DIAGNOSTYKA OBRAZOWA NOWOTWORÓW … · 2 Rekomendacje GEP-NET ENETS – ENETS Consensus Guidelines...
81
DIAGNOSTYKA OBRAZOWA NOWOTWORÓW NEUROENDOKRYNNYCH NEN/NET Jarosław B. Ćwikła UWM; Olsztyn; CMKP Warszawa [email protected]
Transcript of DIAGNOSTYKA OBRAZOWA NOWOTWORÓW … · 2 Rekomendacje GEP-NET ENETS – ENETS Consensus Guidelines...

DIAGNOSTYKA OBRAZOWA
NOWOTWORÓW NEUROENDOKRYNNYCH
NEN/NET
Jarosław B. Ćwikła
UWM; Olsztyn; CMKP Warszawa

2
Rekomendacje GEP-NET
ENETS – www.enets.org
ENETS Consensus Guidelines for the Standard of Care for
Patients with Digestive Neuroendocrine Tumors
Neuroendocrinology 2009;90:159
TNM staging
• Virchows Arch 2006;449:395-401 foregut
• Virchows Arch 2007:451:757-762 – midgut and hindgut
UK&I NET
Guidelines for the management of gastroenteropancreatic
neuroendocrine (including carcinoid) tumours.
Ramage et al. Gut 2011;54:1-16

3
GUZY NEUROENDOKRYNNE (NET)
Guzy NET wywodzą się z wyspecjalizowanych komórek
gruczołów wydzielania wewnętrznego: pheochromocytoma,
adenoma – przysadki; Komórki C tarczycy – MTC;
Paraganglioma, ganglioneuroma - elementów nerwowych;
• Diffuse neuroendocrine system – gastroenteropacreatic
endocrine tumor GEP/NET (2% guzów p. pokarmowego);
– wyspy komórkowe w narządach – trzustka;
– rozproszone komórki najczęściej w przewodzie pokarmowym.

Epidemiologia GEP-NET
• 21/mln/rok;
• 15 typów wyspecjalizowanych komórek;
• 55/mln/rok w badaniach autopsyjnych;
• 70% NET to GEP-NET;
• 2% wszystkich nowotworów układu pokarmowego.
• Najczęściej „rakowiak” 15/mln/rok.
4

5
KLASYFIKACJA KLINICZNA
• Pochodzenie (na podstawie pierwotnej lokalizacji):
– Foregut – przedni odcinek prajelita;
– Midgut – środkowy odcinek prajelita;
– Hindgut – tylny odcinek prajelita.
• Właściwości sekrecyjne:
– Wydzielające:
• Czynne (określony zespół kliniczny, z. rakowiaka, ZES, hipersekrecja
insuliny - insulinoma, gastrinoma, glucagonoma, VIP-oma,
somatostatinoma, z ektopowym wydzielaniem ACTH, 20-32%);
• Nieczynne (bez określonego zespołu klinicznego);
– Niewydzielające (najczęściej niskozróżnicowane raki NET.

6
KLASYFIKACJA WHO GEP-NET
• Nowotwory neuroendokrynne (GEP-NEN/NET):
– NETG1 dobrze zróżnicowane Ki-67 < 2%;
– NETG2 średnio zróżnicowane 2 < Ki-67 < 20%
– NECG3 rak neuroendokrynny nisko zróżnicowany Ki-67> 20%
– Mieszana postać raka NEC i raka gruczołowego gdzie komponent
neuroendokrynny stanowi > 50% objętości guza;
– Zmiany guzo-podobne tumour like lesions.
7
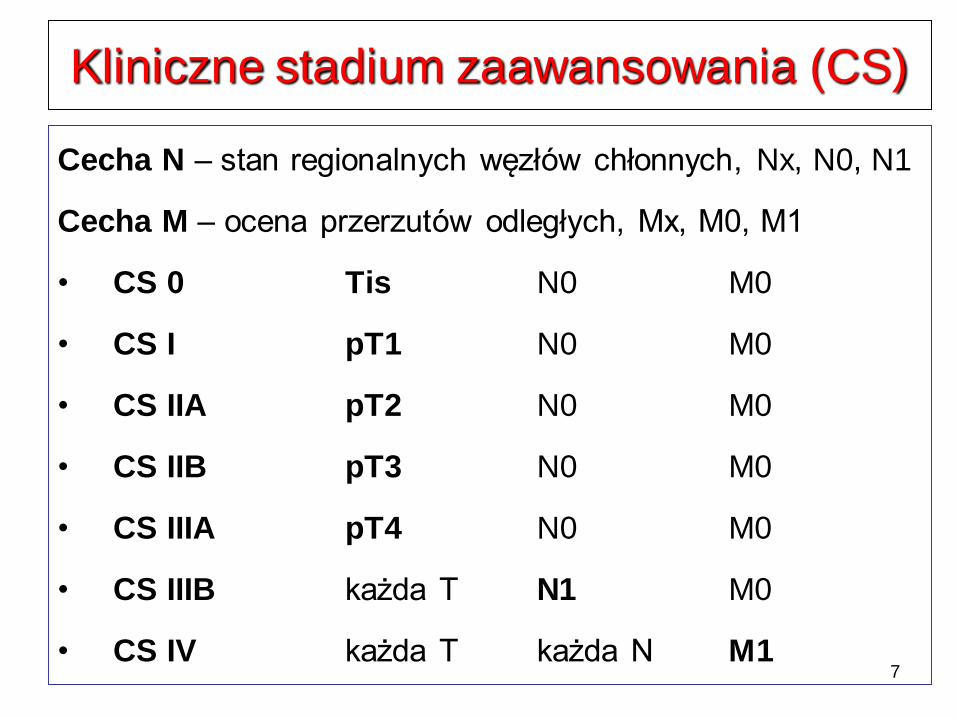
Kliniczne stadium zaawansowania (CS)
Cecha N – stan regionalnych węzłów chłonnych, Nx, N0, N1
Cecha M – ocena przerzutów odległych, Mx, M0, M1
• CS 0 Tis N0 M0
• CS I pT1 N0 M0
• CS IIA pT2 N0 M0
• CS IIB pT3 N0 M0
• CS IIIA pT4 N0 M0
• CS IIIB każda T N1 M0
• CS IV każda T każda N M1

8
IV.2.1 DIAGNOSTYKA OBRAZOWA NET
• USG, najbardziej popularne badanie jamy brzusznej;
• EUS - podstawowe badanie dla guzów WHO gr. 1;
• Spiralna wielorzędowa TK po i.v. śr. Kontrastowego;
• MR przed i po i.v. Gd-DTPA, lub np. Gd-EOB-DTPA;
• Scyntygrafia receptorów somatostatynowych – SRS (99mTc TOC -
Tektrotyd, 99mTc TATE, 99mTc NeoSPECT);
• PET Ga68 DOTATATE (SRS); PET – 18FDG PET;
• Scyntygrafia receptorów adrenergicznych – 123I mIBG;
• Scyntygrafia kości lub MR kręgosłupa.
• WB – DWI (rezonans magentyczny całego ciała.

IV.2.1 Kryteria wyboru metod obrazowych
• Detekcja zmiany pierwotnej (TK > SRS > USG > MR);
• Ocena stopnia lokalnego zaawansowania (TK = MR);
• Stosunek do otaczających tkanek i narządów (TK = MR);
• Obecność przerzutów w regionalnych w. chłonnych i przerzutów
odległych (TK = MR);
• Ocena aktywności procesu chorobowego – DWI i ADC (MR > TK);
• Ocena efektów leczenia: RECIST, pomiar objętości, DWI i ADC
(MR >> TK);
• Obrazowy follow-up z oceną wznowy i/lub progresji (MR > TK).
9

10
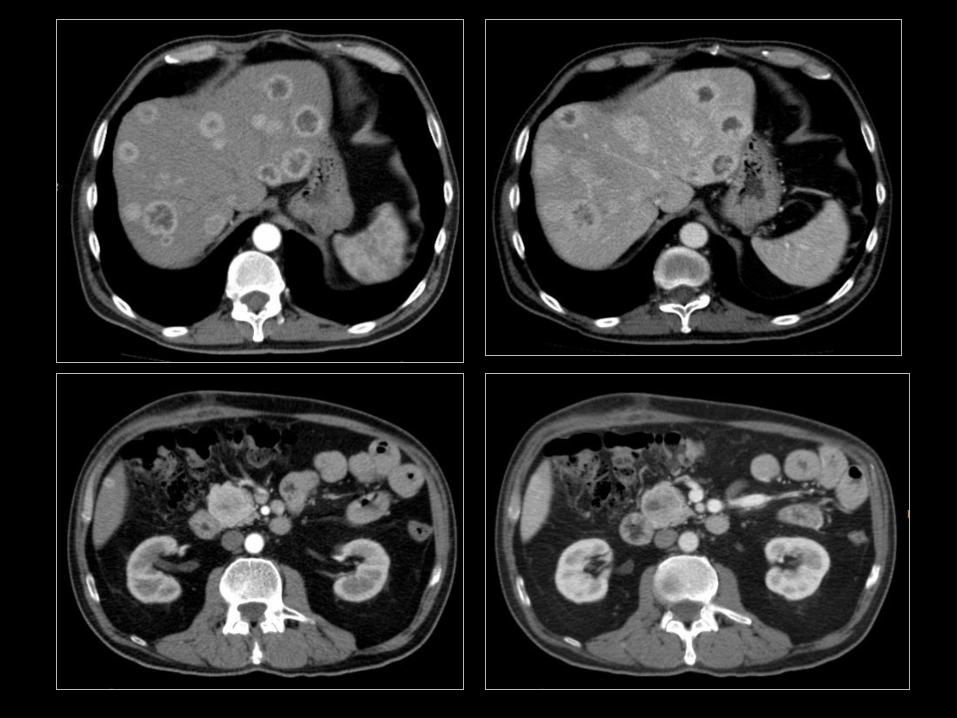
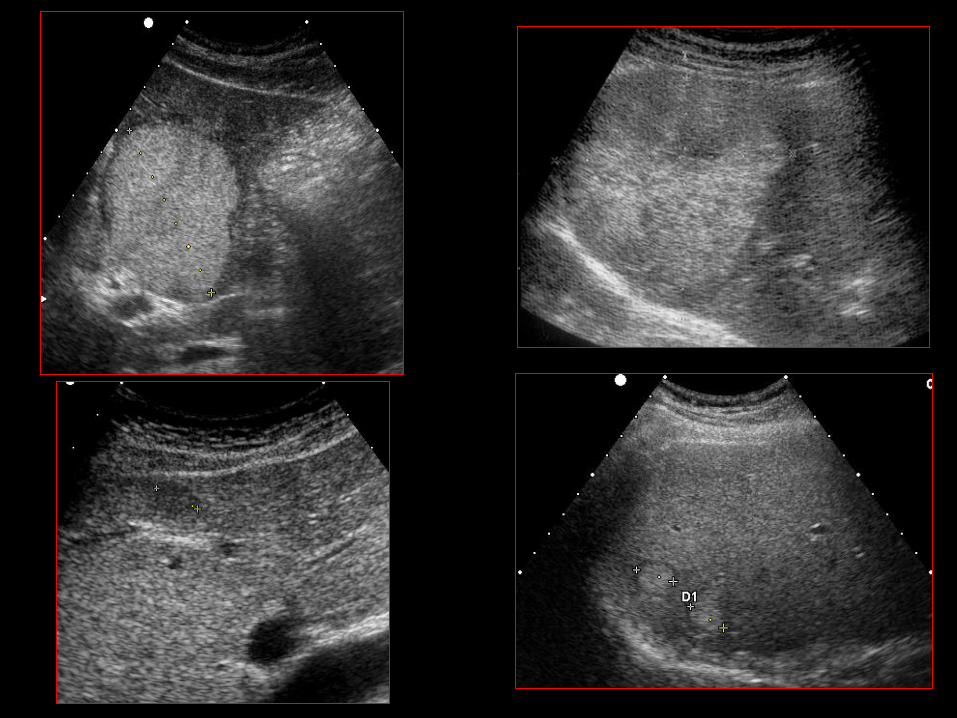
IV.2.1 ROLA DIAGNOSTYKI OBRAZOWEJ
• Potwierdzenie lub wykluczenie obecności guza o typie NET;
• Ustalenie punktu wyjścia NET;
• Ocena stadium zaawansowania nowotworu;
• Precyzyjne ustalenie położenia nowotworu i jego
potencjalnego nacieku/ucisku na struktury przyległe
(informacja dla chirurga);
• prognostyka;
• odpowiedź na leczenie;
• radiologiczny „follow-up”.
11
12
13

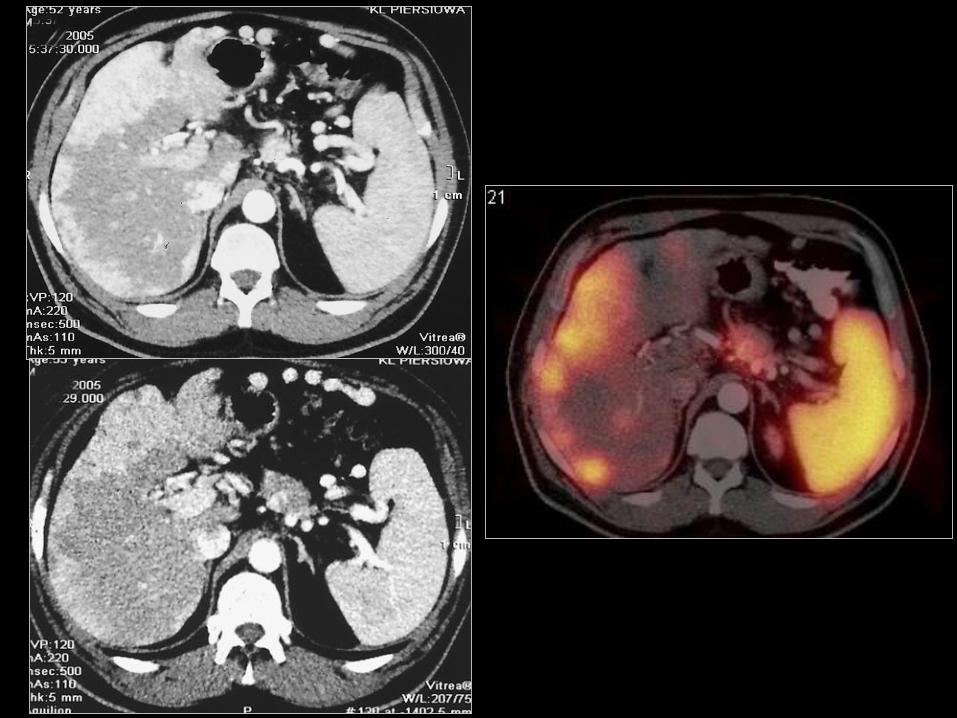
Guzy o typie GEP-NET przedniego odcinka
prajelita (foregut tumours)
• Guzy których punktem wyjścia są:
– Żołądek;
– Dwunastnica z brodawką Vatera;
– proksymalny odcinek jelita cienkiego;
– Trzustka;
– Oskrzele i grasica*.
14

Epidemiologia – żołądek (ECL)
• 1-2/mln/rok;
• 8-9% wszystkich GEP-NET (10%);
• bez przewagi płci;
• Ostatnio 8-9 krotny wzrost występowania,
poprawa wykrywalności oraz możliwość coraz
bardziej częstego użycia IPP;
• 1% wszystkich nowotworów żołądka?
15

GEP/NET – „foregut” -żołądek
• Dominują dobrze zróżnicowane guzy (typu ECL – komórki
enterochromofilno-podobne, np. rakowiaki);
• Typ I najbardziej powszechny 70-85%, drobny pojedynczy polip,
może być wieloogniskowy, WHO grupa 1, wtórny do atroficznego
zapalenia bł. Śluzowej żołądka i następowej hipergastrinemii;
– Bardzo rzadko tworzy przerzuty 1-3%, odsetek przeżyć 5 letnich 100%
• Typ II rzadki (5-7% przypadków), guz pierwotnej hipergastrinemii,
związany z zespołem MEN-1, charakteryzuje się bardziej złośliwym
przebiegiem, odsetek przeżyć 5-o letnich 60-90%.
– Najczęściej guzy WHO grupa 1, bardzo rzadko WHO grupa 2.
16

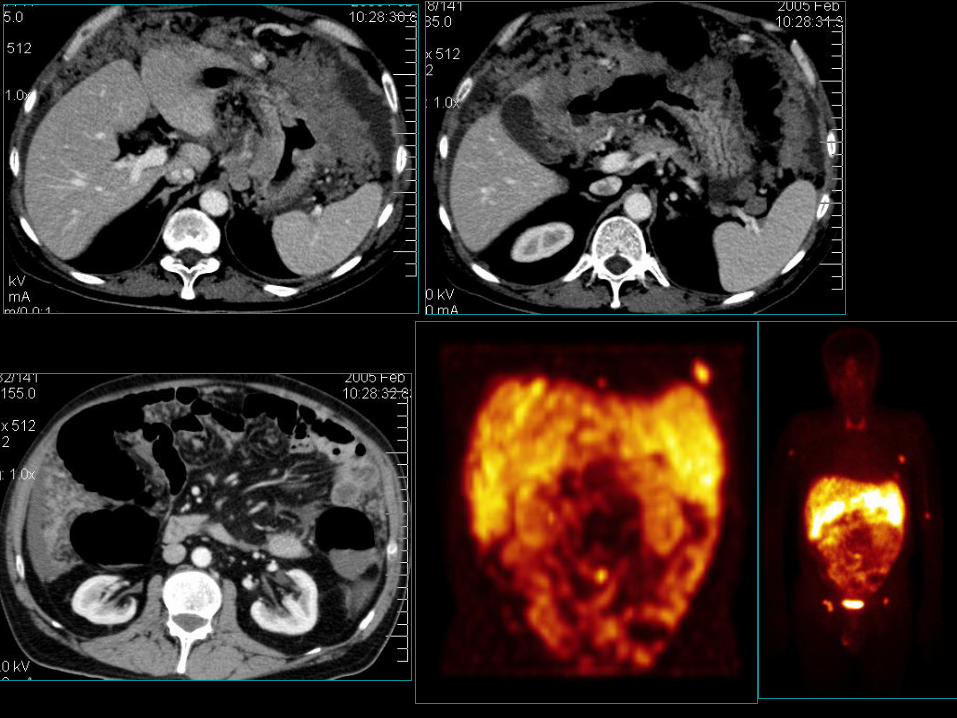
GEP/NET – „foregut” –żołądek, typ III
• Typ 3 sporadyczny,
• 2-i co do częstości 13-20%,
• częściej WHO 2 niż WHO 3,
• Bez towarzyszącego atroficznego zapalenia bł. śluzowej żołądka;
• Występuje w warunkach normogastrynemii;
• Naturalny przebieg przypomina raka gruczołowego;
• Dający przerzuty do okolicznych węzłów chłonnych oraz wątroby;
• Ponad 50% chorych w momencie diagnozy ma przerzuty odległe;
• Przeżycie 5-o letnie <50% chorych;
• W 5% może być przyczyną atypowego zespołu rakowiaka.17
18

Dwunastnica – 5 typów guzów NET
• 27-58% - dwunastniczy gastrinoma (najczęściej);
• 23-45% - somatostatinoma (niewydzielający);
• 27% - niefunkcjonujący zawierający serotoninę;
• 9% - niefunkcjonujący zawierający kalcytoninę;
• Gangliocytowy przyzwojak (gangliocytic-
paraganglioma);
• Niskozróżnicowany rak neuroendokrynny PDNEC.19

Dwunastnica NET - lokalizacja
• 58% - D1 – opuszka;
• 33% - D2 cz. Zstępująca;
• 9% z D3 i D4;
• 20% w okolicach brodawki Vatera, charakterystyka raka
brodawki Vatera.
20

Epidemiologia - Dwunastnica
• 2-4% wszystkich GEP-NET (3%);
• 0,03-0,05% w badaniach autopsyjnych;
• 1-3% wszystkich pierwotnych guzów
dwunastnicy;
• 50-70% wysoko zróżnicowane NET WHO grupa
1 i 2.
21

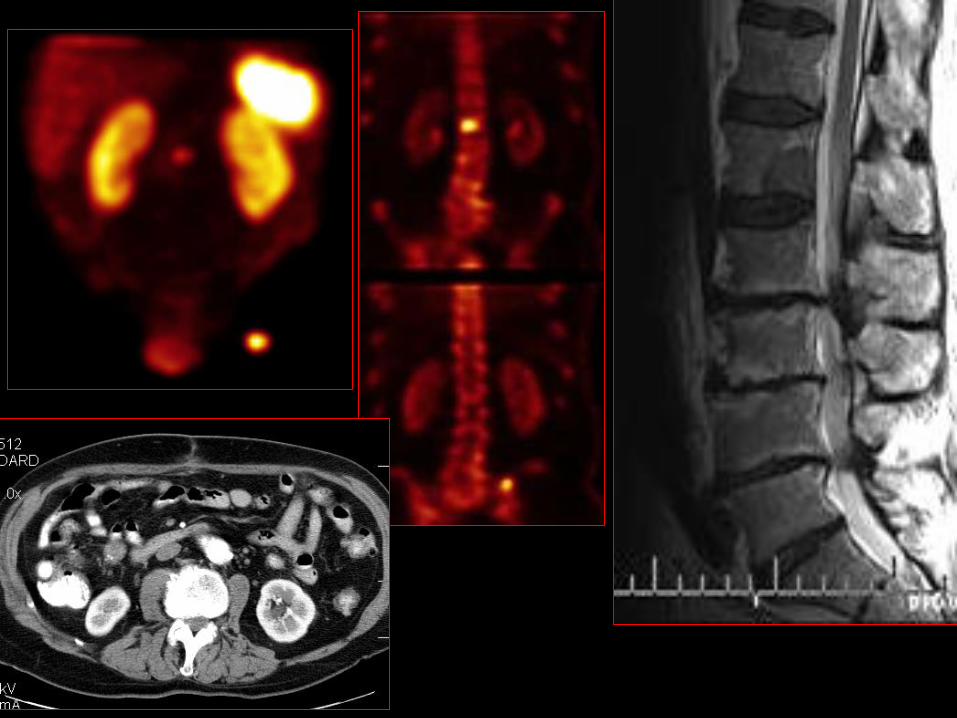
Dwunastniczy GEP-NET
• Zwykle małe tu o śr. 1,2-1,5cm w 75% <2cm;
• Mts do w. chłonnych w 40-60%;
• Mts do wątroby <10%;
• 90% pojedyncze zmiany;
• 10% wieloogniskowe najczęściej w zespole MEN-1;
• 6,5% chorych z dwunastniczym GEP-NET ma MEN-1;
• 20-30% chorych z ZES i dwunastniczym NET ma MEN-1;
• Złe rokowanie jeśli >2cmi nacieka warstwę mięśniową oraz G2 lub
G3.
22
23

Trzustka wszystkie NET-y
• 4-12/mln/rok;
• 2-10% nowotworów trzustki;
• 68-80% guzy nieczynne hormonalnie.
24

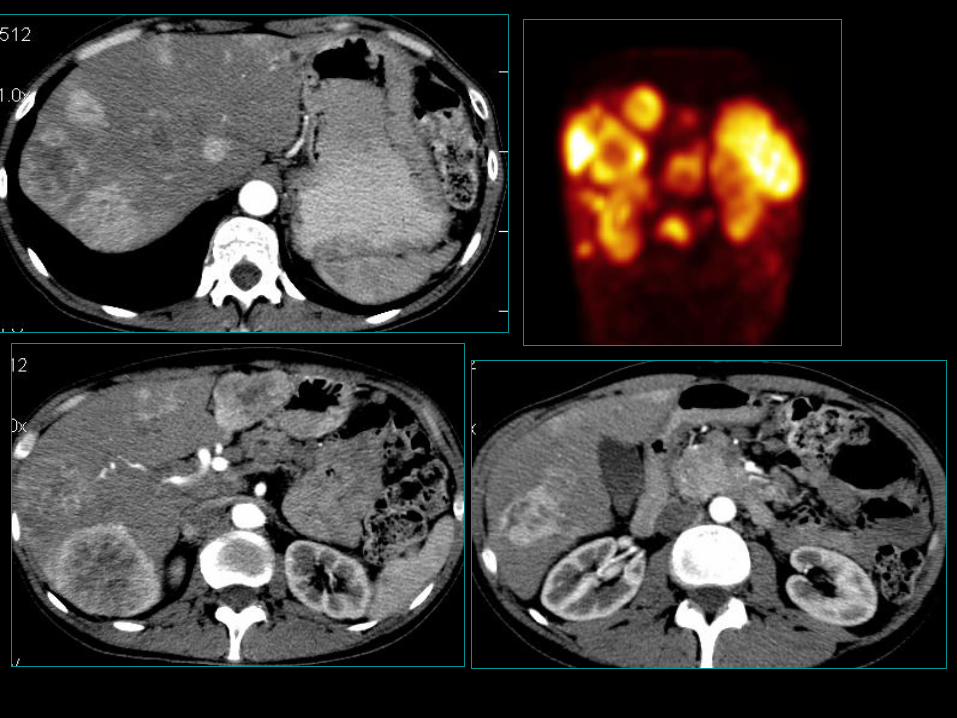
Gastrinoma trzustkowy i XII-czy
• 0,5-3/mln/rok; pik zapadalności pomiędzy 5 a 6 dekadą życia;
• Najczęstszy złośliwy guz wydzielający trzustki (30%);
• 20-25% trzustkowych ZES ma mts do wątroby w momencie diagnozy;
• Przeżycie 10 letnie bez przerzutów 90-100% jeśli przerzuty 10-20%;
• 55-88% dwunastniczy gastrinoma w sporadycznym ZES;
• 70-100% ZES w MEN-1;
• W rzadkich przypadkach inna lokalizacja, żołądek, przewody żółciowe,
wątroba, jajnik, serce oraz w przypadku SCLC.
25
26

Trzustkowy - insulinoma
• 1-3/mln/rok;
• Najczęściej występujący guz hormonalnie czynny
trzustki (17%), kolejny (15%) gastrinoma;
• Pik zapadalności w 5 dekadzie życia;
• Nieco częściej kobiety niż mężczyźni;
• 10% są wielogoniskowe;
• 10% są złośliwe;
• 5-10% towarzyszą MEN-1 z tej grupy 25% może być
złośliwa;27
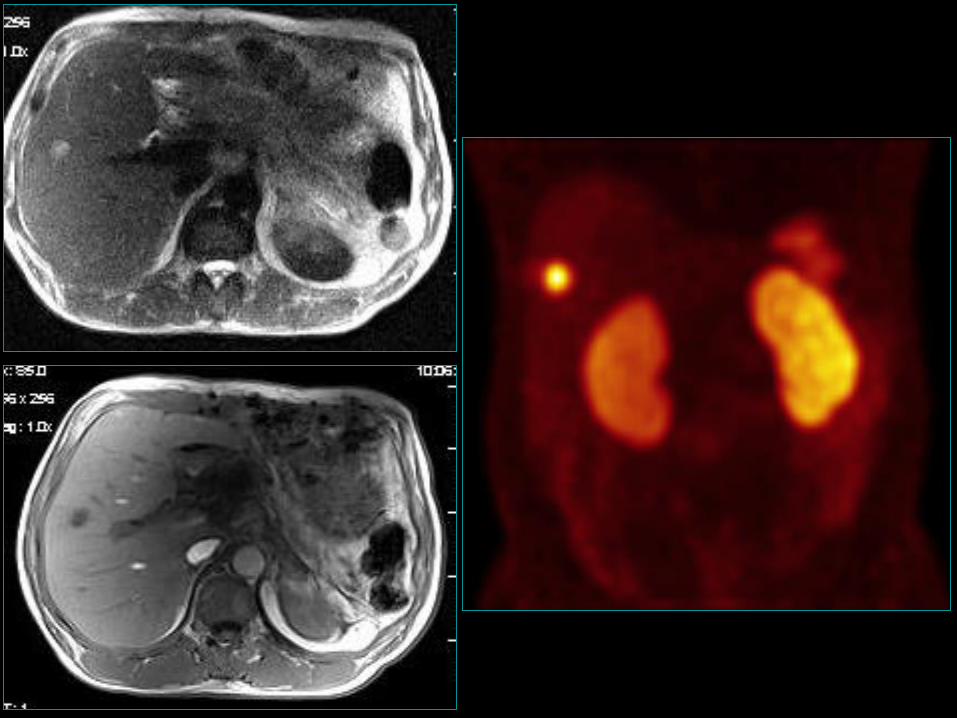
28

Rzadkie guzy sekrecyjne <10%
• Ekstremalnie rzadko, większość WHO grupa 2;
• 2% - VIP-oma;
• 1% - glucagonoma, carcinoid, somatostatinoma,
calcytonin producing tumour, ACTH-omas, GRFomas
• 5 lat przeżycia:
– 60 - 100% dla ograniczonej choroby;
– 40% zajęcie w. chłonnych regionalnych;
– 29% przerzuty odległe;
– 80% dla wszystkich stadiów zaawansowania;
29

Trzustka –niefunkcjonujące NETG1 i NETG2
• 4-8/mln/rok; (5 dekada szczyt zapadalności);
• 2-10% guzów trzustki;
• 68-80% wszystkich GEP-NET trzustki;
• Bez preferencji płci;
NECG3 (klasyfikacja WHO 2010);
• 5-o letnie przeżycie 30-63%;
• 2-3% wszystkich guzów endokrynnych tego narządu,
niedoszacowanie ok. 10%;
• M/K 4:1. 30
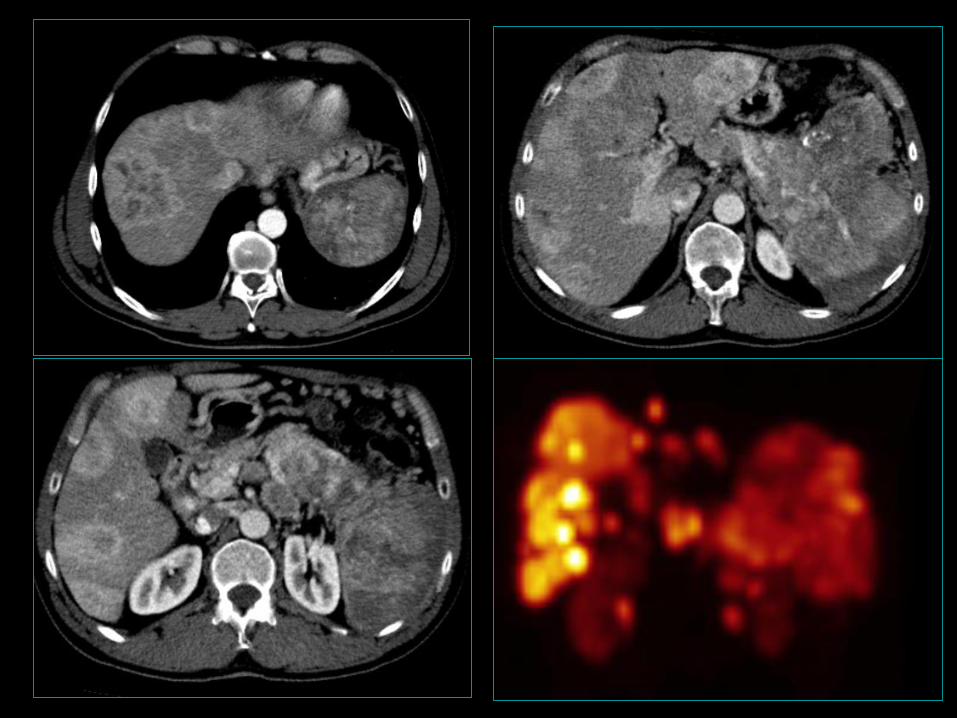
31
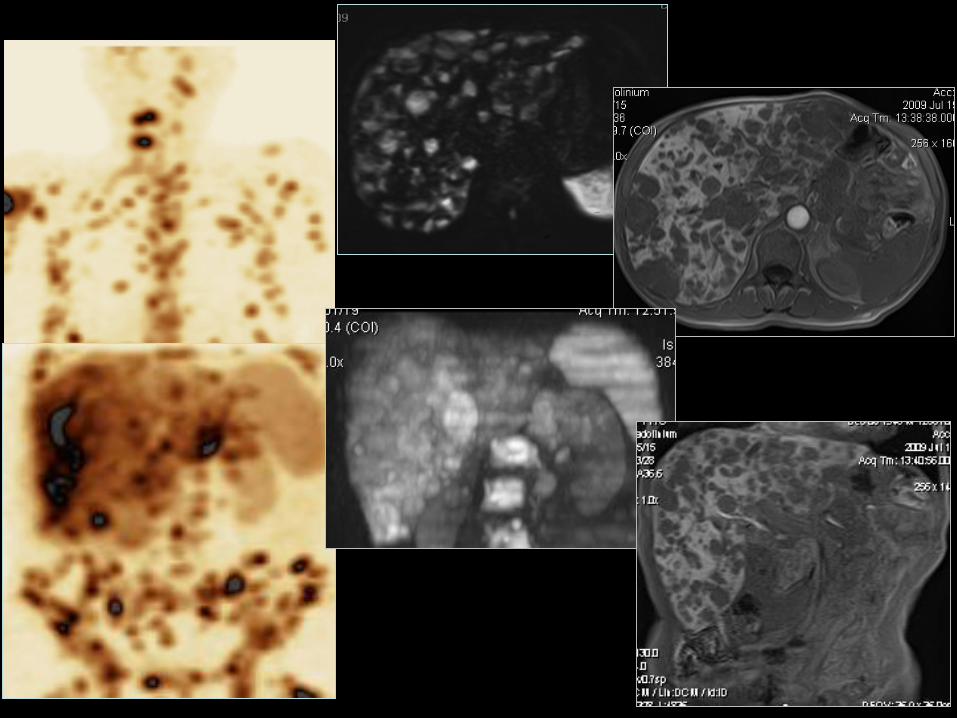
32
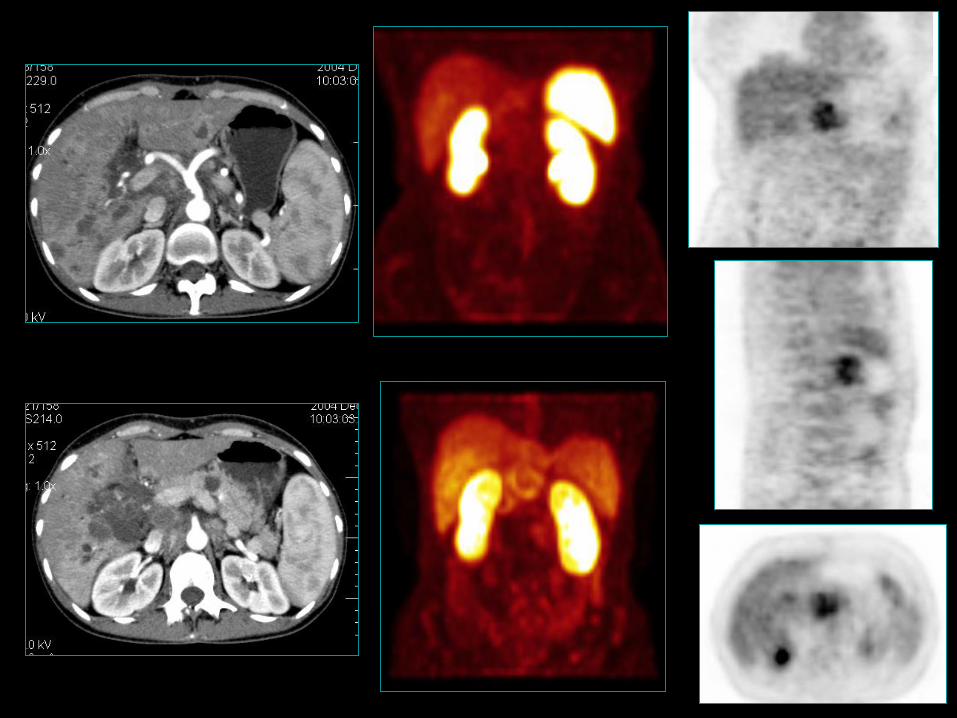
33

Jelito cienkie (midgut)
• Epidemiologia - 3-8/mln/rok,
• 1/150 w badaniach autopsyjnych;
• 23% same jelito cienkie, 28% łącznie z wyrostkiem,
• 33,4% - wszystkich GEP-NET jako midgut;
• Zwykle WHO grupa 2, mniej często grupa 1 i 3
• Jednakowa częstość występowania u kobiet i mężczyzn;
• Szczyt zachorowań pomiędzy 6 i 7 dekadą życia
• Dystalny odcinek jelita krętego, w pobliżu zastawki krętniczo-kątniczej.
• Możliwy wzrost wieloogniskowy, spotykany w 26-30%.34

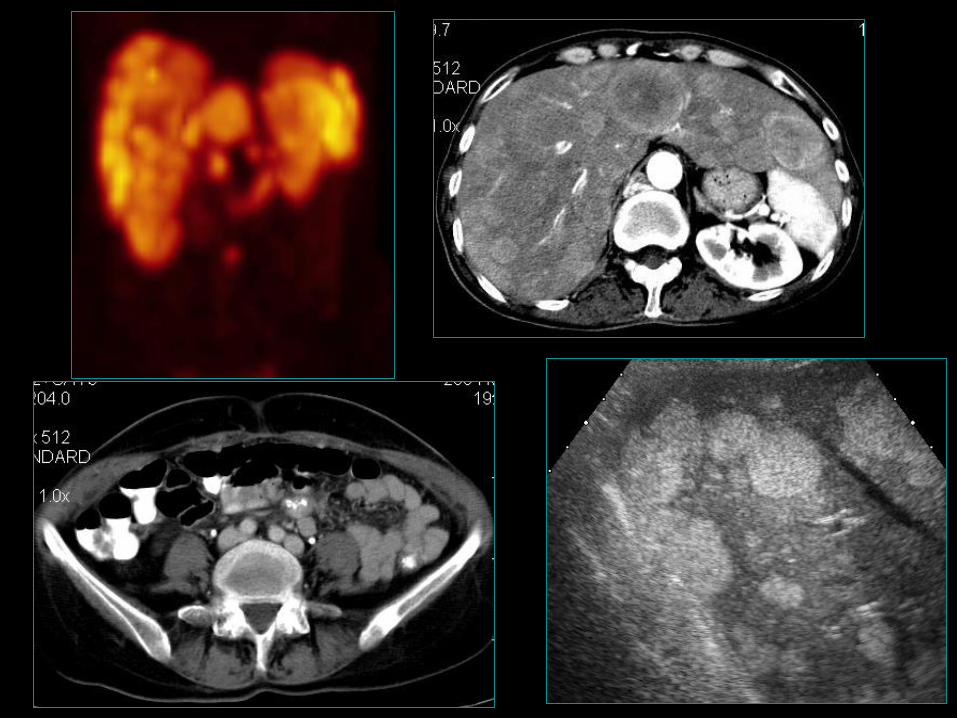
Jelito cienkie (midgut)
• Pierwsze objawy choroby pojawiają się zwykle w zaawansowanych
postaciach procesu nowotworowego;
• Stosunkowo nieduży odsetek chorych przeżywających 5 lat (60,5%);
• Wyjściowy stopień zaawansowania, a nie wielkość pierwotnego
guza jest czynnikiem prognostycznym;
• Obecność przerzutów odległych w momencie diagnozy (szczególnie
wątroba) – ważny czynnik prognostyczny;
• Obecność z. rakowiaka i zajęcie serca (prawego) z jego
niewydolnością, główną przyczyną zgonów wśród chorych z tym
zespołem.
35

Zespół rakowiaka
• „flushing” – 90%, (foregut – „purpura” szyja i twarz, midgut –
róż-czerwień, szyja twarz górna połowa ciała);
• Biegunka sekrecyjna - 70%;
• Ból brzucha – 40%;
• Duszność spastyczna - 15%;
• Telangiektazje - 25%;
• Duszność sercowa, prawe serce - 30% i lewe serce – 10%;
• Pellagra – 5%.36
37
38

Wyrostek robaczkowy
• Epidemiologia 2-3/mln/rok;
• 2/1 kobiety/mężczyźni, bez predyspozycji co do rasy;
• Pik zapadalności w 2 dekadzie życia dla kobiet oraz w 3 dekadzie
dla mężczyzn;
• 0,63/ml/rok złośliwa forma NET, wyrostka na podstawie danych
SEER z pikiem występowania pomiędzy 38-49 rokiem życia;
• Najczęstszy (35-85%) nowotwór wyrostka robaczkowego;
• Diagnozowany od 3 do 9-u wykonanych appendektomii na 1000
zabiegów;
• Częsta lokalizacja w 1/3 dystalnej (90% przypadków przebiega bez
objawów klinicznych); 39

Wyrostek robaczkowy (postacie NET)
• Postacie histologiczne nowotworów neuroendokrynnych wyrostka
robaczkowego:
– Rakowiak;
– Rakowiak cewkowy (zwykle bardzo powolny wzrost);
– Dobrze zróżnicowany rak neuroendokrynny (WDNEC);
– Dobrze zróżnicowany guz neuroendokrynny (WDNET);
– Atypowy rakowiak (liczba mitoz 2-10 HPF i ogniska martwicy;
– Nowotwór mieszany (rakowiako-gruczolak, adenocarcinoid);
• Rakowiak z komórek kubkowych (Goblet cell carcinoid), bardziej
agresywny nowotwór w porównaniu z pozostałymi rakowiakami
wyrostka, może nie tworzyć typowego obrazu guza i przez ciągłość
naciekać kątnice, kluczowy element ocena resekcyjności (R).40

Wyrostek robaczkowy - rokowanie
• Korzystniejsza prognoza niż GEP-NET jelita cienkiego;
• Wielkość guza czynnik rokowniczy - zależność między wielkością
nowotworu, a przerzutami (węzły chłonne i wątroba);
• Wielkość krytyczna 2 cm – około 90% NET-ów wyrostka
robaczkowego ma średnicę poniżej 2 cm i nie daje przerzutów;
• Naciekanie krezki wyrostka oraz naciekanie pni nerwowych
znaczenie rokownicze nie jest jasne;
• Brak wyraźnego stadium neo nienaciekającego (in situ), może
rozwijać głęboko w obrębie błony śluzowej lub warstwy podśluzowej;
• Oznaczenie CgA tylko w przypadku rozsiewu nowotworu.
41

Goblet cell carcinoid
• GCC, występowanie 0,5/1mln/rok;
• 1/10 GEP-NET wyrostka robaczkowego,
• 0,3% wszystkich appendectomii;
• 35-58% wszystkich nowotworów wyrostka
robaczkowego;
• Pik zapadalności 5 dekada oraz ponownie 7-8 dekada;
• Bez preferencji płci, większość chorych – biali;
• Bez hormonalnie czynnego zespołu.
42

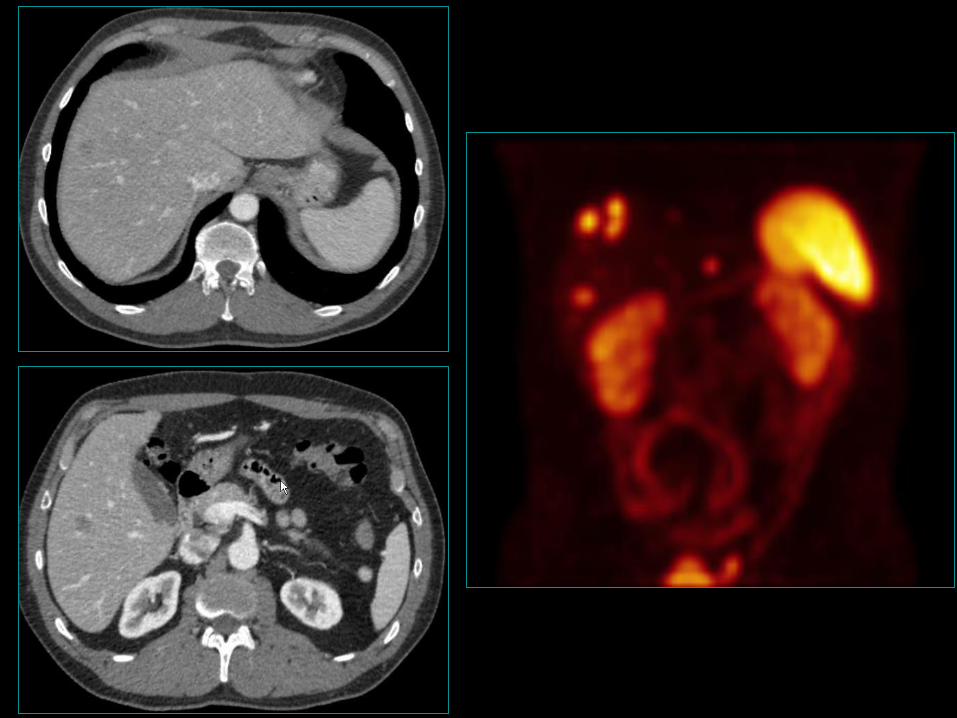
Jelito grube (całe)
• 20% wszystkich GEP-NET;
• 58% przypadków w odbytnicy;
• Odbytnica 13,7% oraz okrężnica 7,8% wszystkich
GEP-NET;
• Z okrężnicy najczęściej kątnica - 18%;
• 30% z przerzutami w momencie diagnozy.
43

Odbytnica/odbyt
• 4,2/mln/rok;
• 3 co do częstości po jelicie cienkim i wyrostku;
• 1,1-1,3% wszystkich guzów odbytnicy;
• 1x1000-2000 badań endoskopowych;
• Bez preferencji płci;
• Czarna/biała 3-4x
• 6 dekada życia szczyt zachorowania;
• 2,2% z mts do w. chłonnych 1,7% mts odległe;
• 90% 5-o letnie przeżycie.44

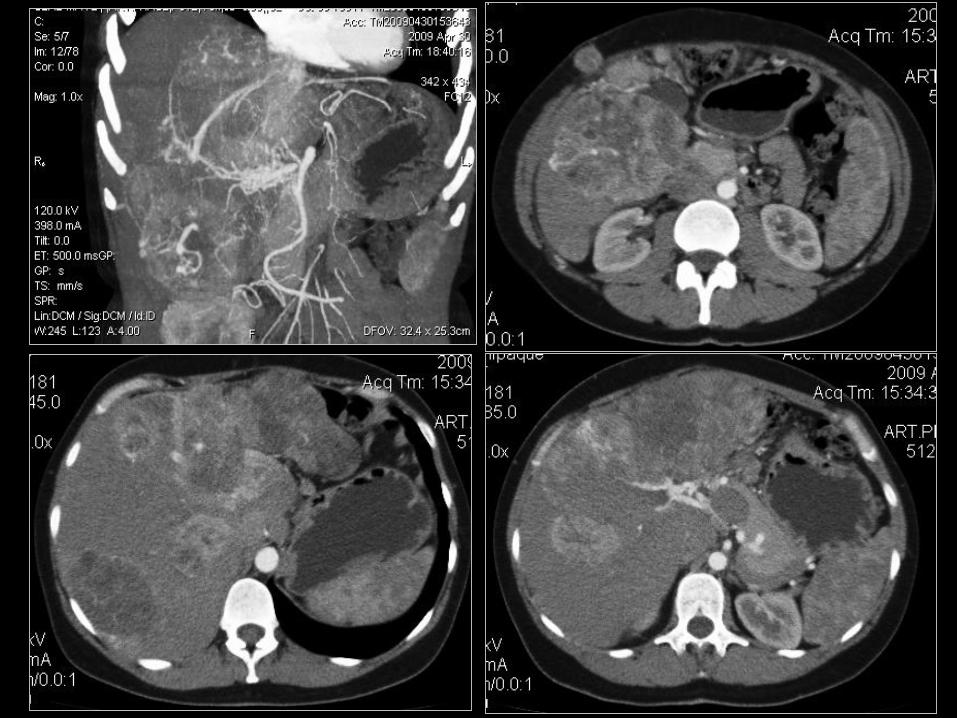
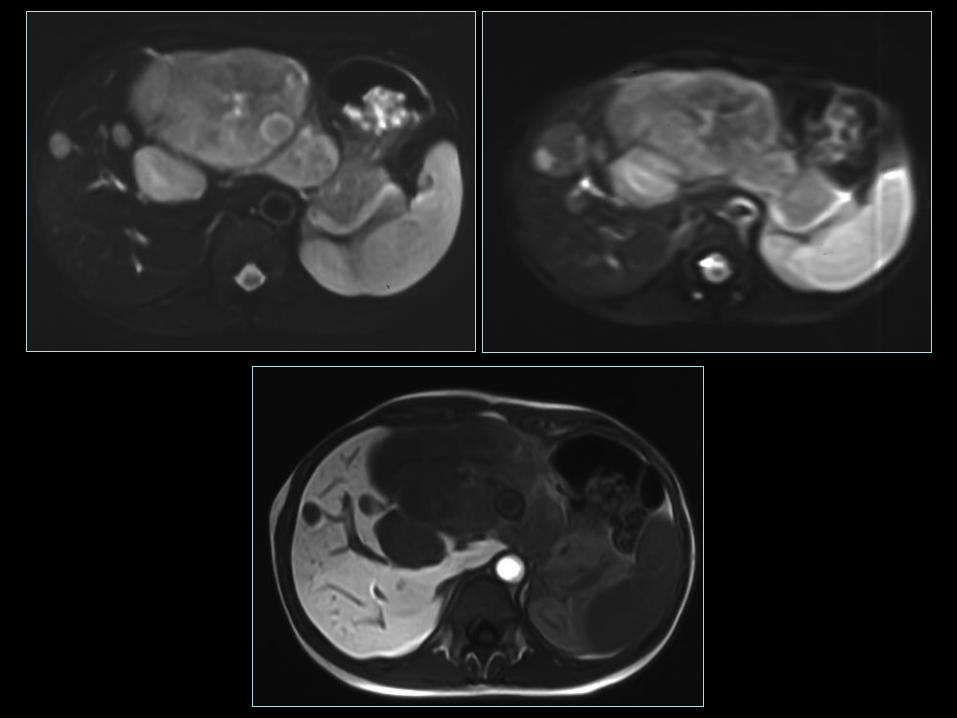
Jelito grube - okrężnica
• 33-60% przeżycia 5-o letniego, najbardziej niekorzystna prognoza;
• 40% chorych z przerzutami odległymi w momencie diagnozy;
• Guzy bez czynności hormonalnej, bez użycia „zimnych” analogów
somatostatyny w leczeniu systemowym;
• W przypadkach zaawansowanych (WHO grupa 2), z progresją
użycie chemioterapii: streptozotocyny + 5FU, zwykle odpowiedź
<25% lub użycie PRRT 90Y lub 177Lu DOTATATE;
• W przypadkach z zajęciem wątroby możliwość leczenia
radioembolizacją (RE) np. 90Y SIR-Spheres.
45
46
47
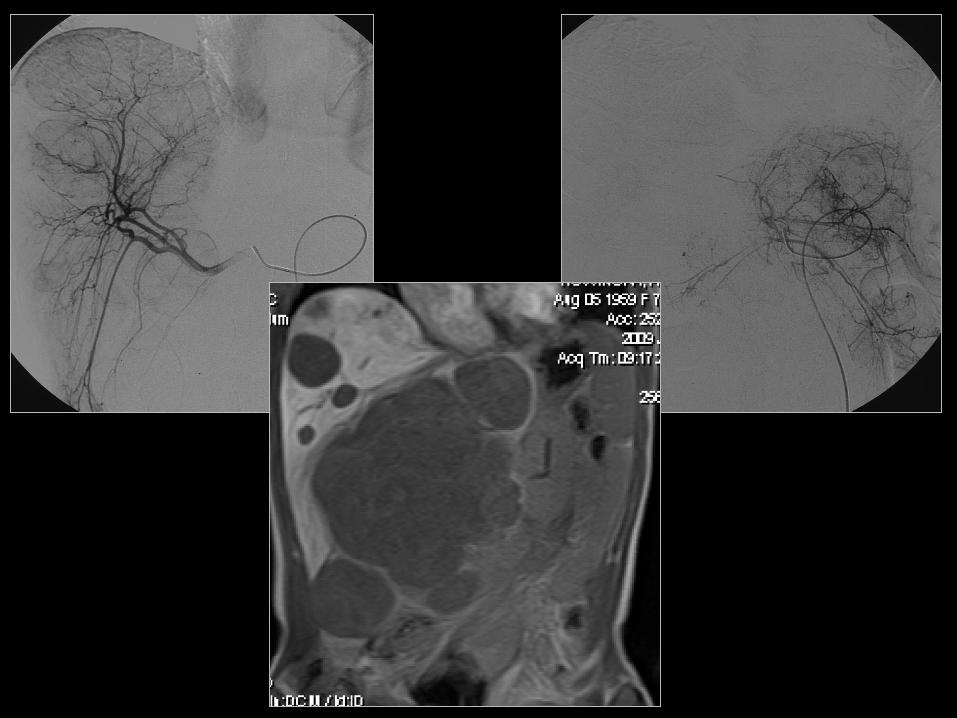
48

FPI – NET
• Pochodzenie - najczęściej odcinek środkowy prajelita
(midgut);
• 10 % wszystkich przypadków GEP-NET;
• Bardzo wysokie prawdopodobieństwo pochodzenia
„midgut” przy współistniejącym typowym „zespole
rakowiaka”;
• Większość guzów (raków) WHO grupa 2 (NETG2 wg
klasyfikacji WHO z 2010), mniejsza część raków WHO
grupa 3 (NECG3; WHO 2010) z złą prognozą.49
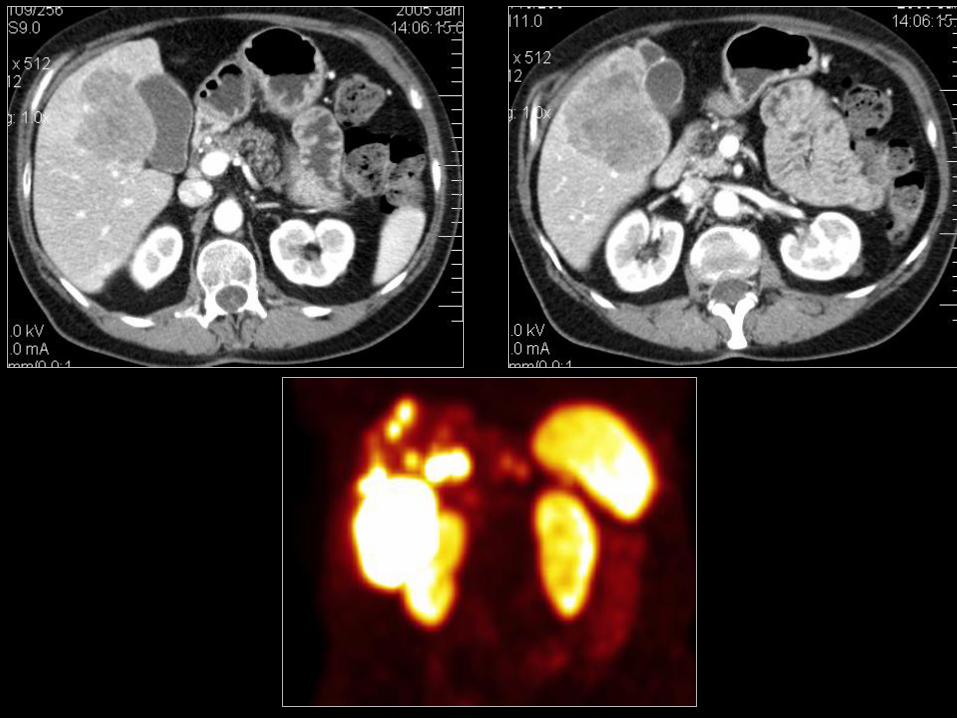
50
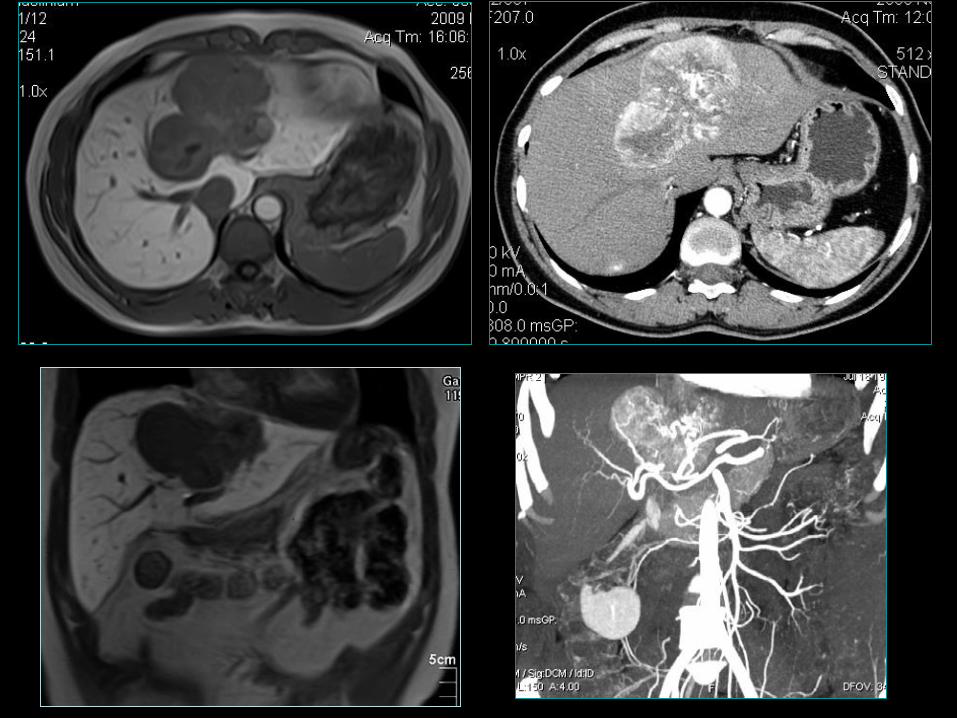
51
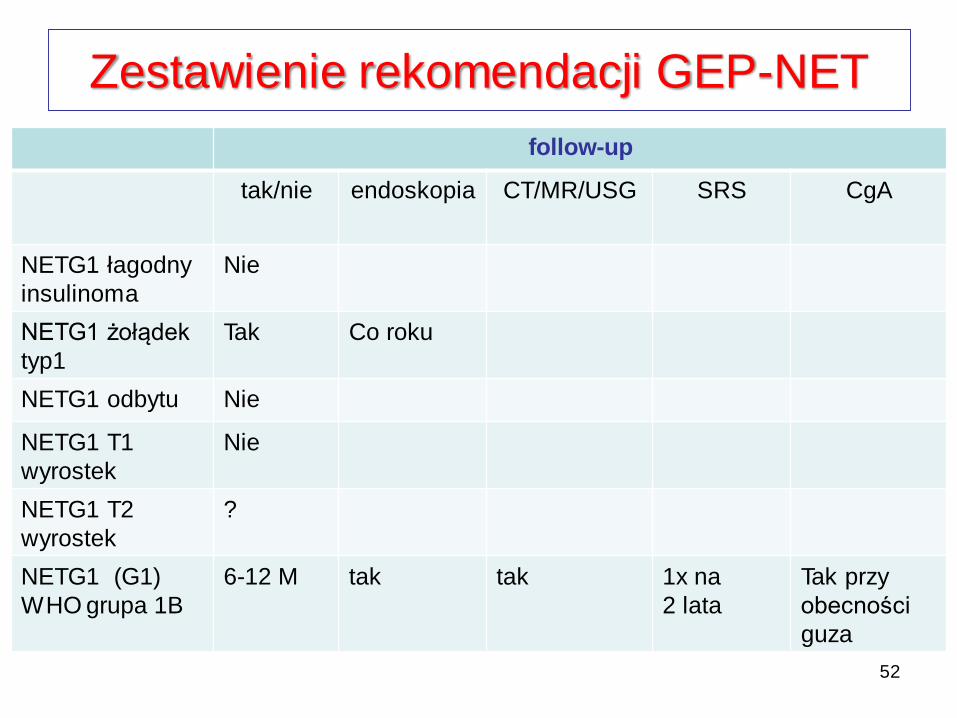
Zestawienie rekomendacji GEP-NET
follow-up
tak/nie endoskopia CT/MR/USG SRS CgA
NETG1 łagodny
insulinoma
Nie
NETG1 żołądek
typ1
Tak Co roku
NETG1 odbytu Nie
NETG1 T1
wyrostek
Nie
NETG1 T2
wyrostek
?
NETG1 (G1)
WHO grupa 1B
6-12 M tak tak 1x na
2 lata
Tak przy
obecności
guza
52
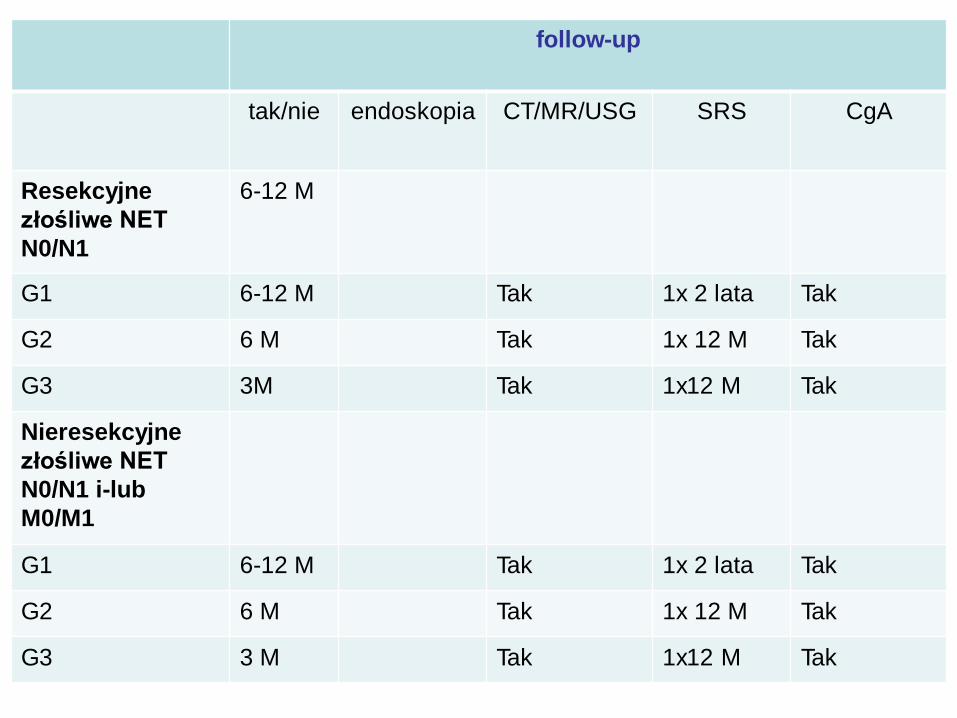
53
follow-up
tak/nie endoskopia CT/MR/USG SRS CgA
Resekcyjne
złośliwe NET
N0/N1
6-12 M
G1 6-12 M Tak 1x 2 lata Tak
G2 6 M Tak 1x 12 M Tak
G3 3M Tak 1x12 M Tak
Nieresekcyjne
złośliwe NET
N0/N1 i-lub
M0/M1
G1 6-12 M Tak 1x 2 lata Tak
G2 6 M Tak 1x 12 M Tak
G3 3 M Tak 1x12 M Tak

54
Inne NET o pochodzeniu foregut
• Układ oddechowy:
– Rakowiaki (typowy, atypowy);
– Rak olbrzymiokomórkowy (LCLC);
– Rak drobnokomórkowy płuca (SCLC);
• Rak rdzeniasty tarczycy - MTC;
• Rakowiak grasicy, pierwotny oraz w przebiegu
MEN-1 (8% przypadków zespołu MEN-1).

NET oskrzeli i płuc
• Rakowiaki płuc i oskrzeli stanowią ok. 25% wszystkich „rakowiaków”;
• Wzrost częstości rozpoznawania rakowiaków płuc wiąże się z
postępem technologicznym w obrazowaniu oraz rozpoznaniu
histopatologicznym wraz z oceną immunohistochemiczną;
• Stanowią tylko 1-2% wszystkich nowotworów płuc;
• Podział wg Travis’a 1991:
– Typowy rakowiak, o niskim stopniu złośliwości;
– Atypowy rakowiak, o średnim stopniu złośliwości;
– Rak neuroendokrynny olbrzymiokomórkowy (LCNEC);
– Rak drobnokomórkowy (SCLC);
Travis WD, et al. Am J Surg Pathol 1991;15:529–553 55

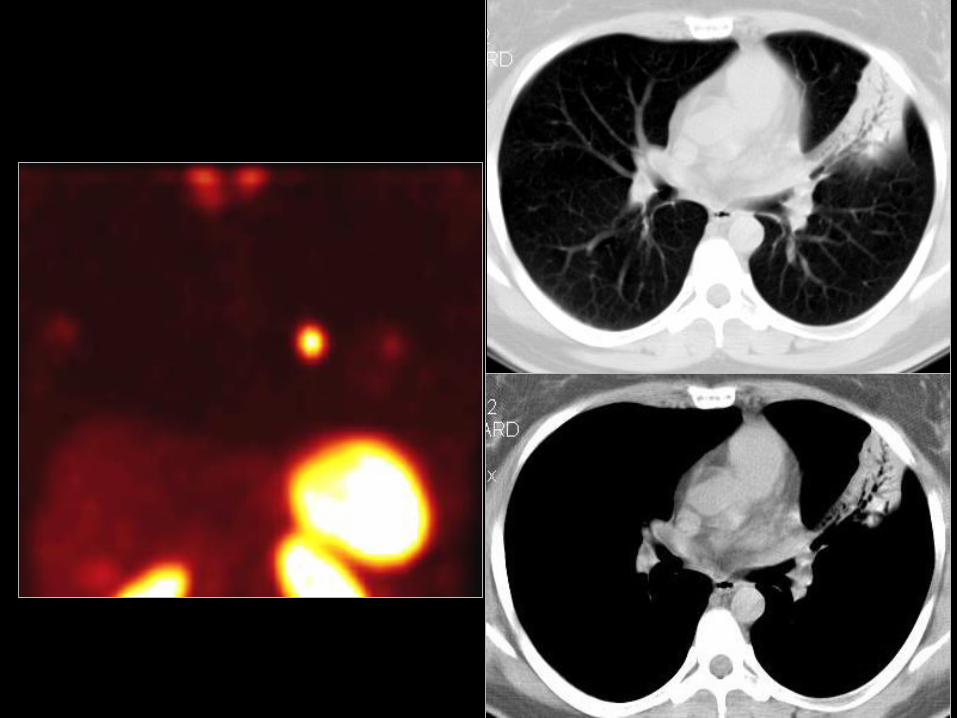
Rakowiaki płuc i oskrzeli
• 80-90% typowe rakowiaki;
• 60-70% zajęcie płatowego lub segmentalnego
oskrzela;
• Kobiety/mężczyźni >1;
• Młodsi niż z rakiem płuca, średnia wieku:
– typowy rakowiak - 46 lat;
– atypowy rakowiak - 56 lat;
• Przeżycie 5-o letnie;
– Typowy rakowiak 87%;
– Atypowy rakowiak 56%.56
57

Rakowiaki układu oddechowego
• Typowy rakowiak niezwiązany z paleniem papierosów, atypowy tak;
• lokalne stadium zaawansowania w momencie diagnozy:
– typowy rakowiak - 80-90%;
– atypowy rakowiak - 50%;
• Przerzuty z rakowiaków;
– Węzły chłonne, regionalne i dystalne, kości, wątroba, nadnercza,
OUN.
• W badaniach obrazowych zwykle niewielkie wymiary;
• Niektóre umieszczone wewnątrzoskrzelowo niewidoczne w
konwencjonalnym rtg, dlatego badanie TK w każdym przypadku.
Travis WD, et al. Am J Surg Pathol 1998;22:934–944 58

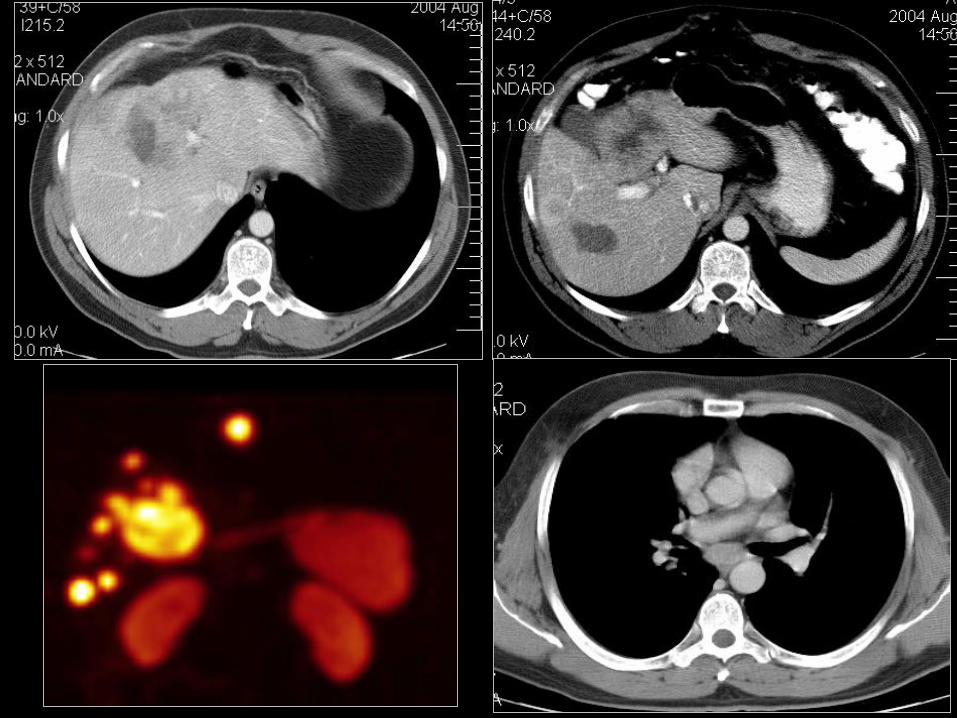
Rakowiaki układu oddechowego-diagnostyka
• Rozpoznanie na podstawie bronochskopii (najczęściej);
• Rozpoznanie z BAC guzka obwodowego, często nie pozwala
rozpoznać stopnia złośliwości i odróżnić od raka drobnokomórkowego
płuca (DRP);
• Badania obrazowe strukturalne TK, MR w przypadku braku możliwości
wykonania TK po podaniu śr. Kontrastowego;
• Badania czynnościowe jak w pozostałych NET, generalnie SRS,
metodą z wyboru w diagnostyce czynnościowej;
• FDG-PET w przypadku zmian średniozróżnicowanych (atypowy
rakowiak z wysokim indeksem proliferacyjnym >10% , w przypadku
badania SRS (FU) i guzy niskozróżnicowane np. LCNEC. 59
60

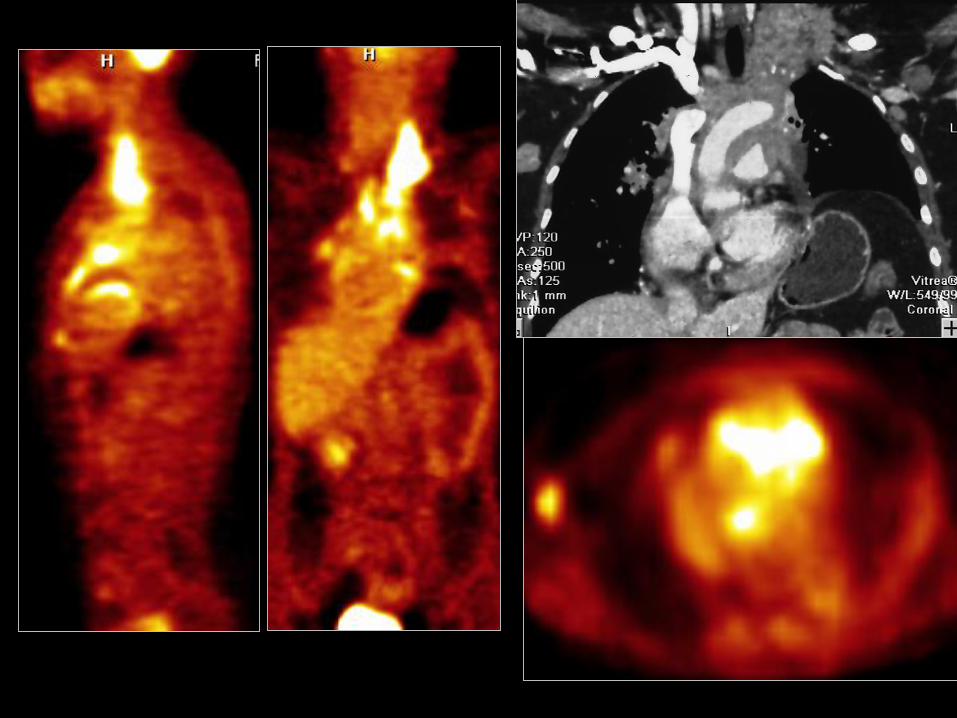
LCNEC
• LCNEC słabo zróżnicowany, o wysokiej złośliwości rak
neuroendokrynny;
• Średnia wieku 60 lat;
• Mężczyźni/kobiety>2,5;
• W 60% palacze;
• 2,9% raków płuc;
• 19% guzów neuroendokrynnych płuc;
• 5 letnie przeżycie 13-45%, obecnie 21%;
Paci M et al. Ann Thorac Surg 2004;77:1163–1167
62
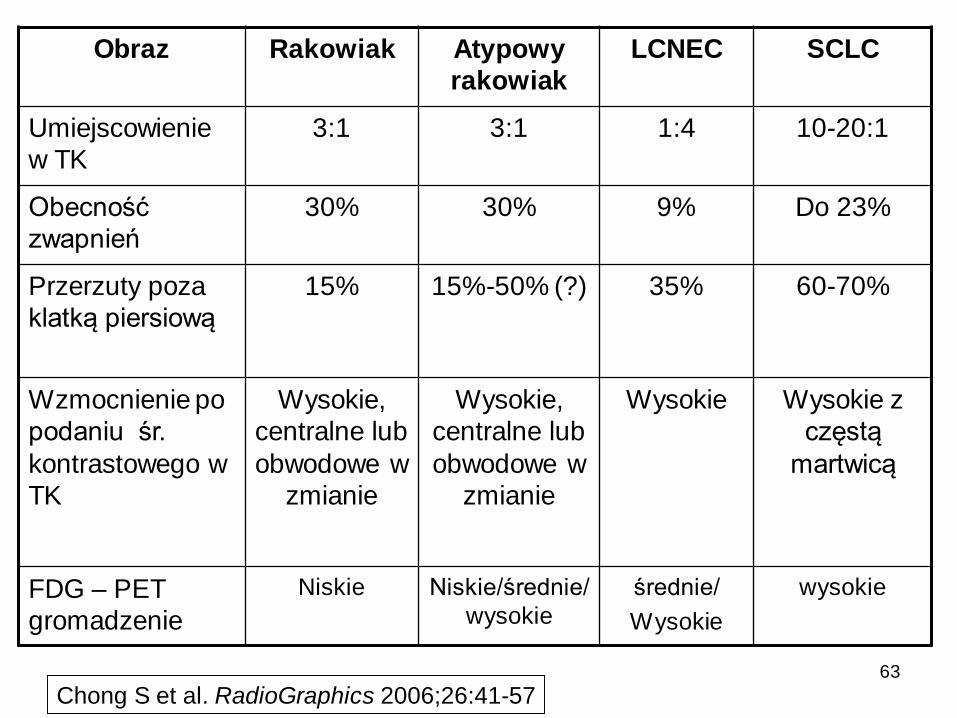
Obraz Rakowiak Atypowy
rakowiak
LCNEC SCLC
Umiejscowienie
w TK
3:1 3:1 1:4 10-20:1
Obecność
zwapnień
30% 30% 9% Do 23%
Przerzuty poza
klatką piersiową
15% 15%-50% (?) 35% 60-70%
Wzmocnienie po
podaniu śr.
kontrastowego w
TK
Wysokie,
centralne lub
obwodowe w
zmianie
Wysokie,
centralne lub
obwodowe w
zmianie
Wysokie Wysokie z
częstą
martwicą
FDG – PET
gromadzenie
Niskie Niskie/średnie/
wysokie
średnie/
Wysokie
wysokie
Chong S et al. RadioGraphics 2006;26:41-5763

64
Inne guzy NET
Guzy pochodzące z systemu sympatyko-adrenergicznego:
• Guz chromochłonny (pheochromocytoma);
• Neuroblastoma;
• Ganglioneuroblastoma;
• Ganglioneuroma;
• Paraganglioma;
• Chemodectoma;
• PPS;
• VHL;
• MEN 2a, MEN 2b;
• Guz z komórek Merkel’a.

Pheo sporadyczny i uwarunkowany genetycznie
• Występowanie guza chromochłonnego i /lub
paraganglioma sporadyczne – 68%;
• Związany z zespołami genetycznymi – 32%;
– MEN 2A (MTC, pheochrmocytoma, gruczolaki przytarczyc);
– MEN 2B (MTC, phochromocytoma, ganglioneurinoma);
– SDH-B, -D, -C (PGL);
– VHL – Choroba Von Hippel-Lindau;
– NF1 - Choroba von Recklinghausena;
– Zespół Sturge'a-Webera.
Neumann, et al. N Engl J Med 2002;346:1459-1466 65
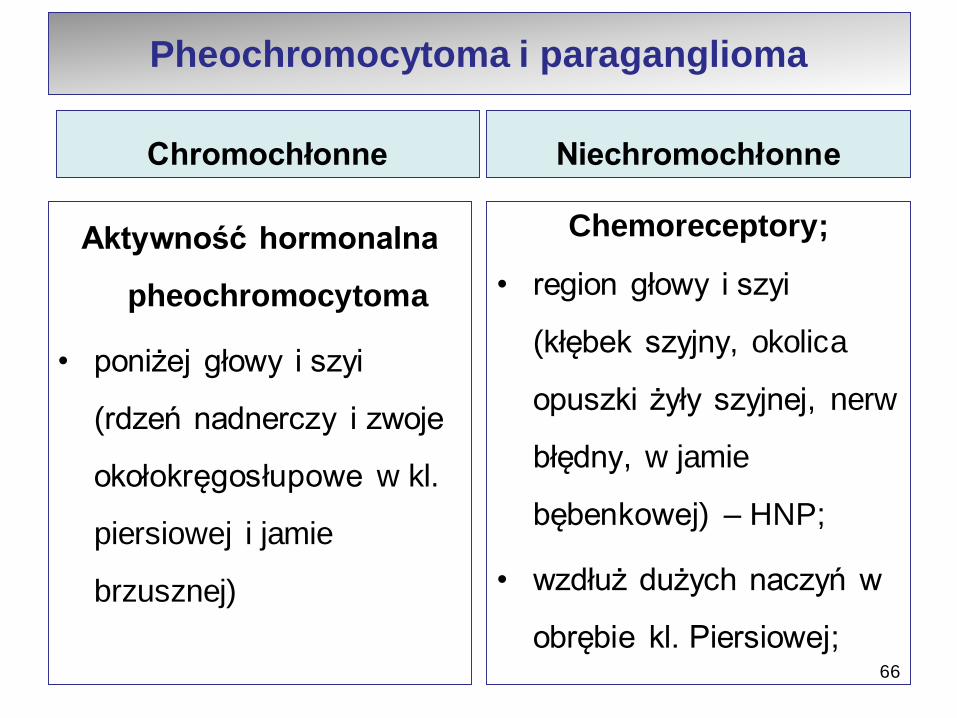
Chromochłonne
Aktywność hormonalna
pheochromocytoma
• poniżej głowy i szyi
(rdzeń nadnerczy i zwoje
okołokręgosłupowe w kl.
piersiowej i jamie
brzusznej)
Niechromochłonne
Chemoreceptory;
• region głowy i szyi
(kłębek szyjny, okolica
opuszki żyły szyjnej, nerw
błędny, w jamie
bębenkowej) – HNP;
• wzdłuż dużych naczyń w
obrębie kl. Piersiowej;66
Pheochromocytoma i paraganglioma

Guz chromochłonny
• 10-15% pheo guzy złośliwy;
• Wielkość guzów nadnercza:
– Adenoma <<< pheo < przerzuty;
– Pheo sekrecyjny << pheo niesekrecyjny;
• Małe pheo zwykle homogenne;
• Duże pheo heterogenne (częste wylewy i martwica);
• Możliwość zawartości tłuszczu oraz zwyrodnienie
torbielowate (wyniki FU w TK);
• Obecność zwapnień w 29% przypadków;67

69
Złośliwy pheo/paraganglioma
• Ocena złośliwości na podstawie obecności mts;
– wątroba, regionalne węzły chłonne, kości w 50%;
– płuca w 30%;
• Guzy złośliwe z czynnością sekrecyjną, podobne
objawy jak w przypadku formy łagodnej;
• Dodatkowo objawy zaawansowanej choroby
nowotworowej (wyniszczenie, nudności,
duszności, etc).
Goldstein et al. Annals of Surgery 1999;229:755–766

Diagnostyka obrazowa
• spiralna wielorzędowa TK po podaniu i.v. środka
kontrastowego;
• MRI przed i po podaniu i.v. środka kontrastowego;
• 123I mIBG układ transportowy i gromadzenia amin
biogennych (VMAT1 i VMAT2);
• SRS scyntygrafia receptorów somatostatynowych z
użyciem 99mTc (TOC/TATE) lub 68Ga DOTATATE.
70
71

Badania czynnościowe – 123I mIBG
• Pheochromocytoma (położenie nadnerczowe wg.
Klasyfikacji WHO);
• Paraganglioma o położeniu poza nadnerczowym z
objęciem tułowia oraz podstawy czaszki;
• Złośliwa postać pheochromocytoma, zróżnicowane
gromadzenie, w znaczącym procencie brak lub niskie
gromadzenie mIBG;
• Przerzuty, szczególnie do kości do w. chłonnych.
72

Badania czynnościowe - SRS
• Detekcja HNP – (chemodectoma-kłębczak) szyja oraz
podstawa czaszki, wraz z penetracją do jam czaszki
(metoda z wyboru);
• Oponiaki OUN;
• Paraganglioma o dowolnej lokalizacji w obrębie tułowia
oraz szyi i podstawy czaszki;
• Pheochromocytoma, szczególnie postać złośliwa;
• Przerzuty, szczególnie w mutacjach SDHB i -D.
73

Złośliwy pheochromocytoma
• 10-15% wszystkich pheo;
• 2 odmiany złośliwe pheo:
– zajęcie tylko tk. Miękkich;
– Zajęcie tk. Miękkich i kości (gorsze rokowanie);
• Diagnostyka biochemiczna:
– mocz :VMA, katecholaminy, metanephryny,
– osocze: wolne metanefryny (specyficzność 84-89%);
• Wielkość pheo w badaniu TK:
– adenoma < pheo < mts; 74

MEN 2A i 2B
• MEN typ 2 zespół autosomalny, dominujący (RET-95%);
• 90% chorych na MEN 2 ma MTC;
• MEN 2A, MTC 90%, jednostronny lub obustronny pheo
50% oraz gruczolak przytarczyc w 20-30%;
• MEN 2A 75% wszystkich MEN 2;
• Inne warianty MEN 2: rodzinny MTC (FMTC), oraz
FMTC łącznie z chorobą Hirschsprunga;
• MEN 2B bardziej agresywny wariant MEN 2.
Brandi ML et al. J Clin Endocrinol & Metabol. 2002;12:5658-567175

76
Zespół PPS
• Występowanie: (dane szacunkowe 12-24% chorych ze
stwierdzonym guzem chromochłonnym bez wywiadu
rodzinnego i wcześniej rozpoznanego zespołu
genetycznego ma mutacje genów VHL, RET, SDH (-D, -B i
–C);
• Dotyczy młodszych pacjentów, częściej guzy
pozanadnerczowe i wieloogniskowe w porównaniu z
chorymi bez stwierdzanych mutacji.
*Pęczkowska M et al. Nature Endocrinol Metabol. 2008;4;111-115

77
Mutacje SDH
• Heterozygotyczne mutacje SDHB, SDHC oraz SDHD (bez SDHA) są
związane z występowaniem PPS;
• Mutacje SDHD towarzyszą występowaniu przyzwojaków o położeniu w
obrębie tułowia, szyi z objęciem podstawy czaszki (chemodektoma –
kłębczaki, ang. head and neck paragangliomas - HNPs) oraz guzami
chromochłonnymi nadnerczy;
• Mutacje SDHB towarzyszą zwykle zmianom wewnątrzbrzusznym,
pozanadnerczowym z możliwym złośliwym charakterem zmian o typie
przyzwojaków;
• Mutacje SDHC są obecnie najmniej poznane i wiedza o towarzyszących
schorzeniach jest ograniczona.

Lokalizacja paraganglioma
• przestrzeń zaotrzewnowa, w okolicy nadnerczy, wnęk i
biegunów nerek;
• w pobliżu aorty;
• w pobliżu narządu Zukerkandla (odejścia IMA);
• pęcherz moczowy;
• klatka piersiowa – śródpiersie;
• Szyja, podstawa czaszki jama bębenkowa, piramida k.
skroniowej.
78
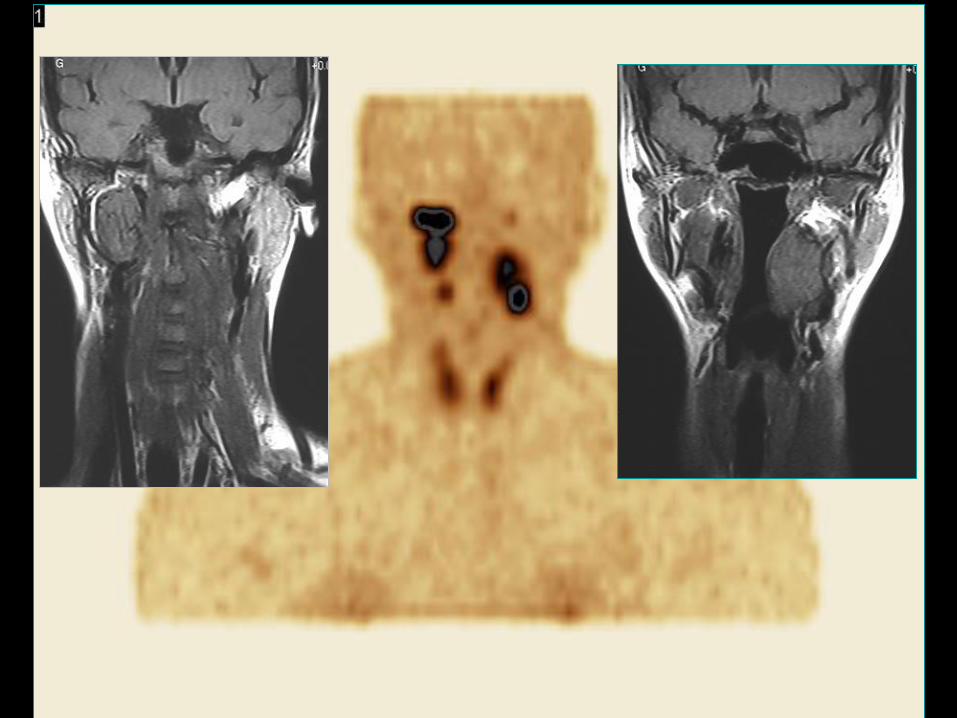

Choroba Von Hippel-Lindau - VHL
• Progresywna, autosomalna, dominująca
choroba związana z mutacją genu VHL;
• Hemangioblastoma siatkówki oraz OUN;
• Rak jasno-komórkowy nerki (RCC);
• Pheochromocytoma;
• GEP-NET, wyspiaki trzustki, średnica < 3cm –
najczęściej łagodny.
80

Dziękuję za uwagę81