
Niepełnosprawność intelektualna - ibb.waw.pl‚nosprawność... · •Niepełnosprawność...
16
Niepełnosprawność intelektualna – stan badań a możliwości diagnostyki molekularnej Agnieszka Charzewska Zakład Genetyki Medycznej Instytut Matki i Dziecka
Transcript of Niepełnosprawność intelektualna - ibb.waw.pl‚nosprawność... · •Niepełnosprawność...

Niepełnosprawność intelektualna – stan badań a możliwości
diagnostyki molekularnej
Agnieszka Charzewska Zakład Genetyki Medycznej Instytut Matki i Dziecka

-umiejętność porozumiewania się,
-sprawność w zakresie samoobsługi,
-radzenie sobie z czynnościami dnia codziennego
-sprawności interpersonalnych
-umiejętności podejmowania decyzji (kierowanie sobą)
-dbania o własne zdrowie i bezpieczeństwo
-radzenie sobie w szkole
-organizowanie czasu wolnego
-radzenie sobie w pracy
Niepełnosprawność intelektualna (NI, ID)
zaburzenie rozwojowe ujawniające się przed 18 rokiem życia,
polegające na znacznym
obniżeniu ogólnego poziomu funkcjonowania intelektualnego,
któremu towarzyszy
deficyt w zakresie zachowań adaptacyjnych:

NI Częstość występowania - 2% populacji generalnej
Częściej chorują mężczyźni niż kobiety (1.4/1) - NI sprzężona z chromosomem X (XLID)
W populacjach zachodnich przeważa autosomalna dominująca NI (mutacje de novo,
częściej na chromosomie odojcowskim), w populacjach krewniaczych (Bliski Wschód)
przeważa autosomalna recesywna postać NI
NI towarzyszy iloraz inteligencji poniżej 70 punktów na skali IQ Wechslera:
69 – 55 - NI w stopniu lekkim – poziom intelektualny charakterystyczny
dla 10 - 12 roku życia. Ta forma deficytu intelektualnego stanowi najwięcej,
ok. 85% rozpoznań.
54 – 35 - NI w stopniu umiarkowanym - funkcjonowanie intelektualne
na poziomie 6-9 roku życia. Częstotliwość występowania – ok. 10% z wszystkich
4 typów upośledzenia.
34 – 20 - NI w stopniu znacznym - poziom rozwoju 3-6-latka. Ok. 4- 5 roku
życia zauważalne opóźnienie rozwoju psychofizycznego.
poniżej 20 - NI w stopniu głębokim - poziom funkcjonowania
odpowiadający max. 3 roku życia.

Genetyczne podłoże NI
Aberacje chromosomowe
i submikroskopowe
• Aneuploidie – trisomia 21,13,18; 45X0,47XXY,47XXX
• Zespoły mikrodelecyjne: z.DiGeorge’a (22q11.2), Wiliamsa, Smitha-Magenisa, Millera-Diekera, Rubinsteina-Taybi’ego
• Pradera-Willi’ego i Angelmana
• Subtelomerowe 1p36; 22q13.3
Monogenowe autosomalne (700 genów)
• Dominujące nerwiakowłókniakowatość typu 1 z. Noonan stwardnienie guzowate
• Recesywne fenyloketonuria galaktozemia homocystynuria choroba Taya-Sachsa
Monogenowe
sprzężone z chromosomem X (XLID - 100 genów)
• Specyficzne z. Westa z.Retta z.Coffin-Lowry’ego
• Niespecyficzne - NSXLID
• Z. łamliwego chromosomu X (FraX) 2% NI, 10% chłopców z NI
Dysregulacja szlaków komórkowych związanych z:
•Synaptogenezą, funkcją synaps
•Neurogenezą, migracją neuronalną
•Regulacją ekspresji genów (transkrypcji, translacji, remodelingu chromatyny)
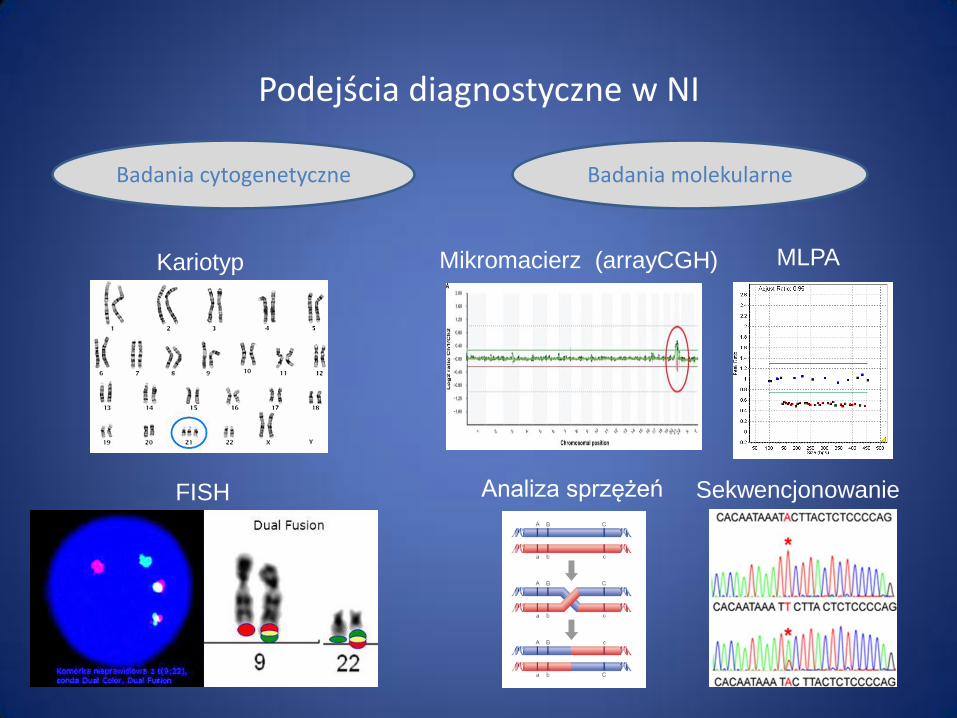
Kariotyp
FISH
Podejścia diagnostyczne w NI
Sekwencjonowanie
Badania cytogenetyczne Badania molekularne
MLPA Mikromacierz (arrayCGH)
Analiza sprzężeń
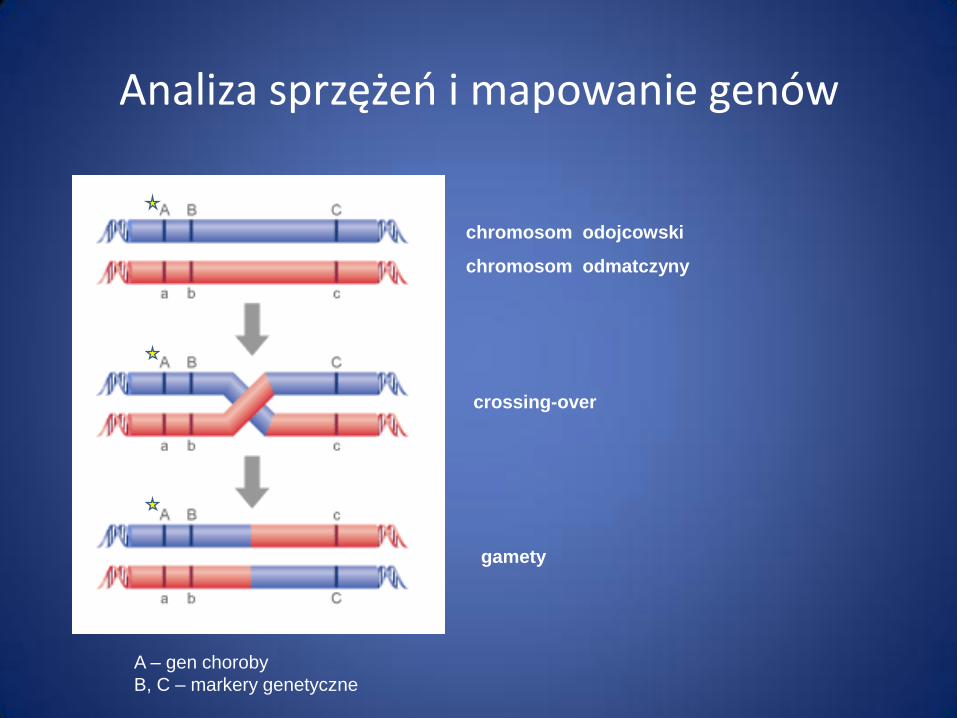
Analiza sprzężeń i mapowanie genów
chromosom odojcowski
chromosom odmatczyny
crossing-over
gamety
A – gen choroby
B, C – markery genetyczne
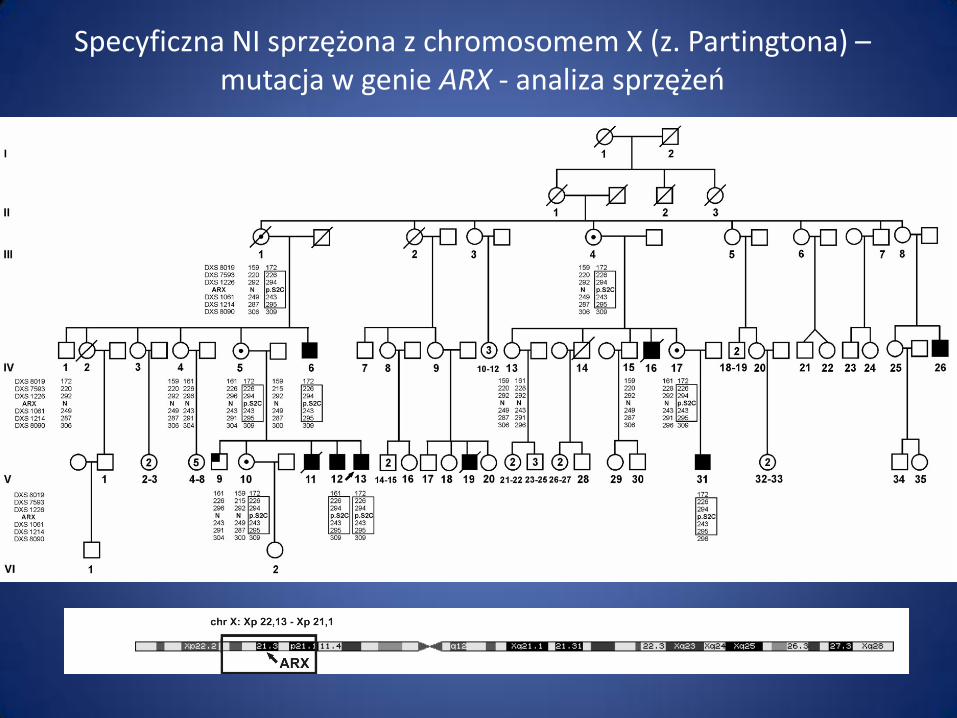
Specyficzna NI sprzężona z chromosomem X (z. Partingtona) – mutacja w genie ARX - analiza sprzężeń
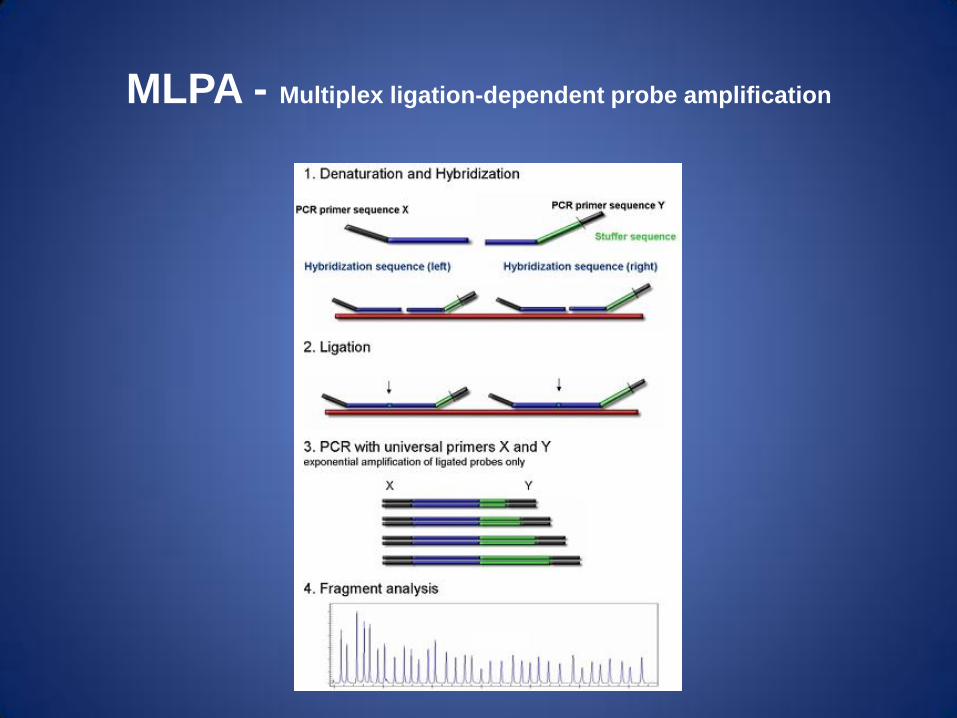
MLPA - Multiplex ligation-dependent probe amplification
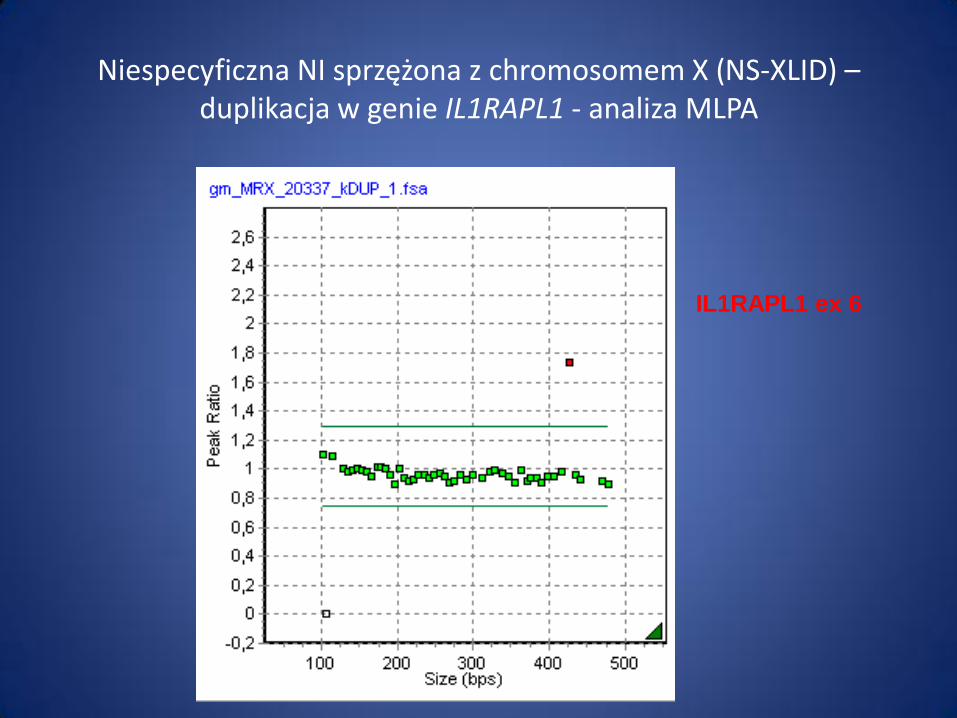
Niespecyficzna NI sprzężona z chromosomem X (NS-XLID) – duplikacja w genie IL1RAPL1 - analiza MLPA
IL1RAPL1 ex 6
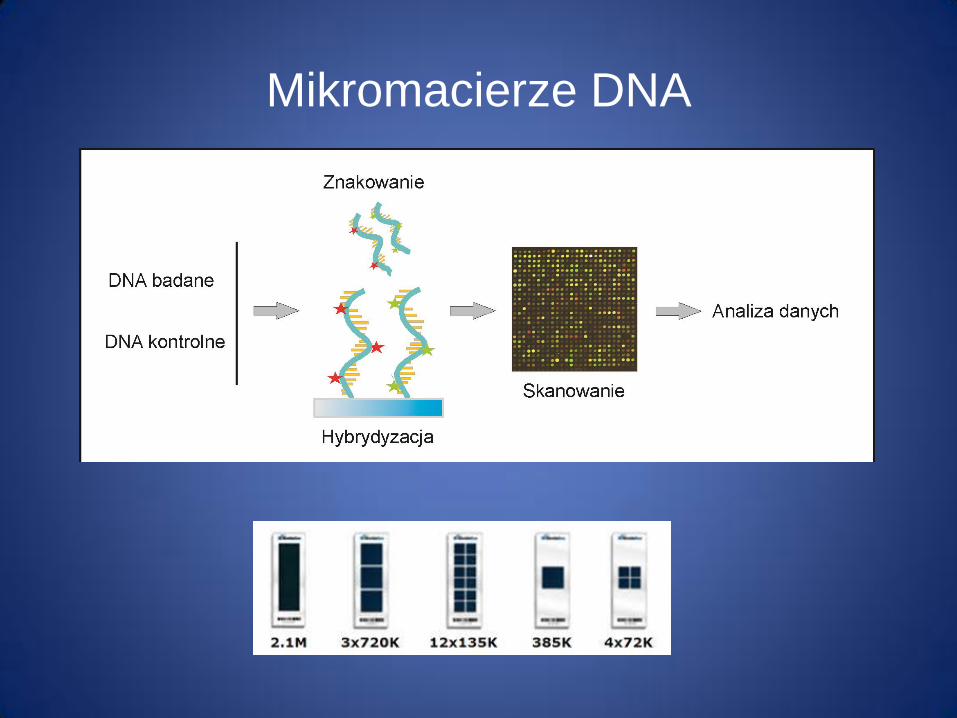
Mikromacierze DNA
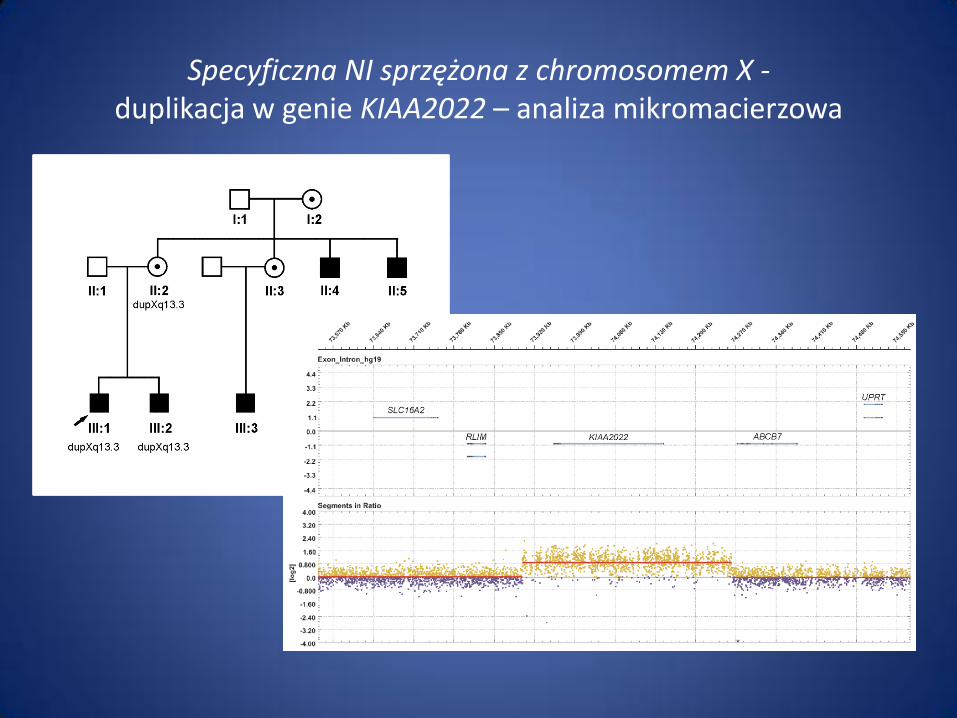
Specyficzna NI sprzężona z chromosomem X - duplikacja w genie KIAA2022 – analiza mikromacierzowa
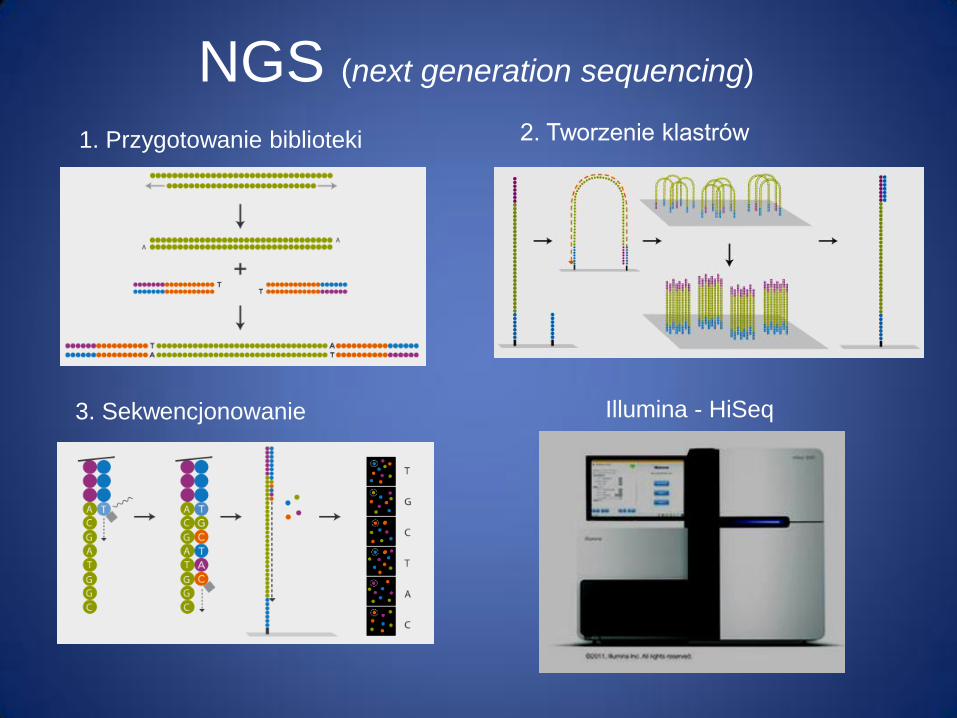
NGS (next generation sequencing)
1. Przygotowanie biblioteki 2. Tworzenie klastrów
3. Sekwencjonowanie Illumina - HiSeq
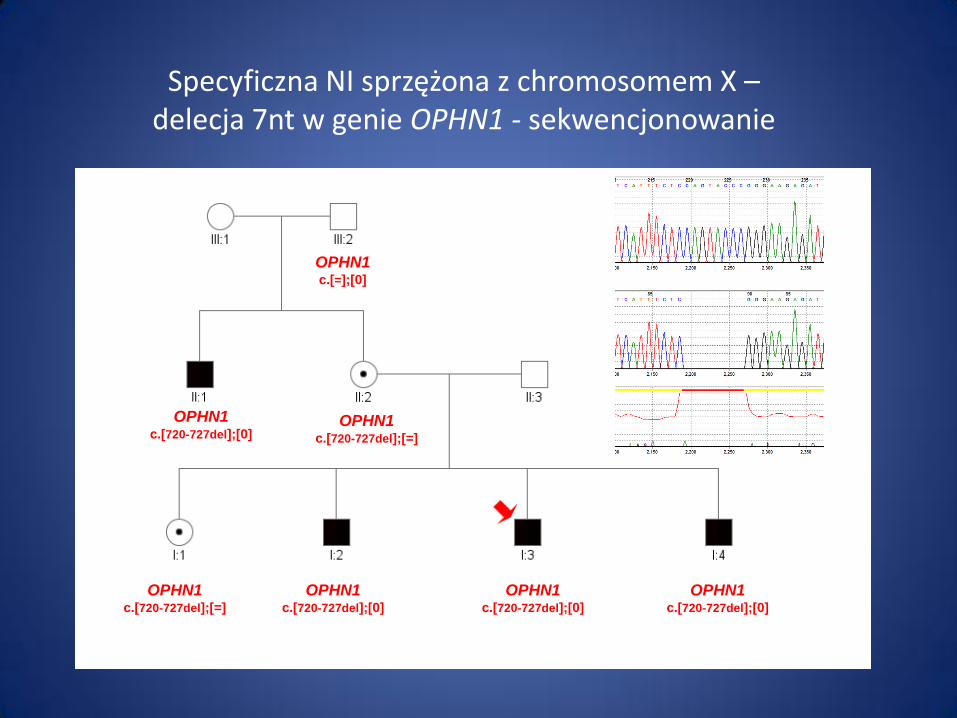
OPHN1 c.[720-727del];[=]
OPHN1 c.[720-727del];[=]
OPHN1 c.[=];[0]
OPHN1 c.[720-727del];[0]
OPHN1 c.[720-727del];[0]
OPHN1 c.[720-727del];[0]
OPHN1 c.[720-727del];[0]
Specyficzna NI sprzężona z chromosomem X – delecja 7nt w genie OPHN1 - sekwencjonowanie
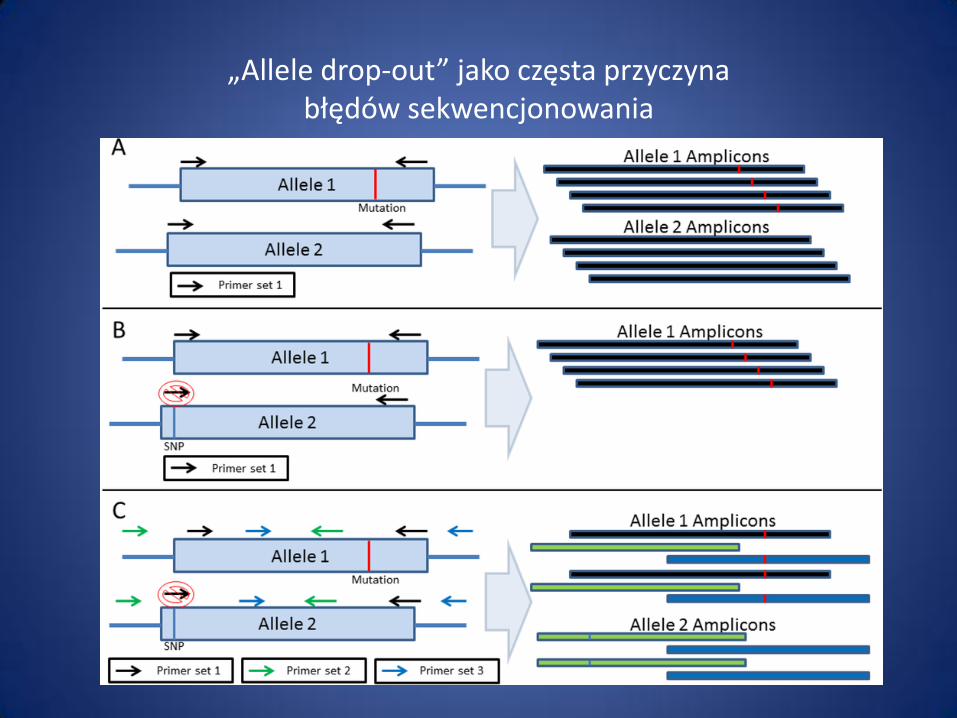
„Allele drop-out” jako częsta przyczyna błędów sekwencjonowania
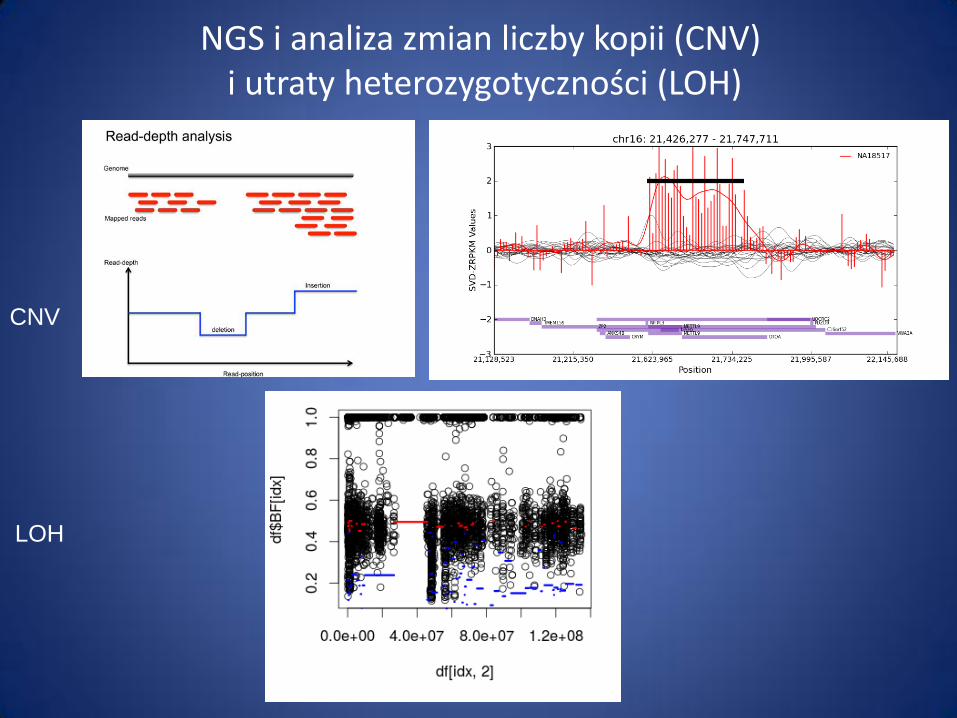
NGS i analiza zmian liczby kopii (CNV) i utraty heterozygotyczności (LOH)
CNV
LOH

Podsumowanie •Niepełnosprawność intelektualna (NI) występuje z częstością 2% w populacji. Jest to
heterogenna grupa chorób neurorozwojowych, którą należy traktować jako zbiór
chorób rzadkich
•Na NI częściej chorują mężczyźni niż kobiety, co związane jest z występowaniem NI
sprzężonej z chromosomem X
NI jest skutkiem dysregulacji szlaków komórkowych związanych z:
- synaptogenezą i funkcją synaps
- neurogenezą i migracją neuronalną
- regulacją ekspresji genów
•Podłoże genetyczne NI stanowią zmiany w DNA na różnych poziomach organizacji
genomu - od aberacji chromosomowych, przez zespoły mikrodelecyjne, aż do chorób
monogenowych.
•Najczęstszą przyczyną NI jest zespół Downa, natomiast najczęstszą monogenową
przyczyną NI jest zespół FraX. Zaleca się przesiewowe badanie w kierunku FraX u
wszystkich pacjentów z NI
•NGS jest obecnie najbardziej efektywną techniką diagnozowania NI i umożliwia
kompleksowe badanie genomu, obok mutacji punktowych także rearanżacje (delecje i
duplikacje)


















