
ERMODYNAMIKA UKŁADÓW BIOLOGICZNYCH · termodynamika jest niezależna od poziomu naszej wiedzy o...
26
Część III: Termodynamika układów biologicznych MATERIAŁY POMOCNICZE DO WYKŁADÓW Z PODSTAW BIOFIZYKI IIIr. Biotechnologii prof. dr hab. inż. Jan Mazerski TERMODYNAMIKA UKŁADÓW BIOLOGICZNYCH Niezwykle cenną metodą badania zjawisk przyrodniczych, zupełnie niezależną od naszej wiedzy o strukturze obiektu na poziomie cząsteczkowym, jest podejście termodynamiczne . Może ono być stosowane do obiektów o różnej skali (od pojedynczego atomu do całego Wszechświata) i różnym stopniu złożoności (od prostego układu kilku atomów do żywej komórki, a nawet całego organizmu wielokomórkowego). Nazwa termodynamika mogłaby sugerować, że chodzi tu o badanie przepływu ciepła. Takie były rzeczywiście początki tej dziedziny. Obecnie termodynamika zajmuje się wszystkimi aspektami energetycznymi badanego układu. Termodynamika jest konstrukcja logiczną o wielkiej elegancji. Trzy zwięzłe stwierdzenia, trzy zasady termodynamiki stanowią podsumowanie naszej wiedzy o wszelkich postaciach energii i prawach rządzących jej przemianami. Prawdziwość termodynamiki zależy jedynie od prawdziwości tych 3 zasad oraz od logicznej poprawności wyciąganych wniosków. Dzięki temu termodynamika jest niezależna od poziomu naszej wiedzy o strukturze wewnętrznej układu oraz od koncepcji czy modeli opisujących tą strukturę. Jest to bardzo istotne w naukach biologicznych, gdzie np. stopień wiedzy o strukturze wewnętrznej komórki zmienia się z roku na rok w miarę postępu technik instrumentalnych i informatycznych. Nie ma to jednak wpływu na opis termodynamiczny komórki. Podstawowe definicje Aby poprawnie posługiwać się podejściem termodynamicznym należy opanować specyficzne słownictwo termodynamiki. Definiowanie pojęć rozpoczniemy od pojęcia układu . Jest to w termodynamice pojęcie pierwotne, w zasadzie niedefiniowalne, tak jak punkt w geometrii. Dla naszych potrzeb wystarczy, jeżeli będziemy pamiętać, że układ termodynamiczny, to zbiór elementów, między którymi istnieją określone relacje. Jeżeli relacje te sprowadzają się do oddziaływania jednych elementów na inne, to mamy do czynienia z układem dynamicznym. Ponieważ z punktu widzenia termodynamiki nie jest istotna struktura elementów, a jedynie relacje pomiędzy nimi, więc układy termodynamiczne mogą być tworzone w dowolnej skali. Jest przy tym charakterystyczne, że obiekt rozpatrywany jako układ na jednym poziomie może być traktowany jako element układu na wyższym poziomie. Możliwa jest również sytuacja odwrotna: element
Transcript of ERMODYNAMIKA UKŁADÓW BIOLOGICZNYCH · termodynamika jest niezależna od poziomu naszej wiedzy o...

Część III: Termodynamika układów biologicznych
MMAATTEERRIIAAŁŁYY PPOOMMOOCCNNIICCZZEE DDOO WWYYKKŁŁAADDÓÓWW ZZ PPOODDSSTTAAWW BBIIOOFFIIZZYYKKII
IIIIIIrr.. BBiiootteecchhnnoollooggiiii pprrooff.. ddrr hhaabb.. iinnżż.. JJaann MMaazzeerrsskkii
TTEERRMMOODDYYNNAAMMIIKKAA UUKKŁŁAADDÓÓWW BBIIOOLLOOGGIICCZZNNYYCCHH
Niezwykle cenną metodą badania zjawisk przyrodniczych, zupełnie niezależną od naszej
wiedzy o strukturze obiektu na poziomie cząsteczkowym, jest podejście termodynamiczne. Może
ono być stosowane do obiektów o różnej skali (od pojedynczego atomu do całego Wszechświata) i
różnym stopniu złożoności (od prostego układu kilku atomów do żywej komórki, a nawet całego
organizmu wielokomórkowego). Nazwa termodynamika mogłaby sugerować, że chodzi tu o
badanie przepływu ciepła. Takie były rzeczywiście początki tej dziedziny. Obecnie termodynamika
zajmuje się wszystkimi aspektami energetycznymi badanego układu.
Termodynamika jest konstrukcja logiczną o wielkiej elegancji. Trzy zwięzłe stwierdzenia,
trzy zasady termodynamiki stanowią podsumowanie naszej wiedzy o wszelkich postaciach energii i
prawach rządzących jej przemianami. Prawdziwość termodynamiki zależy jedynie od
prawdziwości tych 3 zasad oraz od logicznej poprawności wyciąganych wniosków. Dzięki temu
termodynamika jest niezależna od poziomu naszej wiedzy o strukturze wewnętrznej układu oraz od
koncepcji czy modeli opisujących tą strukturę. Jest to bardzo istotne w naukach biologicznych,
gdzie np. stopień wiedzy o strukturze wewnętrznej komórki zmienia się z roku na rok w miarę
postępu technik instrumentalnych i informatycznych. Nie ma to jednak wpływu na opis
termodynamiczny komórki.
Podstawowe definicje Aby poprawnie posługiwać się podejściem termodynamicznym należy opanować
specyficzne słownictwo termodynamiki. Definiowanie pojęć rozpoczniemy od pojęcia układu. Jest
to w termodynamice pojęcie pierwotne, w zasadzie niedefiniowalne, tak jak punkt w geometrii. Dla
naszych potrzeb wystarczy, jeżeli będziemy pamiętać, że układ termodynamiczny, to zbiór
elementów, między którymi istnieją określone relacje. Jeżeli relacje te sprowadzają się do
oddziaływania jednych elementów na inne, to mamy do czynienia z układem dynamicznym.
Ponieważ z punktu widzenia termodynamiki nie jest istotna struktura elementów, a jedynie relacje
pomiędzy nimi, więc układy termodynamiczne mogą być tworzone w dowolnej skali. Jest przy tym
charakterystyczne, że obiekt rozpatrywany jako układ na jednym poziomie może być traktowany
jako element układu na wyższym poziomie. Możliwa jest również sytuacja odwrotna: element

Część III: Termodynamika układów biologicznych
układu na jednym poziomie może być traktowany jako cały układ na poziomie niższym. Ta
hierarchiczna natura układów termodynamicznych wymaga za każdym razem bardzo precyzyjnego
określenia, co dany układ zawiera. Elementy rzeczywistości nie wchodzące w skład układu noszą w
termodynamice nazwę otoczenia.
W zależności od relacji pomiędzy układem a otoczeniem wyróżnia się w termodynamice 4
podstawowe typy układów:
izolowane: nie wymieniają z otoczeniem masy ani żadnej formy energii
adiabatyczne: nie wymieniają masy i ciepła, mogą wymieniać pozostałe formy energii
izotermiczne: swobodnie wymieniają wszystkie formy energii, nie wymieniają masy
otwarte: wymieniają z otoczeniem masę i energię.
Układy izolowane, adiabatyczne i izotermiczne określa się wspólnym terminem układy zamknięte
(nie wymieniające masy z otoczeniem).
Stan układu i jego parametry Stan układu można opisać za pomocą pewnej liczby zmiennych tzw. parametrów stanu.
Parametry mogą być intensywne i ekstensywne. Parametry intensywne nie zależą od wielkości
układu i przy łączeniu elementów w układ nie są addytywne. Typowymi parametrami
intensywnymi są: temperatura, stężenie, ciśnienie. Parametry ekstensywne zależą od wielkości
układu i są addytywne. Do parametrów tego typu należą np. objętość i masa.
Stan układu określony jest przez parametry stanu i zmienia się wraz z ich zmianą. Zależność stanu
układu od wartości parametrów określa tzw. równanie stanu. Np. dla układu termodynamicznego
takiego jak gaz doskonały mamy równanie stanu gazu doskonałego postaci:
pV = nRT
W równaniu tym występuje jedna stała (stała gazowa R) oraz 4 parametry: p - ciśnienie,
V - objętość, n - liczba moli i T - temperatura bezwzględna. Tylko 3 z tych parametrów mogą
przyjmować dowolne wartości - wartość czwartego wynika z równania stanu gazu doskonałego.
Jeżeli nasze rozważania ograniczymy do 1 mola gazu doskonałego, to równanie stanu sprowadzi się
do postaci:
pV = RT
Rozpatrując jaki wpływ na wartość jednego z parametrów stanu mają niewielkie zmiany
pozostałych parametrów wygodnie jest posługiwać się zapisem różniczkowym. Jeżeli
g = f(x,y,z)
to różniczkę funkcji g definiujemy jako:
dg = Ldx + Mdy +Ndz
22

Część III: Termodynamika układów biologicznych
gdzie L, M, N są pochodnymi cząstkowymi funkcji g odpowiednio po x, y i z:
xgL∂∂
= xgL∂∂
= xgL∂∂
=
Ostatecznie różniczka parametru (zmiennej stanu) g przyjmuje postać:
dzzgdy
ygdx
xgdg
∂∂
+∂∂
+∂∂
=
Funkcje stanu Jeżeli wartość pewnego parametru nie zależy od drogi, po jakiej została osiągnięta, a
jedynie od stanu układu, to parametr taki nazywamy funkcją stanu. Opis stanu układu przy pomocy
funkcji stanu ma szereg zalet. Przede wszystkim:
wartości funkcji stanu w stanie równowagi termodynamicznej zależą tylko od parametrów
zewnętrznych, czyli dających się zmierzyć
wartości funkcji stanu w dowolnym stanie można obliczyć korzystając z różniczki tej funkcji.
Istnieje prosty sposób ustalenia, czy dany parametr jest funkcja stanu korzystając z równania stanu.
Można wykazać na gruncie rachunku różniczkowego, że w przypadku funkcji stanu jej różniczka
musi być tzw. różniczką zupełną. Różniczkę nazywamy zupełną, jeżeli dla każdej pary parametrów
x i y zachodzi równość:
xyg
yxg
∂
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
∂=
∂
⎟⎠⎞
⎜⎝⎛∂∂
∂
Sprawdźmy, czy objętość jest funkcją stanu gazu doskonałego. Przekształćmy najpierw równanie
stanu gazu doskonałego tak, aby dla 1 mola gazu uzyskać zależność objętości od ciśnienia i
temperatury:
p
RTV =
WWyyzznnaacczzmmyy tteerraazz rróóżżnniicczzkkęę oobbjjęęttoośśccii::
dppRTdT
pRdp
pVdT
TVdV 2−=
∂∂
+∂∂
=
i sprawdźmy czy jest to różniczka zupełna:
TpV
pTV
∂
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
∂=
∂
⎟⎠⎞
⎜⎝⎛∂∂
∂
TpRT
ppR
2
∂
⎟⎟⎠
⎞⎜⎜⎝
⎛−∂
=∂
⎟⎟⎠
⎞⎜⎜⎝
⎛∂
22 pR
pR
−=−
33

Część III: Termodynamika układów biologicznych
Tak więc objętość jest funkcją stanu gazu doskonałego. W analogiczny sposób można wykazać, że
temperatura i ciśnienie są również funkcjami stanu gazu doskonałego.
Sprawdźmy teraz czy praca mechaniczna wykonywana przez gaz doskonały jest funkcją
stanu tego gazu. Najpierw musimy znać różniczkę pracy gazu doskonałego. Z fizyki i chemii
fizycznej wiemy, że różniczka pracy równa jest:
dpp
RTRdTdppRTdT
pRppdVdW 2 +−=⎟⎟
⎠
⎞⎜⎜⎝
⎛−−=−=
Sprawdźmy teraz czy jest to różniczka zupełna:
( )Tp
RT
pR
∂
⎟⎟⎠
⎞⎜⎜⎝
⎛∂
=∂−∂
pR0 ≠
Wniosek: praca mechaniczna nie jest funkcja stanu gazu doskonałego, chociaż jest parametrem
stanu.
I. zasada termodynamiki Pierwsze sformułowanie tej zasady zawdzięczamy Juliuszowi Robertowi Mayerowi (1942).
Uzupełnił ja Helmholtz w roku 1947. Mówi ona, że suma wszystkich rodzajów energii w układzie
zamkniętym pozostaje stała. Po opublikowaniu teorii Einsteina Henri Poincare sformułował ja w
1908 roku w nieco przekorny sposób: „Istnieje coś, co jest stałe w układzie zamkniętym”.
Ale co to jest to „coś”? Jest ono stałe w układzie zamkniętym, tzn. nie wymieniającym materii z
otoczeniem. Jest funkcja stanu, bo jej różniczka w układzie zamkniętym jest równa 0. Nie jest to
praca, bo praca nie jest funkcja stanu, ale ma z pracą i energią coś wspólnego. Przyjęto nazywać to
„coś” energia wewnętrzną i oznaczać symbolem U.
Współcześnie I zasadę termodynamiki zapisujemy w postaci:
dU = dQ + dW,
a więc przyrost energii wewnętrznej jest sumą przyrostu ciepła i pracy wykonanej nad układem. W
tej postaci nie można jeszcze wykazać, że różniczka energii wewnętrznej jest różniczką zupełną,
ale wykażemy to w toku dalszego wykładu.
I. zasada termodynamiki była badana na gruncie biologii jeszcze przed jej precyzyjnym
sformułowaniem. Już w roku 1777 Lavoisier zastanawiał się nad związkiem ilości wydzielanego
przez zwierzą ciepła z ciepłem uzyskanym przy spalaniu pożywienia w trakcie oddychania. W kilka
lat później Lavoisier i Laplace przeprowadzili pierwsze ilościowe badania kalorymetryczne na
świnkach morskich. Potwierdziły one w pełni stosowalność zasady zachowania energii również w
44

Część III: Termodynamika układów biologicznych
układach biologicznych. Od tego czasu badania takie prowadzone są z zastosowaniem coraz
doskonalszej aparatury.
Samorzutność procesów II.. zzaassaaddaa tteerrmmooddyynnaammiikkii nniiee ookkrreeśśllaa kkiieerruunnkkuu pprrzzeeppłłyywwuu eenneerrggiiii.. DDooppuusszzcczzaa wwiięęcc mmoożżlliiwwoośśćć
zzaajjśścciiaa zzaarróówwnnoo::
ooggrrzzeewwaanniiaa ssiięę cciiaałłaa zziimmnniieejjsszzeeggoo kkoosszztteemm ssttyyggnniięęcciiaa ggoorrąącceeggoo,, jjaakk ii
ooggrrzzeewwaanniiee ssiięę cciiaałłaa ggoorrąącceeggoo kkoosszztteemm ddaallsszzeeggoo oozziięębbiiaanniiaa ssiięę cciiaałłaa zziimmnneeggoo
jjeeżżeellii ttyyllkkoo iilloośśćć eenneerrggiiii ppoobbrraanneejj rróówwnnaa jjeesstt iilloośśccii eenneerrggiiii ooddddaanneejj..
ZZ ddoośśwwiiaaddcczzeenniiaa wwyynniikkaa jjeeddnnaakk,, żżee ww pprraakkttyyccee pprroocceessyy bbiieecc mmooggąą ttyyllkkoo ww jjeeddnnyymm
kkiieerruunnkkuu.. PPrroocceess ww ppeełłnnii ooddwwrraaccaallnnyy jjeesstt ttyyllkkoo mmooddeelleemm tteeoorreettyycczznnyymm.. AAllee ssttooppiieeńń
nniieeooddwwrraaccaallnnoośśccii pprroocceessuu bbyywwaa rróóżżnnyy.. NNpp.. ddoobbrree wwaahhaaddłłoo mmoożżee wwaahhaaćć ssiięę bbaarrddzzoo ddłłuuggoo ppoo
kkaażżddyymm kkoolleejjnnyymm wwaahhnniięęcciiuu wwrraaccaajjąącc pprraakkttyycczznniiee ddoo tteeggoo ssaammeeggoo ppoołłoożżeenniiaa.. AA jjeeddnnaakk ii oonnoo
rroozzpprraasszzaa eenneerrggiięę ii ppoo ppeewwnnyymm cczzaassiiee zzaattrrzzyymmaa ssiięę..
ZZ oobbsseerrwwaaccjjii wwiieemmyy,, żżee ww nniieeooddwwrraaccaallnnoośśćć pprroocceessóóww „„zzaapplląąttaannee”” jjeesstt cciieeppłłoo.. AAllee cciieeppłłoo nniiee jjeesstt
ffuunnkkccjjąą ssttaannuu.. PPrrzzeeppłłyyww cciieeppłłaa ooddbbyywwaa ssiięę zzggooddnniiee zz ggrraaddiieenntteemm tteemmppeerraattuurryy.. MMoożżnnaa wwiięęcc
pprrzzyyppuusszzcczzaaćć żżee zz nniieeooddwwrraaccaallnnoośścciiąą pprroocceessuu ww jjaakkiiśś ssppoossóóbb mmuussii bbyyćć zzwwiiąązzaannaa tteemmppeerraattuurraa
uukkłłaadduu.. OOkkaazzaałłoo ssiięę,, żżee rróóżżnniicczzkkaa cciieeppłłaa zzrreedduukkoowwaanneeggoo
dST
dQ≡
zzwwaannaa rróóżżnniicczzkkąą eennttrrooppiiii jjeesstt ttąą ppoosszzuukkiiwwaannąą mmiiaarrąą ooddwwrraaccaallnnoośśccii pprroocceessuu..
IIII.. zzaassaaddaa tteerrmmooddyynnaammiikkii mmóówwii,, żżee rróóżżnniicczzkkaa eennttrrooppiiii nniiee mmoożżee bbyyćć uujjeemmnnaa.. OOzznnaacczzaa ttoo,, żżee
eennttrrooppiiaa nniiee mmoożżee mmaalleećć.. WW pprroocceessaacchh ooddwwrraaccaallnnyycchh eennttrrooppiiaa nniiee uulleeggaa zzmmiiaanniiee ((ddSS == 00)),, aa ww
pprroocceessaacchh nniieeooddwwrraaccaallnnyycchh eennttrrooppiiaa rroośśnniiee ((ddssSS >> 00))..
DDeeffiinniiccjjęę rróóżżnniicczzkkii eennttrrooppiiii mmoożżnnaa ttaakk pprrzzeekksszzttaałłcciićć,, aabbyy wwyyzznnaacczzyyćć rróóżżnniicczzkkęę cciieeppłłaa
.. TdSdQ =
RRóóżżnniicczzkkęę ttąą mmoożżnnaa tteerraazz wwssttaawwiićć ddoo rróówwnnaanniiaa II.. zzaassaaddyy tteerrmmooddyynnaammiikkii::
dWTdSdWdQdU +=+=
PPoozzoossttaajjee jjeesszzcczzee pprroobblleemm rróóżżnniicczzkkii pprraaccyy.. ZZ II.. zzaassaaddyy tteerrmmooddyynnaammiikkii wwyynniikkaa,, żżee mmuussiimmyy
uuwwzzggllęęddnniićć wwsszzyyssttkkiiee rrooddzzaajjee pprraaccyy jjaakkiiee mmoożżee uukkłłaadd wwyykkoonnyywwaaćć lluubb jjaakkiiee mmooggąą bbyyćć nnaadd uukkłłaaddeemm
wwyykkoonnyywwaannee.. WW pprrzzyyppaaddkkuu ooggóóllnnyymm rróóżżnniicczzkkęę pprraaccyy mmoożżnnaa pprrzzeeddssttaawwiićć jjaakkoo::
∑=n
iidrFdW
ggddzziiee:: FF –– wwssppóółłcczzyynnnniikk pprraaccyy
55

Część III: Termodynamika układów biologicznych
rr –– wwssppóółłrrzzęęddnnaa pprraaccyy..
NNaalleeżżyy tteerraazz ddllaa kkaażżddeeggoo rrooddzzaajjuu pprraaccyy ookkrreeśślliićć wwssppóółłcczzyynnnniikk ii wwssppóółłrrzzęęddnnąą pprraaccyy.. ZZ ppuunnkkttuu
wwiiddzzeenniiaa bbiiooffiizzyykkii iinntteerreessoowwaaćć nnaass bbęęddzziiee::
pprraaccaa oobbjjęęttoośścciioowwaa:: pdVdWV −= ,, ggddzziiee wwssppóółłcczzyynnnniikkiieemm pprraaccyy jjeesstt cciiśśnniieenniiee aa
wwssppóółłrrzzęęddnnąą pprraaccyy oobbjjęęttoośśćć
pprraaccaa mmiięęśśnniioowwaa:: FdldWl = ,, FF –– ssiiłłaa mmeecchhaanniicczznnaa,, ll –– ddłłuuggoośśćć mmiięęśśnniiaa lluubb
wwłłóókknnaa mmiięęśśnniioowweeggoo
pprraaccaa cchheemmiicczznnaa:: dndWn µ= ,, µµ -- ppootteennccjjaałł cchheemmiicczznnyy,, nn –– lliicczzbbaa mmoollii
pprraaccaa jjoonnoowwaa:: dqdWq ψ= ,, ψψ -- ppootteennccjjaałł eelleekkttrryycczznnyy,, qq -- łłaadduunneekk
TTaakk wwiięęcc jjeeżżeellii ttrraannssppoorrttoowwaannee jjeesstt mm rróóżżnnyycchh ssuubbssttaannccjjii,, ttoo rróóżżnniicczzkkęę pprraaccyy pprrzzeeddssttaawwiićć mmoożżnnaa ww
ppoossttaaccii::
∑∑ µ+ψ++−=+++=m
iim
nqlV dndqFdlpdVdWdWdWdWdWi
zzwwaanneejj rróówwnnaanniieemm GGiibbbbssaa.. RRóówwnnaanniiee ttoo mmoożżnnaa rroozzsszzeerrzzaaćć lluubb sskkrraaccaaćć ww zzaalleeżżnnoośśccii oodd
ooppiissyywwaanneeggoo uukkłłaadduu..
PPrrzzyykkłaadd::ł RRoozzwwaażżmmyy qquuaassiiooddwwrraaccaallnnyy pprroocceess kkuurrcczzeenniiaa ssiięę wwłłóókknnaa mmiięęśśnniioowweeggoo.. EEnneerrggiiaa tteeggoo pprroocceessuu ppoocchhooddzzii zz pprroocceessóóww ttrraannssppoorrttuu.. PPrroocceess jjeesstt ccyykklliicczznnyy.. NNaappiisszzmmyy rróówwnnaanniiee II.. zzaassaaddyy tteerrmmooddyynnaammiikkii ww ffoorrmmiiee ccaałłkkoowweejj::
∫∑∫∫∫∫∫ µ+ψ++−=m
iidndqFdlpdVTdSdU
PPoonniieewwaażż ww uukkłłaaddzziiee nniiee mmaa zzmmiiaannyy oobbjjęęttoośśccii,, ddVV == 00,, aannii rruucchhuu łłaadduunnkkóóww,, ddqq == 00,, aa ppoonnaaddttoo pprroocceess mmaa bbyyćć ooddwwrraaccaallnnyy,, ddSS == 00,, wwiięęcc 33 cczzłłoonnyy tteeggoo rróówwnnaanniiaa zzeerruujjąą ssiięę::
∫∑∫∫ µ+=m
iidnFdldU
PPoonniieewwaażż UU jjeesstt ffuunnkkccjjąą ssttaannuu,, wwiięęcc ccaałłkkaa ppoo ddrrooddzzee zzaammkknniięętteejj jjeesstt rróówwnnaa 00:: 0dU =∫ ..
OOssttaatteecczznniiee mmaammyy wwiięęcc::
0dnFdlm
ii =µ+ ∫∑∫ ,, cczzyyllii
0dnFdlm
ii =µ+ ∫∑∫ ..
ZZ uuzzyysskkaanneejj zzaalleeżżnnoośśccii wwiiddaaćć wwyyrraaźźnniiee,, żżee aabbyy mmooggłłaa bbyyćć wwyykkoonnaannaa jjaakkaakkoollwwiieekk pprraaccaa mmiięęśśnniioowwaa mmuussii uulleecc zzmmiiaanniiee ssttęężżeenniiee cchhoocciiaażż jjeeddnneejj ssuubbssttaannccjjii..
Inne użyteczne funkcje stanu PPoozzaa eenneerrggiiąą wweewwnnęęttrrzznnąą uukkłłaadduu iissttnniieejjee jjeesszzcczzee kkiillkkaa iinnnnyycchh,, bbaarrddzzoo uużżyytteecczznnyycchh ffuunnkkccjjii
ssttaannuu zzwwiiąązzaannyycchh zz pprrzzeemmiiaannaammii eenneerrggeettyycczznnyymmii.. NNaalleeżżąą ddoo nniicchh::
eennttaallppiiaa ((HH)) ddeeffiinniioowwaannaa zzaalleeżżnnoośścciiaa:: HH == UU ++ ppVV
66

Część III: Termodynamika układów biologicznych
eenneerrggiiaa sswwoobbooddnnaa ((FF)) zzwwaannaa ttaakkżżee ffuunnkkccjjaa HHeellmmhhoollttzzaa ddeeffiinniioowwaannaa wwzzoorreemm:: FF == UU –– TTSS
eennttaallppiiaa sswwoobbooddnnaa ((GG)) zzwwaannaa ttaakkżżee ffuunnkkccjjąą GGiibbbbssaa:: GG == HH –– TTSS == UU ++ ppVV -- TTSS
IIcchh zznnaacczzeenniiee ddllaa ooppiissuu pprroocceessóóww cchheemmiicczznnyycchh ii bbiioocchheemmiicczznnyycchh ppoozznnaammyy sszzcczzeeggóółłoowwoo ww
nnaassttęęppnnyycchh eettaappaacchh wwyykkłłaadduu..
Potencjał chemiczny OOmmaawwiiaajjąącc pprraaccęę ww uukkłłaaddaacchh tteerrmmooddyynnaammiicczznnyycchh ssttwwiieerrddzziilliiśśmmyy,, żżee jjeeddnnąą zz jjeejj rrooddzzaajjóóww
jjeesstt pprraaccaa cchheemmiicczznnaa zzwwiiąązzaannaa zzee zzmmiiaannąą lliicczzbbyy mmoollii ppoosszzcczzeeggóóllnnyycchh ssuubbssttaannccjjii ww uukkłłaaddzziiee..
WWssppóółłcczzyynnnniikkiieemm pprraaccyy cchheemmiicczznneejj jjeesstt wwiieellkkoośśćć zzwwaannaa ppootteennccjjaałłeemm cchheemmiicczznnyymm,, µµ.. SSpprróóbbuujjmmyy
tteerraazz ppoowwiiąązzaaćć ttąą wwiieellkkoośśćć zz ppoozznnaannyymmii ddoottyycchhcczzaass ffuunnkkccjjaammii ssttaannuu..
RRoozzwwaażżmmyy uukkłłaadd tteerrmmooddyynnaammiicczznnyy,, ww kkttóórryymm mmoożżlliiwwee ssąą ttyyllkkoo 22 rrooddzzaajjee pprraaccyy:: pprraaccaa
oobbjjęęttoośścciioowwaa ii pprraaccaa cchheemmiicczznnaa.. DDllaa ttaakkiieeggoo uukkłłaadduu II.. zzaassaaddaa tteerrmmooddyynnaammiikkii pprrzzyyjjmmuujjee ppoossttaaćć::
∑≠
µ+µ+−=ij
jjii dndnpdVTdSdU
JJeeżżeellii zzaałłoożżyymmyy,, żżee ww uukkłłaaddzziiee nniiee zzmmiieenniiaa ssiięę eennttrrooppiiaa,, ddSS == 00,, oobbjjęęttoośśćć,, ddVV == 00,, oorraazz lliicczzbbaa mmoollii
ssuubbssttaannccjjii iinnnnyycchh nniiżż ii ttoo rróówwnnaanniiee uupprroośśccii ssiięę ddoo ppoossttaaccii::
iidndU µ=
AA wwiieecc ww uukkłłaaddzziiee ttaakkiimm zzmmiiaannaa lliicczzbbyy mmoollii ii--tteejj ssuubbssttaannccjjii,, ddnnii,, zzwwiiąązzaannaa jjeesstt bbeezzppoośśrreeddnniioo zzee
zzmmiiaannąą eenneerrggiiii wweewwnnęęttrrzznneejj uukkłłaadduu.. PPoozzwwaallaa ttoo zzddeeffiinniioowwaaćć ppootteennccjjaałł cchheemmiicczznnyy ii--tteejj ssuubbssttaannccjjii
jjaakkoo::
'n,V,Si
i nU⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=µ
cczzyyllii jjaakkoo ppoocchhooddnnąą eenneerrggiiii wweewwnnęęttrrzznneejj ppoo zzmmiiaanniiee lliicczzbbyy mmoollii ii--tteejj ssuubbssttaannccjjii pprrzzyy ssttaałłeejj eennttrrooppiiii,,
oobbjjęęttoośśccii ii lliicczzbbiiee mmoollii ppoozzoossttaałłyycchh ssuubbssttaannccjjii oobbeeccnnyycchh ww uukkłłaaddzziiee..
JJeeżżeellii ddoo ooppiissuu uukkłłaadduu cchhcceemmyy uużżyyćć eennttaallppiiii zzaammiiaasstt eenneerrggiiii wweewwnnęęttrrzznneejj,, ttoo mmuussiimmyy
nnaajjppiieerrww oobblliicczzyyćć jjeejj rróóżżnniicczzkkęę zzuuppeełłnnąą::
VdppdVdUdH ++=
aa nnaassttęęppnniiee wwyyzznnaacczzyyćć rróóżżnniicczzkkęę eenneerrggiiii wweewwnnęęttrrzznneejj::
VdppdVdHdU −−=
kkttóórrąą tteerraazz mmoożżnnaa ppooddssttaawwiićć ddoo rróówwnnaanniiaa II.. zzaassaaddyy tteerrmmooddyynnaammiikkii::
∑≠
µ+µ+−=−−ij
jjii dndnpdVTdSVdppdVdH
PPoo uupprroosszzcczzeenniiuu wwyyrraazzóóww ppooddoobbnnyycchh oottrrzzyymmuujjeemmyy::
77

Część III: Termodynamika układów biologicznych
∑≠
µ+µ+−=ij
jjii dndnVdpTdSdH
JJeeżżeellii zzaałłoożżyymmyy,, żżee ww uukkłłaaddzziiee nniiee zzmmiieenniiaa ssiięę eennttrrooppiiaa,, ddSS == 00,, cciiśśnniieenniiee,, ddpp == 00,, ii lliicczzbbaa mmoollii
ssuubbssttaannccjjii iinnnnyycchh nniiżż ii,, ttoo oottrrzzyymmaammyy::
iidndH µ=
PPoozzwwaallaa ttoo zzddeeffiinniioowwaaćć ppootteennccjjaałł cchheemmiicczznnyy jjaakkoo::
'n,p,Si
i nH⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=µ
AA wwiięęcc ww uukkłłaaddzziiee oo ssttaałłeejj eennttrrooppiiii ii ssttaałłyymm cciiśśnniieenniiuu ppootteennccjjaałł cchheemmiicczznnyy ssuubbssttaannccjjii ii jjeesstt
ppoocchhooddnnąą eennttaallppiiii ppoo lliicczzbbiiee mmoollii tteejj ssuubbssttaannccjjii.. JJeesstt ttoo pprrzzyy ttyymm tteenn ssaamm ppootteennccjjaałł cchheemmiicczznnyy,, ccoo
wwee wwzzoorrzzee ppoopprrzzeeddnniimm,, ttyyllkkoo ddllaa uukkłłaadduu ww kkttóórryymm ssttaałłee ssąą iinnnnee ppaarraammeettrryy..
WW uukkłłaaddaacchh tteerrmmooddyynnaammiicczznnyycchh ssppoottyykkaannyycchh ww pprraakkttyyccee cchheemmiicczznneejj cczzyy bbiiooffiizzyycczznneejj
uuttrrzzyymmaanniiee ssttaałłoośśccii eennttrrooppiiii jjeesstt pprraakkttyycczznniiee nniieemmoożżlliiwwee.. DDllaatteeggoo ppoosszzuukkiiwwaannoo ttaakkiicchh ddeeffiinniiccjjii
ppootteennccjjaałłuu cchheemmiicczznneeggoo,, ww kkttóórryycchh wwaarruunneekk tteenn nniiee mmuussiiaałłbbyy bbyyćć ssppeełłnniioonnyy.. SSkkuuppmmyy nnaasszząą uuwwaaggęę
zz kkoolleeii nnaa eenneerrggiiii sswwoobbooddnneejj.. JJeejj rróóżżnniicczzkkaa zzuuppeełłnnaa mmaa ppoossttaaćć::
SdTTdSdUdF −−=
SSttoossuujjąącc ppoossttęęppoowwaanniiee aannaallooggiicczznnee jjaakk ddllaa eennttaallppiiii oottrrzzyymmaammyy rróówwnnaanniiee::
∑≠
µ+µ+−−=ij
jjii dndnpdVSdTdF
JJeeżżeellii zzaałłoożżyymmyy,, żżee ww uukkłłaaddzziiee nniiee zzmmiieenniiaa ssiięę tteemmppeerraattuurraa,, ddTT == 00,, oobbjjęęttoośśćć,, ddVV == 00,, ii lliicczzbbaa mmoollii
ssuubbssttaannccjjii iinnnnyycchh nniiżż ii,, ttoo oottrrzzyymmaammyy::
iidndF µ=
PPoozzwwaallaa ttoo zzddeeffiinniioowwaaćć ppootteennccjjaałł cchheemmiicczznnyy jjaakkoo::
'n,V,Ti
i nF⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=µ
AAnnaallooggiicczznnee ppoossttęęppoowwaanniiee zz eennttaallppiiąą sswwoobbooddnnąą pprroowwaaddzzii ddoo ddeeffiinniiccjjii::
'n,p,Ti
i nG⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=µ
TTaakk wwiięęcc ww zzaalleeżżnnoośśccii oodd ssttaannuu uukkłłaadduu rróóżżnnee ffuunnkkccjjee ssttaannuu ddeeffiinniiuujjąą ppootteennccjjaałł cchheemmiicczznnyy
uukkłłaadduu.. PPoonniieewwaażż ww uukkłłaaddaacchh bbiioollooggiicczznnyycchh mmaammyy nnaajjcczzęęśścciieejj ddoo cczzyynniieenniiaa zzee ssttaałłąą tteemmppeerraattuurraa ii
ssttaałłyymm cciiśśnniieenniieemm,, wwiięęcc kkoorrzzyyssttaaćć bbęęddzziieemmyy zz oossttaattnniieejj zz wwyypprroowwaaddzzoonnyycchh ddeeffiinniiccjjii..
MMoożżnnaa wwyykkaazzaaćć,, żżee ppootteennccjjaałł cchheemmiicczznnyy uukkłłaadduu zzaalleeżżyy oodd tteemmppeerraattuurryy ii ssttęężżeenniiaa ((lliicczzbbyy
mmoollii)) ssuubbssttaannccjjii.. ZZaalleeżżnnoośśćć ttaa mmaa ppoossttaaćć::
88

Część III: Termodynamika układów biologicznych
lluubb i0ii nlnRT+µ=µ i
0ii clnRT+µ=µ
ggddzziiee:: µµ00 –– ppootteennccjjaałł cchheemmiicczznnyy pprrzzyy jjeeddnnoossttkkoowwyymm ssttęężżeenniiuu lluubb lliicczzbbiiee mmoollii
Równowaga chemiczna UUkkłłaadd zznnaajjdduujjee ssiięę ww ssttaanniiee rróówwnnoowwaaggii cchheemmiicczznneejj,, ggddyy pprrzzeebbiieeggaajjąą ww nniimm ttyyllkkoo pprroocceessyy
ooddwwrraaccaallnnee,, aa lliicczzbbaa mmoollii wwsszzyyssttkkiicchh ssuubbssttaannccjjii ((lluubb iicchh ssttęężżeenniiee)) nniiee uulleeggaa zzmmiiaanniiee ww cczzaassiiee.. JJaakk
jjuużż wwiieemmyy,, tteerrmmooddyynnaammiicczznnyymm wwaarruunnkkiieemm ooddwwrraaccaallnnoośśccii pprroocceessuu jjeesstt zzeerroowwaa wwaarrttoośśćć rróóżżnniicczzkkii
eennttrrooppiiii,, ddSS == 00..
ZZ wwaarruunnkkuu ssttaałłoośśccii sskkłłaadduu ww ssttaanniiee rróówwnnoowwaaggii wwyynniikkaa,, żżee bbrraakk jjeesstt ww nniimm ssiiłł nnaappęęddoowwyycchh
pprroowwaaddzząąccyycchh ddoo zzmmiiaannyy sskkłłaadduu.. SSiiłłąą nnaappęęddoowwąą zzmmiiaannyy ssttęężżeenniiaa ddaanneejj ssuubbssttaannccjjii jjeesstt jjeejj ppootteennccjjaałł
cchheemmiicczznnyy.. WWyynniikkaa ssttąądd wwpprroosstt,, żżee ww ssttaanniiee rróówwnnoowwaaggii ppootteennccjjaałłyy cchheemmiicczznnee wwsszzyyssttkkiicchh
ssuubbssttaannccjjii ssąą rróówwnnee zzeerroo.. PPaammiięęttaajjąącc,, żżee ppootteennccjjaałł cchheemmiicczznnyy ii--tteejj ssuubbssttaannccjjii jjeesstt ppoocchhooddnnąą eennttaallppiiii
sswwoobbooddnneejj ppoo ssttęężżeenniiuu tteejj ssuubbssttaannccjjii ddoocchhooddzziimmyy ddoo wwnniioosskkuu,, żżee ww ssttaanniiee rróówwnnoowwaaggii eennttaallppiiaa
sswwoobbooddnnaa uukkłłaadduu oossiiąąggaa mmiinniimmuumm.. JJeeżżeellii eennttaallppiiaa jjeesstt wwiięękksszzaa nniiżż ttoo mmiinniimmuumm,, ttoo uukkłłaadd zznnaajjdduujjee
ssiięę ppoozzaa ssttaanneemm rróówwnnoowwaaggii ii bbęęddzziiee zzmmiieenniiaałł sswwóójj sskkłłaadd ddąążżąącc ddoo mmiinniimmuumm eennttaallppiiii sswwoobbooddnneejj..
WWyynniikkaa ssttąądd cciieekkaawwyy wwnniioosseekk:: rreeaakkccjjee cchheemmiicczznnee ww wwaarruunnkkaacchh iizzootteerrmmiicczznnoo--iizzoobbaarryycczznnyycchh
pprrzzeebbiieeggaajjąą ssaammoorrzzuuttnniiee ww kkiieerruunnkkuu oobbnniiżżeenniiaa eennttaallppiiii sswwoobbooddnneejj.. Rozważmy odwracalną reakcję
chemiczną:
A + B <==> C + D
PPrrzzeebbiieegg tteejj rreeaakkccjjii nnaajjwwyyggooddnniieejj jjeesstt śślleeddzziićć wwpprroowwaaddzzaajjąącc ppoojjęęcciiee wwssppóółłrrzzęęddnneejj rreeaakkccjjii,, ξξ.. GGddyy ww
uukkłłaaddzziiee zznnaajjdduujjąą ssiięę wwyyłłąącczznniiee ssuubbssttrraattyy ξξ == 00,, aa ggddyy rreeaakkccjjaa pprrzzeebbiieeggnniiee ccaałłkkoowwiicciiee ii ww uukkłłaaddzziiee
bbęęddąą wwyyłłąącczznniiee pprroodduukkttyy ξξ == 11.. WWpprroowwaaddźźmmyy jjeesszzcczzee jjeeddnnoo ppoojjęęcciiee:: zzmmiiaannaa eennttaallppiiii sswwoobbooddnneejj,,
∆∆GG rróówwnnee zz ddeeffiinniiccjjii::
p,T
GG ⎟⎟⎠
⎞⎜⎜⎝
⎛ξ∂
∂≡∆
NNaalleeżżyy zzwwrróócciićć uuwwaaggęę nnaa nniieeccoo mmyylląąccyy ssyymmbbooll zzmmiiaannyy eennttaallppiiii
sswwoobbooddnneejj.. WWbbrreeww ffoorrmmiiee zzaappiissuu nniiee jjeesstt ttoo rróóżżnniiccaa eennttaallppiiii
sswwoobbooddnneejj mmiięęddzzyy ddwwoommaa ssttaannaammii,, aallee ppoocchhooddnnaa,, cczzyyllii sszzyybbkkoośśćć
zzmmiiaann eennttaallppiiii sswwoobbooddnneejj.. TTaa wwłłaaśśnniiee wwiieellkkoośśćć ddeeccyydduujjee oo
kkiieerruunnkkuu pprrzzeebbiieegguu rreeaakkccjjii::
współrzędna reakcji, ξ 0 1
enta
lpia
swob
odna
, G
∆G < 0 ∆G > 0
∆G = 0 ∆∆GG << 00 –– pprrzzeebbiieegg ssppoonnttaanniicczznnyy:: pprrzzyybbyywwaa pprroodduukkttóóww
∆∆GG == 00 –– ssttaann rróówwnnoowwaaggii
99

Część III: Termodynamika układów biologicznych
∆∆GG >> 00 –– rreeaakkccjjaa pprrzzeebbiieeggaa ssppoonnttaanniicczznniiee,, aallee ww ooddwwrroottnnyymm kkiieerruunnkkuu
PPrrzzeebbiieegg rreeaakkccjjii mmoożżnnaa tteeżż pprrzzeeddssttaawwiićć ggrraaffiicczznniiee ww ffoorrmmiiee wwyykkrreessuu wwaarrttoośśccii eennttaallppiiii sswwoobbooddnneejj
uukkłłaadduu,, GG,, ww ffuunnkkccjjii wwssppóółłrrzzęęddnneejj rreeaakkccjjii,, ξξ,, jjaakk nnaa rryyssuunnkkuu ppoowwyyżżeejj..
NNaa ggrruunncciiee cchheemmiiii ffiizzyycczznneejj mmoożżnnaa wwyypprroowwaaddzziićć zzaalleeżżnnoośśćć zzmmiiaann eennttaallppiiii sswwoobbooddnneejj oodd ssttęężżeeńń
ssuubbssttrraattóóww ii pprroodduukkttóóww::
BA
DC0
cccclnRTGG⋅⋅
+∆=∆
ggddzziiee:: ∆∆GG00 –– ssttaannddaarrddoowwaa zzmmiiaannaa eennttaallppiiii sswwoobbooddnneejj,, cczzyyllii ddllaa jjeeddnnoossttkkoowwyycchh ssttęężżeeńń ssuubbssttrraattóóww ii
pprroodduukkttóóww..
PPoonniieewwaażż ww ssttaanniiee rróówwnnoowwaaggii ∆∆GG == 00,, wwiięęcc::
'B
'A
'D
'C0
cccclnRTG⋅⋅
−=∆
ggddzziiee:: ccii’’ –– ssttęężżeenniiee ii--tteejj ssuubbssttaannccjjii ww ssttaanniiee rróówwnnoowwaaggii.. JJeeżżeellii wwpprroowwaaddzziimmyy tteerraazz ppoojjęęcciiee ssttaałłeejj
rróówwnnoowwaaggii,, KK,, zzddeeffiinniioowwaanneejj jjaakkoo::
'B
'A
'D
'C
ccccK⋅⋅
=
to otrzymamy znane zależności:
ii KlnRTG0 −=∆ KlnRTcccclnRTG
BA
DC −⋅⋅
=∆
Drugie z tych równań nosi nazwę izotermy van’t Hoffa i może być zastosowane do wyznaczania
wartości ∆G0 i K.
Należy bardzo wyraźnie zwrócić uwagę, że o kierunku przebiegu reakcji decyduje ∆∆GG,, aa nniiee
∆∆GG00,, cczzyyllii zzmmiiaannaa eennttaallppiiii sswwoobbooddnneejj pprrzzyy aakkttuuaallnnyycchh ssttęężżeenniiaacchh ssuubbssttrraattóóww ii pprroodduukkttóóww.. JJeesstt ttoo
sszzcczzeeggóóllnniiee wwaażżnnee ww uukkłłaaddaacchh bbiioollooggiicczznnyycchh,, ww kkttóórryycchh ssttęężżeenniiaa nniieekkttóórryycchh ssuubbssttrraattóóww lluubb
pprroodduukkttóóww mmooggąą bbyyćć uuttrrzzyymmyywwaannee nnaa ssttaałłyymm ppoozziioommiiee zzddeeccyyddoowwaanniiee rróóżżnnyymm nniiżż iinnnnyycchh
ssuubbssttaannccjjii bbiioorrąąccyycchh uuddzziiaałł ww rreeaakkccjjii..
PPrrzzyykkłaadd::ł PPrroocceessoowwii pprrzzeemmiiaannyy gglluukkoozzyy ww 22 cczząąsstteecczzkkii kkwwaassuu mmlleekkoowweeggoo ((rreeaakkccjjaa gglliikkoolliizzyy)) CC66HH1122OO66 ==>> 22CC33HH66OO33
ooddppoowwiiaaddaa ssttaannddaarrddoowwaa zzmmiiaannaa eennttaallppiiii sswwoobbooddnneejj rróówwnnaa ∆∆GG00 == --113388 kkJJ//mmooll.. WW kkoommóórrccee ssttęężżeenniiaa gglluukkoozzyy ii kkwwaassuu mmlleekkoowweeggoo ssąą ddoossyyćć śścciiśśllee rreegguulloowwaannee ii uuttrrzzyymmuujjąą ssiięę nnaa ppoozziioommiiee 00,,000055 MM ddllaa gglluukkoozzyy ii 00,,000011 MM ddllaa kkwwaassuu mmlleekkoowweeggoo.. PPoo pprroossttyycchh pprrzzeelliicczzeenniiaacchh wwyynniikkaa,, żżee zzmmiiaannaa eennttaallppiiii sswwoobbooddnneejj tteejj rreeaakkccjjii ww wwaarruunnkkaacchh wwnnęęttrrzzaa kkoommóórrkkii wwyynnoossii ∆∆GG == --115599 kkJJ//mmooll.. TTaakk wwiięęcc rreeaakkccjjaa ttaa pprrzzeebbiieeggaa ww ttyycchh wwaarruunnkkaacchh ssppoonnttaanniicczznniiee ii mmoożżee bbyyćć źźrróóddłłeemm eenneerrggiiii ddllaa kkoommóórrkkii.. NNiieekkttóórree bbaakktteerriiee zzddoollnnee ssąą ddoo wwiiąązzaanniiaa ggaazzoowweeggoo aazzoottuu ii uuttlleenniiaanniiaa ggoo ddoo jjoonnóóww aazzoottaannoowwyycchh::
1100

Część III: Termodynamika układów biologicznych
22NN22 ++ 55OO22 ++ 22HH22OO ==== 44HH++ ++ 44NNOO33--
SSttaannddaarrddoowwaa zzmmiiaannaa eennttaallppiiii sswwoobbooddnneejj tteejj rreeaakkccjjii wwyynnoossii ∆∆GG00 == ++77,,77 kkJJ//mmooll.. TTaakk wwiięęcc ww wwaarruunnkkaacchh ssttaannddaarrddoowwyycchh rreeaakkccjjaa ttaa nniiee mmoożżee zzaacchhooddzziićć ssaammoorrzzuuttnniiee.. JJeeddnnaakkżżee ww kkoommóórrccee ttyycchh bbaakktteerriiii ssttęężżeenniiee jjoonnóóww aazzoottaannoowwyycchh wwyynnoossii ookk.. 1100--44 MM,, ppHH ≈≈ 77,, aa cciiśśnniieenniiaa ppaarrccjjaallnnee ttlleennuu ii aazzoottuu wwyynnoosszząą ooddppoowwiieeddnniioo 220000 ii 880000 hhPP.. WW ttyycchh wwaarruunnkkaacchh zzmmiiaannaa eennttaallppiiii sswwoobbooddnneejj wwyynnoossii ∆∆GG == --3322,,66 kkJJ//mmooll.. TTaakk wwiięęcc rreeaakkccjjaa mmoożżee pprrzzeebbiieeggaaćć ssppoonnttaanniicczznniiee,, oo iillee ttyyllkkoo oobbeeccnnee ssąą ooddppoowwiieeddnniiee kkaattaalliizzaattoorryy ((eennzzyymmyy)).. RRóówwnniieeżż ww pprrzzyyppaaddkkuu hhyyddrroolliizzyy AATTPP ddoo AADDPP ii ffoossffoorraannuu bbęęddąącceejj ppooddssttaawwoowwyymm źźrróóddłłeemm eenneerrggiiii ww kkoommóórrccee zznnaacczzeenniiee wwaarruunnkkóóww llookkaallnnyycchh jjeesstt kklluucczzoowwee.. SSttaannddaarrddoowwaa zzmmiiaannaa eennttaallppiiii sswwoobbooddnneejj tteejj rreeaakkccjjii wwyynnoossii ∆∆GG00 == --55,,44,, kkJJ//mmooll.. MMoożżee wwiięęcc oonnaa pprrzzeebbiieeggaaćć ssaammoorrzzuuttnniiee jjeeddnnaakk nniiee mmoożżee bbyyćć źźrróóddłłeemm eenneerrggiiii.. WW wwaarruunnkkaacchh wwnnęęttrrzzaa kkoommóórrkkii zzmmiiaannaa eennttaallppiiii sswwoobbooddnneejj jjeesstt pprraawwiiee 1100 rraazzyy wwiięękksszzaa,, ∆∆GG == --4477,,77 kkJJ//mmooll,, ccoo uuzzaassaaddnniiaa zznnaacczzeenniiee tteejj rreeaakkccjjii jjaakkoo źźrróóddłłaa eenneerrggiiii..
Termodynamika procesów nieodwracalnych Klasyczna termodynamika opisuje w pełni jedynie procesy odwracalne, a więc takie, w
których dS = 0. Badanie procesów nieodwracalnych, zwłaszcza w układach otwartych, ograniczało
się do niedawna do analizy występujących w nich stanów równowagi. Bowiem w stanach
równowagi dS = 0. Ponadto, w stanach równowagi nie dochodzi do zmian stężeń lub ilości
substancji, a więc w pewnym sensie stan równowagi tworzy układ „zamknięty”.
Układy biologiczne nie znajdują się w stanie równowagi termodynamicznej i są zawsze układami
otwartymi:
wymieniają materię z otoczeniem
produkują entropię
Do układów takich opis klasyczny jest nieprzydatny. Ale nie jest to cecha charakterystyczna
układów ożywionych. Również martwe układy znajdujące się z dala od stanu równowagi nie dają
się opisać na gruncie termodynamiki klasycznej.
Układy otwarte Pierwszej udanej próby wyjścia poza opis stanów równowagowych dokonano dla układów
otwartych. Rozważmy zmiany entropii w układzie otwartym. Całkowitą szybkość zmian entropii
układu dS/dt można opisać równaniem:
dtdS
dtdS
dtdS ie +=
gdzie: Se – entropia wymieniana z otoczeniem,
Si – entropia produkowana przez układ.
Drugi z członów powyższego równania, dSi/dt, opisujący szybkość produkcji entropii w układzie
oznacza się często symbolem σ. Wtedy równanie przyjmuje postać:
σ+=dt
dSdtdS e
1111

Część III: Termodynamika układów biologicznych
Do opisu wymiany entropii z otoczeniem wygodnie jest użyć pojęcia strumienia. W fizyce, chemii
fizycznej i biofizyce strumień J definiujemy jako:
ilość materii lub energii przechodząca w jednostce czasu przez jednostkę
powierzchni prostopadłą do kierunku strumienia.
Warunkiem istnienia strumienia jest występowanie w układzie różnicy jakiegoś potencjału
termodynamicznego, x. Wymianę entropii z otoczeniem można opisać jako strumień entropii:
xJ
dtdS Se
∂∂
−=
Ostatecznie równanie opisujące zmiany entropii w układzie otwartym przyjmie więc postać:
σ+∂∂
−=xJ
dtdS S
Stan stacjonarny Wielkością determinującą stan układu jest przy tym szybkość produkcji entropii σ. Z II. zasady
termodynamiki wynika, że σ nie może być mniejsze od 0. Jeżeli σ = 0, to mamy do czynienia z
równowagą termodynamiczną, stanem nieciekawym z punktu widzenia biofizyki. Gdy σ > 0 w
układzie biegnie samorzutnie jakiś proces i wytwarzana jest entropia. Z punktu widzenia biofizyki
interesujący jest pewien przypadek szczególny procesów samorzutnych w układach otwartych:
przypadek, gdy cała produkowana w układzie entropia σ opuszcza układ w postaci strumienia
entropii:
SJx
∂σ =
∂
Podstawiając ten warunek do powyższego równania dochodzimy do zależności: dS/dt = 0. Taki
stan układu nazywamy stanem stacjonarnym: układ produkuje entropię (biegnie proces
samorzutny), lecz jego entropia nie wzrasta.
Okazuje się, że wiele układów ożywionych można z dobrym przybliżeniem traktować jako układy
stacjonarne. Dla termodynamicznego opisu takich układów musimy jeszcze określić siłę
napędzającą strumień entropii, x, oraz zaproponować sposób wyznaczania szybkości produkcji
entropii w układzie, σ.
Entropia układu jest typowa funkcją wewnętrzną. Wartości funkcji tego typu określone są
przez zmienne zewnętrzne (dające się wyznaczyć z pomiaru) jedynie w stanie równowagi. Również
jedynie w stanie równowagi entropia jest funkcja stanu i tylko wtedy posiada różniczkę zupełną.
Jednakże my z definicji rozpatrujemy układ nie znajdujący się w stanie równowagi. Wygląda to na
ślepą uliczkę.
1122

Część III: Termodynamika układów biologicznych
Przez dłuższy czas poszukiwano wyjścia z tego problemu. Udało się je znaleźć dzięki podziałowi
układu na podukłady tak małe, że każdy z nich można potraktować jako lokalnie znajdujący się w
stanie równowagi. Podejście takie jest możliwe, gdy układ jako całość znajduje się w pobliżu stanu
równowagi. Można wtedy wykazać, że produkcja entropii σ musi spełniać równanie:
∑=
=σ⋅n
1iiixJT
gdzie: Ji – i-ty strumień
xi – siła napędowa i-tego strumienia
Okazuje się więc, że podejście takie wyjaśnia od razu dwie sprawy: wielkość produkcji entropii w
stanie stacjonarnym i siły napędowe strumienia entropii wypływającego z układu: siłami tymi są
siły napędowe innych strumieni obecnych w układzie.
Wyrażenie Tσ zwane jest funkcją dyssypacji. Określa ona zużycie energii w układzie, czyli jak
szybko układ przekształca użyteczne formy energii w zdegenerowaną, bezużyteczną energię
cieplną i przekazuje ją (dyssypuje) do otoczenia.
Siły napędowe i strumienie Ponieważ pojęcie strumienia obecnie jest kluczowe przy opisie układów nieodwracalnych,
poświęćmy trochę uwagi siłom napędowym typowych strumieni występujących w układach
biofizycznych. Tabela 0-1 zawiera zestaw takich sił napędowych i odpowiadających im strumieni.
Tabela 0-1 Strumienie i odpowiadające im siły napędowe
Siła napędowa Strumień
różnica potencjału elektrycznego prąd elektryczny
różnica ciśnień hydrostatycznych przepływ cieczy
różnica potencjału chemicznego dyfuzja cząsteczek obojętnych
różnica potencjału elektrochemicznego ruch jonów
Dla izolowanych strumieni wielkość strumienia, J, jest proporcjonalna do wartości siły
napędowej x:
LxJ =
W takich strumieniach kierunek przepływu musi być zgodny z kierunkiem siły napędowej.
Okazuje się jednak, że w rzeczywistych układach mamy często do czynienia ze strumieniami
sprzężonymi, czyli takimi gdzie strumień zależy od kilku sił napędowych.
ddllaa ii oodd 11 ddoo nn ( ni1i x,,x,,xfJ KK= )
1133

Część III: Termodynamika układów biologicznych
W przypadku strumieni sprzężonych niektóre strumienie mogą płynąć „pod prąd” odpowiadających
im sił napędowych o ile napędzają je inne strumienie.
Pojawia się teraz problem, jaka jest postać funkcji f(x1,..,xn) w powyższym równaniu. Jak
dotychczas przyjmuje się najczęściej, że jest to zależność liniowa:
nnn22n11nn
nn22221212
nn12121111
xLxLxLJ
xLxLxLJxLxLxLJ
+++=
+++=+++=
L
M
L
L
Taki układ równań można zapisać w zapisie macierzowym w dużo bardziej zwartej formie jako:
LXJ =
gdzie: J – kolumnowa macierz strumieni
L – kwadratowa macierz współczynników
X – kolumnowa macierz sił napędowych
Ponadto, w 1931 roku Onsager wykazał, że Lij = Lji, czyli że macierz współczynników jest
macierzą symetryczną.
Siłę sprzężenia pomiędzy strumieniami opisuje wielkość:
jjii
ijij LL
Lq =
zwana stopniem sprzężenia. Gdy qij = 0, czyli Lij = 0, mamy do czynienia z brakiem sprzężenia
strumieni i oraz j. Gdy qij = 1 mamy do czynienia ze sprzężeniem całkowitym.
Przykład 1. Rozważmy układ składający się z dwóch roztworów tej samej substancji o różnych, lecz niedużych stężeniach. Roztwory te znajdują się w dwóch różnych częściach naczynia rozdzielonych błoną (rys. obok). W układzie występują dwie siły napędowe: i) różnica potencjału chemicznego substancji rozpuszczonej wynikająca z różnicy stężeń, oraz ii) różnica ciśnienia hydrostatycznego. Przy niskich stężeniach roztworu potencjał chemiczny rozpuszczalnika jest praktycznie jednakowy w obu częściach układu. Tak więc w układzie pojawić się powinny dwa strumienie: JD - strumień dyfuzyjny substancji oraz JV - strumień objętościowy roztworu. Potencjał chemiczny substancji rozpuszczonej wynosi: clnRT=µ .. RRóóżżnniiccaa ppootteennccjjaałłuu cchheemmiicczznneeggoo,, ∆∆µµ,, ddaannaa jjeesstt wwiięęcc wwzzoorreemm::
2
121 c
clnRTclnRTclnRT =−=µ∆
Zakładając występowanie sprzężenia pomiędzy strumieniami otrzymujemy zależności:
JJVV
JJDD
cc11 >> cc22
pLLJpLLJ
VVDVV
DVDDD
∆+µ∆=∆+µ∆=
gdzie: LDD – współczynnik dyfuzji substancji w błonie LVV – współczynnik filtracji błony (przepuszczalność hydrauliczna)
1144

Część III: Termodynamika układów biologicznych
LDV – współczynnik ultrafiltracji lub osmozy Z faktu występowania sprzężeń pomiędzy strumieniami wynika istnienie dwóch ciekawych zjawisk: ultrafiltracji – przepływu substancji pod wpływem różnicy ciśnienia hydrostatycznego oraz osmozy – przepływu rozpuszczalnika pod wpływem różnicy stężenia substancji rozpuszczonej. Przykład 2. Rozważmy układ otwarty, do którego może wnikać z otoczenia substancję M. Wewnątrz układu substancja ta przekształcana jest z szybkością v w substancję N. Substancja N może dyfundować do otoczenia. Ponadto przez granicę układu może dyfundować substancja O, z która nic się w układzie nie dzieje. Strumienie poszczególnych substancji nie są ze sobą sprzężone i można je opisać zależnościami:
OOOO
NNNN
MMMM
LJLJLJ
µ∆=µ∆=µ∆= JM
JN
JO
M N
WW ssttaanniiee ssttaaccjjoonnaarrnnyymm iilloośśćć ppoosszzcczzeeggóóllnnyycchh ssuubbssttaannccjjii ww uukkłłaaddzziiee nniiee uulleeggaa zzmmiiaanniiee,, ccoo mmoożżeemmyy zzaappiissaaćć jjaakkoo::
0Jdt
dn
0vJdt
dn
0vJdt
dn
OO
NN
MM
==
=+=
=−=
Z dwóch pierwszych równań wynika, że strumień substancji M wpływający do układu równy jest co do wartości strumieniowi substancji N wypływającemu z układu, a oba te strumienie równe są liczbowo szybkości powstawania substancji N. Z ostatniego równania wynika ponadto, że strumień substancji O jest zerowy. Podstawiając wyrażenia na strumienie otrzymujemy:
0L0vL0vL
OOO
NNN
MMM
=µ∆=+µ∆=−µ∆
Można teraz wyznaczyć różnice potencjałów chemicznych poszczególnych substancji w stanie stacjonarnym:
( )
( )( ) 0
Lv
Lv
sO
NNsN
MMsM
=µ∆
−=µ∆
=µ∆
Tak więc różnice potencjałów chemicznych substancji M i N zależą od szybkości przemiany i od współczynników dyfuzji poszczególnych substancji. Potencjał chemiczny substancji O jest identyczny w układzie i w otoczeniu.
Przykład 3. Rozważmy teraz jak będzie wyglądał stan stacjonarny takiego samego układu, jeżeli pomiędzy strumieniami substancji M i O występuje sprzężenie.
1155
JM
JN
JO
M N
OOOMOMO
NNNN
OOMMMMM
LLJLJ
LLJ
µ∆+µ∆=µ∆=
µ∆+µ∆=
WW ssttaanniiee ssttaaccjjoonnaarrnnyymm iilloośśćć ppoosszzcczzeeggóóllnnyycchh ssuubbssttaannccjjii ww uukkłłaaddzziiee nniiee uulleeggaa zzmmiiaanniiee,, ccoo mmoożżeemmyy zzaappiissaaćć jjaakkoo::

Część III: Termodynamika układów biologicznych
0Jdt
dn
0vJdt
dn
0vJdt
dn
OO
NN
MM
==
=+=
=−=
NNaa uuwwaaggęę zzaassłłuugguujjee ffaakktt,, żżee wwyyssttęęppoowwaanniiee sspprrzzęężżeenniiaa nniiee mmaa wwppłłyywwuu nnaa wwaarrttoośśćć ii kkiieerruunneekk ssttrruummiieennii.. WWyyzznnaacczzmmyy tteerraazz rróóżżnniiccee ppootteennccjjaałłóóww cchheemmiicczznnyycchh ppoosszzcczzeeggóóllnnyycchh ssuubbssttaannccjjii ww ssttaanniiee ssttaaccjjoonnaarrnnyymm::
( )
( )
( )
OO
2OM
MM
OO
OM
sO
NNsN
OO
2OM
MM
sM
LLL
vLL
Lv
LLL
v
−−=µ∆
−=µ∆
−=µ∆
RRzzuuccaa ssiięę ww oocczzyy,, żżee ww uukkłłaaddzziiee zzee sspprrzzęężżeenniieemm ssttrruummiieennii wwyyssttęęppuujjee rróóżżnniiccaa ppootteennccjjaałłuu cchheemmiicczznneeggoo ssuubbssttaannccjjii OO ii ttoo ppoommiimmoo zzeerroowweeggoo ssttrruummiieenniiaa tteejj ssuubbssttaannccjjii.. RRóóżżnniiccaa ppootteennccjjaałłuu cchheemmiicczznneeggoo ssuubbssttaannccjjii NN nniiee uulleeggłłaa zzmmiiaanniiee,, aa ssuubbssttaannccjjii MM uulleeggłłaa zzwwiięękksszzeenniiuu ((mmnniieejjsszzyy mmiiaannoowwnniikk uułłaammkkaa))..
Entropia w stanie stacjonarnym Przypomnijmy: stan stacjonarny to stan układu niezmienny w czasie, w którym układ
produkuje entropię w tempie σ, ale entropia układu nie ulega zmianie: dS/dt = 0. Można wykazać,
że stan taki jest możliwy tylko wtedy, gdy strumienie i siły są stałe w czasie:
0t
xtJ ii =
∂∂
=∂∂
Przy czym niektóre z nich mogą być równe 0, a reszta przyjmuje niezerowe, chociaż stałe wartości.
W zależności od liczby niezerowych strumieni mamy stany stacjonarne odpowiednich rzędów:
rząd 0 : wszystkie strumienie zanikają, σ = 0 czyli układ nie wytwarza entropii, równowaga
termodynamiczna,
rząd > 0: dodatnia szybkość produkcji entropii.
Na przełomie lat 50 i 60 XXw. Prigogine wykazał, że w stanie stacjonarnym układu
produkcja entropii osiąga minimum lokalne. Oznacza to m.in., że układ samorzutnie wraca do stanu
stacjonarnego po niewielkich odchyleniach.
Poprawność tego wniosku ograniczona jest tylko do najbliższego otoczenia minimum. Wynika to z
kilku istotnych powodów:
definicja funkcji dyssypacji w podanej powyżej formie jest poprawna jedynie dla stanów
niezbyt odległych od stanu równowagi. Jaka jest postać tej funkcji w znacznej odległości od
stanu równowagi nie zostało w przypadku ogólnym wykazane.
1166

Część III: Termodynamika układów biologicznych
przyjęliśmy liniową definicję funkcji sprzęgającej strumienie. Jest to opis poprawny jedynie
w najbliższym otoczeniu stanu równowagi.
w skomplikowanych układach występować może wiele stanów stacjonarnych, w tym niektóre
blisko siebie. Znaczne wychylenie z jednego z takich stanów może doprowadzić do tego, że
układ znajdzie się w obszarze „przyciągania” innego stanu stacjonarnego.
Pozostaje jednak prawdą, że każdy stan stacjonarny posiada swego rodzaju strefę bezpieczeństwa.
Jeżeli na skutek oddziaływania zewnętrznych czynników zakłócających układ zostanie wytrącony
ze stanu stacjonarnego, ale nie przekroczy granic tej strefy, to będzie samorzutnie starał się
powrócić do stanu stacjonarnego. Im większa jest ta strefa, tym stabilniejszy (mniej wrażliwy na
zkłócenia) jest stan stacjonarny.
1177

Część III: Termodynamika układów biologicznych
POMIARY KALORYMETRYCZNE
Wstęp Pomiary kalorymetryczne to jedyna technika badawcza pozwalająca na bezpośrednie
wyznaczenie wielkości termodynamicznych. Wszystkie inne techniki, np. spektroskopowe, są
metodami pośrednimi: wyznaczanie wielkości termodynamicznych możliwe jest tylko dla układów
spełniających określone, czasami bardzo trudne do spełnienia, warunki. Pomiary kalorymetryczne
dokonywać można przy tym bezpośrednio w warunkach równowagi bez potrzeby izolacji
substratów lub produktów.
Dzięki rozwojowi technik instrumentalnych i komputeryzacji aparatury współczesne
kalorymetry są w stanie wyznaczyć ilość wydzielanego lub pobieranego ciepła z dokładnością do
1 µJ.
Izotermiczne miareczkowanie kalorymetryczne Z punktu widzenia biofizyki najważniejszą techniką kalorymetryczną jest izotermiczne
miareczkowanie kalorymetryczne, ITC (ang. Isotermal Titration Calorymetry). Na podstawie
wyników uzyskanych w trakcie pojedynczej serii pomiarów można wyznaczyć:
- ∆H - zmianę entalpii molowej związanej z badanym oddziaływaniem
- K – stałą tworzenia kompleksu
- n – współczynnik opisujący stechiometrię oddziaływania
Pomiędzy wielkościami termodynamicznymi opisującymi badane oddziaływanie zachodzą przy
tym następujące zależności:
KlnRTSTHG −=∆−∆=∆
Ponieważ wielkości ∆H i K wyznaczane są niezależnie z danych doświadczalnych, więc można
również obliczyć ∆G i ∆S.
Wykonując pomiary w dwóch różnych temperaturach można ponadto wyznaczyć
pojemność cieplną (molowe ciepło właściwe przy stałym ciśnieniu, ∆Cp):
1
2
12
12
12p
TTln
SSTT
HHC ∆−∆=
−∆−∆
=∆
BBuuddoowwaa aappaarraattuu Układ pomiarowy składa się z dwóch naczynek umieszczonych we wspólnej osłonie
cieplnej. Naczynka mają pojemność 0,5÷1,5 ml. Osłona termiczna jest chłodzona do temperatury
1188

Część III: Termodynamika układów biologicznych
niższej niż zadana temperatura doświadczenia T0, więc dla
utrzymania zadanej temperatury naczynek należy je podgrzewać.
Podczas całego doświadczenia oba naczynka są utrzymywane w
równowadze termicznej, ∆T = 0, w zadanej temperaturze. Jedno z
naczynek spełnia przy tym rolę naczynka odniesienia, a drugie
naczynka pomiarowego. W obu naczynkach znajdują się
jednakowe objętości tego samego rozpuszczalnika. W naczynku
pomiarowym znajduje się ponadto jeden ze składników
oddziaływującego układu (składnik A). Do naczynka
pomiarowego dodawany jest niewielkimi porcjami roztwór
drugiego składnika, B, w takim samym rozpuszczalniku.
T = T0 T = T0∆T = 0
PPrrzzeebbiieegg ppoommiiaarruu Podstawą metody jest pomiar ilości energii cieplnej w jednostce czasu (mocy) jaką należy
dostarczyć do naczyńka pomiarowego, aby zachować zerową różnicę temperatur pomiędzy obu
naczynkami. Moc tą wyraża się najczęściej w µW, chociaż µcal/s są również w użyciu.
Właściwy pomiar rozpoczyna się, gdy do naczynka pomiarowego dodajemy w postaci serii
wstrzyknięć niewielkie, 5 ÷ 25 µl, porcje roztworu drugiego składnika. Całkowita objętość
dodawanego roztworu wynosi zwykle 50 ÷ 250 µl.
Dodanie drugiego składnika powoduje zachodzenie analizowanego oddziaływania i zmianę entalpii
układu. Gdy oddziaływanie ma charakter egzotermiczny, to do naczynka pomiarowego dla
zachowania zadanej temperatury wystarczy dostarczyć mniej ciepła niż do naczynka
referencyjnego. W przypadku oddziaływań endotermicznych sytuacja jest odwrotna.
W obu naczyńkach znajdują się bardzo czułe termopary włączone w układ regulacyjny sprzężenia
zwrotnego. Taki układ potrafi wykryć różnice temperatury rzędu 10-4 deg i dostosować ilość ciepła
dostarczanego do obydwu naczyniek tak, aby tę różnicę wyeliminować. Energia dostarczana jest do
układu w postaci krótkich impulsów prądu, po ok. 20 µJ każdy. Moc dostarczana do każdego z
naczyniek regulowana jest częstością impulsów, a nie ich energią jednostkową.
Zawartość naczyńka pomiarowego jest mieszana dla przyśpieszenia oddziaływania obu
składników. Jako mieszadła używa się często igły strzykawki dozującej roztwór składnika B. W
przypadku typowych oddziaływań fizykochemicznych wystarcza zwykle ok. 10 s, aby układ
przeszedł do nowego stanu równowagi. W układach biofizycznych w których składnikami są
makrocząsteczki potrzeba na to ok. 1 min. Zwykle po kilku minutach wprowadzamy kolejną porcję
1199

Część III: Termodynamika układów biologicznych
roztworu składnika B i proces się powtarza. W miarę jak zmienia się stopień przereagowania
(obsadzenia dla makrocząsteczek) zmianie ulega również efekt cieplny związany z pojedynczym
nastrzykiem roztworu.
Typowe doświadczenie ITC trwa zwykle od 1 do 2 godzin. W tym czasie dokonuje się od
kilkunastu do dwudziestu kilku nastrzyków i związanych z nimi pomiarów ciepła oddziaływania.
Surowe wyniki pomiarów przedstawia się w postaci zależności różnicy mocy dostarczonej do
obydwu naczynek w funkcji czasu. Ilość ciepła niezbędnego dla zrównoważenia układu po każdym
kolejnym nastrzyku oblicza się całkując powierzchnię pod pikiem.
-35
-30
-25
-20
-15
-10
-5
0
5
0 5 10 15 20 25
Czas [min]
Moc
P(t)
[ µW
]
PPoommiiaarryy kkoonnttrroollnnee Współczesne kalorymetry izotermiczne są na tyle czułe, że dla uzyskania poprawnych
wyników należy uwzględnić efekty cieplne związane ze zmianą stężeń reagentów podczas
miareczkowania. Aby wyznaczyć te tzw. ciepła rozcieńczania należy wykonać dwie dodatkowe
serie pomiarów.
Dla wyznaczenia ciepła rozcieńczania składnika A do naczyńka pomiarowego dodajemy
rozpuszczalnik bez składnika B w takich samych porcjach jak podczas właściwego
miareczkowania. Ponieważ stopień rozcieńczania składnika A jest niewielki (rzędu 1,01) zdarza się
często, że ciepło rozcieńczania jest praktycznie takie samo we wszystkich etatach miareczkowania.
Można wtedy obliczyć średnie ciepło rozcieńczania składnika A.
Stopień rozcieńczania składnika B jest z reguły duży i wynosi ok. 100 na początku miareczkowania
spadając do ok. 10 w końcowych etapach. Powoduje to, że ciepło rozcieńczania tego składnika jest
2200

Część III: Termodynamika układów biologicznych
zwykle znaczne i zmienia się systematycznie podczas miareczkowania. Dlatego też wyznaczamy to
ciepło dla każdego etapu miareczkowania podczas serii pomiarów w której dodajemy roztwór
składnika B do naczyńka pomiarowego zawierającego tylko rozpuszczalnik (bez składnika A).
Porcje dodawanego roztworu jak i objętość rozpuszczalnika na początku pomiaru powinny być
identyczne jak podczas właściwego miareczkowania.
OOpprraaccoowwaanniiee wwyynniikkóóww Po zgromadzeniu wszystkich danych surowe wyniki przelicza się na wielkości molowe:
molową zmianę entalpii i stosunek moli obydwu składników. Dane te wystarczają do wyznaczenia
∆H, K oraz n.
Załóżmy, że składniki oddziałują ze sobą zgodnie z równaniem:
A + nB == C
Roztwór składnika A o stężeniu [A] moli/litr i objętości V znajduje się w naczyńku pomiarowym, a
składnik B dodawany jest jako roztwór w tym samym rozpuszczalniku w porcjach o jednakowej
objętości v. Wprowadźmy ponadto pojęcie stopnia obsadzenia cząsteczek substancji A
definiowanego wzorem:
[ ][ ]AAf b≡
Stopień obsadzenia mówi jaka część cząsteczek A zaangażowana jest w danych warunkach w
oddziaływanie z cząsteczkami B. Wartość stopnia obsadzenia waha się od 0 (cały składnik A w
formie wolnej) do 1 (cały składnik A obsadzony cząsteczkami składnika B). Korzystając z tej
wielkości można obliczyć stężenie formy związanej [A]b i wolnej [A]f:
[ ] ]A[fA b = [ ] ( ) ]A[f1A f −=
Przyjmując, że z jedną cząsteczką A oddziaływać może n cząsteczek B można wyznaczyć stężenie
formy związanej i wolnej składnika B:
[ ] ]A[nf]A[nB bb == f iB [B ] nf[A]= −
gdzie: [Bi] - ogólne stężenie składnika B po i-tym nastrzyku.
W badaniach biofizycznych stosuje się zwykle zamiast termodynamicznej stałej równowagi tzw.
stałą tworzenia kompleksu. Jest ona równa:
[ ]
[ ] [ ] ( )[ ] [ ] [ ]( )b
if f
A f[A]KA B 1 f A B nf A
= =− −
Po kilku prostych przekształceniach otrzymujemy:
[ ] [ ] [ ] [ ]2i iK B fK B nfK A nf K A f− − + =
2211

Część III: Termodynamika układów biologicznych
Porządkując człony równania i dzieląc je przez nK[A] otrzymujemy:
[ ][ ] [ ]
[ ][ ]
i i2 B B1f 1 fn A nK A n A
⎧ ⎫⎪ ⎪− + + + =⎨ ⎬⎪ ⎪⎩ ⎭
0
czyli równanie kwadratowe ze względu na f. Jeden z jego pierwiastków jest zawsze większy od 1 i
nie ma sensu fizycznego, ale drugi pozwala na obliczenie współczynnika obsadzenia:
[ ][ ]
i2 4 Bf X X
n A= − −
gdzie: [ ][ ] [ ]
iB 1X 1n A nK A
= + +
Ilość energii cieplnej produkowana lub pobierana przez układ podczas pojedynczego aktu
miareczkowania, Qi, zależy od różnicy entalpii produktów i substratów, ∆H, oraz przyrostu liczby
moli substancji B która związała się z substancją A, (∆nB)b:
( )bBi nHQ ∆∆=
Ten przyrost liczby moli można wyznaczyć zgodnie ze wzorem:
( ) [ ] [ ]VAfnVBn bbB ∆=∆=∆
gdzie: ∆f – przyrost stopnia obsadzenia po i-tym miareczkowaniu,
czyli ostatecznie:
[ ] HVAfnQi ∆⋅∆=
Podstawiając do tego wzoru wartości fi i fi-1 z równania kwadratowego otrzymujemy:
[ ] ( )1iii ff2
HVAnQ −−∆
=
Teraz można wyznaczyć ∆Hi, czyli zmianę entalpii molowej w i-tym miareczkowaniu. Jest ona
równa ilości ciepła Qi podzielonego przez ogólny przyrost liczby moli składnika B:
(*) [ ]0i
B
ii Bv
Qn
QH =∆
=∆
gdzie: v – objętość roztworu składnika B dodawana podczas jednego miareczkowania
[B]0 – stężenie dodawanego roztworu
Z drugiej strony wartość Qi dla i-tego miareczkowania wyznaczyć można z danych
doświadczalnych:
( )∫∆+
−=tt
tii qdttPQ
gdzie: qi – aktualne ciepło rozcieńczania.
2222
Część III: Termodynamika układów biologicznych
Doświadczalne wartości Qi można również przeliczyć na zmiany entalpii molowej:
[ ]0i
B
ii Bv
Qn
QH =∆
=∆
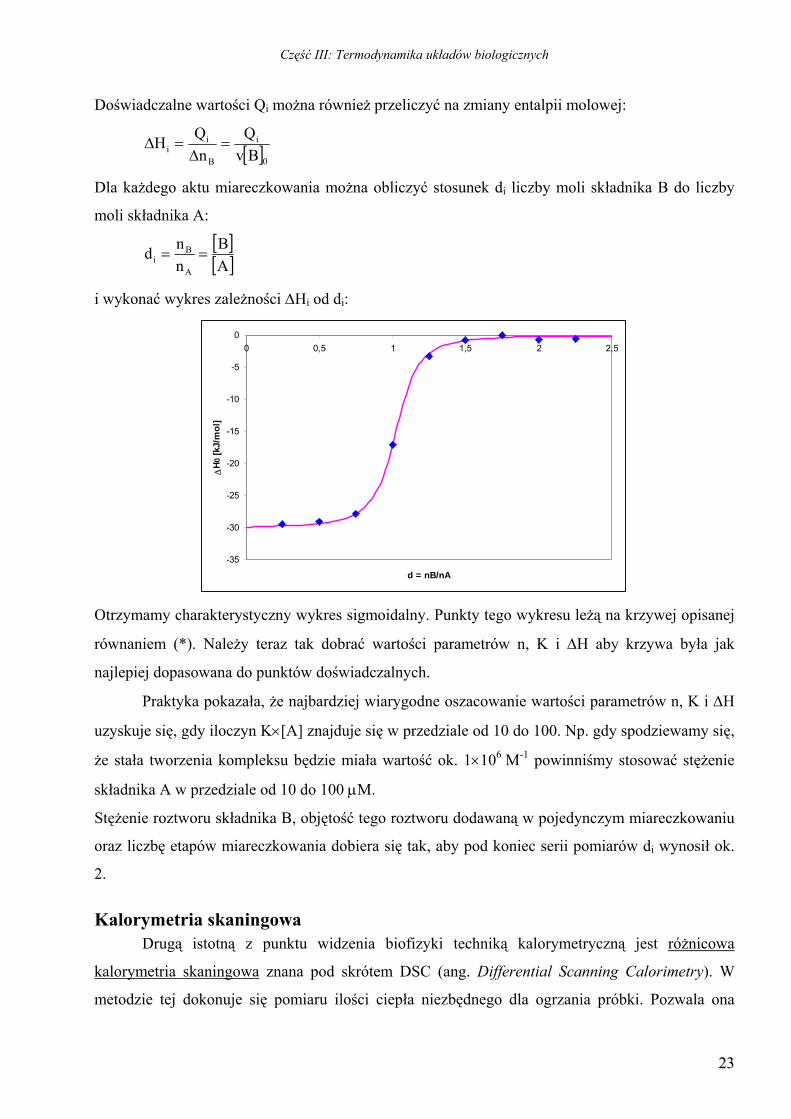
Dla każdego aktu miareczkowania można obliczyć stosunek di liczby moli składnika B do liczby
moli składnika A:
[ ][ ]AB
nnd
A
Bi ==
i wykonać wykres zależności ∆Hi od di:
-35
-30
-25
-20
-15
-10
-5
00 0,5 1 1,5 2 2,5
d = nB/nA
∆H 0
[kJ/
mol
]
Otrzymamy charakterystyczny wykres sigmoidalny. Punkty tego wykresu leżą na krzywej opisanej
równaniem (*). Należy teraz tak dobrać wartości parametrów n, K i ∆H aby krzywa była jak
najlepiej dopasowana do punktów doświadczalnych.
Praktyka pokazała, że najbardziej wiarygodne oszacowanie wartości parametrów n, K i ∆H
uzyskuje się, gdy iloczyn K×[A] znajduje się w przedziale od 10 do 100. Np. gdy spodziewamy się,
że stała tworzenia kompleksu będzie miała wartość ok. 1×106 M-1 powinniśmy stosować stężenie
składnika A w przedziale od 10 do 100 µM.
Stężenie roztworu składnika B, objętość tego roztworu dodawaną w pojedynczym miareczkowaniu
oraz liczbę etapów miareczkowania dobiera się tak, aby pod koniec serii pomiarów di wynosił ok.
2.
Kalorymetria skaningowa Drugą istotną z punktu widzenia biofizyki techniką kalorymetryczną jest różnicowa
kalorymetria skaningowa znana pod skrótem DSC (ang. Differential Scanning Calorimetry). W
metodzie tej dokonuje się pomiaru ilości ciepła niezbędnego dla ogrzania próbki. Pozwala ona
2233

Część III: Termodynamika układów biologicznych
zmierzyć zmiany molowego ciepła właściwego przy stałym ciśnieniu, Cp, w funkcji temperatury.
Jeżeli w układzie w określonym przedziale temperatury zachodzi proces przemiany fazowej
wydzielający lub pobierający ciepło, to wartość Cp ulegnie gwałtownej zmianie. Technika DSC
pozwala ustalić temperaturę tego przejścia fazowego, Tm, oraz ocenić związane z nim parametry
termodynamiczne: ∆H i Cp. Na gruncie biofizyki interesują nas przede wszystkim przemiany
pseudofazowe związane ze zmianami w strukturze biopolimerów (białka i kwasy nukleinowe) lub
układów supramolekularnych (błony lipidowe).
BBuuddoowwaa aappaarraattuu Układ pomiarowy składa się z dwóch naczynek
umieszczonych we wspólnej osłonie termicznej. Do obydwu
naczynek doprowadzane jest ciepło ze wspólnego grzejnika
(czerwony) powodującego powolny wzrost temperatury obu
naczynek w czasie. Podczas całego doświadczenia oba naczyńka
są utrzymywane w równowadze termicznej dzięki pracy
dodatkowych grzejników (brązowe). Temperatura obu naczyniek
jest kontrolowana przez czułe termometry, a praca grzejników sterowana jest przez elektroniczny
układ kontrolny.
T = f(t) T = f(t)∆T = 0
Jedno z naczynek spełnia rolę naczynka odniesienia, a drugie naczynka pomiarowego. W obu
naczynkach znajdują się jednakowe objętości tego samego roztworu (buforu). W naczynku
pomiarowym znajduje się ponadto badana substancja lub mieszanina oddziałujących z sobą
substancji.
PPrrzzeebbiieegg ppoommiiaarruu Po napełnieniu naczynek odpowiednimi roztworami doprowadza się je do równowagi
termicznej w zadanej temperaturze początkowej. Właściwy pomiar odbywa się podczas powolnego
ogrzewania układu z zadaną z góry szybkością τ (zwykle ok. 1°/min). Jeżeli w naczynku
pomiarowym nie zachodzą żadne przemiany fazowe ani zmiany konformacyjne, to i tak należy do
niego dostarczać nieco większą moc cieplną (ilość ciepła w jednostce czasu), gdyż znajduje się w
nim rozpuszczona substancja badana i potrzeba dodatkowej energii, aby doprowadzić ją do żądanej
temperatury. Ta dodatkowa ilość energii pozwala wyznaczyć ciepło molowe badanej substancji, Cp,
zgodnie ze wzorem:
nPCp τ
∆=
2244

Część III: Termodynamika układów biologicznych
gdzie: ∆P – różnica pomiędzy mocą dostarczaną do naczynka pomiarowego i naczynka odniesienia
τ - szybkość zmian temperatury w stopniach na jednostkę czasu
n- liczba moli badanej substancji w naczynku pomiarowym
Zwykle wraz ze wzrostem temperatury wartość Cp nieznacznie wzrasta (czarna linia na rysunku
poniżej).
0
2
4
6
8
10
12
14
16
18
10 20 30 40 50 60 70 80 90 100
Temperatura [C]
Cp [k
J/m
ol/d
eg]
Jeżeli w określonej temperaturze rozpoczyna się jakiś proces pochłaniający lub
wydzielający ciepło, to wartość Cp zmienia się gwałtownie. W przypadku procesów
endotermicznych takich jak denaturacja białka lub „topnienie” DNA do dalszego ogrzewania
układu potrzeba będzie więcej ciepła, więc wartość Cp zacznie gwałtownie wzrastać wraz ze
wzrostem temperatury. Wzrost ten osiągnie maksymalną wartość w temperaturze, w której
przemianie ulega najwięcej cząsteczek. Przy dalszym wzroście temperatury ilość cząsteczek
ulegająca przemianie maleje i wartość Cp wraca prawie do początkowej wartości (linia niebieska).
Entalpia przemiany równa jest przy tym polu pomiędzy krzywą a aproksymowaną linią zmiany Cp.
Przy pomocy techniki DSC badać można nie tylko stabilność termiczną biopolimeru, np.
białka, ale również wpływ na nią liganda tworzącego kompleks z badanym biopolimerem. Można
również wyznaczyć entalpię kompleksowania ligandu. W tym celu należy wykonać dwa niezależne
pomiary: jeden dla samego biopolimeru i drugi dla kompleksu (linia czerwona na rysunku
powyżej). Ponieważ wiązanie ligandu z biopolimerem zachodzi jedynie wtedy, gdy procesowi temu
towarzyszy obniżenie entalpii swobodnej, więc stabilność termiczna kompleksu jest zwykle
większa niż samego biopolimeru (Tmc > Tm). Entalpię wiązania można wyznaczyć z porównania
entalpii przemiany kompleksu z entalpią przemiany samego biopolimeru.
2255

Część III: Termodynamika układów biologicznych
WWyymmaaggaanniiaa ii ooggrraanniicczzeenniiaa Aby otrzymać wiarygodne wyniki pomiarów wielkości termodynamicznych układ badany
techniką DSC musi spełniać szereg wymagań:
i) przemiana musi mieć charakter odwracalny. Występowanie jakichkolwiek procesów
nieodwracalnych komplikuje analizę wyników. Niestety, procesy całkowicie odwracalne są
w biofizyce rzadkością. Problem ten można rozwiązać lub ominąć dobierając odpowiednio
szybki wzrost temperatury układu tak, aby zwykle powolne procesy nieodwracalne nie
zdążyły zajść w znaczącym stopniu.
ii) z drugiej strony szybkość wzrostu temperatury musi być na tyle powolna, aby układ
zawsze znajdował się w stanie równowagi termodynamicznej
iii) proces przemiany powinien być czystym procesem jednoetapowym. Jeżeli przemiana
jest kilkuetapowa i występują stany pośrednie, to analiza wyników jest dużo bardziej
skomplikowana i wymaga dodatkowych założeń.
Jeżeli chcemy wyznaczyć techniką DSC entalpię wiązania ligandu, to pojawiają się jeszcze
dodatkowe wymagania:
iv) stężenie ligandu musi być na tyle duże, aby praktycznie wszystkie cząsteczki
biopolimeru występowały w formie kompleksu
v) ligand nie powinien tworzyć kompleksu z biopolimerem po przemianie (denaturacji).
Jeżeli takie zjawisko zachodzi, to wyznaczona entalpia wiązania jest zaniżona.
Nawet jeżeli wszystkie powyższe wymagania są spełnione pojawić się mogą jeszcze dodatkowe
ograniczenia wiarygodnego stosowania techniki DSC do wyznaczania entalpii wiązania. Ma to
miejsce w szczególności, gdy porównujemy powinowactwo różnych ligandów do biopolimeru i
ligandy te w różnym stopniu stabilizują biopolimer (duże różnice w wartościach Tmc). Wtedy każda
entalpia wiązania wyznaczana jest w innej temperaturze. Aby móc porównać wyniki uzyskane dla
różnych ligandów należy je ekstrapolować do stałej temperatury, zwykle 25°C. Taka odległa
ekstrapolacja może być źródłem znacznych niedokładności.
2266


















