
and Duncan J. Stewart Li L. Yang, Robert Gros, M. Golam Kabir ...
8
and Duncan J. Stewart Li L. Yang, Robert Gros, M. Golam Kabir, Almuktafi Sadi, Avrum I. Gotlieb, Mansoor Husain Cardiomyopathy in Mice Conditional Cardiac Overexpression of Endothelin-1 Induces Inflammation and Dilated Print ISSN: 0009-7322. Online ISSN: 1524-4539 Copyright © 2004 American Heart Association, Inc. All rights reserved. is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231 Circulation doi: 10.1161/01.CIR.0000105701.98663.D4 2004;109:255-261; originally published online January 12, 2004; Circulation. http://circ.ahajournals.org/content/109/2/255 World Wide Web at: The online version of this article, along with updated information and services, is located on the http://circ.ahajournals.org//subscriptions/ is online at: Circulation Information about subscribing to Subscriptions: http://www.lww.com/reprints Information about reprints can be found online at: Reprints: document. Permissions and Rights Question and Answer this process is available in the click Request Permissions in the middle column of the Web page under Services. Further information about Office. Once the online version of the published article for which permission is being requested is located, can be obtained via RightsLink, a service of the Copyright Clearance Center, not the Editorial Circulation in Requests for permissions to reproduce figures, tables, or portions of articles originally published Permissions: by guest on February 23, 2013 http://circ.ahajournals.org/ Downloaded from
Transcript of and Duncan J. Stewart Li L. Yang, Robert Gros, M. Golam Kabir ...

and Duncan J. StewartLi L. Yang, Robert Gros, M. Golam Kabir, Almuktafi Sadi, Avrum I. Gotlieb, Mansoor Husain
Cardiomyopathy in MiceConditional Cardiac Overexpression of Endothelin-1 Induces Inflammation and Dilated
Print ISSN: 0009-7322. Online ISSN: 1524-4539 Copyright © 2004 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation doi: 10.1161/01.CIR.0000105701.98663.D4
2004;109:255-261; originally published online January 12, 2004;Circulation.
http://circ.ahajournals.org/content/109/2/255World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circ.ahajournals.org//subscriptions/
is online at: Circulation Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialCirculationin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on February 23, 2013http://circ.ahajournals.org/Downloaded from

Conditional Cardiac Overexpression of Endothelin-1 InducesInflammation and Dilated Cardiomyopathy in Mice
Li L. Yang, MD, MSc; Robert Gros, PhD; M. Golam Kabir, MD; Almuktafi Sadi, MD, PhD;Avrum I. Gotlieb, MD; Mansoor Husain, MD*; Duncan J. Stewart, MD*
Background—Myocardial expression of endothelin-1 (ET-1) and its receptors ETA and ETB is increased in heart failure.However, the role of ET-1 and its signaling pathways in the pathogenesis of myocardial diseases is unclear.
Methods and Results—Human ET-1 cDNA was placed downstream of a promoter responsive to a doxycycline(DOX)-regulated transcriptional activator (tTA). This line (ET�) was bred with one harboring cardiac myocyte–restricted expression of tTA (�MHC-tTA). Myocardial ET-1 peptide levels were significantly increased in binarytransgenic (BT, ET�/tTA�) compared with nonbinary transgenic (NBT, ET�/tTA�; ET�/tTA�; ET�/tTA�) orDOX-treated BT littermates (40.1�4.7 versus 2.6�1.2 fmol/mL, P�0.003). BT mice demonstrated progressivemortality between 5 and 11 weeks after DOX withdrawal, associated with left ventricular dilatation and contractiledysfunction (peak �dP/dT, 4673�468 versus 5585�658 mm Hg/s, P�0.05). An interstitial inflammatory infiltrate,including macrophages and T lymphocytes, was evident in the myocardium of BT mice, associated with sequentialincreases in nuclear factor-�B translocation and expression of tumor necrosis factor-�, interferon-�, interleukin-1 andinterleukin-6. Significant prolongation of survival was observed with the combined ETA/ETB antagonist LU420627(n�8, P�0.05) in BT mice but not the ETA-selective antagonist LU135252 (n�5, P�0.9), consistent with an importantrole for ETB in this model.
Conclusions—These are the first data to demonstrate that cardiac overexpression of ET-1 is sufficient to cause increasedexpression of inflammatory cytokines and an inflammatory cardiomyopathy leading to heart failure and death.(Circulation. 2004;109:255-261.)
Key Words: endothelin � myocarditis � hypertrophy � heart failure
Endothelin-1 (ET-1) has been implicated in the progres-sion of congestive heart failure (CHF).1 Plasma ET-1
levels are elevated in patients2 and experimental animalmodels of CHF.3 The expression of ET-1 and its receptorsETA and ETB is increased in myocardium of rats withpostinfarction CHF4 and in patients with idiopathic dilatedcardiomyopathy (DCM).5 In vitro and/or pharmacologicalstudies have suggested that ET-1 exerts trophic effects incardiac myocytes6 and stimulates collagen synthesis in fibro-blasts.7 ET-1 also modulates leukocyte adhesion to endothe-lial cells8 and enhances production of cytokines inmonocytes/macrophages.9
However, the role of ET-1 in the pathogenesis of CHFremains controversial. The combined ETA/ETB receptor an-tagonist bosentan improved survival in experimental modelsof CHF10 but not in heart failure patients.11 Similar studiesusing selective ETA antagonists have demonstrated bothbeneficial12 and deleterious effects.13
Previous attempts to modulate ET-1 expression by trans-genic approaches have been complicated by embryonic le-thality in knockout mice14,15 and low levels of ET expressionin viable progeny of gain-of-function mutants.16,17 Indeed,nontargeted and nonconditional ET-1 overexpression studieshave resulted in limited numbers of founder mice, modestelevations in ET-1 levels, and no obvious cardiac pheno-type.16,17 We hypothesized that this was caused by a selectionbias arising from the embryonic lethal consequences of ET-1overexpression in the heart. To overcome this limitation, wegenerated transgenic mice exhibiting conditional cardiac-restricted ET-1 overexpression using the �-myosin heavychain (�-MHC) promoter-dependent cardiac-specifictetracycline-regulated gene expression system (Tet-OFF)(Figure 1).18
In the present study, we demonstrate that postnatal cardiacoverexpression of ET-1 induced inflammation and hypertro-phy, leading to a rapid deterioration in cardiac function anddeath.
Received November 6, 2002; de novo received July 29, 2003; revision received September 11, 2003; accepted September 12, 2003.From the Heart and Stroke Richard Lewar Centre of Excellence (L.L.Y., A.I.G., M.H., D.J.S.); the Departments of Medicine (M.H., D.J.S.) and
Laboratory Medicine and Pathobiology (L.L.Y., A.I.G., M.H., D.J.S.), University of Toronto; Cellular and Molecular Biology, Toronto General Hospital(L.L.Y., R.G., M.G.K., A.S., M.H.); and the Terrence Donnelley Heart Center, St Michael’s Hospital (D.J.S.), Toronto, Canada.
*Drs Husain and Stewart contributed equally to this work.Correspondence to Mansoor Husain, Toronto General Hospital, EN 12-221, 200 Elizabeth St, Toronto, Ontario, Canada, M5G 2C4. E-mail
[email protected]© 2004 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/01.CIR.0000105701.98663.D4
255 by guest on February 23, 2013http://circ.ahajournals.org/Downloaded from
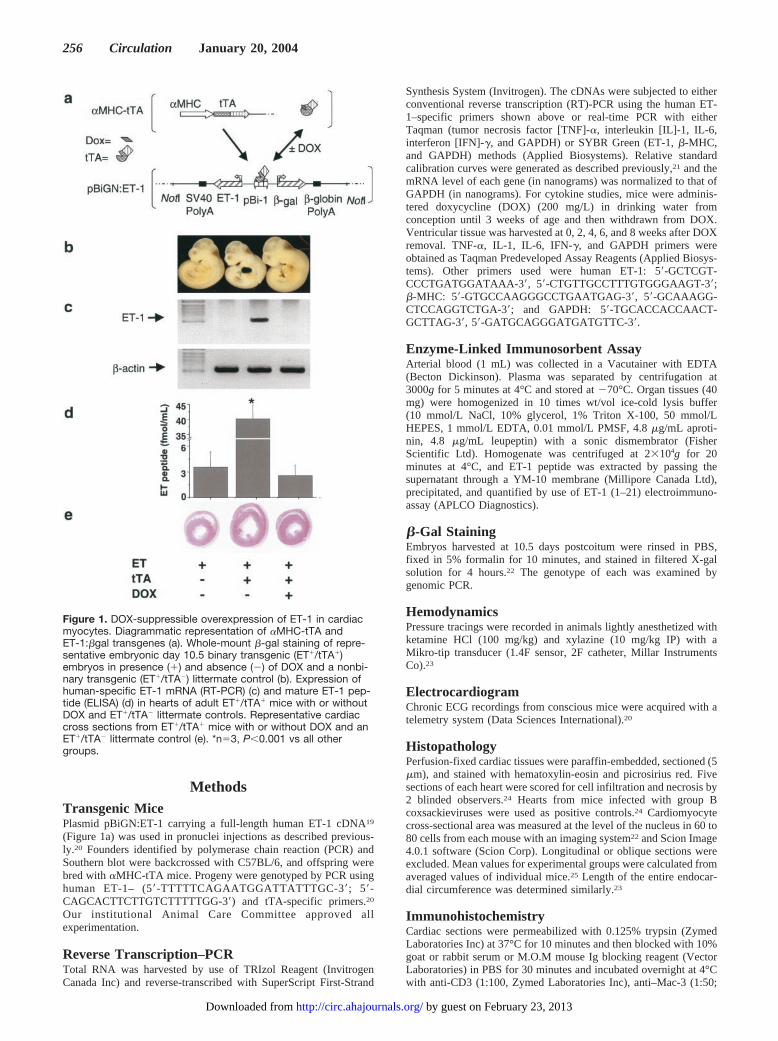
MethodsTransgenic MicePlasmid pBiGN:ET-1 carrying a full-length human ET-1 cDNA19
(Figure 1a) was used in pronuclei injections as described previous-ly.20 Founders identified by polymerase chain reaction (PCR) andSouthern blot were backcrossed with C57BL/6, and offspring werebred with �MHC-tTA mice. Progeny were genotyped by PCR usinghuman ET-1– (5�-TTTTTCAGAATGGATTATTTGC-3�; 5�-CAGCACTTCTTGTCTTTTTGG-3�) and tTA-specific primers.20
Our institutional Animal Care Committee approved allexperimentation.
Reverse Transcription–PCRTotal RNA was harvested by use of TRIzol Reagent (InvitrogenCanada Inc) and reverse-transcribed with SuperScript First-Strand
Synthesis System (Invitrogen). The cDNAs were subjected to eitherconventional reverse transcription (RT)-PCR using the human ET-1–specific primers shown above or real-time PCR with eitherTaqman (tumor necrosis factor [TNF]-�, interleukin [IL]-1, IL-6,interferon [IFN]-�, and GAPDH) or SYBR Green (ET-1, �-MHC,and GAPDH) methods (Applied Biosystems). Relative standardcalibration curves were generated as described previously,21 and themRNA level of each gene (in nanograms) was normalized to that ofGAPDH (in nanograms). For cytokine studies, mice were adminis-tered doxycycline (DOX) (200 mg/L) in drinking water fromconception until 3 weeks of age and then withdrawn from DOX.Ventricular tissue was harvested at 0, 2, 4, 6, and 8 weeks after DOXremoval. TNF-�, IL-1, IL-6, IFN-�, and GAPDH primers wereobtained as Taqman Predeveloped Assay Reagents (Applied Biosys-tems). Other primers used were human ET-1: 5�-GCTCGT-CCCTGATGGATAAA-3�, 5�-CTGTTGCCTTTGTGGGAAGT-3�;�-MHC: 5�-GTGCCAAGGGCCTGAATGAG-3�, 5�-GCAAAGG-CTCCAGGTCTGA-3�; and GAPDH: 5�-TGCACCACCAACT-GCTTAG-3�, 5�-GATGCAGGGATGATGTTC-3�.
Enzyme-Linked Immunosorbent AssayArterial blood (1 mL) was collected in a Vacutainer with EDTA(Becton Dickinson). Plasma was separated by centrifugation at3000g for 5 minutes at 4°C and stored at �70°C. Organ tissues (40mg) were homogenized in 10 times wt/vol ice-cold lysis buffer(10 mmol/L NaCl, 10% glycerol, 1% Triton X-100, 50 mmol/LHEPES, 1 mmol/L EDTA, 0.01 mmol/L PMSF, 4.8 �g/mL aproti-nin, 4.8 �g/mL leupeptin) with a sonic dismembrator (FisherScientific Ltd). Homogenate was centrifuged at 2�104g for 20minutes at 4°C, and ET-1 peptide was extracted by passing thesupernatant through a YM-10 membrane (Millipore Canada Ltd),precipitated, and quantified by use of ET-1 (1–21) electroimmuno-assay (APLCO Diagnostics).
�-Gal StainingEmbryos harvested at 10.5 days postcoitum were rinsed in PBS,fixed in 5% formalin for 10 minutes, and stained in filtered X-galsolution for 4 hours.22 The genotype of each was examined bygenomic PCR.
HemodynamicsPressure tracings were recorded in animals lightly anesthetized withketamine HCl (100 mg/kg) and xylazine (10 mg/kg IP) with aMikro-tip transducer (1.4F sensor, 2F catheter, Millar InstrumentsCo).23
ElectrocardiogramChronic ECG recordings from conscious mice were acquired with atelemetry system (Data Sciences International).20
HistopathologyPerfusion-fixed cardiac tissues were paraffin-embedded, sectioned (5�m), and stained with hematoxylin-eosin and picrosirius red. Fivesections of each heart were scored for cell infiltration and necrosis by2 blinded observers.24 Hearts from mice infected with group Bcoxsackieviruses were used as positive controls.24 Cardiomyocytecross-sectional area was measured at the level of the nucleus in 60 to80 cells from each mouse with an imaging system22 and Scion Image4.0.1 software (Scion Corp). Longitudinal or oblique sections wereexcluded. Mean values for experimental groups were calculated fromaveraged values of individual mice.25 Length of the entire endocar-dial circumference was determined similarly.23
ImmunohistochemistryCardiac sections were permeabilized with 0.125% trypsin (ZymedLaboratories Inc) at 37°C for 10 minutes and then blocked with 10%goat or rabbit serum or M.O.M mouse Ig blocking reagent (VectorLaboratories) in PBS for 30 minutes and incubated overnight at 4°Cwith anti-CD3 (1:100, Zymed Laboratories Inc), anti–Mac-3 (1:50;
Figure 1. DOX-suppressible overexpression of ET-1 in cardiacmyocytes. Diagrammatic representation of �MHC-tTA andET-1:�gal transgenes (a). Whole-mount �-gal staining of repre-sentative embryonic day 10.5 binary transgenic (ET�/tTA�)embryos in presence (�) and absence (�) of DOX and a nonbi-nary transgenic (ET�/tTA�) littermate control (b). Expression ofhuman-specific ET-1 mRNA (RT-PCR) (c) and mature ET-1 pep-tide (ELISA) (d) in hearts of adult ET�/tTA� mice with or withoutDOX and ET�/tTA� littermate controls. Representative cardiaccross sections from ET�/tTA� mice with or without DOX and anET�/tTA� littermate control (e). *n�3, P�0.001 vs all othergroups.
256 Circulation January 20, 2004
by guest on February 23, 2013http://circ.ahajournals.org/Downloaded from

BD PharMingen), or anti–nuclear factor (NF)-�B p65 (1:200, SantaCruz Biotechnology Inc) antibodies. Sections were incubated for 20minutes with biotinylated secondary antibody (goat anti-rabbit1:200, goat anti-rat 1:200, or M.O.M anti-mouse Ig reagent), washedwith PBS, and counterstained with hematoxylin.
ET Receptor AntagonismThe ETA-selective antagonist LU135252, with Ki values of 1.4 and140 nmol/L for ETA and ETB,26 or the combined ETA/ETB antagonistLU420627 (both courtesy Michael Kirchengast, Knoll Pharmaceu-ticals Ltd, Ludwigshafen, Germany), with Ki values of 2 and 6nmol/L for ETA and ETB,27 were given in the drinking water (400 mg· kg�1 · d�1). Body weight and water intake were monitored to ensurethat the animals received the daily dose.
Data AnalysisUnless otherwise stated, data were expressed as mean � SD. Tocompare survival between groups, a Kaplan-Meier analysis wasused.28 To compare differences among groups, �2 test or ANOVAfollowed by Student-Newman-Keuls comparisons were used. Allstatistical tests were conducted with SPSS 10.0. Statistical signifi-cance was defined as a value of P�0.05.
ResultsGeneration of Cardiac-Restricted ET-1Transgenic MiceSingle-cell pronuclei injections with pBiGN:ET-1 produced 3ET-1 founder mice out of 40 births (7.5%). All 3 were bredinto the C57BL/6 background and transmitted the transgenein autosomal fashion. All 3 were then bred with �-MHC-tTAmice18 to generate binary transgenic mice (BT: ET�/tTA�).Of the 3 BT lines, one (101 ET�/tTA�) demonstrated noET-1:�-gal expression in the heart using �-gal staining.Another (403 ET�/tTA�) showed low levels of cardiactransgene overexpression and demonstrated no overt heartfailure over a 6-month period of observation. The third (9ET�/tTA�), which demonstrated high levels of myocardialhuman ET-1 expression during development and adulthood,was characterized in detail.
To localize ET-1:�gal transgene overexpression duringdevelopment, we performed whole-mount �-gal staining onembryonic day 10.5 embryos. The DOX-suppressible ET:�-gal expression was confined to hearts of BT embryos and wasnot observed in non-BT embryos (NBT: ET�/tTA�, ET�/tTA�, and ET�/tTA�) or noncardiac tissues of BT embryos(Figure 1b). Crossing line-9 ET� with �-MHC-tTA micegenerated significantly fewer ET�/tTA� offspring than ex-pected (7% versus expected 25%, n�116, P�0.001), indi-cating that overexpression of ET-1 in the developing heartresulted in fetal loss. DOX administration restored the ex-pected mendelian frequency (25%) of ET�/tTA� progeny(26%, n�160, P�NS). To focus on the consequences ofET-1 overexpression in adult myocardium, DOX was admin-istered to maternal mice to suppress transgene expressionduring development. ET-1 overexpression was then inducedby removal of DOX in the postnatal period.
Targeted and conditional expression of ET-1 in adult micewas confirmed. Human ET-1 mRNA was expressed exclu-sively in hearts of BT and not those of NBT littermates(Figure 1c). Total ET-1 peptide level in the heart was�10-fold higher in BT than NBT littermates at 5 weeks afterDOX removal (40.1�4.7 versus 2.6�1.2 fmol/mL,
P�0.001). No increase in ET-1 expression was detected inplasma or any other organs examined. In BT mice, DOXtreatment completely suppressed the expression of humanET-1 mRNA and brought ELISA-determined peptide levelsdown to background levels (3.5�1.8 fmol/mL) (Figure 1d).
Phenotype of Mice With Cardiac-RestrictedOverexpression of ET-1BT mice released from DOX at birth initially displayednormal appearance, behavior, and oral intake. However,starting at �5 weeks of age, BT mice began to exhibitdecreased activity, hunched posture, and labored breathing,which invariably led to death within days of symptom onset.Pathological examination of symptomatic BT mice revealedpulmonary and hepatic congestion not observed in asymp-tomatic or NBT mice (data not shown). More than half of themice released from DOX died within 8 weeks. By contrast,BT maintained on DOX remained normal in phenotype andshowed no difference in survival compared with NBT litter-mates (Figure 2).
Invasive hemodynamic assessments made before the onsetof overt morbidity or significant mortality (ie, 5 weeks afterDOX) revealed decreased peak arterial systolic blood pres-sure in BT versus NBT littermates (83.6�7.3 versus95.1�10.9 mm Hg, n�5, P�0.05). Peak left ventricularsystolic pressure (74.3�5.9 versus 84.3�5.1 mm Hg, n�5,P�0.05) and �dP/dT (4673�468 versus 5585�658 mm Hg/s,n�5, P�0.05) were also significantly lower in BT than NBTlittermates.
To determine whether arrhythmias contributed to the mor-tality of BT mice, ECG telemetry was monitored for 5 weeksin 8-week-old BT mice withdrawn from DOX for 5 weeksand in DOX-treated age-matched BT controls. PR intervals inBT remained unchanged until the terminal stage. However,QRS complexes lengthened progressively over time, becom-ing significantly wider in overtly ill BT mice compared withDOX-treated controls (31.7�3.6 versus 13.1�0.7 ms, n�3,P�0.05). Large P waves were also observed in BT but not
Figure 2. Kaplan-Meier survival curves in ET�/tTA� mice. DOXwas given before conception and maintained until offspring wereborn. Pups were randomized and treated with or without variousdrugs as described in Methods. No events occurred before day30 or after day 90. *P�0.001, untreated (black line) vs DOX-treated ET�/tTA� (dotted line); †P�0.05, untreated vsLU420627-treated ET�/tTA� (dashed line); ‡P�0.78, untreatedvs LU135252-treated ET�/tTA� (gray line).
Yang et al Endothelin-1 Overexpression Causes Cardiomyopathy 257
by guest on February 23, 2013http://circ.ahajournals.org/Downloaded from
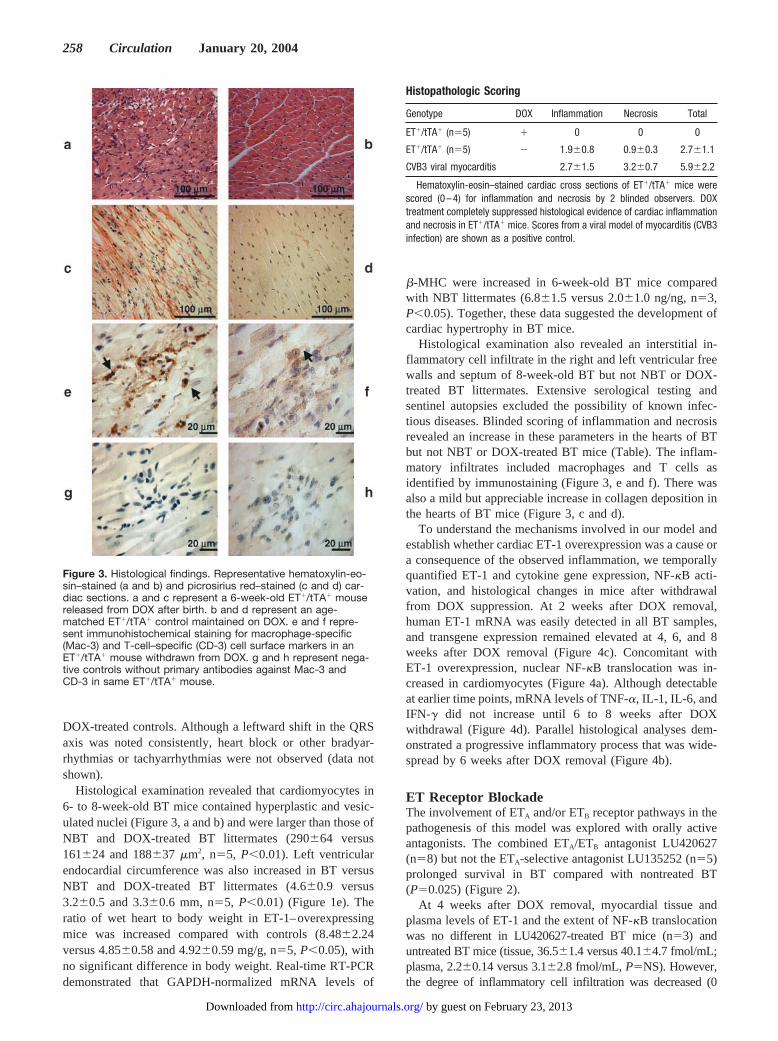
DOX-treated controls. Although a leftward shift in the QRSaxis was noted consistently, heart block or other bradyar-rhythmias or tachyarrhythmias were not observed (data notshown).
Histological examination revealed that cardiomyocytes in6- to 8-week-old BT mice contained hyperplastic and vesic-ulated nuclei (Figure 3, a and b) and were larger than those ofNBT and DOX-treated BT littermates (290�64 versus161�24 and 188�37 �m2, n�5, P�0.01). Left ventricularendocardial circumference was also increased in BT versusNBT and DOX-treated BT littermates (4.6�0.9 versus3.2�0.5 and 3.3�0.6 mm, n�5, P�0.01) (Figure 1e). Theratio of wet heart to body weight in ET-1–overexpressingmice was increased compared with controls (8.48�2.24versus 4.85�0.58 and 4.92�0.59 mg/g, n�5, P�0.05), withno significant difference in body weight. Real-time RT-PCRdemonstrated that GAPDH-normalized mRNA levels of
�-MHC were increased in 6-week-old BT mice comparedwith NBT littermates (6.8�1.5 versus 2.0�1.0 ng/ng, n�3,P�0.05). Together, these data suggested the development ofcardiac hypertrophy in BT mice.
Histological examination also revealed an interstitial in-flammatory cell infiltrate in the right and left ventricular freewalls and septum of 8-week-old BT but not NBT or DOX-treated BT littermates. Extensive serological testing andsentinel autopsies excluded the possibility of known infec-tious diseases. Blinded scoring of inflammation and necrosisrevealed an increase in these parameters in the hearts of BTbut not NBT or DOX-treated BT mice (Table). The inflam-matory infiltrates included macrophages and T cells asidentified by immunostaining (Figure 3, e and f). There wasalso a mild but appreciable increase in collagen deposition inthe hearts of BT mice (Figure 3, c and d).
To understand the mechanisms involved in our model andestablish whether cardiac ET-1 overexpression was a cause ora consequence of the observed inflammation, we temporallyquantified ET-1 and cytokine gene expression, NF-�B acti-vation, and histological changes in mice after withdrawalfrom DOX suppression. At 2 weeks after DOX removal,human ET-1 mRNA was easily detected in all BT samples,and transgene expression remained elevated at 4, 6, and 8weeks after DOX removal (Figure 4c). Concomitant withET-1 overexpression, nuclear NF-�B translocation was in-creased in cardiomyocytes (Figure 4a). Although detectableat earlier time points, mRNA levels of TNF-�, IL-1, IL-6, andIFN-� did not increase until 6 to 8 weeks after DOXwithdrawal (Figure 4d). Parallel histological analyses dem-onstrated a progressive inflammatory process that was wide-spread by 6 weeks after DOX removal (Figure 4b).
ET Receptor BlockadeThe involvement of ETA and/or ETB receptor pathways in thepathogenesis of this model was explored with orally activeantagonists. The combined ETA/ETB antagonist LU420627(n�8) but not the ETA-selective antagonist LU135252 (n�5)prolonged survival in BT compared with nontreated BT(P�0.025) (Figure 2).
At 4 weeks after DOX removal, myocardial tissue andplasma levels of ET-1 and the extent of NF-�B translocationwas no different in LU420627-treated BT mice (n�3) anduntreated BT mice (tissue, 36.5�1.4 versus 40.1�4.7 fmol/mL;plasma, 2.2�0.14 versus 3.1�2.8 fmol/mL, P�NS). However,the degree of inflammatory cell infiltration was decreased (0
Figure 3. Histological findings. Representative hematoxylin-eo-sin–stained (a and b) and picrosirius red–stained (c and d) car-diac sections. a and c represent a 6-week-old ET�/tTA� mousereleased from DOX after birth. b and d represent an age-matched ET�/tTA� control maintained on DOX. e and f repre-sent immunohistochemical staining for macrophage-specific(Mac-3) and T-cell–specific (CD-3) cell surface markers in anET�/tTA� mouse withdrawn from DOX. g and h represent nega-tive controls without primary antibodies against Mac-3 andCD-3 in same ET�/tTA� mouse.
Histopathologic Scoring
Genotype DOX Inflammation Necrosis Total
ET�/tTA� (n�5) � 0 0 0
ET�/tTA� (n�5) � 1.9�0.8 0.9�0.3 2.7�1.1
CVB3 viral myocarditis 2.7�1.5 3.2�0.7 5.9�2.2
Hematoxylin-eosin–stained cardiac cross sections of ET�/tTA� mice werescored (0–4) for inflammation and necrosis by 2 blinded observers. DOXtreatment completely suppressed histological evidence of cardiac inflammationand necrosis in ET�/tTA� mice. Scores from a viral model of myocarditis (CVB3infection) are shown as a positive control.
258 Circulation January 20, 2004
by guest on February 23, 2013http://circ.ahajournals.org/Downloaded from
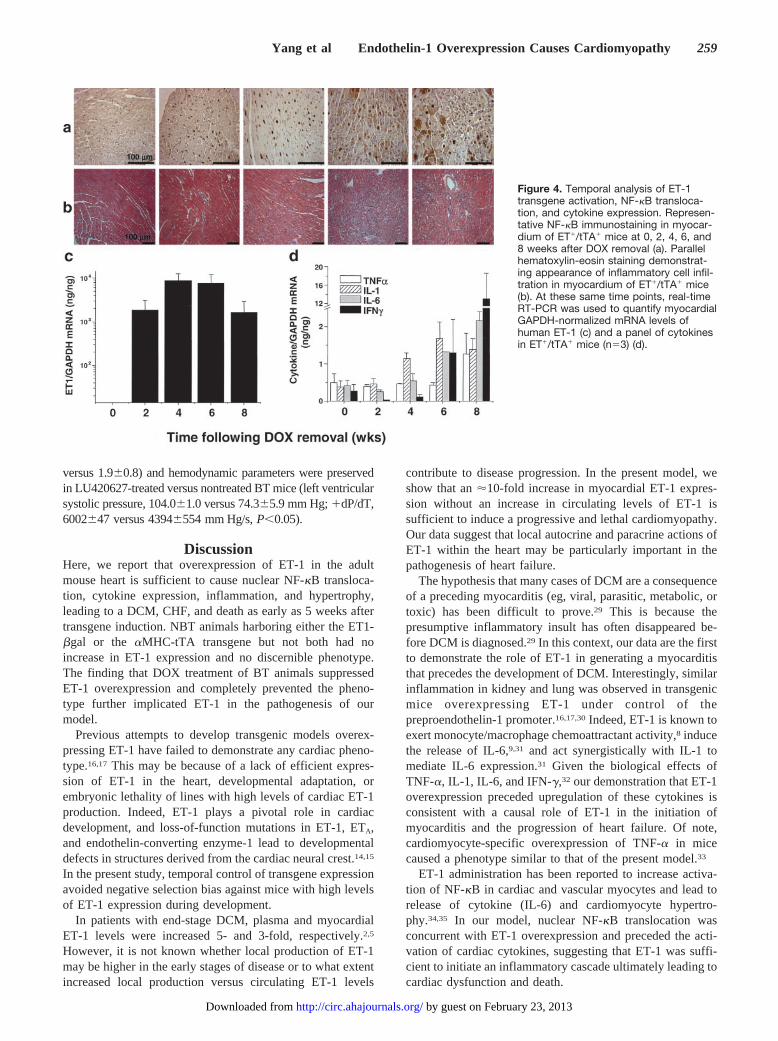
versus 1.9�0.8) and hemodynamic parameters were preservedin LU420627-treated versus nontreated BT mice (left ventricularsystolic pressure, 104.0�1.0 versus 74.3�5.9 mm Hg; �dP/dT,6002�47 versus 4394�554 mm Hg/s, P�0.05).
DiscussionHere, we report that overexpression of ET-1 in the adultmouse heart is sufficient to cause nuclear NF-�B transloca-tion, cytokine expression, inflammation, and hypertrophy,leading to a DCM, CHF, and death as early as 5 weeks aftertransgene induction. NBT animals harboring either the ET1-�gal or the �MHC-tTA transgene but not both had noincrease in ET-1 expression and no discernible phenotype.The finding that DOX treatment of BT animals suppressedET-1 overexpression and completely prevented the pheno-type further implicated ET-1 in the pathogenesis of ourmodel.
Previous attempts to develop transgenic models overex-pressing ET-1 have failed to demonstrate any cardiac pheno-type.16,17 This may be because of a lack of efficient expres-sion of ET-1 in the heart, developmental adaptation, orembryonic lethality of lines with high levels of cardiac ET-1production. Indeed, ET-1 plays a pivotal role in cardiacdevelopment, and loss-of-function mutations in ET-1, ETA,and endothelin-converting enzyme-1 lead to developmentaldefects in structures derived from the cardiac neural crest.14,15
In the present study, temporal control of transgene expressionavoided negative selection bias against mice with high levelsof ET-1 expression during development.
In patients with end-stage DCM, plasma and myocardialET-1 levels were increased 5- and 3-fold, respectively.2,5
However, it is not known whether local production of ET-1may be higher in the early stages of disease or to what extentincreased local production versus circulating ET-1 levels
contribute to disease progression. In the present model, weshow that an �10-fold increase in myocardial ET-1 expres-sion without an increase in circulating levels of ET-1 issufficient to induce a progressive and lethal cardiomyopathy.Our data suggest that local autocrine and paracrine actions ofET-1 within the heart may be particularly important in thepathogenesis of heart failure.
The hypothesis that many cases of DCM are a consequenceof a preceding myocarditis (eg, viral, parasitic, metabolic, ortoxic) has been difficult to prove.29 This is because thepresumptive inflammatory insult has often disappeared be-fore DCM is diagnosed.29 In this context, our data are the firstto demonstrate the role of ET-1 in generating a myocarditisthat precedes the development of DCM. Interestingly, similarinflammation in kidney and lung was observed in transgenicmice overexpressing ET-1 under control of thepreproendothelin-1 promoter.16,17,30 Indeed, ET-1 is known toexert monocyte/macrophage chemoattractant activity,8 inducethe release of IL-6,9,31 and act synergistically with IL-1 tomediate IL-6 expression.31 Given the biological effects ofTNF-�, IL-1, IL-6, and IFN-�,32 our demonstration that ET-1overexpression preceded upregulation of these cytokines isconsistent with a causal role of ET-1 in the initiation ofmyocarditis and the progression of heart failure. Of note,cardiomyocyte-specific overexpression of TNF-� in micecaused a phenotype similar to that of the present model.33
ET-1 administration has been reported to increase activa-tion of NF-�B in cardiac and vascular myocytes and lead torelease of cytokine (IL-6) and cardiomyocyte hypertro-phy.34,35 In our model, nuclear NF-�B translocation wasconcurrent with ET-1 overexpression and preceded the acti-vation of cardiac cytokines, suggesting that ET-1 was suffi-cient to initiate an inflammatory cascade ultimately leading tocardiac dysfunction and death.
Figure 4. Temporal analysis of ET-1transgene activation, NF-�B transloca-tion, and cytokine expression. Represen-tative NF-�B immunostaining in myocar-dium of ET�/tTA� mice at 0, 2, 4, 6, and8 weeks after DOX removal (a). Parallelhematoxylin-eosin staining demonstrat-ing appearance of inflammatory cell infil-tration in myocardium of ET�/tTA� mice(b). At these same time points, real-timeRT-PCR was used to quantify myocardialGAPDH-normalized mRNA levels ofhuman ET-1 (c) and a panel of cytokinesin ET�/tTA� mice (n�3) (d).
Yang et al Endothelin-1 Overexpression Causes Cardiomyopathy 259
by guest on February 23, 2013http://circ.ahajournals.org/Downloaded from

Given the lack of increase in systemic arterial pressures,cardiac hypertrophy of BT mice was probably a result of adirect effect of ET-1 on cardiomyocytes. The hyperplasticand vesiculated nuclei, �-MHC expression, and increasedcardiomyocyte and ventricular size and cardiac mass of ourmodel are consistent with the known effects of chronic ET-1stimulation in vitro.6 The relative decrease in tTA-drivenhuman ET-1 expression at 8 weeks after DOX removal(Figure 4) is also consistent with the downregulation of�-MHC promoter activity that accompanies cardiachypertrophy.36
Although ECG recordings suggested the development ofatrial and ventricular enlargement, they did not demonstratethe arrhythmias noted in another �-MHC-tTA–dependentmodel of cardiac disease.20 Furthermore, concurrent histolog-ical analyses suggested that the ECG findings (ie, nonspecificconduction delays) were attributable to a generalized cardiachypertrophy and/or inflammatory cell infiltration and colla-gen deposition, rather than a specific effect on cardiacconduction tissue.
The present system provided an opportunity to compare theefficacy of ET-receptor antagonists in a model dependent onincreased ET-1 expression. Interestingly, ETA-selective an-tagonism had no effect, whereas combined ETA/ETB antago-nism caused an appreciable delay in mortality. Given thelevels of cardiac ET-1 overproduction in our model, coupledwith the nearly irreversible interaction between ET-1 and itsreceptors,1 it may be difficult for an orally administeredantagonist to compete effectively at the tissue level. Indeed,the partial response to combined ETA/ETB antagonism in ourmodel may provide insight into the limited efficacy of thisclass of agents in clinical trials.11
The mechanisms of how ETA/ETB antagonism amelioratedthe phenotype may be complicated. Reduced inflammatorycell infiltration and improved hemodynamic function ofLU420627-treated BT mice suggested that combined ETA/ETB blockade inhibited leukocyte recruitment and preservedcardiac function. However, the persistence of nuclear NF-�Btranslocation and eventual mortality despite LU420627 treat-ment suggested a partial and transient response. Because the2 compounds tested possess similar Ki for ETA,26,27 thesalutary effect of the combined ETA/ETB antagonist suggesteda more important role for ETB. Indeed, the importance of ETB
in myocardial fibrosis37 and inflammatory models in athero-sclerosis38 and skin39 has been suggested by other studies.Future studies that use more selective ETB receptor antago-nists and temporal analyses of ETA/ETB signaling pathwaysmay directly address this question in our model.
In summary, we have generated transgenic mice withrobust cardiac-specific expression of human ET-1, which wassufficient to induce cardiac inflammation, hypertrophy, dila-tation, dysfunction, and death. Our study has demonstratedthe potential role of ET-1 as a proinflammatory molecule inthe heart and suggests that this effect may be mediated in partby ET-1/ETB interactions. Our model also provides insightsinto the therapeutic potential of blocking ET-1 signalingpathways in heart failure.
AcknowledgmentsDr Yang was supported by a Heart and Stroke Richard Lewar Centreof Excellence/Ontario Graduate School Science and TechnologyStudentship and a Heart and Stroke Foundation of Ontario (HSFO)Doctoral Award. Dr Gros was supported by an HSFO PostdoctoralFellowship. Dr Husain is a recipient of a Clinician Scientist Awardfrom the Canadian Institute of Health Research (CIHR). Dr Stewartholds the Dexler H.C. Man Chair in Cardiology at the University ofToronto. This study was supported in part by CIHR operating grant11620 (Drs Stewart and Husain).
References1. Yanagisawa M, Kurihara H, Kimura S, et al. A novel potent vasocon-
strictor peptide produced by vascular endothelial cells [see comments].Nature. 1988;332:411–415.
2. Stewart DJ, Cernacek P, Costello KB, et al. Elevated endothelin-1 in heartfailure and loss of normal response to postural change. Circulation.1992;85:510–517.
3. Sakai S, Miyauchi T, Sakurai T, et al. Endogenous endothelin-1 partic-ipates in the maintenance of cardiac function in rats with congestive heartfailure: marked increase in endothelin-1 production in the failing heart[see comments]. Circulation. 1996;93:1214–1222.
4. Picard P, Smith PJ, Monge JC, et al. Coordinated upregulation of thecardiac endothelin system in a rat model of heart failure. J CardiovascPharmacol. 1998;31:S294–S297.
5. Pieske B, Beyermann B, Breu V, et al. Functional effects of endothelinand regulation of endothelin receptors in isolated human nonfailing andfailing myocardium. Circulation. 1999;99:1802–1809.
6. Shubeita HE, McDonough PM, Harris AN, et al. Endothelin induction ofinositol phospholipid hydrolysis, sarcomere assembly, and cardiac geneexpression in ventricular myocytes: a paracrine mechanism for myo-cardial cell hypertrophy. J Biol Chem. 1990;265:20555–20562.
7. Rizvi MA, Katwa L, Spadone DP, et al. The effects of endothelin-1 oncollagen type I and type III synthesis in cultured porcine coronary arteryvascular smooth muscle cells. J Mol Cell Cardiol. 1996;28:243–252.
8. Helset E, Ytrehus K, Tveita T, et al. Endothelin-1 causes accumulation ofleukocytes in the pulmonary circulation. Circ Shock. 1994;44:201–209.
9. McMillen MA, Huribal M, Cunningham ME, et al. Endothelin-1increases intracellular calcium in human monocytes and causes produc-tion of interleukin-6. Crit Care Med. 1995;23:34–40.
10. Fraccarollo D, Hu K, Galuppo P, et al. Chronic endothelin receptorblockade attenuates progressive ventricular dilation and improves cardiacfunction in rats with myocardial infarction: possible involvement ofmyocardial endothelin system in ventricular remodeling. Circulation.1997;96:3963–3973.
11. Mylona P, Cleland JG. Update of REACH-1 and MERIT-HF clinicaltrials in heart failure. Cardio.net Editorial Team. Eur J Heart Fail.1999;1:197–200.
12. Yamauchi-Kohno R, Miyauchi T, Hoshino T, et al. Role of endothelin indeterioration of heart failure due to cardiomyopathy in hamsters: increasein endothelin-1 production in the heart and beneficial effect ofendothelin-A receptor antagonist on survival and cardiac function. Cir-culation. 1999;99:2171–2176.
13. Nguyen QT, Cernacek P, Calderoni A, et al. Endothelin A receptorblockade causes adverse left ventricular remodeling but improves pulmo-nary artery pressure after infarction in the rat. Circulation. 1998;98:2323–2330.
14. Kurihara Y, Kurihara H, Oda H, et al. Aortic arch malformations andventricular septal defect in mice deficient in endothelin-1 [see comments].J Clin Invest. 1995;96:293–300.
15. Yanagisawa H, Hammer RE, Richardson JA, et al. Role of endothelin-1/endothelin-A receptor-mediated signaling pathway in the aortic archpatterning in mice [see comments]. J Clin Invest. 1998;102:22–33.
16. Hocher B, Thone-Reineke C, Rohmeiss P, et al. Endothelin-1 transgenicmice develop glomerulosclerosis, interstitial fibrosis, and renal cysts butnot hypertension. J Clin Invest. 1997;99:1380–1389.
17. Shindo T, Kurihara H, Maemura K, et al. Renal damage and salt-dependent hypertension in aged transgenic mice overexpressingendothelin-1. J Mol Med. 2002;80:105–116.
18. Yu Z, Redfern CS, Fishman GI. Conditional transgene expression in theheart. Circ Res. 1996;79:691–697.
19. Benatti L, Bonecchi L, Cozzi L, et al. Two preproendothelin 1 mRNAstranscribed by alternative promoters. J Clin Invest. 1993;91:1149–1156.
260 Circulation January 20, 2004
by guest on February 23, 2013http://circ.ahajournals.org/Downloaded from

20. Mungrue IN, Gros R, You X, et al. Cardiomyocyte overexpression ofiNOS in mice results in peroxynitrite generation, heart block, and suddendeath. J Clin Invest. 2002;109:735–743.
21. Afroze T, Yang LL, Wang C, et al. Calcineurin-independent regulation ofplasma membrane Ca2� ATPase-4 in the vascular smooth muscle cellcycle. Am J Physiol. 2003;285:C88–C95.
22. You XM, Mungrue IN, Kalair W, et al. Conditional expression of adominant-negative c-Myb in vascular smooth muscle cells inhibits arte-rial remodeling after injury. Circ Res. 2003;92:314–321.
23. Gros R, You X, Baggio LL, et al. Cardiac function in mice lacking theglucagon-like peptide-1 receptor. Endocrinology. 2003;144:2242–2252.
24. Opavsky MA, Penninger J, Aitken K, et al. Susceptibility to myocarditisis dependent on the response of �� T lymphocytes to coxsackieviralinfection. Circ Res. 1999;85:551–558.
25. Shioi T, McMullen JR, Tarnavski O, et al. Rapamycin attenuatesload-induced cardiac hypertrophy in mice. Circulation. 2003;107:1664 –1670.
26. Battistini B, Dussault P. Blocking of the endothelin system: thedevelopment of receptor antagonists. Pulm Pharmacol Ther. 1998;11:97–112.
27. Nguyen QT, Cernacek P, Sirois MG, et al. Long-term effects of nonse-lective endothelin A and B receptor antagonism in postinfarction rat:importance of timing. Circulation. 2001;104:2075–2081.
28. Glantz SA. Primer of Biostatistics. 5th ed. New York, NY: McGraw-HillMedical Publishing Division; 2002.
29. Shabetai R. Dilated cardiomyopathy. In: Kanu Chatterjee MDC, KarlinerJ, Parmley WW, et al, eds. Cardiology (An Illustrated Text/Reference).New York, London: JB Lippincott Co; 1999:10.2.
30. Hocher B, Schwarz A, Fagan KA, et al. Pulmonary fibrosis and chroniclung inflammation in ET-1 transgenic mice. Am J Respir Cell Mol Biol.2000;23:19–26.
31. Stankova J, D’Orleans-Juste P, Rola-Pleszczynski M. ET-1 induces IL-6gene expression in human umbilical vein endothelial cells: synergisticeffect of IL-1. Am J Physiol. 1996;271:C1073–C1098.
32. Testa M, Yeh M, Lee P, et al. Circulating levels of cytokines and theirendogenous modulators in patients with mild to severe congestive heartfailure due to coronary artery disease or hypertension. J Am Coll Cardiol.1996;28:964–971.
33. Bryant D, Becker L, Richardson J, et al. Cardiac failure in transgenicmice with myocardial expression of tumor necrosis factor-�. Circulation.1998;97:1375–1381.
34. Hirotani S, Otsu K, Nishida K, et al. Involvement of nuclear factor-�Band apoptosis signal-regulating kinase 1 in G-protein-coupled receptoragonist-induced cardiomyocyte hypertrophy. Circulation. 2002;105:509–515.
35. Browatzki M, Schmidt J, Kubler W, et al. Endothelin-1 inducesinterleukin-6 release via activation of the transcription factor NF-kappaBin human vascular smooth muscle cells. Basic Res Cardiol. 2000;95:98–105.
36. Sheridan DJ, Autelitano DJ, Wang B, et al. Beta(2)-adrenergic receptoroverexpression driven by alpha-MHC promoter is downregulated inhypertrophied and failing myocardium. Cardiovasc Res. 2000;47:133–141.
37. Hocher B, George I, Rebstock J, et al. Endothelin system-dependentcardiac remodeling in renovascular hypertension. Hypertension. 1999;33:816–822.
38. Babaei S, Picard P, Ravandi A, et al. Blockade of endothelin receptorsmarkedly reduces atherosclerosis in LDL receptor deficient mice: role ofendothelin in macrophage foam cell formation. Cardiovasc Res. 2000;48:158–167.
39. Griswold DE, Douglas SA, Martin LD, et al. Endothelin B receptormodulates inflammatory pain and cutaneous inflammation. MolPharmacol. 1999;56:807–812.
Yang et al Endothelin-1 Overexpression Causes Cardiomyopathy 261
by guest on February 23, 2013http://circ.ahajournals.org/Downloaded from


















