
Kardiomiopatia przerostowa ze 2 śródkomorowym zawężaniem ... · Centrum Urazowe Medycyny...
5
271 Przegląd Lekarski 2015 / 72 / 5 Wstęp Kardiomiopatia przerostowa (HCM, hypertrophic cardiomyopathy) jest definio- wana jako przerost miokardium o nieznanej przyczynie, który jest zazwyczaj (ale nie zawsze) asymetryczny i związany z obec- nością zmian mikroskopowych włókien mię- śnia sercowego [1]. Opublikowane w 2014 roku wytyczne Europejskiego Towarzystwa Kardiologicznego na temat kardiomiopatii przerostowej podają, że rozpoznanie HCM można ustalić jeśli grubość ściany lewej komory w jednym lub więcej segmentów wy- nosi ≥ 15 mm mierzona przy użyciu dowolnej techniki diagnostycznej, tj. echokardiografii, rezonansu magnetycznego lub tomografii komputerowej. Warunkiem koniecznym do rozpoznania jest, aby przerostu nie można było wytłumaczyć jedynie nadmiernym ob- ciążeniem lewej komory. Wartości pośrednie grubości ściany lewej komory (13-14 mm) także nie wykluczają rozpoznania HCM, ale wówczas diagnostyka wymaga uzupełnienia o dodatkowe elementy, takie jak wywiad PRACE KAZUISTYCZNE Kardiomiopatia przerostowa ze śródkomorowym zawężaniem lewej komory (MVO) - opis przypadku Hypertrophic cardiomyopathy with midventricular obstruction of the left ventricle (MVO)- case report 1 Szpital Uniwersytecki w Krakowie, Centrum Urazowe Medycyny Ratunkowej i Katastrof, Oddział Obserwacyjno-Internistyczny Kierownik: Lek. med. Janusz Kąkol 2 Uniwersytet Jagielloński w Krakowie, I Oddział Kliniczny Kardiologii i Elektrokardiologii Interwencyjnej oraz Nadciśnienia Tętniczego Kierownik: Prof. dr hab. med. Danuta Czarnecka 3 Szpital Uniwersytecki w Krakowie, Oddział Kliniczny Hematologii Kierownik: Prof. dr hab. med. Aleksander B. Skotnicki Dodatkowe słowa kluczowe: kardiomiopatia przerostowa śródkomorowe zaciskanie Additional key words: hypertrophic cardiomyopathy midventricular obstruction Adres do korespondencji: Centrum Urazowe Medycyny Ratunkowej i Katastrof, Oddział Obserwacyjno-Internistyczny Szpital Uniwersytecki w Krakowie 31-501 Kraków, ul. Kopernika 50, tel. 12-351-66-73 e-mail: [email protected] Tomasz PIZOŃ 1 Marek RAJZER 2 Wiktoria WOJCIECHOWSKA 2 Marek JASTRZĘBSKI 2 Agnieszka OLSZANECKA 2 Marta ROJEK 2 Artur JURCZYSZYN 3 Danuta CZARNECKA 2 76-letnia kobieta z wywiadem nadciśnienia tętniczego w stopniu 3, napadowego migotania przedsionków, hipercholesterolemii i cukrzycy typu 2. Pierwszą kliniczną manifestacją choroby był częstoskurcz komorowy. W badaniu echokardiograficznym stwierdzono kardiomiopatię przerosto- wą z wysokim wewnątrzkomorowym gradientem sięgającym 47 mmHg i śródkomorowym zaciskaniem na poziomie mięśni brodawkowatych (wielkość jamy lewej komory wyno- siła 1-2 mm w okresie skurczu). Nie uwidoczniono tętniaka lewej komory, natomiast komora była wydłużona i poszerzona w części okołokoniusz- kowej, której czynność była obniżona, ale zsynchronizowana w czasie. Obraz naczyń wieńcowych w koronarografii był prawidłowy. W ramach prewencji nagłej śmierci sercowej implantowano jednojamowy kardiowerter-defibrylator (ICD, implantable cardioverter-defibril- lator). W leczeniu antyarytmicznym zastosowano metoprolol o przedłu- żonym uwalnianiu oraz amiodaron. Obecny przypadek przedstawia rzadki rodzaj kardiomiopatii przerostowej u starszej kobiety charakteryzujący się śródkomorowym zaciskaniem. A 76-year old woman with a history of stage 3 arterial hypertension, parox- ysmal atrial fibrillation, hypercholeste- rolemia and type 2 diabetes mellitus. Ventricular tachycardia was the first clinical manifestation of the disease. Echocardiography revealed hyper- trophic cardiomyopathy with a high in- traventricular gradient of 47 mmHg and midventricular obstruction at the level of the papillary muscles (the lumen of the left ventricle was 1-2 mm during systole). No ventricular aneurysm was found but the ventricle was elongated and dilated in the periapical part where systolic function was decreased but synchronized in time. Coronary an- giograms showed no narrowing of coronary arteries. A single-chamber cardioverter-defibrillator (ICD, implant- able cardioverter-defibrillator) was implanted to prevent sudden cardiac death. Modified-release metoprolol and amiodarone were administered in antiarrhythmic therapy. This case represents a rare kind of hypertrophic cardiomyopathy in an elderly woman which is characterized by midventricu- lar obstruction. rodzinny, występowanie objawów poza- sercowych, nieprawidłowości w zakresie elektrokardiogramu, badań laboratoryjnych czy obrazowych [2]. Wewnątrzkomorowe zawężanie w zakresie lewej komory jest istotnym patofi- zjologicznym elementem HCM i klasycznie występuje w odcinku podaortalnym, głównie w wyniku skurczowego ruchu przedniego płatka mitralnego (SAM, systolic anterior motion). To subaortalne zawężanie, na- zywane zawężaniem drogi odpływu lewej komory, występuje u ok. 25% pacjentów z HCM i jest niezależnym predyktorem niekorzystnych następstw klinicznych. Mię- śniowe zawężanie może być także obecne w odcinku śródkomorowym (MVO, midven- tricular obstruction) jako wynik przylegania przerośniętych mięśni brodawkowatych do przegrody międzykomorowej lub nieprawi- dłowego połączenia mięśnia brodawkowa- tego z płatkiem mitralnym [3,4]. Klasyczna (sarkomerowa) postać HCM cechuje się bezładnym ułożeniem prze-
Transcript of Kardiomiopatia przerostowa ze 2 śródkomorowym zawężaniem ... · Centrum Urazowe Medycyny...

271Przegląd Lekarski 2015 / 72 / 5
WstępKardiomiopatia przerostowa (HCM,
hypertrophic cardiomyopathy) jest definio-wana jako przerost miokardium o nieznanej przyczynie, który jest zazwyczaj (ale nie zawsze) asymetryczny i związany z obec-nością zmian mikroskopowych włókien mię-śnia sercowego [1]. Opublikowane w 2014 roku wytyczne Europejskiego Towarzystwa Kardiologicznego na temat kardiomiopatii przerostowej podają, że rozpoznanie HCM można ustalić jeśli grubość ściany lewej komory w jednym lub więcej segmentów wy-nosi ≥ 15 mm mierzona przy użyciu dowolnej techniki diagnostycznej, tj. echokardiografii, rezonansu magnetycznego lub tomografii komputerowej. Warunkiem koniecznym do rozpoznania jest, aby przerostu nie można było wytłumaczyć jedynie nadmiernym ob-ciążeniem lewej komory. Wartości pośrednie grubości ściany lewej komory (13-14 mm) także nie wykluczają rozpoznania HCM, ale wówczas diagnostyka wymaga uzupełnienia o dodatkowe elementy, takie jak wywiad
PRACE KAZUISTYCZNE
Kardiomiopatia przerostowa ze śródkomorowym zawężaniem lewej komory (MVO) - opis przypadkuHypertrophic cardiomyopathy with midventricular obstruction of the left ventricle (MVO)- case report
1Szpital Uniwersytecki w Krakowie, Centrum Urazowe Medycyny Ratunkowej i Katastrof, Oddział Obserwacyjno-InternistycznyKierownik: Lek. med. Janusz Kąkol
2Uniwersytet Jagielloński w Krakowie, I Oddział Kliniczny Kardiologii i Elektrokardiologii Interwencyjnej oraz Nadciśnienia TętniczegoKierownik: Prof. dr hab. med. Danuta Czarnecka
3Szpital Uniwersytecki w Krakowie, Oddział Kliniczny HematologiiKierownik: Prof. dr hab. med. Aleksander B. Skotnicki
Dodatkowe słowa kluczowe:kardiomiopatia przerostowaśródkomorowe zaciskanie
Additional key words:hypertrophic cardiomyopathymidventricular obstruction
Adres do korespondencji:Centrum Urazowe Medycyny Ratunkoweji Katastrof, Oddział Obserwacyjno-Internistyczny Szpital Uniwersytecki w Krakowie31-501 Kraków, ul. Kopernika 50, tel. 12-351-66-73e-mail: [email protected]
Tomasz PIZOŃ1
Marek RAJZER2
Wiktoria WOJCIECHOWSKA2
Marek JASTRZĘBSKI2
Agnieszka OLSZANECKA2
Marta ROJEK2
Artur JURCZYSZYN3
Danuta CZARNECKA2
76-letnia kobieta z wywiadem nadciśnienia tętniczego w stopniu 3, napadowego migotania przedsionków, hipercholesterolemii i cukrzycy typu 2. Pierwszą kliniczną manifestacją choroby był częstoskurcz komorowy. W badaniu echokardiograficznym stwierdzono kardiomiopatię przerosto-wą z wysokim wewnątrzkomorowym gradientem sięgającym 47 mmHg i śródkomorowym zaciskaniem na poziomie mięśni brodawkowatych (wielkość jamy lewej komory wyno-siła 1-2 mm w okresie skurczu). Nie uwidoczniono tętniaka lewej komory, natomiast komora była wydłużona i poszerzona w części okołokoniusz-kowej, której czynność była obniżona, ale zsynchronizowana w czasie. Obraz naczyń wieńcowych w koronarografii był prawidłowy. W ramach prewencji nagłej śmierci sercowej implantowano jednojamowy kardiowerter-defibrylator (ICD, implantable cardioverter-defibril-lator). W leczeniu antyarytmicznym zastosowano metoprolol o przedłu-żonym uwalnianiu oraz amiodaron. Obecny przypadek przedstawia rzadki rodzaj kardiomiopatii przerostowej u starszej kobiety charakteryzujący się śródkomorowym zaciskaniem.
A 76-year old woman with a history of stage 3 arterial hypertension, parox-ysmal atrial fibrillation, hypercholeste-rolemia and type 2 diabetes mellitus. Ventricular tachycardia was the first clinical manifestation of the disease. Echocardiography revealed hyper-trophic cardiomyopathy with a high in-traventricular gradient of 47 mmHg and midventricular obstruction at the level of the papillary muscles (the lumen of the left ventricle was 1-2 mm during systole). No ventricular aneurysm was found but the ventricle was elongated and dilated in the periapical part where systolic function was decreased but synchronized in time. Coronary an-giograms showed no narrowing of coronary arteries. A single-chamber cardioverter-defibrillator (ICD, implant-able cardioverter-defibrillator) was implanted to prevent sudden cardiac death. Modified-release metoprolol and amiodarone were administered in antiarrhythmic therapy. This case represents a rare kind of hypertrophic cardiomyopathy in an elderly woman which is characterized by midventricu-lar obstruction.
rodzinny, występowanie objawów poza-sercowych, nieprawidłowości w zakresie elektrokardiogramu, badań laboratoryjnych czy obrazowych [2].
Wewnątrzkomorowe zawężanie w zakresie lewej komory jest istotnym patofi-zjologicznym elementem HCM i klasycznie występuje w odcinku podaortalnym, głównie w wyniku skurczowego ruchu przedniego płatka mitralnego (SAM, systolic anterior motion). To subaortalne zawężanie, na-zywane zawężaniem drogi odpływu lewej komory, występuje u ok. 25% pacjentów z HCM i jest niezależnym predyktorem niekorzystnych następstw klinicznych. Mię-śniowe zawężanie może być także obecne w odcinku śródkomorowym (MVO, midven-tricular obstruction) jako wynik przylegania przerośniętych mięśni brodawkowatych do przegrody międzykomorowej lub nieprawi-dłowego połączenia mięśnia brodawkowa-tego z płatkiem mitralnym [3,4].
Klasyczna (sarkomerowa) postać HCM cechuje się bezładnym ułożeniem prze-

272 T. Pizoń i wsp.
rośniętych kardiomiocytów, które zwykle otaczają ogniska włóknienia mięśnia ser-cowego (disarray). Wśród mechanizmów patofizjologicznych główną rolę odgrywa dysfunkcja rozkurczowa lewej komory i niedokrwienie mięśnia sercowego, rzadziej dysfunkcja skurczowa [5].
Rys historyczny:Historia nowoczesnych opisów kardio-
miopatii przerostowej ma już ponad 50 lat. Na podstawie badań hemodynamicznych, cewnikowania serca i postępowania opera-cyjnego Brock w roku 1957 [6], a na podsta-wie badań autopsyjnych Teare w roku 1958 [7] przedstawili swoje opisy tej jednostki cho-robowej. Obecnie HCM jest rozpoznawana jako najpowszechniejsza choroba serca występująca rodzinnie, charakteryzująca się heterogennością obrazu i przebiegu klinicz-nego oraz ekspresji fenotypu, podobnie jak szerokie jest spektrum strategii leczniczych wobec tak różnorodnego obrazu klinicznego [8]. W 1976 roku Falicov i wsp. ogłosili opisy przypadków dwóch pacjentów, u których zaciskanie w obrębie lewej komory było obecne w odcinku śródkomorowym (MVO), a nie na łączu dróg napływu i odpływu [9]. Ponad połowa pacjentów z HCM ma jedno-znaczny dodatni wywiad rodzinny choroby i/lub nagłego zgonu sercowego, który jest zgodny z genetyczną cechą autosomalnie dominującą, co wskazuje na genetyczne tło kardiomiopatii przerostowej. Etiologia HCM była nieustalona aż do roku 1990, kiedy to poprzez ocenę sprzężeń genetycznych z na-stępową analizą genów kandydatów, wykry-to mutację genu MYH7 kodującego łańcuch ciężki sercowej β-miozyny u rodziny obcią-żonej HCM. Następnie, stosując tę samą metodologię, podjęto wysiłki zmierzające do ustalenia mutacji wywołujących chorobę, co zaowocowało wykryciem setek mutacji w kilkunastu różnych genach [10]. Najczęściej występują mutacje genu łańcucha ciężkiego β-miozyny (30% rodzinnych postaci HCM), genu białka C wiążącego miozynę (ok. 30% rodzinnych postaci HCM), genu sercowej troponiny (obciążone dużym ryzykiem na-głego zgonu; niekiedy nie stwierdza się przerostu, tylko zaburzenia układu włókien mięśniowych) [5]. Oprócz postaci choroby uwarunkowanej genetycznie, HCM może wystąpić także u starszych pacjentów, u których często jest związana z przerostem lewej komory wynikającym z nadciśnienia tętniczego i/lub mieć związek ze zmianami serca implikowanymi wiekiem (przegroda sigmoidalna). Należy jednak zauważyć, że część przypadków choroby rozpoznawa-nych u starszych pacjentów będzie jednak miała tło genetyczne, np. w sytuacji mutacji białka C wiążącego miozynę, charakteryzu-jącej się opóźnioną penetracją i początkiem choroby (nawet po 60 roku życia) [1].
Obserwuje się przebieg choroby od w pełni asymptomatycznego do poważnych sercowo-naczyniowych manifestacji , takich jak niewydolność serca, omdlenia, udar mózgu, czy nagły zgon. Częstość choroby wynosi 1:500 w populacji ogólnej. Niemniej jednak znaczna ilość osób chorych pozosta-je bez rozpoznania z uwagi na bezobjawowy przebieg. U tych osób ustalenie rozpoznania często jest przypadkowe i odbywa się pod-
czas rutynowych badań [11].
Opis przypadku:Kobieta lat 76 z wywiadem nadciśnie-
nia tętniczego pierwotnego 3 stopnia, napadowego migotania przedsionków, hipercholesterolemii oraz cukrzycy typu 2. We wrześniu 2013 roku hospitalizowana w oddziale kardiologicznym najbliższym miejsca zamieszkania z powodu często-skurczu komorowego umiarowionego za pomocą kardiowersji elektrycznej (Ryc. 1).
Na podstawie przeprowadzonej wówczas diagnostyki ustalono rozpoznanie kar-diomiopatii przerostowej z zawężaniem śródkomorowym. Do I Kliniki Kardiologii i Elektrokardiologii Interwencyjnej oraz Nad-ciśnienia Tętniczego przyjęta została celem kwalifikacji do wszczepienia ICD. Przy przy-jęciu w stanie ogólnym dobrym, ciśnienie tętnicze krwi 130/80 mmHg, osłuchowo nad płucami szmer pęcherzykowy symetryczny, akcja serca miarowa 60/min. W badaniu elektrokardiograficznym stwierdzono rytm
Rycina 1 Zapis EKG w trakcie występowania objawów u Pacjentki: obraz częstoskurczu komorowego.Electrocardiogram in symptomatic period: vantricular tachycardia.
Rycina 2Zapis 12-odprowadzeniowego EKG w okresie bezobjawowym.Electrocardiogram in asymptomatic period.
273Przegląd Lekarski 2015 / 72 / 5
zatokowy miarowy o częstości 80/min, oś serca nieodchylona, odstęp PQ 200 msek, ujemne załamki T w odprowadzeniach V1-V6, przetrwałe uniesienie odcinka ST w odprowadzeniach III, aVF, V4-V6 (Ryc. 2).
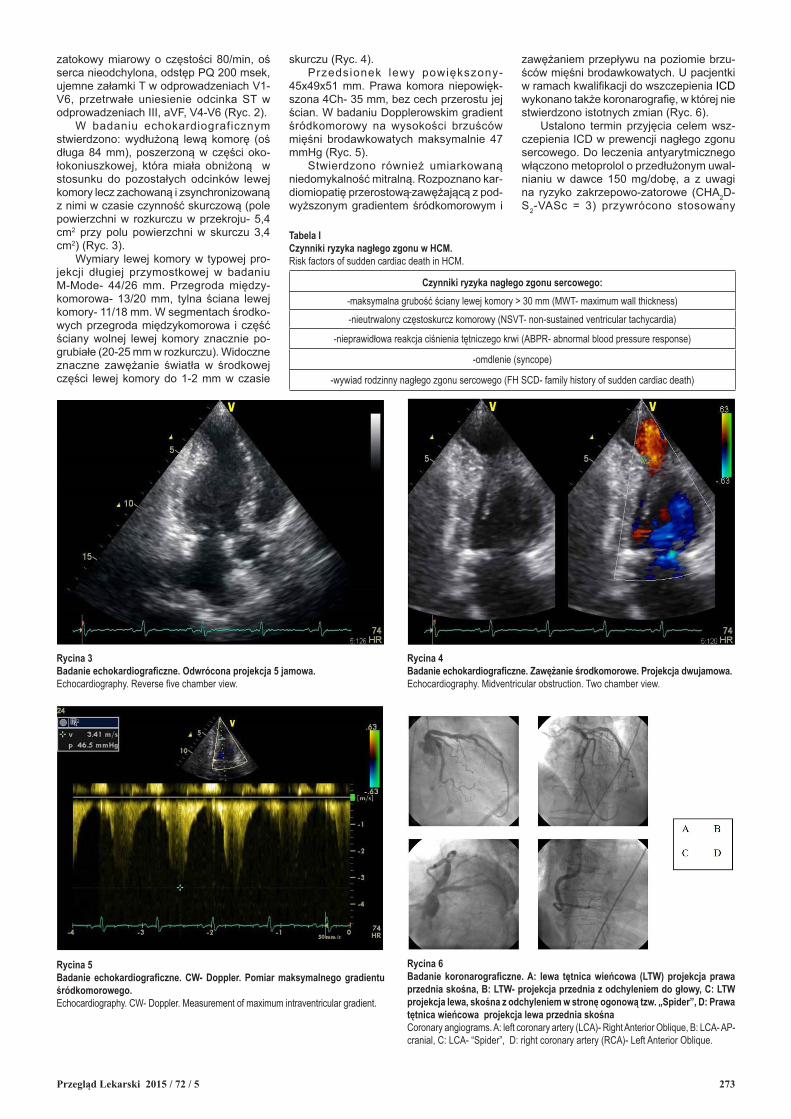
W badaniu echokardiograficznym stwierdzono: wydłużoną lewą komorę (oś długa 84 mm), poszerzoną w części oko-łokoniuszkowej, która miała obniżoną w stosunku do pozostałych odcinków lewej komory lecz zachowaną i zsynchronizowaną z nimi w czasie czynność skurczową (pole powierzchni w rozkurczu w przekroju- 5,4 cm2 przy polu powierzchni w skurczu 3,4 cm2) (Ryc. 3).
Wymiary lewej komory w typowej pro-jekcji długiej przymostkowej w badaniu M-Mode- 44/26 mm. Przegroda między-komorowa- 13/20 mm, tylna ściana lewej komory- 11/18 mm. W segmentach środko-wych przegroda międzykomorowa i część ściany wolnej lewej komory znacznie po-grubiałe (20-25 mm w rozkurczu). Widoczne znaczne zawężanie światła w środkowej części lewej komory do 1-2 mm w czasie
Tabela ICzynniki ryzyka nagłego zgonu w HCM.Risk factors of sudden cardiac death in HCM.
Czynniki ryzyka nagłego zgonu sercowego:-maksymalna grubość ściany lewej komory > 30 mm (MWT- maximum wall thickness)-nieutrwalony częstoskurcz komorowy (NSVT- non-sustained ventricular tachycardia)
-nieprawidłowa reakcja ciśnienia tętniczego krwi (ABPR- abnormal blood pressure response)
-omdlenie (syncope)
-wywiad rodzinny nagłego zgonu sercowego (FH SCD- family history of sudden cardiac death)
Rycina 3Badanie echokardiograficzne. Odwrócona projekcja 5 jamowa.Echocardiography. Reverse five chamber view.
Rycina 4Badanie echokardiograficzne. Zawężanie środkomorowe. Projekcja dwujamowa.Echocardiography. Midventricular obstruction. Two chamber view.
Rycina 5Badanie echokardiograficzne. CW- Doppler. Pomiar maksymalnego gradientu śródkomorowego.Echocardiography. CW- Doppler. Measurement of maximum intraventricular gradient.
Rycina 6Badanie koronarograficzne. A: lewa tętnica wieńcowa (LTW) projekcja prawa przednia skośna, B: LTW- projekcja przednia z odchyleniem do głowy, C: LTW projekcja lewa, skośna z odchyleniem w stronę ogonową tzw. „Spider”, D: Prawa tętnica wieńcowa projekcja lewa przednia skośnaCoronary angiograms. A: left coronary artery (LCA)- Right Anterior Oblique, B: LCA- AP-cranial, C: LCA- “Spider”, D: right coronary artery (RCA)- Left Anterior Oblique.
skurczu (Ryc. 4).Przedsionek lewy powiększony-
45x49x51 mm. Prawa komora niepowięk-szona 4Ch- 35 mm, bez cech przerostu jej ścian. W badaniu Dopplerowskim gradient śródkomorowy na wysokości brzuśców mięśni brodawkowatych maksymalnie 47 mmHg (Ryc. 5).
Stwierdzono również umiarkowaną niedomykalność mitralną. Rozpoznano kar-diomiopatię przerostową zawężającą z pod-wyższonym gradientem śródkomorowym i
zawężaniem przepływu na poziomie brzu-śców mięśni brodawkowatych. U pacjentki w ramach kwalifikacji do wszczepienia ICD wykonano także koronarografię, w której nie stwierdzono istotnych zmian (Ryc. 6).
Ustalono termin przyjęcia celem wsz-czepienia ICD w prewencji nagłego zgonu sercowego. Do leczenia antyarytmicznego włączono metoprolol o przedłużonym uwal-nianiu w dawce 150 mg/dobę, a z uwagi na ryzyko zakrzepowo-zatorowe (CHA2D-S2-VASc = 3) przywrócono stosowany

274 T. Pizoń i wsp.
uprzednio doustny antykoagulant. W okresie oczekiwania na zabieg, w listopadzie 2013 r. ponownie wystąpił epizod częstoskurczu komorowego wymagający kardiowersji elektrycznej. Do Kliniki Pacjentka została ponownie przyjęta w styczniu 2014 roku- wykonano zabieg wszczepienia ICD jedno-jamowego. Zabieg przebyła bez powikłań. W terapii antyarytmicznej zastosowano skojarzenie beta-blokera i amiodaronu, utrzymano leczenie acenocumarolem.
DyskusjaOpisany przypadek 76-letniej kobiety
z kardiomiopatią przerostową z obecnym śródkomorowym zaciskaniem powodują-cym istotny wewnątrzkomorowy gradient i z wydłużeniem oraz poszerzeniem części koniuszkowej lewej komory, z nawracają-cymi epizodami częstoskurczu komorowego wymagającego zastosowania kardiowersji elektrycznej ostatecznie został zakwalifiko-wany do wszczepienia ICD. W literaturze opisano kilka wariantów kardiomiopatii prze-rostowej z obecnością atypowych cech u starszych kobiet- jeden z nich charakteryzo-wał się ciężkim koncentrycznym przerostem lewej komory ze współistniejącym wywia-dem nadciśnienia tętniczego i obecnością nieprawidłowej funkcji rozkurczowej lewej komory [12]; drugi cechował się obecnością zaciskania z podmitralną pierścieniowatą kalcyfikacją [13]. Nasz zaprezentowany przypadek nie odpowiada jednak żadnemu z tych wariantów.
HCM ze śródkomorowym zaciskaniem jest rzadką postacią kardiomiopatii przero-stowej (1%), częściej występującą u kobiet i skojarzoną z obecnością tętniaka koniusz-kowego jako następstwem niedokrwienia mięśnia sercowego pomimo prawidłowego obrazu angiograficznego naczyń wień-cowych [14]. Shah i wsp. przeprowadzili badanie obserwacyjne 444 pacjentów z HCM, wśród których 22 osoby (5%) cha-rakteryzowały się postacią śródkomorową lub koniuszkową. U czterech pacjentów występowało zaciskanie śródkomorowe. Głównym wnioskiem wypływającym z w/w badań było stwierdzenie, że poważnie ob-jawowi pacjenci wykazują śródkomorowe pogrubienie z towarzyszącą ekspansją w kierunku jamy lewej komory, co w konse-kwencji wiedzie do zmniejszenia jej objęto-ści, z następową komplikacją w formie śród-komorowego zaciskania [15]. Inni autorzy wykazali korelację występowania objawów z małą objętością końcoworozkurczową u pacjentów z niewydolnością serca w prze-biegu HCM z towarzyszącym nadciśnieniem tętniczym, szczególnie u kobiet [15-17]. W sumie, płeć żeńska wiąże się z obecnością bardziej poważnych objawów i wyższych gradientów [18]. Efthimiadis i wsp. wykazali, że pacjenci z obecnością śródkomorowego zaciskania są częściej płci żeńskiej i wyka-zują większe nasilenie objawów podczas wstępnego badania. Omdlenie - jeden z czynników ryzyka nagłego zgonu- występo-wało istotnie częściej u pacjentów ze śród-komorowym zaciskaniem niż w pozostałych postaciach HCM. Ponadto obecność ≥ 2 czynników ryzyka nagłej śmierci sercowej występowała istotnie częściej w przypadku śródkomorowego zaciskania niż w sytuacji jego nieobecności (29.4% vs. 18.1%). Jak
należy się spodziewać, leczenie przy użyciu leków o działaniu inotropowo ujemnym, szczególnie β-blokerów i disopiramidu, było stosowane najpowszechniej w przypadku MVO. W okresie obserwacji wynoszącym przeciętnie 84 miesiące (zakres 6-480 mie-sięcy), 5 z 34 pacjentów z MVO (14.7%) przebyło miektomię z powodu obecności objawów opornych na leczenie farmakolo-giczne. Kluczowym wnioskiem wynikającym z powyższych obserwacji jest stwierdzenie relatywnie wysokiej częstości występowania nagłej śmierci i towarzyszących zaburzeń rytmu serca w przypadku obecności MVO, niezależnie od powstania tętniaka koniusz-kowego. Śródkomorowe zaciskanie okazało się być niezależnym predyktorem ryzyka arytmii/zgonu, i to o wiele silniejszym niż uznane kliniczne czynniki ryzyka nagłego zgonu [19]. Czynniki ryzyka nagłego zgonu w kardiomiopatii przerostowej zestawiono w Tabeli I (Tab. I). Można wnioskować, że przerośnięte i w różnym stopniu zwłókniałe segmenty środkomorowe stanowią poten-cjalny substrat arytmogenny ułatwiający wystąpienie mechanizmu re-entry wcześniej lub niezależnie od wytworzenia tętniaka [19-21]. W takim przypadku MVO należy traktować jako czynnik ryzyka nagłego zgo-nu, a interwencje terapeutyczne w tej grupie powinny być ukierunkowane na pierwotną prewencję nagłego zgonu i poważnych arytmii, za pomocą implantowanego kardio-wertera-defibrylatora [22].
Bazując na ocenie ryzyka w oparciu o powyższą tabelę Maron i wsp. poddali obserwacji grupę 506 pacjentów o wysokim ryzyku nagłej śmierci sercowej, u których wszczepiono system do automatycznej kardiowersji-defibrylacji. Interwencje ICD skutecznie przerwały częstoskurcz komo-rowy/migotanie komór u 103 pacjentów (20%). Częstość interwencji wyniosła 10.6% na rok w prewencji wtórnej po zatrzymaniu krążenia oraz 3.6% na rok w przypadku prewencji pierwotnej. W konkluzji autorzy zauważają, że w grupie wysokiego ryzyka HCM, wyładowania ICD w przypadku za-grażających życiu tachyarytmii są częste i wysoce skuteczne w przywracaniu prawidło-wego rytmu. Istotny odsetek interwencji ICD wystąpił w przypadku prewencji pierwotnej u pacjentów, którzy przebyli wszczepienie z powodu jednego czynnika ryzyka. Dlatego też pojedynczy czynnik ryzyka nagłego zgonu może być uznany jako istotny dla roz-ważenia profilaktycznej implantacji defibryla-tora u wybranych pacjentów z HCM [23]. W wytycznych ESC z 2014 roku podkreślono, że pacjenci ze śródkomorowym zaciskaniem powinni być leczeni za pomocą dużych da-wek β-blokerów, werapamilu lub diltiazemu, ale odpowiedź na takie postępowanie nie jest optymalna. Nie ma też wątpliwości, że w przypadku HCM i przebytego migotania komór lub częstoskurczu komorowego ryzyko wystąpienia epizodu śmiertelnej arytmii jest tak duże, że implantacja ICD jest konieczna [2].
W podsumowaniu można posłużyć się zaleceniami ACCF/AHA z 2011 roku mówiącymi, że chociaż całkowita częstość nagłej śmierci sercowej u pacjentów z HCM wynosi przeciętnie 1% w skali roku, to moż-na wyselekcjonować w tej grupie chorych o
zwiększonym ryzyku, dla których postępo-wanie profilaktyczne może być wskazane. Nie udowodniono, aby leczenie farmakolo-giczne zabezpieczało przed nagłym zgonem sercowym, udowodniono natomiast że ICD jest skuteczną formą terapii w przerywaniu zagrażających życiu tachyarytmii komo-rowych w przebiegu HCM, zmieniającą naturalny przebieg choroby i wpływającą na wydłużenie życia. Przetrwały częstoskurcz komorowy znajduje się w czołówce wskazań do implantacji urządzenia [3].
Piśmiennictwo1. Wigle ED: The diagnosis of hypertrophic cardiomy-
opathy. Heart 2001; 86: 709-714.2. Elliott PM, Anastasakis A, Borger MA, Borggrefe
M, Cecchi F. et al: 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Europ Heart J. 2014; 35: 2733-2779.
3. Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA. et al: 2011 ACCF/AHA Guideline for the diagno-sis and treatment of hypertropic cardiomyopathy. JACC 2011; 58: 212-260.
4. Minami Y, Kajimoto K, Terajima Y, Yashiro B, Okay-ama D et al: Clinical implications of midventricular obstruction in patients with hypertrophic cardiomy-opathy. J Am Coll Cardiol. 2011; 57: 2346-2355.
5. Chojnowska SL: Kardiomiopatia przerostowa, w: Szczeklik A, Tendera M (red.), Kardiologia Tom I. Medycyna Praktyczna, Kraków 2009: 628.
6. Brock RC: Functional obstruction of the left ventricle (acquired aortic subvalvar stenosis). Guys Hosp Rep. 1957; 106: 221.
7. Teare D: Asymmetrical hypertrophy of the heart in young adults. Br Heart J. 1958; 20: 1-8.
8. Maron BJ, Maron MS, Wigle ED, Braunwald E: The 50-year history, controversy, and clinical impli-cations of left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009; 54: 191-200.
9. Falicov RE, Resnekov L, Bharati S, Lev M: Mid-ventricular obstruction: a variant of obstructive cardio-myopathy. Am J Cardiol. 1976; 37: 432-437.
10. Otsuka H, Arimura T, Abe T, Kawai H, Aizawa Y. et al: Prevalence and distribution of sarcomeric gene mutations in Japanese patients with familial hypertro-phic cardiomyopathy. Circ J. 2012; 76: 453-461.
11. Efthimiadis GK, Parcharidou D, Pagourelias ED, Meditskou S, Spanos G. et al: Prevalence and clinical outcomes of incidentally diagnosed hypertrophic cardiomyopathy. Am J Cardiol. 2010; 105: 1445-1450.
12. Topol EJ, Trail TA, Fortuin NJ: Hypertensive hyper-trophic cardiomyopathy of the elderly. N Engl J Med. 1985; 312: 277-283.
13. Lewis JF, Maron BJ: Elderly patients with hypertro-phic cardiomyopathy: A subset with distinctive left ventricular morphology and progressive clinical cour-se late in life. J Am Coll Cardiol. 1989; 13: 36-45.
14. Harada K, Shimizy T, Sugishita Y, Yao A, Suzuki J. et al: Hypertrophic cardiomyopathy with midventricu-lar obstruction and apical aneurysm- a case report. Jpn Circ J. 2001; 65: 915-919.
15. Shah A, Duncan K, Winson G, Chaudhry FA, Sherrid MV: Severe symptoms in mid and apical hypertrophic cardiomyopathy. ECHOCARDIOGRA-PHY: A Jrnl of CV Ultrasound & Allied Tech. 2009; 26: 922-933.
16. Gandhi SK, Powers JC, Nomeir AM, Fowle K, Kitzman DW. et al: The pathogenesis of acute pulmonary edema associated with hypertension. N Engl J Med. 2001; 344: 17-22.
17. Nienaber CA, Hiller S, Spilmann RP, Geiger M, Kuck K-H: Syncope in hypertrophic cardiomyopathy: Multivariate analysis of prognostic determinants. J Am Coll Cardiol. 1990; 15: 948-955.
18. Olivotto I, Maron MS, Adabag AS, Casey SA, Var-giu D. et al: Gender-related differences in the clinical presentation and outcome of hypertrophic cardiomy-opathy. J Am Coll Cardiol. 2005; 46: 480-487.
19. Efthimiadis GK, Efstathios D, Pagourelias D, Par-charidou D, Gossios T. et al: Clinical characteristics

275Przegląd Lekarski 2015 / 72 / 5
and natural history of hypertrophic cardiomyopathy with midventricular obstruction. Cir J. 2013; 77: 2366-2374.
20. Adabag AS, Maron BJ, Appelbaum E, Harrigan CJ, Buros JL. et al: Occurrence and frequency of arrhythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular
magnetic resonance. J Am Coll Cardiol. 2008; 51: 1369-1374.
21. Gerstenfeld EP: Hypertrophic cardiomyopathy with midventricular obstruction: Another substrate for ventricular tachycardia? J Cardiovasc Electrophysiol. 2010; 21: 1000-1001.
22. Maron BJ: Risk stratification and role of implantable
defibrillators for prevention of sudden death in patients with hypertrophic cardiomyopathy. Circ J. 2010; 74: 2271-2282.
23. Maron BJ, Spirito P, Shen W-K, Haas TS, Formisa-no F. et al: Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA 2007; 298: 405-412.
















