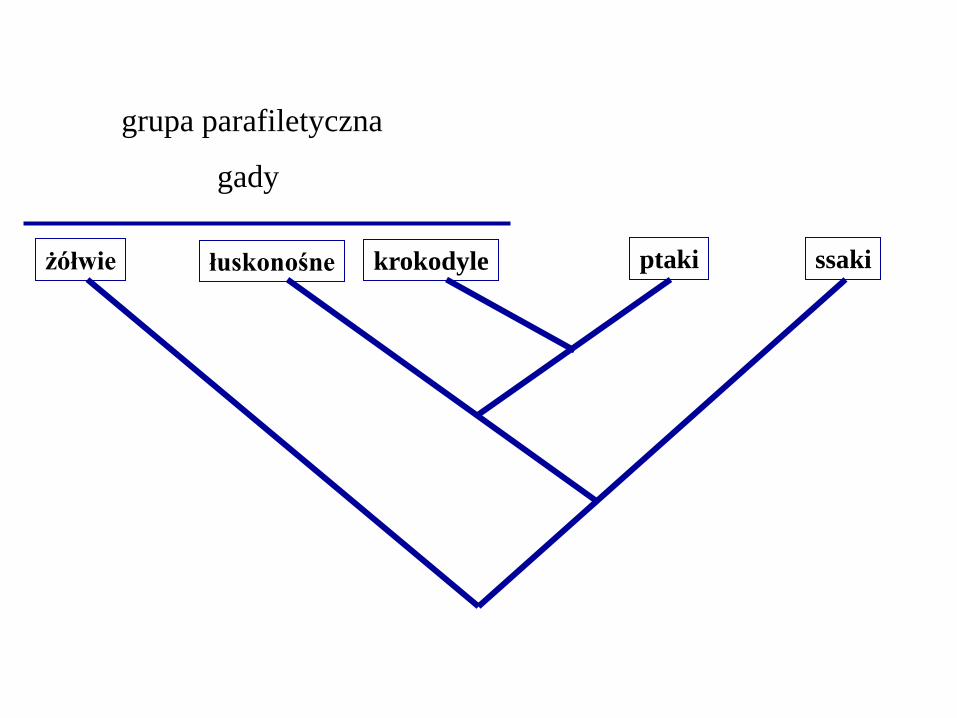
żółwie łuskonośne krokodyle ptaki ssaki
gady
grupa parafiletyczna
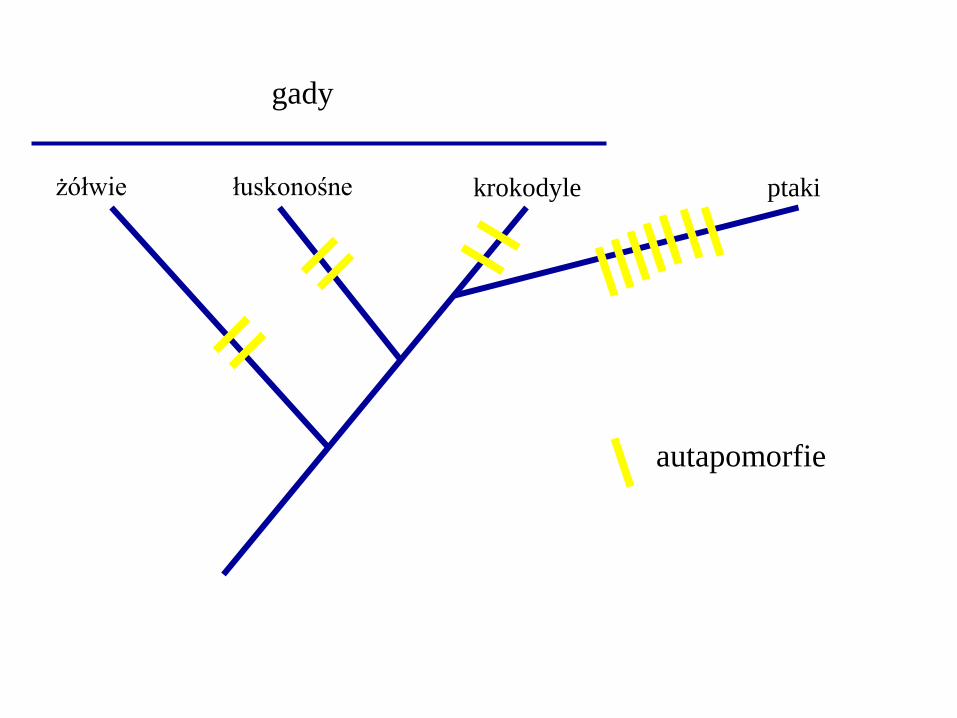
żółwie łuskonośne krokodyle ptaki
autapomorfie
gady

Metody molekularne
w taksonomii

Dwie szkoły w systematyce
Tradycyjna
Relacje pomiędzy organizmami opisywane są na podstawie
cech fenotypowych: morfo-anatomicznych, fizjologicznych,
behawioralnych itp...
Nowoczesna
Relacje pomiędzy organizmami opisywane są na podstawie
informacji pozyskanych bezpośrednio lub pośrednio z
sekwencji kwasów nukleinowych (lub protein).

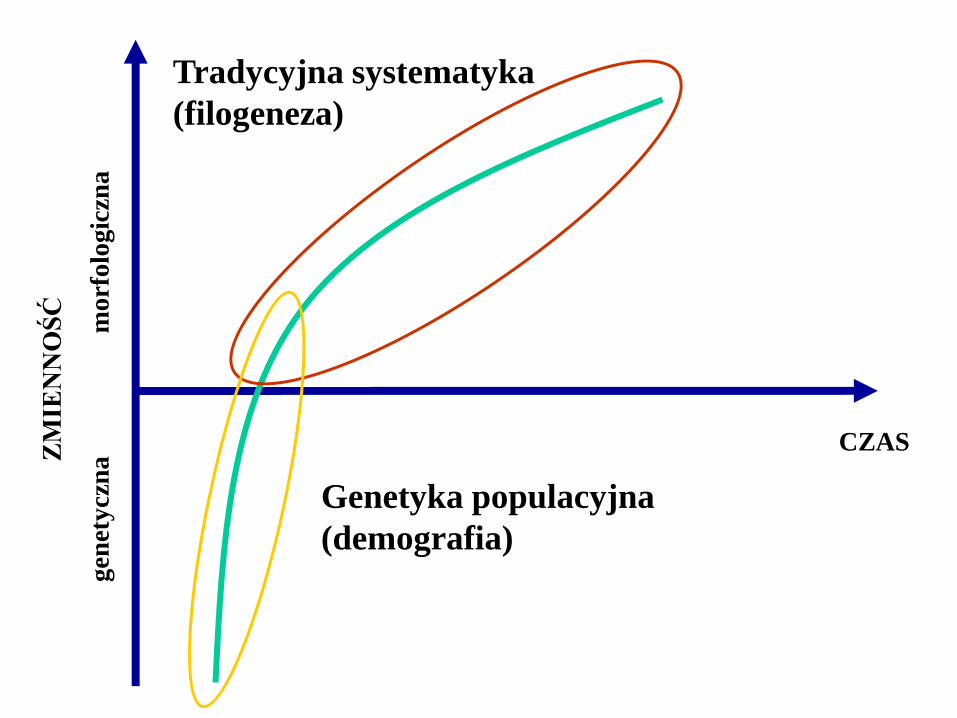
Systematyka tradycyjna
• Markery morfologiczne użyteczne
na wyższych poziomach systematycznych
• Zmiany mikroewolucyjne – domena genetyki
populacyjnej (demografii)
• Zmienność morfologiczna jest pochodną
nagromadzonej zmienności genetycznej
• Stosowane w zakresie zmian
makroewolucyjnych

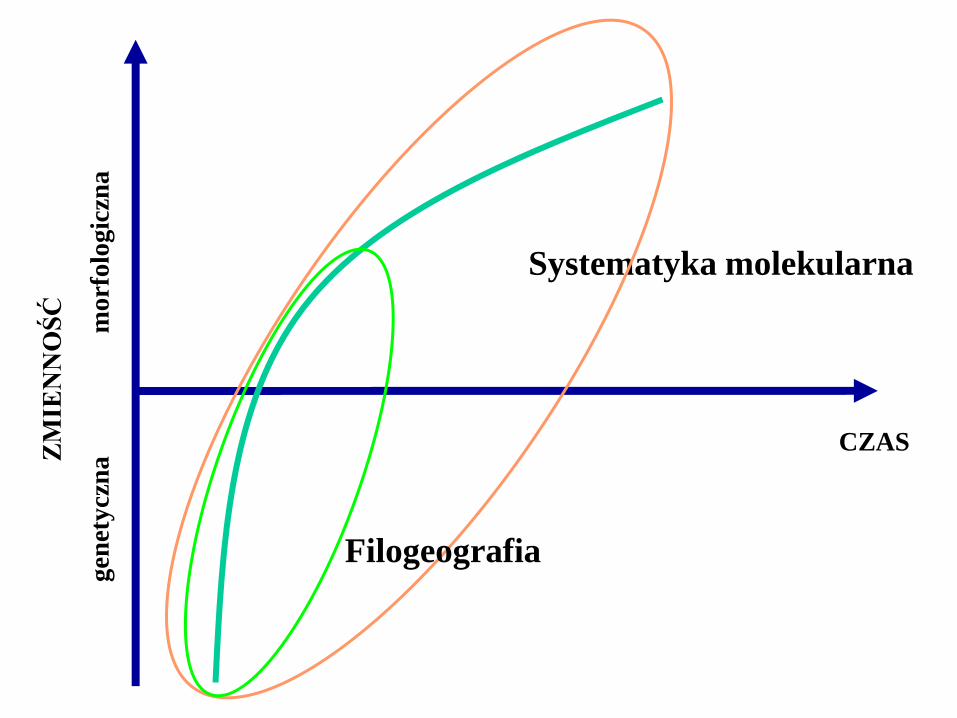
Systematyka molekularna
Pozwala badać relacje filogenetyczne
na poziomie wewnątrzgatunkowym
i wewnątrzpopulacyjnym
Filogeografia
Jest pomostem łączącym genetykę populacji i systematykę
Bada filogenezę organizmów na poziomie gatunku i populacji

Określa zasady i procesy leżące
u podstaw geograficznego rozmieszczenia
linii genealogicznych organizmów
(Avise i in. 1987)
Filogeografia
CZAS
morf
olo
gic
zna
Tradycyjna systematyka
(filogeneza)
Genetyka populacyjna
(demografia)
gen
ety
czn
a ZM
IEN
NO
ŚĆ
CZAS
morf
olo
gic
zna
Systematyka molekularna
gen
ety
czn
a ZM
IEN
NO
ŚĆ
Filogeografia

Dlaczego markery molekularne ?
genetyczny charakter danych
uniwersalna metodyka
praktycznie nieograniczona zmienność

Genomy organizmów
0,6 x 106 – 300 x 109 bp = 18 x
Różnorodność genetyczna
3 x 109 bp
3/100 bp różne
1 000 000 różnic
10 000 000 000 000 000 000 000 000 000 000 000 000 000 000 000 000 000 000
000 000 000 000 000 000 000 000 000 000 000 000 000
genotypów

Dlaczego markery molekularne ?
genetyczny charakter danych
uniwersalna metodyka
praktycznie nieograniczona zmienność
rozróżnienie cech homologicznych i analogicznych
„uniwersalna linijka” w ewolucji molekularnej
podstawa mechanistycznego poglądu na ewolucję
wyzwanie intelektualne !!!

• ocena dystansu genetycznego
• topologia dendrogramów hierarchicznego
podobieństwa genetycznego
• analiza wariancji molekularnych (AMOVA)
• topologia dendrogramów parsymonicznych
• analiza binarna rozmieszczenia haplotypów
Analiza danych molekularnych


Rekonstrukcja filogenezy

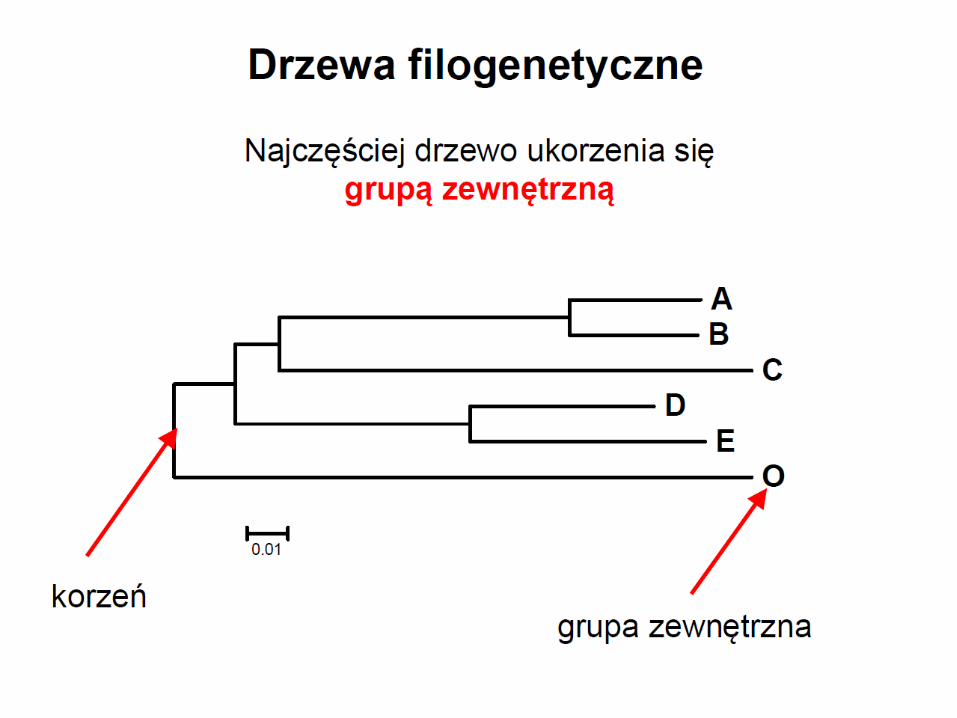
OTU = Operational Taxonomic Units = molekularne jednostki analizy
filogenetycznej
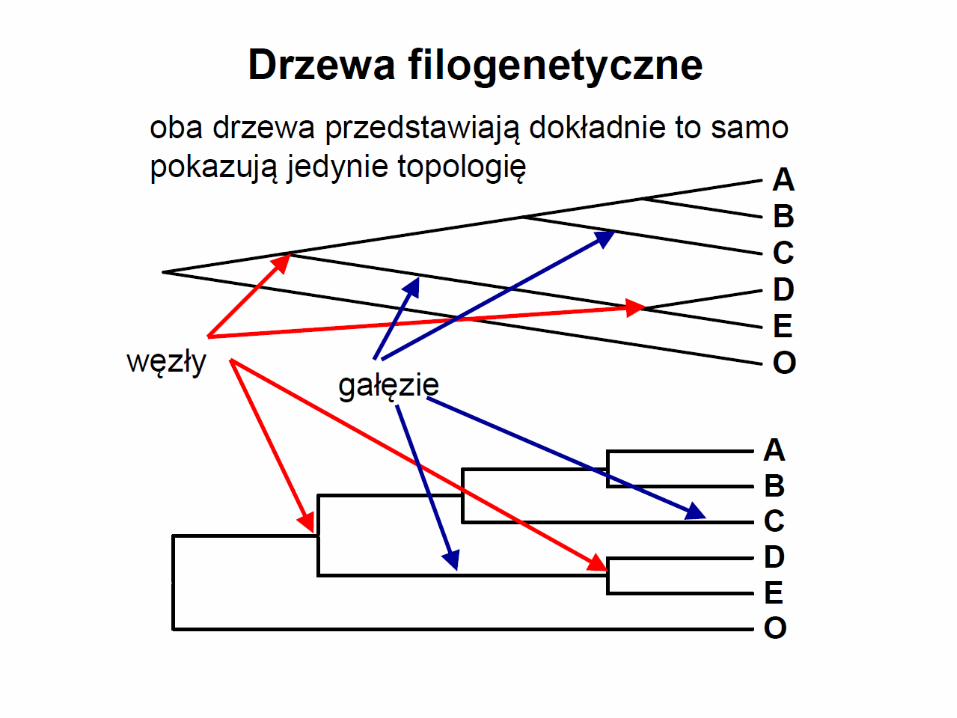
• zewnętrzne węzły na drzewie to współcześnie żyjące OTU
• wewnętrzne węzły to hipotetyczni przodkowie współczesnych OTU
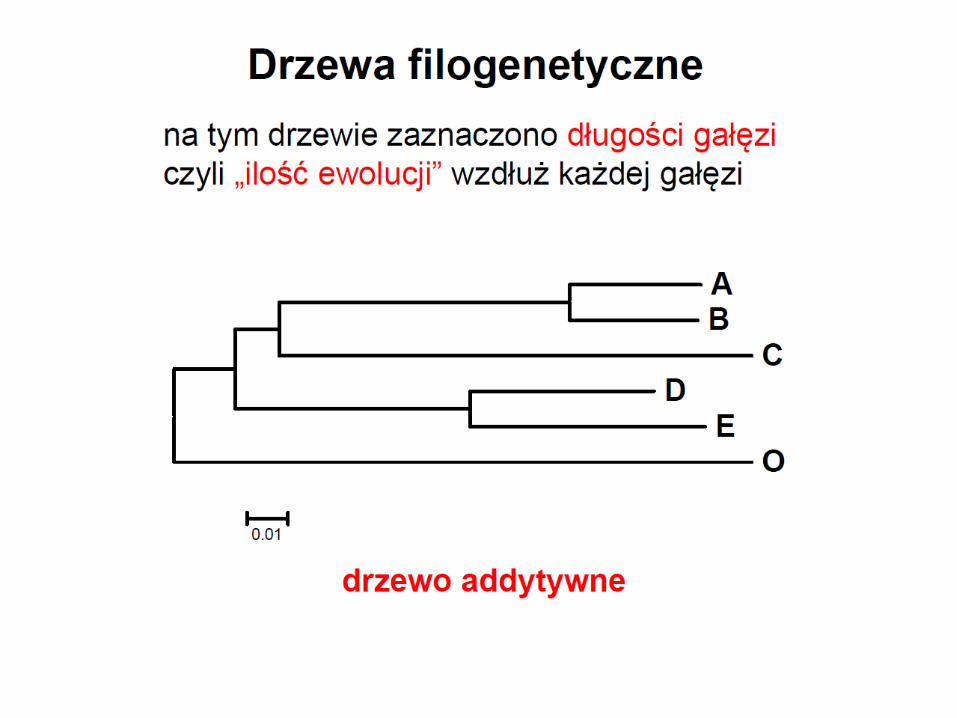
• drzewo jest wyskalowane jeśli długości gałęzi odpowiadają liczbie zmian jakie zaszły
w toku ewolucji między węzłami
• warunkiem użyteczności drzew jest brak połączeń między ich gałęziami
• odtworzenie jest możliwe pod warunkiem bifurkacyjnego rozgałęziania drzew
• rozgałęzienia multifurkacyjne uniemożliwiają wnioskowanie o pokrewieństwie

Wyszukiwanie właściwej (najbardziej prawdopodobnej topologii drzewa)
• liczba wersji rozgałęzień drzewa ilustrujących pokrewieństwa między
taksonami jest ogromna
• dla N = 10 to od 10 milionów do ponad 30 milionów
• do wyszukwiania służą skomplikowane algorytmy matematyczne
• kalkulacja możliwa jedynie przy użyciu komputerów
• algorytmy wyszukują najbardziej prawdopodobne topologie drzew zgodnie z
zadanym kryterium optymalizacji
• zwykle nie znamy prawdziwej filogenezy dlatego wszelkie prezentowane
drzewa są jedynie jej przybliżeniem
• metody oparte na odległościach (dystansie genetycznym)
• analiza skupień (UPGMA)
• metoda łączenia sąsiadów (Neighbor-Joining)
• metody oparte na stanach cech
• największej oszczędności (MP = Maximum Parsimony)
• największej wiarygodności (ML = Maximum Likelihood)
• analiza Bayesowska (Bayesian analysis)
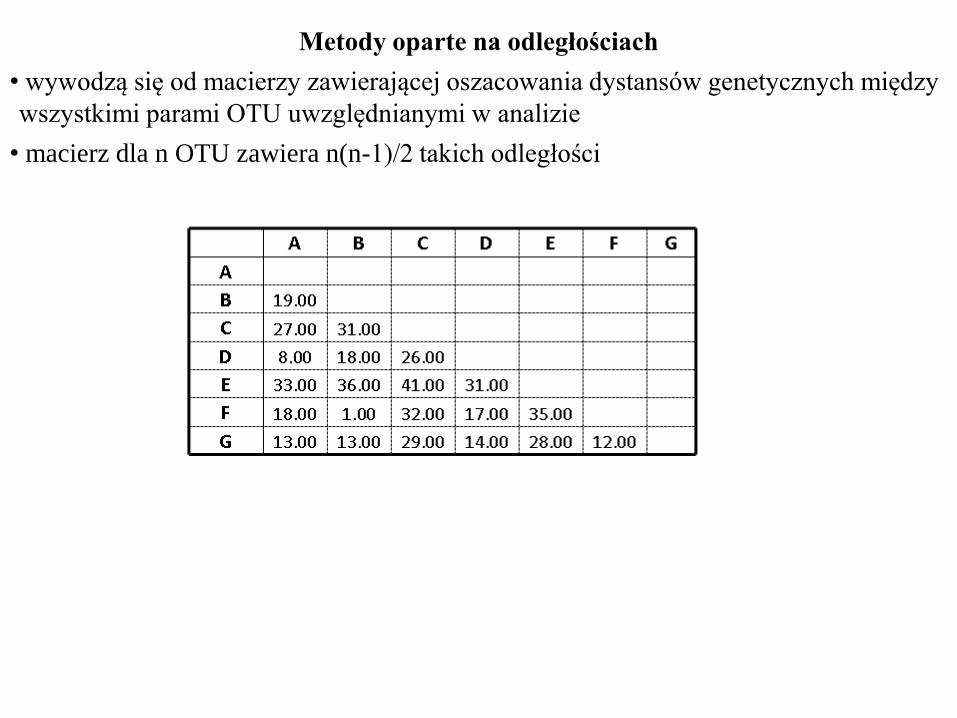
Metody oparte na odległościach
• wywodzą się od macierzy zawierającej oszacowania dystansów genetycznych między
wszystkimi parami OTU uwzględnianymi w analizie
• macierz dla n OTU zawiera n(n-1)/2 takich odległości

UPGMA
Unweighted Pair Group Method with Arithmetic Averages
Nieważona metoda grupowania par średnimi arytmetycznymi
nieważona – wszystkie dystanse są tak samo ważne
grupowanie par – liczą się tylko bifurkacje
średnie arytmetyczne – odległości pomiędzy kladami są średnimy arytmetycznymi odległości pomiędzy wszystkim członkami obu kladów
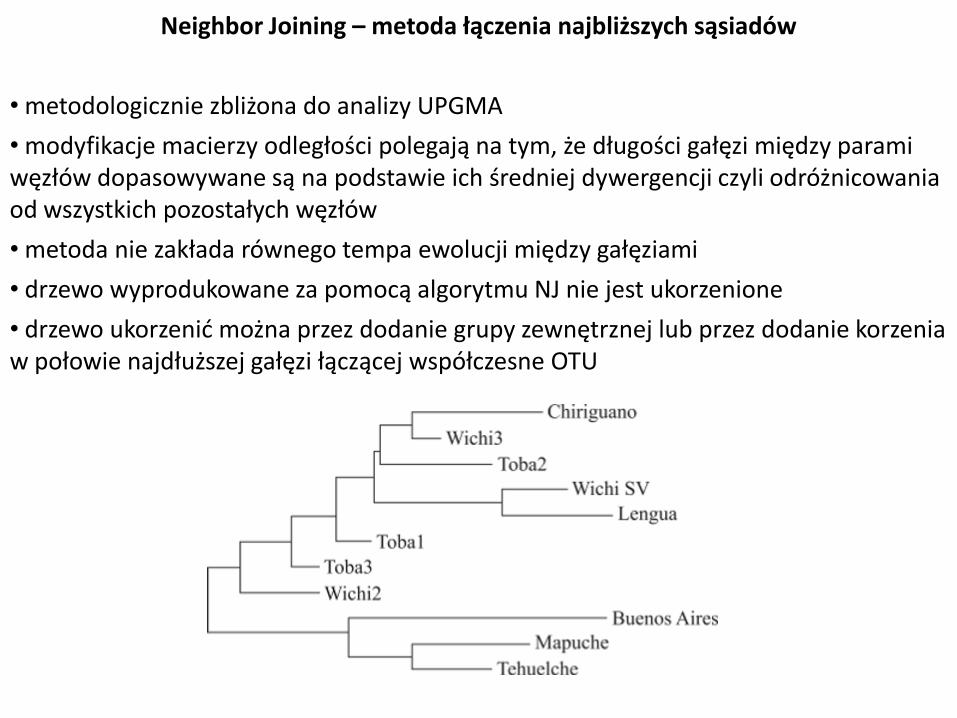
Neighbor Joining – metoda łączenia najbliższych sąsiadów
• metodologicznie zbliżona do analizy UPGMA
• modyfikacje macierzy odległości polegają na tym, że długości gałęzi między parami węzłów dopasowywane są na podstawie ich średniej dywergencji czyli odróżnicowania od wszystkich pozostałych węzłów
• metoda nie zakłada równego tempa ewolucji między gałęziami
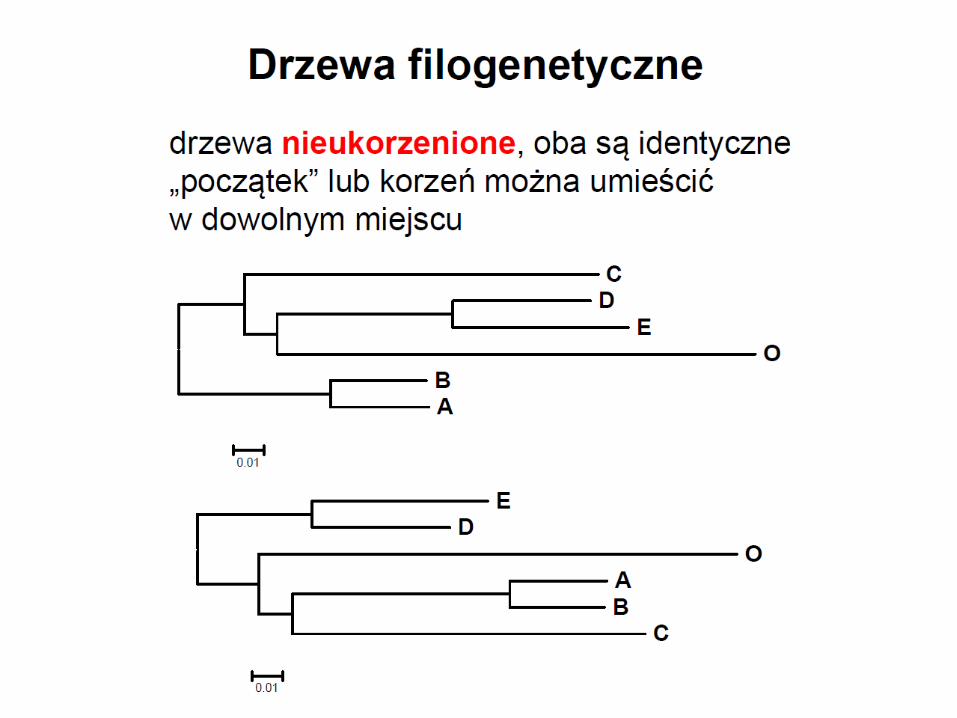
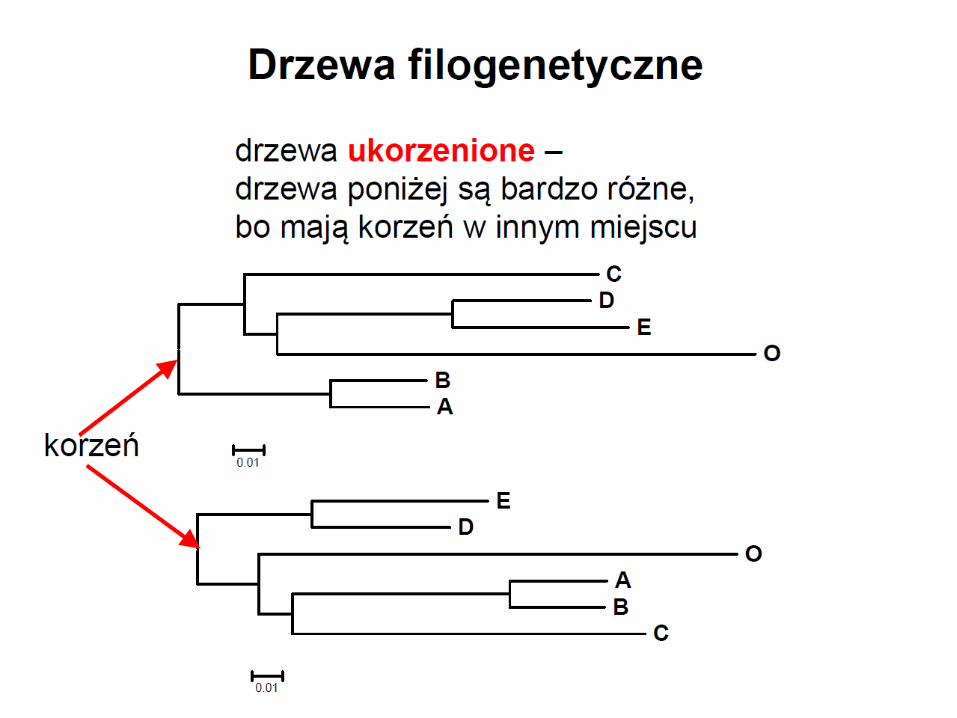
• drzewo wyprodukowane za pomocą algorytmu NJ nie jest ukorzenione
• drzewo ukorzenić można przez dodanie grupy zewnętrznej lub przez dodanie korzenia w połowie najdłuższej gałęzi łączącej współczesne OTU

Metody oparte na stanach cech
• nie wymagają tworzenia macierzy dystansów genetycznych
• analizują prawdopodobieństwa przejścia między dyskretnymi stanami cech
(literami kodu genetycznego) = prawdopodobieństwo substytucji
• prawdopodbieństwo tranzycji (A↔G lub C↔T) jest większe niż
prawdopodobieństwo transwersji (A,G↔C,T)
• metody oparte o stanach cech poszukują drzew o topologii będącej wynikiem
sumy najbardziej prawdopodobnych zdarzeń (mutacji) dzielących analizowane
sekwencje DNA
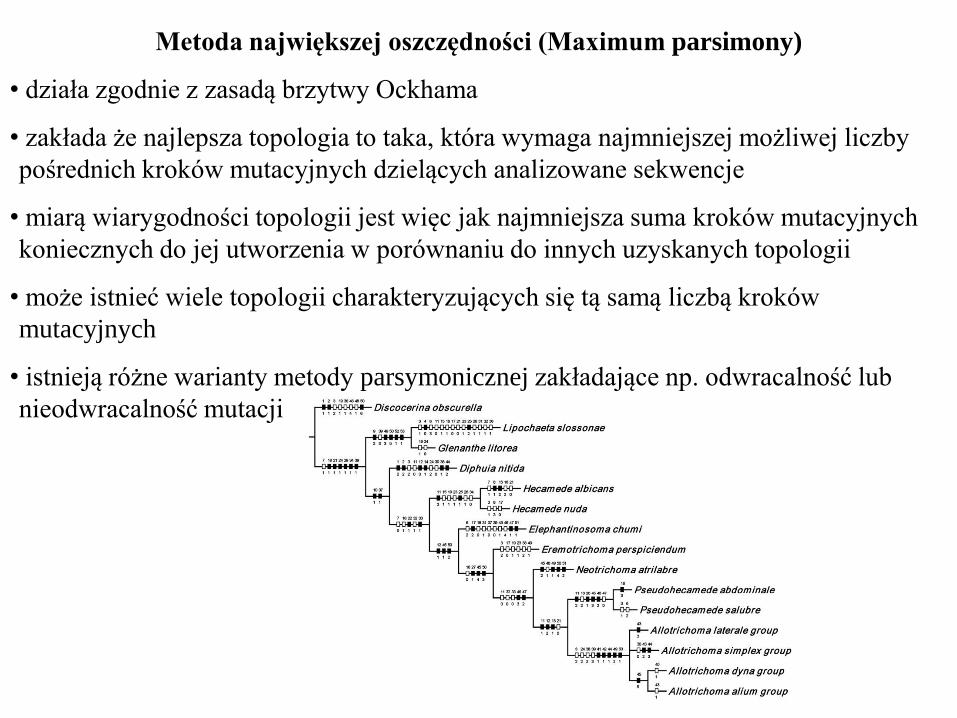
Metoda największej oszczędności (Maximum parsimony)
• działa zgodnie z zasadą brzytwy Ockhama
• zakłada że najlepsza topologia to taka, która wymaga najmniejszej możliwej liczby
pośrednich kroków mutacyjnych dzielących analizowane sekwencje
• miarą wiarygodności topologii jest więc jak najmniejsza suma kroków mutacyjnych
koniecznych do jej utworzenia w porównaniu do innych uzyskanych topologii
• może istnieć wiele topologii charakteryzujących się tą samą liczbą kroków
mutacyjnych
• istnieją różne warianty metody parsymonicznej zakładające np. odwracalność lub
nieodwracalność mutacji

Metoda największej wiarygodności (Maximum Likelihood)
• zakłada określony model ewolucji sekwencji DNA w oparciu o dane empiryczne
• częstość tranzycji/transwersji lub określonych mutacji wyliczone ze zbioru
sekwencji
• częstości te opisywane są matematycznie
• tworzenie matematycznego modelu procesu ewolucyjnego skutkującego
obserwowaną zmiennością w zestawie analizowanych danych
• topologia drzewa konstruowana jest w taki sposób aby uwiarygodniała z jak
największym prawdopodobieństwem zakładany model ewolucyjny (obserwowany
rozkład zmienności danych)
• tworzonych jest wiele topologii drzew i wyliczane są prawdopodobieństwa
wystąpienia zakładanego modelu ewolucji przy danej topologii
• topologia, dzięki której prawdopodobieństwo wystąpienia danego modelu jest
najbliższe 1 zostaje wybierana jaka prawdziwa
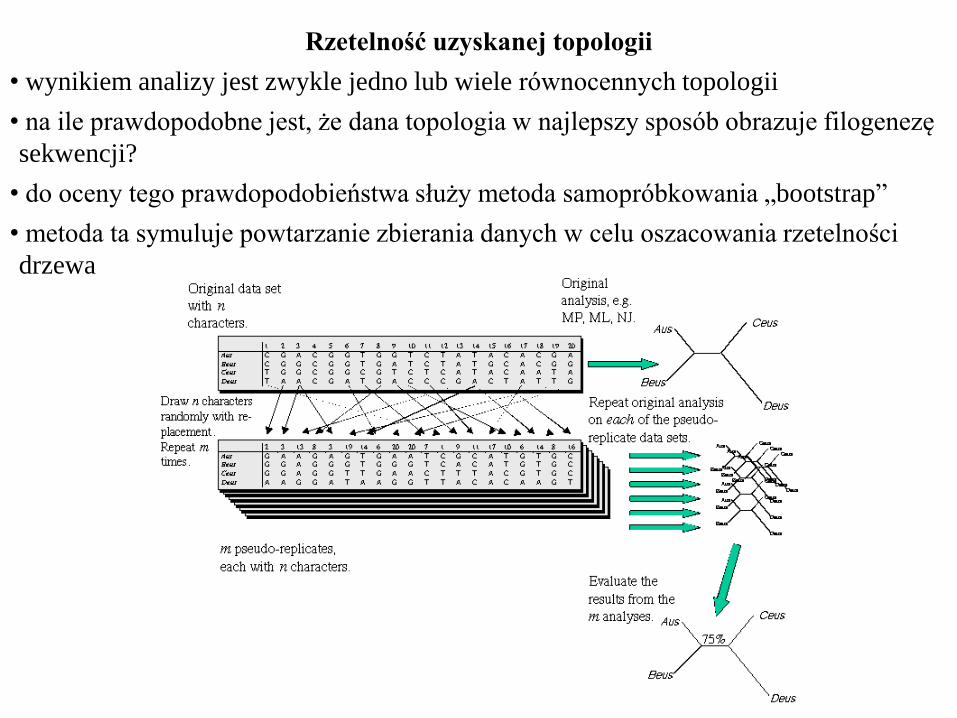
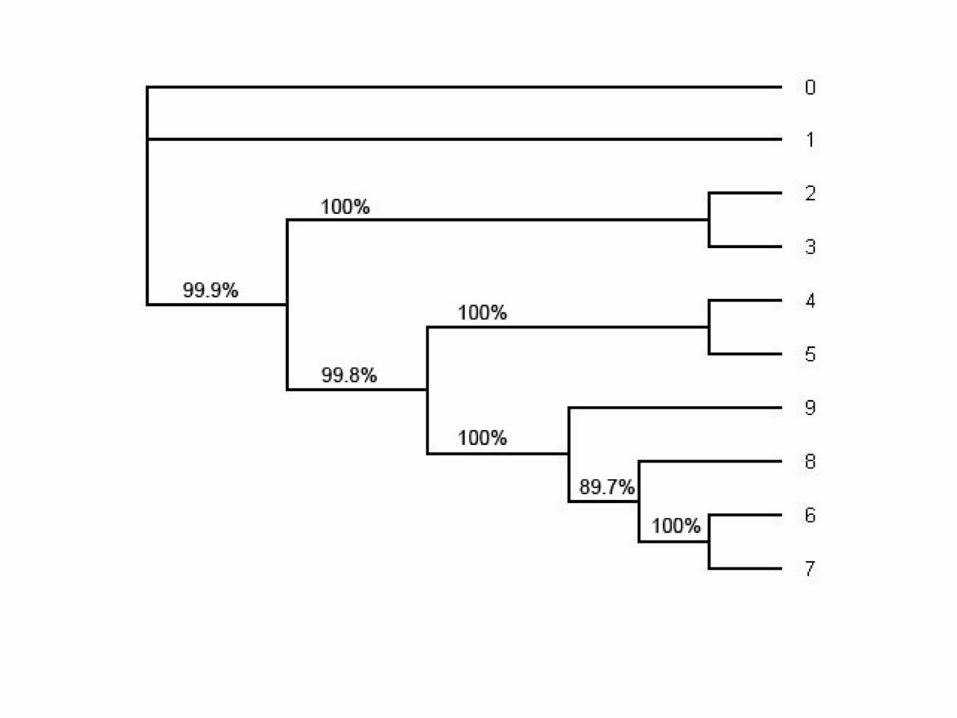
Rzetelność uzyskanej topologii
• wynikiem analizy jest zwykle jedno lub wiele równocennych topologii
• na ile prawdopodobne jest, że dana topologia w najlepszy sposób obrazuje filogenezę
sekwencji?
• do oceny tego prawdopodobieństwa służy metoda samopróbkowania „bootstrap”
• metoda ta symuluje powtarzanie zbierania danych w celu oszacowania rzetelności
drzewa

Dziękuję bardzo
Top Related