
Janusz Zimowski Zakład Genetyki - ipin.edu.pl · deformacje stóp – tak zwana stopa wydrążona...
13
Janusz Zimowski Zakład Genetyki
-
Upload
trinhxuyen -
Category
Documents
-
view
220 -
download
0
Transcript of Janusz Zimowski Zakład Genetyki - ipin.edu.pl · deformacje stóp – tak zwana stopa wydrążona...

Janusz Zimowski
Zakład Genetyki

Wiotkość,
brak postępu w rozwoju ruchowym dziecka,
niewydolność oddechowa lub też
postępujące osłabienie mięśni obręczy biodrowej
Sposób dziedziczenia
– recesywny, autosomalny (5q11.2-13.2)
Częstość zachorowania
– 1 : 6-7 tys.
Częstość nosicielstwa
– 1 : 38-40 osób

SMA1 choroba Werdniga Hoffmanna
SMA2 postać pośrednia
SMA3 choroba Kugelberga Welander
Utrata neuronów ruchowych rdzenia kręgowego
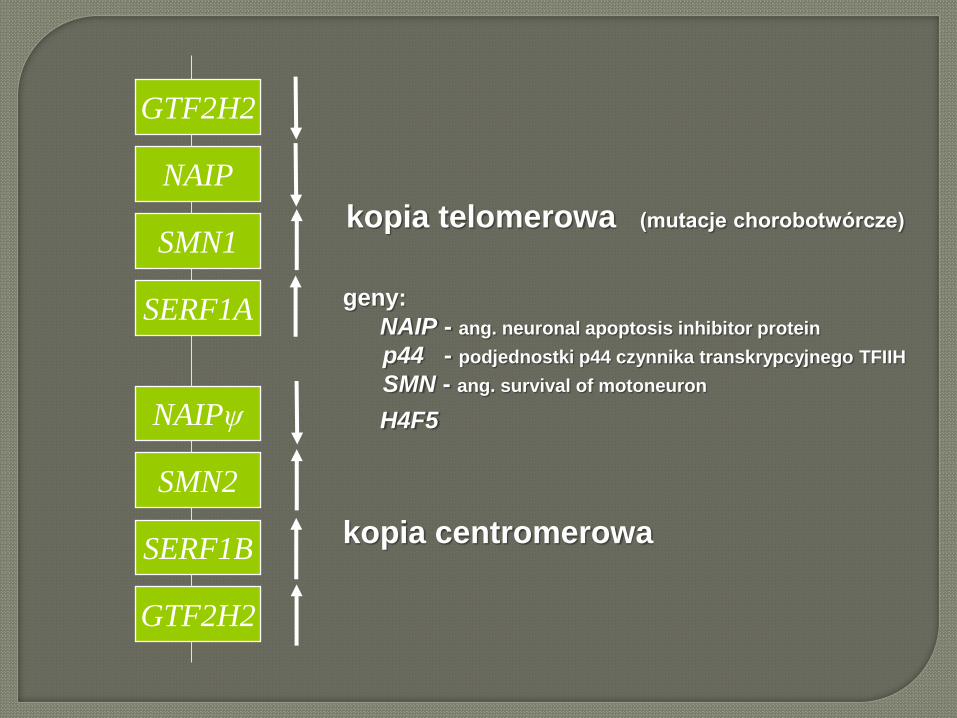
GTF2H2
NAIP
SMN1
SERF1A
SERF1B
SMN2
NAIPψ
GTF2H2
kopia telomerowa (mutacje chorobotwórcze)
geny:
NAIP - ang. neuronal apoptosis inhibitor protein
p44 - podjednostki p44 czynnika transkrypcyjnego TFIIH
SMN - ang. survival of motoneuron
H4F5
kopia centromerowa
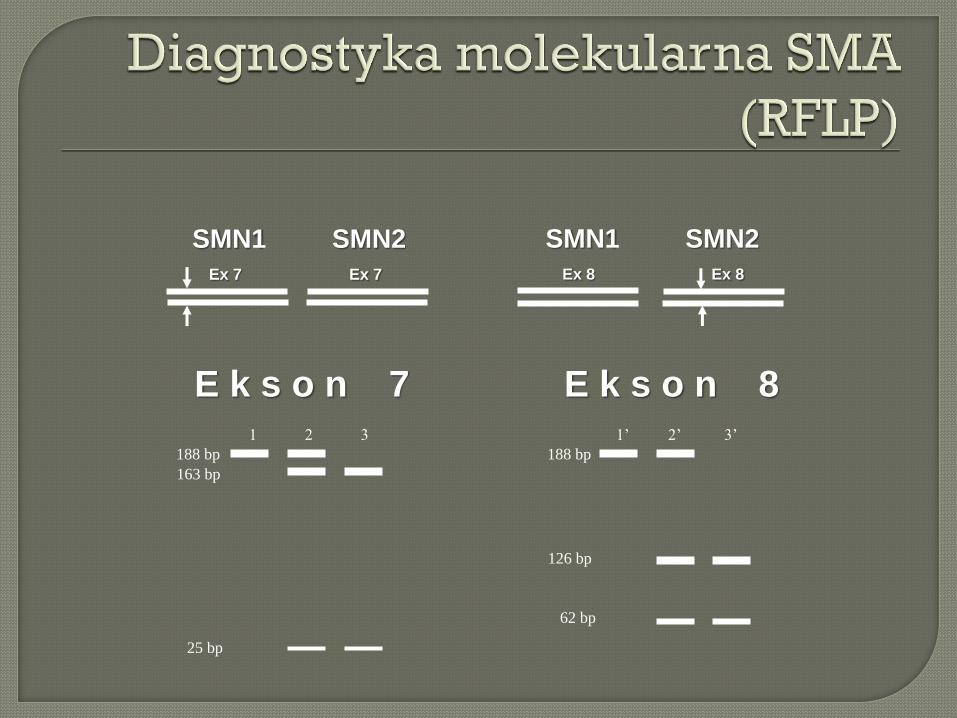
E k s o n 7 E k s o n 8
1 2 3 1’ 2’ 3’
188 bp 188 bp
163 bp
126 bp
62 bp
25 bp
SMN1 SMN2
Ex 7 Ex 7
SMN1 SMN2
Ex 8 Ex 8

SMN2
SMN1
1 2 3 4 5 6 Kn
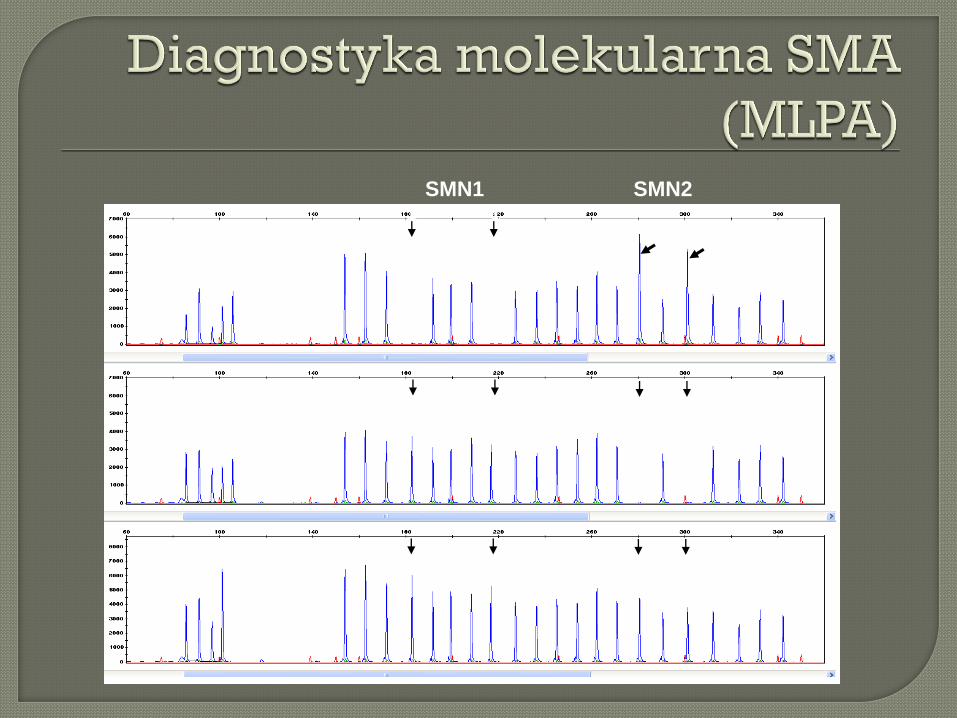
SMN1 SMN2

Choroba objawia się w dzieciństwie albo
wczesnej młodości i ma powolny, postępujący
przebieg.
Charakterystyczne dla CMT są: deformacje stóp – tak zwana stopa wydrążona (pes excavatum)
zanik mięśni strzałkowych
opadanie stopy przy chodzeniu – chód brodzący
osłabienie odruchów skokowych.
Chorzy zwykle zachowują zdolność chodzenia,
ale poszczególne postaci mogą mieć cięższy
przebieg i powodować poważne ograniczenie
sprawności ruchowej.

Neuropatia motoryczna i czuciowa
(zmniejszona szybkość przewodzenia
impulsów przez nerwy).
o CMT1 (typ demielinizacyjny, 5
genów),
o CMT2 (typ aksonalny, 17 genów), o CMT3 (nie wpływa na szybkość przewodzenia, 4 geny)
o CMT4 (typ rdzeniowy, 12 genów),
o CMTX (sprzężony z chr. X, 6 genów)

o Dziedziczenie:
autosomalne dominujące CMT1, CMT2;
autosomalne recesywne CMT4;
sprzężone z chr. X CMTX
o Częstość występowania 1/10000

80-85% przypadków to:
o CMT1A Gen PMP22 60-70%
o CMT1B Gen MPZ 5-10%
o CMTX1 Gen GJB1 ~10%
Mutacje: duplikacje, mutacje punktowe

Dawka genu
Gen PMP22
1 kopia genu – neuropatia dziedziczna z
predyspozycją do porażenia uciskowego
nerwu (HNPP)
2 kopie genu – osoba zdrowa
3 kopie genu – CMT1A
4 kopie genu – bardzo poważna neuropatia
z obwodową demielinizacją (model zwierzęcy)

o Gen PMP22 4 eksony - MLPA
o Gen MPZ 6 eksonów - sekwencjonowanie
o Gen GJB1 1 ekson – sekwencjonowanie
Analiza tych genów pozwala na
wykrycie do 90% mutacji w
CMT


















