
Politechnika Gdańska - mif.pg.gda.pl · katedra Fizyki Teoretycznej i Informatyki Kwantowej ... 1...
110
Politechnika Gdańska wydział Fizyki Technicznej i Matematyki Stosowanej katedra Fizyki Teoretycznej i Informatyki Kwantowej Piotr Michał Łobacz nr albumu 92014 Obliczanie przekroju czynnego na absorpcję promieniowania dla cząsteczki dwuatomowej na przykładzie Li 2 Calculation of absorption cross section for diatomic molecule on example of Li 2 Praca magisterska wykonana pod kierunkiem Prof. dr hab. Józefa E. Sienkiewicza, prof. zw. PG Gdańsk, Czerwiec 2008
Transcript of Politechnika Gdańska - mif.pg.gda.pl · katedra Fizyki Teoretycznej i Informatyki Kwantowej ... 1...

Politechnika Gdańska
wydział Fizyki Technicznej i Matematyki Stosowanej
katedra Fizyki Teoretycznej i Informatyki Kwantowej
Piotr Michał Łobacz
nr albumu 92014
Obliczanie przekroju czynnego na absorpcję
promieniowania dla cząsteczki dwuatomowej
na przykładzie Li2
Calculation of absorption cross section for diatomic molecule
on example of Li2
Praca magisterska wykonana pod kierunkiem
Prof. dr hab. Józefa E. Sienkiewicza, prof. zw. PG
Gdańsk, Czerwiec 2008


Spis treści
1 Wstęp 5
1.1 Rys historyczny . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
1.2 Podstawowe pojęcia - opis fotodysocjacji . . . . . . . . . . . . . . . . 7
1.2.1 Ogólna definicja fotodysocjacji . . . . . . . . . . . . . . . . . . 7
1.2.2 Rodzaje fotodysocjacji . . . . . . . . . . . . . . . . . . . . . . 9
1.2.3 Przekrój czynny - wpływ zasady nieoznaczoności . . . . . . . 14
1.2.4 Funkcja dystrybuanty przestrzeni fazowej . . . . . . . . . . . . 28
1.2.5 Klasyczny przekrój czynny absorpcji oraz fotodysocjacji . . . . 31
1.2.6 Fotodysocjacja bezpośrednia - reguła odbicia . . . . . . . . . . 33
1.3 Reguły wyboru . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36
1.3.1 Spektrometria – rodzaje widm . . . . . . . . . . . . . . . . . . 36
1.3.2 Widmo rotacyjne . . . . . . . . . . . . . . . . . . . . . . . . . 39
1.3.3 Widmo oscylacyjne . . . . . . . . . . . . . . . . . . . . . . . . 42
1.3.4 Przejścia elektronowe . . . . . . . . . . . . . . . . . . . . . . . 45
1.3.5 Zanik wzbudzenia w cząsteczce . . . . . . . . . . . . . . . . . 51
2 Opis modelu fizycznego 54
2.1 Kondensat Bosego-Einsteina . . . . . . . . . . . . . . . . . . . . . . . 56
2.1.1 Rozwinięcie Einsteina . . . . . . . . . . . . . . . . . . . . . . . 57
2.1.2 Równanie Gross-Pitaevskii . . . . . . . . . . . . . . . . . . . . 59
2.1.3 Historyczne odkrycie . . . . . . . . . . . . . . . . . . . . . . . 59
2.1.4 Pierwszy polski kondensat Bosego-Einsteina . . . . . . . . . . 69
2.2 Techniki numeryczne . . . . . . . . . . . . . . . . . . . . . . . . . . . 80
3

2.2.1 Pochodna funkcji . . . . . . . . . . . . . . . . . . . . . . . . . 80
2.2.2 Przykład analitycznego poszukiwania pochodnej
funkcji w punkcie . . . . . . . . . . . . . . . . . . . . . . . . . 82
2.2.3 Implementacja metod matematycznych . . . . . . . . . . . . . 85
3 Obliczanie przekroju czynnego na absorpcję promieniowania 92
3.1 Jednostki . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101
3.2 Wyniki i dyskusja . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 103
3.3 Zakończenie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 104
Dodatek A: Płyta CD 107
Literatura 108
4

Rozdział 1
Wstęp
1.1 Rys historyczny
Gdy 12 marca 1824 roku, w Królewcu urodził się Gustav Robert Kirchhoff, świat
jeszcze wtedy nie przypuszczał, iż jest to nowy rozdział w opisie zjawisk przyrodni-
czych. Był on młodym zdolnym uczonym, który początkowo interesował się mate-
matyką. Wkrótce jednak zainteresowała go chemia, a potem, głównie pod wpływem
profesora Franza Neumanna, zdecydował się na fizykę. Od tego momentu poświęcił
się zwłaszcza fizyce dokonując przy okazji wielu osiągnięć, jak chociażby uogólnił
prawo Ohma. Znalazł dwa ważne prawidła - w szkole średniej poznajemy je jako
prawa Kirchhoffa.
W 1854 r. objął katedrę fizyki na uniwersytecie w Heidelbergu. Na ten okres wła-
śnie przypadają największe jego odkrycia. Zainteresowany zagadnieniem promienio-
wania, odkrył, że zdolność pochłaniania promieniowania przez ciała jest proporcjo-
nalna do ich zdolności wysyłania promieniowania: tzn. im więcej dane ciało pochła-
nia padającej nań energii, tym więcej musi jej emitować. Pomogło to Kirchoffowi
wyjaśnić istnienie jasnych i ciemnych linii widmowych w tych samych położeniach.
Razem z Bunsenem udowodnił, że układ linii widmowych stanowi niepowtarzalną
charakterystykę danego pierwiastka, na podstawie której obu badaczom udało się
jednocześnie odkryć dwa nowe pierwiastki - cez oraz rubid.
Innym razem, obserwując pewnej nocy przez okna swego laboratorium szalejący
5

pożar w pobliskim mieście Mannheim, Bunsen wraz z Kirchhoffem za pomocą spek-
troskopu zbadali, że w płonącym materiale występował bar oraz stront. W owej chwi-
li wpadli na pomysł, aby podobną metodą poznać skład chemiczny Słońca. Wkrótce
mogli potwierdzić, że na Słońcu znajdują się takie same pierwiastki jak na Ziemi.
Tak oto obaj uczeni przyczynili się do stworzenia podstaw, za pomocą których,
po dziś dzień bada się skład chemiczny ciał niebieskich, tj. analiza widmowa tudzież
spektroskopia.
Pod koniec XIX w. fizykę uważano za najbardziej kompletną ze wszystkich nauk
ścisłych. Istniało jedynie zaledwie kilka słabo zbadanych problemów, których rozwią-
zanie spodziewano się wkrótce osiągnąć. Nie przypuszczano jednak, by te rezultaty
miały znaczący wpływ na fizyczny obraz świata. Bardzo niewielu ludzi zdawało so-
bie sprawę z wagi nierozwiązanych problemów, do których w szczególności należał
problem promieniowania ciała doskonale czarnego. Bliższe badania promieniowania
ciała doskonale czarnego, zjawiska fotoelektrycznego a także zjawiska Comptona
sprawiły, że całkowicie zmieniło się nasze postrzeganie świata.
Pionierem fizyki kwantowej był Max Planck. Przewidywania, na podstawie jego
teorii, pokrywały się z wynikami eksperymentalnymi. Uzasadnienie wyników tej teo-
rii na gruncie bardziej podstawowych modeli wymagało jednak założenia, że emisja
promieniowania elektromagnetycznego jest kwantowość wartości takich wielkości,
jak energia; przynajmniej w odniesieniu do procesu emisji i absorpcji światła.
W roku 1859 niemiecki fizyk Gustav Kirchhoff sformułował prawo promieniowa-
nia temperaturowego, które prowadziło do wniosku, że każde ciało doskonale czarne
jest w równowadze termicznej z promieniowaniem cieplnym. Pojęcie ciała doskonale
czarnego wprowadził Kirchhoff w roku 1862 próbując wyjaśnić rozkład widma pro-
mieniowania cieplnego emitowanego przez ciała stałe (np. ogrzany do „czerwoności”
kawałek metalu) lub ciecze (płynne żelazo, stal).
Próby wyjaśnienia rozkładu promieniowania ciała doskonale czarnego na gruncie
termodynamiki klasycznej doprowadziły do sformułowania prawa Rayleigha-Jeansa.
Okazało się jednak, że między przewidywaniami teoretycznymi opartymi na zależno-
ści Rayleigha-Jeansa, a danymi empirycznymi istnieją znaczne rozbieżności. Z teorii
wynikało, że ilość wypromieniowanej energii jest proporcjonalna do czwartej potęgi
6

częstości promieniowania, a to oznaczało, że ciało powinno promieniować znacznie
więcej energii w pasmie ultrafioletu niż w zakresie światła widzialnego i to nieza-
leżnie od temperatury. Rozbieżność ta, nazwana przez Paula Ehrenfesta katastrofą
w nadfiolecie, była głównym motywem do poszukiwania nowej teorii opisującej mi-
kroświat.
14 grudnia 1900 r. Max Planck przedstawił uzasadnienie wzoru przedstawionego
19 października tego roku i będącego poprawną wersją wzoru Wiena. Poprawka
Plancka polegała na odjęciu od mianownika ułamka liczby 1. W uzasadnieniu Planck
przyjął, że oscylatory wytwarzające promieniowanie cieplne mogą przyjmować tylko
pewne wybrane stany energetyczne, a emitowane przez nie promieniowanie może
być wysyłane tylko określonymi porcjami [1].
1.2 Podstawowe pojęcia - opis fotodysocjacji
1.2.1 Ogólna definicja fotodysocjacji
Fotodysocjacja czy też fotoliza, jest procesem chemicznym, w wyniku którego
związek chemiczny (np. sól kuchenna - NaCl, czy też woda - H2O) pod wpływem
bombardowania jednego lub n - fotonów, ulega rozpadowi. Proces fotodysocjacji
nie ogranicza się jedynie do dostarczenia (bombardowania fotonem lub fotonami)
energii do naszego związku, ale także do uzyskania (dostarczenia naszemu związko-
wi chemicznemu) dostatecznej energii aby ten mógł ulec rozpadowi. Jak powszechnie
wiadomo, fotonami nazywamy cząstki elementarne, odpowiedzialne za zjawisko od-
działywań elektromagnetycznych i są postrzegane one jako fala elektromagnetyczna.
Innymi słowy, rozchodzące się w przestrzeni zaburzenie pola elektromagnetycznego,
które ma charakter fali poprzecznej. W takiej fali - występujące obie składowe - elek-
tryczna i magnetyczna, prostopadłe do siebie i kierunku ruchu - ulegają wzajemnym
przekształceniom, a zmieniające się pole elektryczne wytwarza pole magnetyczne
i na odwrót. Fale elektromagnetyczne uzależnione są od długości (częstotliwość fal)
a od tej to uzależniona jest energia fotonów. Mianowicie im krótsza fala tym wyższa
7

jest częstotliwość z jaką ona propaguje:
λ =vw
ν, (1.2.1)
gdzie λ – długość fali, vw – prędkość propagacji fali natomiast ν – częstotliwość.
A ponieważ promieniowanie elektromagnetyczne jest wysyłane i pochłaniane w por-
cjach energii – kwantach, które są zależne od częstotliwości promieniowania – ν,
to można zapisać:
E = hν, (1.2.2)
gdzie h – stała Plancka. Mając zatem powyższe dwa wzory (ze wzoru 1.2.1 wypro-
wadzamy wzór na częstotliwość – ν i podstawiamy do wzoru 1.2.2) otrzymujemy
zależność energii od długości fali:
E =hc
λ. (1.2.3)
Zatem aby móc kontrolować energię emitowanej przez nas wiązki fotonów, musimy
mieć możliwość regulacji długości naszych fal. Co na pozór jest bardzo trywialnym
problemem. Dla przykładu fale radiowe, należące do fal bardzo długich posiadają
bardzo niską energię. Nadają się one do emisji tak powszechnie przez nas słucha-
nych transmisji radiowych czy też telewizyjnych, a to przede wszystkim ze względu
właśnie na niski pobór mocy (nie potrzebują one wzmacniaczy). Natomiast weźmy
pod uwagę powszechne i ogólnodostępne w dzisiejszych czasach urządzenie, tj. mi-
krofalówka. Niebanalne urządzenie służące nam w życiu codziennym, jako „pod-
grzewacz” pokarmu lub wody. W istocie swoją funkcjonalność opiera ono właśnie
o mikrofale, które to posiadają bardzo wysoką energię.
W związku z powyższym energia fotonów (propagowanej przez nas fali elektro-
magnetycznej) zależna jest od długości fali. A zatem fotodysocjacja nie zachodzi
jedynie pod wpływem dostarczonej energii w postaci światła widzialnego (słonecz-
nego), ale także termin ten obejmuje dysocjację pod wpływem promieniowania ul-
trafioletowego, rentgenowskiego a także promieni gamma.
Fotoliza stanowi początkowy etap fotosyntezy. Główny przebieg reakcji fotosyn-
tezy w fotodysocjacji można przedstawić według następującego schematu:
H2A+ 2::::(światło) → 2e− + 2H+ + A.
8

Chemiczna natura naszego „A” zależna jest od rodzaju organizmu. Dla przykła-
du istnieje bakteria, która jest w stanie przeprowadzić reakcję utleniania (zabiera-
nie elektronów) siarkowodoru do siarki. W procesie fotosyntezy zawierającej tlen,
woda jest substratem w reakcji fotolizy charakteryzującym się produkcją czystego
tlenu. Proces ten jest odpowiedzialny za produkcję większości czystego (zdatne-
go) tlenu w atmosferze Ziemskiej. Fotoliza wody ma miejsce w takich strukturach,
jak chociażby błony tylakoid (pęcherzykowata struktura), sinic czy też cyjanobak-
terii, a także w chloroplastach roślin domowych lub zielenic (algi zielone). Zatem,
gdy weźmiemy pod uwagę nasze wcześniejsze rozważania dotyczące fotodysocjacji
oraz główny przebieg reakcji fotosyntezy, możemy przedstawić formalny zapis foto-
dysocjacji w sposób następujący:
AB +Nfotonhν → (1) → (AB)∗ → (2) → A(α) +B(β),
gdzie hν jest energią pojedynczego fotonu o częstotliwości ν natomiast Nfoton to licz-
ba zaabsorbowanych (wchłoniętych) fotonów przez związek AB. (AB)∗ reprezentu-
je pobudzony związek złożony zanim ulegnie on rozpadowi a etykiety α oraz β
określają szczegółowe dane wewnętrznych stanów kwantowych dla nowo powstałych
produktów. Przejście pierwsze wskazuje absorpcję fotonów przez molekułę macierzy-
stą natomiast drugie przejście reprezentowane jest przez fragmentację pobudzonego
już związku (AB)∗.
1.2.2 Rodzaje fotodysocjacji
Jak we wstępie zostało zasygnalizowane do przeprowadzenia fotodysocjacji nie-
zbędne jest źródło fotonów. Przy użyciu takiego źródła bombardujemy interesu-
jący nas obiekt badań (związek chemiczny) jednym lub n - fotonami, który na-
stępnie ulega rozpadowi. Oczywiście w zależności od długości fali takiego fotonu,
posiada on różną energię. Energię tą nazywamy energią dysocjacji D0. Zakres tej
energii mieści się w przedziale kilku meV (dla cząsteczek połączonych wiązaniami
van der Waals’a) do kilkunastu eV (dla cząsteczek posiadających wiązania chemicz-
ne). Cząsteczki związane ze sobą siłami van der Waals’a są bardzo słabe. Ich energia
9

dysocjacji zatem jest bardzo mała. Typowe przykłady:
He - hel...HF – fluorowodór(D0 = 0, 88 meV [2])
Ar – argon...HCl – chlorowodór(D0 = 0, 88 meV [3])
Przykładowymi przedstawicielami cząsteczek z wiązaniami chemicznymi mogą być
np.:
ClNO(D0 = 1, 62 eV [4])
H2O2(D0 = 2, 12 eV [5])
H2O(D0 = 5, 11 eV [6])
Tak ogromy rozrzut możliwych energii dysocjacji wymaga użycia różnych rodza-
jów źródeł światła aby rozbić wiązania chemiczne molekuły. Ponieważ Wiązania
van der Waals’a są słabymi wiązaniami, do rozpadu takowej cząsteczki wystarczą
pojedyncze fotony podczerwone (IR - z ang. infrared photons), podczas gdy do rozsz-
czepienia silnych wiązań chemicznych potrzeba pojedynczego fotonu ultrafioletowego
(UV ) lub bardzo wielu fotonów IR. Taki stan rzeczy powoduje pewne komplikacje.
Rozpatrzmy zatem oba przypadki, jak się będzie zachowywać nasza cząsteczka pod-
V(R
AB)
RAB
AB Ei
P(E)
Rysunek 1.1: Bombardowanie n-fotonami IR
10

czas bombardowania pojedynczym fotonem UV , a jak w przypadku bombardowania
wieloma fotonami IR. Rozważania nasze przedstawione są na rysunkach 1.1 oraz 1.2.
Na przedstawionej ilustracji 1.1 bombardujemy n – fotonami IR. Jak wiadomo
pojedynczy foton IR jest w stanie rozerwać tylko bardzo słabe wiązania chemicz-
ne. Co się zatem może dziać z naszą cząsteczką chemiczną podczas, gdy za każdym
razem absorbuje ona niedostateczną energię do zajścia fotodysocjacji? Do zajścia fo-
todysocjacji potrzebna jest nam odpowiednia ilość energii. Jeżeli natomiast energia
ta będzie za niska, nasza cząsteczka zmieni swój stan kwantowy. Sytuacja ta miałaby
miejsce np. w przypadku bombardowania pojedynczym fotonem IR, jednakże my
mamy sytuację taką gdy bombardujemy n - fotonami IR. W efekcie takiego bom-
bardowania otrzymalibyśmy nieporównywalnie szerszy zespół stanów z dystrybucją
energii. Dysocjację taką nazywamy multifotonową. Ma ona zwykle swoje miejsce
w podstawie elektronicznej, chociażby w nadprzewodnikach. A ponieważ dokład-
V(R
AB)
RAB
AB Ei
(AB)*Ef=Ei+
−hω
−hω
A+B
Rysunek 1.2: Bombardowanie pojedynczym fotonem UV
na ilość absorbowanych fotonów nie może być kontrolowana, laser tworzy zespół
stanów kwantowych wyższych wartości progowej dysocjacji z dystrybucją energii
P (Ef), gdzie Ef jest sumą energii, która wyraża się wzorem:
Ef = Ei +NEfoton. (1.2.4)
11

Zmienna Ei jest energią macierzystą naszej cząsteczki, natomiast NEf jest to nasza
energia n – fotonów jaką dostarczamy do układu. Konsekwencją tejże wielofotonowej
dysocjacji jest konieczność uśredniania bardzo wielu stanów kwantowych, co wymaga
zupełnie innych podstawowych narzędzi teoretycznych oraz modeli niż dysocjacja
z pojedynczym fotonem. Zatem interesującym dla nas modelem fotodysocjacji będzie
ten przedstawiony na wykresie 1.2.
Przyglądając się powyższemu rysunkowi zauważyć można, iż nawiązuje on tutaj
do efektu fotoelektrycznego. Zjawisko to polega na emisji elektronów z powierzchni
przedmiotu (tzw. efekt zewnętrzny) lub na przeniesieniu nośników ładunku elek-
trycznego pomiędzy pasmami energetycznymi (tzw. efekt wewnętrzny), po naświe-
tleniu jej promieniowaniem elektromagnetycznym (na przykład światłem widzial-
nym) o odpowiedniej częstotliwości, zależnej od rodzaju przedmiotu. Wyjaśnienie
tego efektu na gruncie fizyki klasycznej jest niemalże niemożliwe. Zaproponowane
przez Alberta Einsteina wyjaśnienie zjawiska i jego opis matematyczny oparty jest
na założeniu, że energia wiązki światła pochłaniana jest w postaci porcji (kwantów)
równych hν, gdzie h jest stałą Plancka, a ν oznacza częstotliwość fali. Kwant ener-
gii może być zamieniony na energię tylko w całości, na zasadzie wszystko lub nic.
Einstein założył dalej, że usunięcie elektronu z metalu (substancji) wymaga pewnej
pracy zwanej pracą wyjścia, która jest wielkością charakteryzującą daną substan-
cję, pozostała energia rozprasza się częściowo w substancji a częściowo pobiera ją
emitowany elektron. Z tego wynika wzór:
hν = W + Ek, (1.2.5)
gdzie h - stała Plancka, ν - częstotliwość padającego fotonu, W – praca wyjścia, zaś
Ek - maksymalna obserwowana energia kinetyczna emitowanych elektronów. W efek-
cie fotoelektrycznym wewnętrznym energia fotonu też jest całkowicie pochłaniana
przez elektron. Zjawisko to zachodzi jedynie na elektronach, przy spełnionym wa-
runku, że energia fotonu będzie większa niż energia wiązania elektron – atom, w tej
sytuacji atom zostaje zjonizowany. Fotony o dużej energii wybijają elektrony o więk-
szej energii wiązania, więc przy dostatecznie wysokiej energii fotonów wybijane są
nawet elektrony z powłoki K atomu. Miejsca po wybitych elektronach, zapełniają
12

elektrony z wyższych powłok w wyniku czego emitowane jest mono energetyczne
promieniowanie rentgenowskie.
Według teorii pasmowej ciał - elektron, przejmując energię fotonu jest przeno-
szony między dozwolonymi poziomami energetycznymi.
W naszym przykładzie cząsteczkę bombardujemy pojedynczym fotonem,
który pobudza ją ze stanu spoczynkowego od razu do wyższego poziomu elektryczne-
go (posiada wyższą energię). Jeżeli potencjał tego wyższego stanu będzie odpychają-
cy wzdłuż współrzędnej oddziaływań między cząsteczkowych RAB, nasza pobudzona
molekuła (AB)∗ ulegnie natychmiastowemu rozpadowi. Część energii fotonu Efoton
wyrażona we wzorze 1.2.2 zostaje zużywana do rozbicia więzi łączących związek
A − B. Zapisać zatem możemy, iż energia pobudzenia Epob jest to różnica ener-
gii dostarczonej przez foton i energii potrzebnej aby zaszedł proces fotodysocjacji,
i wyraża się ona wzorem:
Epob = Efoton −D0. (1.2.6)
Jeżeli porównamy otrzymany wzór 1.2.6 ze wcześniejszym wzorem 1.2.5, to w tym
przypadku naszą pracą wyjścia będzie energia dysocjacji, natomiast maksymalną
energią kinetyczną energia pobudzenia – Epob. Zatem, można zapisać, iż nasza ener-
gia kinetyczna (pobudzenia) jest to nic innego jak suma dwóch energii. Energia
translacji pomiędzy atomami naszej molekuły oraz energia oddziaływań wewnątrz
atomowych (wliczając w to energię drgań, obrotową a nawet elektryczną). Wyraża
się to wzorem:
Epob = Etrans + Ewew. (1.2.7)
Tak otrzymane równania 1.2.6 i 1.2.7 możemy przyrównać do siebie, i otrzymać
zależność:
Epob = Efoton −D0 = Etrans + Ewew, (1.2.8)
Efoton −D0 = Etrans + Ewew, (1.2.9)
D0 = Efoton − Etrans −Ewew (1.2.10)
na energię dysocjacji.
Fotodysocjacja UV jest zwykle przeprowadzana z wykorzystaniem długich im-
pulsów świetlnych o niskim natężeniu oraz ograniczonym zakresie. Takie warunki
13

gwarantują – przynajmniej z reguły – że foton utworzy pojedynczy stan kwanto-
wy w wyższym kanale elektrycznym z odpowiadającą mu energią wyrażającą się
we wcześniejszym wzorze 1.2.4. Tym razem mamy do dyspozycji pojedynczy foton
zamiast N . Możemy zatem zapisać:
Ef = Ei + Efoton. (1.2.11)
W tak ściśle zdefiniowanych warunkach powinniśmy w sposób trywialny wykazać
jak główne obserwable, spektrum absorpcji oraz dystrybucje stanu produktu, w spo-
sób jednoznaczny „odbijają” molekularną funkcję falową poszczególnych stanów
kwantowych cząsteczki macierzystej przed wzbudzeniem. Jednym z naszych głów-
nych zagadnień będzie zastanowienie się w jakim stopniu dynamika na wyższych
stanach energetycznych pośredniczy w takim rodzaju „odbicia”.
1.2.3 Przekrój czynny - wpływ zasady nieoznaczoności
Jak powszechnie wiadomo świat można podzielić na dwa układy. Makroskopo-
wy, który opisują równania Newtona, oraz mikroskopowy opisany prawami mechani-
ki kwantowej. Teoria praw ruchu obiektów świata mikroskopowego poszerza zakres
mechaniki na odległości czasoprzestrzenne i energie, dla których przewidywania me-
chaniki klasycznej nie sprawdzały się. Teoria ta opisuje przede wszystkim obiekty
o bardzo małych masach i rozmiarach – np. atom, cząstki elementarne itp. Jej grani-
cą dla średnich rozmiarów lub średnich energii czy pędów jest mechanika klasyczna.
Oscylator harmoniczny
W naukach ścisłych jest to model teoretyczny opisujący układ w parabolicznym
potencjale — potencjał oscylatora harmonicznego, bądź krócej potencjał harmo-
niczny, czyli kwadratowa zależność potencjału od odległości V ∼ r2, gdzie r jest
odległością w N -wymiarowej przestrzeni, N zależy od konkretnej realizacji modelu.
Ze względu na skalę modelowanych zjawisk wyróżnia się klasyczny oscylator harmo-
niczny oraz kwantowy oscylator harmoniczny.
Z matematycznego punktu widzenia potencjał paraboliczny jest najprostszym
14

potencjałem lokalizującym, który warto rozważać teoretycznie. Prostsze potencjały
nie są interesujące, gdyż:
1. Potencjał stały to cząstka swobodna.
2. Liniowa zależność:
• w mechanice klasycznej oznacza stałą siłę,
• w mechanice kwantowej potencjał liniowy wymaga doprecyzowania,
gdyż bez określenia warunków brzegowych problem jest źle postawiony
(odpowiednie rozwiązanie równania Schrödingera bez warunków brzego-
wych ma nieograniczone z dołu widmo).
Innym powodem, dla którego model oscylatora harmonicznego jest tak często
eksploatowany w naukach ścisłych wynika z tego, że istnieje bardzo wiele funkcji
potencjału, które można przybliżyć wokół minimum zależnością kwadratową. Ma-
tematycznym warunkiem byłaby istniejąca i nieznikająca druga pochodna funkcji
potencjału w minimum. W praktyce oznacza to, że wiele zagadnień świata realnego
daje się sprowadzić do zagadnienia oscylatora harmonicznego. Przykładami takich
zagadnień są:
1. Mechanika klasyczna:
• wahadło matematyczne,
• wahadło fizyczne,
• masa na sprężynie,
• małe drgania harmoniczne.
2. Mechanika kwantowa
• drgania sieci krystalicznej,
• potencjał jądrowy,
• kropka kwantowa.
15

Zagadnienie oscylatora harmonicznego jest ściśle rozwiązywalne zarówno w me-
chanice klasycznej (klasyczny oscylator harmoniczny) jak i mechanice kwantowej
(kwantowy oscylator harmoniczny).
Drgania inne niż harmoniczne (tzn. dla potencjałów opisywanych innymi zależno-
ściami niż kwadratowymi, bądź nie dające się sprowadzić do potencjału harmonicz-
nego) określa się drganiami anharmonicznymi. Poprawki do ruchu harmonicznego
wynikające z innych zależności potencjału niż kwadratowa nazywa się poprawkami
anharmonicznymi.
W związku z tym, że oscylator harmoniczny jest obecny we wszystkich dziedzi-
nach fizyki, to bardzo często przez oscylator harmoniczny rozumie się konkretną
realizację modelu. Nazwa ta jest używana wszędzie tam, gdzie nie budzi ona wąt-
pliwości, a wyjaśnieniem jest kontekst, w jakim się pojawia.
Kwantowy oscylator harmoniczny
Oscylator harmoniczny jest układem fizycznym, który ma duże zastosowanie
i znaczenie w wielu działach fizyki.
Jest to ciało o masie m, na które działa siła proporcjonalna do wychylenia z prze-
ciwnym zwrotem F = −kx. Ponieważ siła F = −∂U∂x
to układ opisany jest przez po-
tencjał:
U(x) =1
2kx2 =
1
2mω2x2. (1.2.12)
W klasycznej mechanice, mω2 = k nazywamy współczynnikiem sztywności sprę-
żyny lub też stałą siłową, a ω częstotliwością kołową.
Hamiltonian cząstki jest wyrażony przez:
H =p2
2m+
1
2mω2x2, (1.2.13)
gdzie x jest operatorem pozycji, a p jest operatorem pędu(
p = −i~ ddx
)
. Pojęcie
pierwsze reprezentuje energię kinetyczną cząstki, natomiast pojęcie drugie przedsta-
wia energię potencjalną cząstki. Aby znaleźć poziomy energetyczne i odpowiadające
im stany, musimy rozwiązać czasowo niezależne równanie Schrödingera,
H |ψ〉 = E |ψ〉 . (1.2.14)
16

Możemy rozwiązać równanie różniczkowe we współrzędnych podstawowych, używa-
jąc do tego celu metody mocy zbioru. Okazuje się, że istnieje rodzina rozwiązań,
〈x|ψn〉 =
√
1
2n n!·(mω
π~
)1/4
·exp
(
−mωx2
2~
)
·Hn
(√
mω
~x
)
n = 0, 1, 2, . . . (1.2.15)
Pierwsze sześć rozwiązań (od n = 0 do 5) przedstawionych jest po prawej stronie.
Funkcje Hn są wielomianami Hermite’a:
Hn(x) = (−1)nex2 dn
dxne−x2
. (1.2.16)
Nie wolno mylić ich z Hamiltonianem, który także oznaczany jest literą H . Odpo-
wiadające poziomy energii wyrażają się wzorem:
En = ~ω
(
n+1
2
)
. (1.2.17)
Widmo takiej energii jest osobliwe z trzech powodów. Po pierwsze energie są „skwan-
towane”, i mogą przyjmować jedynie dyskretne wartości ~ω pomnożone przez 12, 3
2,
52, i tak dalej. Jest to własność wielu układów mechaniki kwantowej. W poniższej
sekcji przy operatorach kreacji i anihilacji, zajmiemy się bardziej szczegółowym zba-
daniem tego zjawiska. Po drugie najniższą możliwą energią nie jest zero, tylko 12~ω,
które nazywamy „dyskretnym poziomem energii” lub też energią poziomu zerowego.
W dyskretnym poziomie, zgodnie z zasadami mechaniki kwantowej, oscylator wyko-
nuje mało znaczące oscylacje a jego średnia energia kinetyczna ma wartość dodatnią.
Nie jest to oczywiste, że fakt ten jest jednoznaczny, ponieważ normalnie wartość ze-
rowa energii nie jest wymowną wielkością fizyczną, jedynie różnicą energii. Niemniej
jednak, dyskretny poziom energii posiada wiele implikacji, szczególnie w przypadku
kwantowej grawitacji. Ostatnim powodem jest, że poziomy energetyczne są w równej
mierze rozdzielone, inaczej niż w przypadku modelu Bohr’a czy też cząstki w pudeł-
ku.
Zauważmy iż, prawdopodobieństwo gęstości poziomu dyskretnego skoncentrowa-
ne jest w źródle. Oznacza to mniej więcej tyle, że cząstka spędza większość swo-
jego czasu na dnie studni potencjałów, czego spodziewamy się dla stanu o najniż-
szej z możliwych energii. W miarę wzrostu energii, prawdopodobieństwo gęstości
koncentruje się w „klasycznych punktach zwrotu”, gdzie energia stanu zbiera się
17

z energią potencjalną. Jest to zgodne z klasycznym oscylatorem harmonicznym,
w którym cząstka spędza większość swojego czasu (i jest dlatego też najbardziej
prawdopodobne jej wystąpienie) w punktach zwrotu, w których to jest najwolniej-
sza. Zgodność tej zasady jest zatem zadowalająca.
Rozwiązanie mocy zbioru, w sposób bezpośredni jest raczej dość żmudne. Meto-
da „operatorów kreacji i anihilacj”, powstała dzięki Paul’owi Dirac’owi. Umożliwia
ona wyodrębnienie wartości własnych energii bez konieczności bezpośredniego roz-
wiązywania równań różniczkowych. Ponadto z łatwością uogólnia się do bardziej
skomplikowanych problemów, zwłaszcza kwantowej teorii pola.
Twierdzenie 1.2.1 Metoda operatorów kreacji i anihilacji.
Zgodnie z tym rozumowaniem, definiujemy następujące operatory a oraz jego sprzę-
żenie hermitowskie a†
a =√
mω2~
(
x+ imωp)
,
a† =√
mω2~
(
x− imωp)
.(1.2.18)
Operatory x oraz p przestrzegają następującą zbieżność, znaną jako kanoniczną re-
lację komutacyjną:
[x, p] = i~. (1.2.19)
Nawiasy kwadratowe w równaniu tym są powszechnie stosowane narzędziem nota-
cyjnym, powszechnie zwanym jako komutator, i zdefiniowanym przez:
[A,B]def= AB − BA. (1.2.20)
D ow ó d.
Stosując powyższe równania możemy udowodnić tożsamości:
H = ~ω(
a†a + 1/2)
, (1.2.21)
[
a, a†]
= 1. (1.2.22)
Teraz, niech |ψE〉 oznacza tożsamościowy stan energii z energią E. Wewnętrzny
produkt jakiegokolwiek ket z samym sobą musi być nie ujemny. Zatem można zapisać
nierówność:
(a |ψE〉 , a |ψE〉) = 〈ψE| a†a |ψE〉 ≥ 0. (1.2.23)
18

Wyrażając a†a w znaczeniu Hamiltonianu:
〈ψE|H
~ω− 1
2|ψE〉 =
(
E
~ω− 1
2
)
≥ 0, (1.2.24)
tak żeby E ≥ ~ω/2. Zauważmy, iż wtedy kiedy (a |ψE〉) jest zero ket (np. ket o ze-
rowej długości), nierówność jest nasycona, tak że E = ~ω/2. W sposób bezpośredni
można sprawdzić, że istnieje stan potwierdzający ten warunek; jest to dyskretny
(n = 0) stan zadany w poprzedzającej sekcji.
Stosując powyższe tożsamości, możemy wykazać, że relacje komutacji a oraz a†
z H są:
[H, a] = −~ωa,[
H, a†]
= ~ωa†.(1.2.25)
Zatem, warunek (a |ψE〉) nie jest zerowym ket,
H(a |ψE〉) = ([H, a] + aH) |ψE〉= (−~ωa+ aE) |ψE〉= (E − ~ω)(a |ψE〉).
(1.2.26)
Podobnie, możemy wykazać że
H(a† |ψE〉) = (E + ~ω)(a† |ψE〉). (1.2.27)
Innymi słowy, a działa na wartości własne energii E w celu produkcji, aż do sta-
łej mnożnej, kolejny stan własny energii E − ~ω, oraz a† działa na stany własne
energii E w celu wyprowadzenia stanu własnego energii E + ~ω. Z tego powodu,
a nazywamy „operatorem anihilacji”, oraz a† „operatorem kreacji”. Obydwa opera-
tory razem nazywamy „operatory kreacji i anihilacji”. Operatory a oraz a† nazywa-
ne są tak w kwantowej teorii pola tak, ponieważ „niszczą” oraz „tworzą” cząstki,
które odpowiadają naszemu kwantowi energii.
Mając jakikolwiek specyficzny poziom energii, możemy oddziaływać na niego
operatorem zmniejszającym a, ażeby wyprowadzić kolejny specyficzny stan energii
z ~ω - mniejszą energią. Poprzez powtarzanie działania operatorem anihilacji, okazu-
je się że możemy uzyskać specyficzny stan energii, aż do poziomu E = −∞. Jednak,
może to przeczyć naszemu wcześniejszemu postulatowi E ≥ ~ω/2. A więc, musi być
19

taki dyskretny stan energii stanów własnych, który oznaczamy |0〉 (nie powinno się
mylić z zerowym ket), takich jak
a |0〉 = 0(zero ket). (1.2.28)
W takim przypadku, dalsze użycie operatora anihilacji będzie produkowało zerowe
kety, zamiast dodatkowych stanów własnych energii. Ponadto, wykazaliśmy to po-
wyżej, że
H |0〉 = (~ω/2) |0〉 . (1.2.29)
Na koniec, w wyniku działania na |0〉 operatorem kreacji mnożąc przez odpowied-
nie czynniki normalizujące, możemy stworzyć nieskończony zestaw stanów własnych
energii |0〉 , |1〉 , |2〉 , ..., |n〉 , ..., takich jak
H |n〉 = ~ω(n+ 1/2) |n〉 , (1.2.30)
co odpowiada widmu energii, które przedstawiliśmy na początku.
Kwantowy oscylator harmoniczny posiada naturalne zakresy długości oraz ener-
gii, które mogą zostać wykorzystane w celu uproszczenia problemu. Określa się
to mianem bezwymiarowości. W efekcie jeśli mierzymy energię w jednostkach ~ω
oraz odległość w jednostkach(
~
mω
)12 , wtedy równanie Schrödinger’a przyjmuje po-
stać:
H = −1
2
d2
du2+
1
2u2, (1.2.31)
i funkcje własne energii oraz wartości własne przyjmują
〈x|ψn〉 =1√2nn!
π−1/4exp(−u2/2)Hn(u), (1.2.32)
En = n +1
2. (1.2.33)
Żeby uniknąć nieporozumienia, nie zapożyczymy tych naturalnych jednostek w na-
szej pracy, jednakże one często są pomocne podczas wykonywania obliczeń.
Zasada nieoznaczoności Heisenberga
W świecie kwantowym nie ma możliwości dokładnego pomiaru jednocześnie po-
łożenia i pędu cząstki, gdyż każdy pomiar z samej swojej natury wpływa na ba-
20

dany obiekt, zmieniając jego właściwości. Można przewidywać jedynie średnie wy-
niki z serii wielu pomiarów. Ważne jest by podkreślić, że ∆x, itd. nie są błędami
pomiarowymi wynikającymi z niedoskonałości urządzeń lub metod pomiarowych,
ale niepewnościami wyników (wariancją) wynikających z istoty samego pomiaru.
Sam pomiar bowiem w przeważającej większości przypadków zmienia stan ukła-
du. Przykładowo, obserwując dany obiekt oświetlamy go fotonami. Im dokładniej
chcemy zbadać położenie obiektu, tym krótsza musi być długość fali fotonów używa-
nych do obserwacji. Fotony o krótszej długości fali niosą większą energię i pęd, a przez
to bardziej zaburzają badany układ Stała Plancka h = 6.626×10−34 Js wyznacza tu
pewną charakterystyczną skalę. Obiekty, dla których długość fali jest zbliżona do jej
wielkości, nabierają na skutek działania niesamowite własności. Przykładem może
być tu elektron, który na skutek tunelowania, może przejść przez odpowiednio wąską
barierę potencjału, mimo że jego energia jest mniejsza od wysokości tej bariery.
Jednak obiekty fizyczne znacznie większe od długości Plancka nie mają takich
własności. Przykładowo, mrówka o masie 0, 1 g i długości 1 mm, która w czasie 1 s
pokonuje drogę 1 mm ma pęd równy 0, 1 gmms
. Zgodnie z zasadą nieoznaczoności jej
pozycję i pęd można równocześnie zmierzyć z dokładnością nie większą niż do 10
miejsca po przecinku. Taka dokładność jest zupełnie wystarczająca w codziennych
doświadczeniach, dlatego efekty kwantowe nie są tu możliwe do zaobserwowania.
Wielu uczonych spiera się co do skutków zasady nieoznaczoności. Jedną z jej im-
plikacji jest istnienie pewnej elementarnej długości Plancka, która wyznacza granice
pomiarów. Jej wartość szacuje się na 10−35 metra. Wartość tę można interpretować
w ten sposób, że każda inna długość jest jej wielokrotnością. Idąc dalej, niektórzy
naukowcy uważają, że czas też nie płynie w sposób ciągły, lecz zmienia się skokowo.
Na jedną sekundę przypada ok. 5× 1044 elementarnych kroków, w których zmienia
się stan naszego otoczenia. Odwrotność tej liczby określa się jako czas Plancka. Jed-
nak długość Plancka i czas Plancka znajduje się daleko poza zasięgiem dokładności
pomiarów nawet w największych akceleratorach cząstek.
Zasada nieoznaczoności wydaje się mieć niewielki wpływ na codzienne życie. Po-
stęp techniki i nauki zdaje się dowodzić, jak precyzyjnie fizyka determinuje wszystkie
cechy świata wokół nas. Jednak obraz ten jest bardzo zwodniczy. Osiągnięcia w tech-
21

nice sprowadzają się do zdolności budowania maszyn, których konstrukcja redukuje
do minimum element niepewności. Jeżeli jakieś zjawisko, jest zależne od kwantowych
fluktuacji, to nie jest ono stosowane w technice. Technologia nie wymaga całkowitego
zrozumienia otaczającego nas wszechświata, żeby budować działające maszyny.
Istnienie elementu losowego powoduje jednak, że ogromna ilość zjawisk wokół nas
jest bardzo nieprzewidywalna. Niektóre wytwory natury są bardzo podatne na efekt
motyla. Oznacza to, że małe przypadkowe zdarzenie w kwantowym świecie prowadzi
do bardzo dużych zmian.
Twierdzenie 1.2.2 Ogólna postać zasady.
Jeżeli w danym stanie kwantowym |ψ〉 wektory A|ψ〉 i B|ψ〉 są prawidłowo określo-
nymi wektorami stanu, to zachodzi:
σ2(A)σ2(B) ≥ 1
2[〈ψ|1
i[A|B]|ψ〉]2, (1.2.34)
gdzie σ - wariancja, natomiast A, B - dowolne obserwable.
D ow ó d.
Z nierówności Schwartza w przestrzeni Hilberta
〈ψ|A2|ψ〉〈ψ|B2|ψ〉 ≥ |〈ψ|AB|ψ〉|2. (1.2.35)
Z nierówności trójkąta dla liczb zespolonych
4|〈AB〉|2 = (|〈ψ|AB|ψ〉| + |〈ψ|BA|ψ〉|)2 ≥ |〈ψ|[A,B]|ψ〉|2 =
= |〈|AB|ψ〉 − |〈|BA|ψ〉|2. (1.2.36)
Wtedy również dla
A→ A− 〈A〉,
B → B − 〈B〉.
ponieważ liczba komutuje z operatorem, co dowodzi zasadę nieoznaczoności.
Przekrój czynny jest to wielkość fizyczna stosowana w statystycznym opisie zde-
rzeń cząstek bądź obiektów. Określa prawdopodobieństwo wystąpienia zderzenia,
a zdefiniowana jest jako pole powierzchni, mierzone na płaszczyźnie prostopadłej
do kierunku ruchu pocisku, w które musi on trafiać, aby doszło do zderzenia.
22

Pojęcie przekroju czynnego może być stosowane w statystycznej analizie każdego
typu zderzeń, jest jednak rzadko stosowane do opisu zderzeń obiektów makroskopo-
wych. Głównymi użytkownikami pojęcia przekroju czynnego są fizyka jądrowa i fizy-
ka cząstek elementarnych. Jest tak, ponieważ w zderzeniach obiektów mikroskopo-
wych nie jesteśmy z zasady w stanie określić precyzyjnie toru pocisku ani położenia
tarczy – co wynika bezpośrednio z zasady nieoznaczoności. Możemy więc operować
jedynie prawdopodobieństwem zderzenia.
Przekrój czynny jest zwykle oznaczany grecką literą σ (mała sigma).
Przekrój czynny a prawdopodobieństwo zderzenia
Mając dany przekrój czynny możemy wyliczyć prawdopodobieństwo rozprosze-
nia. Dla pojedynczej cząstki padającej na tarczę wynosi ono:
P =nσ
S= ρσd, (1.2.37)
gdzie n – liczba centrów rozpraszających w tarczy, S – powierzchnia tarczy, ρ – gę-
stość centrów rozpraszania (liczba centrów na jednostkę objętości), d– grubość tar-
czy. Wzór ten jest poprawny tylko przy założeniu „cienkiej” tarczy, tzn. takiej,
w której możemy zaniedbać wzajemne przesłanianie się centrów rozpraszania.
W sytuacjach, gdy efekty zderzenia mogą być różne (na przykład w zderzeniu
cząstek elementarnych mogą, ale nie muszą, powstać nowe cząstki) możemy uży-
wać pojęcia przekroju czynnego na określony proces. Definiuje się ono tak samo,
jak całkowity przekrój czynny, z tą różnicą, że uwzględniamy wyłącznie zderzenia,
w wyniku których zaszedł ten właśnie proces. Dla nas interesującym przekrojem
będzie przekrój czynny na pochłonięcie.
Przekrój czynny na pochłonięcie
Używany w fizyce jądrowej, przekrój czynny na to, że padająca cząstka
(np. neutron) zostanie pochłonięta przez jądro tarczy. Przekroje czynne mogą zale-
żeć (i na ogół zależą) od energii cząstki padającej. W tym przypadku naszą cząstką
padającą jest foton. Zatem będziemy mówili tutaj o zdolności (prawdopodobień-
stwie), że nasz układ jest w stanie wchłonąć daną porcję energii. Przyjrzyjmy się
23

na przykład układowi istniejącemu na naszej planecie. W wyższych warstwach at-
mosfery istnieje tzw. tarcza ozonowa. Jak wiadomo ozon czy też tritlen (O3) w at-
mosferze spełnia funkcję filtra pochłaniającego promieniowanie ultrafioletowe (UV ),
które emitowane jest przez Słońce. Pochłanianie to polega na reakcji rozszczepienia
cząsteczki ozonu na tlen i rodnik tlenowy, która jest odwróceniem reakcji syntezy
ozonu. Zatem mówiąc bardziej generalnie, przekrój czynny procesu absorpcji, jest
to zdolność cząsteczki do tego aby pochłonąć energię fotonu o osobliwej długości
fali oraz polaryzacji. Analogicznie jest w przypadku bombardowań neutronami ją-
dra atomu. Aczkolwiek jednostki podane są jako powierzchnia, nie odnoszą się one
do rzeczywistego rozmiaru powierzchni, przynajmniej częściowo, ponieważ objętość
lub też stan docelowej molekuły wpłynie na prawdopodobieństwo absorpcji . Ilo-
ściowo, liczba dN zaabsorbowanych fotonów, pomiędzy odległościami x oraz x+ dx
wzdłuż wiązki promieni jest produktem N fotonów przenikających na głębokość x
pomnożona przez liczbę n pochłaniających molekuł przez jednostkę objętości po-
mnożoną przez przekrój czynny absorpcji σ:
dN
dx= −Nnσ. (1.2.38)
Pojęcie przekroju czynnego jest ściśle powiązane z masowym współczynnikiem osła-
bienia i dla zadanej cząstki i jej przekroju czynnego energii docelowej materii i może
zostać obliczony z wykorzystaniem masowego współczynnika absorpcji z wykorzy-
staniem:dN
dx=µ
ρuma, (1.2.39)
gdzie µρ
– masowy współczynnik osłabienia, ma – masa jest masa molowa w g/mol,
u – masa atomowa (unit) (1/12 masy atomu węgla 12C ) i wynosi ona 1.6605402×10−24 g
Widmo absorpcyjne
Weźmy dla przykładu świat widzialny dla oka ludzkiego. Jest to świat w skali
makroskopowej. Chociaż obowiązujące w nim warunki nie mają prawa bytu w świe-
cie mechaniki kwantowej, to zachodzą w nim zjawiska w swej charakterystyce bardzo
24

podobne do zjawisk znanych nam ze świata mikro. Nasz świat jest w sumie reprezen-
towany przez funkcje falowe, które są odbijane przez te przedmioty. Zatem widzimy,
że wszystko to co nas otacza posiada jakąś barwę. Dla przykładu liście na drzewach
są koloru zielonego. Wynika to z prostej przyczyny, posiadania przez liście barwnika
zwanego chlorofil, który posiada charakterystyczne dla siebie cechy przepuszczania
tylko określonych dla swojej gęstości długości fal. W związku z powyższym istnieje
widmo absorpcji charakterystyczne (jedyne w swoim rodzaju) dla danej materii. Po-
kazuje ono część przypadków promieniowania elektromagnetycznego, pochłoniętych
przez materię na pewnym przedziale częstotliwości. Widmo absorpcji jest w swo-
im znaczeniu przeciwieństwem widma emisyjnego (widmo spektroskopowe). Każdy
pierwiastek chemiczny posiada pozycje absorpcji dla kilkunastu szczególnych długo-
ści falowych odpowiadających różnicą pomiędzy poziomami energetycznymi orbitali
atomowych. Na przykład, przedmiot który pochłania widzialne światło niebieskie,
zielone oraz żółte ukaże się nam w kolorze czerwonym. W związku z powyższym
spektrum światła może być wykorzystywane do identyfikacji pierwiastków obecnych
w gazie lub cieczy. Metoda ta jest wykorzystywana w dedukcji obecności pierwiast-
ków w gwiazdach oraz innych obiektów postaci gazowej, które nie mogą zostać zmie-
rzone w sposób bezpośredni.
Atomy oraz cząsteczki mogą zmieniać swoje stany podczas, gdy pochłoną spe-
cyficzne porcje energii. Stany atomowe są określone przez uporządkowane elektrony
na orbitalach. Elektron znajdujący się na pewnej orbitali może zostać pobudzony
do przejścia na wyższą, bardziej energetyczną orbitalę, poprzez zaabsorbowanie do-
kładnie jednego fotonu, którego energia jest równa różnicy energii pomiędzy tymi
dwoma orbitalami.
Stany molekularne określane są przez rodzaje drgań oraz rotacji cząsteczki. Te ro-
dzaje drgań oraz rotacji są skwantowane, bliźniaczo podobne do orbitali, i mogą być
pobudzone poprzez absorpcję pojedynczego fotonu.
W obydwu przypadkach – atomowym oraz cząsteczkowym, stany pobudzenia
nie są trwałe: po jakimś czasie, atomy oraz molekuły wracają z powrotem do ich
stanu początkowego. W przypadku atomów pobudzony elektron wraca na swoją
niższą powłokę, emitując przy tym foton. W cząsteczkach stany wibracyjne lub ro-
25

tacyjne ulegają rozpadowi, także emitując przy tym foton.
Kiedy następuje taki rozpad, wyprodukowany foton niekoniecznie emitowany jest
w tym samym kierunku co oryginał. Wykazane zostało na podstawie doświadczeń,
że najbardziej powszechnym kątem, w którym zachodzi zjawisko emisji fotonu jest
45 stopni, w stosunku do wektora prędkości fotonu pochłoniętego przez układ. Sytu-
acja taka ma miejsce w przypadku jakiejkolwiek sytuacji, w której gazy znajdują się
pomiędzy źródłem światła a obserwatorem (np. chmury gazów w kosmosie): obser-
wator zobaczy luki w widmie światła odpowiadającego długością falowym fotonów
zaabsorbowanych. Te luki pojawiają się mimo reemisji fotonów, ponieważ są w stanie
równie prawdopodobnie podróżować we wszystkich kierunkach, i statystycznie jest
to nieprawdopodobne, aby mogły one podróżować wzdłuż oryginalnej ścieżki do ob-
serwatora. Luki te występują w postaci czarnych linii na wykresie przedstawiającym
widmo.
Fotodysocjacja na przykładzie wody. Od widma do fotodysocjacji - w za-
leżności od stanu
W przestrzeni kosmicznej fotodysocjacja jest jednym z głównych procesów w wy-
niku, których molekuły ulegają rozpadowi (ale powstają za to nowe cząsteczki).
W związku z tym, iż międzygwiazdowe pole promieniowania znajduje się w próżni,
cząsteczki oraz wolne rodniki mogą istnieć przez bardzo długi czas. Wyniki fotody-
socjacji są bardzo istotne przy poznawaniu składu chmur gwiezdnych. Typowymi
przykładami fotodysocjacji w międzygwiazdowych ośrodkach są:
H2O + hν → H +OH, (1.2.40)
CH4 + hν → CH3 +H. (1.2.41)
Dla nas interesująca jest reakcja 1.2.40. Skoro pod wpływem bombardowania foto-
nami widzialne jest dla oka ludzkiego spektrum emisji, a luki na tymże spektrum
charakteryzują długości fali jakie zostały wchłonięte, w bardzo łatwy sposób można
zmierzyć przekroje czynne dla pochłaniania. Zostały one zmierzone dla większości
stabilnych molekuł [7, 8, 9, 10, 11, 12]. Zastanówmy się zatem jak może wyglądać
26

zajście takiego procesu na schemacie, na przykładzie wody. Wiemy, że spektrum
absorpcji dla wody zostało zmierzone i mieści się ono w przedziale 5 − 11 eV [13].
Natomiast maksima energetyczne to 7, 5 eV (λ ≈ 165 nm – pierwszy poziom absorp-
cji) oraz 9, 7 eV (λ ≈ 128 nm – drugi poziom absorpcji). Można zatem narysować
wykres przekroju czynnego dla absorpcji wody jako funkcję zależną od energii fotonu
1.3.
Zmienne Ã, B oraz C przedstawiają trzy najniższe poziomy absorpcji. Strzał-
ki natomiast wskazują maksima (długości fali), przy których dystrybuanty stanów
rotacyjnych grupy hydroksylowej (produktu OH uzyskanego w wyniku fotodysocja-
cji) zostały zmierzone. Z całkowitego kształtu widma oraz możliwych struktur moż-
na wyciągnąć pewne wnioski odnośnie wpływu dynamiki na dysocjację. Podczas
0
5
10
15
5 6 7 8 9 10 11
σto
t [M
b]
Efoton [eV]
H2O
A
B
C
˜
˜
˜
157nm
122nm
Rysunek 1.3: Całkowity przekrój czynny na absorpcje na przykładzie wody H2O
jako funkcja energii fotonu
gdy dla poziomu à dysocjacja wody zachodzi niemalże natychmiastowo, to już
dla poziomu B nie jest tak ładnie. Dzieje się tak, ponieważ w układzie zaobserwo-
wać można występowanie słabych aczkolwiek mających miejsce drgań w cząsteczce.
Drgania te mają bezpośredni związek z długością życia układu. Jednakże, obszer-
ne tło wskazuje, iż dysocjacja w stanie B także zachodzi praktycznie bezpośrednio.
27

H2O(X 1A1)+−hω
H2O(A1B1) H2S+OH(X2Π,nj)
H2O(B1A1) H2S+OH(A2Σ,n’j’)
˜
˜
˜
˜
˜
9.7 eV
7.5 eV
2. seria
1. seria
fluorescencja
przemiana
nieadiabatyczna
Rysunek 1.4: Schemat przedstawiający reakcje chemiczne zachodzące podczas bom-
bardowania H2O fotonem o odpowiedniej energii
Ostatecznie cząsteczka w stanie C składa się z wyraźnych struktur, które natych-
miastowo mówią nam, że pobudzona cząsteczka wody żyje pod wpływem przynaj-
mniej kilkunastu wewnętrznych wibracji. Jakkolwiek spektrum absorpcji jest silnie
uśrednioną wielkością zawiera szereg informacji dotyczących dynamiki występującej
w cząsteczce. Można zatem przedstawić taki schemat procesów zachodzących pod-
czas pochłaniania pewnych porcji energii przez cząsteczkę wody i wygląda on mniej
więcej tak jak na przedstawionym rysunku 1.4.
1.2.4 Funkcja dystrybuanty przestrzeni fazowej
W matematyce oraz fizyce istnieje pewna przestrzeń, która jest przestrzenią
wszystkich możliwych stanów w jakich może znajdować się badany układ, a każ-
dy stan układu jest jednym punktem tej przestrzeni. Przestrzenią taką nazywamy
przestrzeń fazową. W mechanice klasycznej przestrzeń fazowa zwykle zawiera do-
puszczalne wartości pozycji i prędkości poszczególnych obiektów. Wykres pokazujący
zmiany tych wartości w czasie nazywany jest wykresem fazowym.
Zwykle przestrzeń taka jest wielowymiarowa i każdy stopień swobody układu jest
reprezentowany jako jej osobny wymiar. Kombinacja parametrów układu w danej
chwili odpowiada więc położeniu punktu w tej przestrzeni. Jeśli ewolucja układu
jest w pełni zdeterminowana przez te parametry, można wyznaczyć w przestrzeni
28

trajektorię złożoną z kolejnych stanów w jakich będzie się znajdował układ. Kształty
tych trajektorii pozwalają dokładnie opisywać różne własności układu.
Dla prostych układów, takich jak cząstka poruszająca się w jednym kierunku,
przestrzeń fazowa może mieć mało wymiarów, np. dwa – położenie i prędkość.
W ogólności wymiar przestrzeni fazowej może być bardzo duży. Przykładowo, po-
jemnik z gazem może być opisywany przez przestrzeń, w której każdej molekule
odpowiadają wymiary związane z położeniem i prędkością wzdłuż każdej osi, oscy-
lacjami itp.
W mechanice klasycznej opisuje się układy za pomocą współrzędnych położenia
i prędkości. W takim przypadku trajektorie w przestrzeni fazowej spełniają twier-
dzenie Liouville’a, mówiące że objętość dowolnego regionu przestrzeni nie zmienia
się w trakcie jego ewolucji (o ile nie następują straty energii). Dodatkowo, ponie-
waż każdy punkt przestrzeni fazowej leży na dokładnie jednej trajektorii, trajektorie
nie mogą się przecinać.
Natomiast w mechanice kwantowej, współrzędne w przestrzeni fazowej stają się
operatorami hermitowskimi w przestrzeni Hilberta. Każda obserwabla odpowiada
jakiejś dystrybucji na przestrzeni fazowej.
Przekroje czynne absorpcji oraz fotodysocjacji szacowane są w klasycznym zasię-
gu poprzez zbliżające się roje fotonów o indywidualnych trajektoriach na pobudzone
stany płaszczyzn energii potencjalnych (w zależności od stanu pobudzenia cząsteczka
posiada charakterystyczne dla siebie pole potencjalne). Każdy tor pocisku (fotonu)
ma wpływ na wygląd przekroju czynnego ze szczególną masą P(i)(τ0) która odpowia-
da dystrybucji wszystkich współrzędnych i pędów przed przekształceniem pionowym
z położenia w stanie podstawowym do pobudzonego. P(i)(τ0) powinna być specy-
ficzną w zależności od stanu, funkcją dystrybuanty kwantowej, która odzwierciedla
jak tylko to możliwe, podstawowy stan kwantowy (wskazany przez wykładnik i)
molekuły macierzystej zanim ulegnie ona pobudzeniu. Tak w skrócie przedstawiona
teoria jest mieszanką mechaniki klasycznej oraz kwantowej: cząsteczka macierzysta
w stanie podstawowym jest traktowana w oparciu o teorię mechaniki kwantowej,
podczas gdy dynamika w stanie dysocjacji opisana jest przy użyciu narzędzi mecha-
niki klasycznej.
29

W mechanice kwantowej, współrzędne dystrybucji podane są jako moduł do kwa-
dratu funkcji falowej we współrzędnych przestrzennych - |Ψi(Ei)|2. Jednakże, jak mó-
wi zasada nieoznaczoności Heisenberga, niemożliwym jest określenie jednocześnie
współrzędnych oraz pędu. Zatem możemy jedynie dokładnie poznać wartość pędu
cząsteczki lub jej tor ruchu. Sytuacja ta nie ma miejsca w przypadku mechaniki
klasycznej. Możemy poznać jednakowoż wartość pędu oraz znać tor ruchu. W szcze-
gólnym przypadku, obie wartości – pęd oraz współrzędne – muszą być w chwili
t = 0, aby móc zapoczątkować tor ruchu. Tak więc, mamy problem z opisaniem
funkcji dystrybucji w pojęciu klasycznej przestrzeni fazowej, która jednocześnie ob-
ciąża współrzędne oraz pęd, i która, w tym samym czasie, powinna - najdokładniej
jak to tylko możliwe - odwzorowywać dystrybuantę kwantową.
Problem ten jest szczególnie powszechny we wszystkich teoriach, które są łączo-
ne w oparciu o mechanikę klasyczną oraz kwantową, jednakże ten jest szczególnie
trudny w przypadku fotodysocjacji. Ponieważ ruch w pobudzonej cząsteczce zaczyna
się na krótkich odległościach, gdzie oddziaływania pomiędzy rozmaitymi poziomami
są zwykle najsilniejsze - odpowiednia definicja funkcji dystrybucji P(i)(τ0) jest nie-
zwykle ważna. Drobna zmiana w początkowej funkcji dystrybucji może prowadzić
do znaczących zmian w końcowym stanie dystrybucji. Sytuacja ta różni się od peł-
nych zderzeń, podczas których odczynniki są - na początku trajektorii - całkowicie
odseparowane. Zatem drobne zmiany w warunkach początkowych są prawdopodob-
nie „zapominane” i po upływie czasu nie mają wielkiego wpływu na układ.
Kwantowa definicja funkcji dystrybucji w klasycznej przestrzeni fazowej jest sta-
rym zagadnieniem w fizyce teoretycznej. Bardzo często pojęcie to nazywamy funkcją
dystrybucji Wigner’a [14, 15]. Rozważmy jednowymiarową przestrzeń o współrzęd-
nej R oraz odpowiadającym mu klasycznym pędzie P . Funkcja dystrybucji Wignera
przedstawiona jest wzorem w postaci:
PW (R,P ) = (π~)−1 − 1
∫
dηψ(R+ η)ψ(R− η)e2ηP
~ , (1.2.42)
gdzie ψ(R) jest funkcją falową we współrzędnych sferycznych. Takie założenie speł-
30

nia poniższe warunki:∫
dPPW (R,P ) = |ψ(R)|2, (1.2.43)∫
dR
∫
dPPW (R,P ) = 1, (1.2.44)
które intuicyjnie oczekują na możliwą do przyjęcia funkcje dystrybucji przestrzeni
fazowej. Jednakże, PW (R,P ) niekoniecznie została pozytywnie zdefiniowana, zwłasz-
cza dla pobudzonych stanów wibracyjnych i dlatego też nie może być interpretowana
jako funkcja dystrybucji prawdopodobieństwa.
Dla oscylatora harmonicznego n-ty stan wibracyjny o masie m oraz pulsacji ωOH,
całka w równaniu 1.2.42 może zostać oszacowana analitycznie:
P(n)W (R,P ) = (−1)n(π~n!)−1Ln(ρ2)e−
ρ2
2 , (1.2.45)
gdzie Ln jest n-tym wielomianem Laguerre’a natomiast ρ wyraża się wzorem:
ρ2 =4
~ωHO
[
P 2
2m+
1
2mω2
HO(R −Re)2
]
. (1.2.46)
Dla początkowego stanu wibracyjnego (n = 0). Zatem równanie 1.2.45 skraca się
do postaci:
P(0)W (R,P ) = (π~)−1e
−2α(R−Re)2
~ e−ρ2
2α~ , (1.2.47)
z nową zmienną α zdefiniowaną wzorem:
α =mωHO
2. (1.2.48)
Wobec tego rozwiązaniem funkcji dystrybucji Wignera dla stanu początkowego oscy-
latora harmonicznego będą dwie funkcje Gauss’a – zwane gaussianami. Jeden scen-
tralizowany w przestrzeni R w odległości Re, natomiast drugi zlokalizowany w prze-
strzeni P przy P = 0.
1.2.5 Klasyczny przekrój czynny absorpcji oraz fotodysocja-
cji
Możemy zdefiniować całkowity przekrój absorpcji w ujęciu klasycznym, zacho-
wując niepotrzebną normalizację stałej, jako:
σ(i)tot(ω) ∝ Efoton
∫
dτ0P(i)W (τ0)[µ
(e)fi ]2δ[Hf(τ0) − Ef ], (1.2.49)
31

gdzie P (i)W (τ0) – funkcja dystrybucji dla podstawowego stanu elektronowych (zależna
od współczynnika i), Hf – funkcja Hamiltona (hamiltonian) na wyższych stanach
elektronowych (zależne od współczynnika f), µ(e)fi – składnik funkcji przekształceń
dipolowych w kierunku wektora pola elektrycznego, Ef = Ei + Efoton – energia
rezonansu na wyższym stanie.
(a)
Ei
V(R)
Re
(b)
Ei
V(R)
Re
Rysunek 1.5: Ilustracja przedstawiająca pojęcie przekroju czynnego w ujęciu me-
chaniki a-klasycznej i b-kwantowej we współrzędnych funkcji dystrybucji
Integracja rozciąga się na całym obszarze przestrzeni fazowej ze zmienną dτ0 jako
odpowiednia składowa objętości. Funkcja delta σ[Hf − Ef ] wybiera tylko
te punkty (odpowiadające torowi ruchu) w wielowymiarowej przestrzeni fazowej,
które posiadają odpowiednią energię Ef . To zapewnia, że stan rezonansu kwanto-
wego Ef = Ei + Efoton jest spełniony.
Zatem formalna definicja obejmuje następujące wnioski:
1. Absorpcja fotonu ma miejsce tylko jeżeli energia w stanie pobudzonym,Hf(τ0),
wynosi dokładnie Ef = Ei + Efoton.
2. Prawdopodobieństwo absorpcji fotonu w szczególnym punkcie przestrzeni fa-
zowej jest proporcjonalne do [µ(e)fi ]2.
3. Każdy punkt przestrzeni fazowej posiada ciężar P (i)W (τ0), który odzwierciedla
szczególny stan kwantowy molekuły macierzystej w podstawowym stanie ener-
getycznym.
32

1.2.6 Fotodysocjacja bezpośrednia - reguła odbicia
Można zatem podzielić fotodysocjację jako proces zachodzący w sposób bezpo-
średni lub pośredni. Bezpośredni proces dysocjacji cząsteczki macierzystej dysocjuje
natychmiastowo po tym jak foton przenosi ją na wyższy poziom energetyczny. Żadna
bariera ani ograniczenia dynamiki nie utrudniają fragmentacji a „czas życia” pobu-
dzonego związku jest bardzo krótki, mniejszy niż okres wibracji wewnątrz związku.
Dla porównania, okres wewnętrznych wibracji mieści się w przedziale 30 do 50 fs.
Przykład fotodysocjacji wody w stanie à jest typowym przykładem bezpośredniej
dysocjacji. Tor ruchu lub wystrzelona kwantowa paczka falowa w cząsteczkę wody
w stanie tym prowadzi niemalże natychmiastowo do dysocjacji i otrzymania produk-
tów H oraz OH .
Z drugiej strony, w przypadku pośredniej fotofragmentacji, jakaś bariera poten-
cjału lub siła dynamiczna utrudnia bezpośrednią fragmentację pobudzonego związku
a czas życia składa się przynajmniej z kilkunastu wewnętrznych okresów wibracji.
Bardzo dobrym przykładem będzie tutaj fotoliza wody w stanie C. Zanim ulegnie
ona rozpadowi wykona najpierw kilkanaście drgań na płyciźnie studni potencjałów,
aż w końcu zostanie dostarczona dostateczna ilość energii z wiązania wibracyjnego
do poziomu dysocjacji, która jest konieczna aby przedostać się przez małą barierę.
Bezpośrednia dysocjacja jest zasadniczo klasycznym przykładem procesu, pod-
czas gdy pośrednia dysocjacja wymaga całkowitego opisu w oparciu o mechanikę
kwantową.
Z powodu bardzo krótkiego „czasu życia” w bezpośrednim procesie dysocjacji
energia zależna jest od spektrum absorpcji, tak samo jak ostateczny stan dystrybu-
ant produktów jest bezpośrednio powiązany z początkową dystrybuantą współrzęd-
nych molekuły macierzystej w podstawowym stanie. Zasadę tą nazywamy regułą
odbicia.
Jednowymiarowa reguła odbicia
Rozważmy dysocjację cząsteczki dwuatomowej o odległości między jądrowej R,
momencie pędu P oraz zredukowanej masie m. Funkcja Hamiltona dla takiego przy-
33

kładu w stanie dysocjacji będzie wyglądała następująco:
H(R,P ) =P 2
2m+ V (R). (1.2.50)
Zakładamy, że potencjał w stanie spoczynkowym jest harmoniczny, a macierzysta
cząsteczka znajduje się w najniższym stanie wibracyjnym. Pomimo prostoty takiego
układu, powinniśmy opisać jednowymiarową regułę odbicia w detalach, ponieważ
zasadę tę można w łatwy sposób rozwinąć potem do więcej niż tylko jednego wymiaru
na tej samej zasadzie.
Zgodnie z równaniami 1.2.47 oraz 1.2.49 klasyczna aproksymacja przekroju czyn-
nego absorpcji, jako funkcji rozkładu energii w stanie pobudzenia, podana jest wzo-
rem:
σ(E) ∝∫
dP
∫
dReαR(R−Re)2
~ e− P2
2αR~σ[H − E], (1.2.51)
gdzie założyliśmy że współrzędnie niezależne przekształcenie funkcji dipolowej
oraz wszystkie czynniki, zawierające energię fotonu, zostaną pominięte. Wykładni-
czy parametr jest bliskim związku z pulsacją ωHO oscylatora harmonicznego w stanie
podstawowym wyrażonym przez równanie:
αR =mωHO
2, (1.2.52)
a Re jest odległością, w której molekuła macierzysta znajduje się w stanie równowagi.
Korzystając z własności funkcji delty Dirac’a:
σ[f(y)] = Σβdf
dy
−1
y=βy
δ[y − βy] (1.2.53)
oraz wykorzystując fakt,że P przyjmuje głównie wartość równą 0, co wpływa na wy-
gląd całki, i redukuje się ona do postaci:
σ(E) ≈ e−2αR(Rt−Re)2
~
dV
dR
−1
R=Rt(E)′, (1.2.54)
gdzie Rt(E, P ) ustawia się w klasycznym punkcie zwrotnym zdefiniowanym przez:
H(Rt, P ) = E. (1.2.55)
Aproksymując potencjał do postaci:
V (R) ≈ Ve − VR(R− Re), (1.2.56)
34

R
E
V(R)
Re
|Ψi|2
∆R
(Re,Ve)
Rysunek 1.6: Ilustracja przedstawiająca jednowymiarową zasadę odbicia - przed od-
biciem
z potencjałami Ve = V (Re) oraz VR = −dVdR R=Re
, przekrój czynny przyjmuje postać:
σ(E) ≈ e−2β(E−Ve)2
~
VR
, (1.2.57)
gdzie β =(
V 2R
αR
)−1
. Klasyczne spektrum absorpcji na granicy potencjału liniowego
jest gaussowską funkcją energii mającą swój punkt centralny w Ve = V (Re).
Zagadnienie jednowymiarowej reguły odbicia można w bardzo łatwy sposób wy-
jaśnić na przedstawionych schematach 1.6 oraz 1.7. Na schemacie 1.6 zaobserwować
można potencjał stanu podstawowego dla cząsteczki |Ψi|2 oraz studnię potencjałów
tejże cząsteczki. Nad krzywą potencjału stanu podstawowego znajduje się krzywa
stanu wzbudzonego oraz styczna do niej prosta w punkcie (Re, Ve). Punkt ten jest
miejscem, w którym energia wzbudzenia jest na tyle odpowiednia aby pobudzo-
na cząsteczka uległa procesowi fotodysocjacji. A ponieważ Ve = V (Re) jest liczbą,
35

σ(E)
E
V e ∆E (Re,Ve)
Rysunek 1.7: Ilustracja przedstawiająca jednowymiarową zasadę odbicia - po odbiciu
którą znamy, możemy w bardzo łatwy sposób obliczyć potencjał VR = −dVdR R=Re
z definicji pochodnej (co z resztą wykazane zostanie doświadczalnie w podrozdziale
2.2). Następnie rzut przejścia ze stanu podstawowego do stanu wzbudzonego zostaje
odbity przy przejściu przez prostą styczną w punkcie (Re, Ve) co widać na rysunku
1.7. I tak oto z rzutu otrzymujemy krzywą przedstawiającą przekrój czynny na ab-
sorpcję, która jest w postaci funkcji gaussowskiej (gaussian).
1.3 Reguły wyboru
1.3.1 Spektrometria – rodzaje widm
XIX wiek był bardzo owocnym okresem pracy i osiągnięć dla wielu fizyków.
W tamtym czasie wiele problemów znalazło swoje rozwiązanie, ale też wiele z nich
przyczyniło się do jakże dzisiaj powszechnej a zarazem bardzo różnej od fizyki kla-
36

sycznej, nauki zwanej mechaniką kwantowa. To właśnie w tamtym okresie, w roku
1859 niemiecki uczony Gustav Robert Kirchhoff wraz ze swoim kolegą Robertem
Bunsenem odkryli, że pierwiastek w stanie lotnym, w określonych warunkach pobu-
dzony do świecenia, daje tylko sobie właściwe widmo liniowe. W roku 1861 podczas
badania składu chemicznego Słońca na podstawie sygnatury spektralnej odkryli dwa
nowe pierwiastki – cez oraz rubid.
Można zatem pokusić się o stwierdzenie, że widmo atomu jest jego „liniami pa-
pilarnymi”, które pozwalają na jego bezbłędną identyfikacje i klasyfikację. Każdy
pierwiastek daje pewną, większą albo mniejszą liczbę linii widmowych, w określo-
nych barwach. Tak na przykład, wodór atomowy pobudzony do świecenia daje cztery
linie widmowe widzialne: czerwoną, niebieską i dwie fioletowe. Niektóre pierwiast-
ki dotychczas nieznane zostały odkryte właśnie dzięki metodzie analizy widmowej.
Historia taka miała miejsce w przypadku odkrycia helu. Najpierw zaobserwowano
linie widmowe w widmie Słońca, a dopiero później znaleziono ów pierwiastek na Zie-
mi. Tą metodą odkryto też prawie wszystkie gazy szlachetne, a także gal, ind i tal,
których istnienie jest trudne do stwierdzenia na drodze chemicznej.
Taki złożony charakter widm cząsteczkowych wynika z faktu istnienia bardzo
wielu stopni swobody już w pojedynczej cząsteczce. Dla nas bardzo pomocnym bę-
dzie użycie już wcześniej użytych przez nas pojęć dotyczących energii. Jak wiadomo
cząsteczka w stanie podstawowym posiada już jakąś energię, a ponieważ cząsteczka
składa się z mniejszych elementów, dla których także istnieje jakaś charakterystyczna
energia. Takie przybliżenie pozwala przedstawić energię naszego układu jako sumę
energii ruchu elektronów Eel, energii drgań jąder Eosc oraz energii rotacji cząsteczki
Erot:
E = Eel + Eosc + Erot. (1.3.1)
Odległości pomiędzy sąsiadującymi ze sobą poziomami rotacyjnymi są wielokrotnie
mniejsze niż odległości w porównaniu do poziomów oscylacyjnych, te natomiast są
wielokrotnie mniejsze od odległości stanów elektronowych, możemy zatem podzielić
widma elektronowe w zależności od odległości. Wyróżniamy następujące rodzaje
widm:
37

1. Rotacyjne – związane są jedynie ze zmianą ruchu obrotowego cząsteczki, miesz-
czą się w obszarze centymetrowych długości fali 0.1−10 cm – zakres mikrofali.
2. Oscylacyjno-Rotacyjne – inaczej rotacyjne, odpowiadają za jednoczesną zmia-
nę stanu drgań oraz rotacji cząsteczki, mieszczą się w obszarze 1 do 100 µm –
zakres podczerwieni.
3. Elektronowo-Oscylacyjno-Rotacyjne (widma elektronowe) – związane są
ze zmianą stanu chmury elektronowej, której towarzyszy też zmiana oscylacji
i rotacji, mieszczą się w obszarze 100 nm do 1 µm – zakres obszaru widzialnego
oraz nadfioletu.
Niezależnie od rodzaju obserwowanego widma, oddziaływanie pola elektromagne-
tycznego z cząsteczką polega w pierwszym przybliżeniu na jego oddziaływaniu z elek-
trycznym momentem dipolowym cząsteczki:
µ = −Σieri + ΣNeZNRN , (1.3.2)
gdzie µel i µj są to przyczynki elektronów i jąder do całkowitego momentu dipolo-
wego. A ponieważ prawdopodobieństwo przejścia między dwoma stanami cząsteczki
w mechanice kwantowej, jest proporcjonalne do kwadratu elementu macierzowego
momentu dipolowego, który jest traktowany niejako operator kwantowy:
P ∼ |〈ψ′|µ|ψ′′〉|2. (1.3.3)
Zgodnie z przytoczonym w rozdziale 1 przybliżeniem Borna-Oppenheimera, możemy
przedstawić funkcje falowe obydwu stanów w postaci iloczynów funkcji elektronowej,
oscylacyjnej oraz rotacyjnej w postaci całki:
〈ψ′|µ|ψ′′〉 =
∫
ψ′∗elχ
′∗oscχ
′∗rot(µel + µj)ψ
′′
elχ′′
oscχ′′
rotdτeldτj , (1.3.4)
gdzie dτel oraz dτj są reprezentantami objętości w przestrzeni współrzędnych elek-
tronowych oraz jądrowych, a ponieważ moment dipolowy nie jest zależny od współ-
rzędnych elektronów, wzór 1.3.4 można sprowadzić do postaci:
〈ψ′ |µ|ψ′′〉 =
∫
[
∫
ψ′∗elµelψ
′′
eldτel]χ′∗oscχ
′∗rotχ
′′
oscχ′′
rotdτj
+
∫
[
∫
ψ′∗elψ
′′
eldτel]χ′∗oscχ
′∗rotµjχ
′′
oscχ′′
rotdτj. (1.3.5)
38

Dla uproszczenia zapiszmy I = I1 + I2.
W zależności od rodzaju widma cząsteczkowego spośród wyżej wymienionych, po-
wyższe równanie ulega znaczącemu uproszeniu. Dla przykładu mamy do czynienia
z widmem elektronowym (przejście elektronów z orbitali niższej na wyższą) to w rów-
naniu I2:∫
ψ′∗elψ
′′
eldτel = 0, (1.3.6)
ponieważ funkcje falowe różnych stanów elektronowych są wzajemnie ortogonal-
ne. Zatem I2 = 0, więc o prawdopodobieństwie przejścia decyduje tylko I1. Jeżeli
natomiast będziemy mieli do czynienia z widmem oscylacyjnym bądź rotacyjnym
(przejście elektronów w ramach tego samego stanu), to wtedy w równaniu I1:
∫
ψ′∗elµelψ
′′
eldτel = 0, (1.3.7)
dlatego że jest to całka z wyrażenia nieparzystego względem współrzędnych elek-
tronowych. Zatem tym razem I1 = 0 a o prawdopodobieństwie przejścia decyduje
jedynie I2. Zastanówmy się więc nad analizą poszczególnych widm.
1.3.2 Widmo rotacyjne
Jako, że w powyższej pracy dyplomowej, zajmujemy się cząsteczką litu dwuato-
mowego, to do naszych badań posłuży nam najprostszy model takiego widma – dwu-
atomowa cząsteczka. Traktujemy ją jako sztywny rotator (analogicznie postępuje się
w przypadku liniowo połączonych ze sobą cząsteczek – łańcuchem). Funkcje falowe
rotacji takiej cząsteczki tzw. harmoniki sferyczne (można je otrzymać w wyniku
rozdzielenia zmiennych dla równania Schrödingera w przypadku trójwymiarowym)
YJM(Θ,Φ), natomiast poziomy energetyczne wyrażone są wzorem:
EJrot =
~J(J + 1)
2µR2=
~J(J + 1)
2I. (1.3.8)
A ponieważ układ atom – atom jest symetryczny, wektor jądrowej części dipolowego
momentu µj skierowany jest wzdłuż osi układu wirując wraz z nim. Biorąc pod uwa-
gę, że badany układ jest sztywny, więc moment dipolowy zawsze jest prostopadły
39

do osi obrotu – jeżeli natomiast wektor ten zmieni swoje położenie podczas obro-
tu to zmiana taka wiąże się z absorpcją lub emisją w układzie. Współrzędne tego
wektora można zatem zapisać w postaci następujących równań (dla układu XY Z):
µjX = µ0 sin Θ cosΦ,
µjY = µ0 sin Θ sin Φ, (1.3.9)
µjZ = µ0 cos Φ,
gdzie kąty Φ oraz Θ opisują orientację przestrzenną cząsteczki. W związku, iż w po-
wyższym przykładzie nie mamy do czynienia ze zmianami stanów elektronowych
oraz oscylacyjnych (nasze funkcje falowe dla tych stanów przed i po są identyczne),
możemy więc zapisać ψ′
el ≡ ψ′′
el oraz χ′
osc ≡ χ′′
osc. Zatem nasz element macierzowy
przybierze nową postać:
〈ψ′ |µ|ψ′′〉 = 1 × 1 × µ0
∫
YJ ′M ′
sin Θ cosΦ
sin Θ sin Φ
cos Φ
YJ ′′M ′′
∫
sin ΘdΘdΦ. (1.3.10)
Jak widać pojawiły się we wzorze, po prawej stronie, dwie jedynki. A ponieważ
jak wcześniej wspomniane funkcje falowe stanów elektronowych i oscylacyjnych są
identyczne, są więc one efektem całkowania tych znormalizowanych funkcji. Z wła-
sności harmonik sferycznych wynika, że powyższe całki są różne od zera tylko dla:
∆J ≡ J′ − J
′′
= ±1,
∆M ≡M′ −M
′′
= 0,±1. (1.3.11)
Oczywistym jest fakt, że dla zwykłych widm absorpcyjnych i emisyjnych reguła wy-
boru dla ∆M nie ma wpływu, ponieważ energie poziomów rotacyjnych są niezależne
od liczby kwantowej M . Można zatem zauważyć istotne podobieństwo między całką
1.3.10 a przejściami elektryczno dipolowymi w modelu atomu wodoropodobnego. Za-
tem analogicznie rzecz mówiąc przejścia takie maja miejsce tylko dla sąsiadujących
poziomów, dla których spełniona jest zależność:
∆EJ ≡ E(J + 1) −E(J) = 2B(J + 1). (1.3.12)
40

01
2
3
J = 4Erot
ω2B 4B 6B 8B
Rysunek 1.8: Schemat rotacyjnego widma absorpcyjnego cząsteczki dwuatomowej.
Linie przerywane oznaczają rzeczywiste położenie linii widmowych po uwzgl. siły
odśrodkowej działającej na wirującą cząsteczkę
Jak widać na przedstawionym schemacie 1.8 widmo takiej cząsteczki składa się
z równoległych linii oddalonych od siebie o 2B. Aczkolwiek sytuacja ta jest w po-
staci bardzo przybliżonej, z racji posługiwania się w obliczeniach bardzo prostym
wyrażeniem na energię:
EJrot =
~J(J + 1)
2I,
(co wynika z równania kwantowego oscylatora harmonicznego - harmoniki sferycz-
ne). Wprowadzenie do naszych obliczeń dokładniejszego wzoru na tę energię impli-
kuje na zmianę w bardzo niewielkim wymiarze odległości między liniami.
Jak widać z równania 1.3.10 dodatkowym elementem gwarantującym istnienie
naszego widma dla danej cząsteczki jest istnienie trwałego jądrowego momentu di-
polowego µ0. Ma to poważny wpływ na nasze dalsze obliczenia, ponieważ ogranicza
możliwość przejść elektrycznych dipolowych między poziomami rotacyjnymi, z racji
że dla ogólnej większości cząsteczek o wysokiej symetrii moment dipolowy jest równy
zeru. Oczywiście mowa tutaj o trwałym momencie dipolowym cząsteczki, a nie za-
41

istniałym w wyniku czynników z zewnątrz. Zatem moment dipolowy dla danej czą-
steczki musi być nie zmienny wobec wszystkich przekształceń istniejących dla danej
przez nią grupy symetrii. W związku z powyższym cząsteczki, które cechuje syme-
tria inwersji nie mogą posiadać momentu dipolowego – dotyczy to między innymi
dla nas szczególnie interesujących cząsteczek homojądrowych jak Li2, ale i także
symetrycznych cząsteczek liniowych jak CO2. Momentu dipolowego nie posiadają
również cząsteczki typu bąka sferycznego – CH4. Posiadanie trwałego momentu di-
polowego przez cząsteczkę można wykazać tylko jeśli jedna z jej funkcji x,y bądź
z podlega przekształceniom zgodnie z reprezentacją jednostkową jej grupy syme-
trii. Wyprowadzenie dokładnych reguł wyboru dla takich cząsteczek jest możliwe.
Jednak zajmowanie się tym zagadnieniem ze względu na zakres pracy i złożoność
wyprowadzenia dowodu jest nieco ograniczone.
Zatem jak widać dwuatomowe cząsteczki homojądrowe nie posiadają widma
czysto rotacyjnego. I tak jak w przypadku dwuatomowych cząsteczek heterojądro-
wych, jądra posiadają spiny połówkowe i stosują się do statystyki Fermiego-Diraca,
co w ogólności oznacza, że funkcja falowa dla takiej cząsteczki musi być antysyme-
tryczna przy zamianie miejscami takich identycznych jąder, to dla dwuatomowej
cząsteczki homojądrowej (jądra posiadają parzystą liczbę masową) występują spiny
całkowite i podlegają statystyce Bosego-Einsteina. Dla takich cząsteczek odpowied-
nia funkcja falowa powinna być symetryczna przy ich permutacji.
1.3.3 Widmo oscylacyjne
Zmiana poziomu oscylacyjnego
Podczas gdy nasza cząsteczka drga, zaobserwować można zmiany zachodzące
w rozłożeniu ładunku elektrycznego. Zatem i również przypuszczać można o istnie-
niu zmian periodycznych zachodzących dla momentu dipolowego cząsteczki. Według
praw rządzących w świecie elektrodynamiki klasycznej fakt ten prowadzi do absorp-
cji lub emisji promieniowania. A ponieważ cały czas interesuje nas to zjawisko tylko
dla cząsteczki dwuatomowej, to zajmiemy się jedynie omawianiem problemu na jej
przykładzie, drgającej ruchem harmonicznym. W świetle fizyki oznacza to jedynie,
42

że funkcje falowe dla takiego oscylatora (patrz kwantowy oscylator harmoniczny
w rozdziale pierwszym) χνosc(R) wyrażone są za pomocą wielomianów Hermite’a,
energie własne zaś wyrażone są wzorem 1.2.30. O możliwości przejścia elektrycz-
no dipolowego między poziomami oscylacyjnymi ν′
oraz ν′′
(w ramach konkretnego
stanu elektronowego) decyduje całka I2 w wyrażeniu 1.3.5:
〈ψ′|µ|ψ′′〉 =
∫
χν′∗oscχ
′∗rotµjχ
ν′′
oscχ′′
rotdτjdRdΩ. (1.3.13)
Natomiast symetria dla cząsteczki dwuatomowej wymaga, żeby moment dipolowy
był zawsze skierowany wzdłuż jej osi, występuje tylko jedna składowa µz – pozostałe
wynoszą 0 – a jej wartość jest funkcją odległości międzyjądrowej R. Podczas wystę-
pujących drgań, wychylenia jąder od położenia równowagi są w czasie ich trwania
bardzo niewielkie. Można zatem zapisać przybliżoną postać momentu dipolowego:
µz = µz(Re) +
(
dµz
dR
)
e
(R−Re) = µ0 +
(
dµz
dR
)
e
(R−Re), (1.3.14)
przy czym moment dipolowy µz w położeniu równowagi jąder został utożsamiony
z trwałym momentem jądrowym µ0 . Gdy przeprowadzimy podstawienie równań
1.3.14 → 1.3.13, otrzymamy:
〈ψ′|µ|ψ′′〉 =
∫
∫
χν′∗osc
[
µ0 +
(
dµz
dR
)
e
(R− Re)
]
χν′′
oscdR
χ′∗rotχ
′′
rotdΩ. (1.3.15)
W drugiej całce równania 1.3.15 wyrażenie µ0
∫ ∫
χν′∗oscχ
ν′′
oscdR równe jest zeru wte-
dy gdy ν ′ 6= ν ′′ z wynikającej ortogonalności w oscylacyjnych funkcjach falowych.
Zatem prawdopodobieństwo przejścia w stanach oscylacyjnych jest niezależne
od trwałego momentu dipolowego cząsteczki, aczkolwiek decydującym jest dla czy-
stego widma rotacyjnego. Natomiast drugi element w całce 1.3.15:
Mz =
(
dµz
dR
)
e
∫
χν′∗osc(R− Re)χ
ν′′
oscdR, (1.3.16)
nazywamy oscylacyjnym momentem przejścia. Przejście zatem między stanami ν ′
a ν ′′ jest możliwe tylko w sytuacji gdy podczas drgania cząsteczki zachodzi również
zmiana jej momentu dipolowego, a całka∫
χν′∗osc(R − Re)]χ
ν′′
oscdR jest różna od ze-
ra. Ma to wielkie dla nas znaczenie, ponieważ pierwszy z tych warunków wyklucza
43

istnienie widm oscylacyjnych dla badanych przez nas dwuatomowych cząsteczek
homojądrowych. A ponieważ cząsteczki takie posiadają środek symetrii (symetria
inwersyjna), nie mogą posiadać trwałego momentu dipolowego µ0. Środek ten także
jest zachowawczy podczas występujących drgań oscylacyjnych, gdyż przemieszcze-
nia jąder i występują w przeciwnych kierunkach. Dla układu oznacza to tylko tyle,
że przez cały okres drgań moment dipolowy jest równy zeru, zatem i jego pochodna(
dµz
dR
)
e= 0. W przypadku drugiego elementu składowego dla naszej całki w równa-
niu (2.14), który określiliśmy jako oscylacyjny moment przejścia w równaniu (2.15),
istnieje dla funkcji oscylatora harmonicznego wtedy i tylko wtedy, gdy ν ′ i ν ′′ różnią
się o jedność, a przejścia między stanami oscylatora zachodzą tylko dla:
∆ν = ν ′ − ν ′′ = ±1, (1.3.17)
przy czym:
∆Eν = E(ν − 1) −E(ν) = ωe. (1.3.18)
Z powyższych równań wynika więc równość pomiędzy częstością absorbowanego pro-
mieniowania a klasycznymi drganiami cząsteczki. Natomiast przejście ν ′′ = 0 →ν ′ = 1 nazywane jest przejściem podstawowym cząsteczki, a natężenie uzależnione
jest od momentu dipolowego podczas oscylacji. A ponieważ obsadzenia poziomów
oscylacyjnych określone są przez rozkład Boltzmana:
Nν ∼ eωe(ν+ 1
2 )
kT , (1.3.19)
(w przeciwieństwie do poziomów rotacyjnych, poziomy oscylacyjne dla cząsteczki
dwuatomowej nie są zdegenerowane), więc zauważalnym jest że to właśnie przejście
podstawowe dominuje w widmie absorpcyjnym. Przykładowo dla cząsteczki tlen-
ku węgla CO stosunek obsadzeń tych poziomów ν ′′ = 0, ν ′ = 1 w temperaturze
pokojowej wynosi jak 33000 do 1 [17]. Oczywiście wszystkie te obliczenia zostały
wykonane w oparciu o założenia drgań ściśle harmonicznych, a także wynikającej
z liniowej zależności pomiędzy odległością miedzyjądrową a momentem dipolowym.
Aczkolwiek w rzeczywistości istnieją odstępstwa od założonych przez nas warunków,
gdzie występują przejścia z ∆ν = ±2,±3, . . . to jednak ich natężenie szybko maleje
i nie mają dla nas wielkiego znaczenia.
44

Przejścia oscylacyjno-rotacyjne
Dotychczas widmo oscylacyjne opisane zostało bez ingerencji widma rotacyjnego
– tak jakby podczas zmiany stanu zmieniała się tylko energia drgań bez uwzględ-
nienia energii rotacyjnej. W rzeczywistości jest jednak inaczej. Podczas gdy naj-
niższe stany oscylacyjne można tylko traktować jako najniższe poziomy (J = 0)
dla stanów rotacyjnych z danym stanem drgań jąder, a dla każdego z tych pozio-
mów istnieją dwie liczby kwantowe ν oraz J , to dla każdego przejścia kwantowego
towarzyszy zmiana obydwu tych liczb. Oczywiście istnieją takie przejścia, dla któ-
rych dobrze przybliżone reguły wyboru ν oraz J są nawzajem niezależne. Jednakże
jak w przypadku przejść czysto rotacyjnych, prawdopodobieństwo przejścia było
określone za pomocą trwałego jądrowego momentu dipolowego µ0, tak w tym przy-
padku mamy do czynienia z oscylacyjnym momentem dipolowym M 1.3.16. Innymi
słowy mamy tutaj do czynienia ze zmianami naszego momentu dipolowego, podczas
gdy układ podlega oscylacji.
Jednakowoż celem naszych badań jest w dalszym ciągu dwuatomowa cząsteczka,
zatem w ogólności będziemy brali pod uwagę tylko jedną składową oscylacyjnego
momentu dipolowego Mz , wzdłuż jej osi. A ponieważ badana przez nas dwuatomo-
wa cząsteczka litu jest cząsteczką homojądrową, posiada zatem bardzo duży stopień
symetrii co w efekcie powoduje, że nie posiada ona trwałego momentu dipolowe-
go. W związku z powyższym oscylacyjny moment przejścia dla takiej cząsteczki
Mz = 0. Jedynie dla dwuatomowych cząsteczek heterojądrowych moment ten jest
różny od zera i tylko takie cząsteczki mają widmo oscylacyjne.
1.3.4 Przejścia elektronowe
Jak zostało przedstawione we wstępie tego rozdziału, za zmiany stanu elektro-
nowego cząsteczki, za natężenie przejścia elektrycznego dipolowego odpowiedzialna
jest całka I2 we wzorze 1.3.5. A ponieważ w przypadku przejść pomiędzy różnymi
stanami elektronowymi zachodzi zależność 1.3.6, to nasze równanie możemy skrócić
45

do następującej postaci:
〈ψ′|µ|ψ′′〉 =
∫[∫
ψ′∗elψ
′′
eldτel
]
χ′∗oscχ
′∗rotµjχ
′′
oscχ′′
rotdτj . (1.3.20)
Oczywiście interesuje nas zachodzenie takiego zjawiska tylko dla cząsteczki dwuato-
mowej, zatem i tym razem przeprowadzimy właśnie w tym kierunku naszą analizę.
Jak wiadomo elektronowe funkcje falowe zależą parametrycznie od odległości mię-
dzyjądrowej R. Całkując równanie 1.3.20 po współrzędnych elektronowych otrzymu-
jemy funkcję zależną od R, którą analogicznie tak jak w przypadku rozpatrywanego
wcześniej widma oscylacyjnego, będziemy nazywali elektronowym momentem przej-
ścia:
Mel(R) =
∫
ψ′∗el(r, R)µelψ
′′
el(r, R)dτel. (1.3.21)
Aczkolwiek bardzo często elektronowy moment przejścia ulega bardzo niewielkim
zmianom, które są nieznaczne w przedziale wartości R, dla których oscylacyjne
funkcje falowe przyjmują wartości istotnie różne od zera. Dlatego też, możemy za-
pisać w przybliżeniu następujące równanie:
〈ψ′|µ|ψ′′〉 =
∫
χ∗ν′χν′′dR
∫
〈Mel〉χ′∗rotχ
′′
rotdτj, (1.3.22)
gdzie 〈Mel〉 jest wartością średnią elektronowego momentu przejścia. Zatem
prawdopodobieństwo przejścia elektronowego jest proporcjonalne do wyrażenia:
P ∼∣
∣
∣
∣
∫
χ∗ν′χν′′dR
∣
∣
∣
∣
2
, (1.3.23)
które nazywamy współczynnikiem Francka-Condona dla pasma (słowo „pasmo” uży-
wane jest tutaj z racji występowania wielu linii rotacyjnych wchodzących w skład
stanu) ν ′′ → ν ′ dla konkretnego przejścia elektronowego. A ponieważ w sytuacji
takiej mamy do czynienia z dwoma różnymi poziomami oscylacyjnymi ν ′ oraz ν ′′,
dla których są całkowicie różne stany elektronowe, to drgania odbywają się w dwóch
różnych studniach potencjału. Natomiast oscylacyjne funkcje falowe dla tych drgań
nie są wzajemnie ortonormalne. Z powyższych ustaleń wynika zatem, iż współczyn-
niki Francka-Condona nie mają żadnego wpływu na reguły wyboru wobec zmiany
oscylacyjnej liczby kwantowej ν i mogą przybrać każdą wartość ≤ 1. Muszą one
46

natomiast spełniać regułę sum, z której wynika, że prawdopodobieństwo przejścia
dla przejścia z poziomu ν ′′ do jakiegokolwiek poziomu ν ′:
Σν′
∣
∣
∣
∣
∫
χ∗ν′χν′′dR
∣
∣
∣
∣
2
= 1. (1.3.24)
Oznacza to tylko tyle, że zdarzenie takie jest pewne. W szczególności możliwe jest
także przejście powyżej granicy fotodysocjacji stanu elektronowego, które nie jest
skwantowane. Aczkolwiek przejścia takie są możliwe to jednak równie mało
prawdopodobne i można je zaniedbać.
Tabela 1.1: Charakterystyka wybranych reprezentacji dla dwuatomowej cząsteczki
homojądrowej
D∞h E 2Cφ∞ . . . ∞σv i 2Sφ
∞ . . . ∞C2
σ+g 1 1 . . . 1 1 1 . . . 1
σ−g 1 1 . . . −1 1 1 . . . −1 Rz
σ+u 1 1 . . . 1 −1 −1 . . . −1 z
σ−u 1 1 . . . −1 −1 −1 . . . 1
πg 2 2 cosφ . . . 0 2 −2 cosφ . . . 0 (Rz, Ry)
πu 2 2 cosφ . . . 0 −2 2 cosφ . . . 0 (x, y)
δg 2 2 cos 2φ . . . 0 2 2 cos 2φ . . . 0
δu 2 2 cos 2φ . . . 0 −2 −2 cos 2φ . . . 0
. . . . . . . . . . . . . . . . . . . . . . . . . . .
Przyjrzyjmy się teraz regułom wyboru dla elektronowego momentu przejścia
1.3.21. Żeby zawarty w tym równaniu element macierzowy spełniał założenie µel 6= 0,
prosty iloczyn reprezentacji Γ(ψ′
el) × Γ(µel) × Γ(ψ′′
el) musi zawierać reprezentację
jednostkową. A ponieważ nasza cząsteczka jest homojądrowa, reprezentacja ta musi
być opisana przez grupę symetrii D∞h, jest to reprezentacja σ+g . Nieskończona grupa
D∞h, jest szczególną grupą symetrii. Posiada ona nieskończenie wiele reprezentacji
nieprzewidywalnych, z czego cztery są jednowymiarowe a pozostałe dwuwymiaro-
we. Oznacza to tylko tyle, że poziomy energetyczne mogą być jedynie dwukrotnie
47

zdegenerowane. Na powyższej tabeli 1.1 przedstawione zostały wybrane reprezen-
tacje. Gdyż składowe (µel)x,y oraz (µel)z operatora elektronowego momentu dipo-
lowego transformują się jak widać na załączonej tabelce zgodnie z reprezentacjami
πu oraz σ+u w naszej grupie D∞h. Możemy więc nasz iloczyn prosty zapisać w po-
staci Γ(ψ′
el) × Γ( πu
σ+g) × Γ(ψ
′′
el), który musi zawierać pełnosymetryczną reprezentację
dla σ+g . Dla większości cząsteczek homojądrowych stan podstawowy posiada sy-
metrię σ+g i w związku z tym dozwolone przejście elektryczno dipolowe z poziomu
podstawowego do stanu wzbudzonego, które opisane jest przez funkcję falową ψel,
jest wtedy gdy Γ(ψ′
el) × Γ( πu
σ+u) × σ+
g zawiera reprezentację σ+g . Aby ten warunek
był spełniony reprezentacją funkcji falowej Γ(ψ′
el) musi być σ+u albo πu. Idąc dalej
tym tropem na podstawie badań wielu innych iloczynów prostych stanów elektro-
nowych o dowolnej symetrii, dochodzimy do uzyskania następujących reguł wyboru
dla naszej homojądrowej cząsteczki dwuatomowej:
∆Λ = 0,±1,
Σ+ ↔ Σ+,Σ− ↔ Σ−, ale Σ+ = Σ−,
u ↔ g, ale u = u, g = g. (1.3.25)
W dalszym ciągu nie uwzględniamy statystyki jądrowej. Aczkolwiek cząstki danego
atomu posiadają własne momenty pędu (spiny) to jednak nie mają one dużego wpły-
wu na nasze obliczenia. Tak się składa, iż współrzędne spinowe cząsteczki nie mają
żadnego wpływu na ogólną postać naszego operatora momentu dipolowego. Można
zatem dojść do kolejnej reguły wyboru ∆S = 0 obowiązującej przy przejściu elektro-
nowym. W ogólności dotyczy wszystkich cząsteczek (także wieloatomowych), jeżeli
wypadkowy spin elektronów został dobrze określony. Zatem praktycznie wszystkie
lekkie układy (w szczególności gazy składające się z cząsteczek homojądrowych –
np. H2, N2, Li2 czy też O2), a więc takie dla których oddziaływania spin-orbita
można zaniedbać.
Oczywiście zostało wspomniane wcześniej, iż w cząsteczkach homojądrowych
widma czysto rotacyjne nie istnieją. Jednakże przy przejściach elektronowych wid-
ma takie są zauważalne. Wynika to z bardzo wielkiego stopnia złożoności, z powodu
braku reguł wyboru, która ograniczałaby liczbę przejść. I tak na strukturę widma ta-
48

kiego nakłada się dodatkowo struktura widma rotacyjnego, na której zmiany wpływa
kwantowa liczba J cząsteczki. W związku z powyższym, gdy prawdopodobieństwo
a posteriori (częstość) konkretnego przejścia elektronowego oraz oscylacyjnego ozna-
czymy sobie przez:
ωeν = (E′
el −E′′
el) + (E′
osc − E′′
osc), (1.3.26)
mamy możliwość obliczenia energii dla przejść ze stanów rotacyjnych J ′′ oraz J ′
naszej cząsteczki, która wyraża się wzorem w postaci:
∆E = ωeν +Bν′J ′(J ′ + 1) −Bν′′J ′′(J ′′ + 1). (1.3.27)
I tym razem istotne dla nas jest równanie 1.3.22 określająca dopuszczalne zmiany
w ∆J = J ′ − J ′′. Mianowicie zauważyć można, że w skład powyższej całki wchodzi
wartość średnia elektronowego momentu przejścia 〈Mel〉. Wygląd oraz charaktery-
styka widm rotacyjnych dla takiego widma zależy w sposób szczególny od stanów
elektronowych cząsteczki, dla których istnieje takie przejście elektryczno dipolowe.
Jako, że nie zajmujemy się badaniem jakichś szczególnie skompilowanych cząsteczek,
to w naszej pracy ograniczymy się jedynie do najprostszych możliwych przypadków,
dla których zachodzą przejścia 1Σ −1 Σ oraz 1Π −1 Σ.
W przypadku gdy mamy do czynienia z przejściami 1Σ −1 Σ z zastosowanych
do równania 1.3.22 reguł symetrii wynika, iż elektronowy moment przejścia posiada
(patrz tabela symetrii) tylko jedną składową wzdłuż osi z− (µel)z , która to podlega
transformacją dla reprezentacji σ+u . Idąc dalej tym tropem dojdziemy do kolejnej
reguły wyboru ∆J = ±1. Homojądrowa cząsteczka jako tako nie posiada widm
oscylacyjnych, aczkolwiek zauważalne są oscylacje dla widm elektronowych. Dlatego
też na podstawie układów heterojądrowych i panujących reguł wyboru dla widm
oscylacyjnych dla przejść w gałęziach P (∆J = −1) oraz R(∆J = +1) [17], możemy
zapisać wyrażenia na energię przejść, które w wyniku obliczeń można przedstawić
wspólnym wzorem dla gałęzi P oraz R:
ω = ωeν + (Bν′ +Bν′′)m+ (Bν′ −Bν′′)m2, (1.3.28)
gdzie dla gałęzi R, m = J + 1 = 1, 2, 3, . . . natomiast w przypadku gałęzi P ,
m = −J = −1,−2,−3, . . . . Reprezentację równania 1.3.28 na wykresie nazy-
wamy parabolą Fortrata. Jeżeli mamy do czynienia z elektronowymi przejściami,
49

Rysunek 1.9: Diagram Fortrata i struktura rotacyjna pasma oscylacyjnego w przej-
ściu 1Π-1Σ dla założenia R′ > R′′. Brakujące linie P (1) i Q(0) zaznaczone są linią
przerywaną
dla których międzyjądrowe odległości – Re′ oraz Re′′ – przed i po przejściu są istot-
nie duże, możemy się spodziewać, że i stałe rotacyjne Bν′ oraz Bν′′ będą się bardzo
różnić. Zatem w równaniu 1.3.28 nie możemy zaniedbać czynnika Bν′ − Bν ′′. Z te-
go wynika, że niemożliwym jest aby odległości między sąsiednimi liniami w obu
gałęziach, były co najmniej w przybliżeniu równe. Zauważmy, że równanie 1.3.28
ma charakter równania kwadratowego. Można zatem je rozwiązać w analogiczny
sposób, przy założeniu że ekstremum funkcji dla energii przejścia w punkcie m,
określa warunek dωdm
= 0, dla którego:
m = − Bν′ +Bν′′
2(Bν′ − Bν′′). (1.3.29)
W przypadku większości cząsteczek, wzbudzenie elektronowe wiąże się ze zmianą
odległości międzyjądrowej (elektrony „przeskakują” z orbitali niższych na wyższe).
W efekcie mamy tutaj do czynienia z osłabieniem wiązania, co może nawet prowadzić
do rozpadu cząsteczki. Problem ten został omówiony szerzej w rozdziale pierwszym.
Zatem Bν′ < Bν′′ . Wówczas mamy sytuacje, w której ekstremum w punkcie m jest
50

maksimum i przyjmuje wartości dodatnie (gałąź R) – początkowe linie układają się
w kierunku rosnącej częstości, a ich gęstość rośnie ze wzrostem odległości, tworząc
tzw. głowicę co zresztą widać na wykresie 1.9. Sytuacja ma się nieco inaczej w przy-
padku drugiej gałęzi P . Częstość kolejnych linii po prostu maleje wraz ze wzrostem
J . Jak widać na wykresie punkt początkowy dla obu gałęzi odpowiada przejściu
gdy J ′ = 0 − J ′′ = 0. Zatem znajduje się tam luka. Aczkolwiek tak jak napisałem
„najczęściej” mamy do czynienia z sytuacją, w której Bν′ < Bν′′ , to jednak istnieje
małe prawdopodobieństwo, dla którego zachodzi warunek Bν′ > Bν′′ . W takim przy-
padku głowica pasma tworzy się w gałęzi P , ograniczając przy tym pasmo od strony
małych częstości.
Rozpatrzmy teraz przejścia 1Π −1 Σ, dla których elektronowy moment przej-
ścia (transformacje symetrii podobnie do wcześniej analizowanego przejścia) posiada
w tym przypadku składową prostopadłą do osi cząsteczki, z czego wynika następu-
jąca reguła wyboru ∆J = 0,±1. W związku z powyższym na Diagramie Fortrata,
zaobserwować możemy dodatkową krzywą (gałąź Q), którą także opisuje równanie
1.3.28. Przeanalizujmy przypadek pierwszy Bν′ < Bν′′. Widać wyraźnie, że wyraże-
nia - liniowy oraz kwadratowy - zależne od J są zawsze ujemne. Zatem, podobnie
jak w przypadku gałęzi P aczkolwiek w innym tempie, gałąź Q ulega degradacji
w stronę mniejszych częstości. W przypadku tego przejścia występuje jeszcze jedna
różnica. Mianowicie stan Π ograniczony jest przez liczbę kwantową J ≥ 1. Dla tego
przejścia nie będzie zatem linii Q(0) oraz P (1), co zresztą jest pomocne przy iden-
tyfikacji rodzaju przejścia podczas obserwowania widma.
1.3.5 Zanik wzbudzenia w cząsteczce
Innymi słowy czas relaksacji, jest to czas w którym układ wytrącony z równo-
wagi powraca do stanu równowagi. Dla parametru f opisującego w najprostszym
przypadku zanik opisany jest zależnością:
df
dt=f0 − f
τ, (1.3.30)
gdzie f0 to wartość f , dla której układ jest w stanie równowagi, natomiast τ na-
zywamy czasem relaksacji. Przy takich założeniach parametr f maleje wykładniczo
51

i w czasie relaksacji maleje e razy. Parametr ten używany jest jako parametr drgań
tłumionych gasnących, opisywanych zależnością:
y(t) = Ae−t
τ cos(µt−δ) . (1.3.31)
Pojęcie czasu relaksacji powszechnie stosowane jest do opisu obiektów makroskopo-
wych. Wówczas wynika ono z własności makroskopowych składników. Na przykład
w termodynamice gazu jest to czas potrzebny do ustalenia się równowagi termody-
namicznej. Innymi słowy czas, po którym wartość energii dla układu spadnie „e”
razy. W sensie mikroskopowym czas życia atomu w stanie pobudzonym, np. E ′,
po którym przejdzie do E ′′. W myśl zasady chemii kwantowej, układ primowany ca-
ły czas nazywamy układem pobudzonym natomiast bisowy w stanie podstawowym.
E
a b a d e
F
∆E
M
C
Rysunek 1.10: Uproszczony schemat zjawisk luminescencyjnych (uproszczony sche-
mat Jabłońskiego poziomów energetycznych w luminoforze): C – poziom podstawo-
wy, F – poziom wzbudzony, M – poziom metatrwały, a – absorpcja, b – fluorescencja,
d – fluorescencja długożyciowa, e – fosforescencja
I tak w momencie gdy pobudzimy naszą cząsteczkę do oscylacji w stanie elektro-
nowym powyżej podstawowego stanu w jakim zwykle znajduje się cząsteczka, dalsze
losy naszej molekuły zależne są już jedynie od jej wymiarów oraz fazy w jakiej
52

się znajduje. A ponieważ celem naszym badań jest w dalszym ciągu homojądrowa
dwuatomowa cząsteczka litu (w fazie gazowej) przyjrzymy się tylko jej przypadko-
wi. Weźmy na ten przykład wzbudzenie do stanu A1Σu+ , dla ν = 1. Na podstawie
wcześniejszej analizy dla widm cząsteczek wieloatomowych, jak na przykład miało
to miejsce w przypadku analizowanej w rozdziale 1 cząsteczki wody, która jest w fa-
zie ciekłej widma te były nieco skomplikowane. Wynikało to z szeregu nieudogodnień
oraz procesów w wyniku których istniała możliwość utraty znaczącej energii wzbu-
dzenia (większość trwałych chemicznie cząsteczek posiada parzystą liczbę elektro-
nów, a ich stan podstawowy posiada symetrię singletową). W związku z powyższym
stany wzbudzone mogą być w postaci singletowej lub tripletowej, a zatem w wyniku
dużej liczby drgań w takiej cząsteczce istnieje bardzo duża kombinacja występowa-
nia poziomów oscylacyjnych dla każdego stanu elektronowego, a ich zagęszczenie jest
zdecydowanie większe niż w przypadku molekuł dwuatomowych. Natomiast nasza
cząsteczka w tym przypadku jest homojądrowa, a także występuje w postaci gazo-
wej. To bardzo wiele ułatwia i w tym przypadku możemy spodziewać się wyłącznie
przejść promienistych1 do stanu podstawowego, co zresztą widać na diagramie 1.10.
W efekcie taki stan rzeczy prowadzi do uzyskania statystyki Bosego-Einsteina
oraz możliwości otrzymania kondensatu Bosego-Einsteina, o czym zresztą mowa
w rozdziale 2 tej pracy.
1Przejście promieniste, zmiana stanu układu kwantowego związana z emisją lub absorpcją pro-
mieniowania elektromagnetycznego (fotonów). Przejście promieniste charakteryzowane jest przez
własną multipolowość promieniowania.
53

Rozdział 2
Opis modelu fizycznego
W poprzednim rozdziale doszliśmy do wniosku, że prawdopodobieństwo przej-
ścia ze stanu podstawowego do poziomu pobudzenia zależy od wielkości cząsteczki
oraz stanu, w jakim się ona znajduje (faza). Aczkolwiek, jak na wymiary naszej
cząsteczki nie mamy wielkiego wpływu, to jednak już na fazę w jakiej się znajduje
możemy wywierać wpływ czynnikami z zewnątrz. Dla tego przykładu rozpatrzmy
maszynę losującą lotto. W maszynie tej znajduje się n piłeczek. Między tymi piłecz-
kami nie występują żadne oddziaływania dalekozasięgowe, a nawet jeśli to są one
bardzo słabe i nie mają żadnego wpływu na nasz układ. Natomiast każdą z piłe-
czek reprezentuje charakterystyczna tylko dla niej energia a zatem i indywidualny
dla niej pęd (druga zasada dynamiki Newtona). Wynika z tego następujący fakt.
Kuleczki w maszynie losującej wykazują pełną swobodę ruchu na przestrzeni ogra-
niczonej przez maszynę. Wszystkie one przemieszczają się w przestrzeni zajmowanej
i nigdy nie zatrzymują się w jednym miejscu. Idąc dalej tym tropem sformułujmy
pytanie, które brzmi następująco: „Jaki stan skupienia przypomina nam następujący
przypadek wzięty z życia?” Właśnie identycznymi prawami fizyki charakteryzuje się
gaz.
Cząsteczki gazu przemieszczają się z różną szybkością, a rozkład tych szybko-
ści ma charakter całkowicie statystyczny (gaussowski). Średnia szybkość poruszania
się cząsteczek w gazie jest zależna wyłącznie od ich masy molowej i temperatury.
Podczas obniżania temperatury gazu maleje średnia szybkość cząsteczek, zaś zwięk-
54

szanie ciśnienia powoduje zmniejszenie średniej odległości między nimi. Obniżanie
temperatury lub zwiększanie ciśnienia prowadzi w końcu do skroplenia lub resubli-
macji gazu. Zamiana gazu w ciecz lub ciało stałe wynika z faktu, że w pewnym
momencie energia oddziaływań międzycząsteczkowych (sił van der Waalsa, wiązań
wodorowych itp.) staje się większa od energii kinetycznej cieplnego ruchu cząsteczek.
W fizyce przyjmuje się często prosty model gazu doskonałego, w którym czą-
steczki gazu nie przyciągają się i nie mają objętości własnej. Teorie i zależności
termodynamiczne wywiedzione z założeń gazu doskonałego sprawdzają się dość do-
brze (na ogół) w przypadku niezbyt dużych ciśnień oraz temperatur powyżej punktu
krytycznego. W innych przypadkach prawa te jednak zawodzą i wtedy stosuje się
bardziej złożone modele gazów i tworzy dokładniejsze teorie i zależności wykorzy-
stując równanie van der Waalsa lub wirialne równanie stanu.
Interesującą cechą gazu (a ściślej gazu doskonałego) jest to, że objętość przez nie-
go zajmowana (w danej temperaturze i ciśnieniu) jest stała, niezależnie od rodzaju
cząsteczek, jakie są w gazie, i zależy wyłącznie od liczby tych cząsteczek. Innymi
słowy, jeśli weźmiemy np. 1 litr wodoru i 1 litr tlenu (oba przy tym samym ciśnieniu
i temperaturze), to w obu objętościach będzie dokładnie taka sama liczba cząsteczek.
Jest to tzw. prawo Avogadro.
Aby jednoznacznie określić stan gazu, poza składem chemicznym (ułamki wago-
we lub molowe) i temperaturą należy podać gęstość gazu lub jego ciśnienie. Zamiast
gęstości można podać równoważnie objętość molową lub stężenie gazu. Dla dowol-
nego gazu:
• objętość jednego mola gazu w warunkach normalnych wynosi: V = 22, 4dm3,
• liczność gazu w (liczba moli): n = NNA
= mM
,
• stężenie molowe gazu: Cm = nV
,
• objętość molowa gazu: Vm = Vn,
gdzie m nazywamy masą gazu, V objętością gazu, N liczbą cząsteczek, NA liczbą
Avogadra, natomiast M masą molową. I tak dla gazu doskonałego mamy równanie:
p = CmRT, (2.0.1)
55

które możemy zapisać dalej jako:
p = ρ
(
RT
M
)
, (2.0.2)
gdzie R to uniwersalna stała gazowa a T to temperatura, w której znajduje się nasz
gaz.
2.1 Kondensat Bosego-Einsteina
I tak w roku 1925 indyjski uczony Satyendra Nath Bose przesłał Einsteinowi
swoje odkrycie. Mianowicie w swojej pracy wyprowadził on prawo Plancka dla pro-
mieniowania ciała doskonale czarnego traktując układ fotonów jako gaz identycz-
nych cząstek. Na jego podstawie Einstein uogólnił teorię Bosego, traktując układ
jako gaz doskonały identycznych atomów lub cząsteczek, dla którego to liczba czą-
steczek jest zachowana. W tym samym roku zauważył także istnienie nowego sta-
nu skupienia, który istniałby w wystarczająco niskich temperaturach, dla których
wszystkie te cząstki obsadziłyby najniższy stan kwantowy układu. To właśnie zjawi-
sko nazywamy kondensatem Bosego-Einsteina. Zachodzi ono tylko dla „bozonów”. Są
to cząsteczki dla których całkowity spin równy jest całkowitej wielokrotności (stałej
Plancka podzielonej przez 2π):
~ =h
2π, (2.1.1)
gdzie ~ jest kwantem momentu pędu, a więc tym samym i spinu. Z tego też po-
wodu przez wielu uważana za stałą bardziej podstawową niż sama stała Plancka.
Oznaczenie to wprowadził brytyjski fizyk Paul A. M. Dirac.
Oczywiście jak napisałem wcześniej zjawisko to występuje w bardzo niskich tem-
peraturach. Mianowicie poniżej temperatury krytycznej, dla której jednolity trójwy-
miarowy gazu składający się z nieinterakcyjnych cząstek z jawnymi wewnętrznymi
stopniami swobody, można wyrazić wzorem:
TC =
[
n
ζ(32)
]23[
2πh2
mkB
]
, (2.1.2)
gdzie TC nazywamy temperaturą krytyczną, n gęstością cząstki, m masą na bo-
zon, h stałą Plancka, kB stałą Boltzmanna natomiast ζ funkcją zeta Riemanna,
56

która ζ(32) ≈ 26124 [18].
2.1.1 Rozwinięcie Einsteina
Rozpatrzmy układN cząstek wzajemnie na siebie nie oddziałujących, które mogą
znajdować się w dwóch różnych stanach kwantowych, |0〉 oraz |1〉 (np. spin elektro-
nów). Jeżeli obydwa te stany posiadają równą energię, można powiedzieć że konfigu-
racje obydwu tych cząstek są niemalże podobne. Jeżeli możemy określić, która z tych
cząstek jest która, istnieje 2N różnych konfiguracji, dopóki każda z tych cząstek mo-
że znajdować niezależnie w stanie |0〉 > lub |1〉. W większości takich konfiguracji,
około połowa cząstek znajduje się w stanie |0〉, z kolei druga połowa jest w stanie
|1〉. Równowaga jest efektem statystycznym – liczba konfiguracji jest większa kiedy
cząstki są równo podzielone.
Jeżeli natomiast cząstki są nierozróżnialne, aczkolwiek istnieje N + 1 różnych
konfiguracji, z czego K cząstek znajduje się w stanie |0〉, a N − K w stanie |1〉,nie da się określić czy cząstka znajduje się w stanie |0〉 lub |1〉. Zatem wartość K
określa ściśle określony stan kwantowy dla całego układu. Jeżeli wszystkie te stany
są równie podobne, nie istnieje statystyczne rozłożenie – sytuacja ta jest równie
podobna dla wszystkich cząstek do pozostania w stanie |0〉, jak ulec podziałowi
w połowie na stan |0〉 a druga połowa dla stanu |1〉.Przypuśćmy teraz, że energia dla stanu |1〉 nieznacznie większa niż energia w sta-
nie |0〉 przy sumie energii E. W temperaturze T , cząstka będzie miała mniejsze
prawdopodobieństwo być w stanie |1〉 z powodu funkcji wykładniczej e(ET). W przy-
padku kiedy możemy rozróżnić cząstki, dystrybucja cząstki nieznacznie skierowana
w stronę stanu |0〉 i będzie nieznacznie różniła się od przypadku „pół na pół”. Nato-
miast w drugim przypadku nierozróżnialnym, ponieważ nie istnieje nacisk ze strony
statystyki, w kierunku jednakowych liczb, najbardziej prawdopodobne rozwiązanie
jest takie, że wszystkie cząstki przejdą do stanu |0〉.W tym przypadku, dla dużych wartości N , ułamek w stanie |0〉 może zostać
obliczony (metodami numerycznymi). Przypadek ten jest identyczny jak w sytuacji
rzutu monetą, które określa prawdopodobieństwo p = exp(−ET), że wypadnie reszka.
57

Ułamek, że wypadnie orzeł jest jak 11+p
, który jest równą funkcją p - prawdopodo-
bieństwa, dla energii.
Zatem każda wartość dla K jest pojedynczym stanem, który posiada swoje wła-
sne oddzielne Boltzmannowskie prawdopodobieństwo. Więc dystrybuanta prawdo-
podobieństwa jest ekspotencjalna i wyraża się wzorem:
P (K) = Ce−KET = CpK. (2.1.3)
Dla dużych N , normalizacją stałej C jest (1 − p). Spodziewaną całkowitą liczbą
cząstek, które nie są w najniższym stanie energetycznym, nie rośnie kiedy N jest
duże. Osiąga natomiast stan 11−p
. Ułamek ten jest bez znaczenia dla całkowitej sumy
cząstek. Zatem zbiór wystarczającej ilości cząstek bosego w równowadze termicznej,
w większości będzie w stanie podstawowym, z niewielką ilością jakichkolwiek pobu-
dzonych stanów, nie ważne z jak wielkimi różnicami energii.
Rozważmy teraz gaz cząstek, które mogą posiadać różne stany pędu oznaczone
|k〉. Jeżeli liczba cząstek jest mniejsza niż termicznie dostępnych stanów, dla dużych
temperatur i niskich gęstości, wszystkie cząstki będą posiadały inne stany. W takich
ograniczeniach gaz zachowuje się w oparciu klasycznym. Jeżeli natomiast gęstość
rośnie lub temperatura maleje, liczba wszystkich dostępnych stanów na cząsteczkę
zaczyna maleć, i w pewnym momencie większość cząstek zostanie zmuszona do przej-
ścia do pojedynczego stanu niż dozwolona dla niego większość przez statystyczny
„dodatek cenowy” równoważący wyższe koszty utrzymania takiego stanu. Od tego
momentu, jakakolwiek nadprogramowa cząstka dodana, przejdzie do dyskretnego
poziomu.
Aby obliczyć temperaturę przejścia dla jakiejkolwiek gęstości, całka na przestrze-
ni wszystkich stanów pędu przy wyrażeniu na maksymalną liczbę 11−p
:
N = V
∫[
d3k
(2π)3
]
1
1 − p(k). (2.1.4)
Po obliczeniu całki z mnożnikami kB oraz ~, zrekonstruowanymi przy użyciu analizy
wymiarowej, otrzymujemy formułę na temperaturę krytyczną przekroju poprzednie-
go.
58

2.1.2 Równanie Gross-Pitaevskii
Stan kondensatu Bosego-Einsteina może być opisany przy pomocy funkcji falowej
dla kondensatu ψ(~r). Dla systemu o takiej naturze, współczynnik |ψ(~r)|2 rozumiany
jest jako gęstość cząstki, więc całkowita liczba atomów wynosi:
N =
∫
d~r|ψ(~r)|2. (2.1.5)
Przy zasadniczym założeniu wszystkie atomy znajdują się w kondensacie (krótko
mówiąc, kondensowały do dyskretnego poziomu) i traktując bozony wykorzystując
teorię pól uśrednionych, energię (E) składająca się ze stanu ψ(~r), wyrażamy wzorem:
E =
∫
d~r
[
~2
2m| ψ(~r)|2 + V (~r)| ψ(~r)|2 +
1
2U |ψ(~r)|4
]
. (2.1.6)
Minimalizując tę energię (respektując nieskończenie małe rozbieżności w ψ(|r),i utrzymując stałą liczbę atomów), otrzymujemy równanie Gross-Pitaevski (GPE),
znane również jako nieliniowe równanie Schrödingera:
i~δψ(~r)
d~r=
(
−~22
2m+ V (~r) + U0|ψ(~r)|2
)
ψ(~r), (2.1.7)
gdzie m to masa bozonów, V (~r) jest potencjałem zewnętrznych oddziaływań, przed-
stawicielskim dla U0 wzajemnych wewnątrz-cząstkowych oddziaływań.
2.1.3 Historyczne odkrycie
Kiedy Einstein przewidział istnienie takiego stanu skupienia, wszystkich ogar-
nęła fala poruszenia. Prawdopodobnie z powodu niemożliwości osiągnięcia w owych
czasach tak niskiej temperatury potrzebnej do osiągnięcia przejścia w stan konden-
sacji przez atomy bosego – mniejszej niż jedna milionowa kelwina – wówczas hipo-
tetyczny gazowy kondensat traktowano raczej jako ciekawostkę naukową, do której
nie należy przywiązywać większej wagi. Dla porównania fakt istnienia kondensatu
nie ma potwierdzenia nawet w świecie realnym. Temperatury w przestrzeni kosmicz-
nej nie osiągają stanu krytycznego potrzebnego do przejścia. Jest ona milion razy
za wysoka dla zaistnienia kondensacji.
59

Jednak kilka lat później znów powróciła moda na kondensację Bosego-Einsteina.
Uczeni fizycy Pyotr Kapitsa, John Allen oraz Don Misener w roku 1938 odkryli,
że izotop helu4 staje się w temperaturze poniżej 2.17 kelwina (punkt lambda) no-
wym rodzajem cieczy – nadcieczą. Nadciekłość helu, czyli efekt który pojawia się
w temperaturze niższej niż kondensacja Bosego w gazie. Taki nadciekły hel posiada
szereg niespotykanych dotąd dla cieczy własności, chociażby zerowy opór zewnętrzny
(tarcie wynikające z istnienia zjawiska lepkości w cieczach jest zerowe). Zatem nad-
ciecz zdolna jest płynąć (nie tracąc przy tym energii) ze skwantowaną prędkością.
Szybko jednak zdano sobie sprawę, iż fakt istnienia nadciekłości wynika z konden-
sacji Bosego-Einsteina atomów izotopu helu4, które są bozonami. W rzeczywistości
wiele charakterystycznych cech dla nadciekłego helu również występuje w przypad-
ku gazowych kondensatów Bosego-Einsteina, aczkolwiek izotop ten jest raczej cieczą
niż gazem , co znaczy że oddziaływania międzyatomowe są relatywnie wysokie. Ory-
ginalna teoria kondensatu Bosego-Einsteina musi być mocno uściślona aby możliwe
było opisanie jej. Jednakże kondensacja pozostaje nadal fundamentalna dla nadcie-
kłych własności helu4.
Pierwszy „czysty” kondensat Bosego-Einsteina (Bose–Einstein condensa-
te (BEC))
Przez 70 lat metody chłodzenia nie były na tyle zaawansowane, abyśmy byli
w stanie osiągnąć temperaturę, przy której możliwe byłoby przejście kondensacji
Bosego. Zjawisko te zatem przez tak długi czas było skazane na porażkę, naukow-
cy zaś traktowali je jako czysto naukową ciekawostkę, aż do lat dziewięćdziesiątych
dwudziestego wieku. Fizycy z laboratoriów na całym świecie, z tak prestiżowych
uczelni jak: MIT, University of British Columbia, Universiteit le Amsterdam a tak-
że Cornell University musieli stawić czoła tak fundamentalnym problemom, aby móc
otrzymać BEC w gazie. Do uzyskania kondensatu niezbędne było schłodzenie gazu
znacznie poniżej temperatury, w której dochodzi do jego zamarznięcia. A ponieważ
możemy kontrolować tylko warunki początkowe (na to co dzieje się później nie mamy
większego wpływu) koniecznym było stworzenie przesyconego gazu. Bardzo wielkie
nadzieje pokładano w cząsteczkach wodoru, ponieważ atomy tego pierwiastka opie-
60

rają się procesowi łączenia w grupy zanim ulegną zamrożeniu [19].
Aczkolwiek naukowcom, którzy badali proces kondensacji dla wodoru, nie udało
się tego zjawiska uzyskać, to w bardzo dużej mierze przyczynili się oni do zrozumienia
trudności z jakimi się borykali oraz udało się im sporo z nich rozwiązać. Na podsta-
wie ich doświadczeń kolejne grupy badawcze rozpoczęły swoje prace. I tak w roku
1989, na podstawie wyników badań chwytania i następnego schładzania atomów
metali alkalicznych z wykorzystaniem laserów, zaczęto podejrzewać, że najlepszy-
mi próbkami do schłodzenia będą właśnie te atomy. Czyli wszystkie te pierwiastki
(metale) z pierwszej grupy okresowej (oprócz wodoru) tj. cez, rubid a nawet lit,
który jest przedmiotem badań niniejszej pracy magisterskiej. Jednak w przeciwień-
stwie do wodoru posiadają bardzo wielką zdolność do łączenia się w grupy; to jednak
zdecydowanie szybciej potrafią one przejść do fazy skondensowanej. W przeciwień-
stwie do atomów wodoru, atomy metali mają o wiele większe rozmiary, a zatem
dużo częściej mogą napotkać na swojej drodze sąsiadów i ulec zderzeniom, dzieląc
się przy tym energią kinetyczną. Procesy zderzenia zachodzą tak szybko (często),
że zanim gaz ulegnie zamarznięciu osiągnie on stan kondensatu.
To właśnie dzięki badaczom z MIT a głównie jednym z głównych liderów grupy
badawczej Danielem Kleppnerem, próbujących uzyskać kondensat w gazowym wo-
dorze, udało się połączyć wspaniałe metody chłodzenia i pułapkowania metali alka-
licznych wykorzystując światło promieni laserowych z wypracowanymi przez zespół
Daniela technikami chwytania w zewnętrznym polu magnetycznym i odparowywa-
niu gorących cząstek. Ostatecznie hipoteza uzyskania kondensatu z wykorzystaniem
metali alkalicznych okazała się być słuszną i już w roku 1995 zespół pod przewod-
nictwem Erica A. Cornella oraz Carla Wiemana na University of Colorado at Boul-
der NIST_JILA lab, uzyskali kondensat wykorzystując do tego gaz atomów rubidu
schłodzony do 170 nanokelwinów (nK). Zaledwie w parę miesięcy, po tym udało
się to grupie Wolfganga Ketterlego z MIT, wykorzystując do tego atomy sodu [20].
Grup z MIT udało się otzrymać kondensat składający się z 10 milionów atomów.
W chwili gdy Eric Cornell wraz z Carem Wiemanem opublikowali artykuł doty-
czący kondensacji, co najmniej siedem innych zespołów wytworzyło już kondensa-
ty. Badania dotyczące kondensacji z atomami rubidu prowadzą Daniel J. Heinzen
61

z University of Texas, Gerhard Rempe z Universität Konstanz w Niemczech i Mark
Kasevich z Yale University. Z atomami sodu natomiast pracuje niezależnie od grupy
Keterllego z MIT zespół pod kierunkiem Lene Vestergaard Hau z Rowland Institute
for Science w Cambridge (Massachusetts). Z kolei dla nas interesujący jest bardzo
lit (w końcu dotyczy on tej pracy magisterskiej). I tak dla atomów litu także uda-
ło się otrzymać kondensat przez Randalla G. Hulleta z Rice University. Oczywiście
w Polsce nie tak dawno, bo w tym roku, także udało się otrzymać kondensację.
Aczkolwiek dla atomów rubidu o czym będzie mowa w dalszej części tego rozdziału.
Według wielu teorii w temperaturze zera bezwzględnego zanika wszelki ruch.
Zatem najprostszym rozumowaniem wydaje się, ażeby osiągnąć temperaturę bliską
zera bezwzględnego jest wyhamowywanie atomów cząsteczek, poprzez odbieranie
ciepła z układu oraz izolowanie od otoczenia chłodzonej próbki. A ponieważ proces
uzyskania kondensatu możemy podzielić na dwa etapy, warunki konieczne do zaj-
ścia procesu muszą być spełnione dla obydwu etapów. W pierwszym z nich światło
laserowe izoluje atomy jednocześnie je schładzając. Z kolei w drugim etapie pułapka
magnetyczna izoluje układ, a chłodzenie odbywa się przez odparowywanie.
Laserowe chłodzenie i pułapkowanie
Do naszych badań posłuży nam szklane pudełko (tzw. serce) otoczone cewka-
mi magnetycznymi. Z pudełka tego odpompujemy całkowicie powietrze. Postępując
w taki sposób otrzymamy próżnię, która będzie się zachowywała jak termos. Po czym
wprowadzimy do wnętrza owego pudełka niewielką ilość par metalu alkalicznego w fa-
zie gazowej. Punkt w przestrzeni jednoznacznie wyznaczony jest przez trzy wektory.
A ponieważ do opisu wektora potrzebne są dwa punkty, zatem potrzebować będzie-
my aż sześć wiązek światła laserowego, które przecinać się będą w środku komórki,
otaczając zawarty wewnątrz gaz. W tym celu światło laserowe nie musi być inten-
sywne, możemy zatem do doświadczenia wykorzystać najprostsze półprzewodnikowe
lasery wykorzystywane na szeroką skale w przemyśle elektronicznym w czytnikach
CD.
W rozdziale pierwszym napisałem o zdolnościach absorpcyjnych atomów,
które zależą od budowy atomów. Innymi słowy atomy pierwiastków odznaczają
62

się charakterystycznymi tylko dla siebie częstotliwościami (energiami) jakie chęt-
nie pochłaniają przechodząc w stań pobudzenia a następnie wypromieniowując je,
ażeby następnie wrócić do stanu podstawowego. Dlatego też częstotliwość promie-
niowania laserowego dobieramy tak aby atomy naszego metalu alkalicznego chętnie
pochłaniały i emitowały fotony. Ponieważ atom jest w stanie w ciągu jednej sekundy
pochłonąć i wyemitować wiele milionów fotonów, dokonując przy tym niewielkiego
odrzutu w kierunku ruchu zaabsorbowanego fotonu, wykorzystujemy aż sześć wiązek
laserowych. Odrzut ten natomiast nazywamy ciśnieniem promieniowania, w wyniku
którego następuje zmiana kierunku prędkości, w stronę przeciwną do początkowego
kierunku wektora prędkości. Cała zatem sztuka takiego chłodzenia laserowego pole-
ga na tym, aby zmusić atom do absorpcji jak największej ilości fotonów biegnących
w kierunku przeciwnym do wektora prędkości tych atomów. W efekcie spowalniamy
prędkości tych atomów; innymi słowy wraz ze spadkiem prędkości atomów spada
ich temperatura. I właśnie dzięki dobrze dobranej częstotliwości laserów, która od-
powiada częstotliwości najchętniej pochłanianego światła laserowego przez atomy,
osiągamy nasz cel. Oczywiście lasery służą nie tylko do ochładzania chmury atomów
metalu alkalicznego, ale także do ich więzienia. Tworzą taki bardzo prosty układ
zamykający w pułapce naszą chmurę, utrzymując ją z dala od ścianek pudełka,
które mają temperaturę pokojową.
Światło laserów musi przeciwstawiać się skłonności atomów do dryfowania poza
środek pułapki. W tym celu istnieje słabe pole magnetyczne wytwarzane przy pomo-
cy cewek magnetycznych dookoła kostki, które dostraja rezonansową częstotliwość
atomów, tak aby chętniej pochłaniały one fotony podążające w kierunku środka
pułapki (punkt przecięcia się laserów w środku komory). W efekcie wszystkie ato-
my popychane są w kierunku jednego niewielkiego, punktowego obszaru, w którym
utrzymywane są jedynie przy użyciu siły światła laserowego. Wykorzystując omówio-
ną metodę w ciągu jednej minuty napełniamy naszą pułapkę laserową 10 milionami
atomów takiego metalu, które wcześniej znajdowały się w temperaturze pokojowej
wypełniając kostkę. Niestety w dalszym ciągu temperatura potrzebna do przejścia
w stan kondensatu jest około 100 razy za wysoka. A wykorzystując do tego celu jedy-
nie lasery, nie będziemy w stanie wyeliminować przypadkowych szturchnięć atomów
63

przez wyemitowane fotony. Komplikuje to dalsze chłodzenie, a zatem otrzymanie
większej gęstości atomów w punktowym obszarze.
Pułapka magnetyczna oraz chłodzenie przez odparowywanie
Na tym poziomie wyłączamy wszystkie lasery i przechodzimy do drugiego etapu
chłodzenia. Jak sam tytuł akapitu brzmi, etap ten polega na wykorzystaniu pułapki
magnetycznej i schładzaniu naszej chmury przez odparowywanie. Sama pułapka ma-
gnetyczna wykorzystuje fakt, iż na poziomie mikroskopowym atomy zachowują się
jak niewielkie magnesiki, którymi możemy sterować wykorzystując pole magnetycz-
ne. Proces ten więc polega na wykorzystaniu sił magnetycznych, którym podlegają
atomy. W tym celu musimy starannie dobrać geometryczny kształt takiego pola ma-
gnetycznego, które musi być stosunkowo silne. Wówczas atomy będą się poruszały
jak toczące się piłeczki pingpongowe po wewnętrznych ściankach misy na kształt le-
ja. Następnie w procesie odparowywania atomy o największej energii uciekają z tak
skonstruowanej magnetycznej misy, schładzając przy tym pozostałe atomy – ato-
my które uciekają unoszą ze sobą więcej energii niżby się im to średnio należało,
a zatem pozostałe atomy stają się zimniejsze.
Efekt ten można w bardzo prosty sposób wyjaśnić na przykładzie stygnięcia
kawy czy też innego ciepłego napoju. Mianowicie w procesie stygnięcia cząsteczki
wody o największej energii uciekają z kubka. Innymi słowy ulatniają się z niego
w fazie gazowej (para wodna). W ten sposób obniżają one średnią energię cieczy.
Tymczasem wewnątrz kubka zaistniałe zderzenia prowadzą do równego podziału
pozostałej energii, pomiędzy pozostałymi cząsteczkami wody. Oczywistym faktem
jest, że w przypadku naszej chmury atomowej złapanej w pułapce magnetycznej, jej
gęstość jest znacznie mniejsza niż gęstość wody w kubku. A zatem rewolucja w tym
niezwykłym osiągnięciu naukowym nie polegała na jakimś przełomowym odkryciu,
tylko na drodze wieloletnich drobnych ulepszeń, z którymi zmagali się naukowcy.
Te i inne „drobne”, jakkolwiek bardzo znaczące, udoskonalenia były wielce po-
mocne przy metodzie pułapkowania, to jednak w dalszym ciągu zespołowi Cornela
i Wiemana nie udawało się otrzymać satysfakcjonującej gęstości, przy której możli-
we byłoby efektowne schładzanie metodą odparowywania. Nagminnym problemem
64

była skuteczność pułapki magnetycznej. Pomimo że zespół był w stanie osiągnąć
„magnetyczną miskę” posiadającą na tyle silne pole, to jednak w świecie mikrosko-
powym atom jest bardzo słabym magnesikiem. Ostatecznie przełomowym czasem
dla badaczy był rok 1994, w którym naukowcy stanęli przed koniecznością zbudo-
wania nowego rodzaju pułapki magnetycznej, znacznie głębszej i wyższej w prze-
ciwieństwie do pierwowzoru. Obecnie ekipy badawcze prześcigają się w tworzeniu
coraz to nowych lepszych pułapek. Mówi się nawet, że istnieje tyle ich rodzajów ile
zespołów zajmujących się zjawiskiem BEC.
Rzeczywisty fakt istnienia „superatomu” – migawkowa fotografia
Aby rzeczywiście zaobserwować dowód istnienia grupy schłodzonych atomów, ro-
bimy migawkowe zdjęcie cienia rzucanego przez oświetlony błyskiem lasera konden-
sat. W miarę schładzania, aż do momentu otrzymania kondensatu, rozmiar chmu-
ry staje się stosunkowo niewidoczny dla ludzkiego oka. Dzieje się tak, gdyż wraz
ze spadkiem temperatury atomów, zaczynają się one skupiać w punktowym obsza-
rze, w pobliżu dna misy. W efekcie niemożliwym jest abyśmy byli w stanie zaobser-
wować wymiary rzędu wielkości atomowej. W tym celu następuje zwolnienie „supe-
ratomu” z pułapki magnetycznej (pole magnetyczne zostaje wyłączone), co umożli-
wia rozprzestrzenianie się atomów w dowolnym kierunku. Po upływie 0.1 sekundy
rozszerzoną chmurę naświetlamy krótkim błyskiem światła laserowego. W wyniku
rozproszenia się światła przez atomy na boki, zauważyć możemy cień, który rejestro-
wany jest przy użyciu kamery wideo. Cień ten reprezentuje kształt chmury. Może
on nam wskazać początkowy rozkład prędkości uwięzionych atomów. Na podstawie
pomiaru prędkości możemy określić temperaturę w jakiej znajduję się układ.
W celu przeprowadzenia analizy kondensatu, przyjrzyjmy się schematowi 2.1.
Widzimy, że wykres ten przedstawia obraz cienia rzuconego przez kondensat. Został
on poddany obróbce komputerowej, tak abyśmy mogli w bardzo przejrzysty sposób
zaobserwować, rozkład prędkości chmury atomowej. Na wykresie kolory odpowia-
dają liczbom atomów w danym zakresie prędkości – czerwony oznacza najmniejszą
liczbę, biały natomiast największą, ledwie poruszających się atomów. Na pierwszym,
po lewej stronie, widzimy zanim kondensat się utworzy, rozkład prędkości chmury
65

Rysunek 2.1: Rozkład prędkości kondensatu Bosego-Einsteina powstałego z gazu
składającego się z atomów rubidu. Lewy: tuż przed pojawieniem się kondensatu
Bosego-Einsteina. Środkowy: zaraz po otrzymaniu kondensatu. Prawy: Po dalszym
parowaniu pozostała próbka prawie czystego kondensatu.
o temperaturze około 200 nK (stosunkowo niewielki i łagodny pagórek). Po dalszym
schładzaniu do temperatury bliskiej 100 nK, zauważyć można powstawanie prawie
czystego kondensatu. Aż w ostateczności otrzymany zostaje praktycznie czysty kon-
densat.
Oczywiście błąd określenia pozycji atomów jest niewielki i dlatego błąd pomiaru
pędu (prędkości) musi być odpowiednio większy, aby ich iloczyn był większy niż sta-
ła. Zatem wysunąć można wniosek, że w świetle praw mechaniki klasycznej dzieje się
coś złego. Kondensat powstaje w stanie o najniższej z możliwych energii. W świetle
mechaniki klasycznej, energia najniższego poziomu, to taka energia dla której atomy
spoczywają nieruchomo (zanika wszelki ruch). Na wykresie zaś zjawisko to powinno
się przejawiać nieskończonym i bardzo wąskim wręcz punktowym pikiem. My w rze-
czywistości obserwujemy na wykresie zjawisko zupełnie inne, które wynika z faktu
istnienia efektów kwantowych. Można je w skrócie podsumować do przytoczonej
w rozdziale pierwszym zasady nieoznaczoności Heisenberga.
66

Im dokładniej znamy położenie atomów wewnątrz pułapki, tym znacznie trudniej
jest nam określić ich prędkości i odwrotnie. To właśnie z powodu tej zasady widzimy
na wykresie, iż wierzchołek nie posiada kształtu nieskończonego piku. Gdyby nato-
miast tak było, wiedzielibyśmy, że atomy chmury znajdują się dokładnie w jednym
punktowym miejscu a ich prędkości wynosiłyby dokładnie zero.
Podczas gdy uzasadnienie teorii Einsteina wymaga, aby wszystkie atomy chmury
gazowej w tak niskiej temperaturze osiągnęły stan o najniższej z możliwych energii,
to zasada nieoznaczoności Heisenberga zabrania pozostawać tym atomom na dnie le-
ja. Dlatego też prawa mechaniki kwantowej rozwiązują ten problem w bardzo prosty
sposób. Mianowicie wartość energii atomu znajdującego się w jakimkolwiek pojemni-
ku, chociażby w takiej pułapce magnetycznej, może należeć do pewnego dyskretnego
zbioru dozwolonych wartości dla tej energii. Tak jak w rozdziale pierwszym przed-
stawiony został przypadek oscylatora harmonicznego, który schematycznie rozwią-
zuje nasz problem. A najniższą energię dla tego przykładu nazywamy energią drgań
zerowych i wynosi ona E = 12~ν. Możemy zatem powiedzieć, że kondensat Bosego-
Einsteina jest wręcz rzadkim przykładem, w którym rządzi zasada nieoznaczoności
Heisenberga w świecie obiektów makroskopowych. Ale także jest akademickim, prak-
tycznym i podstawowym przykładem w myśli wyjaśnienia praw mechaniki kwanto-
wej.
Własności oraz możliwe zastosowanie BEC
Atomy które tworzą BEC pod wieloma względami przypominają fotony ze świa-
tła laserowego. Jak powszechnie wiadomo laser charakteryzuje się jednorodną wiąz-
ką fotonów o tej samej częstotliwości a tym samym i energii (te zagadnienie zostało
szeroko omówione w rozdziale pierwszym) oraz fazie oscylacyjnej. To właśnie dzięki
tym właściwością można w bardzo precyzyjny sposób manipulować światłem la-
serowym, chociażby przy użytkowaniu tak powszechnie stosowanych w przemyśle
fonograficznym płyt kompaktowych. Identycznie zachowuje się kondensat, tylko tak
jak w przypadku lasera mamy fotony, to w tym przypadku są to atomy. Idąc dalej
tym tokiem rozumowania dochodzimy do faktu stworzenia lasera atomowego, a fa-
le materii takiego lasera ulegają podobnym zjawiskom jak odbicie, zogniskowanie,
67

dyfrakcja. Można również modulować ich częstotliwość oraz amplitudę. Po tak dłu-
gim okresie badań (około 70 lat minęło zanim naukowcom udało się przeprowadzić
doświadczalny dowód Bosego oraz Einsteina) w ręce uczonych trafiła niezwykła zdo-
bycz naukowa. Zdolna do niewyobrażalnej precyzji w kontrolowaniu ruchu atomów,
chociażby w mierzeniu czasu przy użyciu nowszych zegarów atomowych. Przyczyni
się to zapewne do rozwoju elektroniki. Wyobraźmy sobie coraz to nowsze rozwiąza-
nia, w których wiązka atomów zogniskowana w obszarze o rozmiarach poprzecznych
równej jednej milionowej metra, „napylająca” tranzystor wprost na układ scalony.
Oczywiście, to tylko niewielka garść innowacyjnych zastosowań jaką niesie ze sobą
przyszłość, wykorzystując zjawisko kondensacji Bosego-Einsteina. Zjawisko to cha-
rakteryzuje się jeszcze wieloma innymi, a jakże bardzo interesującymi własnościa-
mi. Interesująca jest chociażby lepkość kondensatu. Tak jak wcześniej wspomnia-
łem jest on nadciekły – zaniedbywana jest zatem jego lepkość, a w konsekwencji
nie opiera się on zjawisku tarcia. Również ciekawym zjawiskiem jest, iż wiązki lasera
atomowego oddziaływają na siebie – w przypadku światła laserowego skrzyżowa-
ne wiązki nie wpływają wzajemnie na siebie. Materiał ten zatem, który nie dość,
że jest cieczą to jeszcze przejawia się w postaci spójnej fali, na pewno charakteryzuje
szereg innych nie poznanych przez uczonych właściwości i zapewne minie wiele lat
(tak jak w przypadku dowodu doświadczalnego) zanim je w pełni zrozumiemy.
Tymczasem wiele grup zajmuje się badaniem cech kondensatu. Tak jak grupa
Carla Wiemana, która podczas wzmocnienia coraz bardziej sił magnetycznych w pu-
łapce, nagle zauważyła, że proces kondensacji odwrócił się w kierunku przyciągania,
a następnie implodował i skurczył się poza zakres detekcji. Następnie eksplodował,
zdmuchując przy tym dwie trzecie atomów spośród 10 tysięcy. Około połowa ato-
mów w kondensacie wydawała się zniknąć całkowicie z eksperymentu, nie będąc
widocznym ani w schłodzonej pozostałości ani rozprzestrzeniającej się chmurze ga-
zowej. Sam Wieman wyjaśnił, iż w świetle obecnej teorii atomowej, charakterystyka
kondensatu Bosego-Einsteina nie może zostać wyjaśniona, ponieważ energia stanu
atomu w temperaturze bliskiej zera absolutnego, nie powinna spowodować implozji.
Jednakże, w następstwie teorii pól uśrednionych, zostało zaproponowane rozwiąza-
nie.
68

Z powodu faktu, że eksplozjami supernowej są implozje, eksplozja zapadającego
się kondensatu Bosego-Einsteina została nazwana „bosenową”. Kalambur, odnoszący
się do stylu muzycznego zwanego bossa nova.
I tak w świetle współczesnych odkryć w dziedzinie fizyki również i w Polsce
naukowcy są niezwykle zainteresowani kondensacją Bosego-Einsteina.
2.1.4 Pierwszy polski kondensat Bosego-Einsteina
Dokładnie 2 marca 2007 roku, grupie polskich fizyków z kilku ośrodków pracują-
cej w Krajowym Laboratorium Fizyki Atomowej, Molekularnej i Optycznej w Toru-
niu pod przewodnictwem Wojciecha Gawlika udało się - wykorzystując do tego celu
atomy izotopu rubidu - stworzyć pierwszy w kraju kondensat Bosego-Einsteina.
W skład zespołu wchodzili tacy uczeni jak Wojciech Gawlik, Andrzej Noga, Je-
rzy Zachorowski i Michał Zawada (ten ostatni od niedawna znajduje się na UMK)
z Uniwersytetu Jagiellońskiego, Franciszek Bylicki oraz Michał Zawada z Uniwersy-
tetu Mikołaja Kopernika w Toruniu, a także Włodzimierz Jastrzębski z Instytutu
Fizyki PAN w Warszawie oraz Jacek Szczepkowski z Pomorskiej Akademii Peda-
gogicznej w Słupsku i Marcin Witkowski z Uniwersytetu Opolskiego. Do wstępnej
fazy projektu duży wkład wnieśli także Maria i Tomasz Brzozowscy z Instytutu Fi-
zyki Uniwersytetu Jagiellońskiego a także Paweł Kruk, który pierwotnie pracował
w Instytucie Fizyki Doświadczalnej Uniwersytetu Warszawskiego. Obecnie pracuje
w Instytucie Fizyki Uniwersytetu Jagiellońskiego [21].
Fakt dokonanego przez polski zespół osiągnięcia stawia nasz kraj w światowej
czołówce (spośród wszystkich krajów na całym globie tylko 16 zajmuje się bada-
niem 6 stanu skupienia), a nasz kraj jest jednym z nich i jedynym pomiędzy Łabą
a Pekinem.
Prace na projektem rozpoczęły się - niespełna w 3 lata po tym jak zespołowi
z MIT udało się to przedsięwzięcie - na Instytucie Fizyki na krakowskim Uniwersy-
tecie Jagiellońskim. W owym czasie uruchomiona została pierwsza pułapka magne-
tooptyczna, przy pomocy której udało się osiągnąć temperaturę rzędu 100 mikro-
kelwinów [22]. W roku 2001 dzięki pomocy finansowej Komitetu Badań Naukowych
69

oraz niezwykłej współpracy całego polskiego środowiska naukowego zostało stworzo-
ne Krajowe Laboratorium Fizyki Atomowej, Molekularnej i Optycznej (KL FAMO)
przy UMK w Toruniu. Obecnie laboratorium to reprezentuje światowy poziom na-
ukowy w dziedzinie fizyki atomowej, zwłaszcza w zastosowaniu pułapek jonowych
oraz fizyki ultrazimnych atomów. To właśnie dzięki KL FAMO do projektu dołączył
Włodzimierz Jastrzębski z IF PAN w Warszawie, który wówczas jako jedyny polski
fizyk eksperymentował z kondensatem za granicą.
Wymagania fizyczne oraz przyjęte rozwiązania
Ponieważ samo zagadnienie kondensatu zostało omówione w podrozdziałach tego
rozdziału, dlatego nie będziemy się zgłębiać ponownie w ten problem. Aczkolwiek sa-
mo osiągnięcie kondensacji wymagało od polskich fizyków nie lada wyczynu, z uwagi
na konieczność posiadania bardzo skomplikowanej aparatury oraz wyrafinowanego
przebiegu doświadczenia.
Samo doświadczenie nie odbiega od tego przeprowadzonego przez zespół z MIT.
Do eksperymentu wykorzystano również atomy rubidu, które w wyniku manipula-
cji wiązkami promieniowania laserowego oraz pól magnetycznych są pułapkowane
a następnie schładzane do temperatury rzędu kilkuset nanokelwinów. Cały proces
prowadzący do otrzymania kondensatu, można podzielić na poszczególne etapy:
1. Wychwyt atomów z chmury termicznej o temperaturze pokojowej (ok. 300 K)
i ich wstępne schłodzenie w pierwszej pułapce magnetooptycznej (MOT1).
2. Transfer atomów z MOT1 za pomocą światła do drugiej, wysoko próżniowej
komory.
3. Przechwycenie przetransferowanych atomów w drugiej pułapce magnetoop-
tycznej (MOT2).
4. Przeładowanie atomów z drugiej pułapki magnetooptycznej (MOT2), już do czy-
stej pułapki magnetycznej (MT).
5. Proces schładzania przez odparowywanie z wykorzystaniem oscylującego pola
magnetycznego o radiowej częstotliwości (RF).
70

6. Detekcja i diagnostyka, dzięki której możemy na podstawie temperatury (roz-
kładu prędkości) stwierdzić zaistnieje kondensacji atomów.
Bezsprzecznym jest fakt, że na każdy z etapów procesu głównego składa się sze-
reg innych skomplikowanych kroków sekwencyjnych, podczas których analizowanych
jest wiele operacji, jak chociażby dobranie częstotliwości laserów, a także ich włą-
czanie i wyłącznie. Oczywiście, aby wszystko było dokładnie wykonane, zajmuje
się tym uprzednio skonfigurowany system komputerowy, dzięki któremu zapewniona
jest szczególnie potrzebna nam precyzja.
1. W etapie tym, tak jak omówione zostało to na początku tego rozdziału,
pułapka optyczna składa się z sześciu punktowych laserów, na których prze-
cięciu opisany jest punkt w przestrzeni oraz pułapka magnetyczna o niejedno-
rodnym polu magnetycznym o symetrii kwadrupolowej. Tak właśnie wygląda
nasza pułapka magnetooptyczna MOT1. Aby proces był dostatecznie krót-
ki, w naszej komorze ciśnienie pary rubidu powinno wynosić ok. 10−8 mbar.
I w tym momencie istotny dla nas jest przekrój czynny na absorpcję. Każ-
dy pierwiastek charakteryzuję się swoją własną częstotliwością. Izotop rubi-
du 87Rb charakteryzuje się liniami widmowymi przejścia 52S1/2 − 52P3/2 –
linia D2 o długości fali 780 nm. Dodatkowo laser transferujący musi zapo-
biegać przy przepompowywaniu stratom w wyniku ucieczki atomów do in-
nych stanów. Zwykle w skład chmury wchodzi 109 atomów (średnica wynosi
ok. 3 mm a gęstość ok. 1011 cm−3) przy temperaturze rzędu 0, 1 − 1 mK.
2. Schłodzone w ten sposób atomy są transportowane do drugiej komory(MOT2),
przy pomocy lasera o niemalże rezonansowej częstotliwości (odstrojenie wynosi
ok. 12 MHz, natomiast natężenie 1 mW ).
3. Następnie przetransportowane atomy zostają przechwycone w drugiej pułap-
ce magnetooptycznej (MOT2). W tym momencie możemy wyłączyć pułapkę
MOT1. W ciągu około 30 sekund w drugiej pułapce przechwyconych zostaje
ok. 109 atomów poniżej temperatury 100 µK. W celu dostatecznie długiego
(powyżej 1.5 minuty) przechowywania na tym obszarze atomów, potrzebna
71

jest niezwykle doskonała próżnia (ciśnienie rzędu 10−11 mbar). Z tego faktu
między obydwiema pułapkami MOT1 oraz MOT2 istniejąca rurka transferowa
musi być bardzo wąska. W chwili wyłączenia MOT1, kładziony jest szczególny
nacisk na racjonale gospodarowanie atomami. Nie można sobie pozwolić nawet
na znikome straty, ponieważ w dalszych etapach uczestniczą tylko te atomy.
Wszelkie zaś obserwacje czy to w MOT1, czy też w MOT2 prowadzone są
przez rejestrację fluorescencji atomów Rb kamerą CCD oraz fotodiodą, wyka-
librowaną tak aby opomiarować liczbę atomów.
4. Na tym etapie niemożliwe już jest dalsze schładzanie przy pomocy pułapkowa-
nia magnetooptycznego. Przechodzimy więc do etapu schładzania przez odpa-
rowywanie. W tym momencie następuje wyłączenie pułapki magnetooptycz-
nej. Po czym następuje włączenie zwykłej pułapki magnetycznej, która jest
pułapką „ciemną”. W celu zapewnienia braku możliwości absorpcji fotonów re-
zonansowych, należ starannie zaciemnić układ. W przeciwieństwie do pułap-
ki magnetooptycznej, zwykła magnetyczna jest bardzo płytka, zachowawcza
i gromadzi wyłącznie te atomy o określonej orientacji atomowego momentu
magnetycznego [wyjaśnienie tego problemu jest w podrozdziale 2.1.3]. A po-
nieważ możliwe jest stworzenie w wolnej przestrzeni lokalnego minimum pola
magnetycznego, zatem wyłapywane przez nas atomy będą w takich stanach
szukających małego pola, a w szczególności dla atomów rubidu w stanie pod-
stawowym 52S1/2 są to takie stany |F = 2, mF = +2〉, |F = 2, mF = +1〉oraz |F = 1, mF = −1〉, podczas gdy atomy w pozostałych stanach są wypy-
chane z minimum pola. Cały etap przejścia z MOT2 do MT możemy podzielić
na poniższe procedury:
a) Skoro wypadkowy potencjał (moc pułapki MOT jest wyższa niż MT)
uwzględniający wpływ grawitacji ma minimum w innym miejscu, to na-
leży przesunąć chmurę atomów ze środka pułapki MIT na środek pułapki
MT.
b) Odstrojenie laserów do 35 MHz powoduje dodatkowy wzrost schłodze-
nia i kompresji – tzw. faza zimnego MOT’a: w wyniku odłączenia pułapki
72

magnetycznej osiągamy chłodzenie subdoplerowskie. W efekcie ochłodze-
nie atomów jest rzędu 20 µK.
c) Wyłączenie laserów pułapkującego/chłodzącego i repompującego w tej ko-
lejności przesądza, w którym z nadsubtelnych stanów (F = 1 lub F = 2)
pozostaną atomy. Polski zespół wyłącza laser repompujący w 500 µs
po wyłączeniu pułapkującego, czego efektem jest transfer atomów w sta-
nie 52S1/2(F = 2). Aczkolwiek w dalszym ciągu są wszystkie zeemanow-
skie poziomy, w związku z czym układ nie jest gotowy do włączenia zwy-
kłej pułapki magnetycznej.
d) W tym celu następuje polaryzacja gazu atomów przez przepompowanie
do stanu |F = 2, mF = +2〉. A zatem włączane jest słabe pole ma-
gnetyczne (ok. 1 Gs) a atomy naświetlane są bardzo słabymi impulsami
światła (celem uniknięcia podgrzania ok. 1 ms i 35 µW ) o polaryzacji
σ+ z przejściami 52S1/2|F = 2〉 → 52P3/2|F = 2〉 oraz 52S1/2|F = 1〉 →52P3/2|F = 2〉 (drugi z błysków powinien trwać dłużej, celem zapobie-
gnięcia utraty atomów przy przepompowaniu do stanu 52S1/2|F = 1〉).W ten sposób większość przepompowanych atomów znajduje się w stanie
|F = 2, mF = +2〉 i można przystąpić do pułapkowania samym polem
magnetycznym.
e) Trwające ok. 2 s „adiabatyczne” włączenie pułapki zapobiega zmianie
temperatury przez chmurę, a następnie ok. 80% atomów jest transfe-
rowanych z MOT2 do MT.
5. W pułapce magnetycznej zaś dochodzi do schładzania przez wymuszone odpa-
rowywanie (efekt stygnięcia kubka gorącej kawy): następuje wyrzucanie ato-
mów z pułapki pod wpływem oscylującego pola magnetycznego z częstotliwo-
ścią rezonansową momentów magnetycznych. W tym celu precyzyjnie dobiera-
na jest częstość oscylacji, dzięki której usuwane są atomy o największej ener-
gii kinetycznej. Pozostałe zaś atomy normalizują swoje poziomy energetyczne
przywracając układ do stanu równowagi. Usuwanie te odbywa się przy ob-
niżaniu z 30 MHz do 800 kHz, do momentu pozyskania chmury o ok. 105
73

atomów przy temperaturze bliskiej 200 nK. Sam proces odparowywania musi
zachodzić na tyle wolno aby termalizacja atomów zachodziła na tyle wydajnie,
że mimo spadku liczby atomów gęstość w przestrzeni fazowej wzrastała, co jest
niezwykle niezbędne do zajścia procesu kondensacji.
6. W ostatnim etapie przedstawiony już zostaje efekt obrazowania chmury ato-
mów w wyniku rejestracji. Etap ten jest niemalże identyczny jak w przypadku
przedstawionym przez grupę MIT.
Omówienie niektórych elementów oraz procedur zastosowanych w do-
świadczeniu
Do przeprowadzenia doświadczenia głównymi elementami aparatury były:
1. Lasery – podobnie do grupy Wolfganga Keterllego czy też Carla Wiemana
do doświadczenia wykorzystywane są proste lasery półprzewodnikowe i muszą
one spełniać następujące wymagania:
a) szerokość linii musi być mniejsza od naturalnej linii przejścia optycznego,
b) absolutna stabilność częstości na poziomie 1 MHz, o względnej stabilno-
ści ok. 3 × 10−9 ,czego efektem jest wymóg stabilizacji częstości promie-
niowania laserowego względem częstości przejścia atomowego,
c) moc rzędu 100 mW − 1 W , wykazująca się dużą stabilnością,
d) wiązki laserowe kierowane są do doświadczenia jednomodowymi odcinka-
mi włókna światłowodowego celem zapewnienia jednorodnego poprzecz-
nego kształtu tychże wiązek, co wiąże się z dużymi stratami mocy.
2. Układ próżniowy – celem rozgraniczenia obszarów zbierania atomów oraz ich
wstępnego schłodzenia poniżej 1 mK oraz obszaru kondensacji z ciśnieniami
10−8 oraz 10−11 następuje konieczność zastosowania komór połączonych cienką
rurką oraz różnicowego pompowania obu stref.
3. Transfer atomów między pułapkami – z racji, że nie możemy sobie pozwolić
na duże straty podczas przejść atomów z pułapki MOT1 do MOT2 (grawita-
74

cyjny spadek trwałby zbyt długo i wiązałby się ze znaczną ekspansją atomów),
dokonuje się więc przepchnięcia grupy schłodzonych atomów za pomocą wiąz-
ki światła (ciśnienie promieniowania). W tym celu konieczne jest odpowiednie
dobranie profilu promienia, aby miał on odpowiednie natężenie i działał on
z różnymi siłami na atomy w różnych obszarach. Ponieważ w górnej pułapce
działają silne wiązki nasz promień musi być tam zatem porównywalnie silny.
Cel ten osiągamy ogniskując go do średnicy ω1 = 1, 1 mm. Natomiast w dol-
nej natężenie musi być małe, aby nie spowodować destabilizacji dolnej pułapki.
Dlatego też rozszerzona jest ona do ω = 3, 3 mm.
4. Pułapka magnetyczna – jedną z najprostszych pułapek tego typu jest pułapka
kwadrupolowa. Stworzona jest z dwóch cewek o przeciwległych polach – pole
potencjalne wzrasta w niej liniowo wraz ze wzrostem odległości od centrum.
Jest ona bardzo skuteczne. Jednakże w centrum takiej pułapki pole jest zero-
we [23, 24]. W związku z czym powolne atomy przelatujące przez sam środek
Rysunek 2.2: Ilustracja przedstawiająca a) układ cewek pułapki magnetycznej
oraz b) rozkład pola magnetycznego jaki powstaje wewnątrz takiego układu
pułapki mogą doznać odwrócenia momentu magnetycznego w wyniku czego
atomy te przestają być pułapkowane. Dlatego też konfiguruje się pola tak aby
nigdzie nie było zerowego natężenia. Jak już wcześniej zostało wyjaśnione,
rodzajów pułapek magnetycznych jest tyle ile grup badających kondensację.
W układzie badawczym Polaków występują trzy cewki umieszczone blisko sie-
75

bie, które zapewniają wysoki gradient pola oraz dwie cewki wytwarzające pole
jednorodne (rysunek 2.2).
Prąd płynący w cewkach jest o jednakowym natężeniu 40 A (układ strasznie
się nagrzewa). Zatem bardzo trudnym problemem jest, przy tak skonstruowa-
nej pułapce, chłodzenie. Zwłaszcza że wymiary kondensatu są mikroskopijne,
a zatem pułapka też jest bardzo niewielka, a drut nawinięty na cewkach stożko-
wych jest bardzo cienki (1 mm). Trzeba także zapewnić układowi odpowiednie
odprowadzanie ciepła. W tym celu stosuje się skuteczny przepływ wody mię-
dzy uzwojeniami cewek stożkowych. Natomiast w cewkach pola jednorodnego
z racji niewielkich wymiarów użyto zamiast drutu cienkiej rurki miedzianej,
wewnątrz której przepływa ciecz chłodząca.
Ponieważ włączanie (adiabatyczne) i wyłączanie (szybkie) musi być kontrolo-
wane perfekcyjne, niezbędny okazuje się do tego celu układ elektroniczny. Za-
pewnia on w bardzo szybki sposób (ok. 1 ms) przełączanie prądu, co zwłaszcza
w przypadku cewek indukcyjnych jest bardzo trudne.
5. Sterowanie doświadczeniem – tak jak to opisał Carl Wieman problemem prze-
biegu doświadczenia nie jest powstanie jakiejś nowej niezbadanej teorii,
której my fizycy jeszcze nie rozumiemy, a wymóg nieskazitelnej precyzji i do-
skonale zsynchronizowanej regulacji wielu parametrów, wśród których najważ-
niejsze to:
a) częstości modulatorów akustooptycznych (AOM),
b) momenty włączania i wyłączania modulatorów AOM,
c) otwieranie i zamykanie przesłon,
d) natężenia prądów cewek pułapki magnetycznej,
e) częstość pola radiowego,
f) wyzwalanie kamery CCS.
W celu spełnienia powyższych wymogów oraz wielu innych parametrów, stero-
wanie doświadczeniem odbywa się przy użyciu dwóch komputerów, z których jeden
76

zajmuje się przebiegiem doświadczenia (rozdzielczość 25 µs), natomiast drugi reje-
stracją zjawiska oraz dalszą obróbką i analizą widma atomów. Na przedstawionym
poniżej rysunku widzimy kolejność przełączania poszczególnych parametrów.
Rysunek 2.3: Schemat przedstawiający przebieg doświadczenia w czasie
Trudności na jakie napotkał polski zespół
Z praktycznego punktu widzenia proces zajścia kondensacji jest w istocie bar-
dzo trudnym zjawiskiem. Dzieje się tak dlatego, ponieważ bardzo trudno sprowadzić
układ do tak niskiej temperatury a polscy naukowcy nie posiadają układu chłodzą-
cego prostego w swojej konstrukcji. Jest to szereg niezwykle skomplikowanych i jakże
od siebie uzależnionych mniejszych układów. Oczywiście nie tylko stopień skompliko-
wania jest tutaj poważnym problemem. Jak się okazało w przypadku polskiej grupy
naukowców największą przeszkodą była logistyka a raczej silne jej rozproszenie na te-
renie całej polski. Zlokalizowanie KL FAMO w Toruniu spowodowało konieczność
77

dojazdu wielu naukowców z ich rodzimych ośrodków niekiedy oddalonych o setki
kilometrów (Kraków, Warszawa, Opole, Słupsk) a także pogodzenie tego ze stałą
pracą.
Na etap pierwszy budowy, składała się realizacja budowy od podstaw całej apa-
ratury próżniowej oraz zestawienie toru optycznego i uruchomienie pułapek ma-
gnetycznych co miało miejsce w IF UJ w Krakowie. W tym samym czasie w IF
PAN odbywała się budowa pułapki magnetycznej, natomiast w Toruniu przygoto-
wywano laboratorium, które wymagało m.in. odpowiednich pomieszczeń gotowych
na przyjęcie aparatury. Etap ten trwał dwa lata. Całą aparaturę z Krakowa, należało
przetransportować praktycznie bez demontażu. W przeciwnym wypadku dwuletni
wysiłek poszedłby na marne. 12 marca 2004 r. 400 kg stół z całą aparaturą uda-
ło się zapakować do specjalnie przygotowanej skrzyni a następnie ją bezpiecznie
przetransportować. Dalsze prace już odbywały się wyłącznie w Toruniu. W dal-
szym ciągu jednak występowały utrudnienia przedłużające drogę do celu. Chociażby
fakt zniszczenia cewek pułapki magnetycznej podczas omyłkowego włączenie prądu
bez chłodzenia uzwojenia. Oczywiście zdarzenie to było najbardziej dramatyczne,
chociaż najbardziej czasochłonne okazało się zdiagnozowanie i usunięcie problemów
z niewłaściwie pracującym pompowaniem różnicowym (zwiększenie ciśnienia rubidu
w górnej komorze powodowało pogorszenie się próżni w dolnej komorze). Widzi-
my zatem, jak niezwykle precyzyjne muszą być urządzenia do otrzymania czystego
kondensatu.
Trudności były różnorakie. Okazało się ponadto, że to nie wszystko, ponieważ
największym utrudnieniem był dorywczy charakter pracy dojeżdżającego zespołu.
Aż do roku 2006 kiedy Andrzej Noga doktorant IF UJ osiadł w Toruniu na ponad pół
roku a Michał Zawada w październiku tego samego roku został zatrudniony w UMK
i przeprowadził się do Torunia. W tym właśnie czasie nastąpił przełom w systema-
tycznym prowadzeniu eksperymentu i 2 marca 2007 roku o godzinie 20:30 ekspery-
ment został uwieńczony sukcesem. Zarejestrowano po raz pierwszy dane świadczące
o uzyskaniu pierwszego w Polsce kondensatu Bosego-Einsteina.
78

Wymagania fizyczne oraz przyjęte rozwiązania
Do pomiaru ultraniskiej temperatury kondensatu stosowana jest metoda zasto-
sowana po raz pierwszy w roku 1995 przez zespół Carla Wiemana oraz Erica Cor-
nella. W tym celu oświetlamy chmurę atomów wiązką światła laserowego, będącego
w rezonansie z przejściem atomowym rejestrowanie cienia atomów na tle wiązki
przy użyciu kamery CCD. Ponieważ atomy mają swoją charakterystyczną długość
fal jakie pochłaniają, możliwe jest zaobserwowanie cienia. Obraz ten jest odwzoro-
wany na matrycy kamery CCD za pomocą prostego układu optycznego.
Tak obecnie jak i wcześniej wykazano w momencie uwolnienia „superatomu”
z pułapki magnetycznej dojdzie do rozbiegania się atomów na wszystkie strony
pod wpływem grawitacji. Efekt ten odzwierciedla rozkład prędkości, jaki był w chmu-
rze atomów spułapkowanych (temperaturę). Jeżeli założymy dla takiej chmury tylko
rodzaj rozkładu pędu, możliwe będzie z dużą dokładnością wyznaczenie liczby ato-
mów, ich gęstość, temperaturę i rozmiary chmury.
W przypadku klasycznej chmury gazowej mamy zupełnie inny rozkład pędów.
Podczas gdy kondensat - klasyczna chmura termiczna - charakteryzuje się Gaus-
sowskim rozkładem wynikającym z rozkładu Maxwella-Boltzmana kondensat BE
ma rozkład Thomasa-Fermiego (odwrócona parabola). Jest to pierwsza oznaka zaj-
ścia kondensacji, podczas której widzimy wyraźne przejście fazowe pomiędzy dwoma
rozkładami. Kondensat otrzymany przez polaków ma aż 100 tys. atomów i powstaje
on przy temperaturze 250 nK. Temperatura ta jest temperaturą przy której zaczyna
się przejście fazowe. Dopiero poniżej 70 nk zaobserwować można czysty kondensat
BE.
W przeciwieństwie do termicznej chmury atomów (podczas spadku swobod-
nego atomów chmura przyjmuje kształt sferycznie symetryczny), kondensat jako
obiekt mechaniki kwantowej zachowuje się zupełnie inaczej. Kondensat w pułapce
ma kształt leżącego poziomo cygara. W chwili wyłączenia pułapki i następnemu
spadkowi kondensatu w wyniku grawitacji zaobserwować możemy, że zaczyna być
wydłużony w kierunku pionowym. I znowu możemy powiedzieć, jak wielki wpływ
na świat mikroskopowy ma zasada nieoznaczoności Heisenberga. Znaliśmy dokład-
79

ne położenie kondensatu pułapce w kierunku pionowym. Zatem posiadał on dużą
nieokreśloność pędu. Dlatego też rozbieganie się atomów jest znacznie szybsze w kon-
densacie w kierunku pionowym niż poziomym.
Perspektywy na przyszłość
Naukowcy, fizycy, ludzie nauki na przełomie wieków dostali do rąk własnych
niezwykłe osiągnięcie. Na naszych oczach dokonuje się przełomowych odkryć, moż-
liwe także staje się jeszcze lepsze poznanie świata materii w ujęciu praw mecha-
niki kwantowej. Jesteśmy świadkami nowej epoki. Epoki która będzie miała wielki
wpływ na nas ludzi. Dzięki temu odkryciu Polska jako jedno z szesnastu państw,
ma możliwość prowadzenia badań doświadczalnych na światowym poziomie w pręż-
nie rozwijającej się dziedzinie. Niewiadomą jest jak szybko uda się zaznaczyć istnieje
KL FAMO oraz wkładu jakim się odznaczył pozyskując szósty stan skupienia. Za-
leży to od wielu czynników – głównie zaangażowanie młodych ludzi, którzy mogliby
znaleźć ciekawą pracę w KL. Zespół ma w planach wiele badań związanych z ba-
daniami kondensatu a także z możliwością praktycznego zastosowania kondensatu,
jak chociażby powstanie ultrastabilnego zegara atomowego nowej generacji.
2.2 Techniki numeryczne
2.2.1 Pochodna funkcji
Z wiadomości zdobytych w analizie matematycznej wiemy, iż pochodna funkcji
jest narzędziem, które służy nam do badania przebiegu zmienności wartości funk-
cji, określonej na pewnym przedziale o wartościach rzeczywistych, przy zmianie jej
argumentów. Z punktu widzenia analizy funkcjonalnej, pochodna jest operatorem
liniowym. Pojęcie pochodnej było uogólniane, na przykład na przestrzenie unormo-
wane. Proces odnajdywania pochodnej nazywamy różniczkowaniem, a dział mate-
matyki zajmujący się pochodnymi, ich własnościami i zastosowaniami rachunkiem
różniczkowym.
80
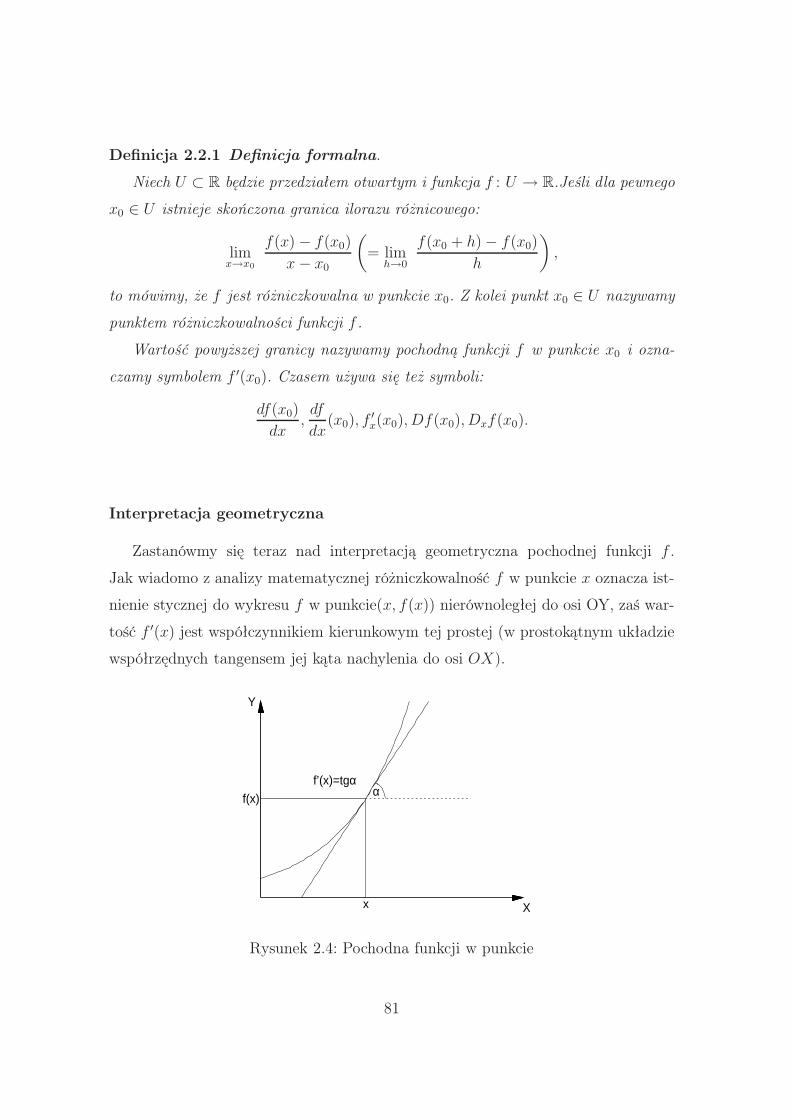
Definicja 2.2.1 Definicja formalna.
Niech U ⊂ R będzie przedziałem otwartym i funkcja f : U → R.Jeśli dla pewnego
x0 ∈ U istnieje skończona granica ilorazu różnicowego:
limx→x0
f(x) − f(x0)
x− x0
(
= limh→0
f(x0 + h) − f(x0)
h
)
,
to mówimy, że f jest różniczkowalna w punkcie x0. Z kolei punkt x0 ∈ U nazywamy
punktem różniczkowalności funkcji f .
Wartość powyższej granicy nazywamy pochodną funkcji f w punkcie x0 i ozna-
czamy symbolem f ′(x0). Czasem używa się też symboli:
df(x0)
dx,df
dx(x0), f
′x(x0), Df(x0), Dxf(x0).
Interpretacja geometryczna
Zastanówmy się teraz nad interpretacją geometryczna pochodnej funkcji f .
Jak wiadomo z analizy matematycznej różniczkowalność f w punkcie x oznacza ist-
nienie stycznej do wykresu f w punkcie(x, f(x)) nierównoległej do osi OY, zaś war-
tość f ′(x) jest współczynnikiem kierunkowym tej prostej (w prostokątnym układzie
współrzędnych tangensem jej kąta nachylenia do osi OX).
X
Y
f(x)
x
f’(x)=tgαα
Rysunek 2.4: Pochodna funkcji w punkcie
81

Pochodną funkcji na przedziale można uważać za liczbową charakterystykę szyb-
kości wzrostu danej funkcji (duża pochodna – stromy wykres, niewielka pochodna –
wykres łagodnie wznoszący się, ujemna pochodna – wykres opadający itp.) co jest
z resztą logiczne gdy przyjrzymy się równaniu prostej.
A zatem, skoro f ′(x) jest współczynnikiem kierunkowym prostej, której wzór
funkcji jest postaci f(x) = ax+ b, to równie dobrze możemy napisać, że f ′(x) = a.
Skoro tak to należałoby najpierw rozwiązać analitycznie jakiś trywialny przykład
celem sprawdzenia w dalszym kroku poprawności naszych metod numerycznych,
które mają nam posłużyć w dalszych rozważaniach tejże pracy.
2.2.2 Przykład analitycznego poszukiwania pochodnej
funkcji w punkcie
Dla przykładu weźmiemy okrąg. Okrąg jest to całkiem nietrywialna krzywa,
dla której istnieje nieskończenie wiele stycznych. Natomiast wybierzemy sobie pro-
sty wzór dla tej krzywej aby nasze obliczenia nie były za bardzo skomplikowane,
ale także aby były one słuszne i żebyśmy mogli później na ich podstawie przepro-
wadzić dowód poprawności naszego algorytmu numerycznego. Zatem,
Definicja 2.2.2 Definicja okręgu.
Niech S = (x0, y0) będzie ustalonym punktem, zaś r odcinkiem o dodatniej dłu-
gości. Okręgiem nazywamy zbiór punktów płaszczyzny euklidesowej spełniającej rów-
ność:
(x, y) : (x− x0)2 + (y − y0)
2 = r2.
Jest to wzór geometrii analitycznej obowiązujący w kartezjańskim układzie współ-
rzędnych. Zmienne x0 oraz y0 opisują położenie środka S = (x0, y0) okręgu względem
układu i są to stałe. W tym samym układzie współrzędnych okrąg może być opisany
również za pomocą równania parametrycznego
x = x0 + r cosα
y = y0 + r sinα,
gdzie parametr α ∈ (0, 2π).
82

dla uproszczenia obliczeń przyjmiemy, że punkt S = (0, 0) a promień r = 5. Wzór
okręgu będzie więc wyglądał w następującej postaci:
(x, y) : x2 + y2 = 25,
a jego wykres tak jak na przedstawionym poniżej rysunku 2.5.
Niestety krzywa x2 + y2 = 25 nie jest funkcją. Zatem należałoby sprowadzić ją
do postaci funkcji, które będą wyglądać w następujący sposób:
f(x) =√
25 − x2
g(x) = −√
25 − x2
, gdzie Df,g ∈ [−5, 5].
-5
-4
-3
-2
-1
0
1
2
3
4
5
-4 -2 0 2 4
f(x)g(x)
Rysunek 2.5: Wykres funkcji f(x) oraz g(x)
Możemy teraz przystąpić do utworzenia dla tego okręgu tabeli, w której będą za-
wierały się (oczywiście nie wszystkie ale możliwie dużo) punkty należące do wykresu
krzywej.
Dla tak stworzonej przez nas tabeli 2.1 z wartościami x oraz odpowiadający-
mi im argumentami f(x) policzymy pochodną funkcji w punkcje z definicji 2.2.1.
83

Tabela 2.1: Przykładowe punkty dla krzywej x2 + y2 = 25
x 0 0, 31712 0, 63296 0, 94625 1, 25574 1, 56017 1, 85831 2, 14897
f(x) 5 4, 98993 4, 95977 4, 90964 4, 83974 4, 75036 4, 64184 4, 51463
I tak zaczniemy nasze obliczenia dla punktu P1 = (x1, f(x1)) = (0, 5):
f ′(x1) = limx→x1
f(x) − f(x1)
x− x1
(
= limh→0
f(x1 + h) − f(x1)
h
)
.
Dla f(x) =√
25 − x2, gdzie y ∈ [0, 5] zatem:
limh→0
√
25 − (x0 + h)2 −√
25 − x20
h= lim
h→0
√
25 − (0 + h)2 −√
25 − 02
h=
= limh→0
√25 − h2 −
√25
h= lim
h→0
√25 − h2 − 5
h= lim
h→0
√25 − h2
h− lim
h→0
5
h=,
a ponieważ limh→0
5h
= 0, więc = limh→0
√25−h2
h=
[
00
]
.
Wychodzi nam więc, że granica jest nieokreślona. Możemy zatem skorzystać
z twierdzenia de l’Hospitala, które brzmi:
Twierdzenie 2.2.1 Twierdzenie de l’Hospitala
Jeżeli funkcje f i g są określone w przedziale (a, b] oraz
1. limx→a
f(x) = 0,
2. limx→a
g(x) = 0,
3. istnieją (skończone) pochodne f ′ i g′, w przedziale (a, b], przy czym g′(a) 6= 0
dla x ∈ (a, b].
wówczas istnieje granica limx→a
f ′(x)g′(x)
= K to wtedy również limx→a
f(x)g(x)
= K.
Zatem:
limh→0
12√
25−h2 · (−2h)
1= lim
h→0
1
2√
25 − h2· (−2h) = lim
h→0
−h√25 − h2
= − 0√25 − 0
=0
5= 0.
84

Tabela 2.2: Tabela pochodnych dla przykładowych punktów P należących do krzy-
wej x2 + y2 = 25
x 0 0, 31712 0, 632962 0, 946256
f(x) 5 4, 98993 4, 95977 4, 90964
f ′(x) 0 −0, 0636526 −0, 127722 −0, 19284
x 1, 25574 1, 56017 1, 85831 2, 14897
f(x) 4, 83974 4, 75036 4, 64184 4, 51463
f ′(x) −0, 259574 −0, 328549 −0, 400464 −0, 476137
Tak więc, możemy zapisać, że pochodna dla f(x) =√
25 − x2 w punkcie P1 = (0, 5)
istnieje i wynosi f ′(0) = 0. Obliczmy zatem pochodne funkcji f(x) dla pozosta-
łych punktów z tabeli 2.1. Oczywiście nie ma sensu rozpisywać się w naszej pracy
na temat obliczeń pochodnej dla pozostałych punktów. Nie to jest celem naszego za-
dania. Dlatego też pominiemy te zapisy i od razu przedstawimy tabele pochodnych
dla poszczególnych punktów P .
Zatem, tak jak widzimy na powyższej tabeli 2.2 obliczone zostały przez nas po-
chodne w wybranych wcześniej punktach P . Następnie tak jak wcześniej pisałem
na podstawie właśnie tych oto dokonanych przez nas obliczeń sprawdzimy popraw-
ność algorytmu numerycznego zastosowanego w naszym programie, którego zada-
niem jest obliczanie pochodnej w punkcie.
2.2.3 Implementacja metod matematycznych
Wyznaczanie pochodnej metodą numeryczną - z definicji pochodnej
Ponieważ celem naszych obliczeń jest pochodna funkcji w punkcie i jest ona dys-
kretna (zadana w postaci punktów) to program nasz będzie liczył styczne do krzywej.
A jak wiadomo z definicji 2.2.1, pochodna funkcji w punkcie f ′(x) jest to nic innego
jak współczynnik kierunkowy prostej, która jest styczna do naszej krzywej. Listing
powyższej metody czyli wyznaczania pochodnej z lewego członu równania występu-
85

jącego w definicji 2.2.1 przedstawiony jest poniżej:
vector<double> X;
vector<double> FX;
...
double a = 0.0;
double b = 0.0;
for(int i=0; i<X.size()-1; i++)
a = ( FX[i+1]-FX[i] ) / (X[i+1] - X[i]);
b = FX[i+1] - a*X[i+1];
...
Jak zauważymy, jest to prosta pętla realizująca część numeryczną algorytmu z for-
malnej definicji pochodnej 2.2.1, można nawet powiedzieć, że jest to dosłowne tłuma-
czenie algorytmu na język C++. W całym naszym programie współrzędne punktów
oraz zmienne a, b zostały zadeklarowane jako wskaźniki na wektory liczb podwójnej
precyzji, natomiast parametr i jest liczbą całkowitą, która ma być liczbą wykonywa-
nego kroku. Oczywiście, dla tak stworzonego algorytmu program będzie liczył pierw-
szą styczną, począwszy dla drugiego punktu, ponieważ skądś musi mieć on deltę.
Nie jest to jednak duże uniedogodnienie zwłaszcza, iż pierwszą testowaną przez nas
krzywą będzie okrąg. Zanim jednak przystąpimy do obliczeń wykorzystamy do tego
celu oprogramowanie zwane Gnuplot, które jest powszechnie dostępne na licencji
GPLv2 dla systemów GNU/Linux. Ja osobiście sam posiadam jedną z dystrybucji
systemu operacyjnego GNU/Linux jakim jest właśnie Gentoo oraz skompilowanego
Gnuplota w wersji 4.4.2. Program ten służy do tworzenia wykresów w 2 i 3 wymia-
rach. My jednak ograniczymy się do dwóch i stworzymy w nim kod, którego listing
przedstawimy poniżej:
set term postscript eps enhanced
set output "circle.eps"
set parametric
set size square
86

set origin 0, 0
set xrange [-10:10]
set yrange [-10:10]
plot [0:2*pi] 5*sin(t),5*cos(t) title "x^2+y^2=25"
set term table
set output "dane.dat"
#set sample 10000
replot
Jak widać w naszym kodzie występuje równanie parametryczne. A ponieważ zgodnie
z definicją okręgu 2.2.2 promień okręgu r sinα, r cosα, to tak napisany kod narysu-
je nam okrąg o promieniu r = 5 i następnie dokona zapisu punktów wchodzących
w skład krzywej do pliku, przy czym zauważmy że linijka set sample 10000 zo-
stała zahaszowana nie bez powodu. Jest to ustawione przez nas próbkowanie. Inny-
mi słowy, jeżeli każemy Gnuplotowi zapisać punkty należące do krzywej do pliku,
to domyślnie wartość próbkowania ustawiona jest na 100. Zatem jeżeli tę czynność
wykonamy przy domyślnej wartości próbkowania, to otrzymamy 100 punktów nale-
żących do naszej krzywej x2 + y2 = 25. Wykonajmy zatem nasz kod w Gnuplocie,
a następnie otrzymane przez nas punkty w pliku dane.dat (po uprzednim wyma-
zaniu przez nas zbędnych danych) poddamy obróbce w napisanym wcześniej przeze
mnie programie liczącym proste styczne do krzywej zadanej w postaci dyskretnej.
Po wykonaniu powyższych czynności program wypisze na naszym ekranie funkcje
określone równaniem y = ax+ b, które są styczne do krzywej dla podanych w pliku
dane.dat punktów, co z resztą powinno wyglądać mniej więcej tak jak na poniższym
listingu:
[0.31712,4.98993] : y = -0.0317545x + 5
[0.632962,4.95977] : y = -0.0954908x + 5.02021
[0.946256,4.90964] : y = -0.160009x + 5.06105
[1.25574,4.83974] : y = -0.22586x + 5.12336
[1.56017,4.75036] : y = -0.293598x + 5.20842
[1.85831,4.64184] : y = -0.36399x + 5.31825
87

[2.14897,4.51463] : y = -0.437659x + 5.45515
[2.43098,4.36925] : y = -0.515514x + 5.62245
[2.7032,4.20627] : y = -0.598707x + 5.82469
[2.96454,4.02635] : y = -0.688452x + 6.06729
[3.21394,3.83022] : y = -0.786407x + 6.35769
[3.4504,3.61867] : y = -0.894654x + 6.70559
[3.67296,3.39255] : y = -1.016x + 7.12426
[3.88073,3.15276] : y = -1.15411x + 7.63156
[4.07288,2.90028] : y = -1.31397x + 8.25194
[4.24863,2.63613] : y = -1.50299x + 9.02177
...
[0,5] : y = 0x + 5
Zapisano w pliku styczne.exp, ilosc: 99
Jak wcześniej wspomniałem, pomimo że w pliku dane.dat znajduje się 100 punk-
tów zawierających się w krzywej, to program liczy styczne tylko dla 99. Niestety
problem ten istnieje, wprawdzie nie jest on znaczący i nie ma on wpływu na dalszą
część naszego zadania. Wracając zatem do naszych obliczeń, widzimy że dla każ-
dego punktu istnieje przyporządkowana do niej styczna. A ponieważ jak zostało
wcześniej napisane w interpretacji geometrycznej pochodnej funkcji, wartość f ′(x)
jest to nic innego, jak współczynnik kierunkowy a naszej stycznej. Zatem weźmy
z naszego listingu, pierwszy z dołu wiersz i widzimy, że pochodna f ′(x) w punkcie
(0, 5) istnieje i wynosi f ′(x) = 0. Nic bardziej trywialnego jak prosta równoległa
do osi OX. Porównując to z naszą tabelą 2.2 wychodzi, że się zgadza. Dla przykła-
du weźmiemy tym razem pierwszy z góry. Widzimy, że pochodna f ′(x) w punkcie
(0, 31712; 4, 98993) istnieje i wynosi f ′(x) = −0, 0317545. Porównując jak poprzed-
nio z wynikami w tabeli 2.2 dochodzimy do wniosku, że nasz program liczy błęd-
nie. Skoro między obydwoma wynikami −0, 0317545, −0, 0636526 występuje różnica
o ponad 0, 03. Nic bardziej mylnego. Różnica ta wynika z faktu, że mamy do dys-
pozycji tylko 100 punktów. Należy zwrócić uwagę na fakt, iż algorytm ten liczy
przybliżone wartości pochodnych, ponieważ aproksymuje od odległości pomiędzy
kolejnymi punktami. I tak jak wcześniej wspomniałem domyśla wartość próbkowa-
88

nia w Gnuplocie wynosi 100. Skoro tak, to ustalmy ją sobie na 10000 i zobaczmy
jak wtedy będą wyglądać nasze wyniki. W tym celu odhaszujemy w naszym wcze-
śniejszym listingu wiersz set sample 10000 i ponownie wykonamy kod w Gnu-
plocie. Po tak wykonanej czynności otrzymamy 10000 zawierających się w równa-
niu naszej krzywej x2 + y2 = 25. Następnie wykonujemy ponownie nasz program,
który tym razem powinien policzyć pochodne z większą dokładnością. Po uruchomie-
niu programu, na ekranie zostaną znowu wypisane równania prostych, które tym ra-
zem przedstawione powinny być zgodnie z poniższym listingiem:
[0.00314191,5] : y = 0x + 5
[0.00628381,5] : y = 0x + 5
[0.00942571,4.99999] : y = -0.00318279x + 5.00002
[0.0125676,4.99998] : y = -0.0031828x + 5.00002
[0.0157095,4.99998] : y = 0x + 4.99998
[0.0188514,4.99996] : y = -0.00636557x + 5.00008
[0.0219933,4.99995] : y = -0.00318279x + 5.00002
[0.0251351,4.99994] : y = -0.00318289x + 5.00002
[0.028277,4.99992] : y = -0.00636557x + 5.0001
...
[0.31712,4.98993] : y = -0.0637755x + 5.01015
...
[0.632962,4.95977] : y = -0.12837x + 5.04102
...
[0.946256,4.90964] : y = -0.194489x + 5.09368
...
[1.25574,4.83974] : y = -0.259868x + 5.16607
...
[1.56017,4.75036] : y = -0.327759x + 5.26172
...
[1.85831,4.64184] : y = -0.402062x + 5.389
...
[2.14897,4.51463] : y = -0.477032x + 5.53976
89

...
Zapisano w pliku styczne.exp, ilosc: 9999
Tabela 2.3: Tabela pochodnych analitycznych oraz numerycznych dla przykładowych
punktów P należących do krzywej x2 + y2 = 25
Źródło
f ′(x) 0 −0, 0636526 −0, 127722 −0, 19284 a)
f ′(x) 0 −0, 0637755 −0, 12837 −0, 194489 b)
Źródło
f ′(x) −0, 259574 −0, 328549 −0, 400464 −0, 476137 a)
f ′(x) −0, 259868 −0, 327759 −0, 402062 −0, 477032 b)
Porównamy znowu nasze pochodne tylko, że tym razem stworzymy sobie tabelę,
na podstawie której porównamy nasze wyniki. Tym razem dokładność naszego al-
gorytmu jest niemalże idealna.1 A zatem dowiedliśmy, iż liczy on poprawnie nasze
pochodne. Z tym jednak wyjątkiem, iż należy zwracać należytą uwagę na próbko-
wanie. Inaczej mogą wychodzić w naszych obliczeniach, tak jak wcześniej błędy.
Obsługa programu
W oparciu o metody obliczeniowe podane na początku tego rozdziału, powstał
nasz program liczący styczne do krzywej dyskretnej. Stąd jego nazwa styczne.cpp.
Jest to bardzo prosty kod napisany w C++ i można go skompilować na dowolnej ma-
szynie zawierającej system operacyjny GNU/Linux oraz oprogramowanie potrzebne
do jego kompilacji jak chociażby powszechnie dostępny pakiet gcc, który jest zwykle
dołączany do każdej dystrybucji systemu GNU/Linux. Wykonuje się go z poziomu
terminalu.
Program nasz odczytuje wartości punktów z pliku dane.dat. My oczywiście mo-
żemy zmienić nazwę odczytywanego pliku na jakiś inny a następnie przekompilować
1Porównując ze sobą źródła, a) i b), gdzie a) jest to pochodna liczona metodą analityczną,
a b) numeryczną, dochodzimy do następujących wniosków.
90

źródła. Oprócz tego program zapisuje wykonane przez niego obliczenia do pliku
styczne.exp. Nazwę tę również możemy zmienić według własnych potrzeb. I tutaj
także należy po dokonanych zmianach w kodzie, przekompilować źródła.
Jak zatem widzimy obsługa programu jest wręcz trywialna i nie powinna przy-
spożyć przeciętnemu użytkownikowi problemów. Przystępujemy zatem do dalszych
obliczeń naszego zagadnienia.
91

Rozdział 3
Obliczanie przekroju czynnego na
absorpcję promieniowania
Celem naszej pracy jest wyznaczenie przekroju czynnego na absorpcję promienio-
wania na przykładzie cząsteczki Li2. Do tego celu wykorzystamy istniejące już ob-
liczenia wykonane przez P. Jasika oraz J.E. Sienkiewicza, które są dostępne w ich
wspólnej pracy [25]. Oczywiście nasze obliczenia zostaną wykonane tylko dla jednego
przykładowego stanu wybranego przez nas. Ponieważ znamy z rozdziału 1 niniejszej
pracy magisterskiej reguły wyboru i wiemy, że dla naszego litu dwuatomowego -
Li2 zachodzą następujące własności Σ+ ↔ Σ+,Σ− ↔ Σ− oraz u ↔ g, to wtedy
11Σ+g ↔ 11Πu. Obydwa stany są singletami z czego pierwszy (stan podstawowy) jest
w stanie 2s + 2s natomiast drugi (stan wzbudzony) znajduje się w stanie 2s + 2p.
Następne stany wzbudzone znajdują się w 2s + 3s, 2p + 2p oraz 2s + 3p. Oczy-
wiście również z reguły wyboru ∆S = 0 wynika, że przejścia nie mogą odbywać
się z sigleta do tripleta i odwrotnie. Dla wygody naszych obliczeń posłużymy się
pierwszym stanem podstawowym oraz wzbudzonym stanem 11Πu. Przystępujemy
więc do etapu, w którym wykorzystany zostanie program liczący pochodną funkcji
w punkcie. Zanim jednak to nastąpi, należy pamiętać o próbkowaniu. Jest to bar-
dzo ważny detal, na który zwracałem uwagę w poprzednim rozdziale przy okazji
sprawdzania poprawności działania algorytmu numerycznego pochodnej funkcji w
punkcie. A ponieważ tabele z wartościami punktów dla krzywych potencjałów ma-
92

ją niewystarczającą łączną ich ilość, to w takim razie zostaną one wygenerowane.
Do tego celu wykorzystamy jakże niezastąpione narzędzie jakim jest nasz Gnuplot.
Nie ma sensu pisać nowych aplikacji, skoro są już znane i sprawdzone. Przy pomo-
cy Gnuplota zostaną zinterpolowane - algorytm interpolacji krzywej metodą cspline
(cubic spline). Jest to jedna z dokładniejszych metod, która pozwoli nam na dal-
sze wyliczenia. Zatem kod Gnuplota dla naszego potencjału (oczywiście w stanie
pobudzonym) będzie wyglądał identycznie jak na następującym listingu:
set term postscript eps enhanced
set output "PI.eps"
set size square
set origin 0, 0
set xlabel "R_AB"
set ylabel "V(R_AB)"
set xrange [0:15]
set yrange [5000:35000]
plot "poPiu.exp" u 1:2 smooth cspline title "1^1/Symbol P_u"
set term table
set output "data.dat"
set sample 100000
replot
Jak można zauważyć, Gnuplot najpierw narysuje nasz wykres zadany dla punktów
w pliku poPi.exp wykorzystując algorytm cspline, a następnie tak narysowaną funk-
cję zapisze w postaci dyskretnej. Oprócz tego należy pamiętać o dopisaniu w tym sa-
mym pliku wartości punktu dla którego szukamy pochodnej (Re, Ve). Jest to bardzo
istotne, abyśmy mieli jak najdokładniejsze dane celem uzyskania najlepszych wyni-
ków. Całość możemy przedstawić na wykresie, który zapisze się nam do pliku PI.eps
i będzie wyglądać, tak jak na schemacie 3.1. Zajmijmy się teraz wykresem krzywej
w postaci dyskretnej. Należy zwrócić szczególną uwagę na próbkowanie. Zostało
ono ustawione tym razem na dziesięć tysięcy punktów. Zauważmy zatem jak bardzo
nietrywialny jest nasz problem i jak dużo obliczeń wykonujemy po drodze. Nie mo-
93

5000
10000
15000
20000
25000
30000
35000
0 2 4 6 8 10 12 14
V(R
AB)
RAB
11Πu
Rysunek 3.1: Wykres krzywej adiabatycznej energii potencjalnej dla stanu wzbudzo-
nego 11Πu
żemy sobie pozwolić nawet na najdrobniejsze potknięcie. Dobrze więc, możemy teraz
przystąpić do obliczenia naszej pochodnej w punkcie (Re, Ve) = (5, 316; y) dla krzy-
wej potencjału w stanie pobudzonym 11Πu. Zauważmy, że Ve = y. Wartość poten-
cjału zostanie wyliczona przez nasz program. Należy również pamiętać o podaniu
dokładnej nazwy pliku wyjściowego (zamiana dane.dat na data.dat) w kodzie pro-
gramu. Przystępujemy zatem do uruchomienia pliku wykonywalnego styczne.out,
który wyświetli na naszym ekranie punkty oraz styczne do krzywej w tych punktach.
Ponieważ nasze próbkowanie jest na poziomie miliona punktów to może to trochę
potrwać. Tak jak w naszym wcześniejszym przykładzie, który wykorzystywaliśmy
do udowodnienia poprawności algorytmu, tak i tym razem wszystkie wyniki zostaną
zapisane do pliku styczne.exp. Niestety jest ich ogromnie dużo, to najprostszym
sposobem będzie skorzystanie z programu grep, który jest dostępny na każdej dys-
trybucji GNU/Linux. W tym celu wykonamy w terminalu następującą komendę
grep 5.316 styczne.exp, w wyniku której na naszym ekranie pojawi się zdecy-
dowanie mniej punktów razem z punktem szukanym przez nas, co widać z resztą
94

na poniższym listingu:
[3.80676,18421.5] : y = -9166.67x + 53316.8
[3.80712,18418] : y = -9166.67x + 53316.6
[3.80747,18414.5] : y = -9166.67x + 53316.3
[5.316,12034.3] : y = -833.333x + 16464.3
[5.31612,12034.3] : y = 0x + 12034.3
[5.31623,12034.2] : y = -909.091x + 16867.1
[5.31635,12034.1] : y = -833.333x + 16464.4
[5.31647,12034.1] : y = 0x + 12034.1
[5.31659,12034] : y = -833.333x + 16464.5
[5.31671,12033.9] : y = -833.333x + 16464.5
[5.31682,12033.9] : y = 0x + 12033.9
[5.31694,12033.8] : y = -833.333x + 16464.6
[9.58316,15257.3] : y = 0x + 15257.3
...
Tych wyników oczywiście wyświetli się nam więcej, jednakże szukany przez nas po-
winien być 4 z góry. Oczywiście my nie znamy jednoznacznie potencjału jaki odpo-
wiada naszej wartości odległości. Aczkolwiek wartości te zostały obliczone w pracy
[25], to są one w postaci dyskretnej. Dlatego też do tego celu wykorzystany zo-
stał algorytm cspline oraz narzucona duża liczba próbkowania. Po tak wykonanych
obliczeniach wartość energii czyli naszego Ve powinna być mniej więcej dokładna
i wynosi Ve(Re) = Ve(5, 316) = 12034, 3. Zatem należałoby sprawdzić czy rzeczywi-
ście szukana pochodna została poprawnie policzona. W tym celu zostanie stworzony
wykres reprezentujący dwie funkcje, prostą y = −833, 333x+ 16464, 3 oraz krzywą
zadaną w pliku data.dat, a listing z kodem wygląda następująco:
set term postscript eps enhanced
set output "pochodnaPI.eps"
set size square
set origin 0, 0
set xlabel "R_AB"
95

set ylabel "V(R_AB)"
set xrange [0:15]
set yrange [5000:35000]
unset key
set multiplot
plot "data.dat" u 1:2 smooth csplines
plot -833.333*x + 16464.3
unset multiplot
Natomiast wykres wygląda tak jak na schemacie 3.2. Widzimy zatem, że pochodna
5000
10000
15000
20000
25000
30000
35000
0 2 4 6 8 10 12 14
V(R
AB)
RAB
5000
10000
15000
20000
25000
30000
35000
0 2 4 6 8 10 12 14
V(R
AB)
RAB
Rysunek 3.2: Wykres krzywej adiabatycznej energii potencjalnej dla stanu wzbudzo-
nego 11Πu oraz styczna do niej prosta y = −833, 333x+ 16464, 3 jako pochodna
w naszym punkcie (Ve, Re) istnieje i wynosi f ′(5, 316) = −833, 333. Skoro tak, to nic
nie stoi na przeszkodzie abyśmy obliczyli w końcu nasz przekrój czynny na absorp-
cję promieniowania. Skorzystamy zatem ze wzoru 1.2.57, przy czym jak wiadomo
β =(
V 2R
αR
)−1
. Zatem podstawiąc do β wzór numer 1.2.52 otrzymamy, że przekrój
96

czynny na absorpcję wynosi odpowiednio:
σ(E) ≈ e−2
2
4
V 2R
mωHO2
!−13
5(E−Ve)2
~
VR, (3.0.1)
gdzie m to masa zredukowana naszej dwuatomowej cząsteczki litu, natomiast ωHO
to pulsacja oscylatora harmonicznego w stanie podstawowym. Ponieważ znamy ma-
sę atomową atomu Li i wynosi ona MALi= 6, 941 u, gdzie u jest to jednostka
masy atomowej mu = 1 u ≈ 1, 6605387313 ·10−27 kg ≈ 931, 494028(23) MeVc2
. Zatem
skoro znana jest masa atomowa Li, obliczona zostanie zatem zredukowana masa
dwuatomowej cząsteczki litu - Li2. A ponieważ cząsteczka nasza jest dwuatomowa,
wykorzystajmy więc wielkość służącą do opisu układu oddziałujących ze sobą ciał,
którą nazywamy masą zredukowaną. W przypadku np. dwóch mas oddziałujących
ze sobą grawitacyjnie przyjmuje ona postać:
1
µ=
1
m1+
1
m2. (3.0.2)
Wykonując przekształcenia matematyczne na powyższym wzorze 3.0.2:
1
µ=
m2
m1m2+
m1
m1m2=m1 +m2
m1m2.
Następnie mnożąc równość na krzyż otrzymamy:
µ(m1 +m2) = m1m2,
i podzielimy obustronnie przez (m1 +m2), wtedy nasza masa zredukowana wynosi:
µ =m1m2
m1 +m2
.
Przy czym zauważyć należy, że nasza cząsteczka dwuatomowa składa się z takich
samych atomów. Zatem należało by napisać, że m1 = m2. Możemy więc zapisać
wzór na masę zredukowaną w następującej postaci:
µ =m2
1
2m1=
1
2m1. (3.0.3)
97

W związku z powyższymi obliczeniami podstawiamy za m1 = MALi= 6, 941 u
i otrzymujemy, że masa zredukowana dla Li2 (dilitu) wynosi µ = 3, 4705 u. Mo-
żemy zatem przystąpić do obliczenia αR podstawiając za m = µ = 3, 4705 u na-
tomiast za ωHO pulsację dla Li2 [26], która wynosi ωLi2 = 352, 41 cm−1. Ponie-
waż wartość omegi jest liczbą falową (z ang. wavelenght), czasem zwana często-
ścią, zostanie ona zamieniona na częstotliwość w celu ułatwienia naszych obliczeń.
Zgodnie z obliczeniami w pracy [27], ω wynosi odpowiednio 2πν = 2π10 THz.
Podstawiając tak otrzymaną omegę do wzoru na αR otrzymamy wartość dla αR,
która zgodnie z równaniem 1.2.52 wynosi αR = 10, 89737 · 10 uTHz, przy czym
1u ≈ 1, 6605387313 · 10−27 kg, zatem:
αR = 18, 095504954306681 · 10−27 · 1013 kgHz =
= 18, 095504954306681 · 10−14 kgHz ≈
≈ 18, 0955 · 10−14 kgHz.
Przejdźmy zatem do obliczenia przekroju czynnego. Do tego celu wykorzystamy
już obliczoną przez nas pochodną potencjału VR = −833, 333 cm−1
ao. Wiemy z pra-
cy [28], że 1 cm−1 = 1, 987 · 10−23 J . Zatem podstawiając to do naszej pochodnej,
otrzymamy:
VR = −833, 333 · 1, 987 · 10−23J
a0= −1, 66 · 10−20 J
a0,
gdzie a0 = 0, 5 Å = 0, 5 · 10−10 m, a Jm
= N . Możemy więc zapisać, że pochodna
VR = −3, 3 · 10−10 N . Oprócz tego stała Plancka ~ = 1, 05457168(18) · 10−34 Js ≈≈ 1 · 10−34 Js, a potencjał Ve(Re) = Ve(5, 316) = 12034, 3 cm−1 = 23, 9121 ·10−20 J . Po tak dokonanych czynnościach możemy przystąpić do obliczenia szu-
kanej przez nas σ(E), podstawiając do wzoru 3.0.1 nasze wartości otrzymamy, że
przekrój czynny wynosi:
σ(E) ≈ e−
2
2
6
4
0
@
3,32·10−20 N2
18,0955·10−27·1013
kgs
1
A
−13
7
5(E−23,9121·10−20 J)2
1,05457168(18)·10−34 Js
|−3, 3 · 10−10 N | ≈∴
98

∴ ≈ e−2
0
@
18,0955·10−14 kgs
3,32·10−20 N2
1
A(E−23,9121·10−20 J)2
1·10−34 Js
3, 3 · 10−10 N≈
≈ e−2
0
@
18,0955·10−14 kgs
10,89·10−20N2
1
A(E−23,9121·10−20 J)2
1·10−34 Js
3, 3 · 10−10 N≈
≈ e−3,3233·10−14
·1020kgs
N2 (E−23,9121·10−20 J)2
1·10−34 Js
3.3 · 10−10 N≈
≈ e−3,3233·106
kgs
N2 (E−23,9121·10−20 J)2
1·10−34 Js
3, 3 · 10−10 N. (3.0.4)
W równaniu występuje podstawa logarytmu naturalnego (eksponent), zatem jed-
nostki w niej zawarte powinny wynieść 1. Na początek przyjmijmy, że tak jest.
Słuszność założenia sprawdzimy w następnym podrozdziale. Skoro tak, to możemy
zapisać dalej, że:
σ(E) ≈ e−3,3233·106 ·(E−23,9121·10−20)
2
1·10−34
3, 3 · 10−10 N,
przy czym w mianowniku tego ułamka obliczona pochodna VR jest w Newtonach.
A ponieważ wcześniej ją przekształciliśmy do takiej postaci to nic nie stoi na prze-
szkodzie aby z powrotem ją zamienić do postaci:
VR = −3, 3 · 10−10 J
m= −3, 3 ·
10−10
1,987·10−23 cm−1
m= −1, 666666 · 1015 1
m2.
Jak widać na powyższych przekształceniach, nasza pochodna wychodzi w 1m2 . Pod-
stawiając ją do wcześniejszych obliczeń dotyczących σ(E) otrzymamy, że:
σ(E) ≈ e−3,3233·106(E−23,9121·10−20)
2
10−34
1, 666666 · 1015m2.
Widzimy zatem, że poszukiwana przez nas funkcja σ(E) ma wymiar pola powierzch-
ni pod wykresem. Co się oczywiście zgadza z teorią dotyczącą przekroju czynnego
przytoczoną w rozdziale pierwszym niniejszej pracy magisterskiej. Natomiast wykres
naszej funkcji powinien być gaussianem. Skoro tak to narysujmy przy pomocy Gnu-
plota naszą funkcję sigma. Zatem kod potrzebny do wykreślenia naszego przekroju
będzie wyglądał tak jak na poniższym listingu:
99
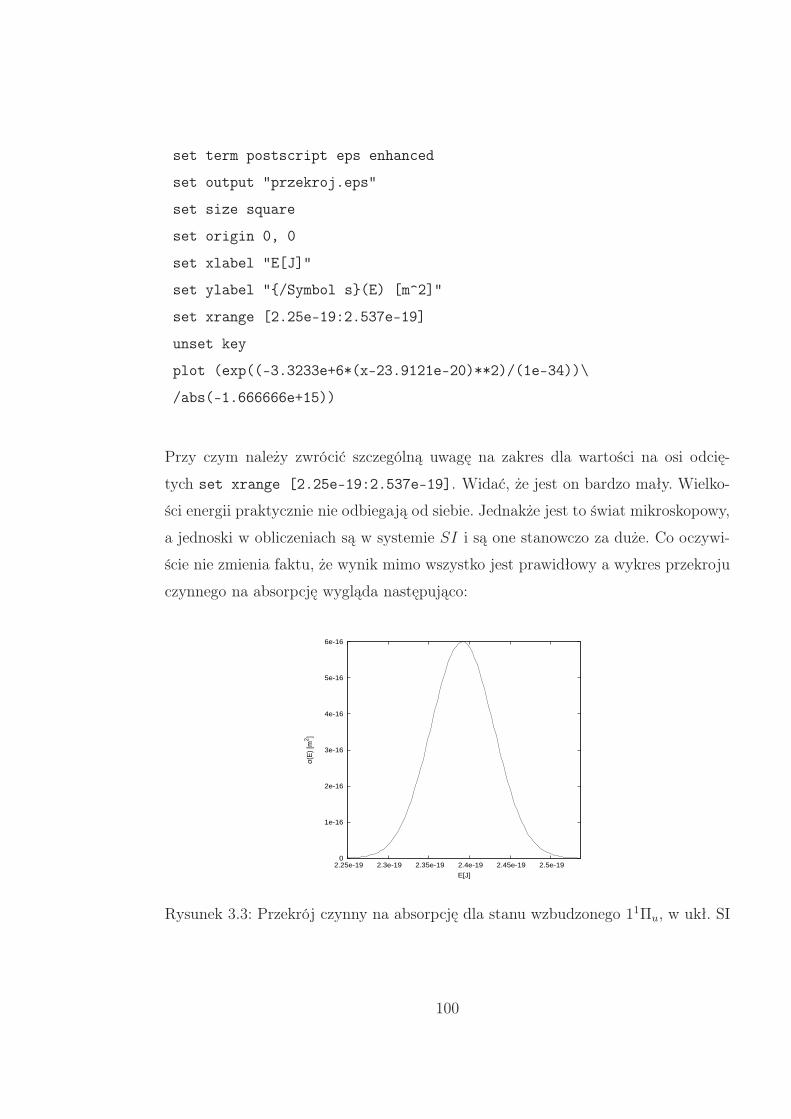
set term postscript eps enhanced
set output "przekroj.eps"
set size square
set origin 0, 0
set xlabel "E[J]"
set ylabel "/Symbol s(E) [m^2]"
set xrange [2.25e-19:2.537e-19]
unset key
plot (exp((-3.3233e+6*(x-23.9121e-20)**2)/(1e-34))\
/abs(-1.666666e+15))
Przy czym należy zwrócić szczególną uwagę na zakres dla wartości na osi odcię-
tych set xrange [2.25e-19:2.537e-19]. Widać, że jest on bardzo mały. Wielko-
ści energii praktycznie nie odbiegają od siebie. Jednakże jest to świat mikroskopowy,
a jednoski w obliczeniach są w systemie SI i są one stanowczo za duże. Co oczywi-
ście nie zmienia faktu, że wynik mimo wszystko jest prawidłowy a wykres przekroju
czynnego na absorpcję wygląda następująco:
0
1e-16
2e-16
3e-16
4e-16
5e-16
6e-16
2.25e-19 2.3e-19 2.35e-19 2.4e-19 2.45e-19 2.5e-19
σ(E
) [m
2 ]
E[J]
Rysunek 3.3: Przekrój czynny na absorpcję dla stanu wzbudzonego 11Πu, w ukł. SI
100

3.1 Jednostki
Sprawdźmy zatem jednostki. Ponieważ wcześniej zostało napisane założenie od-
nośnie jednostek występujących w potędze podstawy logarytmu naturalnego, w wy-
niku którego przyjęliśmy, że muszą one wynieść 1, zostanie przeprowadzony teraz do-
wód tego założenia. Nie bez powodu podczas obliczeń przez cały czas była zwracana
szczególna uwaga na jednostki podstawianych zmiennych. Przyglądając się oblicze-
niom w równaniu 3.0.4 możemy zapisać równanie jednostek w samym wykładniku,
które wygląda następująco:
kg·HzN2 · J2
J · s =kg·Hz
N2 · Js
=∴
a ponieważ J = N ·m oraz Hz = 1s, zatem:
∴=
kg· 1s
N2 ·N ·ms
=
kg· 1s
N·ms
=∴
z kolei N = kg · ms2 , a s
s= 1 więc:
∴=
kg·ms
N· s
s
s=
kg· m
s2
N· s
s=
NN· ss
= 1,
co należało wykazać.
Aby mieć nieco większe wyobrażenie o wymiarach przy jakich prawdopodobień-
stwo absorpcji dla przykładowej cząsteczki zachodzi, przedstawiony wcześniej wy-
kres przekształcony zostanie do nieco bardziej wygodnych jednostek, czyli jednostek
Heaviside’a-Lorentza. Należy więc odwołać się do równania 3.0.4. A ponieważ ekspo-
nent nie wpływa na jednostki i muszą się one skracać, można więc przejść z systemu
SI na system H-L. Do tego celu należy skorzystać z czynników przekształcających
nasze jednostki przedstawionych w tabeli 1.3 w pracy [29]. Podstawiając za 1 kg =
= 2, 84278882·1042 m−1, 1 m = 1 m, 1 s = 3·108 m oraz 1 J = 3, 16302914·1025 m−1
do naszego równania otrzymamy:
σ(E) ≈ e−0,31476505097·10−10 m2(E−75,6346690986·105 m−1)
2
0,9482528
1, 666666 · 1015m2,
101

gdzie m2 zamienimy dla wygody obliczeń na Å2, natomiast m−1 na cm−1. Przy czym
wartości w potędze mnoży się przez ~c, gdzie ~ = h2π
. Podstawiając, otrzymujemy:
σ(E) ≈ e−0,0500964137744·10−6 cm2(E−12,0376314562·103 cm−1)
2
0,150919120421
1, 666666 · 1015· 1020 Å
2. (3.1.1)
Na podstawie powyższego równania tworzy się nowy wykres w układzie H-L,
którego kod wygląda tak:
set term postscript eps enhanced
set output "przekrojH-L.eps"
set encoding iso
set size square
set origin 0, 0
set xlabel "E[cm^-1]"
set ylabel "/Symbol s(E) [\305^2]"
set xrange [7.6e+3:16.6e+3]
plot (exp((-0.05009641377e-6*(x-12.0376314562e+3)**2)/(0.1509191))\
/abs(-1.666666e+15))*(1e+20) title "1^1/Symbol P_u"
a wykres tak:
0
10000
20000
30000
40000
50000
60000
8000 9000 10000 11000 12000 13000 14000 15000 16000
σ(E
) [Å
2 ]
E[cm−1]
11Πu
Rysunek 3.4: Przekrój czynny na absorpcję dla stanu wzbudzonego 11Πu w ukł. H-L
102

Zatem został obliczony przekrój czynny na absorpcję promieniowania dla czą-
steczki dilitu. Wykres wygląda też tak jak powinien.
3.2 Wyniki i dyskusja
Po tak dokonanych obliczeniach należałoby się teraz zastanowić czy są one po-
prawne. Dla porównania przytoczę z rozdziału pierwszego niniejszej pracy,
że dla przykładu wody - H2O, pierwszy stan wzbudzenia występował dla warto-
ści energii w przybliżeniu równej 7, 5 eV . Niemniej jednak woda jest zupełnie in-
nym związkiem. W przypadku dilitu, fale energii powinny być z zakresu fal IR.
Tak jak ma to miejsce dla Rubidu, który to właśnie posłużył za obiekt badaw-
czy polskiej ekipie zajmującej się BEC, o czym z resztą zostało napisane w roz-
dziale 2 niniejszej pracy. Jeśli chodzi o Rb2 fala taka ma długość λ = 780 nm,
czyli E ≈ 1, 59 eV . Zatem zostanie teraz obliczona wartość energii dla najbardziej
prawdopodobnego przejścia ze stanu podstawowego do stanu pobudzonego. Jeże-
li przyjrzeć się równaniu 3.1.1 oraz wykresowi 3.4 zauważyć można, że tzw. liczba
falowa wynosi ν ≈ 12, 0376314562 · 103 cm−1 ≈ 12037, 6314562 cm−1. A ponieważ:
λ =1
ν,
więc:
λ =1
12037, 6314562 cm−1= 8, 30 · 10−5 cm = 8, 30 · 10−7 m = 830 nm.
Zatem korzystając ze wzoru 1.2.3 w rozdziale pierwszym można w bardzo trywialny
sposób obliczyć ile wynosi energia potrzebna do przejścia ze stanu podstawowego
11Σ+g do stanu 11Πu. A więc podstawmy za h = 4, 135667443 · 10−15 eV s, natomiast
c ≈ 3 · 108 ms
oraz obliczoną długość fali λ = 830 nm:
E =4, 135667443 · 10−15 eV s · 3 · 108 m
s
830 · 10−9 m≈ 1, 49 eV. (3.2.1)
Tak więc, aby zaistniało przejście 11Σ+g ↔ 11Πu wystarczy Ve ≈ 1, 49 eV , co się
w przybliżeniu zgadza porównując niniejsze wyniki dla cząsteczki Li2 z wynikami
103

dla cząsteczki Rb2. Widać również wyraźnie, że dla dłuższych fal są mniejsze energie
o czym była mowa w podrozdziale 1.2.1.
Można więc zauważyć, że wiązka promieniowania elektromagnetycznego potrzeb-
na do pobudzenia dilitu jest z zakresu większego niż długości fali dla światła wi-
dzialnego (400 − 700 nm). W związku z powyższym, nasza długość zaliczana jest
do zakresu promieniowania podczerwieni - IR - i mieści się ona na przedziale zakresu
bliskiej podczerwieni (ang. near infrared, NIR) - dla długości fali 0, 7−5 µm. Można
więc napisać, że pierwsze maksimum energetyczne dla dwuatomowej cząsteczki litu
- Li2 wynosi 1, 49 eV przy długości fali elektromagnetycznej λ = 830 nm dla prze-
kroju czynnego σ(E) = 60000 Å2, który jest niczym innym jak polem powierzchni.
Gdyby obliczyć całkę oznaczoną z równania 3.1.1 na przedziale [7600:16600], to po-
winna wyjść dokładnie ta sama wartość. Fakt ten wynika z definicji całki oznaczonej
w sensie Riemanna.
Definicja 3.2.1 Definicja intuicyjna - w sensie Riemanna.
Całka funkcji f na przedziale domkniętym [a, b] jest to pewna liczba, która w przy-
padku funkcji dodatnich mierzy powierzchnię między wykresem funkcji a osią OX.
Przypuśćmy, że f : R −→ [0,∞) oraz a < b. Pytamy się jakie jest pole powierzchni
figury S = (x, y) : a ≤ x ≤ b ∧ 0 ≤ y ≤ f(x). Aby obliczyć to pole, będziemy
przybliżać figurę S za pomocą skończonej, choć dowolnie dużej, liczby prostokątów.
Jeśli ten proces się uda, to otrzymaną wartość nazywamy całką Riemanna z funkcji
f na odcinku [a, b] i oznaczamy przez:
b∫
a
f(x) dx.
3.3 Zakończenie
Wszystko ma jakiś sens, a sens jest tylko wtedy kiedy się wie dlaczego się to
robi. Weźmy chociażby dla przykładu zjawisko zwane „efektem motyla”. Jest to
anegdotyczna ilustracja zjawiska chaosu deterministycznego, tj. wielkiej wrażliwo-
ści zachowania układów nieliniowych na małe zmiany warunków początkowych. W
tytułowej anegdocie trzepot skrzydeł motyla np. w Ohio może po trzech dniach
104

spowodować w Teksasie burzę piaskową. Przykładami efektów nielinowych są zja-
wiska meteorologiczne lub zmiany klimatu. W obecnych czasach zmiany klimatu są
wręcz namacalne. Wszystkim też ludziom coraz drastyczniej daje się odczuć globalne
ocieplenie. Nasze środowisko chroni warstwa ozonowa, jak zostało napisane we wstę-
pie, ozon chroni ludzi przed wysokim promieniowaniem UV. Bunsen z Kirchhoffem
stwierdzili, że widmo absorpcyjne jest jedyną w swoim rodzaju charakterystyką da-
nego związku chemicznego. Zatem skoro ozon ma jedyną dla siebie taką charakte-
rystykę, to czy może istnieć również związek chemiczny o podobnych własnościach,
który byłby w stanie uchronić organizmy żywe na naszej planecie? Niestety nie.
Wszystko ma swój sens w życiu, a jedna niepozornie zaczynająca się reakcja łańcu-
chowa może pozbawić nas tej jedynej ochrony. Nawet najmniejsze szczegóły i naj-
drobniejsze prace mają wielki wpływ na nasz wielki świat. Jeżeli przyjrzymy się ba-
danemu układowi w niniejszej pracy i porównamy ją do efektu motyla to niewątpli-
wie odniesiemy zaskakujące wrażenie, iż to właśnie fizyka kwantowa jest naukowym
udowodnieniem filozofii, która jest uniwersalna we wszystkich krajach, niezależnie
od położenia geograficznego.


Dodatek A: Płyta CD
Do niniejszej pracy dołączona jest płyta CD, która zawiera przedstawione ob-
liczenia oraz źródła programu wykorzystane w pracy. Na płycie tej znajduje się
również sama praca dyplomowa w formacie pdf.
107

Literatura
[1] M. Planck, „On the Law of Distribution of Energy in the Normal Spectrum”
Annalen der Physik, vol. 4, p. 553 ff (1901)
[2] C.M. Lovejoy and D.J. Nesbitt, „Mode specific internal and direct rotational
predissociation in HeHF, HeDF, and HeHCl: van der Waals complexes in the
weak binding limit” Chem. Phys. 93 (1990) 5387
[3] B.J. Howard and A.S. Pine, „Near-infrared spectra of rare gas-HCl complexes”
Chem. Phys. 122 (1985) 1
[4] A.E. Bruno, U. Brühlmannan and J.R. Huber, „Photofragmentation LIF spec-
troscopy of NOCl at dissociation wavelengths > 450 nm” Chem. Phys. 120
(1988) 155-167
[5] P.A. Giguèra, „Revised Values of the O-O and the O-H Bond Dissociation Ener-
gies” Chem. Phys. 30 (1959) 322
[6] G. Herzberg, „Molecular Spectra and Molecular Structure I. Spectra of Poly-
atomic Molecules”, Van Nostrand, New York 1950
[7] G. Herzberg, „Molecular Spectra and Molecular Structure III. Spectra of Poly-
atomic Molecules”, Van Nostrand, New York 1967
[8] J.W. Rabalais, J.M. McDonald, V. Scherr, and S.P. McGlynn, Chem. Rev. 71,
73 (1971)
[9] M.B. Robin, „Higher Excited States in Polyatomic Molecules”, Vol. I, Academic
Press, New York 1974
108

[10] M.B. Robin, „Higher Excited States in Polyatomic Molecules”, Vol. II, Academic
Press, New York 1975
[11] M.B. Robin, „Higher Excited States in Polyatomic Molecules”, Vol. III, Acade-
mic Press, New York 1985
[12] H. Okabe, „Photochemistry of Small Molecules”, Wiley, New York, 1978
[13] P. Gürtler, V. Saile, E.E. Koch „Rydberg series in the absorption spectra of
H2O and D2O in the vacuum Ultraviolet” Chem. Phys. 51 (1977) 386-391
[14] E.P. Wigner, „Über die Operation der Zeitumkehr in der Quantenmechanik /
On the Time Inversion Operation in Quantum Mechanics” Nachr. Akad. Ges.
Wiss. Göttingen 31, p.546 (1932)
[15] M. Hillery, R.F. O’Connel, M.O. Scully, and E.P. Wigner, „Distribution func-
tions in physics: fundamentals” Phys. Rep. 106 (1984) 121–167
[16] R. Schinke, „Photodissociation Dynamics”, Cambridge University Press, Cam-
bridge, 1993
[17] P. Kowalczyk „Fizyka cząsteczek”, Wydawnictwo Naukowe PWN, Warszawa,
2000
[18] http://www.research.att.com/~njas/sequences/A078434 2008
[19] http://nobelprize.org/nobel_prizes/physics/laureates/2001/
cornellwieman-lecture.pdf 2008
[20] C. Towndend, W. Ketterle, S. Strignari, „Bose-Einstein Condensation” Postępy
Fizyki, Tom 48, Zeszyt 4 (1997) 333-349
[21] Postępy Fizyki, Tom 58, Zeszyt 4 (2007)
[22] J.Zachorowski, T. Pałasz, W. Gawlik, Postępy Fizyki 49 (1998) 338
[23] M. Inguscio, Proc. of Science (EMC2006) 008
109

[24] E. Majorana, Nuovo Cimento 9, 43 (1932)
[25] P. Jasik and J.E. Sienkiewicz „Calculation of adiabatic potentials of Li2” Chem.
Phys. 323 (2006) 563-573
[26] Journal of NUCLEAR SCIENCE and TECHNOLOGY, Vol. 42, No. 4, p.
362–367 (April 2005)
[27] http://mlyniec.gda.pl/~chemia/ogolna/energia_cz.htm 2008
[28] http://www.ftj.agh.edu.pl/~fiszer/laser/lasery.htm 2008
[29] G.W.F. Drake, „Springer Handbook of Atomic, Molecular, and Optical Physics”,
Springer Verlag, Berlin 2005
[30] http://en.wikipedia.org/wiki/Bose%E2%80%93Einstein_condensate
2008
[31] http://en.wikipedia.org/wiki/Cross_section_%28physics%29 2008
[32] http://en.wikipedia.org/wiki/Absorption_cross_section 2008
[33] http://en.wikipedia.org/wiki/Dilithium 2008
[34] http://en.wikipedia.org/wiki/Photodissociation 2008


![:VW S G R& - mif.pg.gda.pl · • metody numeryczne, • $ojr u\wp\uyzq r ohjáh • =d vwrvrzdqlhu y * q\fk urgrzl v nn r psxwhurz\fkgrreol f ]h qxp h u\ f ]q \ fk 8zdjlu y * qh](https://static.fdocuments.pl/doc/165x107/5c77a3fe09d3f2a94e8c0b6f/vw-s-g-r-mifpggdapl-metody-numeryczne-ojr-uwpuyzq-r-ohjah.jpg)















