
TEST TOKSYCZNOŚCI
14
1 WYZNACZANIE WSPÓŁCZYNNIKA PODZIAŁU OKTANOL/WODA SUBSTANCJI TOKSYCZNYCH TECHNIKĄ HPLC WPROWADZENIE Aktywność biologiczna związku chemicznego wiąże się w sposób bezpośredni z jego cechami fizykochemicznymi. Najbardziej użytecznym parametrem w przewidywaniu aktywności biologicznej danej substancji, a także prognozowania jej aktywności toksycznej jest lipofilowość - parametr będący wypadkową kilku cech. Lipofilowość charakteryzuje powinowactwo związku chemicznego do fazy lipidowej i wodnej, a jej miarą jest stosunek równowagowych stężeń rozpuszczonej substancji w układzie dwufazowym, składającym się z dwóch niemieszających się rozpuszczalników. Zgodnie z prawem Nernsta, w warunkach równowagi termodynamicznej, w stałej temperaturze i przy stałym ciśnieniu, stosunek stężeń (a dokładniej aktywności) rozpuszczonej substancji w obu takich rozpuszczalnikach jest wielkością stałą, zwaną współczynnikiem podziału. Należy zaznaczyć, iż wartość współczynnika podziału nie zależy od aktywności substancji rozpuszczonej, jest natomiast cechą charakterystyczną dla danego układu dwóch rozpuszczalników i jednej substancji rozpuszczonej, zależną od temperatury i ciśnienia. Lipofilowość jest ważnym parametrem charakteryzującym substancje czynne biologicznie jakimi są leki czy środki ochrony roślin. Działanie substancji chemicznej (leku lub trucizny) na żywy organizm to szereg procesów biofizycznych i biochemicznych, które podzielić można na trzy fazy: farmaceutyczną, na którą składa się forma w jakiej przyjmowana substancja dociera do organizmu, sposób jej podania oraz uwalniania się w organizmie; farmakokinetyczną, czyli transport cząsteczek substancji w organizmie, ich metabolizm i eliminacja; farmakodynamiczną, czyli oddziaływanie cząsteczki substancji z farmakoreceptorem przynoszące pożądany efekt farmaceutyczny.
Transcript of TEST TOKSYCZNOŚCI

1
WYZNACZANIE WSPÓŁCZYNNIKA PODZIAŁU OKTANOL/WODA
SUBSTANCJI TOKSYCZNYCH TECHNIKĄ HPLC
WPROWADZENIE
Aktywność biologiczna związku chemicznego wiąże się w sposób bezpośredni
z jego cechami fizykochemicznymi. Najbardziej użytecznym parametrem
w przewidywaniu aktywności biologicznej danej substancji, a także prognozowania jej
aktywności toksycznej jest lipofilowość - parametr będący wypadkową kilku cech.
Lipofilowość charakteryzuje powinowactwo związku chemicznego do fazy lipidowej i
wodnej, a jej miarą jest stosunek równowagowych stężeń rozpuszczonej substancji w
układzie dwufazowym, składającym się z dwóch niemieszających się rozpuszczalników.
Zgodnie z prawem Nernsta, w warunkach równowagi termodynamicznej, w stałej
temperaturze i przy stałym ciśnieniu, stosunek stężeń (a dokładniej aktywności)
rozpuszczonej substancji w obu takich rozpuszczalnikach jest wielkością stałą, zwaną
współczynnikiem podziału. Należy zaznaczyć, iż wartość współczynnika podziału nie
zależy od aktywności substancji rozpuszczonej, jest natomiast cechą charakterystyczną
dla danego układu dwóch rozpuszczalników i jednej substancji rozpuszczonej, zależną
od temperatury i ciśnienia. Lipofilowość jest ważnym parametrem charakteryzującym
substancje czynne biologicznie jakimi są leki czy środki ochrony roślin.
Działanie substancji chemicznej (leku lub trucizny) na żywy organizm to szereg
procesów biofizycznych i biochemicznych, które podzielić można na trzy fazy:
farmaceutyczną, na którą składa się forma w jakiej przyjmowana substancja
dociera do organizmu, sposób jej podania oraz uwalniania się w organizmie;
farmakokinetyczną, czyli transport cząsteczek substancji w organizmie, ich
metabolizm i eliminacja;
farmakodynamiczną, czyli oddziaływanie cząsteczki substancji z
farmakoreceptorem przynoszące pożądany efekt farmaceutyczny.

2
Choć każdy z tych etapów jest związany z charakterem lipofilowym substancji, to
najistotniejszy wpływ wywiera lipofilowość na procesy dystrybucji związku
chemicznego w organizmie, czyli na fazę farmakokinetyczną. Szczególną uwagę należy
zwrócić na zachowanie się danej substancji podczas przenikania przez lipofilowe błony
komórkowe oddzielające środowiska wodne. Lipofilowość jest opisywana przez procesy
podziałowe między dwiema fazami: niepolarną (organiczną) i polarną (najczęściej
wodną). Przyjęto, że rozpuszczalnikami najlepiej odwzorowującymi układ faz polarnych
i niepolarnych w ustroju biologicznym oraz środowisku naturalnym są n-oktanol i woda.
Rozpuszczalniki te po zmieszaniu tworzą dwie rozseparowane fazy, przy czym ze
względu na wzajemną częściową rozpuszczalność jest to układ zawierający oktanol
nasycony wodą (2,3 mol/L) oraz wodę nasyconą oktanolem (4,5.10-3 mol/L).
Współczynnik podziału P jest wyrażany ilorazem dwóch stężeń substancji
rozpuszczonej:
𝑷 = 𝑪𝒐𝒌𝒕
𝑪𝒘 (1)
gdzie: Cokt (mol/L) stężenie molowe substancji w oktanolu, a Cw (mol/L) stężenie
molowe substancji w wodzie.
Współczynnik taki mierzy się w 25°C, przy stężeniu substancji badanej nie
wyższym niż 0,01 mol/L. W takim układzie, jeżeli większa ilość cząsteczek badanego
związku chemicznego znajdzie się w fazie oktanolowej, to mamy do czynienia ze
związkiem lipofilowym (hydrofobowym). Jeżeli zaś większość pozostanie w fazie
wodnej, będzie to związek lipofobowy (hydrofilowy). W fazie oktanolowej,
dominującymi staną się oddziaływania dyspersyjne pomiędzy hydrofobowymi
fragmentami cząsteczki rozpuszczonego związku a łańcuchem alkilowym n-oktanolu,
podczas gdy nieco mniejsze znaczenie odgrywać tu będą oddziaływania dipol-dipol czy
wiązania wodorowe z grupą hydroksylową alkoholu. Wynika stąd jednak, iż n-oktanol
posiada zarówno właściwości lipo- jak i hydrofilowe, a cechę taką nazywa się
amfifilowością. Zjawisko to przedstawiono schematycznie na rys. 1, gdzie uwidoczniono
cząsteczkę rozpuszczonego benzenu w n-oktanolu (oddziaływania dyspersyjne), w
którym równocześnie rozpuszczona jest woda (oddziaływania dipol-dipol i wiązania
wodorowe). W fazie wodnej z kolei, występować będą niemal wyłącznie wiązania
wodorowe oraz oddziaływania dipol-dipol pomiędzy polarnymi fragmentami cząsteczki
związku rozpuszczonego a wodą.

3
Rys.1. Oddziaływania międzycząsteczkowe substancji rozpuszczonych w n-oktanolu
Wartość współczynnika podziału zależy zarówno od budowy chemicznej
cząsteczki rozpuszczonego związku (liczba i rodzaj różnych grup funkcyjnych, udział
miejsc nienasyconych, fragmentów alkilowych, wartość momentu dipolowego i in.) jak i,
w dużej mierze, od jej wielkości. W trakcie mieszania obu rozpuszczalników (proces
spontaniczny) całkowita entropia takiego układu gwałtownie wzrasta, jednak jej
uprzywilejowany wzrost będzie ograniczany spadkiem entropii w fazie wodnej,
związanym ze stopniowym uporządkowywaniem cząsteczek wody w otoczce
hydratacyjnej solwatowanego związku. Ponieważ większe rozmiary cząsteczki wymagać
będą coraz większej wnęki w rozpuszczalniku (większej otoczki hydratacyjnej)
powodować to będzie nieuprzywilejowany spadek entropii całego układu. W takiej
sytuacji korzystniejsze termodynamicznie staje się obniżenie rozpuszczalności
cząsteczek o dużych rozmiarach w fazie wodnej, co wpływa na zwiększenie
współczynnika podziału. Stąd też nawet stosunkowo duże cząsteczki, ale zawierające w
swojej strukturze układy nienasycone, rozgałęzione i in., ułatwiające zmniejszenie
rozmiaru molekularnego, charakteryzują się dużo niższymi współczynnikami podziału
od swoich analogów pozbawionych tych cech. Współczynnik podziału oktanol – woda
zmierzony dla typowych zanieczyszczeń środowiska mieści się w bardzo szerokim
zakresie od 0,01 dla związków o wysokiej polarności do 1010 dla substancji wysoce
hydrofobowych. Na rysunku 2 przedstawiono zakres wartości współczynnika podziału
dla wybranych toksycznych związków organicznych.

4
Rys.2. Wartości współczynnika podziału P dla wybranych toksycznych związków organicznych
Tak rozpięty zakres wartości współczynnika spowodował, że wyraża się go w
formie logarytmicznej:
𝐥𝐨𝐠 𝑷 = 𝐥𝐨𝐠 𝑪𝒐𝒌𝒕 − 𝐥𝐨𝐠 𝑪𝒘 (2)
Dla tak obliczonego współczynnika podziału przyjmuje się, iż związki których log
P jest mniejsze od jedności, mogą być charakteryzowane jako hydrofilowe, a więc takie,
które nie ulegają bioakumulacji. Związki dla których 1 < log P < 3, charakteryzują się
średnią lipofilowością i mogą ulegać częściowej bioakumulacji. Natomiast związki,
których log P > 3, charakteryzują się wysoką lipofilowością i wysokim potencjałem
bioakumulacji. Jest to typowa wartość współczynnika podziału dla większości tzw.
trwałych zanieczyszczeń organicznych (chlorowane węglowodory, PCB, WWA,
polichlorowane dibenzodioksyny i furany).
Tak jak przedstawiono wcześniej, oddziaływania międzycząsteczkowe
zachodzące pomiędzy substancją rozpuszczoną a n-oktanolem i wodą zależą od jej
budowy chemicznej i wielkości ale też od obecności ugrupowań kwaśnych bądź
zasadowych. Mając do czynienia z kwasami bądź zasadami organicznymi, kolejnym
bardzo ważnym czynnikiem wpływającym na podział staje się również wartość pH
układu, która determinuje stopień zjonizowania substancji rozpuszczonej. W przypadku
kwasów i zasad podziałowi pomiędzy fazy ulegają nie tylko związki neutralne, ale
również formy zjonizowane oraz formy zasocjowane tych związków. Dodatkowo,
występowanie związku w formie zjonizowanej może stać się poważnym ograniczeniem

5
technicznym w wyznaczeniu współczynnika podziału. Jeżeli związek taki w swojej
strukturze zawierałby także obszerny fragment hydrofobowy (np. kilku- lub
kilkunastowęglowy łańcuch alkilowy), to swoją budową przypominałby substancję
powierzchniowo czynną. Na ogół, substancje takie ulegają emulsyfikacji na granicy faz
dwóch niemieszających się rozpuszczalników, co utrudnia a w zasadzie uniemożliwia
wyznaczenie dla nich wiarygodnego współczynnika podziału. Na rys. 3 przedstawiono
przykład podziału kwasu octowego pomiędzy oktanol i wodę z uwzględnieniem
wszystkich możliwych form występowania.
Rys.3. Formy występowania kwasu octowego w układzie n-oktanol – woda
W fazie wodnej kwas występować będzie zarówno w formie zjonizowanej jak i
obojętnej. Im większy będzie udział formy zjonizowanej, tym większa ilość cząsteczek
kwasu znajdzie się w tej właśnie fazie. Oczywiście, jeżeli pH układu uległoby obniżeniu,
to przeważałaby forma obojętna, a poniżej wartości 4,8 (wartość pKa dla kwasu
octowego) byłaby formą dominującą. W miarę protonowania grupy karboksylowej część
cząsteczek kwasu przechodzić będzie do fazy oktanolowej. Oprócz formy obojętnej
w fazie n-oktanolu utworzyć się mogą także dimery kwasu octowego powstałe w wyniku
asocjacji. Asocjacja ma miejsce w niepolarnych rozpuszczalnikach i określą ją stałą
równowagi reakcji asocjacji. Polega ona na tworzeniu cząstek zawierających dwie lub
więcej cząsteczek związanych wiązaniami wodorowymi. Strukturę dimeru kwasu
octowego przedstawiono na rys. 4.
Rys.4. Asocjat dwóch cząsteczek kwasu octowego
Jeżeli kwas asocjuje w fazie oktanolowej, to jego współczynnik podziału
pomiędzy fazę oktanolową i wodną przyjmuje następującą postać:

6
𝑷 =√𝑪𝒐𝒌𝒕
𝒏
𝑪𝒘 (3)
gdzie n to liczba asocjujących cząsteczek. Zlogarytmowana postać tego równania to:
𝐥𝐨𝐠 𝑷 = 𝟏
𝒏𝐥𝐨𝐠 𝑪𝒐𝒌𝒕 − 𝐥𝐨𝐠 𝑪𝒘 (4)
Po przekształceniu uzyskujemy równanie liniowe, gdzie log P jest punktem przecięcia
prostej z osią y przy x = 0, a 1/n jest współczynnikiem kierunkowym prostej:
𝐥𝐨𝐠 𝑪𝒘 = 𝟏
𝒏𝐥𝐨𝐠 𝑪𝒐𝒌𝒕 − 𝐥𝐨𝐠 𝑷 (5)
Równanie to pozwala na podstawie wykresu wyznaczyć współczynnik podziału P
oraz liczbę cząsteczek n tworzących asocjaty w fazie oktanolowej. Do obliczenia log P
należy analitycznie określić aktywność substancji rozpuszczonej w obydwóch fazach, co
w przypadku roztworów rozcieńczonych sprowadza się do wyznaczenia stężenia. W tym
celu stosuje się szereg metod np. miareczkowanie, oznaczanie kolorymetrycznie,
chromatografię cieczową lub gazową. Pomiędzy log P a parametrami retencji z RP HPLC
można zauważyć ścisłą zależność jeżeli układ chromatograficzny przypomina układ
podziałowy n-oktanol/woda. Dlatego też współczynnik podziału można wyznaczać
metodami pośrednimi, z użyciem wysokosprawnej chromatografii cieczowej (HPLC) lub
metodami obliczeniowymi.
1. Metoda HPLC
Hydrofobowość można zdefiniować jako tendencję danego związku do
“preferowania” środowiska niewodnego względem wodnego, pojęcie to może być
rozumiane jako tendencja cząsteczek związku rozpuszczonego do agregacji w
roztworach wodnych. Właściwości hydrofobowe są sumą oddziaływań
fizykochemicznych: orientacyjnych, indukcyjnych, dyspersyjnych, wiązania
wodorowego i przeniesienia ładunku. W przypadku oddziaływań hydrofobowych siły te
zależą głównie od właściwości rozpuszczalników, a nie od substancji rozpuszczonych.
Od czasu wprowadzenia chromatografii cieczowej z fazami odwróconymi jest ona
szczególnie odpowiednia do oznaczania hydrofobowości. W przypadku metody z
wykorzystaniem HPLC zakłada się, iż retencja badanej substancji w odwróconym
układzie faz odpowiada jej podziałowi pomiędzy dwie nie mieszające się fazy i jako taka
jest proporcjonalna do współczynnika podziału P. W takim układzie, jedną z faz (faza
stacjonarna) stanowią ziarna krzemionki chemicznie zmodyfikowane warstwą
hydrofobową złożoną z podstawników n-alkilowych o różnej długości łańcucha (C8 –

7
C18), natomiast drugą fazę (faza ruchoma) stanowi polarny rozpuszczalnik, na ogół
mieszanina wody i metanolu. Podstawowa zaleta oznaczania hydrofobowości tą metodą
to możliwość użycia modyfikatorów organicznych jako składników binarnych
(dwuskładnikowych) eluentów. Jednak obecność modyfikatora organicznego powoduje
podniesienie stopnia skomplikowania natury oddziaływań wpływających na rozdział
chromatograficzny. Przy zastosowaniu ustalonego składu eluentu badany związek może
okazywać się bardziej hydrofobowy niż inny związek, podczas gdy sytuacja taka może
przedstawiać się odwrotnie przy innym składzie eluentu. Dlatego też stosuje się metodę
polegającą na wykreśleniu log k względem procentowej objętościowej zawartości
modyfikatora. W trakcie przemieszczania się przez kolumnę chromatograficzną badana
substancja ulega podziałowi pomiędzy hydrofobową warstwą w fazie stacjonarnej a
polarnym rozpuszczalnikiem w fazie ruchomej. Podobnie jak to ma miejsce w metodzie
wyznaczania równowagowego współczynnika podziału pomiędzy oktanolem a wodą, im
bardziej substancja będzie lipofilową tym większe będzie jej powinowactwo do
stacjonarnej fazy hydrofobowej. W praktyce oznacza to, iż w układzie
chromatograficznym substancja taka będzie dłużej zatrzymywana, a więc wzrastać
będzie jej czas retencji. Dla dużej grupy związków chemicznych wykazano liniową
zależność pomiędzy czasem retencji a współczynnikiem podziału wg następującego
równania:
𝐥𝐨𝐠 𝑷 = 𝒂 𝐥𝐨𝐠 𝒕𝑹 + 𝒃 (6)
przy czym a i b są charakterystyczne dla danej grupy związków (homologów,
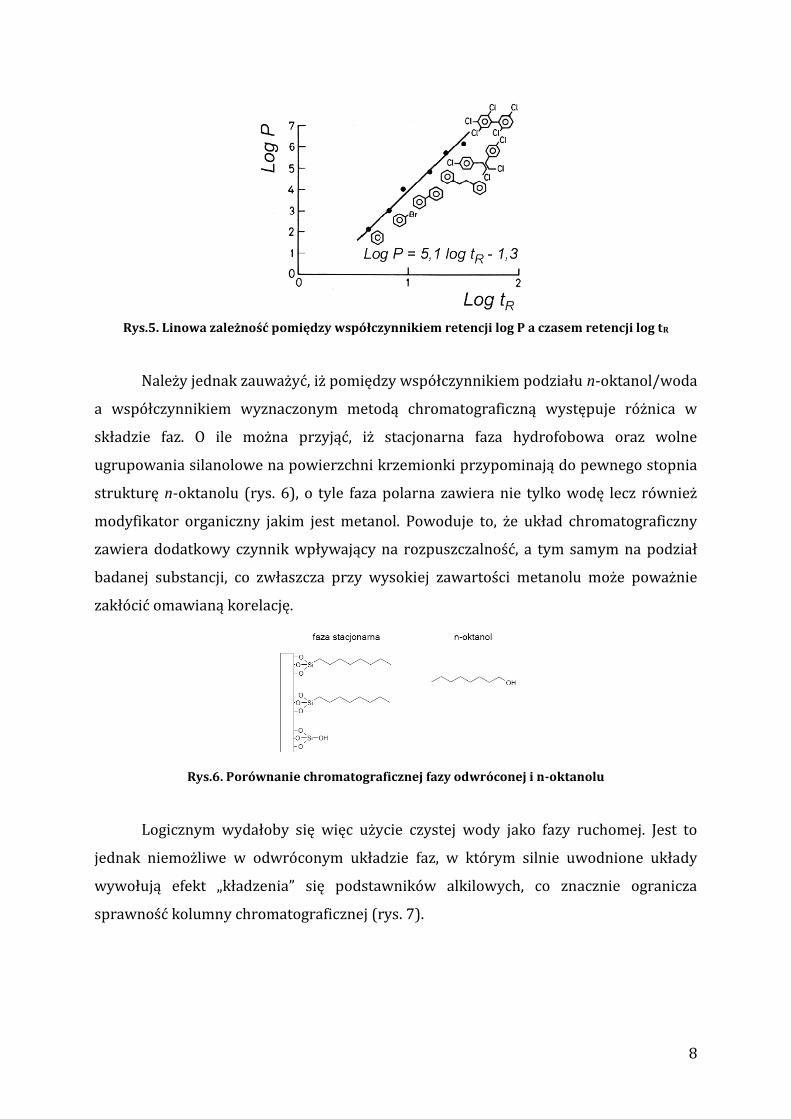
kongenerów, izomerów itp.). Rys. 5 przedstawia tę korelację dla wybranych związków
aromatycznych, których współczynnik podziału mieści się w szerokim zakresie 4
rzędów wielkości. Najprostszym, praktycznym zastosowaniem tej metody jest
wyznaczenie kilkunastu współczynników retencji dla grupy związków referencyjnych, o
znanych współczynnikach podziału a następnie, w oparciu o uzyskaną korelację
szacowanie tych współczynników dla związków nieznanych na podstawie ich czasów
retencji w tych samych warunkach chromatograficznych.
8
Rys.5. Linowa zależność pomiędzy współczynnikiem retencji log P a czasem retencji log tR
Należy jednak zauważyć, iż pomiędzy współczynnikiem podziału n-oktanol/woda
a współczynnikiem wyznaczonym metodą chromatograficzną występuje różnica w
składzie faz. O ile można przyjąć, iż stacjonarna faza hydrofobowa oraz wolne
ugrupowania silanolowe na powierzchni krzemionki przypominają do pewnego stopnia
strukturę n-oktanolu (rys. 6), o tyle faza polarna zawiera nie tylko wodę lecz również
modyfikator organiczny jakim jest metanol. Powoduje to, że układ chromatograficzny
zawiera dodatkowy czynnik wpływający na rozpuszczalność, a tym samym na podział
badanej substancji, co zwłaszcza przy wysokiej zawartości metanolu może poważnie
zakłócić omawianą korelację.
Rys.6. Porównanie chromatograficznej fazy odwróconej i n-oktanolu
Logicznym wydałoby się więc użycie czystej wody jako fazy ruchomej. Jest to
jednak niemożliwe w odwróconym układzie faz, w którym silnie uwodnione układy
wywołują efekt „kładzenia” się podstawników alkilowych, co znacznie ogranicza
sprawność kolumny chromatograficznej (rys. 7).

9
Rys.7. Porównanie struktury hydrofobowej fazy stacjonarnej w przypadku użycia czystej wody (A)
oraz wody z metanolem (B) jako faz ruchomych
Dlatego do chromatograficznego wyznaczania współczynnika podziału stosuje się
metodę ekstrapolacyjną. W skrócie, polega ona na wyznaczeniu czasów
(współczynników) retencji badanego związku dla kilku faz ruchomych o różnej
zawartości metanolu i wykreśleniu powstałej zależności według następującego,
liniowego równania:
𝐥𝐨𝐠 𝒌 = 𝐥𝐨𝐠 𝒌𝒘 − 𝑺𝝓 (7)
nazywanego równaniem Snydera-Soczewińskiego, które jest jedną z podstawowych
zależności w chromatografii w odwróconym układzie faz. W równaniu, k to
współczynnik retencji (lub współczynnik objętościowy) wyznaczony z zależności:
k = (tR – t0)/t0 (8)
gdzie t0 to czas martwy układu; kw to wartość współczynnika retencji odpowiadająca
fazie zawierającej wyłącznie wodę (0% metanolu). Z podanych powyżej ograniczeń
wartość tę wyznacza się ekstrapolując wyniki uzyskane dla faz zawierających metanol
do punktu przecięcia z osią y. Stąd, log kw uznaje się za chromatograficzny współczynnik
podziału (odpowiadający podziałowi pomiędzy wodę a fazę hydrofobową). S to
nachylenie wykreślonej prostej, charakterystyczne dla badanego związku a φ to udział
metanolu w fazie ruchomej (%). Rys. 8 przedstawia opisaną zależność.
Rys.8. Zależność Snydera – Soczewińskiego, z której wyznacza się chromatograficzny współczynnik
podziału log kw

10
W metodzie HPLC substancje o bardzo niskiej rozpuszczalności w n-oktanolu
wykazują tendencję do dawania zaniżonych wartości log P. Piki takich związków czasem
towarzyszą czołu rozpuszczalnika. Dzieje się tak dlatego, iż w takiej sytuacji proces
podziału jest zbyt powolny do uzyskania równowagi w czasie branym pod uwagę przy
zwykłym rozdzielaniu metodą HPLC. Zmniejszenie szybkości przepływu i/lub obniżenie
stosunku metanol/woda może być w tym przypadku pożądane, aby uzyskać rzeczywistą
wartość. Wartość pH eluentu jest krytyczna dla związków ulegających jonizacji. Powinno
być ono w obrębie zakresu roboczego pH kolumny, które zwykle wynosi między 2 i 8.
Zalecane jest buforowanie. Należy zachować ostrożność, aby nie dopuścić do wytrącania
się soli i zniszczenia kolumny, co zachodzi dla niektórych mieszanin faza
organiczna/bufor. Pomiary HPLC faz stacjonarnych opartych na krzemionce powyżej pH
8 nie są polecane, gdyż użycie zasadowej fazy ruchomej powoduje gwałtowne
pogorszenie się wydajności kolumny. Warto dodać, iż prowadzono badania nad użyciem
wypełnień innych, niż oparte na chemicznie związanej krzemionce. Przebadano między
innymi następujące wypełnienia: kopolimery poli(styrenodiwinylobenzenowe),
kopolimer oktadecylopoliwinylowy, tlenek glinowy, tlenek cyrkonowy, oraz niedawno
wprowadzony materiał do pakowania kolumn tzw. immobilizowana sztuczna
membrana (IAM). W przypadku ODS proponuje się modyfikację fazy stacjonarnej bądź
też ruchomej polegającej na np.: zastosowaniu pH od 2,5 do 3,5 i wyższych stężeń
buforu, zastąpienie soli sodowych potasowymi i dodatek modyfikatorów aminowych
(dimetylooktyloamina, trimetyloamina), dodanie oktanolu i decylaminy do fazy
ruchomej składającej się między innymi z MeOH. Jednak nie istnieje sposób który
umożliwiałby uczynienie układu RP HPLC identycznym z układem n-oktanol/woda.
2. Metody obliczeniowe
Wszystkie metody obliczeniowe są oparte o formalną fragmentację cząsteczki na
odpowiednie podstruktury dla których pewne przyrosty wartości log P są znane z
eksperymentu. Wartość log P całej cząsteczki jest następnie obliczana jako suma
wartości odpowiadająca jej fragmentom plus suma składników korekcji dla oddziaływań
wewnątrzcząsteczkowych. Wiarygodność metod obliczeniowych obniża się ze wzrostem
złożoności badanych związków. W przypadku prostych cząsteczek o niskiej masie
cząsteczkowej i jednej lub dwu grupach funkcyjnych, można oczekiwać odchylenia od
0,1 do 0,3 jednostek log P między wynikami różnych metod fragmentaryzacji i

11
wartościami zmierzonymi. Jedną z pierwszych stosowanych półempirycznych metod
obliczeniowych była metoda stałej podstawienia hydrofobowego π, definiowaną jako:
𝝅𝒙 = 𝒍𝒐𝒈 𝑷(𝑷𝒉𝑿) − 𝒍𝒐𝒈𝑷 (𝑷𝒉𝑯) (9)
gdzie P (PhX) jest współczynnikiem podziału pochodnej aromatycznej, a P (PhH)
związku macierzystego.
Metoda stosowana jest głównie dla podstawienia aromatycznego. Wartości dla
większej liczby podstawników zostały stabelaryzowane w licznych pozycjach
literaturowych. Są one stosowane dla obliczeń log P cząsteczek aromatycznych lub
podstruktur. Stosuje się również metodę Rekkera w której wartość log P jest obliczana z
następującej zależności:
𝐥𝐨𝐠 𝑷 = ∑ 𝒂𝒊𝒇𝒊 + ∑ 𝑪𝒋𝒋𝒊 (10)
gdzie fi reprezentuje różne stałe fragmentów cząsteczki, natomiast ai, częstotliwość ich
występowania w badanej cząsteczce. Składniki korekcji są wyrażone jako całkowita
wielokrotność pojedynczej stałej Cj. Stałe fragmentu fi, i Cj zostały ustalone z wykazu z
ponad tysiąca doświadczalnych wartości współczynnika podziału P. Dla prostych
cząsteczek, równanie przyjmuje uproszczoną postać, gdzie suma składników korekcji
wynosi 0,229:
𝐥𝐨𝐠 𝑷 = ∑ 𝒂𝒊𝒇𝒊 + 𝟎, 𝟐𝟐𝟗𝒊 (11)
Najbardziej zaawansowanym sposobem wyznaczania współczynnika podziału
jest metoda Hansch-Leo zgodnie z którą wartość log P jest obliczana z:
𝐥𝐨𝐠 𝑷 = ∑ 𝒂𝒊𝒇𝒊 + ∑ 𝒃𝒋𝒋𝒊 𝑭𝒋 (12)
gdzie fi przedstawia różne stałe fragmentu cząsteczkowego, Fj składniki korekcji oraz ai,
bi odpowiadające częstotliwości występowania. Wyprowadzony z doświadczalnych
wartości współczynników podziału wykaz atomowych i cząsteczkowych grup wartości,
oraz wykaz składników korekcji Fj zostały ustalone metodą prób i błędów. Składniki
korekcji zostały uporządkowane w kilka różnych klas. Wzięcie pod uwagę wszystkich
zasad i składników korekcji jest względnie skomplikowane i zużywające czas. Dlatego
współcześnie istnieje szereg programów komputerowych obliczających lipofilowość
związku na podstawie zadanej struktury.

12
Celem ćwiczenia jest wyznaczanie współczynnika podziału oktanol/woda
wybranych związków organicznych o działaniu toksycznym techniką
wysokosprawnej chromatografii cieczowej oraz metodą obliczeniową.
Odczynniki:
metanol cz. HPLC, woda cz. HPLC, wodny roztwór fenolu (1 mmol/dm3), wodny roztwór
o-nitrofenolu (1 mmol/dm3)
Sprzęt laboratoryjny i aparatura pomiarowa:
chromatograf HP 1050 z detektorem UV-Vis i pętlą nastrzykową o objętości 20 mm3,
kolumna chromatograficzna oktadecylosilanowa ODS (RP-18) 125x4 mm, 5 m.
SPOSÓB WYKONANIA
1. Wyznaczyć czas martwy teoretyczny układu chromatograficznego ze wzoru:
F
Vt m0
,
gdzie:
Vm - martwa objętość kolumny (objętość fazy ruchomej w kolumnie) w cm3,
F - przepływ przez kolumnę w cm3/min.
Martwą objętość kolumny można wyznaczyć z przybliżonego wzoru:
25,0 cm dLV ,
gdzie:
L - długość kolumny w cm,
dc - wewnętrzna średnica kolumny w cm.
2. Przeprowadzić rozdzielenie chromatograficzne roztworów fenolu i o-nitrofenolu o
stężeniach 1 mmol/dm3 w trzech układach elucyjnych metanol–woda: 50:50, 60:40,
70:30 (v/v) przy przepływie 0,8 cm3/min. Detekcję przeprowadzić przy długości fali
278 nm.
Dla każdej z faz ruchomych należy wykonać nastrzyk po 25-30 μL każdego z
roztworów i określić czasy retencji badanych związków. Na ich podstawie wyznaczyć
współczynniki retencji k.
3. W trakcie wykonywania analiz należy obliczyć współczynniki podziału dla
związków z Tabeli 1.
13
W tym celu należy skorzystać z programu EpiSuite, udostępnionego przez US EPA.
Po otwarciu programu wybieramy po lewej stronie okna moduł KOWWIN, w kolejnym
oknie NameLookup, w kolejnym wpisujemy nazwę związku i potwierdzamy OK. W
otwartym oknie wyboru potwierdzamy nasz wybór lub wyszukujemy na liście żądany
związek. W uzupełnionym o dane związku oknie programu KOWWIN wybieramy
Calculate.
Wyniki: przewidywany współczynnik podziału (w czerwonej ramce) oraz wartość
literaturową (Exp Log P) wpisujemy do tabeli. Jeśli brakuje wartości eksperymentalnej,
pozostawiamy wolne miejsce. Program KOWWIN wykorzystuje do obliczeń algorytm
identyczny z opisanym w ćwiczeniu.
Tabela 1. Wartości eksperymentalne i przewidywane dla szeregu homologicznego kwasów
karboksylowych
Liczba atomów
węgla Nazwa
log P
obliczony
log P
eksperymentalny
2 acetic acid
6 hexanoic acid
10 decanoic acid
14 tetradecanoic acid
20 eicosanoic acid
24 tetracosanoic acid
28 octacosanoic acid
32 dotriacontanoic acid

14
OPRACOWANIE WYNIKÓW
1. Należy wykreślić zależność log k od procentowego udziału MeOH w fazie
ruchomej zgodnie z równaniem Snydera-Soczewińskiego i odczytać wartość
log kw.
2. Dla analizowanych związków należy obliczyć współczynniki podziału korzystając
z równania (11) oraz stałych fi fragmentów cząsteczkowych zawartych w Tabeli 2.
Tabela 2. Wartości stałych fi fragmentów cząsteczkowych
Fragment fi
alifatyczny –CH3 0,5473
alifatyczny –CH2 - 0,4911
aromatyczny C lub CH 0,2940
Br- (połączony z aromatem) 0,8900
Cl- (połączony z aromatem) 0,6445
OH- (połączony z aromatem) -0,4802
aromatyczny –NH2 -0,9170
aromatyczny –NO2 -0,1823
3. Na podstawie uzupełnionej Tabeli 1 wykonać na jednym rysunku wykresy
zależności log P eksperymentalnego oraz log P przewidywanego (y) od liczby atomów
węgla w cząsteczce (x). Czy przebieg obu zależności jest liniowy? Dokonaj ekstrapolacji
uzyskanej zależności przewidywanej dla kwasów o 35 i 40 atomach węgla w cząsteczce.
Instrukcja opracowana na podstawie: Chemia fizyczna, Pigoń K., Podstawy chromatografii, Witkiewicz Z.,
Fizykochemiczne metody analizy w chemii środowiska, red. B. Buszewski, P. Kosobucki, Ćwiczenia
laboratoryjne z chemii wody, B. i E. Gomółkowie.
Opracowali: B. Krawczyk, D. Szczukocki


















