Webmobis – platforma informatyczna do analizy białekrobertnowak.pl/publikacje/Webmobis.pdf ·...
88
Politechnika Poznańska, Instytut Informatyki Webmobis – platforma informatyczna do analizy bialek Grzegorz Gębura Rafal Masztalerz Robert Nowak Marek Wronowski Praca inżynierska wykonana pod kierunkiem dr inż. Piotra Lukasiaka Poznań, 2006
Transcript of Webmobis – platforma informatyczna do analizy białekrobertnowak.pl/publikacje/Webmobis.pdf ·...

Politechnika Poznańska,
Instytut Informatyki
Webmobis – platforma informatycznado analizy białek
Grzegorz GęburaRafał MasztalerzRobert NowakMarek Wronowski
Praca inżynierska wykonana pod kierunkiem
dr inż. Piotra Łukasiaka
Poznań, 2006


Spis treści
Spis treści i
1 Wprowadzenie 1
1.1 Bioinformatyka . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
1.2 Cel i zakres pracy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
2 Zagadnienia biologiczne 3
2.1 Istota bioinformatyki w świetle dotychczasowych nauk przyrodniczych . . 3
2.1.1 Analiza białek . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
2.1.2 DNA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
2.1.3 RNA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
2.2 Rola białek w organizmach . . . . . . . . . . . . . . . . . . . . . . . . . . 13
2.2.1 Budowa i struktura białek . . . . . . . . . . . . . . . . . . . . . . 13
2.2.1.1 Struktura pierwszorzędowa . . . . . . . . . . . . . . . . 16
2.2.1.2 Struktura drugorzędowa . . . . . . . . . . . . . . . . . 16
2.2.1.3 Struktura trzeciorzędowa . . . . . . . . . . . . . . . . . 20
2.2.1.4 Struktura czwartorzędowa . . . . . . . . . . . . . . . . 22
2.2.1.5 Podsumowanie . . . . . . . . . . . . . . . . . . . . . . . 22
2.2.2 Metody stosowane do analizy struktur białkowych . . . . . . . . 23
2.2.2.1 Analiza struktur pierwszorzędowych . . . . . . . . . . . 23
2.2.2.2 Analiza struktur drugo-, trzecio- i czwartorzędowych . 25
2.2.3 CASP . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
3 Narzędzia 31
3.1 Narzędzia biologiczne . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
3.1.1 BLAST . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
3.1.1.1 Działanie programu . . . . . . . . . . . . . . . . . . . . 32
3.1.1.2 Algorytm . . . . . . . . . . . . . . . . . . . . . . . . . . 33
3.1.1.3 Parametry . . . . . . . . . . . . . . . . . . . . . . . . . 33
3.1.1.4 Poszukiwanie MSP . . . . . . . . . . . . . . . . . . . . 34
3.1.2 LGScore . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
3.1.2.1 Rodzaje LGScore . . . . . . . . . . . . . . . . . . . . . 35
3.1.2.2 Uruchomienie i wynik działania programu . . . . . . . 35
i

ii Spis treści
3.1.3 Jmol . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
3.1.4 MinRMS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
3.1.5 DSSP . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
3.1.6 MaxSub . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
3.2 Technologie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
3.2.1 Java . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
3.2.2 Eclipse . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43
3.2.3 Tomcat . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
3.2.4 Tapestry . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46
3.2.5 SYSDEO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
3.2.6 Spindle . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
3.2.7 JDO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
3.2.8 JPOX . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
3.2.9 JUnit . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
3.2.10 JMock . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 49
3.2.11 PostgreSQL . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 49
4 Budowa systemu 51
4.1 Funkcjonalność . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51
4.1.1 Współpraca z metaserwerami . . . . . . . . . . . . . . . . . . . . 51
4.1.2 Obsługa użytkowników . . . . . . . . . . . . . . . . . . . . . . . . 52
4.1.3 Wprowadzenie sekwencji białkowej . . . . . . . . . . . . . . . . . 52
4.1.4 Dodawanie własnych plików PDB . . . . . . . . . . . . . . . . . . 55
4.1.5 Projekt i budowa komponentów analizy struktur białkowych . . . 55
4.1.5.1 Pairwise Alignment Table . . . . . . . . . . . . . . . . 55
4.1.5.2 Secondary Structures Table . . . . . . . . . . . . . . . 57
4.1.5.3 PDB Result Table . . . . . . . . . . . . . . . . . . . . . 58
4.1.5.4 Status przetwarzania . . . . . . . . . . . . . . . . . . . 60
4.1.6 Dodawanie plików PDB . . . . . . . . . . . . . . . . . . . . . . . 61
4.1.7 Wizualizacja Jmola . . . . . . . . . . . . . . . . . . . . . . . . . . 61
4.2 Serwer HTTP . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 61
4.2.1 Strona wizualna . . . . . . . . . . . . . . . . . . . . . . . . . . . . 61
4.2.1.1 Ruchome menu . . . . . . . . . . . . . . . . . . . . . . 62
4.2.1.2 System podpowiedzi . . . . . . . . . . . . . . . . . . . 62
4.2.2 Obsługa błędów i wyjątków . . . . . . . . . . . . . . . . . . . . . 63
4.2.3 Opracowanie sposobu przekazywania parametrów pomiędzy
stronami w technologii Tapestry . . . . . . . . . . . . . . . . . . 65
4.3 Zewnętrzne narzędzia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 65
4.3.1 Skrypt instalacyjny . . . . . . . . . . . . . . . . . . . . . . . . . 66
4.3.2 Jmol . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68
4.3.3 Skrypt uruchamiający MaxSub . . . . . . . . . . . . . . . . . . . 70
4.3.4 Moduł plików tymczasowych . . . . . . . . . . . . . . . . . . . . 70
iii
5 Podsumowanie 73
Słownik 75
Indeks 79
Bibliografia 81


Rozdział 1
Wprowadzenie
1.1 Bioinformatyka
Jednym z największych osiągnięć XX wieku był rozwój elektroniki
i informatyki. Dzięki niebywałemu postępowi, jaki dokonał się na obszarze tych
dwóch dziedzin, możliwe stało się skonstruowanie niezwykle złożonych systemów,
pozwalających na przetwarzanie ogromnych ilości danych. Dzięki temu znalazły
one zastosowanie w praktycznie każdej dziedzinie życia, od administracji, przez
przemysł, naukę, a kończąc na dostarczaniu rozrywki. Wśród wielu z nich
szczególnego znaczenia nabrała w ostatnich latach biologia. Na styku tych dwóch
dziedzin – biologii i informatyki – pojawiła się nowa interdyscyplinarna dziedzina
nauki – bioinformatyka (ang. bioinformatics). Wniosła on własne problemy
badawcze i metody ich rozwiązania.
Jednym z najbardziej dynamicznie rozwijających się działów bioinformatyki
jest analiza sekwencji białkowych. W ramach tej analizy wyróżnić możemy:
• porównywanie sekwencji białek,
• przewidywanie struktur drugo–, trzecio– i czwartorzędowych,
• wyszukiwanie domen (podjednostek) i motywów.Istnieje już spora grupa serwerów (np. EsyPred, Fugue, Wurst) oraz
narzędzi informatycznych (m.in. PSIBLAST, LgScore, DSSP, MinRMS, MaxSub)
pozwalających wykonywać podane wyżej operacje na białkach, w znacznym
stopniu wspomagając pracę biologów molekularnych. Z drugiej jednak strony
duża liczba tych narzędzi, brak przystępnego interfejsu (np. wyświetlanie
wyników w konsoli), duża liczba parametrów, które należy określić przed
wywołaniem programów oraz brak wspólnego formatu danych nie sprzyja
szybkiemu i sprawnemu ich wykorzystaniu.
1

2 Wprowadzenie
1.2 Cel i zakres pracy
Celem projektu było stworzenie platformy internetowej integrującej narzędzia
wspomagające pracę biologów. System miał umożliwiać pozyskanie informacji
z serwerów wyspecjalizowanych w analizie struktur i funkcjonalności białek oraz
dalsze ich analizowanie z wykorzystaniem dołączonych do platformy narzędzi.
Wszystkie dane znajdujące się na serwerze, miały być przechowywane wyłącznie
w bazie danych. Pliki tworzone miały być dopiero w chwili żądania ich przez
użytkownika i tylko na określony czas. W celu usprawnienia pracy naukowców do
projektu miała zostać włączona możliwość założenia własnego konta, na którym
zapisywano by ustawienia oraz badane aktualnie struktury razem z wynikami
ich przetwarzania. Poprzez konto użytkownika dostarczane miały być informacje
o wynikach analiz z innych serwerów za pomocą wiadomości e-mail.

Rozdział 2
Zagadnienia biologiczne
2.1 Istota bioinformatyki w świetle dotychczasowych
nauk przyrodniczych
Bioinformatyka jest to dziedzina zajmująca się stosowaniem narzędzi
matematycznych i informatycznych do rozwiązywania problemów biologicznych.
Podstawowe zagadnienia bioinformatyki to:
• katalogowanie informacji biologicznych (bazy danych, wyszukiwaniesekwencji, annotacji, danych numerycznych w bazach danych)
• analiza sekwencji DNA i RNA (składanie sekwencji, annotacja,
wyszukiwanie sekwencji kodujących, regulatorowych i repetytywnych,
motywów, markerów)
• analiza sekwencji genomów, porównywanie genomów
• ustalanie ewolucyjnych relacji pomiędzy zbiorami sekwencji / organizmów
• genotypowanie (używane między innymi do wyszukiwania genówodpowiedzialnych za choroby genetyczne, w ustalaniu ojcostwa,
kryminalistyce)
• analiza ekspresji genów (głównie ang. microarrays analysis)
• analiza sekwencji białek (porównywanie sekwencji, wyszukiwanie domeni motywów, przewidywanie własności drugo– i trzeciorzędowej struktury
białka, lokalizacji w obrębie komórki)
• katalogowanie funkcji genów/białek, analiza dróg metabolicznych(np. metabolizm lipidów) oraz dróg sygnałowych (np. od receptora
na powierzchni komórki poprzez kaskadę kinaz do czynników
transkrypcyjnych)
3

4 Zagadnienia biologiczne
• modelowanie układów biologicznych (np. kinetyka szeregu reakcji
enzymatycznych w komórce)
• wirtualne dokowanie (ang. virtual docking) – np. używając trójwymiarowejstruktury aktywnego centrum enzymu („zamek” albo „kieszonka”,
ang. pocket) uruchamia sie wyszukiwanie w bazach danych tysięcy małych
cząsteczek, z których kilka, kilkanaście („kluczy”) będzie miało kształt
mieszczący się w centrum aktywnym. Zagadnienie to stanowi pierwszy krok
w kierunku odkrywania nowych leków i preparatów leczniczych.
• morfometria – analiza obrazuNa przestrzeni lat bioinformatyka przekształciła się w samodzielną dziedzinę
z własnymi problemami biologiczno–obliczeniowymi[26].
2.1.1 Analiza białek
Rola bioinformatyki, przy dzisiejszym rozwoju informatyki oraz nauk
matematycznych, nie ogranicza się jedynie do zarządzania i operowania
materiałem informacyjnym pochodzącym z analizy DNA i RNA. Wykorzystując
różne narzędzia informatyczne i algorytmy, bioinformatyka stała się jedną
z najważniejszych i najszybciej rozwijających się dziedzin nauki.
Powszechnie stosowanymi metodologiami umożliwiającymi dekodowanie
informacji o sekwencji aminokwasów w białkach, zawartej w łańcuchu DNA,
a nastepnie przewidywanie struktury i funkcji białka, są metody numeryczne.
Ważnym krokiem analiz numerycznych, niezbędnym dla przejścia od informacji
genetycznej do poziomu funkcji biologicznej, jest przewidywanie struktury
przestrzennej dla łańcucha białkowego o danej sekwencji aminokwasowej.
Realizowane jest to z wykorzystaniem baz danych dotyczących białek, których
struktura trójwymiarowa (3D) jest już poznana w oparciu o eksperymenty
krystalograficzne1. Prowadzenie analiz porównawczych danych strukturalnych,
ma na celu znalezienie reguły ogólnej, która pozwoli zbudować model
powstawania struktury przestrzennej białka tylko w oparciu o poznaną sekwencję
aminokwasową. Biorąc pod uwagę liczbę sposobów ułożenia przestrzennego
łańcucha aminokwasów, zadanie to jest bardzo złożone. Z tego powodu poszukuje
się modeli, mogących odtworzyć proces zachodzący w naturze, przyjmując, że
w uwarunkowaniach środowiskowych ukryta jest procedura tworzenia struktur
prawidłowych.
Cząsteczka białka jest strukturą wysoce dynamiczną i wrażliwą na wpływ
środowiska. Dlatego na świecie rośnie zapotrzebowanie na specjalistyczne
1naświetlanie kryształu białka promieniami rentgenowskimi

2.1. Istota bioinformatyki w świetle dotychczasowych nauk przyrodniczych 5
programy, które potrafiłyby przeprowadzić symulację dynamiki molekularnej
w możliwie krótkich okresach obliczeniowych. Dotyczy to zwłaszcza tych zmian
strukturalnych, które są warunkiem koniecznym do tworzenia kompleksów białek.
Możliwość tworzenia kompleksów jest podstawowym warunkiem dla wyrażenia
aktywności biologicznej danego białka. Symulacja dynamiki molekularnej,
weryfikującej możliwość powstawania kompleksu białkowego oraz określenie jego
trwałości, zaliczane są do zagadnień związanych z przewidywaniem funkcji
biologicznej białek. Dla układów biologicznych symulacja dynamiki molekularnej
jest zagadnieniem należącym do tzw. wielkoskalowych równoległych obliczeń
komputerowych, realizowanych przez najnowsze superkomputery. Możliwe jest
ponadto symulowanie reakcji enzymatycznych, w czasie których cząsteczka
substratu (substrat: patrz słownik) pod wpływem określonego enzymu (enzym:
patrz słownik) przekształcana jest w inną, stanowiącą produkt reakcji.
Realizują to nowoczesne wysokowyspecjalizowane programy, budowane w oparciu
o zaawansowane modele chemii oraz chemii kwantowej[22].
2.1.2 DNA
DNA nazywane kwasem deoksyrybonukleinowym, to długi liniowy polimer
(patrz słownik), który przenosi informację przechodzącą z jednego pokolenia
na drugie. Składa się on z ogromnej ilości połączonych ze sobą nukleotydów
(nukleotyd: patrz słownik). Każdy z nich jest złożony z: cukru, kwasu fosforowego
i zasady. Cukry połączone fosforanami tworzą wspólny rdzeń cząsteczki, podczas
gdy zasady mogą się różnić i należą do następujących typów:
• zasady purynowe– adenina – A
– guanina – G
• zasady pirymidynowe– cytozyna – C
– tymina – T
Informacja genetyczna jest przechowywana w sekwencji zasad leżących wzdłuż
łańcucha kwasu nukleinowego, które tworzą specyficzne pary połączone
wiązaniami wodorowymi. Parowanie się zasad powoduje powstawanie podwójnej
helisy, helikalnej struktury składającej się z dwóch nici kwasu nukleinowego,
a także jest podstawą mechanizmu kopiowania informacji genetycznej
z istniejącego łańcucha kwasu nukleinowego na nowo tworzony łańcuch[11].
W 1953 r. Watson i Crick opracowali trójwymiarową strukturę DNA (patrz
rys. 2.1 oraz 2.2). Okres powtarzalności wzdłuż osi helisy wynosi 0,34 nm, co

6 Zagadnienia biologiczne
Rysunek 2.1: Model dwuniciowej helisy DNA zaproponowany przez Watsonai Cricka – widok z boku
Rysunek 2.2: Model dwuniciowej helisy DNA zaproponowany przez Watsonai Cricka – widok z góry, wzdłuż osi helisy
odpowiada 10 nukleotydom w każdym łańcuchu (patrz rys. 2.1). Na podstawie
obrazów dyfrakcji promieni X uzyskanych przez Franklin i Wilkinsa stwierdzili, że
DNA jest zbudowany z dwóch wzajemnie oplatających się nici, razem tworzących
helikalną strukturę, w której zasady skierowane są do wnętrza helisy, a rdzeń
cukrowo–fosforanowy (patrz rys. 2.3) znajduje się na zewnątrz.
Na rysunku pierwszy łańcuch polinukleotydowy został zaznaczony na
niebiesko, natomiast drugi – na różowo. Zasady purynowe i pirymidynowe
zostały przedstawione kolorami o mniejszej intensywności, niż rdzeń
cukrowo-fosforanowy. Dwie nici (łańcuchy) DNA w dwuniciowej helisie
(patrz rys. 2.4) są ułożone antyrównolegle względem siebie (jeśli jedna nić jest

2.1. Istota bioinformatyki w świetle dotychczasowych nauk przyrodniczych 7
Rysunek 2.3: Rdzeń cukrowo-fosforanowy DNA
zorientowana w kierunku 5’ → 3’ (3’ i 5’ są oznaczeniami końców nici DNA),to druga przyjmuje orientację 3’ → 5’). Zasady znajdujące się w przeciwległychłańcuchach tworzą ze sobą wiązania wodorowe:
• A z T
• G z CŁączenie się zasad w pary jest określane jako tworzenie się komplementarnych
par zasad. Większa, dwupierścieniowa zasada purynowa tworzy parę z mniejszą,
jednopierścieniową zasadą pirymidynową. Pary zasad doskonale „pasują” do
odległości pomiędzy łańcuchami cukrowo-fosforanowymi i decydują o utrzymaniu
poprawnego odstępu między nimi. Odległość ta byłaby zbyt mała dla pary
utworzonej z dwóch dużych zasad purynowych i zbyt duża dla pary dwóch
pirymidyn, które znajdowałyby się zbyt daleko od siebie, by utworzyć wiązania
wodorowe. Pomiędzy komplementarnymi zasadami tworzy się maksymalna
(spośród możliwych) liczba wiązań wodorowych. W każdej parze G:C są trzy
wiązania wodorowe, zasady w parze A:T są związane dwoma wiązaniami
wodorowymi. Tak, więc pary zasad A:T i G:C stanowią układy najbardziej
stabilne zarówno pod względem sferycznym (przestrzennym), jak i tworzenia się
maksymalnej liczby wiązań wodorowych[9].
DNA jest replikowane przez enzymy nazywane polimerazami DNA
(polimeraza: patrz słownik), które bardzo precyzyjnie kopiują sekwencje matryc
nukleotydowych z częstotliwością błędów niższą niż 1 na 100 milionów
nukleotydów[11].
Rozwój bioinformatyki, w odniesieniu do DNA, nastąpił w momencie
intensywnego rozwoju oprogramowania komputerowego służącego do analizy
sekwencji nukleotydów. Na początku, w oparciu o teorię informacji, rozwijano
metody poszukiwania podobieństw i różnic sekwencji w obrębie genomu tego
samego oraz różnych gatunków. Doprowadziło to do określenia kryteriów
dla rozróżniania fragmentów kodujących (niosących informacje o funkcjach

8 Zagadnienia biologiczne
Rysunek 2.4: Schemat dwuniciowej helisy DNA
i budowie białek) i niekodujących (nie wykorzystywanych do syntezy
białek). Analiza porównawcza pomiędzy gatunkami pozwoliła na śledzenie
procesów ewolucyjnych, określenie kryterium dla przynależności gatunkowej oraz
identyfikację specyfiki osobniczej.
Automatyzacja i dynamiczny rozwój technik analizy sekwencji DNA,
stworzyły warunki techniczne dla realizacji światowego projektu o nazwie Human
Genom Project (HGP). Projekt polega na analizie sekwencji nukleotydów
w całym ludzkim genomie, a w efekcie na rozszyfrowaniu „programu” tworzenia
i funkcjonowania organizmu człowieka. Jest to ogromne wyzwanie, jeśli weźmie
się pod uwagę rozmiary ludzkiego genomu. Podwójna nić DNA człowieka zawiera
ok. 3 miliardy par nukleotydów. Gdyby cały DNA człowieka wymodelować
za pomocą zamka błyskawicznego, przyjmując, że 1 ząbek odpowiadałby
jednemu nukleotydowi, a 300 ząbków przypadałoby na 1 m długości zamka,
to zamek taki powinien mieć długość ok. 10 000 km (odległość z Warszawy
do Nowego Yorku i z powrotem). W chwili rozpoczęcia realizacji projektu
(w 1986 roku), oznaczenie sekwencji DNA o takich rozmiarach przekraczało
możliwości niejednego laboratorium, a koszty zakupu sprzętu niezbędnego do
realizacji tego projektu, budżet jednego kraju. Przedsięwzięcie to zostało więc
podjęte wspólnie przez Stany Zjednoczone Ameryki Północnej, Japonię oraz
Unię Europejską. W okresie późniejszym dołączyły inne kraje. W trakcie

2.1. Istota bioinformatyki w świetle dotychczasowych nauk przyrodniczych 9
Rysunek 2.5: Rdzeń cukrowo-fosforanowy RNA
realizacji projektu techniki sekwencjonowania kwasów nukleinowych rozwinęły
się tak znacznie, że zakładany początkowo termin zakończenia projektu został
już istotnie przybliżony. Pierwotny plan zakładał oprócz analizy genomu
człowieka (Homo sapiens – 3 000 Mpz (Mpz oznacza jednostkę Mega Par
Zasad) także analizę genomów innych najlepiej poznanych organizmów jak:
bakteria – Ecoli (4,7 Mpz), muszka owocowa – D. melanogaster (165,0 Mpz),
drożdże – S. cerevisiae (13,5 Mpz), nicień – C. elegans (100,0 Mpz), mysz
laboratoryjna – M. musculus (3 000 Mpz). Lista gatunków włączonych w badania
genomu powiększyła się o wiele innych organizmów (w tym także o rośliny –
np. rzodkiewnik Arabidopsis- thaliana)[22].
2.1.3 RNA
RNA (kwas rybonukleinowy) jest długim polimerem (polimer: patrz słownik)
składającym się z nukleotydów (nukleotyd: słownik, strona 77) połączonych
wiązaniami 3’, 5’ – fosfodiestrowymi. Cząsteczki RNA pełnią bardzo ważną rolę,
ponieważ stanowią matrycę dla syntezy białek.
Porównując budowę DNA (patrz podrozdział 2.1.2) i RNA można zauważyć
następujące różnice:
• W skład RNA wchodzą zasady: adenina (A), guanina (G), uracyl (U)i cytozyna (C). Tak więc tymina z DNA jest w RNA zastąpiona przez uracyl
– inną zasadę pirymidynową. Uracyl może jednak, podobnie jak tymina,
tworzyć komplementarną parę z adeniną;
• Cukrem występującym w RNA jest ryboza, natomiast DNA zawieradeoksyrybozę
Rysunek 2.5 przedstawia rdzeń cukrowo-fosforanowy RNA, który podobnie jak
w DNA, utworzony jest z wiązań fosfodiestrowych 3’ do 5’.
Rybonukleozydy (rybonukleozyd: patrz słownik) występujące w RNA
to: adenozyna, guanozyna, cytydyna oraz urydyna. Rybonukleotydami

10 Zagadnienia biologiczne
(rybonukleotyd: patrz słownik) są adenozyno-5’-trifosforan (ATP),
guanozyno-5’-trifosforan (GTP), cytydyno-5’-trifosforan (CTP)
i urydyno-5’-trifosforan (UTP). 5’-Trifosforany nukleozydów są substratami
do syntezy RNA. Ogniwami gotowych łańcuchów RNA są monofosforany
odpowiednich nukleozydów.
Podobnie jak w przypadku DNA, sekwencję nukleotydów RNA zapisuje się
jako sekwencję zasad w kierunku 5’ → 3’. Na przykład GUCAAGCCGGAC jestsekwencją krótkiej cząsteczki RNA[9].
Większość cząsteczek RNA jest jednoniciowa, ale cząsteczka RNA może
zawierać regiony, które tworzą komplementarne pary zasad po zmianie biegu
łańcucha RNA o 180 stopni (tworzą się tak zwane struktury typu „spinki
do włosów”). Cząsteczka RNA może więc zawierać pewne odcinki o budowie
dwuniciowej helisy. Sparowane odcinki RNA stanowią drugorzędową strukturę
tych cząsteczek. Wyróżniamy następujące formy RNA[30]:
Jądrowy RNA (nRNA) – stanowi połączenie wielu rodzajów kwasów
rybonukleinowych. Niektóre z nich, np. rRNA i tRNA, są w jądrze
komórkowym syntetyzowane i przebywają w nim tylko okresowo.
Występujący stale w jądrze komórkowym RNA można podzielić na dwa
rodzaje:
Kwas rybonukleinowy heterogenny pre–RNA – jest bardzo
szybko syntetyzowany i katabolizowany. Jego okres półtrwania
wynosi od kilku minut do kilku godzin. Został nazwany
kwasem rybonukleinowym heterogennym (hnRNA) o dużej masie
cząsteczkowej, obecnie jest okreslany jako prekursorowy RNA lub
pre–RNA. Pewna jego część ulega przeistoczeniu w mRNA.
Kwas metabolicznie stabilny snRNA – stanowi drugi rodzaj RNA
jądrowego. Posiada stosunkowo małe cząsteczki. Zawiera, oprócz
typowych zasad azotowych, ich postacie umetylowane. Kwas ten
został elektroforetycznie (elektroforeza: patrz słownik) rozdzielony na
12 frakcji, którym przypisuje się funkcje regulatorowe.
Transferowy RNA (tRNA) –stanowi 10-13 procent ogólnej ilości kwasów
rybonukleinowych w komórce. Jest on zbudowany z 70-90 nukleotydów.
Jego cechą charakterystyczną wśród innych rodzajów RNA jest to, że
ma najmniejszą masą cząsteczkową, zawartą w granicach od 25 do
30 kDa. tRNA cechuje wysoka specyficzność w stosunku do aminokwasów.
Każdy z aminokwasów syntetyzowanego białka może być transportowany
przez jeden, a niektóre przez kilka różnych tRNA. Cząsteczki tRNA

2.1. Istota bioinformatyki w świetle dotychczasowych nauk przyrodniczych 11
Rysunek 2.6: Budowa kwasu tRNA, bez uwzględnienia skręcenia nici wefragmentach dwuniciowych ramion
w komórkach występują w stanie wolnym bądź też są związane z określonym
aminokwasem.
Cząsteczka tRNA ma budowę palczastą (patrz rys. 2.6). Jest ona zwinięta
spiralnie, a w pewnych miejscach tworzą się pętle. Ramiona tych pętli są
dwuniciowe, skręcone w spiralę. Na tych odcinkach pary zasad mogą łączyć
się wiązaniami wodorowymi. Niektóre fragmenty pętli mają jednakowe
sekwencje nukleotydowe we wszystkich tRNA. Istnieją odcinki wykazujące
znaczne różnice, które decydują o specyficzności tych kwasów.
W cząsteczce tRNA wyróżniono 5 ramion:
• aminokwasowe• antykodonowe• dihydrourydynowe• pseudourydynowe (ramię TΨC)• ramię dodatkowe
Ramię dodatkowe jest cechą charakterystyczną każdego tRNA i stanowi
podstawę klasyfikacji cząsteczek tRNA.
Matrycowy (informacyjny) RNA (mRNA) – powstaje w jądrze
komórkowym w procesie transkrypcji z DNA. Jest syntetyzowany
z trifosforanów nukleozydów. Jego zasady są komplementarne w stosunku
do jednej z nici chromosomowego DNA, na której jest wytwarzany.
Matrycowy RNA przenosi informację genetyczną z DNA do cytoplazmy.

12 Zagadnienia biologiczne
Masa cząsteczkowa mRNA oraz sekwencja nukleotydów zależą do rodzaju
białka, które jest w nim zakodowane. Trójki nukleotydów, czyli kodony
(kodon: patrz słownik), rozmieszczone w jego łańcuchu wyznaczają
kolejność aminokwasów syntetyzowanego białka.
Budowa mRNA pro- i eukariotów wykazuje wyraźne różnice, decydujące
o ich różnych właściwościach. Cząsteczka bakteryjnego mRNA może
dysponować kodem dla całego zespołu białek. Ten mRNA jest zatem
policistronowy. Oprócz kodonów zwykłych, łańcuch mRNA zawiera
tzw. kodony startu i stopu, które są znakami przestankowymi,
umożliwiającymi syntezę wielu białek. Długość łańcucha mRNA
u prokariotów jest zależna od wielkości cząsteczek zakodowanych w nim
białek.
W procesie transkrypcji u eukariotów powstaje najpierw pre–mRNA, jako
składnik frakcji heterogennego jądrowego hnRNA. Dalszym etapem jest
proces modyfikacji, w którym następuje dobieranie i łączenie z sobą
fragmentów łańcucha RNA, aby powstał ostatecznie łańcuch zawierający
informację o ściśle określonym białku.
Rybosomalny RNA (rRNA) – stanowi około 80 procent ilości kwasów
rybonukleinowych komórki. Jest on podstawowym składnikiem rybosomów,
gdzie sięga 65 procent zawartości. Resztę stanowią białka.
Rybosomalny RNA, podobnie jak inne rodzaje RNA, powstaje
w procesie transkrypcji z DNA. Zawiera on typowe zasady
azotowe z niewielką domieszką ich metylowych pochodnych. Jest
pojedynczym łańcuchem, bardzo mocno poskręcanym, tworzącym pętle,
z fragmentami dwuniciowymi, gdzie występują wiązania wodorowe między
komplementarnymi zasadami.
Wszystkie przedstawione powyżej rodzaje RNA, odgrywają ważną rolę
w procesie syntezy białek. Dużym udziałem drugorzędowych struktur,
charakteryzują się cząsteczki rybosomowych RNA (rRNA) i transportujących
RNA (transferowych RNA – tRNA) oraz informacyjnych RNA (mRNA).
Wszystkie formy komórkowego RNA są syntetyzowane przez polimerazy RNA
(polimeraza: patrz słownik), które czerpią instrukcje z matryc DNA. Po tym
procesie „przepisywania informacji”, czyli transkrypcji, następuje jej tłumaczenie
na język białek, czyli translacja, zgodnie z instrukcjami zawartymi w matrycach
mRNA. Przepływ informacji genetycznej, inaczej ekspresja genów, w normalnych
komórkach przebiega w następujący sposób:
DNA→ Transkrypcja→ RNA→ Translacja→ Biako

2.2. Rola białek w organizmach 13
Ten przepływ informacji zależy od kodu genetycznego, który określa związek
pomiędzy sekwencją zasad w DNA (lub mRNA) a sekwencją aminokwasów
w białku. Kod jest niemal jednakowy we wszystkich organizmach: sekwencja
trzech zasad, nazywana kodonem, określa dany aminokwas. Kodony w mRNA są
kolejno odczytywane przez cząsteczki tRNA, które służą jako adaptery w trakcie
syntezy białek. Proces syntezy białka odbywa się na rybosomach, które są
złożonymi kompleksami różnych rRNA i ponad 50 rodzajów białek[11].
Komplementarne pary zasad w zwiniętej pojedynczej nici mogą ulec
rozerwaniu w podwyższonej temperaturze, podobnie jak w przypadku podwójnej
helisy DNA. RNA może tworzyć nawet długą podwójną helisę, jednak
w komórkach sytuacje takie zdarzają się rzadko, ponieważ zazwyczaj nie
ma w nich tak długich nici komplementarnych. W odróżnieniu od replikacji
DNA, efektem syntezy RNA jest tylko jedna nić. W odpowiednich warunkach
pojedyncze nici RNA i DNA o komplementarnych sekwencjach mogą utworzyć
podwójną helisę RNA–DNA. Wtedy uracyle RNA łączą się z adeninami
DNA, zaś adeniny RNA z tyminami DNA. Dzięki powstawaniu takich
hybrydowych połączeń możliwe są najrozmaitsze laboratoryjne manipulacje
kwasami nukleinowymi; na przykład stopień komplementarności sekwencji
wyizolowanych RNA i DNA mierzy się sprawdzając ich zdolność do tworzenia
podwójnej helisy RNA–DNA[14].
2.2 Rola białek w organizmach
2.2.1 Budowa i struktura białek
Białka (ang. proteins) są jednymi z tych związków chemicznych, które
odgrywają najważniejszą rolę w procesach biochemicznych, determinujących
zjawiska życiowe. Stanowią one około 85% wszystkich związków organicznych
występujących w organizmach żywych, a ich właściwości i role są bardzo
zróżnicowane. Białka mogą pełnić następujące funkcje:
• zapasowe,• obronne,• regulujące (np. insulina),• katalizujące (enzymy, np. pepsyna, trypsyna, amylaza),• transportowe, magazynowe (np. hemoglobina),• strukturalne (aktyna, miozyna) oraz wiele innych.Podstawowym elementem, z którego zbudowane jest każde białko, są
aminokwasy. Aminokwasy to związki organiczne zbudowane z grupy aminowej,

14 Zagadnienia biologiczne
grupy karboksylowej, atomu wodoru i łańcucha bocznego, specyficznego dla
każdego aminokwasu (patrz rys. 2.7). W białkach występuje jedynie 20 rodzajów
Rysunek 2.7: Ogólny wzór strukturalny aminokwasów.
aminokwasów, jednak różnorodność tworzonych przez nie struktur białkowych
jest praktycznie nieograniczona. Listę tych aminokwasów oraz ich oznaczenia
przedstawia tabela 2.1.
Tablica 2.1: Aminokwasy występujące w białkach.
Nazwa aminokwasu Skrót Oznaczenie literowealanina ALA Aaspargina ASN Narginina ARG Rcysteina CYS Cfenyloalanina PHE Fglicyna GLY Gglutamina GLN Qhistydyna HIS Hizoleucyna ILE I
kwas asparginowy ASP Dkwas glutaminowy GLU E
leucyna LEU Llizyna LYS Kmetionina MET Mprolina PRO Pseryna SER Streonina THR Ttryptofan TRP Wtyrozyna TYR Ywalina VAL V

2.2. Rola białek w organizmach 15
Ważną własnością aminokwasów z punktu widzenia białek jest ich zdolność
do łączenia się, poprzez reakcje pomiędzy grupą karboksylową jednego
aminokwasu, a grupą aminową drugiego. W ten sposób pomiędzy dwoma
resztami aminokwasowymi powstaje wiązanie, nazywane fachowo „wiązaniem
peptydowym”. Schemat takiej reakcji przedstawia rysunek 2.8.
Rysunek 2.8: Tworzenie wiązania (poli)peptydowego.
Cząsteczki aminokwasów mogą łączyć się w ten sposób tworząc niezwykle
długie łańcuchy, zwane peptydami (o masie cząsteczkowej do 10 000u – patrz
słownik) i białkami (o masie cząsteczkowej powyżej 10 000u).Widzimy zatem, że
białka są związkami wielocząsteczkowymi (makromolekularnymi), a pojedynczy
łańcuch takiego białka może zawierać setki, a nawet tysiące aminokwasów. Taki
łańcuch białkowy nie stanowi jednak prostej nitki. W wyniku oddziaływań
pomiędzy cząsteczkami takiego łańcucha, fragmenty łańcucha przybierają
lokalnie różne skomplikowane struktury (helisa α, struktura β, zwrot β).
Dodatkowo, duże białka mogą zawierać kilka takich łańcuchów, co jeszcze
bardziej komplikuje ich budowę, ponieważ dochodzą jeszcze oddziaływania
między niezależnymi łańcuchami.
Biologowie molekularni zajmujący się badaniami nad białkami wyróżnili
cztery poziomy, na których rozważa się budowę białek:
• pierwszy poziom – struktura pierwszorzędowa,
• drugi poziom – struktura drugorzędowa,
• trzeci poziom – struktura trzeciorzędowaregulujące,
• czwarty poziom – struktura czwartorzędowa.

16 Zagadnienia biologiczne
2.2.1.1 Struktura pierwszorzędowa
Struktura pierwszorzędowa określa z jakich reszt aminokwasowych zbudowane
jest białko oraz w jakiej kolejności są one ze sobą powiązane w ramach całego
łańcucha. Skład i kolejność reszt ma istotny wpływ na własności takiego białka.
Zmiana choćby jednej reszty aminokwasowej (tzw. mutacja) może doprowadzić
do poważnych dolegliwości i chorób.
Strukturę pierwszorzędową białka można w łatwy sposób przedstawić,
zapisując ją jako ciąg liter (skrótów nazw), z których każda jednoznacznie
identyfikuje aminokwas tworzący daną resztę aminokwasową. Kolejność reszt
w łańcuchu wyznacza położenie w sekwencji (patrz rys. 2.9).
Rysunek 2.9: Sposób prezentacji struktury pierwszorzędowej białka.
2.2.1.2 Struktura drugorzędowa
W 1951 roku Robert Covey i Linus Pauling doszli do wniosku, że łańcuch
polipeptydowy nie stanowi prostej „nici”, a zawija się tworząc regularnie
powtarzające się struktury, nazwane przez nich helisą α (czyt. helisa alfa)
i harmonijką β (czyt. harmonijka beta). Dopiero sześć lat po dokananiu
tego odkrycia, udało się zaobserwować je doświadczalnie. Był to punkt
zwrotny w biologii molekularnej, ponieważ okazało się, że jeżeli znane są
dokładne parametry łańcucha polipeptydowego, wówczas można przewidzieć
jego konformację(sposób przestrzennego ułożenia atomów w cząsteczce białka).
Z czasem udało się zidentyfikować kolejne struktury, takie jak: zwroty β (czyt.
zwrot beta) i pętlę Ω (czyt. pętla omega), które pomimo tego, że nie występują
okresowo, to są powszechne i razem z helisami α i harmonijkami β tworzą
strukturę drugorzędową białka.
Podstawowym i powtarzającym się cyklicznie elementem w każdym łańcuchu
białkowym jest płaskie wiązanie peptydowe — CO — NH — . Obrót wokół niego
jest jednak ograniczony, ponieważ ma ono charakter wiązania podwójnego i jest
konstrukcją dość sztywną. Swobodny obrót ma jednak miejsce pomiędzy:

2.2. Rola białek w organizmach 17
• Cα (patrz rys. 2.7) i karbonylowym atomem węgla (Cα -– CO),• Cα i atomem azotu (N – Cα ).
Właśnie poprzez obrót wokół tych wiązań łańcuch polipeptydowy lub jego
fragmenty mogą tworzyć takie uporządkowane konformacje, jak helisy α,
harmonijki β, zwroty β, czy pętle Ω.
Helisa α jest prawoskrętną strukturą cylindryczną, powstałą na skutek
tworzenia się wewnątrzcząsteczkowych wiązań wodorowych pomiędzy grupami
NH i CO głównego łańcucha. Grupa CO każdego aminokwasu tworzy takie
wiązanie z grupą NH aminokwasu, który w sekwencji liniowej oddalony jest
o cztery reszty aminokwasowe (patrz rys. 2.10). Łańcuchy boczne aminokwasów
występują na zewnątrz cylindra, a ciasno skręcony łańcuch główny tworzy część
wewnętrzną. Na jeden obrót helisy α przypada 3,6 reszt aminokwasowych, a każda
reszta aminokwasowa przesunięta jest w stosunku do sąsiedniej o 0,15 nm (1nm
= 10−9m) wzdłuż osi helisy i obrócona o kąt 100 wokół osi. Skok helisy α, czyli
odległość między początkami dwóch kolejnych zwojów wynosi 0,54 nm (patrz
rys. 2.11).
Rysunek 2.10: Schemat wiązań wodorowych w helisie α.
Helisa α jest jedną z najlepiej poznanych struktur, a jej zawartość w białkach
może być bardzo różna i wahać się od 0% do niemal 100%. Przykładem może
być ferrytyna, białko magazynujące żelazo, w którym 75% reszt tworzy helisy α.
Udział struktury helikalnej w białkach określa się na podstawie wyników badań
metodami chiralooptycznymi. Okazuje się, że pojedyncze helisy nie przekraczają
długości 4,5 nm, tym samym są strukturami dość krótkimi. Jednak możliwość
splatania się dwóch lub większej ilości helis wokół siebie (tzw. helisy wyższego
rzędu) sprawia, że mogą one osiągnąć długości dochodzące do ponad 100 nm
(0,1 µm, czyli 0,1 * 10−6m)(patrz rys. 2.12). Przykładem może być miozyna
i tropozyna mięśni, keratyna włosów czy fibryna skrzepów krwi.
Obok helisy α istnieją również inne rodzaje helis, zarówno prawo–,
jak i lewoskrętne. O ich cechach charakterystycznych i rodzaju decydują

18 Zagadnienia biologiczne
Rysunek 2.11: Struktura helisy α. (A) Model wstęgowy z zaznaczonymi atomamiwęgla α i łańcuchami bocznymi (kolor zielony). (B) Widok z boku w modelukulkowym przedstawiający wiązania wodorowe (linie przerywane) między grupamiNH i CO. (C) Widok z góry pokazuje zwinięty szkielet znajdujący się w środkuhelisy α i wystające na zewnątrz łańcuchy boczne (kolor zielony). (D) Modelczaszowy rysunku C przedstawiający ściśle upakowany wewnętrzny rdzeń helisyα.
Rysunek 2.12: Helikalnie zwinięte helisy α. Dwie helisy są splecione wokół siebietworząc helisę wyższego rzędu tzw. superhelisę.
łańcuchy boczne aminokwasów występujących w łańcuchu polipeptydowym. Do
aminokwasów, które sprzyjają tworzeniu helis należą: Ala, Val, Phe, Met, Gln
i His, a do aminokwasów destabilizujących taką strukturę należą: Pro, Gly, Asn,
Tyr, czy Thr.
Drugą strukturą zaproponowaną przez Pauling’a i Corey’a była struktura
harmonijki β (patrz rys. 2.13, 2.14), nazywana również strukturą „pofałdowanej
kartki”, „pofałdowanego łańcucha” lub po prostu „strukturą β”. Budowa
takiej struktury, podobnie jak w przypadku helisy α, opiera się na wiązaniach
wodorowych pomiędzy grupami NH i CO. W przeciwieństwie do helisy, gdzie
takie wiązania tworzą się pomiędzy odpowiednimi, blisko leżącymi atomami
tego samego łańcucha, o tyle w harmonijce β wiązanie wodorowe może zostać
utworzone zarówno pomiędzy bliskimi, jak i odległymi częściami tego samego
łańcucha lub różnych łańcuchów. Tym samym harmonijka β jest strukturą prawie

2.2. Rola białek w organizmach 19
całkowicie rozciągniętą, w odróżnieniu od cylindrycznej helisy.
Rysunek 2.13: Białko bogate w struktury typu harmonijka β.
Rysunek 2.14: Struktura nici β. Łańcuchy boczne (kolor zielony) znajdują siępowyżej lub poniżej płaszczyzny nici.
Sąsiadujące ze sobą łańcuchy harmonijki mogą być ułożone w tym samym
kierunku (tzw. równoległa harmonijka β) lub w przeciwnych kierunkach
(tzw. antyrównoległa harmonijka β). Obie struktury oraz sposób połączenia grup
NH i CO przedstawia rys. 2.15 i 2.16.
Rysunek 2.15: Antyrównoległa harmonijka β. Sąsiednie nici β są ułożonew przeciwnych kierunkach. Strukturę stabilizują wiązania wodorowe międzygrupami NH i CO, łączące każdy aminokwas z sąsiadującym aminokwasem na niciantyrównoległej.

20 Zagadnienia biologiczne
Rysunek 2.16: Równoległa harmonijka β. Sąsiednie nici β są ułożone w tym samymkierunku. Wiązania wodorowe łączą każdy aminokwas na jednej nici z dwomaróżnymi aminokwasami na nici sąsiedniej.
Harmonijka β jest ważnym elementem strukturalnym białek. Przykładem
mogą być białka wiążące kwasy tłuszczowe, których budowa opiera się prawie
całkowicie na harmonijce β. Innymi strukturami, dzięki którym łańcuch
polipeptydowy może zmieniać swój kierunek są:
• struktura wstęga–zwrot–wstęga, nazywana również zwrotem β, β–zgięciemlub „zgięciem spinki do włosów”
• oraz struktura pętli.W miejscu, w którym występuje zwrot β, obserwuje się zmianę kierunku
łańcucha o 180 stopni. W strukturach tego typu wiązanie wodorowe tworzy się
najczęściej między grupą –CO reszty aminokwasowej na pozycji i, a grupą NH
reszty aminokwasowej na pozycji i+3. Struktury tego typu nie mają regularnej,
okresowej struktury jak helisy α i harmonijki β, są natomiast dość sztywne
i dobrze zdefiniowane. Najczęściej zwroty β i pętle znajdują się na powierzchni
białek, a to ma wpływ na ich reakcje z innymi białkami i innymi cząsteczkami.
2.2.1.3 Struktura trzeciorzędowa
Struktura trzeciorzędowa obrazuje wzajemne ułożenie w przestrzeni
poszczególnych fragmentów łańcucha peptydowego, a tym samym określa
trójwymiarowy kształt całej cząsteczki białka. Na kształt ten wpływ mają takie
czynniki jak:
• oddziaływanie na siebie zjonizowanych grup funkcyjnych (−COO(−)
i –NH(+)3 ),
• wytworzone wiązania disulfidowe — są to silne wiązania —S–S– (siarka)pomiędzy odległymi od siebie resztami cystern (Cys, C),
• oddziaływania dipol – dipol,

2.2. Rola białek w organizmach 21
• siły dyspersyjne hydrofobowych łańcuchów bocznych.
Struktura trzeciorzędowa białek jest strukturą niejednorodną, ponieważ obok
uporządkowanych struktur, takich jak helisy α, harmonijki β czy zwroty β,
występują również struktury nieuporządkowane. W strukturze białka możemy
ponadto wyróżnić ściśle upakowane części, które są wyraźnie oddzielone od reszty
cząsteczki. Każdą z tych części, których w pojedynczej cząsteczce może być nawet
kilka, nazywamy domeną.
Konformacja cząsteczki białka kształtuje się pod wpływem wielu czynników,
jednak w określonym środowisku decydującą rolę odgrywa sekwencja łańcucha
białkowego. Różnica między energią najbardziej uprzywilejowanej konformacji,
a energią innych struktur uporządkowanych (nawet strukturą rozplecioną) jest
niewielka, rzędu 10 kcal. W efekcie konformacja białek jest bardzo labilna
i zdarza się, że łańcuchy polipeptydowe o bardzo różnej sekwencji przybierają
zbliżoną konformację. Podobieństwo sekwencyjne ludzkiej hemoglobiny, owadziej
erytrokruoryny i leghemoglobiny, białka występującego w brodawkach na
korzeniach roślin motylkowych, jest mniejsze niż 20%, a mimo to wszystkie
te białka pełnią taką samą funkcję – są przenośnikami tlenu. Wszystkie one
zawierają hem jako grupę prostetyczną i mają prawie identyczną budowę
przestrzenną. Ta obserwacja skłoniła do przeprowadzenia badań porównawczych
znanych globin tworzących z hemem białka złożone. Okazało się, że spośród 226
porównywanych globin tylko dwie reszty aminokwasowe, histydyny proksymalnej
(najbliższej) wiążącej tlen i fenyloalaniny bezpośrednio oddziałującej z hemem,
powtarzały się i zajmowały te same pozycje w polipeptydowych łańcuchach.
Rezultaty te świadczą o nieograniczonych wprost adaptacyjnych możliwościach
białek. Z drugiej strony wiadomo, jak dramatyczne skutki może powodować
zmiana reszty chociażby jednego aminokwasu, czego przykładem są choroby typu
anemii sierpowatej i mukowiskocydozy. (patrz rys. 2.17)
Każde białko może zostać przekształcone ze struktury pofałdowanej
w strukturę rozplecioną. W tym stanie białko nie pozostaje jednak długo
i ponownie ulega pofałdowaniu do formy najkorzystniejszej energetycznie, przy
czym nie koniecznie musi to być postać wyjściowa. Cały proces trwa kilka sekund
lub minut, ponieważ fałdowanie ma miejsce w wielu miejscach jednocześnie.
Widać zatem, że tworzenie się takich struktur jak helisy czy harmonijki jest
istotnym elementem w konstrukcji uporządkowanej struktury białka.

22 Zagadnienia biologiczne
Rysunek 2.17: Struktura przestrzenna (trzeciorzędowa) mioglobiny.
2.2.1.4 Struktura czwartorzędowa
Struktura czwartorzędowa odnosi się do sytuacji kiedy białko zbudowane
jest z więcej niż jednego łańcucha polipeptydowego. Każdy z tych łańcuchów
nazywany jest podjednostką (domeną), tym samym struktura czwartorzędowa
odnosi się do wzajemnego ułożenia przestrzennego podjednostek i oddziaływań
jakie między nimi występują. Przykładem białka u którego możemy wyróżnić
strukturę czwartorzędową jest ludzka hemoglobina. Składa się ona z dwóch
podjednostek typu α i dwóch podjednostek typu β (patrz rys. 2.18).
Rysunek 2.18: Struktura czwartorzędowa ludzkiej hemoglobiny. Zbudowanajest z dwóch identycznych podjednostek (domen) α (kolor czerwony) i dwóchidentycznych podjednostek β (kolor żółty).
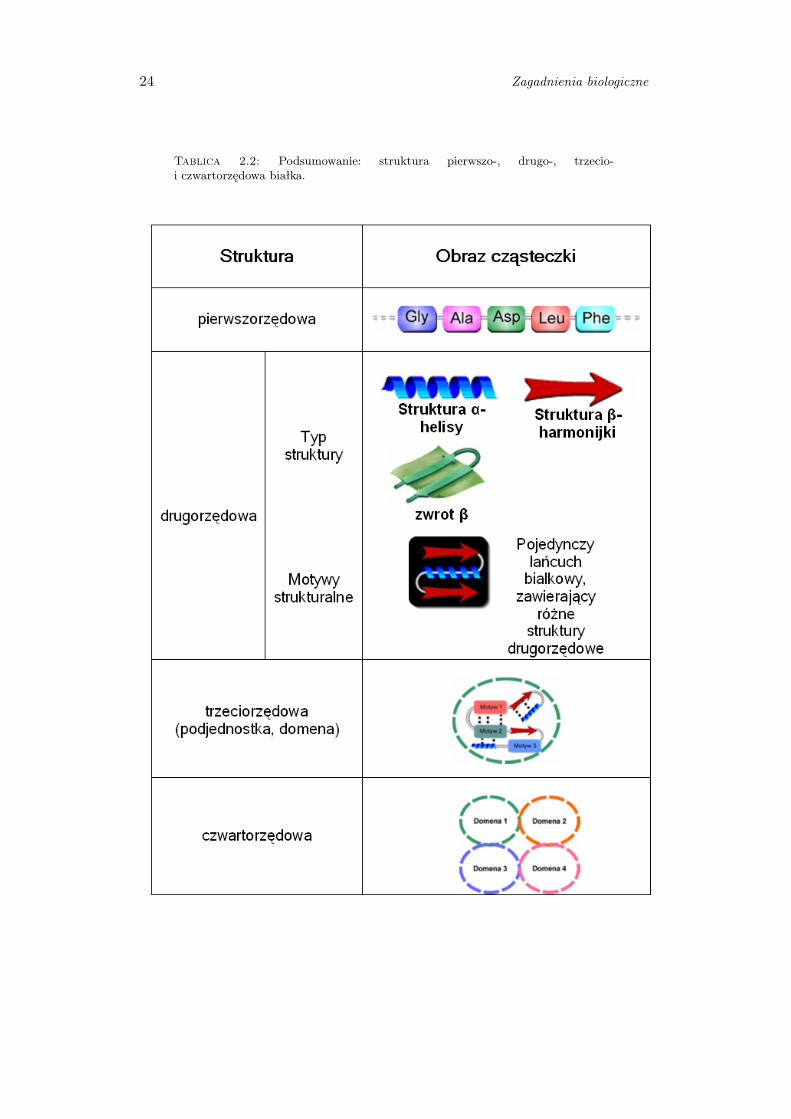
2.2.1.5 Podsumowanie
Analizując budowę białek wyróżnia się cztery poziomy ich organizacji.
Struktura pierwszorzędowa określa sekwencję aminokwasów w łańcuchu
polipeptydowym. Struktura drugorzędowa opisuje wzajemne, przestrzenne

2.2. Rola białek w organizmach 23
ułożenie reszt aminokwasów sąsiadujących ze sobą w sekwencji liniowej. Struktura
trzeciorzędowa odnosi się do powiązań przestrzennych i wzajemnego ułożenia
reszt aminokwasowych, znacznie oddalonych od siebie w sekwencji liniowej,
natomiast struktura czwartorzędowa dotyczy sytuacji kiedy białko zbudowane
jest z więcej niż jednego łańcucha polipeptydowego. Przedstawienie wszystkich
struktur zawiera tabela 2.2.
2.2.2 Metody stosowane do analizy struktur białkowych
2.2.2.1 Analiza struktur pierwszorzędowych
Analiza struktury pierwszorzędowej składa się z dwóch etapów:
• analizy składu aminokwasowego,• analizy sekwencyjnej.Analiza składu aminokwasowego dostarcza nam informacji, z jakich
aminokwasów i w jakich stosunkach zbudowane jest białko. Skład aminokwasowy
określa się po jego hydrolizie, która najczęściej odbywa się w sześciomolowym
kwasie solnym w temperaturze ok. 110C przez 24 godziny. W następnym
kroku określa się jaka jest zawartość poszczególnych aminokwasów w hydrolizacie
(analiza jakościowa i ilościowa). Obecnie cała analiza jest automatycznie
wykonywana przez analizatory aminokwasowe, które działają w oparciu o HPLC
(ang. High Performance Liquid Chromatography). HPLC to metoda analityczna
a także preparatywna, służąca do oczyszczania i identyfikacji substancji
chemicznych.
Analiza sekwencyjna służy do określenia kolejności reszt aminokwasowych
w łańcuchu peptydowym. Polega ona na „odcinaniu” i identyfikacji
każdego aminokwasu z łańcucha białkowego. Najczęściej wykorzystywana
jest tutaj degradacja Edmana, która polega na reakcji analizowanego
peptydu z fenyloizocyjanianem, w wyniku czego N–końcowy aminokwas
zostaje odszczepiony i ostatecznie, na skutek hydrolizy i cyklizacji, powstaje
3–fenylo–2–tiohydantoina odpowiedniego aminokwasu (PTH). PTH następnie
jest analizowane za pomocą metod chromatograficznych2: HPLC, GC (ang. gas
chromatography) bądź spektrometrii mas3. Im łańcuch białkowy jest dłuższy
tym bardziej opisana metoda staje się uciążliwa w stosowaniu. Z tej przyczyny
długie łańcuchy dzieli się na mniejsze fragmenty w różnych miejscach, określa się
2technika analityczna lub preparatywna. Wykorzystywana do rozdzielania i badania składumieszanin związków chemicznych. [27]
3uniwersalna technika analityczna, zaliczana do metod spektroskopowych, której podstawąjest pomiar stosunku masy molowej do ładunku elektrycznego jonów otrzymywanych z atomówlub związków chemicznych. [27]
24 Zagadnienia biologiczne
Tablica 2.2: Podsumowanie: struktura pierwszo-, drugo-, trzecio-i czwartorzędowa białka.

2.2. Rola białek w organizmach 25
sekwencję aminokwasów w każdym fragmencie, a następnie na zasadzie układania
puzzli odtwarza się całą sekwencję łańcucha polipeptydowego.
2.2.2.2 Analiza struktur drugo-, trzecio- i czwartorzędowych
Jedną z najbardziej popularnych metod badania przestrzennej budowy białek
jest rentgenografia. Metoda ta polega na rejestracji i analizie widm dyfrakcyjnych
promieni rentgenowskich, które powstają na skutek interakcji promieniowania
rentgenowskiego z chmurami elektronowymi cząsteczek tworzących analizowany
kryształ. Warunkiem zastosowania tej metody jest zatem otrzymanie białka
w postaci krystalicznej. Udało się to uzyskać jak dotąd zaledwie dla kilkuset
białek, ale dzięki temu ich struktura została dokładnie poznana.
Inną metodą, która pozwala określić udział struktur drugorzędowych
w cząsteczce białka jest spektrometria w nadfiolecie. Polega ona na wykorzystaniu
promieniowania UV z zakresu 170-240 nm. Najczęściej korzysta się tutaj
z tzw. dychroizmu kołowego CD (ang. circular dichroism), dzięki któremu
stosunkowo łatwo oszacować udział helis α w cząsteczce białka. Struktury β mają
niestety znacznie mniejszy wpływ na widmo CD, stąd trudniej określić ich udział
metodami chiralooptycznymi.
Kolejną metodą wykorzystywaną w określaniu konformacji białka jest
spektroskopia magnetycznego rezonansu jądrowego (ang. Nuclear Magnetic
Resonance), zarówno protonowa, jak i 13C, 31P, 15N. Dostarcza ona cennych
informacji o tym jak ułożone są protony w przestrzeni, a tym samym jaka jest
odległość między nimi. To ostatecznie pozwala odtworzyć model przestrzenny
białka. Technika NMR ma tą wadę, że wykorzystuje roztwory, a to ogranicza
pole działania do białek rozpuszczalnych w wodzie.
Podane wyżej metody pozwalają dokładnie określić strukturę przestrzenną
białka. Wymagają jednak wielu godzin pracy wysoko wykwalifikowanej obsługi
technicznej, co przy dużej liczbie sekwencji aminokwasowych białek jest trudne
do zrealizowania. Z tego względu podejmuje się próby przewidywania struktur
przestrzennych i własności białek przy wykorzystaniu metod informatycznych.
Metody te bazują na tym, że właściwości fizyczne i chemiczne 20 aminokwasów
budujących białka są gruntownie poznane, a tym samym można spróbować
określić cechy nieznanych białek na podstawie tych właściwości.
Do przewidywania struktur drugorzędowych wykorzystuje się m.in. sieci
neuronowe. Sieć neuronowa ma możliwość „uczenia się” w oparciu o przykłady,
zamiast ślepego wykonywania instrukcji krok po kroku. Każda taka sieć posiada
warstwę wejściową i wyjściową. Pomiędzy tymi warstwami może znajdować

26 Zagadnienia biologiczne
się więcej warstw pośrednich, w których następuje właściwy proces „uczenia”.
Osiąga się to poprzez dostarczenie takiej sieci zestawu danych treningowych.
W przypadku białek, zestawem danych treningowych są sekwencje aminokwasowe
dla których znane są dokładne drugorzędowe struktury przestrzenne. Przykłady
algorytmów bazujących na sieciach neuronowych to:
nnpredict – wykorzystuje dwuwarstwową sieć neuronową, na wejście której
podawana jest sekwencja w formacie FASTA. Stopień zgodności z danymi
empirycznymi dla najlepszych wyników wynosi około 65% [1];
PHDsec – bazuje na sieciach neuronowych i przypisuje każdą resztę
aminokwasową do określonego typu struktury drugorzędowej. Średni
stopień zgodności z danymi eksperymentalnymi wynosi 72%, najlepsze
wyniki sięgają 90%;
PSIPRED – algorytm rozwinięty na Uniwersytecie w Warwick (Wielka
Brytania), korzysta z dwóch wspomagających się sieci neuronowych. Średni
stopień zgodności wyników z danymi eksperymentalnymi wynosi 76.5%.
Inne metody przewidywania struktur drugorzędowych to:
metoda Garnier-Gibsona-Robson (GOR) – przy analizie struktury
drugorzędowej bierze pod uwagę własności każdego aminokwasu oraz
aminokwasów z nim sąsiadujących, nie skupia się na jednym elemencie
łańcucha polipeptydowego określając czy należy on do helisy α, czy
harmonijki β, a rozważa cały segment aminokwasów budujących białko.
Średni stopień zgodności wyników z danymi eksperymentalnymi wynosi
63%
metoda homologów Levina – bazuje na założeniu, że krótkie, podobne
łańcuchy peptydowe powinny mieć identyczną strukturę drugorzędową. Pod
uwagę brane są tutaj fragmenty białek zawierające 7 reszt aminokwasowych,
które porównywane są ze wszystkimi białkami zgromadzonymi w bazie
danych, przy wykorzystaniu macierzy podobieństwa.
metoda podwójnego przewidywania (G. Deleage, B. Roux) – składa się
z dwóch faz. W pierwszej fazie następuje przewidywanie struktury
drugorzędowej w oparciu o parametry opisane przez Chou i Fasmana, a w
drugiej opierając się na sekwencji aminokwasów w łańcuchu białkowym.
Oba wyniki są następnie analizowane i konstruowana jest ostateczna postać
struktury drugorzędowej białka.
metoda CNRS , znana jako SOMPA – wykorzystuje szereg innych
algorytmów, takich jak: metoda GOR, metoda homologów Levina,
metoda PHD i metoda podwójnego przewidywania do skonstruowania
tzw. „przewidywania integrującego”.

2.2. Rola białek w organizmach 27
W przypadku struktur trzecio– i czwartorzędowych wykorzystuje się inne
metody:
budowanie modelu homologii lub metoda „nawlekania”. Takie metody
poszukują struktur o podobnych elementach zwinięcia łańcucha
z pominięciem kryterium podobieństwa na poziomie sekwencji.
W metodzie tej pobierana jest sekwencja badana, dla
której nie jest znana struktura, a następnie nakłada się ją
na współrzędne strukturalne białka docelowego o strukturze
poznanej w wyniku analizy krystograficznej lub NMR. Sekwencja
jest „nawlekana” z przemieszczeniem co jedną pozycję przez
znaną strukturę wzorcową, która z góry narzuca pewne
ograniczenia fizyczne i przestrzenne. (. . . ) Dla każdego ułożenia
sekwencji w strukturze docelowej obliczane są oddziaływania
między parami reszt oraz oddziaływania hydrofobowe. Te
obliczenia termodynamiczne służą do określenia najbardziej
optymalnego uzgodnienia pod względem energetycznym
i stabilności konformacyjnej dla badanej sekwencji w odniesieniu
do struktury wzorcowej.[1]
Metody te są bardzo skuteczne ale wymagają rozbudowanych programów
i komputerów o dużej mocy obliczeniowej.
Inne metody przewidywania struktur trzecio– i czwartorzędowych to:
algorytm DALI – poszukuje podobnych szablonów kontaktu między dwoma
białkami, przeprowadza optymalizację wyników i zwraca najlepsze wyniki
dla sporządzonego zestawienia dopasowanych struktur (Holm i Sander,
1993).
metoda TOPITS (Rost, 1995) – rozszerzenie metody PHD. W metodzie
TOPITS struktura trzeciorzędowa zawarta w PDB zostaje przetłumaczona
na jednowymiarowe „łańcuchy” struktur drugorzędowych. Następnie za
pomocą PHD określa się dla danej sekwencji strukturę drugorzędową.
Aby uzyskać przewidywaną strukturę dopasowuje się badany łańcuch oraz
łańcuchy docelowe wykorzystując do tego programowanie dynamiczne.
2.2.3 CASP
Z pomysłu organizacji Protein Structure Prediction Center4 narodził się
projekt CASP — Critical Assessment of Techniques for Protein Structure
4Protein Structure Prediction Center, http://predictioncenter.org/, jest częściąCentrum Genomów na Uniwersytecie Kaliforni. PSPC jest wspierana przez Państwowy InstytutZdrowia w Ameryce oraz Amerykańską Państwową Bibliotekę Medyczną.

28 Zagadnienia biologiczne
Rysunek 2.19: Logo CASP.
Prediction, (Krytyczna Ocena Technik Przewidywania Struktury Białka). Miał on
pomóc w poznaniu stopnia rozwoju prac w dziedzinie przewidywania struktury
białek, ocenieniu poczynionych postępów i wskazaniu najbardziej opłacalnych
kierunków rozwoju w tej dziedzinie [6]. O randze konkursu może świadczyć fakt,
że w ostatnich latach w Stanach Zjednoczonych wysokie miejsce w konkursie
CASP stało się prawie wymogiem przy staraniu się o granty naukowe w dziedzinie
biochemii.
Przebieg konkursu jest następujący: organizatorzy ogłaszają listę
kilkudziesięciu sekwencji białek. Ich struktury przestrzenne zostały już wcześniej
poznane w wyniku prac doświadczalnych, które przeprowadziły zespoły
badawcze. Struktury te nie zostają upubliczniane aż do zakończenia konkursu.
Uczestnicy mają za zadanie znalezienie za pomocą symulacji komputerowej
struktur przestrzennych jak najbardziej zbliżonych do oryginalnych (przykład
na rysunku 2.20). Na koniec konkursu odbywa się spotkanie, na którym zostają
podsumowane wszystkie uzyskane wyniki.
Pierwsze takie spotkanie odbyło się w centrum konferencyjnym Asilomar
w San Francisco, w grudniu 1994. Poświęcono mu specjalne wydanie raportu
PROTEINS: Structure, Function, and Genetics (Tom 23, numer 3, 1995). Pod
adresem http://bbrp.llnl.gov/bbrp/structural_bio/asilomar/Meeting.
desc.text.html dostępne jest również streszczenie tego spotkania.
Informacje o strukturach do rozpoznania pozyskiwane są przez organizatorów
za pomocą krystalografii5 lub metody magnetycznego rezonansu jądrowego. We
wszystkich rozpoznaniach cele są uzyskiwane w czterech kategoriach:
• modelowanie homologiczne (comparative modeling)stosowane gdy występuje jasne powiązanie pomiędzy badaną sekwencją
5naświetlanie kryształu białka promieniami rentgenowskimi

2.2. Rola białek w organizmach 29
Tablica 2.3: Popularność CASP
CASP1 CASP2 CASP3 CASP4 CASP5 CASP6
grupy uczestników 35 152 120 160 187 201zgłoszone cele 33 42 43 43 67 87
a jedną lub więcej znaną strukturą (podobne sekwencje→ podobne białka)• identyfikacja foldu wraz z przeciąganiem sekwencji przez struktury (foldrecognition or threading )
sprawdzanie sekwencji pod względem podobieństwa z bazą danych foldów
• składanie od początku (ab initio folding)otrzymywanie struktur przybliżonych poprzez czytanie z sekwencji, zwijanie
na siatkach, składanie z krótkich fragmentów
• dokowanie6 (docking)przewidywanie trybu wiązania się ligandów7 i protein
Włożono sporo wysiłku aby eksperyment zyskał jak największe grono
uczestników. Tabela 2.3 prezentuje ilość uczestników i wykonanych przez nich
zgłoszeń celów na przestrzeni lat.
Warto wspomnieć również o niedawnym udanym udziale polskich naukowców
w eksperymencie CASP6 [19]. Byli to profesor Andrzej Koliński z Wydziału
Chemii UW, dr Krzysztof Ginalski z Interdyscyplinarnego Centrum Modelowania
Matematycznego i Komputerowego UW i BioInfoBank Institute (został po raz
drugi nieoficjalnym liderem CASP) oraz dr Janusz Bujnicki z Międzynarodowego
Instytutu Biologii Molekularnej i Komórkowej. Znaleźli się oni wśród zwycięzców
ostatniej edycji konkursu. Jedno z najlepszych ich dopasowań widać na
rysunku 2.20.
Do tej pory odbyło się sześć eksperymentów.
Rezultaty prac na przestrzeni lat zostały zebrane w internecie:
• CASP1 (1994):ftp://iris4.carb.nist.gov/pub/model_database
• CASP2 (1995):http://predictioncenter.org/casp2/targets.html
• CASP3 (1998):http://predictioncenter.org/casp3/targets/cgi/casp3-view.cgi
• CASP4 (2000):http://predictioncenter.org/casp4/targets/cgi/casp4-view.cgi
6pojawiło się w 1996 roku7cząsteczki rozpoznawane przez receptor i przyłączające się do niego

30 Zagadnienia biologiczne
Rysunek 2.20: Po lewej najdokładniejszy model struktury jednego z białek(wystawiony do konkursu CASP6, opracowany przez dr Krzysztofa Ginalskiego)oraz jego prawdziwy odpowiednik.
• CASP5 (2002):http://predictioncenter.org/casp5/targets/cgi/casp5-view.cgi
• CASP6 (2004):http://predictioncenter.org/casp6/targets/cgi/casp6-view.cgi
Ciekawe adresy związane z CASP:
• http://www.predictioncenter.org/ – strona organizatora konkursu• http://www.forcasp.org/ – dyskusje na temat CASP• http://www.mnii.gov.pl/mnii/index.jsp?place=Lead08&news_cat_id=495&news_id=1669&layout=4&page=text artykuł opisujący sukces
polskiego zespołu
• http://www3.interscience.wiley.com/cgi-bin/jissue/104551827periodyk podsumowujący CASP (dostęp płatny)

Rozdział 3
Narzędzia
3.1 Narzędzia biologiczne
Analizując pojedynczą sekwencję trudno jest wnioskować o pełnej strukturze
i funkcjonalności cząsteczki przez nią opisywanej dopóki nie zostaną odkryte
mechanizmy skręcania białka. Powinna być ona badana przez porównanie
do innych, poznanych już dokładnie sekwencji. Dopiero na tej podstawie
można tworzyć hipotezy dotyczące powiązań i funkcji białek. Być może dzięki
dokładniejszym badaniom ludzkiego genomu, naukowcy będą potrafili znaleźć
lekarstwa na choroby takie jak AIDS czy rak. Algorytmy, które powstały na
przestrzeni ostatnich lat mają za zadanie jak najdokładniej zbadać strukturę
białka. Przy tworzeniu platformy WebMobis wykorzystano gotowe implementacje
najbardziej skutecznych i popularnych algorytmów.
3.1.1 BLAST
Najważniejszym z narzędzi, które dołączono do projektu WebMobis, był
BLAST ( ang. Basic Local Alignment Search Tool ). BLAST dostarcza
narzędzi do szybkiego i sprawnego przeszukiwania baz danych nukleotydów
i białek. Potrafi odnaleźć obszary podobieństwa, czyli fragmenty sekwencji
o podobnej kolejności aminokwasów (patrz rysunek 3.1 na stronie 34) zawarte
w niepowiązanych ze sobą białkach.
Istnieją następujące odmiany BLAST [3]:
BLASTP porównywanie sekwencji białka z bazą sekwencji białek
BLASTN porównywanie sekwencji nukleotydów z bazą sekwencji nukleotydów
31

32 Narzędzia
BLASTX porównywanie („przetłumaczonej”1) sekwencji nukleotydów z bazą
sekwencji białek
TBLASTN porównywanie („przetłumaczonej”) sekwencji białka z bazą
sekwencji nukleotydów
TBLASTX porównywanie (przetłumaczonej) sekwencji nukleotydów z bazą
(przetłumaczonych) sekwencji nukleotydów
MEGABLAST odmiana optymalizowana do porównań długich, niewiele się
różniących sekwencji nukleotydów (szybsza niż BLASTN).
W projekcie użyto odmiany BLASTP.
3.1.1.1 Działanie programu
Działanie programu opiera się na porównaniu podanej przez użytkownika
sekwencji z sekwencjami w istniejącej bazie danych [7]. Porównywane
są zawsze pary sekwencji, a następnie każdemu takiemu dopasowaniu
zostaje przyporządkowana liczba punktów S proporcjonalna do podobieństwa
dostarczonego celu do wzorcowej sekwencji.
Wartość S obliczania jest poprzez zsumowanie punktów dla każdej
dopasowanej pozycji2 i luk. Za każdą dopasowaną pozycję do S dodawany jest
jeden punkt. Do obliczenia punktów ujemnych za wystąpienie luk, w BLAST
użyto „afinicznego kosztu luk”, który obciąża wynik wartością −a za każdewystąpienie luki i wartością −b dla każdej reszty aminokwasowej w luce. Lukazłożona z k reszt otrzyma punktację −(a + b ∗ k), a luka o długości 1 uzyska−(a + b) punktów. Wartości a i b różnią się w zależności od odmiany BLAST.Dla użytego w projekcie BLASTP wynoszą one odpowiedno -11 i -1.
Podobieństwo jest mierzone i pokazywane poprzez uszeregowanie dwóch
sekwencji. Wyniki uszeregowania mogą być globalne lub lokalne (jest to zależne
od algorytmu). Uszeregowanie globalne jest optymalnym uszeregowaniem, które
zawiera wszystkie znaki z każdej sekwencji. Uszeregowanie lokalne zawiera tylko
najbardziej podobne do siebie obszary obu sekwencji [7]. Rozróżnienie pomiędzy
rzeczywistymi i błędnymi dopasowaniami wykonywane jest przy oszacowaniu
prawdopodobieństwa możliwego wystąpienia dopasowania. Oczywiście samo
podobieństwo nie jest wystarczające do wyrokowania o podobnej funkcji
obszarów.
1konwersja potrzebna aby móc porównywać białka z bazą danych nukleotydów2dopasowana pozycja to para tych samych aminokwasów na tych samych pozycjach
względnych w porównywanych sekwencjach

3.1. Narzędzia biologiczne 33
3.1.1.2 Algorytm
Algorytm wyszukiwania odpowiednich uszeregowań można w uproszczeniu
przedstawić w następujących krokach [17]:
1. podzielenie badanej sekwencji na nakładające się słowa
2. dla każdego ze słów określenie słów pokrewnych (neighborhood words), które
jeśli zostaną znalezione w innej sekwencji, mają szansę stać się częścią
MSP (Maximal Segment Pair) – pary o najwyższej punktacji S spośród
wszystkich par w porównywanych sekwencjach.
3. przeszukanie bazy w celu znalezienia słów pokrewnych
4. wydłużenie uszeregowania zaczynając od słowa pokrewnego
5. programowanie dynamiczne wokół MSP
3.1.1.3 Parametry
W pierwszych etapach szczególnie ważne są dwa parametry: długość słowa
i minimum punktowe, które musi uzyskać słowo sparowane z innym, aby
uszeregowanie zostało zainicjowane. Dla białek minimalna długość słowa wynosi
3, ale wymagane są dwa słowa w bliskiej odległości od siebie. Długość
słowa i punktacja powinny być tak dobrane, aby zmaksymalizować prędkość
(zminimalizować czas zajętości procesora) i dokładność przeszukiwania (znaleźć
jak najwięcej homologii w bazie). Od tych parametrów zależy również częstość
występowania dwóch błędów: pojawiania się w wynikach sekwencji, które nie są
prawdziwymi homologiami (ang. false positives) i brak prawdziwych homologii
w wyniku pominięcia ich przy przeszukiwaniu (ang. false negatives).
Działaniem programu można sterować poprzez zmianę wartości progowych
dwóch parametrów: długości słowa i punktacji minimalnej [17]. Gdy długość słowa
zwiększa się dla danej punktacji minimalnej, prawdopodobieństwo znalezienia
słowa pokrewnego w bazie maleje. Również zużycie czasu procesora zmniejsza
się, ale obniża się też skuteczność przeszukiwania. Wzrasta też liczba słów
pokrewnych. Gdy zmniejsza się minimalna punktacja dla danego słowa, liczba
słów pokrewnych zwiększa się – staje się proporcjonalna do e−T (gdzie T to
minimalna punktacja jaką musi otrzymać dane słowo sparowane z innym, aby
dopasowanie zostało zainicjowane), wydłuża się też czas zajętości procesora, który
jest proporcjonalny do liczby słów. Za cenę zwiększenia czułości następuje utrata
specyficzności.

34 Narzędzia
Rysunek 3.1: Przebieg porównywania sekwencji. Na czerwono i niebieskozaznaczono serie zgodnych wartości. Podkreśleniem oznaczono HSP
Rysunek 3.2: Logo programu LGScore
3.1.1.4 Poszukiwanie MSP
W BLAST została zaimplementowana metoda poszukiwania podobieństw
MSP (Maximal Segment Pairs). Przebieg porównywania pary sekwencji został
przedstawiony na rysunku 3.1. Stosuje się tam następującą punktację:
• +1 za każdą zgodność
• -1 za każdą niezgodność
Najniższą wartością jest zero. Na podstawie przydzielonych punktów tworzone
są HSP (ang. High Scoring Pairs),które można traktować jako potencjalne MSP.
HSP to nie tylko serie identycznych aminokwasów w obu strukturach. Stosuje się
też tzw. wydłużanie, które pozwala na wcielenie do HSP również niepasujących
aminokwasów. Wydłużenie jest prowadzone do momentu osiągnięcia punktacji
nie mniejszej od maksymalnej o ustaloną (parametrem x-dropoff ) wartość. Jest
to najbardziej czasochłonny etap pracy BLAST.
W kolejnym etapie na znalezionych HSP wykonywany jest algorytm
Smith – Watermana [13], a istotność szeregowania mierzona jest za pomocą
statystyki Karlina i Altschula [12] i na tej podstawie sekwencje dołączane są
do wyników programu.

3.1. Narzędzia biologiczne 35
3.1.2 LGScore
Program LGScore (logo na rysunku 3.2), użyty w projekcie, jest
implementacją algorytmów porównujących dwa modele cząsteczek i określających
ich podobieństwo. Jeden z modeli powinien być uzyskany przez użytkownika,
drugi natomiast rzeczywistą strukturą. Obie cząsteczki powinny mieć tę samą
numerację reszt aminokwasowych. Algorytm do obliczeń wymaga jedynie
obecności wpółrzędnych położeń węgli Cα.
3.1.2.1 Rodzaje LGScore
LGScore istnieje w dwóch odmianach. Pierwsza z nich jest metodą
zależną od uszeregowania. Próbuje ona odnaleźć jak najdłuższe przybliżone
dopasowanie3 (podobnie jak próbuje to robić MaxSub, patrz punkt 3.1.6)
i oblicza dla niego punktację statystyczną (definicja 2). LGScore2 nie zależy
od uszeregowania, pozwala więc między innymi na porównania nie obciążone
błędami spowodowanymi przez pewne przesunięcia między częściami modeli.
3.1.2.2 Uruchomienie i wynik działania programu
Do uruchomienia programu potrzebne są dwa modele białek w formacie
PDB (Protein Data Bank) (patrz słownik). Pierwszy z nich to plik struktury
użytkownika, drugi – oryginalna struktura.
Użytkownik wpisując
LGscore uzyskany.pdb rzeczywisty.pdb
otrzyma wynik podobny do następującego:
SCORE: 0.88 28 0.9 485.2 1.636783e-04
Oznacza on, że w pierwszym białku został odnaleziony fragment pokrywający
88% rzeczywistego modelu. Długość tego pokrycia wyniosła 28, a współczynnik
rmsd (definicja 1) 0.9. Kolejnym elementem jest punktacja S (definicja 2) równa
w tym przypadku 485, 2. Została również obliczona P-wartość4 według ustaleń
zawartych w [16] o poziomie 1.64e−4.
Definicja 1 RMSD – Root Mean Square Deviation - wyznacza standardowe
odchylenie we współrzędnych kartezjańskich obliczone między zbiorem atomów
3dopasowanie przybliżone to dopasowanie które pozawala na pewne niezgodności pomiędzyporównywanymi sekwencjami
4najmniejszy poziom istotności, przy którym zaobserwowana wartość statystyki testowejprowadzi do odrzucenia hipotezy zerowej

36 Narzędzia
pewnej struktury dynamicznej a odpowiadającym zbiorem struktury odniesienia
(innej cząsteczki lub innej konfiguracji) [29].
Wzór służący do obliczenia współczynnika wygląda następująco [10]:
RMSD (N ;x, y) =
[∑Ni=1wi‖ xi − yi ‖
2
N∑Ni=1wi
] 12
(3.1)
gdzie:
N liczba pozycji atomów dla struktury x i odpowiadających im atomów ze
struktury y
w(i) współczynniki wag atomów ze struktury y
Levitt i Gerstein [16] wprowadzili wartość pomiarową służącą do obliczania
wagi podobieństwa pomiędzy dwoma strukturami będącymi w superpozycji.
Definicja 2 (Levitt and Gerstein) Statystyczna waga podobieństwa pomiędzy
parą białek Sstr
Sstr =M
(∑ 1
1 + (dij/d0)2 −Ngap2
)(3.2)
gdzie M jest równe 20, dij jest odległością między resztami aminokwasowymi i
a j, d0 jest równane do 5A (0.1 nm), a Ngap jest liczbą przerw w uszeregowaniu.
Aby obliczyć rzeczywiste znaczenie wyniku równania w definicji 2, użyto
zbioru uszeregowań strukturalnych niepowiązanych ze sobą białek i wyznaczono
rozkład wartości atrybutu Sstr w zależności od długości uszeregowania l. Z tego
rozkładu obliczono P–wartość zależną od Sstr i l [16].
Wartość S jest obliczana w ten sam sposób co LGScore, ale zamiast używać
P-wartości zastosowano podział pomiaru Sstr przez długość modelu. Początkowo
statystyki były ponownie liczone, aby lepiej radzić sobie z krótkimi fragmentami
białek.
Wyniki uzyskane z LGScore i LGScore2 są równoważne. Aby otrzymać
jak najlepszy rezultat zaleca się przetestowanie plików obiema metodami
i wykorzystanie dokładniejszego wyniku. Zdarzyć się może bowiem, że wynik
uzyskany za pomocą LGScore2 będzie gorszy jeśli wstawiono lub usunięto duże
fragmenty struktury.
LGScore jest wykorzystywany również w konkursie CASP (punkt 2.2.3).
Więcej o LGScore znaleźć można na stronie autorów, http://www.sbc.su.se/
~arne/lgscore/ oraz w dokumencie [5].

3.1. Narzędzia biologiczne 37
3.1.3 Jmol
Jmol jest narzędziem do wizualizacji molekularnej, który miał wspomagać
pracę naukowców, tworząc wysokiej jakości obrazy białek, a także pozwolić
osobom zainteresowanym tą dziedziną nauki na poznanie i zrozumienie
podstawowej budowy związków chemicznych.
Jmol [2] powstał w celu zastąpienia narzędzia Xmol (patrz słownik).
Następujące przyczyny spowodowały powstanie Jmola:
• jakość obrazów tworzonych przez Xmol była dość niskiej jakości;• źródła programu były niedostępne dla użytkowników, a projekt nie byłrozwijany i z biegiem czasu wersje były przestarzałe.
Powyższe argumenty sprawiły, że stworzono narzędzie z otwartymi źródłami,
które działałoby sprawniej i szybciej, generując przy tym lepszej jakości obrazy.
Okazało się jednak, że w miarę powstawania Jmol stał się narzędziem, które
znacznie przewyższało funkcjonalnością Xmola i zdecydowanie wyparło go
z rynku. Wydano kolejne wersje z wieloma dodatkowymi opcjami, sugerowanymi
nawet przez samych użytkowników. Pod koniec 2002 roku zintegrowano Jmola
z Chemical Development Kit (jest to biblioteka javowa wspomagająca chemię
i bioinformatykę), w celu stworzenia pluginu mającego zastąpić plugin Chime.
W miarę upływu czasu powstał nowy engine, który miał zapewnić, że aplet
Jmola będzie działał bez wymagania dodatkowego wsparcia sprzętowego. Engine
ponadto wspierał wizualizację dużego rozmiaru molekuł. Ostatecznie w grudniu
2004 wydano w pełni funkcjonalną wersję Jmola.
Jmol jest apletem napisanym w Javie, który działa na plikach w formacie
PDB (Protein Data Bank). Aplet umieszcza się na stronie internetowej
z wykorzystaniem skryptu JavaScript, który bardziej szczegółowo opisany jest
na stronie http://www.jmol.org/jslibrary. Wykorzystując funkcje ze skryptu,
aplet otwiera i wizualizuje zawartość pliku PDB. Na rysunku 3.3 widoczny jest
przykład wizualizacji aminokwasu.
3.1.4 MinRMS
Istniejące algorytmy dopasowujące struktury białkowe generują sprzeczne
wyniki, które trudno zinterpretować. Potrzebny był algorytm, który mógłby
dostarczyć kontekst interpretacji dopasowań i wykorzystywałby prostą metodę
do scharakteryzowania podobieństw struktur białkowych.
MinRMS [25] jest programem, który znajduje minimalne RMSD (definicja 1
na stronie 35) pomiędzy dwoma białkami jako funkcję ilości dopasowań resztek
par z heurystycznie ograniczoną przestrzenią szukania.

38 Narzędzia
Rysunek 3.3: Przykład wizualizacji Jmol
Algorytm uszeregowania wykorzystuje koordynaty węgli Cα do reprezentacji
każdej reszty kwasowej aminokwasu i ma złożoność obliczeniową O(m3n2
), gdzie
m i n oznaczają długości sekwencji białek. Z przyczyn praktycznych wyszukiwanie
jest obszerne. Metoda jest wystarczająco szybka dla porównania umiarkowanych
rozmiarów białek (mniejszych niż 800 reszt) na konkurencyjnych komputerach.
MinRMS generuje rodzinę uszeregowań jako funkcję ilości dopasowanych par
reszt. Dla dwóch białek o długości sekwencji m i n (n ¬ m), będzie m najlepszychuszeregowań RMSD. Aby ułatwić przeszukiwanie tych wielu dopasowań powstało
narzędzie do wizualizacji, AlignPlot. Ten graficzny interfejs użytkownika pozwala
w łatwy sposób przeszukać przestrzeń dopasowań.
MinRMS jest programem nie posiadającym graficznego interfejsu
użytkownika. Działa na dwóch plikach zapisanych w formacie PDB (Protein
Data Bank). Uruchamiany jest z linii poleceń komendą:
minrms filename1 filename2
Gdzie:

3.1. Narzędzia biologiczne 39
filename1, filename2 - pliki w formacie PDB zawierające strukturę białka
Program można również uruchomić na kilka innych sposobów. Między innymi
z wykorzystaniem pliku konfiguracyjnego lub z filtrami. Bardziej szczegółowe
informacje znajdują się na stronie http://www.cgl.ucsf.edu/chimera/docs/
ContributedSoftware/minrms/minrms.html.
MinRMS zwraca tablicę, w której zawarte są informacje o każdym
uszeregowaniu struktury:
• liczba dopasowanych reszt• RMSD (definicja 1) dla uszeregowania• najdłuższą odległość pomiędzy jakąkolwiek parą dopasowanych
uszeregowań
• wagę podobieństwa pary białek (definicja 2)• macierz tranformacji dla uszeregowań struktur
3.1.5 DSSP
Program DSSP (ang. Database of Secondary Structure in Proteins) [23]
służy do określania elementów struktur drugorzędowych w znanych strukturach
białkowych. Innymi słowy definiuje strukturę drugorzędową oraz cechy
geometryczne białek. Należy jednak zwrócić uwagę, że DSSP nie przewiduje
struktury białka. Program został zaprojektowany przez Wolfgang Kabscha
i Chrisa Sandera. DSSP jest bazą danych struktur drugorzędowych dla wszystkich
białek zawartych w PDB (Protein Data Bank).
Program działa na plikach w formacie PDB (Protein Data Bank). Można go
uruchomić poleceniem:
dsspcmbi filename
Gdzie:
filename - plik w formacie PDB zawierający strukturę białka
Bardziej szczegółowe informacje o programie DSSP dotyczące uruchamiania
można znaleźć na stronie http://swift.cmbi.ru.nl/gv/dssp/descrip.html.
Na rysunku 3.4 zamieszony jest przykładowy, uproszczony wynik działania
programu DSSP.
Każda linia zawiera następującą informację:
RESIDUE – dwie kolumny z numerami reszt. Pierwsza z nich jest
sekwencyjnym numerem reszt DSSP, rozpoczynając od pierwszej reszty
w zbiorze danych, włączając łańcuch przerw. Druga kolumna daje numery
reszt sekwencji, kody wstawienia i identyfikatory łańcucha.

40 Narzędzia
HEADER HYDROLASE (SERINE PROTEINASE) 17-MAY-76 1EST240 1 4 4 0 TOTAL NUMBER OF RESIDUES, NUMBER OF CHAINS,
NUMBER OF SS-BRIDGES(TOTAL,INTRACHAIN,INTERCHAIN) .10891.0 ACCESSIBLE SURFACE OF PROTEIN (ANGSTROM**2)162 67.5 TOTAL NUMBER OF HYDROGEN BONDS OF TYPE O(I)-->H-N(J);PER 100 RESIDUES0 0.0 TOTAL NUMBER OF HYDROGEN BONDS IN PARALLEL BRIDGES;
PER 100 RESIDUES84 35.0 TOTAL NUMBER OF HYDROGEN BONDS IN ANTIPARALLEL BRIDGES;PER 100 RESIDUES...26 10.8 TOTAL NUMBER OF HYDROGEN BONDS OF TYPE O(I)-->H-N(I+2)30 12.5 TOTAL NUMBER OF HYDROGEN BONDS OF TYPE O(I)-->H-N(I+3)10 4.2 TOTAL NUMBER OF HYDROGEN BONDS OF TYPE O(I)-->H-N(I+4)...# RESIDUE AA STRUCTURE BP1 BP2 ACC N-H-->O O-->H-N N-H-->O O-->H-N2 17 V B 3 +A 182 0A 8 180,-2.5 180,-1.9 1,-0.2 134,-0.1
TCO KAPPA ALPHA PHI PSI X-CA Y-CA Z-CA-0.776 360.0 8.1 -84.5 125.5 -14.7 34.4 34.8
....;....1....;....2....;....3....;....4....;....5....;....6....;....7..
.-- sequential resnumber, including chain breaks as extra residues| .-- original PDB resname, not nec. sequential, may contain letters| | .-- amino acid sequence in one letter code| | | .-- secondary structure summary based on columns 19-38| | | | xxxxxxxxxxxxxxxxx recommend columns for secstruc details| | | | .-- 3-turns/helix| | | | |.-- 4-turns/helix| | | | ||.-- 5-turns/helix| | | | |||.-- geometrical bend| | | | ||||.-- chirality| | | | |||||.-- beta bridge label| | | | ||||||.-- beta bridge label| | | | ||||||| .-- beta bridge partner resnum| | | | ||||||| | .-- beta bridge partner resnum| | | | ||||||| | |.-- beta sheet label| | | | ||||||| | || .-- solvent accessibility| | | | ||||||| | || |# RESIDUE AA STRUCTURE BP1 BP2 ACC| | | | ||||||| | || |35 47 I E + 0 0 236 48 R E > S- K 0 39C 9737 49 Q T 3 S+ 0 0 86 (example from 1EST)38 50 N T 3 S+ 0 0 3439 51 W E < -KL 36 98C 6
Rysunek 3.4: Przykładowe wyjście programu DSSP

3.1. Narzędzia biologiczne 41
AA – jednoliterowy kod aminokwasu
S (pierwsza kolumna w bloku strukturalnym) – streszczenie struktury
drugorzędowej. Są to kolumny od 19 do 38, stanowiące główny wynik
działania analizy DSSP na koordynatach atomów.
BP1 BP2 – numer reszty aminokwasowej pierwszego i drugiego mostka
sąsiedztwa
ACC – numer molekuł połączonych wiązaniem wodorowym w kontakcie z resztą
*10
N-H → O – wiązania wodorowe. Dla przykładu -3, -1.4 oznaczają, że jeśli tareszta jest na pozycji i to N–H z i jest powiązane h-wiązaniem z C=O
z i -3 z elektrostatycznym energią H-wiązania równą -1.4 kcal/mol. Są dwie
kolumny na każdy typ H-wiązania.
TCO – cosinus kąta pomiędzy resztą C=O z i a resztą C=O z i -1. Dla α -helis
TCO ma wartość bliską + 1, dla promieni beta TCO jest bliskie -1.
KAPPA – wirtualny kąt wiązania zdefiniowany przez drzewo atomów Cα reszt
i -2, i, i +2. Używane do zdefiniowania odchylenia.
ALPHA – wirtualny kąt skrętu definiowany przez atomy Cα reszt i -1, i, i +1.
PHI PSI – kąty skrętu podstaw peptydowych.
X-CA Y-CA -Z-CA – koordynaty atomów Cα.
3.1.6 MaxSub
Szacowanie dokładności przewidzianych modeli jest krytycznym dla metod
obliczających struktury. Na przestrzeni kilku lat przeprowadzono kilka badań
(CASP1, CASP2, CASP3 - punkt 2.2.3) w celu znalezienia metody, która w szybki
sposób znajdowałaby rozwiązanie, a ponadto wynik tego rozwiązania byłby
bardzo zbliżony do oszacowanego przez naukowca - eksperta.
Algorytm MaxSub powstał w wyniku kolejnych eksperymentów w ramach
CASP3. Jego celem jest zidentyfikowanie największego podzbioru atomów
Cα w modelu, który później produkuje pojedynczy i znormalizowany wynik
reprezentujący jakość modelu. Dalsze badania porównujące działanie MaxSub
z pracą ekspertów pokazały, że wyniki są bardzo podobne. Przyprowadzono sześć
prób dla różnych białek i po obliczeniu uśrednionych wyników nie było właściwie
żadnej znaczącej różnicy pomiędzy pracą naukowców a nowo powstałą metodą
MaxSub.
MaxSub jest programem, który wykorzystuje ten algorytm do oszacowania
jakości struktury modelu białka. Uruchomienie wygląda następująco:

42 Narzędzia
maxsub filename1 filename2 threshold
Gdzie:
filename1 - plik w formacie PDB (Protein Data Bank) zawierający przewidziany
model
filename2 - plik w formacie PDB zawierający strukturę białka
threshold - próg odległości pomiędzy resztami aminokwasowymi modelu
i struktury, domyślne to 3.5.
3.2 Technologie
3.2.1 Java
Java to obiektowy język programowania stworzony w firmie Sun Microsystems
przez grupę roboczą pod kierownictwem Jamesa Goslinga.
Technologia Javy pojawiła się w roku 1996 jako środowisko programistyczne
JDK 1.0 (ang. Java Development Kit) i od tego czasu ewoluowała w trzech
kierunkach:
J2SE (ang. Java 2 Standard Edition) – najbardziej rozpowszechniona wersja,
stosowana w komputerach osobistych,
J2EE (ang. Java 2 Enterprise Edition) – wersja języka używana do tworzenia
rozbudowanych aplikacji biznesowych, wykorzystywana w tzw. serwerach
aplikacji,
J2ME (ang. Java 2 Micro Edition) – okrojona wersja języka Java przeznaczona
dla telefonów komórkowych i innych urządzeń przenośnych (np. PDA).
Kompilator Javy nie generuje kodu wykonywalnego specyficznego dla danej
architektury systemowej, a kod pośredni tzw. B-code, który interpretowany
jest przez maszynę wirtualną (ang. JVM – Java Virtual Machine). JVM
dostosowuje instrukcje zawarte w pseudokodzie do platformy i procesora, na
którym uruchomiony jest program Javy. Dzięki temu programy napisane w Javie
są przenośne i mogą działać na wielu różnych platformach systemowych, o ile
wyposażono je w odpowiednie maszyny wirtualne.
Java jest językiem zorientowanym na zastosowania sieciowe i przetwarzanie
rozproszone. Dzięki obiektowości, dla Javy nie ma znaczenia, czy dane
pobierane są z pliku, czy poprzez połączenie HTTP czy FTP. Wyspecjalizowane
funkcje przetwarzania rozproszonego pozwalają zarówno na komunikację między
aplikacjami Javy (RMI), jak i między aplikacjami napisanymi w innych językach
(CORBA, WebServices). Inne biblioteki pozwalają na pisanie programów

3.2. Technologie 43
działających w przeglądarkach internetowych (aplety) oraz aplikacji działających
po stronie serwera (serwlety).
W zamierzeniu projektantów Java miała zastąpić C++ – obiektową wersję
języka C. Jej projektanci zaczęli od rozpoznania cech języka C++, które są
przyczyną największej ilości błędów programistycznych, by stworzyć język prosty
w użyciu, bezpieczny i niezawodny. O ile po pięciu odsłonach Javy jej prostota
jest dyskusyjna, o tyle język faktycznie robi dużo, by utrudnić programiście
popełnienie błędu. Przede wszystkim Java posiada system wyjątków, czyli
sytuacji, gdy kod programu natrafia na nieprzewidywalne trudności, takie jak
np.:
• czytanie z niedostępnego pliku lub nieprawidłowego adresu URL,
• podanie nieprawidłowych danych przez użytkownika.W innych językach programowania programista oczywiście może wprowadzić
wewnętrzne testy sprawdzające poprawność danych, pozycję indeksu tablicy,
inicjalizację zmiennych itd., ale jest to jego dobra wola i nie jest to jakoś
szczególnie wspierane przez dany język. W Javie jest inaczej – obsługa wyjątków
jest obowiązkowa, bez tego program się nie skompiluje. Przy tym obiekty
wchodzące w skład pakietu standardowego Javy implementują wyjątki w każdym
miejscu kodu, którego wykonanie jest niepewne ze względu na okoliczności
zewnętrzne.
Wszystkie opisane cechy, czyli obiektowość, niezależność od platformy
systemowej, rozwiązanie zagadnień sieciowych, bezpieczeństwo i niezawodność
sprawiły, że Java jest obecnie jednym z najbardziej popularnych języków
programowania na świecie i nic nie zapowiada, żeby miało się to w najbliższym
czasie zmienić.
3.2.2 Eclipse
Eclipse to udostępniona na zasadzie Open Source (patrz słownik) platforma
służąca do integracji różnego typu narzędzi programistycznych [18]. Działają
one w ramach jednego środowiska, co sprawia, że użytkownik przekonany jest,
że ma do czynienia z zaawansowanym, ale pojedynczym produktem. Sam jest
w stanie je skonfigurować dobierając takie moduły (pluginy), które odpowiadają
realizowanym przez niego zadaniom. Standardowo Eclipse wyposażone jest
w zestaw narzędzi do tworzenia aplikacji w języku Java. Oprócz tego, że zapewnia
ono duży komfort pracy i jest wyjątkowo elastyczne, bardzo blisko potrafi
integrować się z innymi modułami. Dostępne są moduły do pisania w języku
C, C++, HTML, a nawet w Cobolu.

44 Narzędzia
Eclipse został zaprezentowany w listopadzie 2001 roku przez IBM, Object
Technology International (OTI) oraz osiem innych firm. Produkt został przyjęty
entuzjastycznie.The New York Times napisał:
Eclipse łamie dotychczasowy wzorzec produktu firmowego i pojawia
się w kluczowym dla całej branży momencie . . .
Filozofia, która przyświecała twórcom narzędzia, polega na tym, aby
do maksimum zautomatyzować wszystkie powtarzalne i przyziemne wręcz
czynności towarzyszące pisaniu programu, czyli zredukować czas potrzebny na ich
wykonanie. Tym samym doprowadzono do sytuacji, gdy programista koncentruje
się na rzeczach naprawdę istotnych. Dlatego uproszczono edycję kodu, sposób
nawigacji, wprowadzono też mechanizmy, które pozwalaj na generowanie wielu
typowych elementów programu. Szczególnymi częściami środowiska Eclipse są:
edytor, brudnopis oraz debugger.
Edytor, ma możliwość wyróżniania składni i uzupełniania kodu. Istnieje
również kilka powiązanych z nim widoków, które służą do prezentacji struktury
kodu, szybkiej w niej nawigacji oraz wskazują wszelkie miejsca, które należy
uzupełnić bądź zweryfikować.
Brudnopis (ang. scrapbook) pozwala na testowanie fragmentów kodu
i budowanie wyrażeń bez konieczności uruchamiania całej aplikacji.
Debugger jest narzędziem pozwalającym programiście na obserwację działania
aplikacji krok po kroku. Daje ponadto możliwość podglądania zmiennych
programu podczas jego działania.
Eclipse to ponadto pewien rodzaj fundamentu, który umożliwia scalenie
aplikacji służących do projektowania i modelowania, programowania, testowania
czy też tworzenia np.plików graficznych. Eclipse osiąga to poprzez dostarczenie
programiście określonej architektury, w której występuje zbiór reguł oraz klas
pozwalających na tworzenie kolejnych modułów w ramach określonego standardu.
Środowisko posiada również mechanizmy pozwalające na pracę zespołową.
W roli systemu kontroli wersji występuje system CVS (patrz słownik).
Dzięki niemu programista ma możliwość korzystania ze współdzielonego
kodu. Natomiast automatycznie nadawane numery wersji pozwalają śledzić
i synchronizować zmiany, a nawet stworzyć kilka gałęzi projektu w repozytorium.
System pomocy w Eclipse jest to dostępna bezpośrednio dokumentacja
oraz pomoc kontekstowa. Istnieje możliwość dołączenia do niej dodatkowych
dokumentów, co nie wymaga żadnych umiejętności programistycznych. Ponadto
można odwołać się do sieciowej wersji systemu pomocy.
Eclipse to platforma, która jest nie tylko otwarta, ale również dostępna
dla maksymalnej liczby osób. Również niepełnosprawnych. Środowisko można

3.2. Technologie 45
Tablica 3.1: Systemy operacyjne, na których można uruchomić platformę Eclipse
System operacyjny Interfejs użytkownika
Microsoft Windows XP, 2000, NT, 98, Me WindowsRed Hat Linux 7.1 x86 Motif, GTK 2.0SuSE Linux 7.1 x86 Motif, GTK 2.0Sun Solaris 8 SPARC MotifIBM AIX 5.1 PowerPC MotifHP-UX 11i HP9000 MotifQNX x86 PhotonMac OS X Carbon
uruchomić na wielu systemach operacyjnych. Pokazuje to tablica 3.1. Dzięki
odpowiednio skonstruowanemu interfejsowi, wszystkie funkcje wykonywane
typowo przy pomocy myszki dostępne są także z poziomu klawiatury. Eclipse
współpracuje z oprogramowaniem, które odczytuje na głos zawartość ekranu.
Dodatkowo można użyć systemu rozpoznawania mowy, do wydawania komend
systemowych. Bogate możliwości w zakresie konfiguracji pozwalają dobrać
odpowiednio duży rozmiar czcionki oraz jej kolor.
Środowisko dostępne jest ponadto w wielu wersjach językowych.
Przetłumaczone zostało na język niemiecki, włoski, hiszpański, francuski,
japoński, koreański, chiński (uproszczony) oraz chiński (tradycyjny).
Tłumaczenia te dostępne są nie tylko dla użytkowników samej platformy,
ale również dla twórców modułów dodatkowych. Dzięki temu proces
przygotowywania wersji narodowych jest uproszczony, a wejście na
międzynarodowe rynki nie jest już tak trudną do pokonania barierą.
3.2.3 Tomcat
Tomcat jest częścią szeroko zakrojonego projektu Jakarta Apache
nadzorowanego przez Apache Software Foundation [15]. W skład Jakarty
wchodzą różne narzędzia programistyczne wykorzystujące Javę – biblioteki,
interfejsy programistyczne API (patrz słownik), programy pomocnicze, szkielety
aplikacji. Tomcat jest to otoczenie (kontener: patrz słownik) dla wykonywanych
programów Javy, zgodne z opracowaną przez Sun specyfikacją serwletów, czyli
odpowiedników apletów Javy (aplet: patrz słownik) wykonywanych po stronie
serwera. Tomcat zajął miejsce zarzuconego projektu JServ i stanowi wzorcową
implementację dwóch specyfikacji programistycznych Java Servlets oraz Java
Server Pages.

46 Narzędzia
Rysunek 3.5: Struktura aplikacji WWW.
Serwer Tomcat może służyć jako niezależny serwer WWW lub współpracować
z innymi serwerami – najbardziej oczywistym partnerem jest Apache, chociaż
możliwa jest integracja innych produktów za pomocą odpowiednich adapterów
(adapter: patrz słownik).
3.2.4 Tapestry
Tapestry to szkielet aplikacji (ang. application framework) przeznaczony do
budowy złożonych aplikacji WWW (ang. web application) przy wykorzystaniu
technologii Java. Aplikacje Tapestry działają w ramach kontenera serwletów Java
(np. Tomcat) lub serwera aplikacji (np. JBoss, WebSphere lub WebLogic), a samo
Tapestry stanowi warstwę pośrednią pomiędzy kontenerem, a tymi aplikacjami
(patrz rys. 3.5).
Większość rozbudowanych aplikacji WWW budowana jest w oparciu o model
MVC (ang. Model – View – Controller), zaproponowany po raz pierwszy przez
firmę Xerox na początku lat osiemdziesiątych. Model ten zakłada podział aplikacji
na trzy niezależne, współpracujące ze sobą jednostki:
• warstwę logiki biznesowej (Model),• warstwę prezentacji (View),• warstwę przetwarzania żądania (Controller).
Tapestry zostało zaprojektowane, aby pełnić rolę warstwy prezentacji, a zatem
jego zadania ograniczają się do prezentowania informacji użytkownikowi
końcowemu (w postaci dokumentów HTML) oraz obsługi zdarzeń takich jak
kliknięcie odnośników na stronie, czy przesłanie formularza. Głównym celem
projektantów było jednak maksymalne uproszczenie procesu konstrukcji samej
warstwy prezentacyjnej, a tym samym odciążenie programisty/projektanta od
takich zadań jak:
• zarządzanie sesją,• zarządzanie adresami URL (parsowanie i konstrukcja adresów URL),• przetwarzanie formularzy.

3.2. Technologie 47
Każda aplikacja Tapestry składa się z takich elementów jak strony
i komponenty. Szkielet Tapestry standardowo dostarcza już pewną liczbę
komponentów, m.in. do obsługi formularzy, przetwarzania odnośników, czy
kontrolowania przepływu sterowania w ramach stron HTML. Pozostałe można
stworzyć samemu, korzystając z dostępnych narzędzi.
3.2.5 SYSDEO
Sysdeo to plugin pozwalający na zarządzanie kontenerem serwletów Tomcat
z poziomu Eclipsa. Plugin pozwala na:
• uruchamianie, zatrzymywanie i restartowanie pracy Tomcata,
• zintegrowanie konsoli Tomcata z konsolą Eclipsa,
• dodawanie projektów Java do ścieżki systemowej Tomcata (ang. classpath),
• eksportowanie projektów do plików *.war,
• wybranie pliku konfiguracyjnego dla Tomcata.Sysdeo w znacznym stopniu upraszcza proces uruchamiania aplikacji WWW
działających pod kontrolą Tomcata, wspierając tym samym proces testowania
projektu i oszczędzając czas pracy programistów.
3.2.6 Spindle
Spindle to pakiet Eclipsa, który automatycznie generuje szkielet aplikacji
Tapestry. Z jego pomocą można w łatwy sposób generować szkielety stron,
komponentów i klas. Spindle automatycznie uzupełnia nazwy znaczników
stosowane w plikach, podkreśla błędy i sugeruje poprawne nazwy znaczników.
3.2.7 JDO
Java Data Objects (JDO) jest to interfejs API dostarczający obiektowego
mechanizmu trwałości i standardowych interfejsów umożliwiających korzystanie
z baz danych [21].
Interfejs JDO API, umożliwia bezpośrednie zapisywanie instancji obiektów
Java do bazy danych w sposób trwały.
Ponadto, korzystanie z JDO przynosi programiście następujące korzyści:
przenośność – aplikacja napisana w oparciu o JDO API może zostać
uruchomiona na wielu implementacjach, bez potrzeby ponownej
rekompilacji lub zmiany kodu źródłowego
niezależność od systemu bazy danych

48 Narzędzia
wygoda w użyciu – programiści nie muszą przejmować się szczegółami
odnośnie realizacji trwałości tworzonych przez nich obiektów, tym
wszystkim zajmuje się właśnie JDO
wysoka wydajność – JDO optymalizuje sposoby dostępu do przechowywanych
obiektów w celu zapewnienia optymalnej wydajności aplikacji
integracja z mechanizmami EJB (ang. Enterprise Java Beans) (EJB: patrz
słownik): aplikacje mogą korzystać z zaawansowanego mechanizmu
EJB w celu zdalnego przekazywania komunikatów, automatycznej
koordynacji rozproszonego przetwarzania oraz zapewnienia pewnego
poziomu bezpieczeństwa
3.2.8 JPOX
JPOX (ang. Java Persistent Object JDO) jest to darmowa i w pełni zgodna
implementacja interfejsu JDO w wersji 1.0 oraz 2.0, która zapewnia trwałość
obiektów Java w stosunku do wszystkich proponowanych na rynku systemów baz
danych [20].
Wspiera ponadto wszystkie główne wzorce obiektowo-relacyjnego mapowania
(ang. Object-Relational Mapping), które pozwalają, przy wykonywaniu zapytań
do bazy, na użycie mechanizmu JDOQL albo czystego SQL’a.
Rozpowszechniany jest na zasadzie licencji open source Apache 2, która
pozwala nie tylko na dostęp do mechanizmu trwałości obiektów, ale także do
kodu źródłowego.
3.2.9 JUnit
JUnit jest to platforma testująca napisana przez Erich’a Gamma’e i Kent’a
Beck’a [24].
Używana przez programistów oprogramowania, którzy wykorzystują
mechanizmy testów jednostkowych (test jednostkowy: patrz słownik). Łatwo
można zbudować testy pojedynczych klas i metod, jak i całe przypadki testowe
dla programów w Javie.
Umożliwia ponadto testowanie dużych aplikacji i automatyczne generowanie
raportów z testów.
JUnit jest projektem open source rozpowszechnianym na zasadzie licencji CPL
(ang. Common Public License) (licencja CPL: patrz słownik).

3.2. Technologie 49
3.2.10 JMock
JMock jest to biblioteka służąca do testowania oprogramowania w języku Java
za pomocą specjalnych obiektów o nazwie mock [8].
Mocki (ang. mock objects) bardzo ułatwiają pisanie testów jednostkowych.
Programista może się wyłącznie skupić na testowaniu właściwej klasy nie
martwiąc się o jej zależności, ponieważ ma gwarancję, że podstawione mocki
zachowają się zawsze zgodnie z jego oczekiwaniami.
Szczególnie wykorzystywane są tzw. mocki dynamiczne (mock dynamiczny:
patrz słownik), które pozwalają ograniczyć liczbę tworzonych klas na potrzeby
testów. Powstają w pamięci i dzięki temu nie obciążają projektu dziesiątkami
dodatkowych klas testowych. Dodatkową zaletą jest proste definiowanie właściwie
dowolnych zachowań i szerokie możliwości weryfikacji rzeczywistych interakcji
testowanej klasy z podstawionym mockiem. Wykorzystanie mocka dynamicznego
przebiega w następujący sposób:
• Stworzenie i zdefiniowanie mocka• Podstawienie mocka do testowanej klasy• Wywołanie testowanej metody• Weryfikacja interakcji testowanej klasy z mockiem
3.2.11 PostgreSQL
PostgreSQL jest to, udostępniany na zasadzie open source, system
obiektowo–relacyjnych baz danych. Stworzony przez Uniwersytet Kalifornijski
i rozwijany od ponad 15 lat, osiągnął bardzo dużą niezawodność, stabilność
i poprawność w działaniu [28].
Posiada wyrafinowane narzędzia takie jak:
• algorytmy kontroli współbieżnego dostępu do danych (ang. Multi–VersionConcurrency Control)
• system znaczników czasowych (ang. timestamp)• mechanizm asynchronicznych replikacji• mechanizm punktów kontrolnych (ang.savepoints)• mechanizm kopii online (ang. backup)• zaawansowany optymalizator zapytań (ang. query optimizer)• dzienniki systemowe (ang. logs)• obsługę złożonych zapytań• procedury składowane• system perspektyw

50 Narzędzia
PostgreSQL jest zgodny z większością standardów dotyczących baz danych.
Implementacja języka zapytań SQL (patrz słownik), zastosowana w systemie,
spełnia standardy ANSI–SQL 92/99 oraz w dużej części standard z 2003 roku.
Dzięki temu język SQL, oprócz standardowych operacji na bazie danych, wspiera
zaawansowane mechanizmy podzapytań (włączając w to także podzapytania
w klauzuli FROM) oraz transakcyjności.
Aby zapewnić spójność danych, system zawiera obsługę szeregu ograniczeń
takich jak: klucze podstawowe (ang. primary keys), klucze obce (ang. foreign
keys) wraz z możliwością kaskadowego usuwania bądź aktualizacji powiązań.
Ponadto zaimplementowana została obsługa różnego rodzaju ograniczeń jakie
użytkownik może nałożyć na tworzone schematy baz danych.
PostgreSQL oferuje także zaawansowany mechanizm indeksowania danych
GiST (ang. Generalized Search Tree), w którym zostały zaimplementowane
algorytmy sortowania i wyszukiwania takie jak: B drzewo, B+ drzewo, R drzewo
i wiele innych. Dzięki zastosowaniu tych algorytmów poprawiono w znaczącym
stopniu czas dostępu, jak i operacji na danych.
Obsługa procedur składowanych może odbywać się w wielu językach
programowania jak np. Java, Perl, Python, Ruby, Tcl, C/C++ i we własnym
języku PostgreSQL PL/pgSQL, który jest podobny do komercyjnego systemy
firmy Oracle PL/SQL.
Jako projekt open source, PostgreSQL daje bardzo duże pole manewru
w tworzeniu:
• nowych typów danych• funkcji• operatorów• funkcji agregujących• algorytmów indeksowania• języków procedurJest rozwijany przez osoby na całym świecie w celach komercyjnych,
prywatnych oraz naukowych.

Rozdział 4
Budowa systemu
4.1 Funkcjonalność
4.1.1 Współpraca z metaserwerami
Użytkownik wprowadzając sekwencję aminokwasową do systemu WebMobis,
może dokonać jej analizy za pomocą biologicznych serwerów obliczeniowych
(patrz lista metaserwerów, punkt 4.1.5). Serwery te umieszczone są w różnych
miejscach na całym świecie. System WebMobis dzięki swojej modułowej
budowie posiada klasy, które pełnią rolę warstwy pośredniej w komunikacji
między systemem a metaserwerami. Stanowią one pewnego rodzaju fundament
w przetwarzaniu i analizie sekwencji aminokwasowych. Lista metaserwerów
pobierana jest z klasy BioServersList za pomocą metody getAll().
Użytkownik, chcąc przeprowadzić analizę konkretnej sekwencji, na stronie
ShowSequence wybiera metaserwery obliczeniowe i naciska przycisk Analyze.
Powoduje to wywołanie metody formSubmit() z klasy ShowSequence. Pobiera ona
listę zaznaczonych metaserwerów a następnie za pomocą metody sendQuery(Map
selectedServers) z klasy BioServerManager wysyła zapytanie z sekwencją
aminokwasową. Użytkownik ma możliwość określenia czasu oczekiwania na
zapytanie, wpisując datę w formacie DD/MM/YYYY do pola o nazwie
Computation deadline.
Może się zdarzyć, że po wysłaniu zapytania do metaserwerów trzeba będzie
poczekać na wyniki, bądź też pojawi się błąd lub komunikat informujący
o problemach, np. niedostępności serwerów.
51

52 Budowa systemu
Rysunek 4.1: Autoryzacja użytkownika.
4.1.2 Obsługa użytkowników
SystemWebmobis może być używany przez wielu użytkowników. Każdy z nich
posiada swoją nazwę (ang.username, login) oraz hasło. Informacje te są potrzebne
podczas procesu autoryzacji użytkowników. Rysunek 4.1 przedstawia główną
stronę portalu, na której użytkownik podaje swoją nazwę i hasło.
Podział na użytkowników wynika z tego, że każdy z nich posiada swoją własną
listę białek, plików pdb i dopasowań (ang. alignmentów). Dzięki oddzieleniu
użytkowników od siebie, białka oraz wyniki analiz są pamiętane oddzielnie dla
każdego użytkownika.
Użytkowników dzielimy na zwykłych i administratorów. Zwykły użytkownik
może tylko zmieniać informacje dotyczące niego samego, natomiast administrator
może zmieniać ustawienia swoje i innych użytkowników, usuwać i tworzyć innych
użytkowników, w tym zwykłych i administratorów.
W momencie pierwszego uruchomienia systemu tworzony jest użytkownik
„admin” z hasłem „biaue3K”. Konto to pozwala administratorowi na szybkie
i sprawnie dodawanie, usuwanie i zmienianie ustawień użytkowników systemu
(patrz rys. 4.2). Należy pamiętać o zmianie hasła zaraz po pierwszym
uruchomieniu systemu Webmobis.
Użytkownik który po raz pierwszy wchodzi na główną stronę portalu może
zarejestrować się samodzielnie. W tym celu musi kliknąć na odnośnik sign up
i wpisać podstawowe informacje na swój temat: nazwę użytkownika, hasło i adres
email (patrz rys. 4.3, 4.4).
4.1.3 Wprowadzenie sekwencji białkowej
Po zalogowaniu się do systemu WebMobis, na stronie Headquater (patrz
rys. 4.5), użytkownik ma możliwość wprowadzenia sekwencji aminokwasowej
w formacie FASTA. Format ten zakłada, że każda sekwencja aminokwasowa
składa się z ciągu literowych oznaczeń aminokwasów budujących łańcuch
4.1. Funkcjonalność 53
Rysunek 4.2: Funkcje dostępne administratorowi.
Rysunek 4.3: Utworzenie nowego konta.
Rysunek 4.4: Okno rejestracji użytkownika.
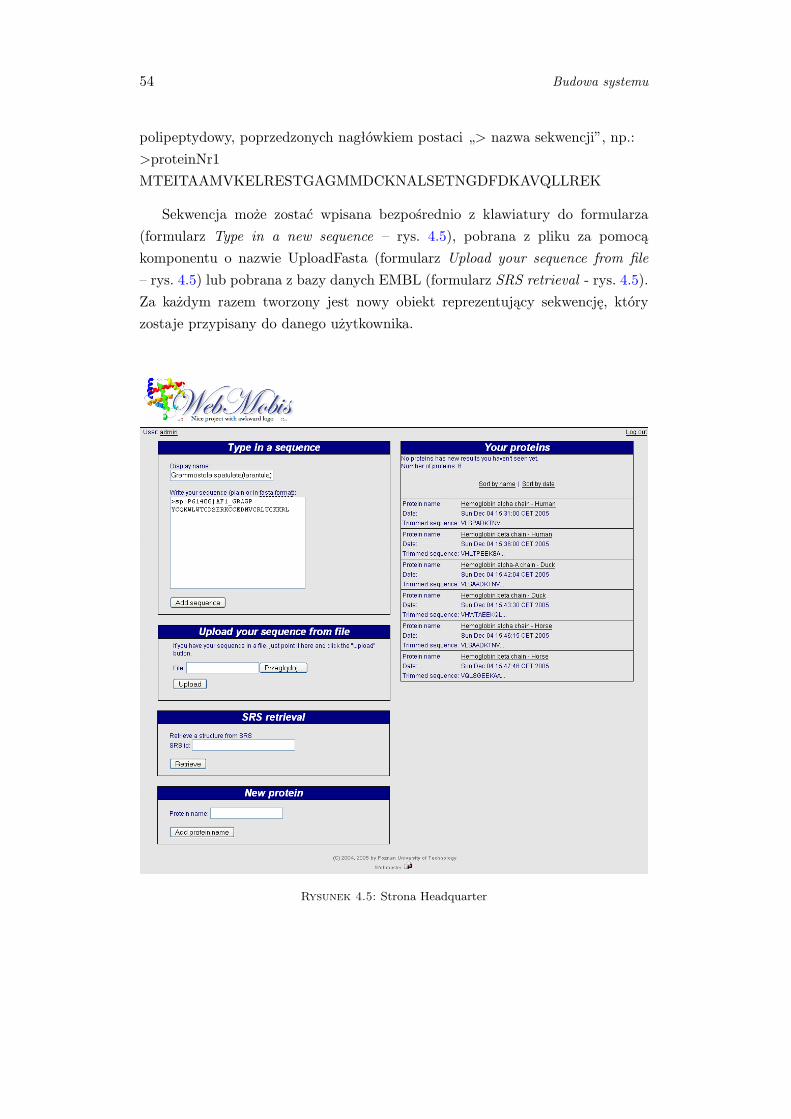
54 Budowa systemu
polipeptydowy, poprzedzonych nagłówkiem postaci „> nazwa sekwencji”, np.:
>proteinNr1
MTEITAAMVKELRESTGAGMMDCKNALSETNGDFDKAVQLLREK
Sekwencja może zostać wpisana bezpośrednio z klawiatury do formularza
(formularz Type in a new sequence – rys. 4.5), pobrana z pliku za pomocą
komponentu o nazwie UploadFasta (formularz Upload your sequence from file
– rys. 4.5) lub pobrana z bazy danych EMBL (formularz SRS retrieval - rys. 4.5).
Za każdym razem tworzony jest nowy obiekt reprezentujący sekwencję, który
zostaje przypisany do danego użytkownika.
Rysunek 4.5: Strona Headquarter

4.1. Funkcjonalność 55
4.1.4 Dodawanie własnych plików PDB
Aby umożliwić użytkownikom wprowadzanie i analizę plików PDB, bez
wprowadzania sekwencji FASTA, stworzono mechanizm dodawania pustej
sekwencji w formularzu o nazwie New Protein (patrz rys. 4.5). Podając
jedynie nazwę sekwencji uzyskuje się możliwość dodawania własnych plików
PDB, a następnie ich analizę za pomocą narzędzi z pakietu „biotools” (patrz
rozdział 3.1).
4.1.5 Projekt i budowa komponentów analizy strukturbiałkowych
Użytkownik wprowadzając sekwencję aminokwasową w formacie FASTA
(patrz słownik) na stronie HeadQuater uzyskuje dostęp do strony ShowSequence
(patrz rys. 4.6), na której może dokonywać analizy wcześniej wprowadzonej
sekwencji za pomocą ośmiu dedykowanych metaserwerów:
• MGen• EasyPred• Wurst• Sparks2• Gen• Fugue2• Sam–T02• FortePlanujemy również dodanie dzięwiątego metaserwera, zaprojektowanego przez
studentów i pracowników Politechniki Poznańskiej.
Metaserwery, jako wyniki swojego działania, przekazują:
• dopasowanie sekwencji aminokwasowych (ang. pairwise alignment)• struktury trzeciorzędowe białek w postaci plików PDBW celu czytelnego pogrupowania i późniejszej analizy wyników, stworzone
zostały trzy komponenty odpowiedzialne za przechowywanie otrzymanych
wyników oraz ich późniejsze przetwarzanie.
4.1.5.1 Pairwise Alignment Table
Komponent, przedstawiony na rys. 4.6, odpowiedzialny jest za
pobieranie i wyświetlenie listy dopasowań dwóch sekwencji aminokwasowych
z poszczególnych metaserwerów. Dostępne dopasowania pobierane są za pomocą
metody getAlignmentResults() z klasy Protein.
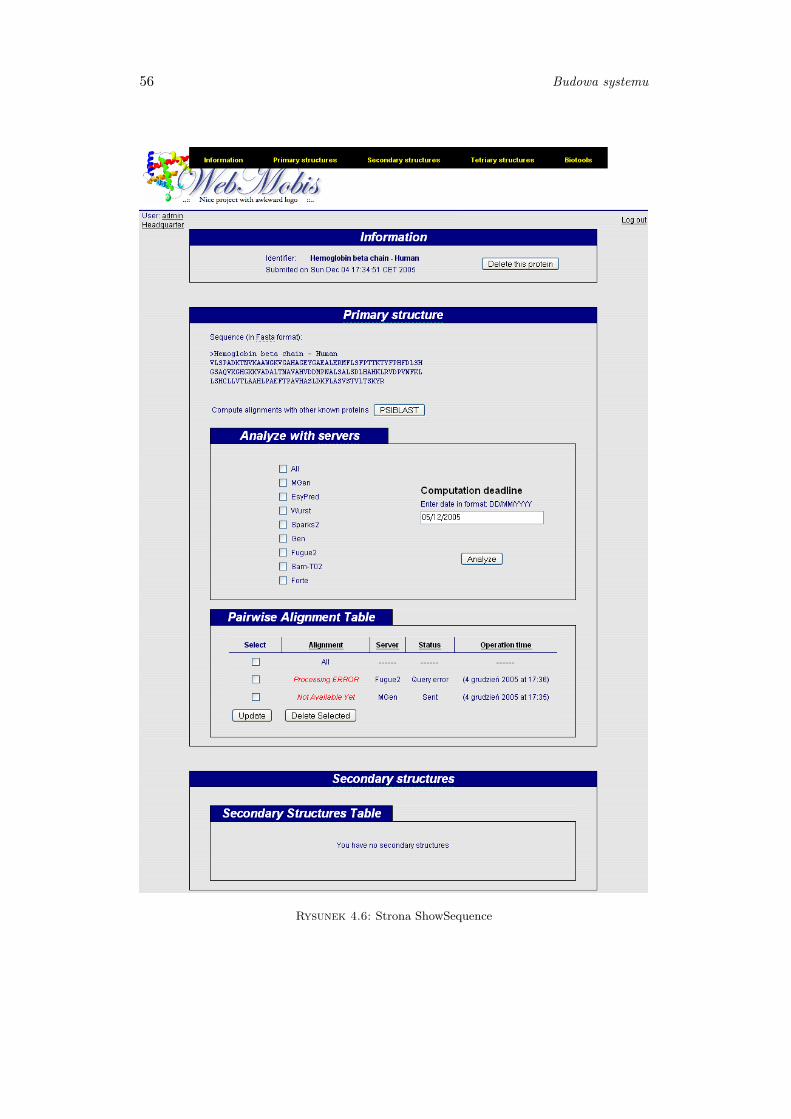
56 Budowa systemu
Rysunek 4.6: Strona ShowSequence

4.1. Funkcjonalność 57
Rysunek 4.7: Zawartość dopasowania przedstawiona na stronie AlignmentView
Elementy z listy dopasowań są obiektami klasy PairwiseAlignmentItem,
posiadającymi następujące składowe:
• nazwa pliku przedstawiającego dopasowanie (ang. fileName) – kolumnaAlignment
• zawartość dopasowania (ang. content)• nazwę przetwarzającego metaserwera (ang. serverDescriptor) – kolumnaServer
• status operacji (ang. status) – kolumna Status• datę zmiany statusu (ang. statusTime) – kolumna Operation timeUzytkownik ma możliwość dokładnego zapoznania się z wyliczonym przez
metaserwer dopasowaniem po naciśnięciu na odpowiadającym mu pliku
z kolumny Aligment. Zawartość dopasownia przedstawiana jest na osobnej stronie
AlignmentView (patrz rys. 4.7).
Metaserwery mogą z pewnym opóźnieniem przesyłać wyniki. Stąd,
do rozróżniania wyników dostępnych od niedostępnych i błędnych,
wprowadzono pole status, które może przyjmować następujące wartości
(opisane szczegółowo 4.1.5.4) Dzięki statusowi uzytkownik może zobaczyć, czy
przetwarzanie zakończyło się pomyślnie, czy też wystąpił określony problem.
Komponent umożliwia usuwanie z listy poszczególnych dopasowań.Odbywa
się to poprzez zaznaczenie jednego, bądź kilku elementów i wywołanie metody
deleteSelected() przy pomocy przycisku o nazwie Delete selected. Dostępny jest
również przycisk ręcznego odświeżenia okna komponentu, w celu sprawdzenia
czy poszczególne elementy z listy dopasowań nie zmieniły statusu na odebrany
(ang. Result received), co oznacza poprawne przetworzenie zapytania. Zadanie to
realizuje przycisk o nazwie Update.
4.1.5.2 Secondary Structures Table
Do reprezentacji otrzymanych struktur drugorzędowych, służy komponent
o nazwie SecondaryStructuresTable przedstawiony na rysunku 4.6. Pobiera on

58 Budowa systemu
i prezentuje struktury drugorzędowe białek otrzymane jako wynik działania
narzedzi z pakietu biotools. Pobranie elementów następuje za pomocą
metody getSecondaryResults(), która pobiera je z kolekcji secondaryResults
w klasie Protein. Poszczególnym strukturom drugorzędowym zostaje
przypisany odpowiedni status, zgodnie ze stanem przetwarzania, w jakim
się znajduje (opisane szczegółowo 4.1.5.4). Następnie już jako obiekty klasy
SecondaryStructuresItem wyposażone w następujące pola:
• zawartość struktury (ang. contentStructure) – kolumna Struct• nazwę przetwarzającego narzędzia z pakietu biotools
(ang. toolDescriptor) – kolumna Tool
• status operacji (ang. status) – kolumna Status• datę zmiany statusu (ang. statusTime) – kolumna Operation time
trafiają do listy obiektów o nazwie list. Korzystając z metody
getSecondStructList() mechanizm Tapestry generuje elementy HTML
komponentu.
Prezentacja budowy określonej struktury drugorzędowej pokazana jest
w kolumnie o nazwie Structure, jeżeli struktura została poprawnie przetworzona.
Jeżeli struktura nie jest jeszcze dostępna, to zgodnie ze statusem z pola
Status, w kolumnie Structure zamiast struktury pojawi się jeden z komunikatów
opisanych w punkcie 4.1.5.4 Podobnie jak w komponencie PairwiseAlignment
wprowadzone zostało pole status pokazujące aktualny stan przetwarzania
struktury, który może zostać odświeżony przez użytkownika, za pomocą
przycisku Update. Szczegółowa specyfikacja wartości tego pola znajduje się
w punkcie 4.1.5.4. Ponadto istnieje możliwość usunięcia z systemu jednej bądź
kilku struktur drugorzędowych, wywołując przyciskiem Delete selected, metodę
deleteSelected().
4.1.5.3 PDB Result Table
Mataserwery, oprócz dopasowań dwóch sekwencji (ang. pairwise alignment),
przekazują rezultaty w postaci plików PDB, które reprezentują struktury
trzeciorzędowe białek. Komponent PdbTable, przedstawiony na rysunku 4.8, ma
za zadanie poprawnie odczytać listę plików PDB z kolekcji elementów o nazwie
pdbresults z klasy Protein, za pomocą metody getPdbResults(). Pobrane wyniki
są dodawane do kolekcji PdbFileList jako obiekty klasy PdbItem, co umożliwia
ich późniejsze wyświetlenie i przetwarzanie. Każdy obiekt klasy PdbItem posiada:
• nazwę pliku przedstawiającego strukturę trzeciorzędową
(ang. file) – kolumna PDB filename

4.1. Funkcjonalność 59
Rysunek 4.8: Strona ShowSequence – komponent PdbTable
• zawartość pliku PDB (ang. content)• nazwę przetwarzającego metaserwera (ang. serverDescriptor) – kolumnaServer
• status operacji (ang. status) – kolumna Status• identyfikator w bazie danych (ang. resultId)• datę zmiany statusu (ang. statusTime) – kolumna Operation timeDo sygnalizacji użytkownikowi różnego typu błędów jak i opóźnień
przetwarzania, służy pole status. Dokładna specyfikacja wartości tego pola
znajduje się w punkcie 4.1.5.4. Jeżeli wszystko odbywa się poprawnie, to
użytkownik otrzymując poprawny wynik z metaserwera w postaci pliku PDB,
może dokonać jego analizy za pomocą wymienionych narzedzi z pakietu biotools.
Każdy plik PDB można obejrzeć w widoku 3D za pomocą narzędzia Jmol (patrz
punkt 3.1.3), dostępnego w kolumnie Options. W momencie wybrania opcji
Jmol, użytkownik uzyskuje dostęp do strony JmolView, na której może obejrzeć
wizualizację danego pliku PDB, korzystając z różnych opcji tego narzędzia.
Ponadto z komponentem są zintegrowane następujące narzędzia:
• LGScore• DSSP• MinRMS

60 Budowa systemu
• MaxSubDostępne są one pod listą plików PDB. Dodatkowo wprowadzona została
kontrola błędów, ponieważ część z wymienionych narzędzi potrzebuje więcej niż
jeden plik PDB do swojego przetwarzania. Konieczne było także rozróżnienie
ilości zaznaczonych przez użytkownika plików PDB. Realizuje to metoda
getSelectedPdbFiles(int numberToGet). Wszelkie błędy związane z zaznaczeniem
niepoprawnej ilości plików PDB do przetwarzania, zgłaszane są w postaci
odpowiedniego komunikatu pod dostępnymi narzędziami.
Użytkownik ma możliwość dołączenia swoich własnych plików PDB, które
posiada u siebie lokalnie, dzieki zintegrowanemu komponentowi Uploads.
Możliwość dodawania własnych plików PDB do konkrentej sekwencji jest bardzo
istotne z punktu użyteczności komponentu PdbTable. Wspomnianą użyteczność
komponentu rozszerza możliwość ściągnięcia pliku PDB i jego zapisania lokalnie
na komputerze uzytkownika. Realizowane jest to poprzez naciśnięcie na nazwie
wybranego pliku PDB w kolumnie PDB filename.
Istnieje ponadto możliwość usuwania zaznaczonej ilości plików PDB za
pomocą metody deleteSelected(), dostępnej pod przyciskiem o nazwie Delete
selected.
Odświeżanie dostępnych wyników PDB następuje w momencie odświeżenia
strony ShowSequence, bądź ręcznie poprzez przycisk o nazwie Update
4.1.5.4 Status przetwarzania
Każdy rezultat przetwarzania posiada pole status, które informuje
użytkownika o stanie przetwarzania. Pole te może przyjmować następujące
wartości:
Success poprawna odpowiedź metaserwera
Sent pomyślne wysłanie zapytania do metaserwera
Sending wysyłanie zapytania do metaserwera w toku
Page error niepoprawny adres strony serwera
Server error niepoprawny adres metaserwera
Query error błąd w treści zapytania
Address error błąd w adresie URL serwera
IO error błąd połączenia z metaserwerem
Pdb mail error brak pliku PDB w wiadomości (ang. mail)
Timeout przekroczenie czasu oczekiwania na wynik przetwarzania
Parsing mail error błąd podczas pobierania pliku PDB z wiadomości
nadesłanej przez metaserwer

4.2. Serwer HTTP 61
Service unavailable metaserwer jest niedostępny
Result received wynik przetwarzania poprawnie odebrany
Quota Error przekroczenie dostępnego miejsca
Ponadto, w kolumnach reprezentujących nazwy przekazanych wyników mogą
zamiast nazw wyników pojawić się dodatkowe komunikaty:
Processing ERROR – występuje dla każdego rodzaju błędu z pola status
Not Available Yet – zapytanie zostało wysłane, ale nie jest jeszcze dostępne
do dalszej analizy (oczekiwanie na rezultat przetwarzania)
4.1.6 Dodawanie plików PDB
W projekcie każdy użytkownik ma możliwość wprowadzania własnych
plików PDB, dołączając je pod już istniejącą sekwencję albo tworząc pustą
(patrz podrozdział 4.1.4). Zadanie to realizuje komponent o nazwie Uploads.
Odpowiedzialny jest on za poprawne odczytanie pliku PDB i utworzenie obiektu
klasy PdbResult, której zadaniem jest przechowywać informacje o wszystkich
plikach PDB w systemie. Dla dodawanego ręcznie pliku PDB, w obiekcie klasy
PdbResult, pamiętana jest nazwa pliku, czas dodania, zawartość oraz status,
który informuje o tym, iż plik nie jest wynikiem automatycznego przetwarznia za
pomocą metaserwerów.
4.1.7 Wizualizacja Jmola
Pliki PDB, dostępne na stronie ShowSequence (rys. 4.8) zawierają opis
struktury białka w przestrzenii 3D. Do wizualizacji tych plików wykorzystujemy
applet Jmol dostępny na stronie JmolView portalu. Szczegółowy opis można
znaleźć w punkcie 4.3.2.
4.2 Serwer HTTP
4.2.1 Strona wizualna
Użytkownik systemu Webmobis komunikuje się z systemem Webmobis
korzystając z przeglądarki internetowej takiej jak np. Internet Explorer czy
Mozilla Firefox. Tym samym strona wizualna projektu została zaimplementowana
za pomocą języka HTML. HTML to hipertekstowy język znaczników
zaprojektowany przez Tima Berners-Lee w 1981 roku. Rewolucyjność HTML’a
polega na tym, że użytkownik może przemieszczać się od jednego dokumentu

62 Budowa systemu
Rysunek 4.9: Overlib: wyjaśnienie funkcji .
do drugie nie wiedząc gdzie fizycznie te dokumenty się znajdują, klikając na
tzw. odnośniki (ang links).
Poza HTML’em wykorzystaliśmy style CSS (ang. Cascading Style Sheets),
które pozwalają na łatwe zarządzanie wyglądem strony. Dzięki temu zmiana
wyglądu np. tła, kolorów, tabel itd. sprowadza się tylko do niewielkiej modyfikacji
odpowiedniego pliku ∗.css, przez co nie trzeba modyfikować plików HTML. Jestto bardzo wygodne narzędzie dla projektanta.
4.2.1.1 Ruchome menu
Aby ułatwić pracę użytkownikom, stworzone zostało menu pozwalające na
prostą i wygodną nawigację na stronie. Wykorzystano do tego celu technologię
HTML, JavaScript (patrz słownik) i CSS. Dużą zaletą menu jest jego stała
pozycja: znajduje się zawsze na górze widocznego obszaru okna przeglądarki,
nawet przy przewijaniu strony. Efekt ten został uzyskany dzięki użyciu opcji
ustalonej, stałej pozycji w przeglądarkach poprawnie interpretujących CSS (m. in.
Mozilla Firefox, Opera, Konqueror). W przypadku Microsoft Internet Explorer
pozycja menu utrzymywana jest dzięki JavaScript.
4.2.1.2 System podpowiedzi
W WebMobis zaimplementowany został system podpowiedzi (ang. hints),
mający na celu ułatwienie osobom rozpoczynającym pracę w serwisie
zorientowanie się w jego funkcjach. Wykorzystana została darmowa biblioteka
OverLib (http://www.bosrup.com/web/overlib/). Podpowiedź ukazuje się,
gdy użytkownik ustawi kursor nad opisanym elementem. Przykładowe
podpowiedzi mogą wyjaśniać działanie obiektu (rysunek 4.9), a także niektóre
pojęcia (rysunek 4.10).

4.2. Serwer HTTP 63
Rysunek 4.10: Overlib: objaśnienie opcji wraz z opisem technicznym (zielonykolor).
4.2.2 Obsługa błędów i wyjątków
Obsługa błędów i wyjątków, które mogą pojawić się podczas pracy
systemu, to jedno z głównych wymagań jakie stawia się przed każdym dobrze
zaprojektowanym systemem informatycznym.
W systemie Webmobis błędy mogą wynikać z następujących przyczyn:
• niepoprawne wprowadzenie nazwy i hasła przy autoryzacji,
• niepoprawne wpisanie sekwencji w formacie Fasta,
• niepoprawne wypełnienie formularza przy rejestracji,
• wygaśnięcie sesji,
• utrata połączenia z bazą danych,
• wybranie większej lub mniejszej liczby plików pdb do analizy.
Tego typu błędy bardzo trudno zlikwidować, ponieważ nie zależą od twórcy
systemu, a od środowiska i użytkowników, którzy z niego korzystają. Aby
rozwiązać ten problem utworzono procedury informujące użytkownika końcowego
o powstałym problemie oraz sugerujące co należy zrobić, aby go rozwiązać.
W sytuacji gdy użytkownik wprowadzi niepoprawne dane przy autoryzacji
wówczas może otrzymać komunikaty pokazane na rysunkach 4.11 i 4.12.
O błędzie wygaśnięcia sesji użytkownik informowany jest w sposób pokazany
na rysunku 4.13. Innego rodzaju błędy, które mogą się pojawić, wyświetlane są
w sposób przedstawiony na rysunku 4.14.
64 Budowa systemu
Rysunek 4.11: Użytkownik nie wprowadził ani hasła, ani loginu, a próbuje sięzalogować.
Rysunek 4.12: Użytkownik niepoprawnie wprowadził login lub hasło.
Rysunek 4.13: Informacja o tym, że sesja użytkownika wygasła.
Rysunek 4.14: Nieoczekiwany błąd wraz opisem.

4.3. Zewnętrzne narzędzia 65
4.2.3 Opracowanie sposobu przekazywania parametrówpomiędzy stronami w technologii Tapestry
Technologia Tapestry znacznie upraszcza zarządzanie projektami aplikacji
WWW. Dotyczy to również aspektu związanego z przekazywaniem parametrów
między kolejnymi stronami. Twórca systemu nie musi przejmować się takimi
zagadnieniami jak konstrukcja sesji, budowa adresów URL do poszczególnych
stron czy dodawaniem do nich parametrów. Tym wszystkim zajmuje się Tapestry.
Programista musi tylko określić, co chce przekazywać między kolejnymi stronami.
Dane mogą być przekazywane w dwojaki sposób. Pierwszy z nich polega
na wykorzystaniu formularza. Użytkownik podając dane, wypełnia wszystkie
pola, po czym zatwierdza je naciskając przykładowo przycisk Add sequence po
wprowadzeniu sekwencji (patrz rys. 4.5). W tym momencie każde wprowadzone
pole zostaje przesłane do serwera, gdzie następuje przetworzenie danych
i zwrócenie wyników w postaci komunikatu błędu lub jako nowa strona. Drugi
sposób przekazywania parametrów związany jest z odnośnikami (ang. links).
W adresie URL obok adresu samego serwera można wprowadzić dodatkowe
parametry np.
http://bio.cs.put.poznan.pl/webmobis/app?protein=Hemoglobine&user=Admin.
W tym przypadku do adresu http://bio.cs.put.poznan.pl/webmobis/app dodane
zostały dwa parametry protein i user. Pierwszy z nich ma wartość Hemoglobine
i oznacza białko, które chcemy analizować, drugi oznacza użytkownika, który
zleca żądanie. Wszystkie parametry oddzielone są od adresu serwera za pomocą
znaku „?”, natomiast same parametry oddzielone są między sobą przy pomocy
znaku „&”
System Webmobis wykorzystuje obie metody przekazywania parametrów.
4.3 Zewnętrzne narzędzia
Większość narzędzi użytych w projekcie obsługiwana jest w podobny sposób.
Są one uruchamiane za pomocą skryptów w trybie tekstowym. Wykorzystanie
skryptów bash’a niesie za sobą także obsługę odpowiednich kodów standardowych
błędów. W związku z tym napisana została ogólna nadklasa, która nadzoruje cały
proces od uruchomienia do zakończenia programu. Uniknięto w ten sposób wielu
powtórzeń w podklasach odpowiedzialnych za pojedyncze narzędzie. Stworzoną
klasę nazwano BioTools. Poza metodą uruchamiania narzędzi i przechwytywania
wyników ich działania, klasę uzupełniono także o metody służące pobieraniu

66 Budowa systemu
ścieżek do zainstalowanych narzędzi (punkt 4.3.1) oraz dodawania ewentualnych
dodatkowych paramaterów dla programów.
W skład całego pakietu BioTools wchodzą następujące klasy:
• BioTools (nadklasa)
• Blast
• MaxSub
• MinRMS
• LGScore
• DSSPWszystkie klasy dziedziczące po BioTools w budowie i działaniu nie różnią
się od siebie w znaczący sposób. Jednak zostały umieszczone osobno ze
względu na różnicę w poleceniach wykonania i liście paramaterów. W każdej
klasie jest konstruktor, który pozwala na stworzenie obiektu, z bezpośrednim
ustawieniem parametrów oraz metody pozwalające na zrobienie tego manualnie.
Wszystkie narzędzia działają na plikach i niestety nie ma możliwości przekazania
struktury do programu jako łańcuch znakowy. W tym celu powstała klasa
TempFileManager do zarządzania tymczasowymi plikami, bardziej szczegółowo
opisana w punkcie 4.3.4. Po stworzeniu obiektu i ustawieniu paramaterów
tworzony jest plik lub pliki ze strukturami, w zależności od tego jak działa
program (więcej szczegółów zostało przedstawionych w punkcie 3.1). Następnie
na danym obiekcie wywoływana jest metoda execute() dziedziczona z nadklasy
BioTools. Po wykonaniu zostaje zwrócony łańcuch wynikowy i usuwane są
pliki tymczasowe. Wszystkie klasy w pakiecie działają dokładnie tak samo,
z wyjątkiem klas Blast i MaxSub. W klasie Blast, metoda execute() jest
przeciążona i po wykonaniu metody z superklasy dodatkowo parsowany jest
łańcuch wynikowy w celu utworzenia kolekcji uszeregowań. Inna sytuacja ma
miejsce w klasie MaxSub. Tutaj różnica polega na tym, iż metoda execute()
nie wykonuje bezpośrednio programu, tylko skrypt uruchamiający MaxSub.
Szczegóły opisane są w punkcie 4.3.3.
Dodatkowo dla potrzeb narzędzi zostały napisane skrypty instalacyjne, które
bardziej szczegółowo opisane są w punkcie 4.3.1.
4.3.1 Skrypt instalacyjny
Aby ułatwić pracę osobom instalującym WebMobis, stworzony został
zestaw skryptów instalacyjnych dla narzędzi biologicznych (3.1). Starano się
maksymalnie uprościć zadanie użytkownika i zwolnić go od obowiązku znajomości

4.3. Zewnętrzne narzędzia 67
adresów internetowych pod którymi pobrać można pakiety z narzędziami, metod
ich kompilacji (MinRMS) czy też dopisywaniu ich do odpowiednich ścieżek.
Struktura katalogów instalatora wygląda następująco:
[bioinstall]
|--[files] - katalog tymczasowy dla plików ściągniętych z sieci
| |--[unpacked] - rozpakowane pliki z internetu
|--[scripts] - zawiera skrypty dla poszczególnych narzędzi
| |--[files] - programy ściągnięte przez użytkownika do instalacji
| |--iBlast.sh - skrypt instalujący blast
| |--iDssp.sh - skrypt instalujący DSSP
| |--iJmol.sh - skrypt instalujący JMol
| |--iLgscore.sh - skrypt instalujący LGScore
| |--iMaxsub.sh - skrypt instalujący MaxSub
| |--iProperties.sh - skrypt ustawiający właściwości
| |--properties.conf - plik z właściwościami instalatora
|--Instal.sh - główny skrypt instalacyjny
Rysunek 4.15: Ekran instalatora biotools
Obsługę instalatora można przedstawić w kilku krokach. Aby rozpocząć
instalację, należy najpierw ustawić opcje w pliku properties.conf . Przykładowe
ustawnienie wygląda następująco:
INSTALL_DIR_PREFIX=home
INSTALL_DIR=biotools
PROJECT_PATH_PREFIX=home
PROJECT_PATH=workspace/webmobis
Opis poszczególnych zmiennych znajduje się poniżej:

68 Budowa systemu
INSTALL DIR PREFIX – wpis może przyjąć wartość home jeśli użytkownik
chce instalować narzędzia w katalogu domowym, lub / jeśli chce użyć
katalogu głównego;
INSTALL DIR – podkatalog INSTALL DIR PREFIX w którym mają znaleźć
się narzędzia;
PROJECT PATH PREFIX – analogicznie do INSTALL DIR PREFIX,
tylko docelowym katalogiem będzie tutaj katalog z projektem WebMobis,
w którego konfiguracji konieczne jest wpisanie ścieżki do bionarzędzi;
PROJECT PATH – analogicznie do INSTALL DIR.
Po zakończeniu ustawiania pliku properties.conf, należy uruchomić instalator
za pomocą skryptu Install.sh. Jeśli w systemie zainstalowano program dialog1,
zostanie wyświetlone okienko dialogowe (rysunek 4.15), gdzie wybrać można
programy, które mają zostać zainstalowane. W przeciwnym wypadku użytkownik
zostanie poproszony o wpisanie nazw narzędzi. Po zakończeniu wyboru,
sterowanie przejmują pomniejsze skrypty, specyficzne dla każdego z narzędzi.
Każdy z nich sprawdza, czy użytkownik nie dograł do katalogu files spakowanej
wersji programu. Jeśli nie, następuje próba ściągnięcia z Internetu najświeższej
wersji. Następnie archiwa są rozpakowywane i wykonywane są czynności
instalacyjne, inne dla każdego programu (np. BLAST wystarczy rozpakować,
natomiast minrms wymaga kompilacji). Katalog docelowy jest pobierany
z properties.conf. Ażeby uniezależnić działanie samego projektu od wersji
programów, w katalogu docelowym tworzone są dowiązania symboliczne.
Po zakończeniu pracy skryptu, wybrane narzędzia powinny się znaleźć
w katalogu wpisanym w konfiguracji, a plik projektu webmobis.properties
wskazywać na ten katalog.
4.3.2 Jmol
Jmol jest jedynym z narzędzi, które nie dziedziczy po klasie BioTools.
Spowodowane jest to różnicą w uruchamianiu, gdyż jak to zostało napisane
w punkcie 3.1.3, Jmol jest apletem javowym. W związku z tym jest dołączony
do projektu i umieszczony na serwerze. Kiedy użytkownik chce wizualizować
strukturę, zostaje utworzona strona HTML z apletem (wcześniej, jest on
ściągnięty lokalnie), dodatkowo wykorzystująca gotowy skrypt napisany w języku
JavaScript.
1Dialog – służy do wyświetlania okienek dialogowych w trybie tekstowym powłoki. Autor:Savio Lam, http://invisible-island.net/dialog/

4.3. Zewnętrzne narzędzia 69
Rysunek 4.16: Strona JmolView
Klasa Jmol powstała właśnie w celu tworzenia kodu HTML strony, na której
umieszczony jest aplet. Jej metody generują nagłówek HTML z danymi odnośnie
skryptu oraz ciało strony (ang. body) . Strona HTML z wykorzystaniem skryptu
umożliwia dodatkowe zarządzanie apletem, tj. rotacje struktury, wyświetlanie
wiązań. Ponadto w kodzie zawarta jest informacja o nazwie pliku, w którym
umieszczona jest nasza struktura. Wadą apletów jest to, że działają tylko po
stronie klienta, a w konsekwencji mogą wykorzystywać dane pobrane tylko
i wyłącznie z serwera, z którego aplet pochodzi. W związku z powyższym należało
napisać aplikację, która przekazałaby do apletu żądany przez użytkownika plik
PDB. Stworzony został serwlet (patrz słownik), którego zadaniem jest pobranie
z bazy danych odpowiedniej struktury i wysłanie jej przez HTTP bez ingerencji
użytkownika. Metoda klasy Jmol generująca stronę HTML, wstawia w ciało
strony przekazany jako parametr identyfikator struktury w bazie danych.
Przykład wizualizacji Jmol pokazany jest na rysunku 4.16.

70 Budowa systemu
4.3.3 Skrypt uruchamiający MaxSub
Większość narzędzi z pakietu BioTools uruchamiana jest bezpośrednio z linii
poleceń, jednak program MaxSub do uruchomienia potrzebuje dodatkowego
skryptu. Wewnątrz niego wywoływane jest polecenie
/home/webmobis/biotools/main.linux $model $pdb $threshold
gdzie
main.linux – nazwa pliku wykonywalnego;
$model – model badanego białka;
$pdb – badane białko w formacie PDB (Protein Data Bank) (patrz słownik);
$threshold – próg odległości.
Należy dodać, że w razie zmiany katalogu z narzędziami, za podmienienie
ścieżki do programu (tutaj: /home/webmobis/biotools/ ) odpowiadają skrypty
instalacyjne (punkt 4.3.1). Wyniki uzyskane z programu, przetwarzane są do
przejrzystej postaci za pomocą kolejnego skryptu w językach BASH i AWK:
set f = ‘grep MAXSUB maxsub.out‘
set ms = ‘echo $f | awk ’print $16;’‘
set n = ‘echo $f | awk ’print $3;’‘
echo $f
set s = ‘echo $n $ms | awk ’
if ($1>=40)
printf("%f\n",$2*10); exit;;
if ($2 < .1)
printf("0\n"); exit; ;
if ($2>=.125 && $1 >=25)
printf("%f\n",$2*10);else printf("0\n");’‘
echo $s
4.3.4 Moduł plików tymczasowych
Zewnętrzne narzędzia dołączone do projektu, są programami uruchamianymi
z linii poleceń. Dlatego większość serwerów udostępniających interfejsy dla nich,
używa technologii CGI-BIN aby móc je wykorzystać na serwerach WWW.
Jako przykład podać można ExPASy (http://www.expasy.org/tools/blast/),
a nawet samych twórców BLAST (http://www.ncbi.nlm.nih.gov/Education/
BLASTinfo/psi1.html).
W WebMobis zdecydowano się na inne rozwiązanie. Programy nie są
uruchamiane za pomocą skryptów cgi, lecz bezpośrednio z kodu Javy, przy

4.3. Zewnętrzne narzędzia 71
użyciu funkcji exec(). Zrodziło to problemy związane z plikami wymaganymi
przez biotools. W WebMobis, pliki pdb i uszeregowania, które są przetwarzane
przez narzędzia, nie są zachowywane w systemie plików – są przechowywane
w bazie danych. W obrębie projektu można je bardzo łatwo przetwarzać czy
też wypisywać na stronie. Jednak napisane w języku C programy, nie radzą sobie
z plikami wejściowymi podawanymi pod postacią łańcucha znaków bezpośrednio
do linii poleceń czy też jako strumienie danych.
Konieczne okazało się utworzenie klasy odpowiedzialnej za tworzenie plików
i wypełnianie ich zawartością pobraną z bazy danych. Ponieważ pliki te potrzebne
były tylko podczas wykonywania obliczeń przez narzędzia, zdecydowano się
stworzyć je przy pomocy mechanizmu plików tymczasowych. Ponieważ tworzenie
i obsługa takich plików są dosyć skomplikowane, powstała osobna klasa,
zmniejszająca nakład pracy osoby korzystającej do utworzenia nowej instancji
(z opcjonalnym podaniem nazwy pliku, w przeciwnym przypadku nazwa jest
losowa) i wypełnienia jej łańcuchem znaków pobranym np. z bazy danych.
Pliki tymczasowe przechowywane są w katalogu wskazywanym przez zmienną
java.io.tmpdir, w podkatalogu webmobis-temp. Tak przygotowany plik lub
pliki można przesłać do skryptu uruchamiającego narzędzie. Po wykorzystaniu
pliki należy skasować przez własnoręczne wywołanie odpowiedniej metody.
Aby zautomatyzować kasowanie podjęto próby umieszczenia kasowania pliku
w destruktorze obiektu, a dokładniej w metodzie finalize(). Jest ona wywoływana
przed zniszczeniem obiektu przez Garbage Collector. Zastosowanie tej techniki
nie powiodło się ze względu na to, że nie każdy obiekt jest niszczony przed
zakończeniem programu przez garbage collection (patrz słownik)[4]. Dzieje się
tak np. w przypadku zakończenia procesu i przejęcie jego zasobów przez zarządcę
pamięci systemu operacyjnego.


Rozdział 5
Podsumowanie
Przyglądając się rozwojowi bioinformatyki wyraźnie widać, że ta dyscyplina
nauki nabrała w ostatnich czasach ogromnego znaczenia. Coraz więcej odkryć
w takich dziedzinach jak np. farmacja zaczyna bazować na wynikach analiz
bioinformatycznych. Tendencja ta z pewnością utrzyma się w najbliższych
latach biorąc pod uwagę fakt, że koszty finansowe i czasowe, przeprowadzenia
doświadczeń eksperymentalnych znacznie przewyższają wykorzystanie metod
bioinformatycznych.
System Webmobis jako jedno z narzędzi bioinformatycznych nastawione jest
na analizę białek i pozwala ją wykonywać łatwo i szybko, integrując szereg
powszechnie wykorzystywanych przez biologów narzędzi informatycznych. Tym
samym system Webmobis jest niezastąpionym narzędziem w pracy każdego
biologa molekularnego.
Zrealizowany projekt ma w znacznym stopniu ułatwić pracę ludziom
zajmującym się bioinformatyką. Położono duży nacisk na wygodę użytkowania
i funkcjonalność systemu. Przystępny interfejs użytkownika, logiczny układ stron
i dodatkowe informacje wyświetlane w momencie przesunięcia kursora myszki
nad wybrany element sprawiają, że nauka i wykorzystanie systemu jest proste
i przyjemne.
Każdy nowy użytkownik po zalogowaniu się może wprowadzić nową sekwencję
na kilka różnych sposobów. Sekwencja białkowa może zostać wprowadzona
z pliku, pobrana z zewnętrznego serwera oraz zwyczajnie wpisana na klawiaturze.
To użytkownik decyduje z którego podejścia skorzysta.
Wprowadzenie sekwencji to jednak dopiero początek całej złożonej analizy
na jaką pozwala system. Wprowadzona sekwencja jest przez cały czas pamiętana
w bazie danych, a użytkownik może wykonywać na niej szereg różnych operacji.
W tym celu system został wyposażony w dwa rodzaje narzędzi. Pierwszy
73

74 Podsumowanie
z nich to sprawdzone i dobrze przetestowane zewnętrzne serwery, z których
każdy implementuje inną metodę analizy białka. System implementuje 8 takich
serwerów stąd dla pojedynczej sekwencji można otrzymać wiele wyników analiz.
O tym jakie serwery zastaną wykorzystane decyduje jednak sam użytkownik.
Drugi rodzaj narzędzi to narzędzia informatyczne wykonujące różne analizy na
wynikach zwróconych przez owe serwery, od prostego porównywania sekwencji
po wyznaczanie różnych współczynników (np. rmsd). Dodatkowo możliwość
wizualizacji otrzymanych wyników w postaci plików pdb sprawia, że użytkownik
może od razu zobaczyć jak wygląda analizowane białko i nie musi korzystać
z zewnętrznych programów do tego celu.
System Webmobis dostarcza zatem kompleksowej platformy, za pomocą
której można szybko i sprawnie wykonywać analizy białek. W czasach kiedy
istnieje duża liczba narzędzi informatycznych analizujących białka, a stopień
ich złożoności wymaga poświęcenia dużej ilości czasu na ich opanowanie,
system Webmobis pozwala wykonywać wszystkie te działania szybko i sprawnie,
stanowiąc tym samym doskonałą alternatywę dla istniejących narzędzi i platform
bioinformatycznych.

Słownik
A
adapter – jest to specjalny interfejs (najczęściej programowy), dzięki któremu dany
produkt potrafi współpracować z innym. (str. 46)
API – (ang. Application Programming Interface) jest to interfejs programu
użytkownika – specyfikacja procedur, funkcji lub interfejsów umożliwiających
komunikację z biblioteką, systemem operacyjnym lub innym systemem
zewnętrznym w stosunku do aplikacji korzystającej z API. Dobry interfejs
API tworzony jest nie tylko z myślą o ułatwieniu procesu tworzenia
oprogramowania programistom, poprzez dobrą dokumentację oraz ukrycie
szczegółów implementacyjnych, ale także z myślą o użytkowniku, dzięki
zagwarantowaniu podobnego interfejsu wszystkim aplikacjom opartym o dany
API. (str. 45)
aplet – (ang. applet) jest to niewielki program ( w języku Java) napisany w taki sposób,
by mógł zostać osadzony w stronie WWW i uruchomiony przez przeglądarkę
internetową. (str. 45)
C
CVS – (ang. Concurrent Versions System) – popularny system kontroli wersji. Jest
oparty o architekturę klient/serwer, został stworzony do pracy grupowej nad
kodem programów lub innych projektów realizowanych w zapisie elektronicznym.
Więcej informacji na stronie http://www.nongnu.org/cvs/. (str. 44)
E
EJB – (ang. Enterprise Java Beans) – są to programowe komponenty, umieszczane w
wielowarstwowym rozproszonym środowisku serwera J2EE. Komponent składa
się z jednego lub wielu obiektów Java. Więcej informacji na stronie http://www.
webdeveloper.pl/enterprise_java_beans__ejb__czesc_1,324,1,1,pl.html.
(str. 48)
elektroforeza – jest to technika analityczna stosowana w chemii i biologii molekularnej,
zwłaszcza w genetyce. Jej istotą jest rozdzielenie mieszaniny związków
chemicznych na możliwie jednorodne frakcje przez wymuszanie wędrówki ich
cząsteczek w polu elektrycznym. (str. 10)
75

76 SŁOWNIK
enzym – rodzaj białka występującego naturalnie w organizmach żywych, którego
działanie sprowadza się do obniżenia energii aktywacji (katalizy) danych reakcji
biochemicznych. (str. 5)
F
FASTA – jeden z pierwszych programów (1985 r.) do przeszukiwania baz w celu znalezienia
sekwencji podobnych. Dużą czułość algorytmu osiągnięto dzięki zoptymalizowaniu
poszukiwania dopasowań lokalnych przy pomocy macierzy substytucji. (str. 55)
G
garbage collection (zbieranie nieużytków) to architektura zarządzania pamięcią, w której
proces zwalniania nieużywanych jej obszarów odbywa się automatycznie.
Mechanizm taki stosuje się na przykład w wysokopoziomowych językach (Python,
Ruby i Java).
Definicja pochodzi z Wikipedii, wolnej encyklopedii.. (str. 71)
J
JavaScript to stworzony przez firmę Netscape zorientowany obiektowo skryptowy język
programowania, najczęściej stosowany na stronach WWW. Implementacja
JavaScriptu stworzona przez firmę Microsoft nosi nazwę JScript. Głównym
autorem języka JavaScript jest Brendan Eich. Pod koniec lat 90. ubiegłego wieku
organizacja ECMA wydała ustandaryzowaną specyfikację tego języka pod nazwą
ECMAScript.
Definicja pochodzi z Wikipedii, wolnej encyklopedii. (str. 62)
K
kodon – jest to sekwencja trzech nukleotydów mRNA warunkująca włączenie jednego
aminokwasu do łańcucha polipeptydowego lub warunkująca zatrzymanie syntezy
polipeptydu. (stop kodon). (str. 12)
kontener – to program, który odpowiada za obsługę i funkcjowanie serwletów. Przykładem
kontenera serwletów jest Tomcat. (str. 45)
L
licencja CPL – (Common Public License) jest jedną z licencji wolnego oprogramowania, która
została sformułowana w 1988 przez IBM. Treść licencji została zatwierdzona przez
Open Source Initiative. Środowisko programistyczne Eclipse jest przykładowym
projektem rozprowadzanym na licencji CPL. CPL jest licencją copyleft, bardzo
podobną do w treści do GNU General Public License. Główną zmianą jest dodanie
klauzuli uniemożliwiającej zmiany w kodzie programu mające na celu czerpanie
korzyści ze sprzedaży zmienionego programu. W takich sytuacjach treść licencji
CPL pozwala jedynie na darmowe rozprowadzanie programu. (str. 48)

SŁOWNIK 77
M
mock dynamiczny -nie jest konkretną klasą, lecz specjalnym obiektem wyprodukowanym przez
odpowiednią bibliotekę (np. jMock); pozwala na proste definiowanie właściwie
dowolnych zachowań i szerokie możliwości weryfikacji rzeczywistych interakcji
testowanej klasy z podstawionym mockiem. (str. 49)
N
nukleotyd – jest to podstawowy składnik budulcowy kwasów nukleinowych (DNA i RNA).
Jest on zbudowany z cukru - pentozy (w DNA wystepuje deoksyryboza, zaś w
RNA ryboza), co najmniej jednej reszty fosforanowej i zasady azotowej (zasady
purynowej, pirymidynowej lub flawinowej). W nukleotydach DNA wystepują takie
zasady azotowe jak: adenina (A), guanina (G), cytozyna (C) i tymina (T). W
nukleotydach RNA występują takie zasady azotowe jak: adenina (A), guanina
(G), cytozyna (C) i uracyl (U). (str. 5)
O
Open Source – (dosłownie Otwarte Źródła) to oprogramowanie, którego licencja pozwala na
legalne i nieodpłatne kopiowane, zarówno kodu wynikowego jak i źródłowego oraz
na dowolne modyfikacje kodu źródłowego. Więcej informacji na stronie http:
//www.opensource.ite.pl/page/main.html. (str. 43)
P
PDB (Protein Data Bank) – jest to archiwum eksperymentalnie zdefiniowanych
trójwymiarowych struktur biologicznych. Składa się z koordynatów atomów,
odnośników do literatury oraz informacji o strukturze pierwszo i drugorzędowej.
(str. 35)
polimer – jest to związek o bardzo dużej masie cząsteczkowej i jednocześnie regularnej,
powtarzalnej budowie. Zbudowany jest z długich łańcuchów utworzonych ze
stale powtarzających się jednostek zwanych merami. Wyróżnia się polimery
naturlane oraz sztuczne. Polimery naturalne są jednym z podstawowych budulców
organizmów żywych. Polimery syntetyczne są podstawowym budulcem tworzyw
sztucznych, a także wielu innych powszechnie wykorzystywanych produktów
chemicznych. (str. 5)
polimeraza – enzym uzywany powszechnie w biologii molekularnej i inzynierii genetycznej.Ma
on zdolność syntezy nici komplementarnej. Może syntetyzować zarowno DNA jak
i RNA (polimeraza DNA i RNA) . (str. 7)
R
rybonukleotyd – nukleotyd, w którego skład wchodzi cukier ryboza. Polimerem
rybonukleotydów jest RNA, czyli kwas rybonukleinowy. (str. 10)

78 SŁOWNIK
rybonukleozyd – jest to glikozoamina powstała w wyniku połączenia zasady azotowej z rybozą.
(str. 9)
S
serwlet – program napisany w Javie, uruchomiony w ramach kontenera serwletów (np.
Tomcata), którego zadaniem jest przetwarzać i odpowiadać na żądania klientów.
Serwlety są alternatywą dla appletów Javy, ponieważ wykonywane są po stronie
serwera, a aplety działają po stronie klienta. (str. 69)
SQL – (ang. Structured Query Language) jest to strukturalny język zapytań używany
do tworzenia, modyfikowania baz danych oraz do umieszczania i pobierania danych
z baz danych. (str. 50)
substrat – jest ogólną nazwą każdej substancji chemicznej, która, biorąc udział w reakcji
chemicznej, ulega przemianie (tj. jest zużywana), (czym różni się od katalizatora,
który biorąc udział w reakcji, odtwarza się po jej zakończeniu). (str. 5)
T
test jednostkowy – sprawdza działanie danej klasy w całkowitej izolacji od reszty systemu;
powstaje równolegle z testowanym kodem. (str. 48)
X
Xmol – jest to narzędzie do tworzenia i wizualizowania obrazów związków chemicznych.
(str. 37)

Indeks
algorytm DALI, 27
algorytm Smith – Watermana, 34
aminokwas, 4, 10, 13–15, 18, 21, 23, 25, 51, 52,
55
analiza sekwencyjna, 23
analiza składu aminokwasowego, 23
awk, 70
bash, 65, 70
białko, 3–5, 8, 11, 13, 14, 17, 20, 22, 25, 27, 31,
33, 35, 37, 39, 42, 52, 55, 61, 65, 70
bioinformatyka, 1, 3, 37
biotools, 55, 65, 70
BLAST, 31, 66
algorytm, 32, 33
odmiany, 31
wydłużanie, 34
CASP, 27, 36
CGI-BIN, 70
CSS, 62
degradacja Edmana, 23
DNA, 3–6, 8, 11, 13
dokowanie, 29
domena, 21, 22
dopasowanie przybliżone, 35
DSSP, 39, 59
dychroizm kołowy CD, 25
Eclipse, 43
FASTA, 26, 52, 55
ferrytyna, 17
harmonijka β, 16, 18
helisa α, 14, 16, 17
High Scoring Pair, 34
HPLC, 23
HSP, 34
HTML, 61, 62, 68
Human Genom Project, 8
identyfikacja foldu, 29
J2EE, 42
J2ME, 42
J2SE, 42
Java, 42
java
destruktor, 71
garbage collector, 71
Java Script, 62, 68
JDO, 47
JMock, 49
Jmol, 37, 59, 68
JPOX, 48
junit, 48
Karlin i Altschul, 34
kodon, 12
konfiguracja projektu, 67
konformacja białka, 16, 17, 21
krystalografia, 4, 28
Levitt i Gerstein, 36
LGScore, 35, 59, 66
liglandy, 29
macierz podobieństwa, 26
Maximal Segment Pair, 33, 34
MaxSub, 35, 41, 59, 66
metaserwer, 51, 55, 58, 61
odpowiedź, 57
status, 57
79

80 Indeks
metoda „nawlekania—hyperpage, 27
metoda chiralooptyczna, 17, 25
metoda CNRS(SOMPA), 26
metoda GOR, 26
metoda homologów Levina, 26
metoda podwójnego przewidywania, 26
metoda TOPITS, 27
metody numeryczne, 4
MinRMS, 37, 38, 59, 66, 67
modelowanie homologiczne, 28
MSP, 33, 34
narzędzia biologiczne, 66
nnpredict, 26
nRNA, 10
obsługa błędów i wyjątków, 63
obszary podobieństwa, 31
P-wartość, 35
Pairwise Alignment Table, 55
pakiet BioTools, 66
PDB, 27, 35, 37–39, 42, 55, 58, 69, 71
PHDsec, 26
pliki tymczasowe, 70
podjednostka, 22
polimeraza, 7, 12
PostgreSQL, 49
PSIPRED, 26
RMSD, 35
RNA, 3, 9, 12
Root Mean Square Deviation, 35
Secondary Structures Table, 57
sekwencja, 32, 51
wysłanie do metaserwera, 51
serwlet, 69
sieć neuronowa, 25
składanie od początku, 29
skrypt MaxSub, 70
Smith – Waterman, 34
SOMPA, 26
spektroskopia magnetycznego rezonansu
jądrowego , 25
Spindle, 47
status przetwarzania, 60
statystyka Karlina i Altschula, 34
strona
Headquater, 52
ShowSequence, 51, 55, 60, 61
struktura
czwartorzędowa, 22
drugorzędowa, 16, 26, 27, 57
pierwszorzędowa, 15
trzeciorzędowa, 20, 27, 55, 58
Sysdeo, 47
Tapestry, 46
tapestry, 65
użytkownik
administrator, 52
konto, 52
uszeregowanie
globalne, 32
lokalne, 32
uzytkownik, 52
węgiel C-α, 35
Xmol, 37
zwrot β, 16

Bibliografia
[1] B.F.F. Ouellette A.D.Baxevanis. Bioinformatyka - Podręcznik do analizy genów ibiałek. Wydawnictwo Naukowe PWN, Warszawa, 2004.
[2] Jmol developers. Jmol history. http://jmol.sourceforge.net/history/, 2004.
[3] dr inż. Paweł Kędzierski. Porównywanie sekwencji. www.mml.ch.pwr.wroc.pl/
mml/en/people/pk/wyklad/wyklad2_slajdy.pdf, 2004. [Online; przeczytane 27
listopada 2005].
[4] Bruce Eckel. Thinking in Java. Rozdział ’Cleanup: finalization and garbagecollection’. Prentice Hall PTR, 1998.
[5] Susana Cristobal. Adam Zemla. Daniel Fischer. Leszek Rychlewski. Arne Elofsson.
A study of quality measures for protein threading models. BMC Bioinformatics 2:5,2001.
[6] Krzysztof Fidelis. Protein structure prediction center. Strona http://
predictioncenter.org, 2005.
[7] Jan Fassler. National Center for Biotechnology Information. Blast - similarity
searching. http://www.ncbi.nlm.nih.gov/Education/BLASTinfo/similarity.
html, 2000. [Online; przeczytane 26 listopada 2005].
[8] Mock A Lightweight Mock Object Library for Java. Jmock. http://www.jmock.
org/.
[9] B.D. Hades . N.M. Hooper. Krótkie wykłady - Biochemia. Wydawnictwo NaukowePWN, 2002.
[10] Dr. Hung-Chung (Joe) Huang. Rmsd: Squared root of mean square deviations.
http://prion.bchs.uh.edu/~jhuang/RMSD.html, 2005. [Online; przeczytane 22
listopada 2005].
[11] Eremy M. Berg . Jon L. Tymoczko . Hubert. Biochemia. Wydawnictwo NaukowePWN, 2005.
[12] Karlin i Altschul. The karlin-altschul statistical theory. http://blast.wustl.edu/
doc/infotheory.html#KAStats, 1981. [Online; przeczytane 27 listopada 2005].
81

82 Bibliografia
[13] Temple F. Smith i Michael S. Waterman. Identification of common
molecular subsequences. http://gel.ym.edu.tw/~chc/AB_papers/03.pdf, 1981.
Opublikowany w Journal of Molecular Biology.
[14] Maxine Singer i Paul Berg. Język genów. Prószyński i S-ka SA, 1997.
[15] Maciej Koziński. Tomcat. http://www.pckurier.pl/archiwum/art0.asp?ID=
5377.
[16] Gerstein M Levitt M. A unified statistical framework for sequence comparison andstructure comparison. Proc Natl Acad Sci U S A, 1998.
[17] Poznańska Letnia Szkoła Bioinformatyki. Wojciech Makalowski. Wyszukiwanie
sekwencji podobnych. http://bioinformatyka.amu.edu.pl/workshop/files/
psb-wyklad.cz2.pdf, 2002. [Online; przeczytane 26 listopada 2005].
[18] Sherry Shavour . Jim Danjou . Scott Fairbrother . Dan Kehn . John Kellerman . Pat
McCarthy. Eclipse - podręcznik programisty. Helion, 2005.
[19] Serwis naukawpolsce.pap.pl. Światowy sukces polskich naukowców w modelowaniu
białek. Strona Ministerstwa Nauki i Edukacji, styczeń 2005.
[20] Java Persistent Object. Jpox. http://www.jpox.org/index.jsp.
[21] Java Data Objects. Jdo. http://java.sun.com/products/jdo/overview.html.
[22] Irena Roterman-Konieczna. Bioinformatyka we współczesnej biologii. http://www3.
uj.edu.pl/alma/alma/29/01/03.html, 2003.
[23] Chris Sander. Dssp: Database of secondary structure in proteins. http://www.
sander.ebi.ac.uk/dssp/, 1998.
[24] JUnit A Regression testing framework. Junit. http://www.junit.org/index.htm.
[25] University of California The Regents. Minrms: A tool for determining
protein similarity. http://www.cgl.ucsf.edu/Research/minrms/, 2004. [Online;
przeczytane 26 listopada 2005].
[26] Wikipedia. Bioinformatyka. http://pl.wikipedia.org/wiki/Bioinformatyka.
[27] Wikipedia. Wikipedia. http://pl.wikipedia.org.
[28] PostgreSQL The world’s most advanced open source database. Postgresql. http:
//www.postgresql.org/about/.
[29] Tomasz Włodarski. Opis programu do wizualizacji struktur makrocząsteczek -
chimera. http://bioinfo.mol.uj.edu.pl/articles/Wlodarski04, 2005. [Online;
przeczytane 22 listopada 2005].
[30] Zagadnienia z biochemii. Rodzaje rna. http://www.biochemia2004.republika.
pl/index.htm, 2004.