
Warstwy azotku tytanu - TiN – początek ery twardych pokryć.
122
Warstwy azotku tytanu - TiN – początek ery twardych pokryć.
Transcript of Warstwy azotku tytanu - TiN – początek ery twardych pokryć.

Warstwy azotku tytanu - TiN
– początek ery twardych pokryć.

!"##
$%
&'(
&(
)
!*+
$&(
,&(-
.#+
/-
0&(#
1&(
2&(
3#
4
! #'#5(
!6#)(# #
73897 &(#
%
!" ' #:;
!!" ((<=
$6')

!
"! ! "# !"$ " %
#$ ! ! #"#
&
' # ! $ !
#"# ! # $
$
& ( ! " $
$! )
#%
(!"$ $ # " ! )
&& "* #
% # & + $
$ ,
! $ "
& &
- # ) # & .
!# /01 $!
$# $ # ! ,
$ $
* $& '
! $! # #!
$&
2 ! $
$ $ $ $
$ , " $
$ ! ! $&
( #
) ! # ,
& ( # ! %
$ " !
$ $! " "
!
! # $ & 3
# ) )$ ! #$
4) "5 "
) ) &
6 $
)$&
2 ! #
$ $

$ 76 7 " $
! #
#" # )
" $!&
2* ! " "$ %
6 # ' $ & 2
$
!" $ , 6
!#
# $ $
! , '&
8$ $ 6
" # "
$& ( $ #
!$ $ $ " %&

4
I Warstwy azotku tytanu.
1. Charakterystyka ogólna
1.1. Dlaczego stosuje si! cienkie warstwy TiN?
Cienkie warstwy azotku tytanu (grubo!": 0,5 µm - 3 µm) na#o$one na powierzchni% narz%dzi i cz%!ci maszyn zwi%kszaj& ich odporno!" na zu$ycie. W rezultacie, zwi%ksza si% czas u$ytkowania narz%dzi, ich $ywotno!", i to od kilku do kilkunastu razy. Tak, najogólniej, mo$na odpowiedzie" na postawione wy$ej pytanie.
Obecne warunki produkcyjne narzucaj& na stosowane narz%dzia kil-ka $&da', których spe#nienie jest niezb%dne dla utrzymania si% w nurcie post%pu technicznego. S& to mianowicie:
• mo$liwo!" wzrostu szybko!ci obróbki,• mo$liwo!" obróbki nowych, trudnoobrabialnych materia#ów,• mo$liwo!" pracy narz%dzia w automatycznych centrach obróbczych, w
zespó# z wieloma innymi narz%dziami - poci&ga to za sob& konieczno!"wzrostu trwa#o!ci, $ywotno!ci i niezawodno!ci narz%dzi.
U$ywane okre!lenie: cienkie warstwy azotku tytanu, zamiennie i równowa$nie z takimi okre!lenieami jak: twarde pokrycia, twarde warstwy jest wynikiem przyzwyczajenia si%nas do korzystania z podzia#u warstw na dwie grupy:
- warstwy cienkie (o grubo!ciach umownie, od kilkudziesi%ciu Å do kilkunastu µm)
- warstwy grube (o grubo!ciach umownie, od kilkunastu do kilkuset µm)
Podzia# ten powsta# we wczesnych etapach rozwoju warstwowej technologii pó#przewodnikowej. Cienkie warstwy otrzymywano zwykle metodami pró$niowymi (naparowywa-nie, rozpylanie), za! warstwy grube mi%dzy innymi metod&sitodruku.
1Å=10-4µm=10-10m

5
Warunki te mog& by" spe#nione poprzez:
• wytwarzanie nowych, doskonalszych materia#ów do konstrukcji narz%-dzi,
• wykorzystanie mo$liwo!ci kszta#towania w#a!ciwo!ci powierzchniowychpod k&tem wzrostu $ywotno!ci narz%dzi,
• w#a!ciwy wybór geometrii cz%!ci roboczych narz%dzi (ostrzenie)• stosowanie odpowiednich !rodków ch#odz&cych i smaruj&cych.
Wspó#czesne materia#y narz%dziowe dzieli si% na cztery grupy:
• stale szybkotn&ce (konwencjonalne i spiekane),• w%gliki spiekane,• ceramik% narz%dziow&,• polikrystaliczne, supertwarde materia#y narz%dziowe.
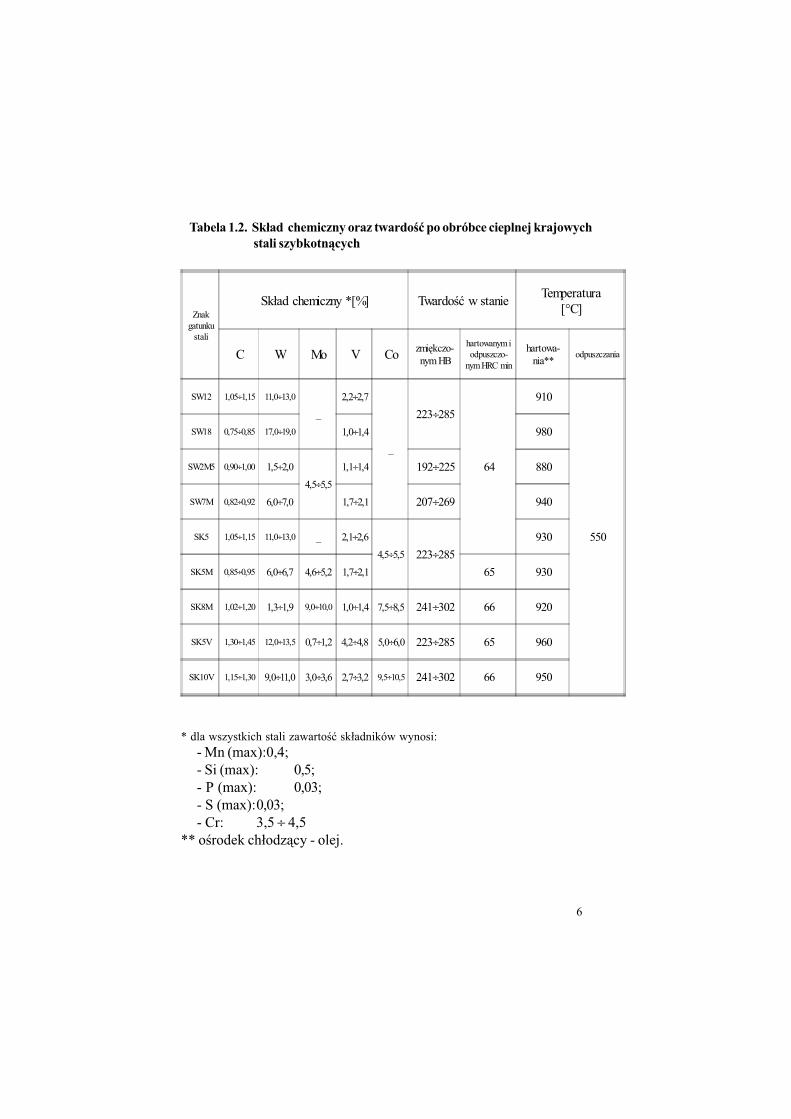
Tabele 1.1 i 1.2 przedstawiaj& wybrane stale szybkotn&ce.
Oznacze-nie stali
Typ stali(W-Mo-V-Co)
Odpowied-nik stali
konwencjo-nalnej
wg PN
Zawarto!", % wag.
C W Mo V Cr Co Ni Si O2
Fe
M2 PM 6-5-2 SW7M 0,87 6,4 5,0 1,9 4,2 - - -
<1000ppm
reszta
M4 PM 6-5-4 1,3 5,7 5,0 4,2 4,5 - - 0,3
M35 PM 6-5-2-5 SK5M 0,9 6,4 5,0 1,9 4,1 4,8 - -
M42 PM 2-10-1-8 1,08 1,5 9,4 1,2 3,9 8,0 - -
T1 PM 18-0-1 SW18 0,96 18 0,2 1,05 4,37 - 0,05 0,16
T15 PM 12-1-5-5 SK5V 1,6 12 <1 5,0 4,0 4,8 - 0,3
T42 PM 10-4-3-10 SK10V 1,4 9,7 4,0 2,4 4,0 10,6 - -
ASP23 PM 6-5-3 1,27 6,4 5,0 3,1 4,2 - - 0,3
ASP30 PM 6-5-3-8 1,28 6,4 5,0 3,1 4,2 8,5 - - 60
ASP60 PM 6-7-6-10 2,3 6,5 7,0 6,5 4,2 10,5 - - 100
M - stal szybkotn&ca molibdenowa, T - stal szybkotn&ca wolframowa, PM - Powder Metallurgy (metalurgia proszkowa)
Tabela 1.1. "redni sk#ad chemiczny typowych spiekanych stali szybkotn$cych.
6
Znakgatunku
stali
Sk#ad chemiczny *[%] Twardo!" w stanieTemperatura
[°C]
C W Mo V Cozmi%kczo-nym HB
hartowanym iodpuszczo-
nym HRC min
hartowa-nia**
odpuszczania
SW12 1,05÷1,15 11,0÷13,0
_
2,2÷2,7
_
223÷285
64
910
550
SW18 0,75÷0,85 17,0÷19,0 1,0÷1,4 980
SW2M5 0,90÷1,00 1,5÷2,0
4,5÷5,5
1,1÷1,4 192÷225 880
SW7M 0,82÷0,92 6,0÷7,0 1,7÷2,1 207÷269 940
SK5 1,05÷1,15 11,0÷13,0 _ 2,1÷2,6
4,5÷5,5 223÷285
930
SK5M 0,85÷0,95 6,0÷6,7 4,6÷5,2 1,7÷2,1 65 930
SK8M 1,02÷1,20 1,3÷1,9 9,0÷10,0 1,0÷1,4 7,5÷8,5 241÷302 66 920
SK5V 1,30÷1,45 12,0÷13,5 0,7÷1,2 4,2÷4,8 5,0÷6,0 223÷285 65 960
SK10V 1,15÷1,30 9,0÷11,0 3,0÷3,6 2,7÷3,2 9,5÷10,5 241÷302 66 950
Tabela 1.2. Sk#ad chemiczny oraz twardo%& po obróbce cieplnej krajowych
stali szybkotn$cych
* dla wszystkich stali zawarto!" sk#adników wynosi:
- Mn (max):0,4;- Si (max): 0,5;- P (max): 0,03;- S (max):0,03;- Cr: 3,5 ÷ 4,5
** o!rodek ch#odz&cy - olej.

7
Wytwarzanie nowych, doskonalszych materia#ów do konstrukcji narz%-dzi zwi&zane jest z ci&g#ym poszukiwaniem, wew&trz wymienionych wy$ejgrup materia#owych, nowych sk#adników, pierwiastków stopowych, opraco-wywaniu w#a!ciwych procedur obróbki cieplnej itd.
Je!li chodzi o drugi z warunków, to dobrym przyk#adem s& cienkiewarstwy TiN nanoszone na cz%!ci robocze powierzchni narz%dzi. Od dawnaby#o wiadomo, $e azotki iw%gliki metali posiadaj& wy-bitn& odporno!" na zu$ycieoraz $e charakteryzuj& si% do-brymi w#a!ciwo!ciami trybo-
logicznymi. Na przeszkodziew ich wykorzystaniu w litej formie stoj&, pomijaj&c cen%, du$a krucho!" i#amliwo!". Znalaz#y one jednak zastosowanie w innej postaci, mianowiciejako sk#adniki proszkowe w grupie narz%dziowej w%glików spiekanych.
Wykorzystanie walorów azotków i w%glików metali przej!ciowych,nie w postaci proszkowej, lecz w formie struktury ci&g#ej, sta#o si% mo$liwedzi%kiopracowaniu technologii nanoszenia ich jako cienkich warstw na ma-teria#y narz%dziowe.
Pierwszym zwi&zkiem, który otrzymano w tej postaci by# TiN. On te$zapocz&tkowa#,pod koniec lat siedemdziesi&tych, prawdziwy przewrót narynku narz%dziowym.
Podstawowe cechy warstw azotku tytanu, które zdecydowa#y o jegosukcesie to:• bardzo du$a twardo!",• odporno!" na !cieranie,• odporno!" na korozj%,• ma#y wspó#czynnik tarcia,• gorsze od stali przewodnictwo cieplne,• z#oty kolor,• niewielka grubo!" (stosuje si% warstwy o grubo!ciach w zakresie od kilku-
dziesi%tnych cz%!ci mikrometra do kilku mikrometrów, co nie stwarzazwykle problemów w tolerancji wymiarów).
Nie bez znaczenia s& równie$ w#a!ciwo!ci chemiczne TiN:• proces utleniania staje si% widoczny po godzinie je!li, warstwa znajduje
si% w atmosferze silnie utleniaj&cej w temperaturze 550°C,• nie reaguje z H2, N2, CO,• jest oboj%tny wobec HCl, HNO3, H2SO4.
TRYBOLOGIA (równie$: tribologia) : (gr. tribo - tr%,pocieram) - nauka o ogóle si# i zjawisk wyst%puj&-cych podczas tarcia.

8
Jednak warstwy TiN:• rozk#adaj& si% w wrz&cych roztworach alkalicznych,• NO oraz O2 dzia#&j& na nie szybko utleniaj&co w temperaturach wy-$szych od 1200°C.
Pokrycia warstwami TiN wykorzystuje si% g#ównie w takich sytuacjachgdzie istniej& problemy z:• tarciem,• erozj&,• korozj&,• zu$yciem !ciernym i adhezyjnym.Podkre!lamy tak$e znaczenie warstw TiN jako pokry" dekoracyjnych.
We(my, jako przyk#ad, zastosowania warstw TiN do narz%dzi skra-waj&cych. Warstwa TiN na#o$ona na ostrza narz%dzi skrawaj&cych spe#nianast%puj&ce role:• redukuje si#y tarcia prowadz&c do lepszego przep#ywu wióra,• zmniejsza zjawisko przypawania obrabianego materia#u (narosty) do ostrza,• zmniejsza zu$ycie kraterowe ostrza,• os#abia mechanizmy mikrop%kni%" ostrza,• dzia#& jako bariera cieplna, której zadaniem jest odprowadzanie ciep#a,
wytworzonego w trakcie obróbki, do wióra - chroni zatem ostrze przedprzegrzaniem, a w konsekwencji przed odpuszczeniem.
Dla wyrobienia sobie opinii na temat korzystnego wp#ywu wy$ejwymienionych w#asno!ci warstw TiN na proces skrawania niech s#u$& po-moc& rysunki 1.1, 1.2, 1.3 i 1.4. Wystarczy rozwa$y" dwie sytuacje: ostrzeniepokryte i ostrze pokryte warstw& TiN oraz porówna" mechanizmy zy$y-cia w tych przypadkach z uwzgl%dnieniem walorów TiN.

9
Rys. 1.1. Ostrze narz!dzia skrawaj$cego oraz opis procesu jego zu'ycia.
Rys. 1.2. Geometryczne wska(niki zu'ycia ostrza no'a tokarskiego (oznaczenie
wg ISO): KT - g#!boko%& maksymalna wy'#obienia powierzchni
natarcia, KM - odleg#o%& najni'ej po#o'onego punktu wy'#obienia od
kraw!dzi skrawaj$cej, KB - szeroko%& wy'#obienia powierzchni
natarcia, KE - skrócenie ostrza, VB - szeroko%& pasma zu'ycia
powierzchni przy#o'enia przy naro'u, VN - maksymalna szeroko%&pasma zu'ycia powierzchni przy#o'enia.
10
Rys. 1.4. Proces skrawania
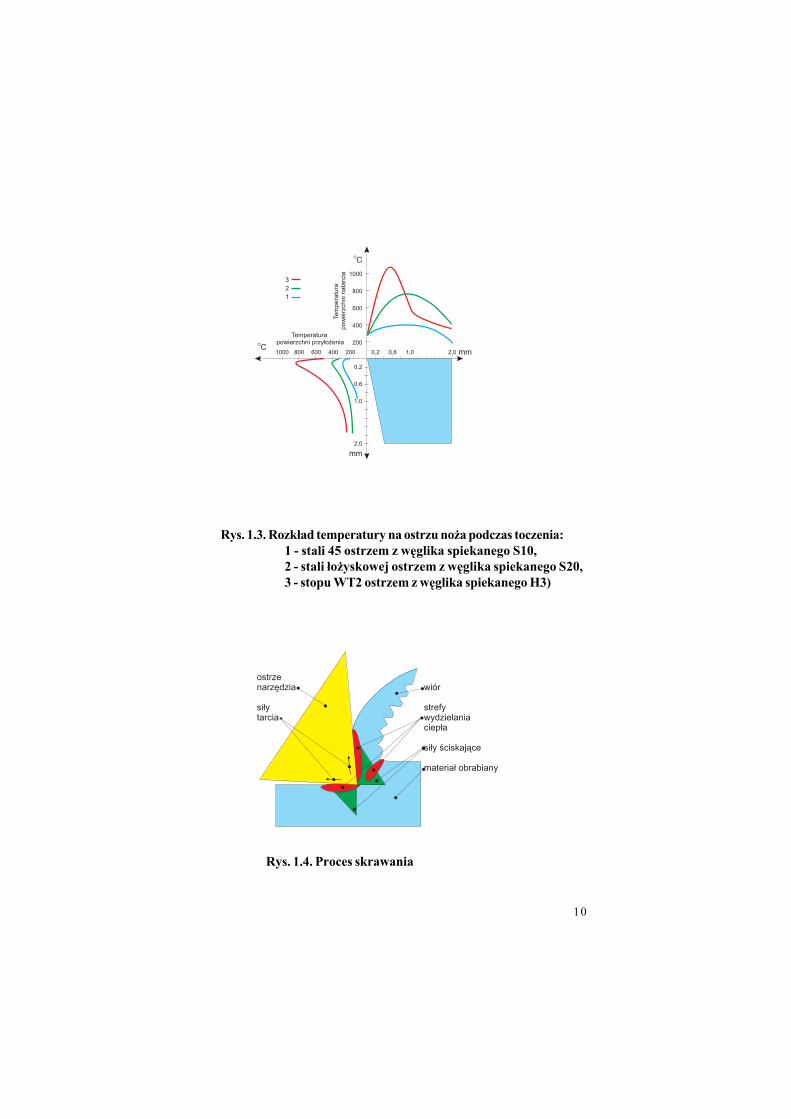
Rys. 1.3. Rozk#ad temperatury na ostrzu no'a podczas toczenia:
1 - stali 45 ostrzem z w!glika spiekanego S10,
2 - stali #o'yskowej ostrzem z w!glika spiekanego S20,
3 - stopu WT2 ostrzem z w!glika spiekanego H3)
11
W wyniku pokrycia warstw& TiN otrzymujemy:• wzrost trwa#o!ci narz%dzia,• mo$liwo!" szybszego skrawania,• lepsz& jako!" obrabianych powierzchni.
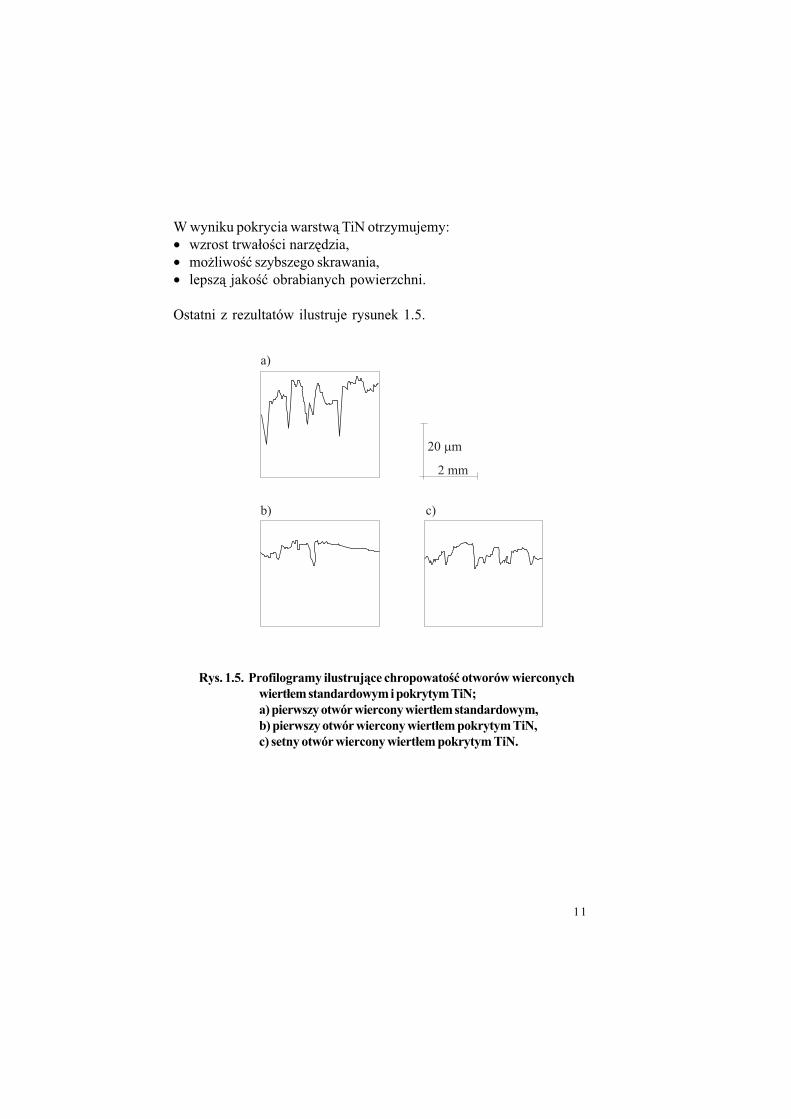
Ostatni z rezultatów ilustruje rysunek 1.5.
Rys. 1.5. Profilogramy ilustruj$ce chropowato%& otworów wierconych
wiert#em standardowym i pokrytym TiN;
a) pierwszy otwór wiercony wiert#em standardowym,
b) pierwszy otwór wiercony wiert#em pokrytym TiN,
c) setny otwór wiercony wiert#em pokrytym TiN.

12
1.2. Troch! ekonomiki
Zestawmy podstawowe zyski i straty p#yn&ce ze stosowania narz%dzipokrytych warstwami TiN.Po stronie strat (wydatków) mamy:• wy$sz& cen% narz%dzi,• konieczno!" stosowania maszyn o wi%kszej sztywno!ci (je!li chcemy zwi%k-
sza" parametry obróbki),• potrzeb% reorganizacji procesu produkcji, zwi&zan& ze znacz&cym wzro-
stem produkcyjno!ci.Po stronie zysków za!:• wi%ksza $ywotno!" narz%dzia (formalnie zast%puje wi%c kilka, kilkana!cie
narz%dzi),• mo$liwo!" zwi%kszania parametrów obróbki,• zmniejszenie ilo!ci ostrze' na jednostk% czasu,• zmniejszenie ilo!ci przerw mi%dzyoperacyjnych w trakcie obróbki zwi&za-
nych z wymian& narz%dzia: ma to szczególne znaczenie gdy narz%dziepracuje jako jednostka (zdarza si%, $e jest "najs#abszym ogniwem") wta!mowym systemie produkcyjnym, lub gdy urz&dzeniem jest centrumobróbcze,
• mo$liwo!" ponownego pokrycia po przeostrzeniu narz%dzia,• mo$liwo!" nieu$ywania ch#odziwa w pewnych operacjach (praca "na
sucho"),• brak konieczno!ci konserwacji przy przechowywaniu narz%dzi,• zwi%kszona jako!" obrabianych powierzchni (w niektórych wypadkach
wyeliminowanie konieczno!ci szlifowania lub rozwiercania ),• wzrost w#asno!ci antykorozyjnych narz%dzi,• zmniejszenie prawdopodobie'stwa tzw. "zimnych napawa'".
Dla zobrazowania korzy!ci p#yn&cych ze stosowania narz%dzi pokry-tych twardymi warstwami rozpatrzmy modelowo diagram op#acalno!ci dlamaszynowej cz%!ci kosztów zmiennych produkcji w przypadku wierceniaotworów na wiertarce kolumnowej.Za#ó$my, $e:1. koszt godziny maszynowej wynosi 50 z#,2. wykorzystujemy dla porównania trzy rodzaje wierte#:
A - wykonane z HSS, szlifowane,B - wykonane z HSS, szlifowane, pokryte TiN,C - wykonane z HSS, szlifowane, pokryte TiCN.
13
3. nie precyzujemy rodzaju obrabianego materia#u,4. wiercenie przy pomocy: wierte# A (HSS) pozwala wykona" 100 otworów,
wierte# B (HSS + TiN) 200 otworów, za! wierte# C (HSS + TiCN) 300otworów w ci&gu jednej godziny,
5. ceny wierte# wynosz& odpowiednio:A - 5 z#,B - 10- z#,C - 20 z#,
6. nie przeostrzamy wierte# (wiert#a pokryte twardymi warstwami po prze-ostrzeniu trac& nieco na walorach, uwzgl%dnianie przeostrzenia wprowa-dzi#oby wi%c nieliniowo!" do rozpatrywanej sytuacji)
7. zwi%kszone ilo!ci otworów otrzymanych w przypadku stosowania wierte#pokrytych TiN czy TiCN w jednostce czasu s& wynikiem zwi%kszonychparametrów obróbki i zmniejszania ilo!ci przerw mi%dzyoperacyjnych.
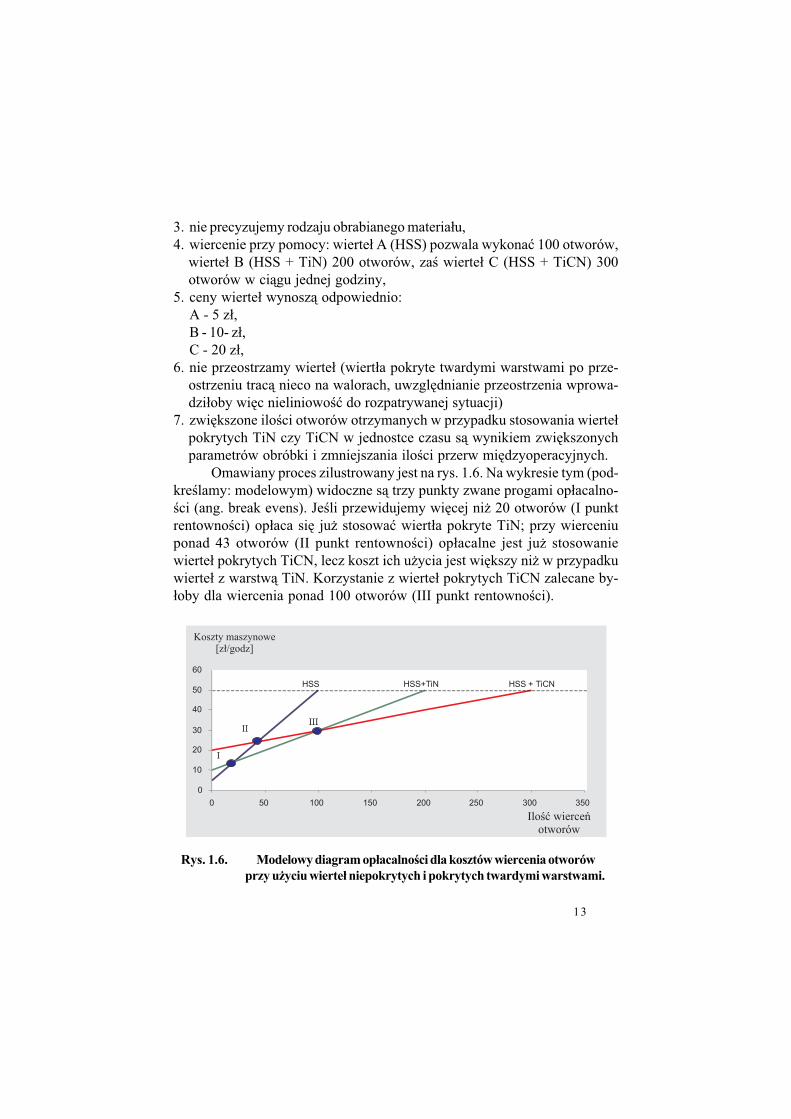
Omawiany proces zilustrowany jest na rys. 1.6. Na wykresie tym (pod-kre!lamy: modelowym) widoczne s& trzy punkty zwane progami op#acalno-!ci (ang. break evens). Je!li przewidujemy wi%cej ni$ 20 otworów (I punktrentowno!ci) op#aca si% ju$ stosowa" wiert#a pokryte TiN; przy wierceniuponad 43 otworów (II punkt rentowno!ci) op#acalne jest ju$ stosowaniewierte# pokrytych TiCN, lecz koszt ich u$ycia jest wi%kszy ni$ w przypadkuwierte# z warstw& TiN. Korzystanie z wierte# pokrytych TiCN zalecane by-#oby dla wiercenia ponad 100 otworów (III punkt rentowno!ci).
Rys. 1.6. Modelowy diagram op#acalno%ci dla kosztów wiercenia otworów
przy u'yciu wierte# niepokrytych i pokrytych twardymi warstwami.

14
W omawianej sytuacji je!li wiercimy na przyk#ad 90 otworów to kosztymaszynowe wynosz& odpowiednio:
dla wierte# typu A - 40 z#,dla wierte# typu B - 25 z#,dla wierte# typu C - 27,5 z#.
Analiza wykresu pozwala sformu#owa" nast%puj&ce spostrze$enia:1. wykres taki jest prosty i #atwy do sporz&dzenia w warunkach produkcyj-
nych dla dowolnego narz%dzia, urz&dzenia i rodzaju obrabianego mate-ria#u.
2. oszcz%dno!ci wynikaj&ce ze stosowania narz%dzi pokrytych twardymiwarstwami rosn& gwa#townie ( co najmniej liniowo ) ze wzrostem ilo!ciwykonywanych przez narz%dzie elementarnych zada' ( wiercenie, frezo-wanie, formowanie itd. ),
3. popularny pogl&d, $e narz%dzia pokryte warto u$ywa" jedynie przy bar-dzo du$ej ilo!ci elementarnych operacji wydaje si% nie by" s#usznym,
4. oszcz%dno!ci rosn&, gdy u$ywa si% urz&dze' o wy$szych kosztach maszy-nowych, np. centra obróbcze,
5. oszcz%dno!ci rosn& w miar% obni$ania cen narz%dzi pokrytych twardymiwarstwami (przesuwanie si% punktów rentowno!ci w lewo).
Zwykle cena narz%dzi pokrytych TiN jest o 40÷80 % wy$sza od na-rz%dzi niepokrytych, za! TiCN tylko nieco wy$sza od pokrytych TiN.Dla wyrazisto!ci przyj%to cen% narz%dzia pokrytego TiN dwukrotniewi%ksz& od niepokrytego, za! pokrytego TiCN czterokrotnie wi%ksz&.Uwzgl%dniaj&c rzeczywiste, ni$sze ceny otrzymamy dalsz& rentowno!".
6. stosuj&c narz%dzia pokryte twardymi warstwami otrzymamy dodatkowokorzy!ci nieuwzgl%dniane przy tworzeniu rysunku, lecz wyszczególnionew zestawieniu zysków.
Zauwa$my na koniec, $e istniej& dwie mo$liwo!ci pracy narz%dziami pokry-tymi twardymi warstwami, obydwie korzystne.Sposób I : pracuj&c tak d#ugo jak narz%dziami niepokrytymi, lecz na zwi%k-
szonych parametrach obróbki wykonujemy w tym samym cza-sie wi%cej detali "nie oszcz%dzaj&c narz%dzia". Sposób ten wy-maga dobrych urz&dze' obróbczych pozwalaj&cych na prac%przy zwi%kszonych parametrach.
Sposób II : pracuj&c na parametrach "normalnych" korzy!ci uzyskujemy"oszcz%dzaj&c narz%dzie" oraz zmniejszaj&c ilo!" przerw mi%dzy-operacyjnych na zmian% narz%dzia.
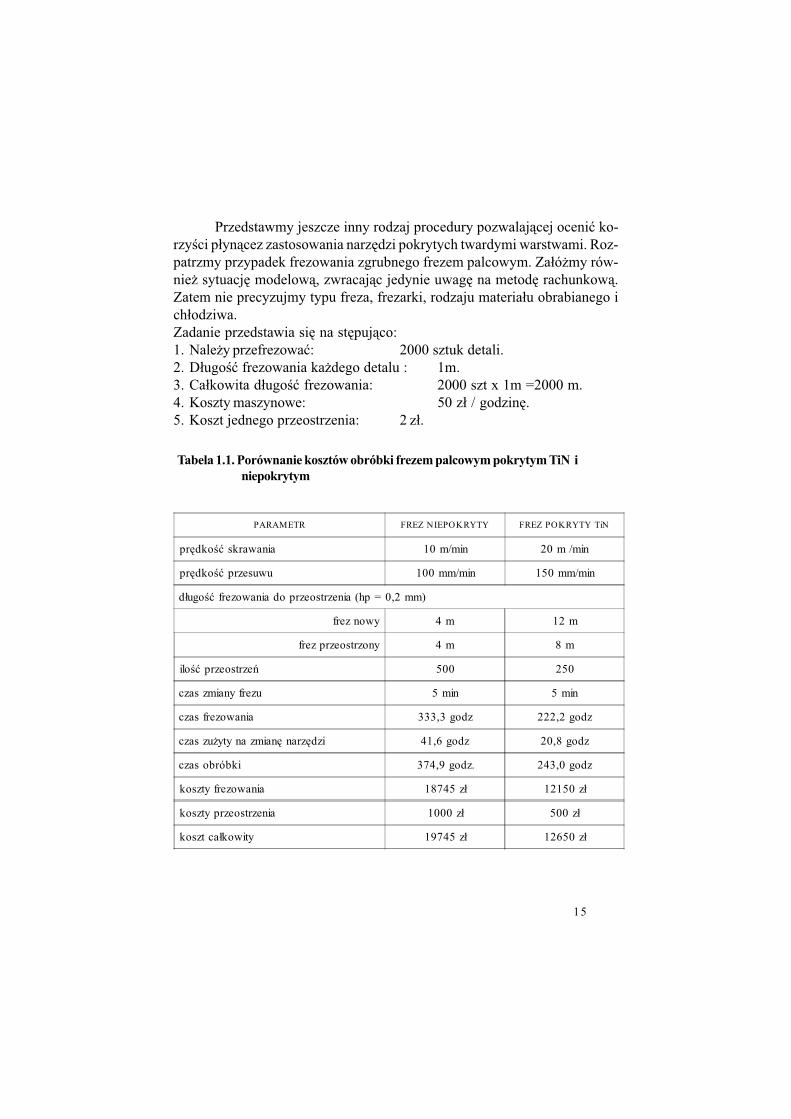
15
Przedstawmy jeszcze inny rodzaj procedury pozwalaj&cej oceni" ko-rzy!ci p#yn&cez zastosowania narz%dzi pokrytych twardymi warstwami. Roz-patrzmy przypadek frezowania zgrubnego frezem palcowym. Za#ó$my rów-nie$ sytuacj% modelow&, zwracaj&c jedynie uwag% na metod% rachunkow&.Zatem nie precyzujmy typu freza, frezarki, rodzaju materia#u obrabianego ich#odziwa.Zadanie przedstawia si% na st%puj&co:1. Nale$y przefrezowa": 2000 sztuk detali.2. D#ugo!" frezowania ka$dego detalu : 1m.3. Ca#kowita d#ugo!" frezowania: 2000 szt x 1m =2000 m.4. Koszty maszynowe: 50 z# / godzin%.5. Koszt jednego przeostrzenia: 2 z#.
PARAMETR FREZ NIEPOKRYTY FREZ POKRYTY TiN
pr%dko!" skrawania 10 m/min 20 m /min
pr%dko!" przesuwu 100 mm/min 150 mm/min
d#ugo!" frezowania do przeostrzenia (hp = 0,2 mm)
frez nowy 4 m 12 m
frez przeostrzony 4 m 8 m
ilo!" przeostrze' 500 250
czas zmiany frezu 5 min 5 min
czas frezowania 333,3 godz 222,2 godz
czas zu$yty na zmian% narz%dzi 41,6 godz 20,8 godz
czas obróbki 374,9 godz. 243,0 godz
koszty frezowania 18745 z# 12150 z#
koszty przeostrzenia 1000 z# 500 z#
koszt ca#kowity 19745 z# 12650 z#
Tabela 1.1. Porównanie kosztów obróbki frezem palcowym pokrytym TiN i
niepokrytym

16
Opis parametrów procesu skrawania frezem pokrytym warstw& TiN i niepo-krytym oraz zestawienie wielko!ci ekonomicznych potrzebnych dla przepro-wadzanego posiadania zestawiono w tabeli (Tab. 1.1.)przy czym parametry okre!lone s& nast%puj&co:• Ilo!" przeostrze' = Ca#kowita d#ugo!" frezowania / D#ugo!" frezowania
do przeostrzeniatutaj: (2000 szt. x 1 m) / 4m = 500
[(2000 szt. x 1 m - 12m)/8]+1 = 250• Czas frezowania = Ca#kowita d#ugo!" frezowania / Pr%dko!" posuwu
tutaj: 2000 m / (100 mm / min x 60) = 333.3• Czas zu$yty na zmian% narz%dzia = Ilo!" ostrze' x Czas zmiany• Koszty frezowania = Czas frezowania x Koszt maszynowy• Koszt przeostrzenia = Ilo!" przeostrze' x Koszt ostrzenia
W omawianym przypadku ró$nica w kosztach wynosi 7095 z#. Pro-centowo za! otrzymujemy warto!" oszcz%dno!ci równ& 56%.
Przedstawiony schemat oblicze', cho" modelowy, zawiera realne pa-rametry. Dla uproszczenia przyj%to jedynie, $e w trakcie pracy nie wymie-niano frezów. St&d du$e ilo!ci przeostrze'. Naturalnie, tak jak poprzednio,warto!ci brane do oblicze' b%d& w ka$dej sytuacji produkcyjnej inne. Sche-mat jest #atwy do sporz&dzenia i mo$e stanowi" podstaw% do decyzji: „u$y-wa" czy nie” narz%dzi pokrytych warstwami TiN.
1.3. Przygotowanie narz!dzi do pokrywania TiN.
Narz%dzia i cz%!ci maszyn, na które ma by" naniesiona cienka war-stwa TiN musz& by" odpowienio przygotowane. Omówmy ten problemprzed przyst&pieniem do opisu technologii warstw.
Poni$ej przedstawiony jest przedruk informacji na ten temat, któredostarczamy potencjalnemu u$ytkownikami narz%dzi pokrytych TiN.
Warunki, jakie powinny spe#ni& narz!dzia i elementy przeznaczone
do pokrywania warstwami TiN
Narz%dzia lub cz%!ci maszyn przeznaczone do pokrywania warstwamiazotku tytanu - TiN - powinny spe#nia" nast%puj&ce warunki:

17
W#a%ciwo%ci materia#u:
• warstwy TiN mog& by" nanoszone na: stale szybkotn&ce, stale narz%-dziowe pracuj&ce na zimno i gor&co, stale nierdzewne, w%gliki, tytan ijego stopy, stopy specjalne, np. monel, NiCr; mo$liwe jest tak$e nak#ada-nie, po specjalnym przygotowaniu warstw TiN na inne pod#o$a,
• mo$na pokrywa" elementy lutowane pod warunkiem, $e lutowie nie za-wiera cynku lub kadmu, jest odporne termicznie do 600°C, nie posiadazamkni%tych por i nie jest zanieczyszczone topnikiem.
Charakterystyka powierzchni:
• powierzchnia elementu powinna by" jasna; w#a!ciwymi metodami ob-róbki ko'cowej s&: szlifowanie, polerowanie, piaskowanie korundem oma#ym ziarnie, elektropolerowanie. Pozosta#o!ci !rodków poleruj&cychnale$y usun&" przy pomocy odpowiednich rozpuszczalników,
• chropowato!" powierzchni roboczych powinna by": Ra < 0,4 µm dlanarz%dzi formuj&cych oraz Ra < 0,2 µm dla narz%dzi skrawaj&cych; za-wsze Rm - minimalne,
• na powierzchni elementów nie powinny wyst%powa" przypalenia szlifir-skie,
• elementy powinny by" pozbawione resztek soli hartowniczych, a tak$epozosta#o!ci materia#u opakowania (kleje, woski, itp.),
• elementy musz& by" pozbawione wszelkich pokry" takich jak np. elek-trolitycznie naniesiony Ni, Cr, Sn czy Zn; winny by" wolne od rdzy,pozosta#o!ci farby czy barwnych oznakowa',
• warstwy TiN mo$na nanosi" na elementy naazotowane,• mo$na pokrywa" powierzchnie wewn%trzne elementów o stosunku sze-
roko!ci do ich g#%boko!ci jak 1:1.
Dane, które nale'y za#$czy& dodatkowo:
• przeznaczenie elementów i sposób ich pracy,• rodzaj i gatunek materia#u oraz jego oznaczenie i temperatur% odpuszcza-
nia,• w przypadku stali naazotowanych nale$y poda" metod% azotowania: k&-
pielow&, gazow& lub plazmow& oraz charakterystyk% azotowania,

18
• wymiary elementów (rysunek lub szkic) wraz ze wskazaniem powierzchni, które musz& by" bezwzgl%dnie pokryte TiN lub te$ pozosta" bezwzgl%dnie niepokryte.
UWAGA:Nale$y w miar% mo$liwo!ci, przewidzie" sposób mocowania elemen-tów w napylarce pró$niowej - konieczno!" wykonania np. otworów, zeszlifowa' technologicznych, itp.
Komentuj&c powy$sz& informacj%, zwró"my uwag% w szczególno!ci na trzy elementy zwi&zane z przygotowaniem powierzchni elementu na któ-ry naniesiona ma by" warstwa TiN, a mianowicie: $&danie podania tempe-ratury odpuszczania dla stali, czysto!" powierzchni oraz jej chropowato!". )&dania te zwi&zane s&, z bodaj czy nie najwa$niejszym problemem w tech-nologii cienkich warstw - przyleganiem warstwy do pod#o$a, inaczej adhe-zj& warstwy. Pomocne przy tym b%d&: rysunek 1.7 opisuj&cy modelowo war-stw% przypowierzchniow& materia#u oraz krótki przypis o sposobach cha-rakteryzowania chropowato!ci powierzchni cia#a sta#ego.
Rys. 1.7. Schematyczne przedstawienie struktury obszaru
przypowierzchniowego.
19
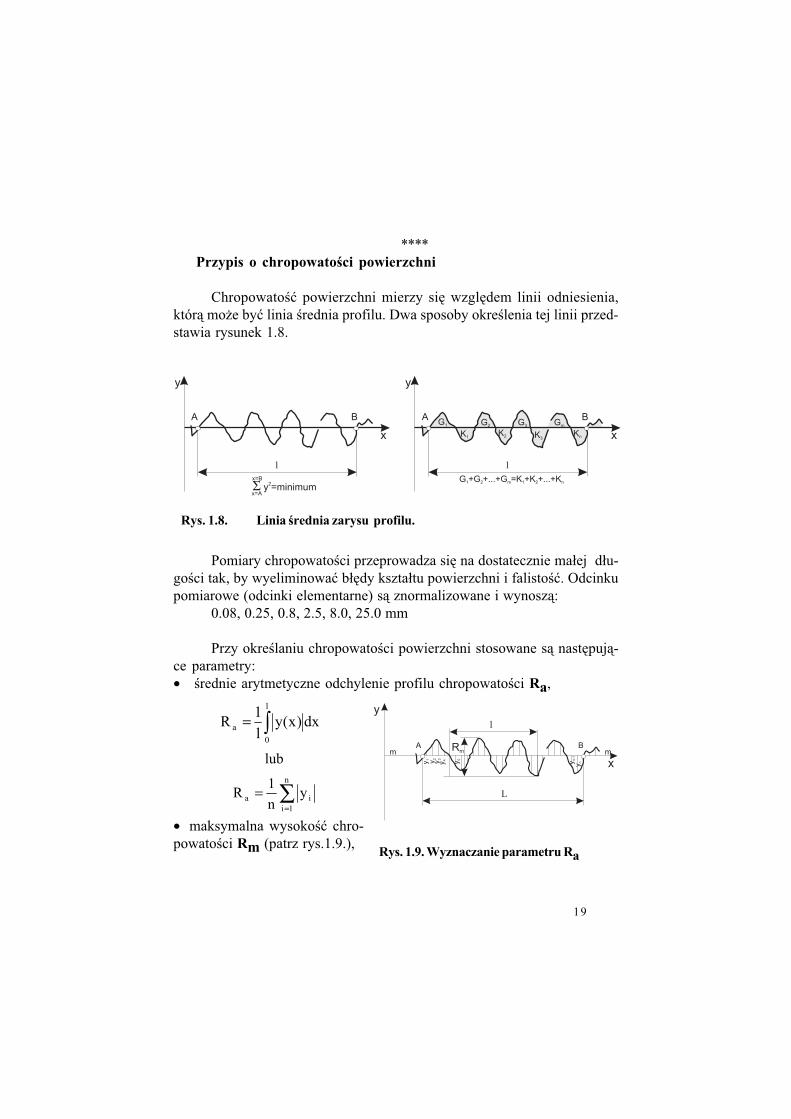
****Przypis o chropowato%ci powierzchni
Chropowato!" powierzchni mierzy si% wzgl%dem linii odniesienia,któr& mo$e by" linia !rednia profilu. Dwa sposoby okre!lenia tej linii przed-stawia rysunek 1.8.
Pomiary chropowato!ci przeprowadza si% na dostatecznie ma#ej d#u-go!ci tak, by wyeliminowa" b#%dy kszta#tu powierzchni i falisto!". Odcinkupomiarowe (odcinki elementarne) s& znormalizowane i wynosz&:
0.08, 0.25, 0.8, 2.5, 8.0, 25.0 mm
Przy okre!laniu chropowato!ci powierzchni stosowane s& nast%puj&-ce parametry:• !rednie arytmetyczne odchylenie profilu chropowato!ci Ra,
∫=l
0
a dx)x(yl
1R
lub
∑=
=n
1iia y
n
1R
• maksymalna wysoko!" chro-powato!ci Rm (patrz rys.1.9.),
Rys. 1.8. Linia %rednia zarysu profilu.
Rys. 1.9. Wyznaczanie parametru Ra
20
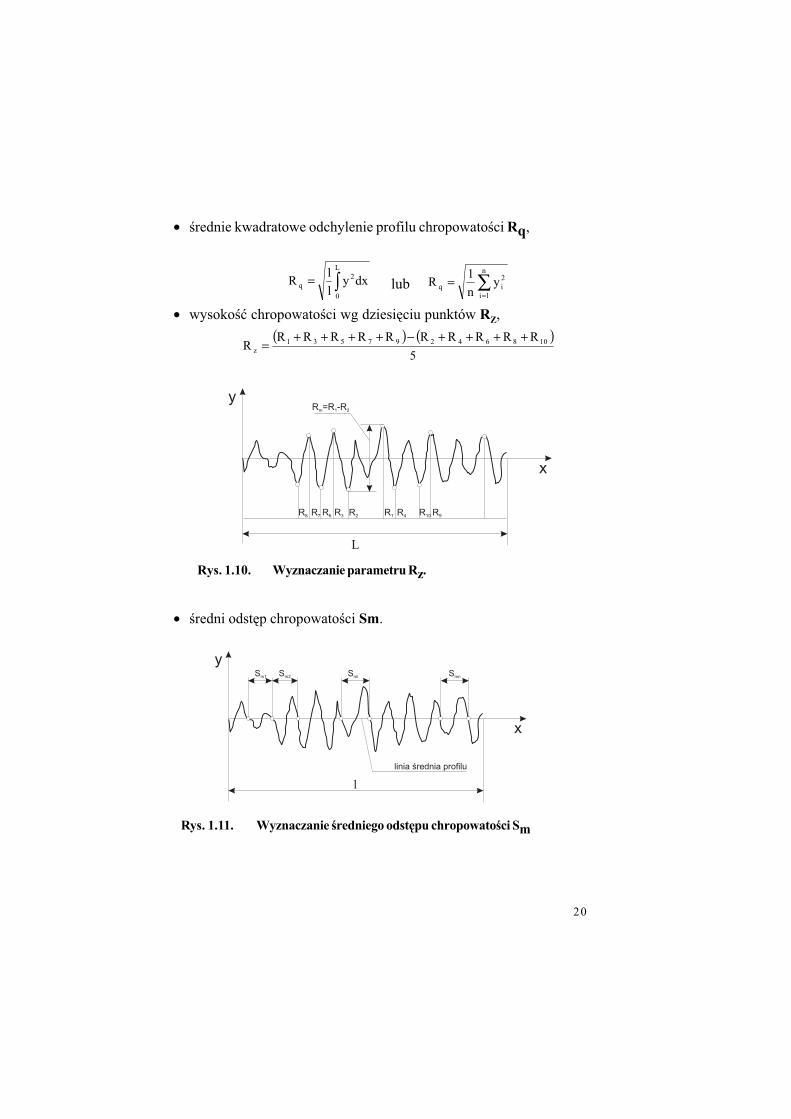
• !rednie kwadratowe odchylenie profilu chropowato!ci Rq,
∫=L
0
2q dxy
l
1R lub ∑
=
=n
1i
2iq y
n
1R
• wysoko!" chropowato!ci wg dziesi%ciu punktów RZ,
( ) ( )
5
RRRRRRRRRRR 10864297531
z
++++−++++=
• !redni odst%p chropowato!ci Sm.
Rys. 1.10. Wyznaczanie parametru Rz.
Rys. 1.11. Wyznaczanie %redniego odst!pu chropowato%ci Sm

21
Do pomiaru chropowato!ci stosuje si% nast%puj&ce me-tody:• metody oceny chropo-
wato!ci za pomoc& wzor-ców,
• metody przekrojów po-wierzchni,
• mikrointerferencyjne metody odwzorowania powierzchni,
• stykowe metody odwzo-rowania lub pomiaru profilu powierzchni.
Oznaczenia stanu powierzchni przedmiotów obrabianych.
Przyk#ad pe#nego oznaczenia stanu powierzchni przedstawia rysunek 1.13. Jako warto!" liczbow& dopuszczalnych chropowato!ci powierzchni przyj-muje si% warto!ci Ra lub Rz.
Rys. 1.12. Idea pomiaru profilografem.
Rys. 1.13. Przyk#ad pe#nego oznaczenia stanu powierzchni przedmiotu
obrabianego.
22
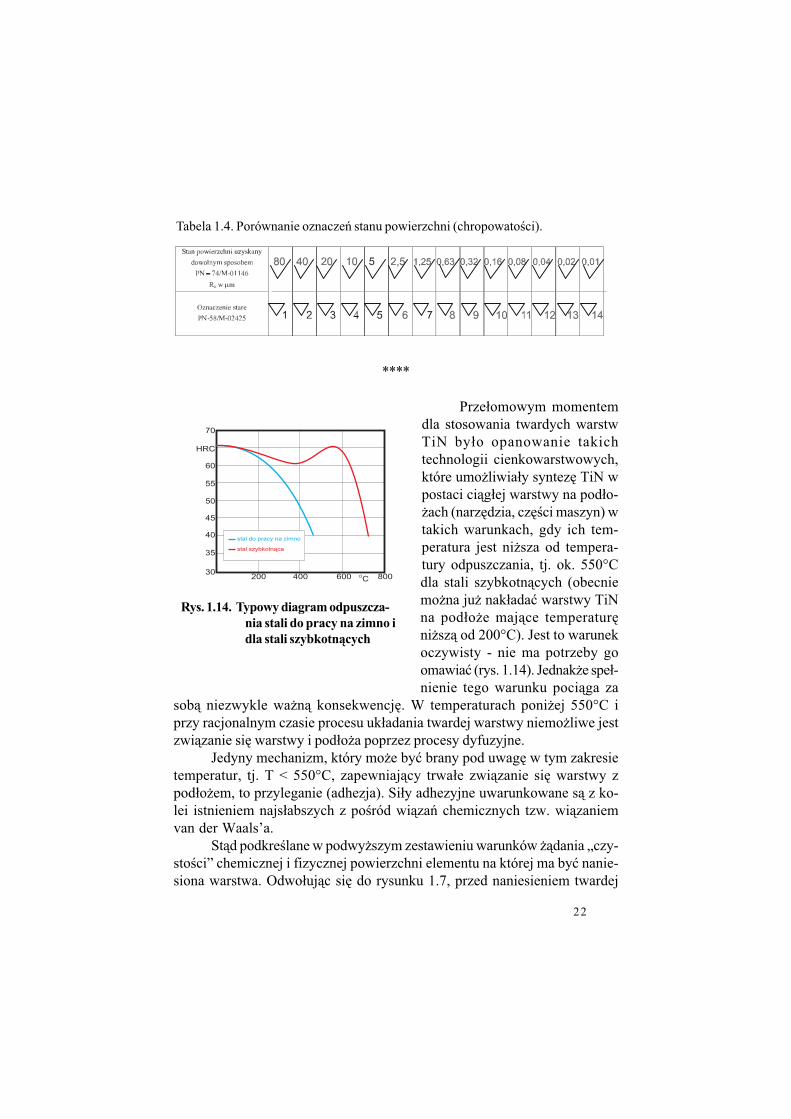
Tabela 1.4. Porównanie oznacze' stanu powierzchni (chropowato!ci).
****
Prze#omowym momentemdla stosowania twardych warstwTiN by#o opanowanie takichtechnologii cienkowarstwowych,które umo$liwia#y syntez% TiN wpostaci ci&g#ej warstwy na pod#o-$ach (narz%dzia, cz%!ci maszyn) wtakich warunkach, gdy ich tem-peratura jest ni$sza od tempera-tury odpuszczania, tj. ok. 550°Cdla stali szybkotn&cych (obecniemo$na ju$ nak#ada" warstwy TiNna pod#o$e maj&ce temperatur%ni$sz& od 200°C). Jest to warunekoczywisty - nie ma potrzeby goomawia" (rys. 1.14). Jednak$e spe#-nienie tego warunku poci&ga za
sob& niezwykle wa$n& konsekwencj%. W temperaturach poni$ej 550°C iprzy racjonalnym czasie procesu uk#adania twardej warstwy niemo$liwe jestzwi&zanie si% warstwy i pod#o$a poprzez procesy dyfuzyjne.
Jedyny mechanizm, który mo$e by" brany pod uwag% w tym zakresietemperatur, tj. T < 550°C, zapewniaj&cy trwa#e zwi&zanie si% warstwy zpod#o$em, to przyleganie (adhezja). Si#y adhezyjne uwarunkowane s& z ko-lei istnieniem najs#abszych z po!ród wi&za' chemicznych tzw. wi&zaniemvan der Waals’a.
St&d podkre!lane w podwy$szym zestawieniu warunków $&dania „czy-sto!ci” chemicznej i fizycznej powierzchni elementu na której ma by" nanie-siona warstwa. Odwo#uj&c si% do rysunku 1.7, przed naniesieniem twardej
Rys. 1.14. Typowy diagram odpuszcza-
nia stali do pracy na zimno i
dla stali szybkotn$cych

23
pow#oki nale$y usun&" wszystkie warstwy zewn%trzne pod#o$a a$ do powierzch-ni czystego fizycznie materia#u.
Je!li chodzi o chropowato!" powierzchni to nale$y zwróci" uwag% nadwa istotne problemy: trwa#o!" po#&czenia pow#oki z pod#o$em oraz trwa-#o!" warstwy w warunkach eksploatacji.
Trwa#o!" po#&czenia warstwy z pod#o$em zale$y nie tylko od energiiwi&zania van der Waals’a mi%dzy atomami materia#ów pod#o$a i warstwy.Zale$y równie$ od, nazwijmy to, „zakleszczania” si% warstwy w nierówno-!ciach pod#o$a. Efekt ten znacznie zwi%ksza odporno!" warstwy na zewn%trz-ne napr%$enia !cinaj&ce. Zatem rozwini%cie powierzchni sprzyja silniejsze-mu powi%kszaniu si% warstwy z pod#o$em. St&d te$ wywodzi si%, do!wiad-czalnie osi&gni%ty warunek Ra < 0,2 µm.
Jednak$e istnienie nawet stosunkowo niewielkiej ilo!ci du$ych nie-równo!ci powierzchni (Ra mo$e pozosta" ma#ym, poniewa$ jest obliczaneprzez ca#kowanie na stosunkowo du$ym odcinku) mo$e doprowadzi" dodegradacji warstwy i ods#oni%cia pod#o$a (ilustruje to rysunek 1.15.) Ods#o-ni%te bowiem pod#o$e staje si% potencjalnym o!rodkiem ataku korozji, cie-p#a, erozji itd., w efekcie czego nast%puje z#uszczanie si% warstwy.
Rys. 1.15. Fragment pod#o'a z naniesion$ tward$ warstw$
24
1.4. Jak otrzymuje si! warstwy TiN
Przed przyst&pieniem do opisu technologii cienkich warstw dokona-my pewnego ogólnego wprowadzenia, którego celem b%dzie przywo#anieniezb%dnego aparatu poj%ciowego. W szczególno!ci zwrócimy uwag% na pro-cesy odbywaj&ce si% na granicy dwóch faz, sta#ej i gazowej.
W istocie swojej bowiem wszystkie technologie cienkowarstwowe wy-korzystywane do nanoszenia warstw TiN to, z fizycznego punktu widzenia,sterowane i kontrolowane procesy oddzia#ywania powierzchni fazy sta#ej -pod#o$a, (a w trakcie nanoszenia warstwy - powierzchni warstwy), z faz&gazow& (!ci!lej z plazm&), Plazma bowiem jest no!nikiem sk#adników war-stwy, a tak$e innych atomów koniecznych dla procesu. Dodajmy od razu,co wa$ne, $e procesy nak#adania cienkich warstw TiN odbywaj& si%, w prze-wa$aj&cej wi%kszo!ci, w warunkach obni$onego ci!nienia - pró$ni.
Zajmijmy si% zatem, pokrótce si#& rzeczy, zapowiadanym aparatempoj%ciowym.
****Pró'nia
Dla naszych potrzeb okre!lmy pró$ni% jako ograniczony obszar, wktórym istnieje gaz lub plazma pod obni$onym, wzgl%dem atmosferyczne-go, ci!nieniem.
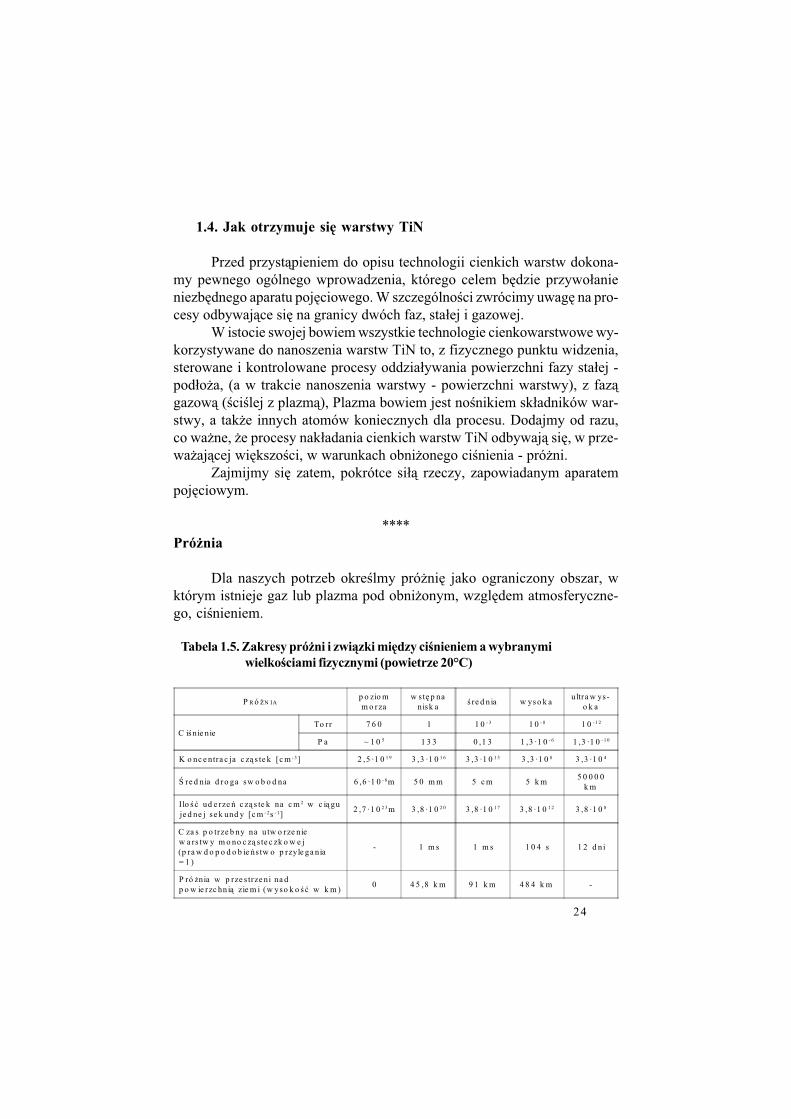
Tabela 1.5. Zakresy pró'ni i zwi$zki mi!dzy ci%nieniem a wybranymi
wielko%ciami fizycznymi (powietrze 20°C)
P R ó $N IAp o zio mm o rza
w st% p nanisk a
! re d n ia w ys o k aultra w ys-
o k a
C i!nie nieTo rr 7 6 0 1 1 0 - 3 1 0 - 8 1 0 - 1 2
P a ~ 1 0 5 1 3 3 0 ,1 3 1 ,3 ·1 0 - 6 1 ,3 ·1 0 - 1 0
K o nc e ntra c ja c z& s te k [c m - 3 ] 2 ,5 ·1 0 1 9 3 ,3 ·1 0 1 6 3 ,3 ·1 0 1 3 3 ,3 ·1 0 8 3 ,3 ·1 0 4
* re d nia d ro ga sw o b o d na 6 ,6 ·1 0 - 8m 5 0 m m 5 c m 5 k m5 0 0 0 0
k m
Ilo ! " ud e rze ' c z& s te k na c m 2 w c i& guje d ne j se k und y [c m - 2s - 1]
2 ,7 ·1 0 2 3 m 3 ,8 ·1 0 2 0 3 ,8 ·1 0 1 7 3 ,8 ·1 0 1 2 3 ,8 ·1 0 8
C za s p o trze b ny na u tw o rze niew a rs tw y m o no c z& s te c zk o w e j(p ra w d o p o d o b ie 's tw o p rzyle ga nia= 1 )
- 1 m s 1 m s 1 0 4 s 1 2 d n i
P ró $nia w p rze s trze ni na dp o w ie rzc hn i& zie m i (w yso k o ! " w k m )
0 4 5 ,8 k m 9 1 k m 4 8 4 k m -
25
Pró$nia jest przedmiotem wielu dzia#ów nauki, techniki oraz technologiistanowi&c zagadnienie tak rozleg#e, $e nie pozostaje nic innego jak tylkoodes#anie zainteresowanego czytelnika do klasycznych pozycji ksi&$kowychcytowanych w za#&czonej bibliografii.
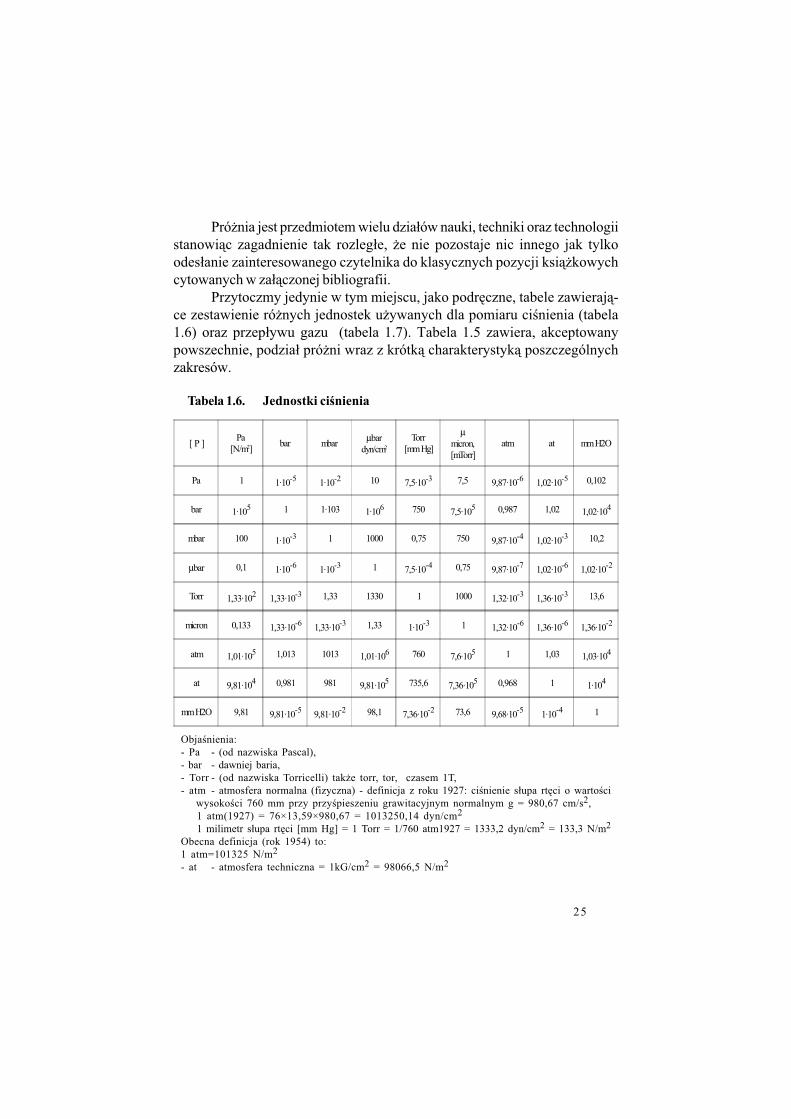
Przytoczmy jedynie w tym miejscu, jako podr%czne, tabele zawieraj&-ce zestawienie ró$nych jednostek u$ywanych dla pomiaru ci!nienia (tabela1.6) oraz przep#ywu gazu (tabela 1.7). Tabela 1.5 zawiera, akceptowanypowszechnie, podzia# pró$ni wraz z krótk& charakterystyk& poszczególnychzakresów.
[ P ]Pa
[N/m2]bar mbar
µbardyn/cm2
Torr[mm Hg]
µmicron,[mTorr]
atm at mm H2O
Pa 1 1·10-5 1·10-2 10 7,5·10-3 7,5 9,87·10-6 1,02·10-5 0,102
bar 1·105 1 1·103 1·106 750 7,5·105 0,987 1,02 1,02·104
mbar 100 1·10-3 1 1000 0,75 750 9,87·10-4 1,02·10-3 10,2
µbar 0,1 1·10-6 1·10-3 1 7,5·10-4 0,75 9,87·10-7 1,02·10-6 1,02·10-2
Torr 1,33·102 1,33·10-3 1,33 1330 1 1000 1,32·10-3 1,36·10-3 13,6
micron 0,133 1,33·10-6 1,33·10-3 1,33 1·10-3 1 1,32·10-6 1,36·10-6 1,36·10-2
atm 1,01·105 1,013 1013 1,01·106 760 7,6·105 1 1,03 1,03·104
at 9,81·104 0,981 981 9,81·105 735,6 7,36·105 0,968 1 1·104
mm H2O 9,81 9,81·10-5 9,81·10-2 98,1 7,36·10-2 73,6 9,68·10-5 1·10-4 1
Tabela 1.6. Jednostki ci%nienia
Obja!nienia:- Pa - (od nazwiska Pascal),- bar - dawniej baria,- Torr - (od nazwiska Torricelli) tak$e torr, tor, czasem 1T,- atm - atmosfera normalna (fizyczna) - definicja z roku 1927: ci!nienie s#upa rt%ci o warto!ci
wysoko!ci 760 mm przy przy!pieszeniu grawitacyjnym normalnym g = 980,67 cm/s2,1 atm(1927) = 76×13,59×980,67 = 1013250,14 dyn/cm2
1 milimetr s#upa rt%ci [mm Hg] = 1 Torr = 1/760 atm1927 = 1333,2 dyn/cm2 = 133,3 N/m2
Obecna definicja (rok 1954) to:1 atm=101325 N/m2
- at - atmosfera techniczna = 1kG/cm2 = 98066,5 N/m2
26
****
Stany materii
Za Empedoklesem z Agrigentum w Grecji (490-430 p.n.e.) uwa$amyobecnie, $e otaczaj&ca nas materia wyst%puje w czterech stanach, fazach -sta#ym, ciek#ym, gazowym oraz jako plazma. W nomenklaturze Empedokla-desa by#y to cztery pierwiastki ($ywio#y): ziemia, woda, powietrze i ogie'.Interesuj&cym jest, $e nowo$ytna nauka zda#a sobie spraw% z istnienia czwar-tego stanu materii dopiero w XIX wieku (M. Faraday, H. Crookes), sam za!termin plazma, na jego okre!lenie, zosta# wprowadzony przez I. Langmu-ir`a w 1923r.Na rysunku 1.16, przytoczony jest tzw. wykres fazowy w osiach: ci!nienie -temperatura.
Na wykresie tym, przesuwaj&c si% wzd#u$ strza#ki, od punktu 1 dopunktu 2 przechodzimy, w miar% wzrostu temperatury, przez wszystkie sta-ny od fazy sta#ej do plazmy. Wzrost temperatury jest to$samy z dostarcza-niem do danej substancji ciep#a, które z kolei okre!la jego energi% kinetycz-n&, jego sk#adników.
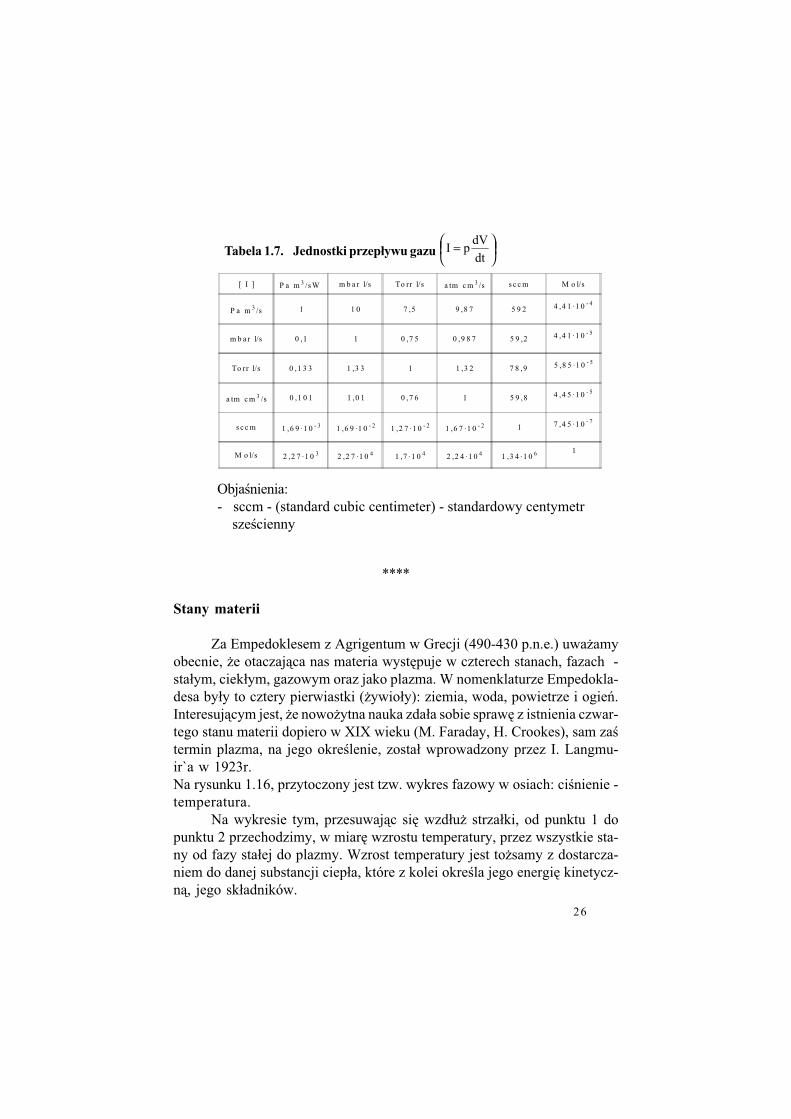
Tabela 1.7. Jednostki przep#ywu gazu
=
dt
dVpI
[ I ] P a m 3 /s W m b a r l/s To rr l/s a tm c m 3 /s s c c m M o l/s
P a m 3 /s 1 1 0 7 ,5 9 ,8 7 5 9 2 4 ,4 1 ·1 0 - 4
m b a r l/s 0 ,1 1 0 ,7 5 0 ,9 8 7 5 9 ,2 4 ,4 1 ·1 0 - 5
To rr l/s 0 ,1 3 3 1 ,3 3 1 1 ,3 2 7 8 ,9 5 ,8 5 ·1 0 - 5
a tm c m 3 /s 0 ,1 0 1 1 ,0 1 0 ,7 6 1 5 9 ,8 4 ,4 5 ·1 0 - 5
s c c m 1 ,6 9 ·1 0 - 3 1 ,6 9 ·1 0 - 2 1 ,2 7 ·1 0 - 2 1 ,6 7 ·1 0 - 2 1 7 ,4 5 ·1 0 - 7
M o l/s 2 ,2 7 ·1 0 3 2 ,2 7 ·1 0 4 1 ,7 ·1 0 4 2 ,2 4 ·1 0 4 1 ,3 4 ·1 0 6 1
Obja!nienia:- sccm - (standard cubic centimeter) - standardowy centymetr
sze!cienny

27
Ka$da zmiana stanu, jest tzw. przej!ciem fazo-wym I rodzaju, i wi&$e si% z dodatkowym poch#anianiem ciep#a.
W przypadku przej-!cia cia#o sta#e - ciecz jest to ciep#o topnienia, dla przej-!cia ciecz - gaz ciep#o paro-wania, za! przej!cie gaz -plazma zwi&zane jest z ko-nieczno!ci& dostarczenia do gazu dodatkowej energii na jego jonizacj%.
Na rysunku 1.17 pokazane s& schematycznie zakresy wyst%powania poszczególnych faz w funkcji temperatury i energii przypadaj&cej na jedn&cz&steczk% (lub atom) w danej fazie. Obszar plazmy na wykresie odpowiada, !ci!le rzecz bior&c, tak zwanej plazmie zimnej.
O tym jak& faz% tworzy dany zbiór atomów lub cz&steczek decyduje stosunek ich energii kinetycznej do energii potencjalnej wzajemnego od-dzia#ywania.
W ciele sta#ym, u#o$one periodycznie w sieci krystalicznej atomy lub cz&steczki oddzia#uj& wzajemnie poprzez si#y kohezji - inaczej spójno!ci.
Termin CZ)STKA obejmuje:cz&steczki (moleku#y), ato-my, jony, elektrony.
Rys. 1.16. Wykres fazowy
Rys. 1.17. Zakresy: temperatury i energetyczny
wyst!powania poszczególnych faz materii

28
Tutaj energia potencjalnaznacznie przewy$sza energi%kinetyczn& jak& te cz&stkiposiadaj&.W cieczy cz&steczki nie s&ju$ u#o$one w regularnej sie-ci, lecz nadal oddzia#ywuj&na siebie s#abymi si#amispójno!ci zwanymi si#amivan der Walls'a. Ich ener-gia kinetyczna jest niecomniejsza od potencjalnej,lecz przy dostatecznie du$ejtemperaturze mo$e j& prze-wy$szy" - ma to miejsce wtemperaturze wrzenia. Wte-dy ciecz przechodzi w gaz.W fazie gazowej (gaz ideal-ny) nie ma $adnego oddzia-#ywania mi%dzy cz&steczka-mi. S& one ca#kowicie nie-
zale$ne. Ich energia kinetyczna wzrasta w miar% wzrostu temperatury.
****
Plazma
Przy dalszym wzro!cie temperatu-ry energia kinetyczna cz&stek gazu (ato-mów, cz&steczek) mo$e by" wystarczaj&-ca do tego by w trakcie zderze' nast&pi-#a ich jonizacja. Powstaj&ce, dodatniona#adowane jony i elektrony oddzia#uj&na siebie si#ami elektrostatycznymi. S&to si#y daleko - zasi%gowe, tak $e ka$da zcz&stek (jon lub elektron) znajduje si% wka$dej chwili w polu elektrycznym wy-twarzanym przez pozosta#e cz&stki. Two-rzy si% w ten sposób pewne uporz&dko-
PLAZMA - #ac. plasma, gr. plásma = co!ulepszonego, twór. S#owo to po razpierwszy wyst&pi#o w biologii jakocz%!" terminu protoplazma, oznacza-j&cego substancj% $yw& komórek ro-!linnych i zwierz%cych. (J.E. Purkine,Czechy 1839r.).PLAZMA - fizjol. - p#ynna cz%!" krwi,sk#adaj&ca si% z 90% wody i zwi&z-ków organicznych g#ównie bia#ek.
Jednostki temperatury, energii, cz%stotliwo!ci i d#u-go!ci fali elektromagnetycznej mo-$emy wzajemnieporównywa" wyra$aj&c jedne przez drugie. Korzy-stamy przy tym z formu#:
λ=ν==
chheUkT ,
deg
J1038054,1k 23−⋅= - sta#a Boltzmana,
sJ106256,6h 34 ⋅⋅= − - sta#a Plancka,
s
m109979,2c 8⋅= - pr%dko!" !wiat#a w pró$ni
C106021,1e 19−⋅= - #adunek elementarny+atwo wyliczy", $e energii termicznej elektronu rów-nej 1 eV odpowiada:
)Hz(s
1104184,2eV1
K11605eV1
14⋅⇒
°⇒

29
wanie systemu o naturze zupe#nie ró$ni&cej si% od tej, która wyst%puje w fazachskondensowanych, gdzie za uporz&dkowanie odpowiadaj& krótkozasi%gowesi#y spójno!ci. Zachowanie si% plazmy w zewn%trznych polach: elektrycznymi magnetycznym przypomina zachowanie si% cieczy. Plazma jest, ze wzgl%duna du$& ruchliwo!" elektronów, doskona#ym przewodnikiem pr&du.Wed#ug obecnych danych dost%pny nam do obserwacji Wszech!wiat sk#adasi% w 99,9% z plazmy.Plazm% dzielimy na nisko- i wysokotemperaturow&. W plazmie niskotempe-raturowej energia termiczna cz&stek jest porównywana z energi& jonizacjiatomów lub cz&steczek; plazma taka jest mieszanin& cz&stek oboj%tnych,zjonizowanych i swobodnych elektronów.Podstawowe procesy elementarne w plazmie niskotemperaturowej to:• jonizacja - proces odrywania elektronu od atomu lub cz&steczki,• rekombinacja - proces odwrotny do procesu jonizacji, tj. #&czenie si% elek-
tronu i jonu dodatniego w oboj%tny elektrycznie atom,• wymiana #adunku - proces, w którym jon i atom zderzaj&c si% wymienia-
j& mi%dzy sob& elektron, w rezultacie czego jon staje si% oboj%tnym ato-mem a atom jonem,
• wzbudzenia - w wyniku zderze' ró$nego rodzaju, np. elektron - atom,atom - atom, jon - atom, elektrony znajduj&ce si% na orbitach atomo-wych lub jonowych przechodz& w wy$sze stany energetyczne; proces od-wrotny zwany jest równie$ rekombinacj& i wi&$e si% z wypromieniowa-niem przez atom lub jon fali elektromagnetyczne.
W plazmie gor&cej - wysokotemperaturowej - energia kinetyczna cz&-stek znacznie przewy$sza energi% jonizacji. W plazmie tej nie ma w zasadziecz&stek oboj%tnych. W najprostszym przypadku sk#ada si% ona z elektro-nów i jonów jednego rodzaju.
Na rysunku 1.18 przedstawione s& zakresy: temperaturowy i energe-tyczny wyst%powania plazmy zimnej (niskotemperaturowej) oraz gor&cej (wy-sokotemperaturowej).
Z kolei rysunek 1.19 przedstawia ró$ne (ród#a ciep#a w zakresie tem-peratury wyst%powania plazmy niskotemperaturowej.
Zaznaczmy od razu, $e w procesach otrzymywania warstw TiN wyko-rzystuje si% plazm% niskotemperaturow&.

30
****
Kohezja, adhezja, adsorpcja, absorpcja, sorpcja, desorpcja.Za spójno!" wewn%trzn& (kohezj%) faz skondensowanych odpowiada-
j& wi&zania chemiczne. Zatem gdy mówimy o si#ach kohezji, mamy na my!liwi&zania mi%dzyatomowe lub mi%dzycz&steczkowe dzia#aj&ce w obr%bie jed-nej fazy. Rozró$niamy nast%puj&ce ich rodzaje (uporz&dkowanie w stron%malej&cych energii wi&zania):• wi&zanie jonowe (heteropolarne),• wi&zanie kowalencyjne (homopolarne, atomowe),• wi&zanie metaliczne,
Rys. 1.19. Ró'ne (ród#a ciep#a w
zakresie temperatury
wyst!powania zimnej
plazmy
Rys. 1.18. Zakresy energii i
temperatury wyst!powa-
nia plazmy nisko- i
wysoko temperaturowej

31
• wi&zanie wodorowe,• wi&zanie van der Waals'a
W zwi&zkach chemicznych rzadko zdarza si% by wyst&pi# jeden, wpostaci czystej, rodzaj wi&zania chemicznego. Przyk#adem jest TiN gdziewi&zanie ma natur% kowalencyjno - metaliczn&.
W przypadku czterech pierwszych wi&za' energia wi&za' jest rz%dukilku elektronowoltów na cz&stk% (przez cz&stk% rozumiemy atom lub mole-ku#% - cz&steczk%). Energia wi&za' van der Waals'a jest o rz&d, dwa rz%dymniejsza.
Jednak$e wi&zanie van der Waals'a jest najbardziej uniwersalnym,wyst%puje we wszystkich bez wyj&tku wi&zaniach mi%dzyatomowych i mi%-dzycz&steczkowych. W postaci czystej wi&zanie to istnieje mi%dzy cz&stecz-kami o nasyconych wi&zaniach chemicznych (02, N2, H2 itd.), jak równie$mi%dzy atomami gazów szlachetnych w fazach skondensowanych.
Przypomnijmy, $e w równaniu stanu gazów rzeczywistych, tzw. rów-nanie van der Waals'a:
RTbVV
ap
2=
+ ,
poprawki a/V2 i b uwzgl%dniaj& si#y przyci&gania i odpychania mi%-dzy cz&stkami gazów rzeczywistych, typowe dla oddzia#ywa' van der Wa-als'a.
Dalej podkre!limy ten rodzaj wi&za', poniewa$ w interesuj&cym nasprzypadku warstw TiN, warunkuj& one przyleganie - adhezj! warstw dopod#o$a.
Ogólnie, przez adhezj% rozumiemy trwa#e po#&czenie dwóch faz skon-densowanych, charakteryzuj&ce si% brakiem wzajemnego przenikania (dy-fuzji) sk#adników faz, bez tworzenia si% nowych faz (np. zwi&zków chemicz-nych) w obszarze kontaktowym. W naszym przypadku chodzi naturalnie opo#&czenie typu faza sta#a (pod#o$e) - faza sta#a (warstwa).
Zwró"my jeszcze uwag% na co! szczególnego. W czterech, wymienio-nych wy$ej jako pierwsze, wi&zaniach chemicznych mamy do czynienia zoddzia#ywaniem elektrycznym #adunków punktowych reprezentowanychprzez elektrony i dodatnio na#adowane jony.
Rzeczywi!cie, we wi&zaniach jonowych wyst%puje transfer, tj. przenie-sienie elektronów z atomów elektrododatnich na elektroujemne, w kowalen-

32
cyjnych - uwspólnienie elektronów, w metalicznych - kolektywne oddzia#ywa-nie elektronów z sieci& zlokalizowanych dodatnio jonów za! w wodorowych -oddzia#ywanie elektronu atomu wodoru z elektroujemnymi atomami.
Wi&zania van der Waals'a maj& charakter oddzia#ywa' dipolowych -natura ich jest wi%c ca#kowicie ró$na od oddzia#ywa' wyst%puj&cych w in-nych wi&zaniach chemicznych.
Rozró$nia si% trzy rodzaje oddzia#ywa' dipolowych, dzi%ki którym po-wstaj& wi&zania van der Waals'a, a mianowicie:• oddzia#ywanie dyspersyjne,
• oddzia#ywanie orientacyjne,• oddzia#ywanie indukcyjne.
W oddzia#ywaniu dyspersyjnym chwilowy, szybkozmienny rozk#ad#adunku w oboj%tnej cz&stce, maj&c charakter dipolu, wchodzi w korelacyj-ne oddzia#ywanie z podobnymi momentami s&siednich cz&stek, powoduj&cich wzajemne przyci&ganie si% i odpychanie. Bardziej prawdopodobny jestefekt przyci&gania, poniewa$ daje on obni$enie energii ca#kowitej zbiorucz&stek.
W oddzia#ywaniu orientacyjnym si#y wi&$&ce (przyci&gania) wyst%pu-j& pomi%dzy cz&stkami o sta#ych dipolach (tzw. cz&stki polarne).
W oddzia#ywaniu indukowanym si#y wi&$&ce powstaj& poprzez od-dzia#ywanie cz&stek posiadaj&cych trwa#y moment dipolowy z innymi cz&st-kami, które polaryzuj& si% (t.j. powstaje w nich indukowany moment dipo-lowy) w polu elektrycznym trwa#ych dipoli.
Wszystkie te oddzia#ywania sumuj& si%, przy czym w moleku#ach po-larnych dominuje oddzia#ywanie orientacyjne, za! w niepolarnych dysper-syjne.
Si#y van der Waals'a bardzo silnie zale$& od wzajemnej odleg#o!ci cz&-stek, mi%dzy którymi wyst%puj&:
Rys. 1.20. Dipol elektryczny

33
6r
AU −= ,
gdzie:U - energia oddzia#ywania,A - pewna sta#a, r - wzajemna odleg#o!" cz&stek
Je$eli dowolna faza skondensowana znajduje si% w kontakcie z jedno-rodn& faz& gazow& lub ciek#& to na jej powierzchni, ujmuj&c rzecz makro-skopowo, nast%puje zag%szczenie si% (zmiana st%$enia) sk#adników tych faz. Mikroskopowo, nast%puje to w wyniku oddzia#ywa' van der Waals'a mi%-dzy cz&steczkami powierzchni fazy skondensowanej, a cz&stkami fazy b%d&-cej z ni& w kontakcie. Oddzia#ywanie to obrazuje rysunek 1.21.
Zbli$aj&ca si% do powierzchni fazy skondensowanej cz&stka (sk#adnik fazy gazowej lub ciek#ej) podlega dzia#aniu si# powierzchniowych o charakte-
Rys.1.21. Energia potencjalna cz$stki w pobli'u powierzchni
cia#a sta#ego.

34
rze van der Waals'a i zostaje na tej powierzchni zwi&zania. O ile cz&stka tajest oboj%tna chemicznie wobec atomów powierzchni to mówimy, $e nast&-pi#o zjawisko adsorpcji fizycznej. Efekt ten wi&$e si% z obni$eniem energiicz&stki o EF.
Je$eli cz&stka mo$e tworzy" zwi&zki chemiczne z atomami powierzch-ni to zwi&zanie jej z powierzchni& ma bardziej niskoenergetyczny charakter(Ea). Zachodzi zjawisko adsorpcji - teraz nazwanej chemiczn$. Wielko!" Ea
nazywa si% energi& aktywacji tego procesu.W sprzyjaj&cych warunkach termodynamicznych cz&stka mo$e wnik-
n&" w faz% sta#&. Jak wida", cz&stka musi wtedy pokona" barier% potencja#uE1 - proces ten musi by" zatem silnie aktywowany. Cz&stka taka ulega, jaksi% powiada, absorpcji przez cia#o sta#e. Nast%puje obni$enie energii cz&stkio warto!" EB.
Zjawiska adsorpcji i absorpcji obejmuje si% wspólnym terminem - sorp-
cja.Istnieje zjawisko odwrotne do sorpcji, mianowicie - desorpcja. Poj%-
ciem tym obejmuje si% uwalnianie z powierzchni cz&stek uprzednio zaadsor-bowanych lub zaabsorbowanych. Potrzebne s& w tym celu, w zale$no!ci odcharakteru zwi&zania cz&stki z cia#em sta#ym, odpowiednio energie EF, Ed,EB+E1.
Fenomenologicznie podej!cie do opisu zjawisk sorpcji i desorpcji po-lega na spostrze$eniu, $e na granicy fazy skondensowanej tj. jej powierzchninast%puje nieci&g#o!" wi&za' wewn&trzfazowych, to jest si# kohezji. Si#ywypadkowe skierowane s& zawsze do wewn&trz fazy i tam kompensuj& si%.
Sytuacja ta sprawia, $e cz&stki przypowierzchniowe fazy znajduj& si%w innym stanie energetycznym ni$ cz&stki wewn&trz fazy. Ta dodatkowawarto!" energii, zwana energi$ powierzchniow$ powoduje szereg szcze-gólnych w#a!ciwo!ci obszaru przypowierzchniowego ró$ni&cych si% od w#a-!ciwo!ci wn%trza fazy.Ca#kowit& energi% fazy dzieli si% na:
•energi% zwi&zan& z jednostk& obj%to!ci fazy UV,•energi% zwi&zan& z jednostk& powierzchni fazy UP.
Je$eli V jest obj%to!ci& fazy, a P jej powierzchni& to energia ca#kowitafazy daje si% zapisa" jako:

35
PUVUUPV
⋅+⋅= .
Po podzieleniu tego wyra$enia przez obj%to!" V otrzymujemy warto!"energii fazy przypadaj&c& na jednostk% obj%to!ci:
PVU
V
PU
V
U⋅+=
Iloraz V
P oznaczaj&cy powierzchni% fazy przypadaj&c& na jednostk%
obj%to!ci nazywamy powierzchni& w#a!ciw&. Dla cia# o ma#o rozwini%tej powierzchni stosunek ten jest ma#y i drugi cz#on wyra$enia na energi% fazy jest nieistotny. Dla cia# o rozwini%tych powierzchniach (sorbenty) sytuacja jest przeciwna i wk#ad energii powierzchniowej odgrywa znacz&c& rol% dla zjawisk zachodz&cych na powierzchni.
****
Oddzia#ywanie wi$zki jonów z powierzchni$ cia#a sta#ego.
Rys.1.22. Zestawienie zjawisk zachodz$cych podczas oddzia#ywania wi$zki
jonów z powierzchni$ cia#a sta#ego.

36
Procesy nanoszenia warstw TiN odbywaj& si% w takich sytuacjach,gdzie pod#o$e na którym tworzy si% warstwa poddawane jest ci&g#emu bom-bardowaniu przez ró$ne jony. Jak oka$e si% pó(niej jest to konieczne dlauzyskania w#a!ciwych warunków szybkiego wzrostu warstwy.
Na rysunku 1.22, przedstawiono obrazowo podstawowe zjawiskamaj&ce miejsce w trakcie oddzia#ywania wi&zk& jonów - cia#o sta#e.
****
Model tworzenia si! cienkiej warstwy na powierzchni cia#a sta#ego.
Rysunek 1.23 przedstawia, w etapach, mo$liwy model tworzenia si%warstwy na powierzchni cia#a sta#ego. Warstwa wzrasta dzi%ki dostarczaniuna powierzchni% atomów z fazy gazowej.
Rys. 1.23. Model tworzenia si! warstwy na powierzchni cia#a sta#ego.

37
Narastaj&ca na pod#o$u sta#ym warstwa mo$e mie", w zale$no!ci odszeroko poj%tych, parametrów technologicznych struktur%:• monokrystaliczn& (tzw. warstwy epitaksjalne),• polikrystaliczn& (posta" t% maj& szeroko stosowane warstwy TiN),• bezpostaciow& (amorficzn&).
Rysunek 1.24 ilustruje wp#yw bombardowania jonowego na charak-ter tworz&cej si% warstwy.
Rysunek ten zosta# uzyskany poprzez symulacj% komputerow& rzeczy-wistego procesu technologicznego. Widoczny jest wyra(ny wp#yw bombardo-wania jonowego na makrostruktur% warstwy, jej g%sto!".
****
Metody CVD i PVD.
Najogólniejszy obecnie podzia# metod otrzymywania warstw TiN (atak$e innych twardych warstw) wydziela dwie klasy technologii cienkowar-stwowych okre!lane mianami CVD i PVD.Skróty te pochodz& od pierwszych liter pe#nych nazw w j%zyku angielskim:
• CVD od Chemical Vapour Deposition,• PVD od Phisical Vapour Deposition,
Rys. 1.24. Warstwa tytanu narastaj$ca bez i z bombardowaniem
jonowym.

38
które w sposób dowolny mo$na przet#umaczy" nast%puj&co:• CVD - nak#adanie chemiczne z fazy gazowej,• PVD - nak#adanie fizyczne z fazy gazowej,
Podstaw& powy$szego podzia#u s& mechanizmy, dzi%ki którym nast%-puje wytwarzanie warstw na pod#o$u.
W technologiach CVD warstwy powstaj& dzi%ki procesom chemicz-nym zachodz&cym w gazach dostarczanych z zewn&trz (pod ci!nieniem at-mosferycznym lub niewiele obni$onym) do komory reakcyjnej.
Istotne jest, $e do aktywacji syntezy TiN konieczna jest bardzo wyso-ka temperatura, w granicach 950 ÷ 1100°C. Ogranicza to rodzaje pod#o$y,na które mog& by" nanoszone warstwy do w%glików spiekanych.
Metody CVD zostan& nieco szerzej omówione dalej, tutaj dla ichilustracji zamieszczony zosta# rysunek 1.25 opisuj&cy ide% metody dla przy-padku nanoszenia warstw w%glika tytanu TiC.
W przypadku metod PVD warstwy powstaj& na pod#o$ach dzi%ki od-parowaniu lub rozpyleniu materia#u b%d&cego w postaci sta#ej, w warun-kach pró$ni, przy czym (ród#o tego materia#u znajduje si% wewn&trz komo-ry reakcyjnej.
Rysunek 1.26 ilustruje prosty przyk#ad jednej z metod PVD, a mia-nowicie nanoszenie warstw aluminium poprzez odparowanie za pomoc&grzejnika oporowego.
Rys. 1.25. Konwencjonalny proces CVD dla warstw TiC

39
Nie wchodz&c w szczegó#y, proces ten mo$na opisa" nast%puj&co: od-powiednio przygotowane pod#o$e umieszcza si% w komorze pró$niowej nad wyparnikiem oporowym, w którym znajduje si% aluminium. Komora zosta-je odpompowana do pró$ni przynajmniej 10-4 mbar. W tych warunkach podnosi si% temperatur% wyparnika w celu stopienia wsadu. Aluminium odparowuje, i je!li w strumieniu par znajduje si% pod#o$e, to tworzy si% na nim warstwa Al o grubo!ci zale$nej od czasu trwania procesu.
Istotne jest, $e metody PVD pozwalaj& na otrzymywanie warstw TiN w temperaturach ni$szych od 550°C, tj. temperatury odpuszczania stali szyb-kotn&cej. Z tego te$ powodu znajduj& one, w chwili obecnej, tak szerokie zastosowanie.
****
Rys. 1.26. Elementrany proces PVD - nanoszenie warstw Al przez odparowanie
grzaniem oporowym sta#ego Al w pró'ni.

40
Przyk#ad technologii PVD - metoda aktywowanego reaktywnego napa-
rowywania.
Teoretycznie, warstwy azotku tytanu mo$na by otrzymywa" poprzez bezpo!rednie odparowanie tego zwi&zku w pró$ni w uk#adzie przedstawio-nym na rysunku 1.26. Praktycznie jednak metoda ta nie jest stosowana, ze wzgl%du na wysok& temperatur% topnienia TiN (ok. 3000°C), ma#& szyb-ko!" wzrostu warstwy oraz brak kontroli jej sk#adu chemicznego podczas narastania.
Cienkie warstwy TiN otrzymuje si% w procesach okre!lanych jako reaktywne nanoszenie cienkich warstw.Ogólnie mówi&c, w komorze pró$niowej zawieraj&cej azot (N2) pod obni$onym ci-!nieniem (10-2 ÷ 10-4 Torr) wytwarza si%pary tytanu i tak ustala parametry tech-nologiczne, by na pod#o$u zachodzi#a syn-teza azotku tytanu, tzn. reakcja:
TiNN2
1Ti 2 →+
Omówmy takie procesy przyjmu-j&c jako (ród#o atomów Ti odparowuj&c&w pró$ni powierzchni% roztopionego metalicznego tytanu - jest to metoda re-aktywnego odparowywania. Ry-sunki 1.28 i 1.29 porównuj&, da-j&ce si% wydzieli" kroki, etapy wy-st%puj&ce w metodzie "normalne-go" naparowywania cienkich warstw oraz metodzie reaktywnej.
W pierwszej z metod atomy Ti wytworzone w (ródle poprzez odparowanie litego materia#u, po-konuj&, w warunkach pró$nio-wych prawie bez zderze', odle-g#o!" (ród#o pod#o$e tworz&c war-stw% metalicznego tytanu.
W metodzie reaktywnego naparowywania (ród#o atomów
Rys. 1.29. Etapy w metodzie
reaktywnego naparowy-
wania cienkich warstw
Rys. 1.28. Etapy w metodzie
naparowywania
cienkich warstw
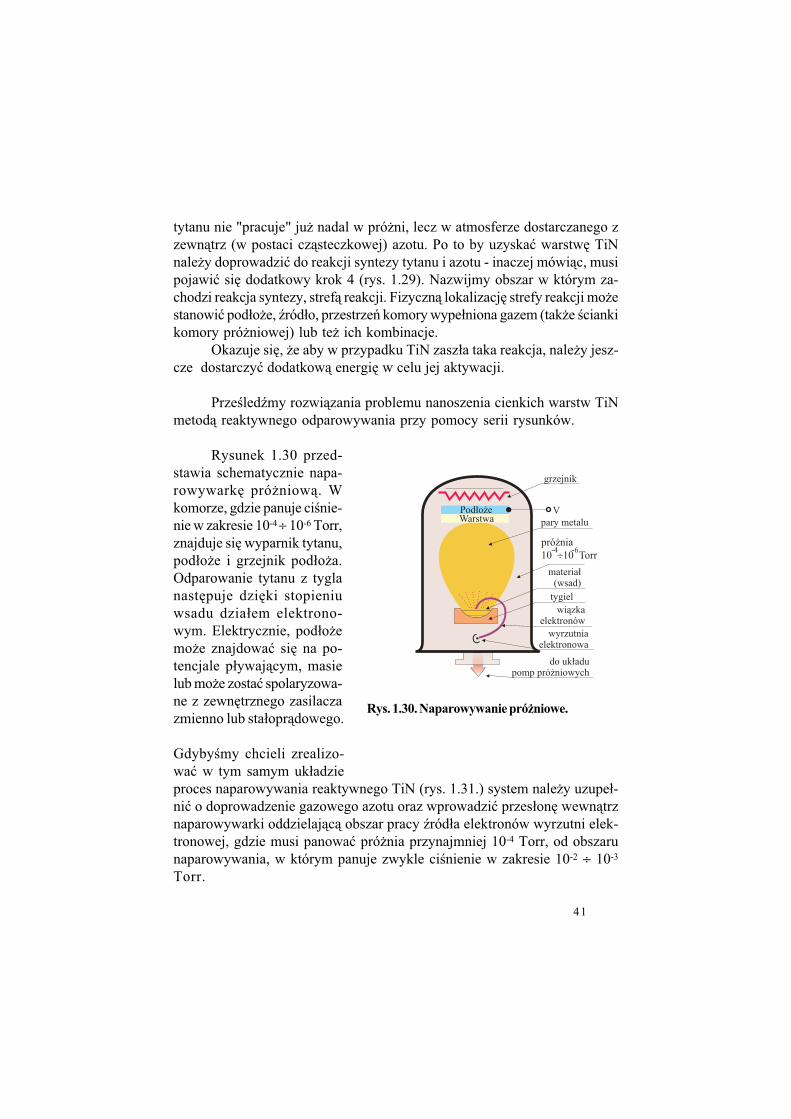
41
tytanu nie "pracuje" ju$ nadal w pró$ni, lecz w atmosferze dostarczanego zzewn&trz (w postaci cz&steczkowej) azotu. Po to by uzyska" warstw% TiNnale$y doprowadzi" do reakcji syntezy tytanu i azotu - inaczej mówi&c, musipojawi" si% dodatkowy krok 4 (rys. 1.29). Nazwijmy obszar w którym za-chodzi reakcja syntezy, stref& reakcji. Fizyczn& lokalizacj% strefy reakcji mo$estanowi" pod#o$e, (ród#o, przestrze' komory wype#niona gazem (tak$e !ciankikomory pró$niowej) lub te$ ich kombinacje.
Okazuje si%, $e aby w przypadku TiN zasz#a taka reakcja, nale$y jesz-cze dostarczy" dodatkow& energi% w celu jej aktywacji.
Prze!led(my rozwi&zania problemu nanoszenia cienkich warstw TiNmetod& reaktywnego odparowywania przy pomocy serii rysunków.
Rysunek 1.30 przed-stawia schematycznie napa-rowywark% pró$niow&. Wkomorze, gdzie panuje ci!nie-nie w zakresie 10-4 ÷ 10-6 Torr,znajduje si% wyparnik tytanu,pod#o$e i grzejnik pod#o$a.Odparowanie tytanu z tyglanast%puje dzi%ki stopieniuwsadu dzia#em elektrono-wym. Elektrycznie, pod#o$emo$e znajdowa" si% na po-tencjale p#ywaj&cym, masielub mo$e zosta" spolaryzowa-ne z zewn%trznego zasilaczazmienno lub sta#opr&dowego.
Gdyby!my chcieli zrealizo-wa" w tym samym uk#adzieproces naparowywania reaktywnego TiN (rys. 1.31.) system nale$y uzupe#-ni" o doprowadzenie gazowego azotu oraz wprowadzi" przes#on% wewn&trznaparowywarki oddzielaj&c& obszar pracy (ród#a elektronów wyrzutni elek-tronowej, gdzie musi panowa" pró$nia przynajmniej 10-4 Torr, od obszarunaparowywania, w którym panuje zwykle ci!nienie w zakresie 10-2 ÷ 10-3
Torr.
Rys. 1.30. Naparowywanie pró'niowe.
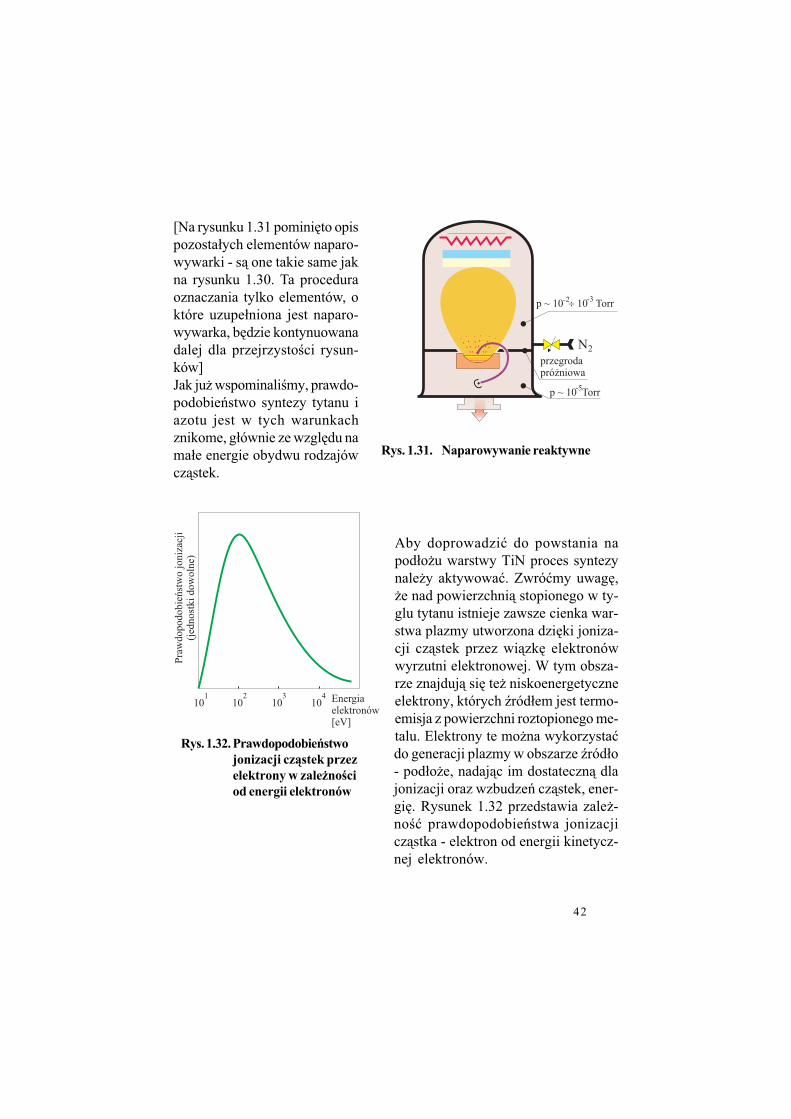
42
[Na rysunku 1.31 pomini%to opis pozosta#ych elementów naparo-wywarki - s& one takie same jak na rysunku 1.30. Ta procedura oznaczania tylko elementów, o które uzupe#niona jest naparo-wywarka, b%dzie kontynuowana dalej dla przejrzysto!ci rysun-ków]Jak ju$ wspominali!my, prawdo-podobie'stwo syntezy tytanu i azotu jest w tych warunkach znikome, g#ównie ze wzgl%du na ma#e energie obydwu rodzajów cz&stek.
Aby doprowadzi" do powstania na pod#o$u warstwy TiN proces syntezy nale$y aktywowa". Zwró"my uwag%, $e nad powierzchni& stopionego w ty-glu tytanu istnieje zawsze cienka war-stwa plazmy utworzona dzi%ki joniza-cji cz&stek przez wi&zk% elektronów wyrzutni elektronowej. W tym obsza-rze znajduj& si% te$ niskoenergetyczne elektrony, których (ród#em jest termo-emisja z powierzchni roztopionego me-talu. Elektrony te mo$na wykorzysta"do generacji plazmy w obszarze (ród#o - pod#o$e, nadaj&c im dostateczn& dla jonizacji oraz wzbudze' cz&stek, ener-gi%. Rysunek 1.32 przedstawia zale$-no!" prawdopodobie'stwa jonizacji cz&stka - elektron od energii kinetycz-nej elektronów.
Rys. 1.31. Naparowywanie reaktywne
Rys. 1.32. Prawdopodobie*stwo
jonizacji cz$stek przez
elektrony w zale'no%ci
od energii elektronów
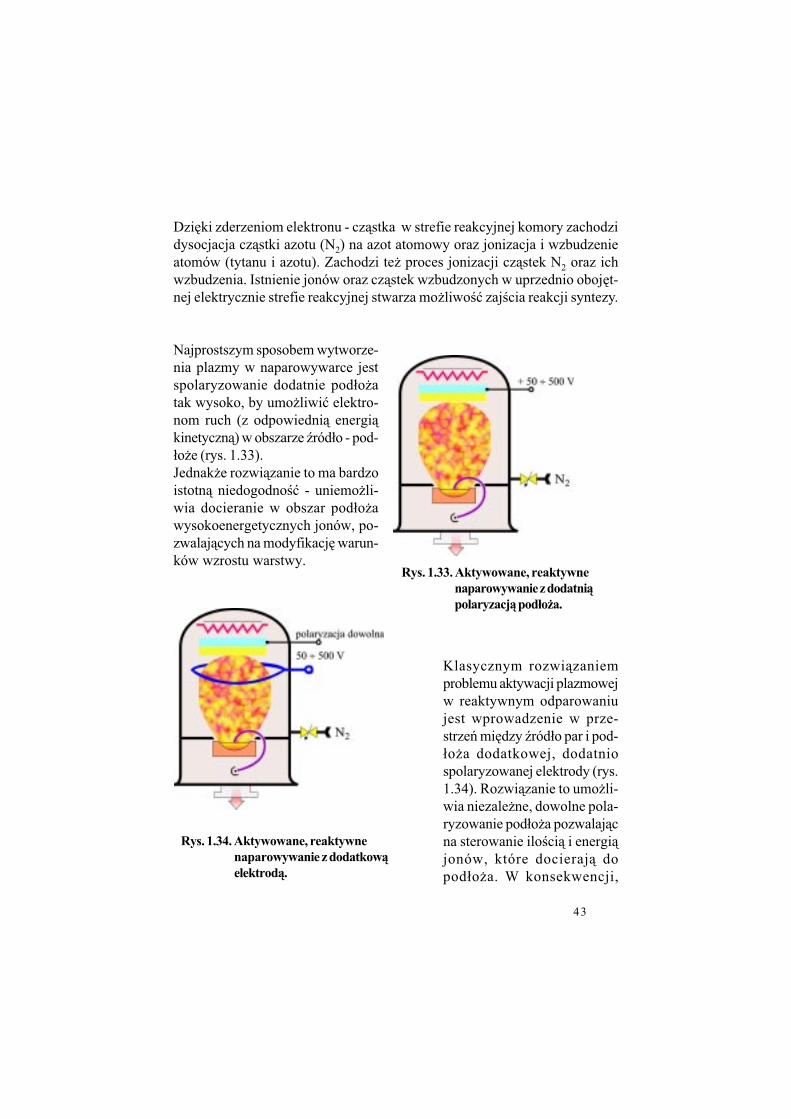
43
Dzi%ki zderzeniom elektronu - cz&stka w strefie reakcyjnej komory zachodzi dysocjacja cz&stki azotu (N2) na azot atomowy oraz jonizacja i wzbudzenie atomów (tytanu i azotu). Zachodzi te$ proces jonizacji cz&stek N2 oraz ich wzbudzenia. Istnienie jonów oraz cz&stek wzbudzonych w uprzednio oboj%t-nej elektrycznie strefie reakcyjnej stwarza mo$liwo!" zaj!cia reakcji syntezy.
Najprostszym sposobem wytworze-nia plazmy w naparowywarce jest spolaryzowanie dodatnie pod#o$a tak wysoko, by umo$liwi" elektro-nom ruch (z odpowiedni& energi&kinetyczn&) w obszarze (ród#o - pod-#o$e (rys. 1.33).Jednak$e rozwi&zanie to ma bardzo istotn& niedogodno!" - uniemo$li-wia docieranie w obszar pod#o$a wysokoenergetycznych jonów, po-zwalaj&cych na modyfikacj% warun-ków wzrostu warstwy.
Klasycznym rozwi&zaniem problemu aktywacji plazmowej w reaktywnym odparowaniu jest wprowadzenie w prze-strze' mi%dzy (ród#o par i pod-#o$a dodatkowej, dodatnio spolaryzowanej elektrody (rys.1.34). Rozwi&zanie to umo$li-wia niezale$ne, dowolne pola-ryzowanie pod#o$a pozwalaj&c na sterowanie ilo!ci& i energi&jonów, które docieraj& do pod#o$a. W konsekwencji,
Rys. 1.33. Aktywowane, reaktywne
naparowywanie z dodatni$polaryzacj$ pod#o'a.
Rys. 1.34. Aktywowane, reaktywne
naparowywanie z dodatkow$elektrod$.
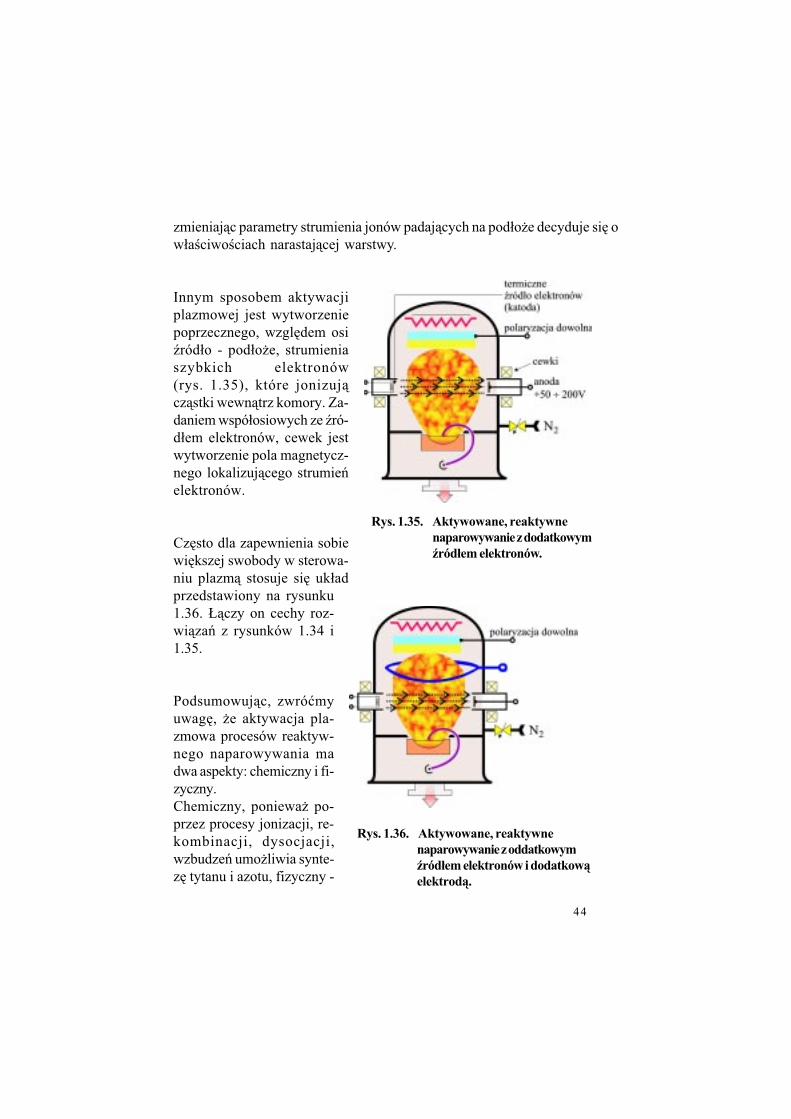
44
zmieniaj&c parametry strumienia jonów padaj&cych na pod#o$e decyduje si% o w#a!ciwo!ciach narastaj&cej warstwy.
Innym sposobem aktywacji plazmowej jest wytworzenie poprzecznego, wzgl%dem osi (ród#o - pod#o$e, strumienia szybkich elektronów (rys. 1.35), które jonizuj&cz&stki wewn&trz komory. Za-daniem wspó#osiowych ze (ró-d#em elektronów, cewek jest wytworzenie pola magnetycz-nego lokalizuj&cego strumie'elektronów.
Cz%sto dla zapewnienia sobie wi%kszej swobody w sterowa-niu plazm& stosuje si% uk#ad przedstawiony na rysunku1.36. +&czy on cechy roz-wi&za' z rysunków 1.34 i1.35.
Podsumowuj&c, zwró"my uwag%, $e aktywacja pla-zmowa procesów reaktyw-nego naparowywania ma dwa aspekty: chemiczny i fi-zyczny.Chemiczny, poniewa$ po-przez procesy jonizacji, re-kombinacji, dysocjacji, wzbudze' umo$liwia synte-z% tytanu i azotu, fizyczny -
Rys. 1.36. Aktywowane, reaktywne
naparowywanie z oddatkowym
(ród#em elektronów i dodatkow$elektrod$.
Rys. 1.35. Aktywowane, reaktywne
naparowywanie z dodatkowym
(ród#em elektronów.

45
poniewa$ umo$liwia sterowa-nie warunkami wzrostu war-stwy na pod#o$u poprzez efekt bombardowania jonowego. Warunki bombardowania jo-nowego pod#o$a mo$na usta-la" w ka$dym z opisanych wy$ej procesach, przede wszystkim przez wybór napi%-cia polaryzuj&cego pod#o$e. Dla dodatkowej, niezale$nej kontroli tego efektu mo$na wprowadzi" do komory reak-cyjnej dzia#o jonowe, wytwa-rzaj&ce zdefiniowany strumie'jonów skierowany na pod#o$e (rys. 1.37).
Padaj&ce na powierzchni% pod#o$a, a potem na powierzchni% nara-staj&cej warstwy jony i atomy (mo$liwa jest bowiem neutralizacja przyspie-szonego w kierunku pod#o$a jonu np. poprzez efekt wymiany #adunku) prze-kazuj& pod#o$u oraz kondensuj&cym na nim atomom swoj& energi% i p%d w rezultacie czego:• wzrasta ruchliwo!" powierzchniowa osadzonych na pod#o$u atomów,• przyspieszone s&: zarodkowanie, wzrost zarodków oraz koalescencja w
pocz&tkowych fazach zarodkowania,• wytwarzane s& oraz aktywowane miejsca zarodkowania,• wspomagane s& efekty rekrystalizacji narastaj&cych ziaren polikrystalicz-
nych,• wzrasta energia wi&zania mi%dzy warstw& a pod#o$em,• zmniejszaj& si% napr%$enia w powstaj&cych warstwach,• usuwane s& przypadkowe (pochodz&ce z atmosfery resztkowej komory
pró$niowej) zanieczyszczenia kondensuj&ce na pod#o$u.Wszystkie idee metod aktywowanego reaktywnego naparowywania opisa-ne wy$ej znajduj& zastosowania w praktycznych systemach nanoszenia warstw TiN.
Rys. 1.37. Dodatkowa aktywacja pod#o'a
przy pomocy wyrzutni (dzia#a)
jonowej.

46
2. Wybrane w!a"ciwo"ci TiN
Ni!ej zostan" przedstawione niektóre w#a$ciwo$ci warstw azotku ty-tanu. Przytoczone dane maj" charakter raczej orientacyjny ni! dokumen-talny; pochodz" z ró!nych %róde#, zawieraj" równie! wyniki prac w#asnych.W zwi"zku z tym nie wprowadzono odsy#aczy literaturowych.
Dane te, mimo to, pozwol" na zorientowanie si& w warto$ciach ró!-nych parametrów charakterystycznych warstwy TiN.
2.1. W!a"ciwo"ci atomowe.
Struktura elektronowa
Tytan:
Ti4822
1s22s22p63s23p63d24s2
Azot:
N147 1s22s22p3
W&giel:1s22s22p2
Promienie atomowe:
Ti 1,47 Å
N 0,70 Å
C 0,77 Å
Energia jonizacji [eV]:
Ti N C
I 6,81 14,51 11,24
II 13,6 29,41 24,28
III 28,39 47,36 47,55
JEDNOSTKA D'UGO(CI stosowana w fizyce ato-mowej
1 Ångstrom - 1 Å = 10-10m = 10-8 cm
JEDNOSTKA ENERGII stosowana w fizyce ato-mowej1 elektronowolt - 1 eV1 Joule - 1 J1 eV = 1,6 × 10-19 J.1 J = 1N × 1mWzór okre$laj"cy energi& cz"stki o #adunku elek-trycznym q przemieszczonej w polu elektrycznymmi&dzy potencja#ami V1 i V2 ma posta):E = q(V
2 - V
1) = qU [C·V]
U = V2 - V
1- napi&cie elektryczne
st"d: 1 eV = |e|·1V = 1,6·10-19 C·1V = 1,6·10-19J.
ENERGIA JONIZACJI atomu to energia potrzebnado oderwania elektronu z pow#oki atomowej iprzesuni&ciu go w obszar, w którym jego oddzia#y-wanie z powsta#ym jonem mo!na zaniedba).Pierwszy stopie* jonizacji (I), drugi, trzeci itd. - tokolejne odrywanie zewn&trznych elektronów z ato-mu. Tworz" si& w ten sposób jony dodatnie, jedno-, dwu-, … warto$ciowe
47
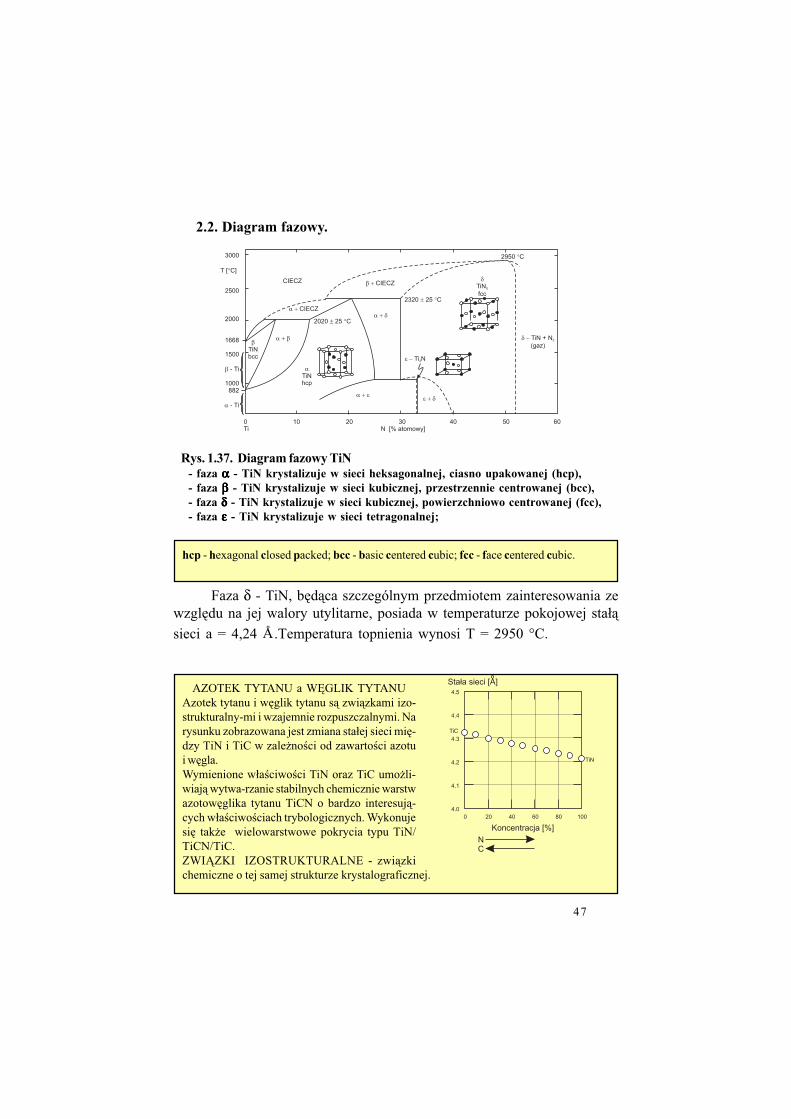
2.2. Diagram fazowy.
Faza δ - TiN, b&d"ca szczególnym przedmiotem zainteresowania zewzgl&du na jej walory utylitarne, posiada w temperaturze pokojowej sta#"
sieci a = 4,24 Å .Temperatura topnienia wynosi T = 2950 °C.
hcp - hexagonal closed packed; bcc - basic centered cubic; fcc - face centered cubic.
AZOTEK TYTANU a W+GLIK TYTANUAzotek tytanu i w&glik tytanu s" zwi"zkami izo-strukturalny-mi i wzajemnie rozpuszczalnymi. Narysunku zobrazowana jest zmiana sta#ej sieci mi&-dzy TiN i TiC w zale!no$ci od zawarto$ci azotui w&gla.Wymienione w#a$ciwo$ci TiN oraz TiC umo!li-wiaj" wytwa-rzanie stabilnych chemicznie warstwazotow&glika tytanu TiCN o bardzo interesuj"-cych w#a$ciwo$ciach trybologicznych. Wykonujesi& tak!e wielowarstwowe pokrycia typu TiN/TiCN/TiC.ZWI,ZKI IZOSTRUKTURALNE - zwi"zkichemiczne o tej samej strukturze krystalograficznej.
Rys. 1.37. Diagram fazowy TiN
- faza ααααα - TiN krystalizuje w sieci heksagonalnej, ciasno upakowanej (hcp),
- faza βββββ - TiN krystalizuje w sieci kubicznej, przestrzennie centrowanej (bcc),
- faza δδδδδ - TiN krystalizuje w sieci kubicznej, powierzchniowo centrowanej (fcc),
- faza εεεεε - TiN krystalizuje w sieci tetragonalnej;

48
2.3. G#sto"$
G&sto$) o$rodka definiuje si& jako:
V
m=ρ ,
gdzie: m - masa próbki, V - obj&to$) próbki.
Na rysunku 1.38 przedsta-wiona jest zale!no$) g&sto$ci TiN
od sk#adu atomowego Ti
N . Linia
przerywana oznacza teoretyczne warto$ci obliczone przy za#o!eniu, !e brak azotu lub tytanu kompensowa-ny jest przez wakansje w odpowiednich podsieciach azotku tytanu.Dla stechiometrycznego, monokrysta-licznego TiN g&sto$) wynosi:
3TiN cm
g2,5=ρ .
Dla warstw polikrystalicznych warto$)g&sto$ci jest mniejsza (zwykle jednak wi&ksza od4 g/cm3) i w sposób wyra%ny zale!y od rodzaju wykorzystanej technologii.
2.4. Wspó!czynnik rozszerzalno"ci liniowej
Wzór definiuj"cy wspó#czynnik termicznej rozsze-rzalno$ci liniowej α:
LT = LTo×[1+ααααα(T-TO)]
Dla TiN (T=20°C): α=9,35×10-6 [deg-1].
ZWI,ZKI STECHIOMETRYCZNE - zwi"zkio sk#adzie zgodnym z opisuj"c" je for-maln" formu#" chemiczn" (np. TiN) Zwi"zki NIESTECHIOMETRYCZNE - (nadste-chiometryczne i podstechiometryczne) - zwi"zki chemiczne posiadaj"ce nadmiar lub niedobór jednego przynajmniej ze sk#adników (np. TiNx, x∈0,6÷1,1, dla x=1 otrzymuje si& zwi"zek stechiometryczny)
Rys. 1.39.
Rys. 1.38. Zale%no"$ g#sto"ci TiN
od sk!adu atomowego

49
Dla porównania wspó#czynnik α innych materia#ów wynosz":
materia#
!|deg |-1
stale
~ 12·10-6 18·10-6 3·10-6 1,2·10-6 14·10-6
mosi"dz szk#o diament z#oto
2.5. W!a"ciwo"ci spr#%yste
Wzory definiuj"ce modu# Youngai wspó#czynnik Poissona.Wyd#u!enie ∆L próbki po przy#o-!eniu si#y rozci"gaj"cej F w przy-bli!eniu wynosi (prawo Hook'a):
S
FLkL
⋅=∆ , k - sta#a spr&!o$ci.
st"d:
Modu# Younga nazywany jest te! modu#em spr&!ysto$ci.
Wspó#czynnik Poissona okre$la stosunek wzgl&dnych zmian wymiarów po-przecznych i pod#u!nych cia#a pod wp#ywem napr&!enia. W sytuacji przed-stawionej na rysunku 1.40 wynosi on:
LL
rr
∆
∆
=ν .
Robert Hook w 1678 r."ut tensio sic vis"
"jakie wyd#u!enie takasi#a"
ε = α ⋅ σ
Rys. 1.40
ε⋅=σ↑↑↑
∆⋅=
E
L
L
k
1
S
Fσ - napr&!enie [N/m2]=[Pa],ε - odkszta#cenie,E = 1/k - modu# Younga.

50
Uwaga
Zauwa!my, !e po rozszerzeniu lewej strony równania definiuj"cegomodu# Younga przez ∆L otrzymujemy prac& (równ" energii zgroma-dzonej w próbce), potrzebn" na deformacj& próbki przypadaj"c" najednostk& obj&to$ci próbki po odkszta#ceniu.
L
LE
LS
LF
.obj
Energia ∆⋅=
∆⋅∆⋅∆
−
Zatem modu# Younga jest miar" energii potrzebnej do jednostkowej obj&-to$ciowo deformacji cia#a.
W tabeli poni!ej podane s" warto$ci wspó#czynnika Younga i wspó#-czynnika Poissona oraz dodatkowo wspó#czynnika rozszerzalno$ci cieplnejdla azotku tytanu. Dla porównania umieszczono warto$ci tych parametrówdla warstw TiC, TiCN oraz stali i miedzi.
Wspó#czynnikrozszerzalno$cicieplnej
2.6. Twardo"$
Przytoczmy warto$ci twardo$ci Vickers»a dla kilku rodzajów pod#o!y i kil-ku twardych warstw:
stal szybkotn"ca HSS
Pod#o!a:
Materia# Twardo$)HV
Warastwy:
w&glik spiekany 94 WC - 6Co
aluminium Al
stal naazotowana
TiN
800 - 1000
1500
30
1000 - 1300
200 - 2500
260 - 3000
300 - 3500
TiCN
TiC
51
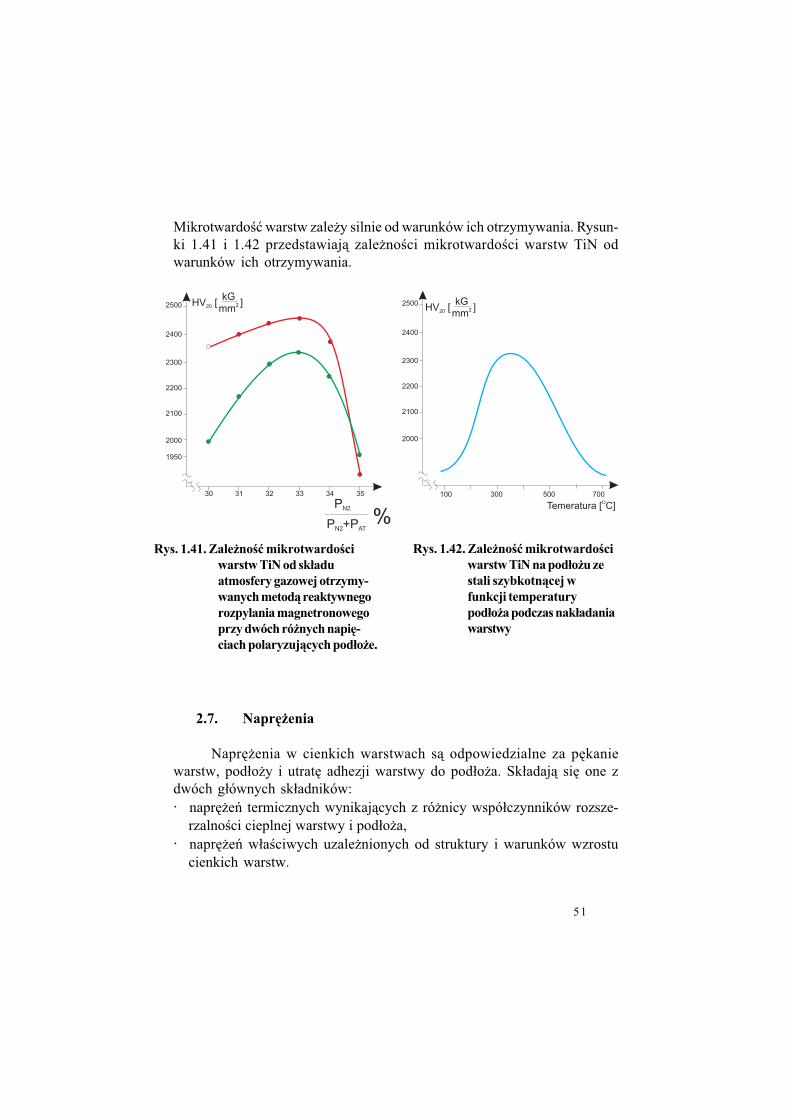
Mikrotwardo$) warstw zale!y silnie od warunków ich otrzymywania. Rysun-ki 1.41 i 1.42 przedstawiaj" zale!no$ci mikrotwardo$ci warstw TiN odwarunków ich otrzymywania.
2.7. Napr#%enia
Napr&!enia w cienkich warstwach s" odpowiedzialne za p&kaniewarstw, pod#o!y i utrat& adhezji warstwy do pod#o!a. Sk#adaj" si& one zdwóch g#ównych sk#adników:· napr&!e* termicznych wynikaj"cych z ró!nicy wspó#czynników rozsze-
rzalno$ci cieplnej warstwy i pod#o!a,· napr&!e* w#a$ciwych uzale!nionych od struktury i warunków wzrostu
cienkich warstw.
Rys. 1.42. Zale%no"$ mikrotwardo"ci
warstw TiN na pod!o%u ze
stali szybkotn&cej w
funkcji temperatury
pod!o%a podczas nak!adania
warstwy
Rys. 1.41. Zale%no"$ mikrotwardo"ci
warstw TiN od sk!adu
atmosfery gazowej otrzymy-
wanych metod& reaktywnego
rozpylania magnetronowego
przy dwóch ró%nych napi#-
ciach polaryzuj&cych pod!o%e.

52
Napr&!enia w#a$ciwe mog" mie) charakter $ciskaj"cy lub rozci"gaj"-cy w zale!no$ci od materia#u warstwy i warunków wzrostu. Powoduj" onedeformacj& cienkich pod#o!y na których znajduje si& warstwa.
Napr&!enia $ciskaj"ce zginaj" pod#o!e, tak !e powierzchnia warstwyjest wypuk#a; na-pr&!enia rozci"gaj"ce powoduj" wkl&s#o$) warstwy.
Jedn" z metod pomiaru napr&!e* w warstwach jest metoda optyczna.W metodzie tej próbka (cienki pod#o!e z warstw") przesuwana jest
prostopadle wzgl&dem wi"zki promieniowania laserowego tzn. normalnado powierzchni próbki jest prawie równoleg#a do wi"zki. W ten sposóbokre$la si& promienie krzywizn próbki przed i po na#o!eniu warstwy.
Je!eli:
∆x - oznacza warto$) prze-suni&cia próbki, ∆d - ozna-cza warto$) przesuni&cia de-tektora, L - odleg#o$) próbkiod detektora,
Poniewa!:L >> ∆x, L >> ∆d,
to promie* krzywizny prób-ki R wynosi:
d
xL2R
∆∆
=
Je!eli prowadzi si& nast&puj"ce oznaczenia:E - modu# Young'a pod#o!a,n - wspó#czynnik Poissona pod#o!a,D - grubo$) pod#o!a,t - grubo$) warstwy,Rp - promie* krzywizny pod#o!a przed naniesieniem warstwy,RW - promie* krzywizny pod#o!a z warstw",
Rys. 1.44. Zale%no"$ napr#%e' w warstwach
TiN otrzymywanych przy ró%nych
napi#ciach polaryzuj&cych
pod!o%e za pomoc& jednej z metod
reaktywnego rozpylania.
53
to napr&!enia w warstwie mo!na otrzyma) przy pomocy równania Stoney'a:
( )
−
⋅ν−⋅
=σPW
2
R
1
R
1
t16
DE
Wymiarem jest:
2m
N , lub
2m
kG , lub
2cm
dyna .
W przypadku azotku tytanu warto$ci napr&!e* podaje si& w GPa ([109
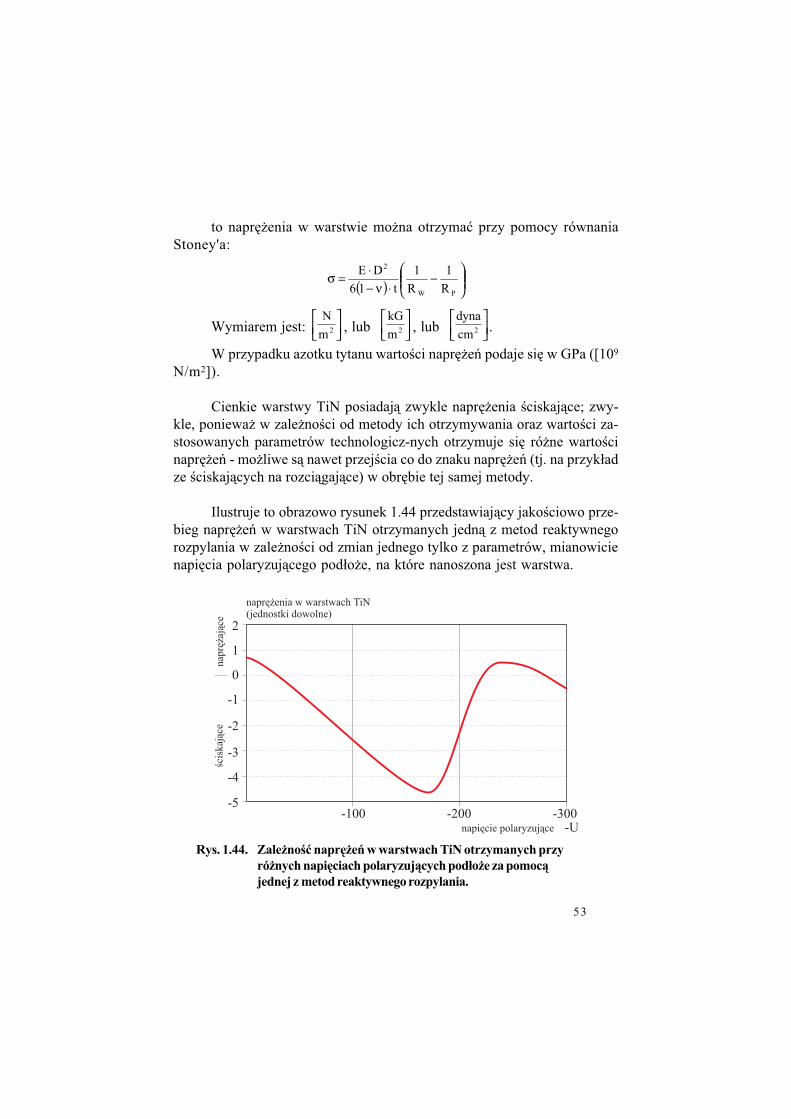
N/m2]).
Cienkie warstwy TiN posiadaj" zwykle napr&!enia $ciskaj"ce; zwy-kle, poniewa! w zale!no$ci od metody ich otrzymywania oraz warto$ci za-stosowanych parametrów technologicz-nych otrzymuje si& ró!ne warto$ci napr&!e* - mo!liwe s" nawet przej$cia co do znaku napr&!e* (tj. na przyk#ad ze $ciskaj"cych na rozci"gaj"ce) w obr&bie tej samej metody.
Ilustruje to obrazowo rysunek 1.44 przedstawiaj"cy jako$ciowo prze-bieg napr&!e* w warstwach TiN otrzymanych jedn" z metod reaktywnego rozpylania w zale!no$ci od zmian jednego tylko z parametrów, mianowicie napi&cia polaryzuj"cego pod#o!e, na które nanoszona jest warstwa.
Rys. 1.44. Zale%no"$ napr#%e' w warstwach TiN otrzymanych przy
ró%nych napi#ciach polaryzuj&cych pod!o%e za pomoc&
jednej z metod reaktywnego rozpylania.

54
2.8. Wspó!czynnik przewodnictwa cieplnego
Zdolno$) o$rodka do przewodzenia cie-p#a charakteryzuje si& przy pomocy parametru zwanego wspó#czynnikiem przewodzenia ciep#a λ. Definiuje si& go nast&puj"co: ilo$) ciep#a, które przep#ywa przez próbk& o d#ugo$ci ∆L i polu przekroju S (pole S jest prostopad#e do kie-runku przep#ywu ciep#a) w czasie ∆t (je$li T2>T1,∆T= T2-T1 - patrz rysunek) wynosi:
L
tTSQ
∆∆∆
λ=
Dla TiN:
⋅
⋅⋅=λ
degm
W4,29
degcms
cal07,0
Dla porównania, wspó#czynniki l dla innych materia#ów wynosz":
Mate-ria#
stal
40 80" ~ 40 110 2300 317 401 429 0,6 0,02 ~ 1,00,7 1,5"
!eliwo mosi"dz szk#o diament z#oto mied% srebro woda powie-trze
beton
#
$%
&'(
)
*degm
W
1 cal = 4,186 J1 J = 1W × 1s
Istniej" trzy mechanizmy przenoszenia ciep#a:1. przewodnictwo (bezpo$redni kontakt),2. promieniowanie cieplne (fala elektromagnetycz-
na - podczerwie*),3. konwekcja
Rys. 1.43. Ilustracja
mechanizmu
przenoszenia
ciep!a.

55
****Przypis - "O formule opisuj&cej przewodnictwo cieplne nieco szerzej."
Aby dok#adniej zapisa) przed-stawiony wy!ej wzór opisuj"cy prze-wodzenie ciep#a we%my pod uwag&nieograniczony, jednorodny o$ro-dek materialny o temperaturze TO,wewn"trz której znajduje si& punk-towe %ród#o ciep#a o temperaturzeT takiej, !e: T > TO. Pocz"wszy odmomentu kreacji %ród#a ciep#a wo$rodku nast"pi przep#yw ciep#a, ze%ród#a w kierunku o$rodka. Zewzgl&du na symetri& przep#yw b&dziemia# charakter radialny, powierzch-nie izotermiczne b&d" powierzchniami kulistymi. Niech zdolno$) o$rodkado przewodzenia ciep#a b&dzie scharakteryzowana wymiarowym wspó#czyn-nikiem λ. Niech S oznacza dowoln" powierzchni& wewn"trz o$rodka z przy-pisanym wektorem jednostkowym nd
ρ prostopad#ym do tej powierzchni wka!dym jej punkcie. Elementarne pole
sdρ wynosi zatem:
dsnsdρρ
= .Ilo$) ciep#a ∆Q przechodz"cego przezdowoln" powierzchni& S w jednostceczasu ∆t nazywamy strumieniemciep#a Φ:
t
Q
∆∆
=Φ
Strumie* ten opisany jest jako :
∫ ∫ ⋅∇−=⋅∇−=ΦS S
sdTsdTρρρρ
λλ
Wielko$) Tq ∇λ=ρρ nazywamy g&sto$ci" strumienia ciep#a, za$
z
Tk
y
Tj
x
TiT
∂∂
+∂∂
+∂∂
=∇ρρρρ
gradientem temperatury.
O$rodek jest, ze wzgl&du na pewn" w#a-$ciwo$) opisan" parametrem χ:
· JEDNORODNY - gdy χ nie jest funk-
cja po#o!enia, (w ka!dym punkcieo$rodka χ ma t& sam" warto$)),
· NIEJEDNORODNY - gdy χ jest funk-
cj" po#o!enia,
· IZOTROPOWY - gdy χ nie zale!y od
kierunku w którym mierzy si& j" wo$rodku.
· ANIZOTROPOWY - gdy χ zale!y od
kierunku pomiarowego.
Rys. 1.45.

56
Operator zk
yj
xi
∂∂
+∂∂
+∂∂
=∇ ρρρρ
nazywany jest operatorem nabla (tu zosta#
zapisany w trójwymiarowym uk#adzie kartezja*skim).Gradient jest wektorow" funkcj" pola skalarnego pokazuj"cy kierunek i szybko$) (po wspó#rz&dnych) wzrostu pola skalarnego (tutaj pola tempera-turowego). Zatem, poniewa! pole temperaturowe mo!e by) w ogólno$ci funk-cj" równie! czasu (T=T(X,Y,Z,t)) mo!na zapisa) nast&puj"ce wyra!enia na ilo$) ciep#a przechodz"cego przez powierzchni& S w okresie od t1 do t2:
tdsTdtdsdqQS
t
1tS
t
1t∫ ∫∫ ∫ ∇λ−=⋅−=
ρρρρ - dla stanów nieustalonych,
∫∫ ⋅∇λ∆−=∆−=SS
sdTtsdqtQ ρρρρ
- dla stanów ustalonych.
Znak minus wyst&puje formalnie w powy!szych wzorach, poniewa! wek-tor T∇
ρskierowany jest
w stron& wzrastaj"cych T, za$ przep#yw ciep#a ma kierunek przeciwny.
****2.9. W!a"ciwo"ci optyczne
STAN USTALONY (STACJONARNY) - stan uk#a-du niezale!ny od czasu.
(tutaj: T jest tylko funkcj" po#o!e*: T=T(x,y,z))
STAN NIEUSTALONY (NIESTACJONARNY) -stan uk#adu zale!ny od czasu
(tutaj: T jest funkcj" po#o!enia i czasu: T=T(x,y,z,t))
Rys. 1.47. Diagram zale%no"ci koloru warstwy od stosunku atomów azotu i
tytanu w ró%nych fazach TiN

57
Z#ota barwa warstw TiN jest jedn" z ich niezwykle atrakcyjnych cech. Zale!yona bardzo silnie od sk#adu chemicznego warstwy. Ilustruje to poni!szy dia-gram (rys. 1.47) wi"!"cy kolor warstwy ze stosunkiem atomów azotu i tytanuw odpowiednich fazach TiN.Takie walory jak po#ysk, odcie*, czy "ciep#o" barwy z#otej warstwy TiNzale!" od tekstury warstwy (uprzywilejowane krystalograficzne kierunki wzro-stu ziaren), mikrostruktury (wielko$ci ziaren) oraz chropowato$ci powierzch-ni.
Na rysunku 1.48, przedstawiona jest zale!no$) wspó#czynnika odbicia TiNod d#ugo$ci padaj"cej fali elektromagnetycznej. W#a$nieta zale!no$) decyduje o kolorze warstw TiN. Dla porów-nania przytoczono wykresy dla z#ota, srebra i miedzi.
WIDMO
ELEKTROMAGNE-TYCZNE
Rys. 1.48. Zale%no"$ wspó!czynnika odbicia od d!ugo"ci
padaj&cej fali elektromagnetycznej

58
2.10. W!a"ciwo"ci elektryczne
· Oporno"$ w!a"ciwa.
Oporno$) w#a$ciwa ρ polikrystalicz-nych warstw azotku tytanu w temperaturzepokojowej zale!y od warunków technologicz-nych ich otrzymywania i waha si& w grani-cach:
ρ ∈ρ ∈ρ ∈ρ ∈ρ ∈ (20÷÷÷÷÷200) µΩµΩµΩµΩµΩcm.
Dla monokrystalicznych warstw osa-dzonych epitaksjalnie na pod#o!u z monokry-stalicznego MgO (dla kierun-ku <111>)oporno$) w#a$ciwa wynosi:
ρρρρρ = 18 µΩµΩµΩµΩµΩcm.
Dla litego, spiekanego TiN ρ zawiera si& w granicach:ρ ∈ ρ ∈ ρ ∈ ρ ∈ ρ ∈ (25÷÷÷÷÷50) µΩµΩµΩµΩµΩcm
Typowe warto$ci oporno$ci w#a$ciwej dla innych materia#ów przedstawiatabela:
materia# !elazo
9,7·10 1,7·10 1,6·10 71·10 6·10 10 10
mied% srebro stalnierdzewna mosi"dz diament szk#o
+ , [ cm] -6 -6 -6 -6 -6 15 14
· Temperaturowy wspó!czynnik oporu αααααρρρρρ
Wzór definiuj"cy: ρρρρρT= ρρρρρ0[1+αααααρρρρρ(T-T0)]
αρ polikrystalicznych warstw TiN wynosi: αααααρρρρρ ∈ ∈ ∈ ∈ ∈ (900÷÷÷÷÷2000) ppm °K-1
αρ monokrystalicznego TiN (warstwa epitaksjalna): αααααρρρρρ = 6200 ppm °K-1
· Oporno"$ resztkowa
Oporno$) resztkowa ρr
jest warto$ci" oporno$ci w#a$ci-wej otrzymanej z przeci&cia eks-trapolowanego do T = 0°K wy-
1µΩcm=10-6Ωcm
EPITAKSJA - wzrost mono-krystalicznej warstwy zwi"zkuchemicznego na monokrysta-licznym pod#o!u z innego ma-teria#u o tej samej strukturzekrystalograficznej i bardzo zbli-!onej (lub tej samej) sta#ej siecikrystalograficznej co dany zwi"-zek.
RT - temperaturapokojowa (RoomTemperature)
RRRR
RT =ρρ

59
kresu ρ=ρ(T) z osi" oporno$ci w#a$ciwej. (patrz rysunek dalej)
Typowe warto$ci oporno$ci resztkowej:ρρρρρr ∈ (9 ÷100) µΩ ∈ (9 ÷100) µΩ ∈ (9 ÷100) µΩ ∈ (9 ÷100) µΩ ∈ (9 ÷100) µΩcm
· Iloraz oporno"ci warstw TiN
Iloraz oporno$ci RRR (Residual Resistance Ratio) to stosunek opor-no$ci w#a$ciwej w temperaturze pokojowej do oporno$ci resztkowej.Typowe warto$ci ilorazu oporno$ci:
RRR ∈(1,8 ÷ 3)∈(1,8 ÷ 3)∈(1,8 ÷ 3)∈(1,8 ÷ 3)∈(1,8 ÷ 3)
· Magnetoopór
Warstwy TiN nie wykazuj" zale!no$ci oporno$ciw#a$ciwej od przy#o!onego z zewn"trz pola magnetycz-nego (zjawisko magnetooporu) do warto$ci pola równej15 kGs (1,5 T), w zakresie temperatur 2 ÷ 300 °K
· Koncentracja no"ników !adunku (elektronów)
Typowa warto$) koncentracji elek-tronów w polikrystalicznych warstwachTiN wynosi:
n = 6,8·1022 cm-3
i jest niezale!na od temperatury.
· Ruchliwo"$ no"ników !adunku
Typowa warto$) ruchliwo$cino$ników w polikrystalicznych war-stwach TiN w temperaturze pokojo-wej jest równa:
µµµµµ = 4,9 cm22222V-1s-1
RUCHLIWO(- definiuje si& jako stosunekpr&dko$ci dryfu elektronów swobod-nych (przewodnictwa) do nat&!enia polaelektrycznego
EEdr ρ
ρρρ υ=µ⇒µ=υ
1 Gs = 10-4 TGs - GaussT - Tesla
KONCENTRACJA:Ilo$) cz"stek / jednostk& obj&to$ci
=
3cm
1
V
Nn

60
· Temperatura krytyczna przej"cia w stan nadprzewodz&cy.
Temperatura krytycznaTC (c - critical - krytyczna) totemperatura poni!ej której ma-teria# przechodzi w stan nad-przewodz"cy. Warto$) jej jestzale!na od zewn&trznego polamagnetycznego.
Warto$) TC warstw azotku ty-tanu zawieraj" si& w granicach: TC ∈ ∈ ∈ ∈ ∈ (4 ÷ ÷ ÷ ÷ ÷ 6) °K
· Magnetyczne pole krytyczne
Magnetyczne pole krytyczne HC to warto$) zewn&trznego pola ma-gnetycznego, które niszczy stan nadprzewodz"cy materia#u znajduj"cego si&w temperaturze T ≤ TC.Magnetyczne pole krytyczne dla polikrystalicznej warstwy TiN o tempera-turze krytycznej TC = 4,7 °K wynosi:
T[°K] 4,5
450 580 1000
4,4 4,3
H [Gs]C
· Zale%no"$ w!asno"ci elektrycznych warstw TiN od parametrów tech-
nologicznych stosowanych w trakcie ich otrzymywania.
Wszystkie wymienione wy!ej parametry warstw TiN zale!" od wa-runków technologicznych ich otrzymywania. Ni!ej zamieszczone przyk#a-dowe wykresy ilustruj" to stwierdzenie.
Skroplanie helu -K. Onnes (1908)Odkrycie nadprze-wodnictwa w rt&ci -K. Onnes (1911)Leida - Holandia

61
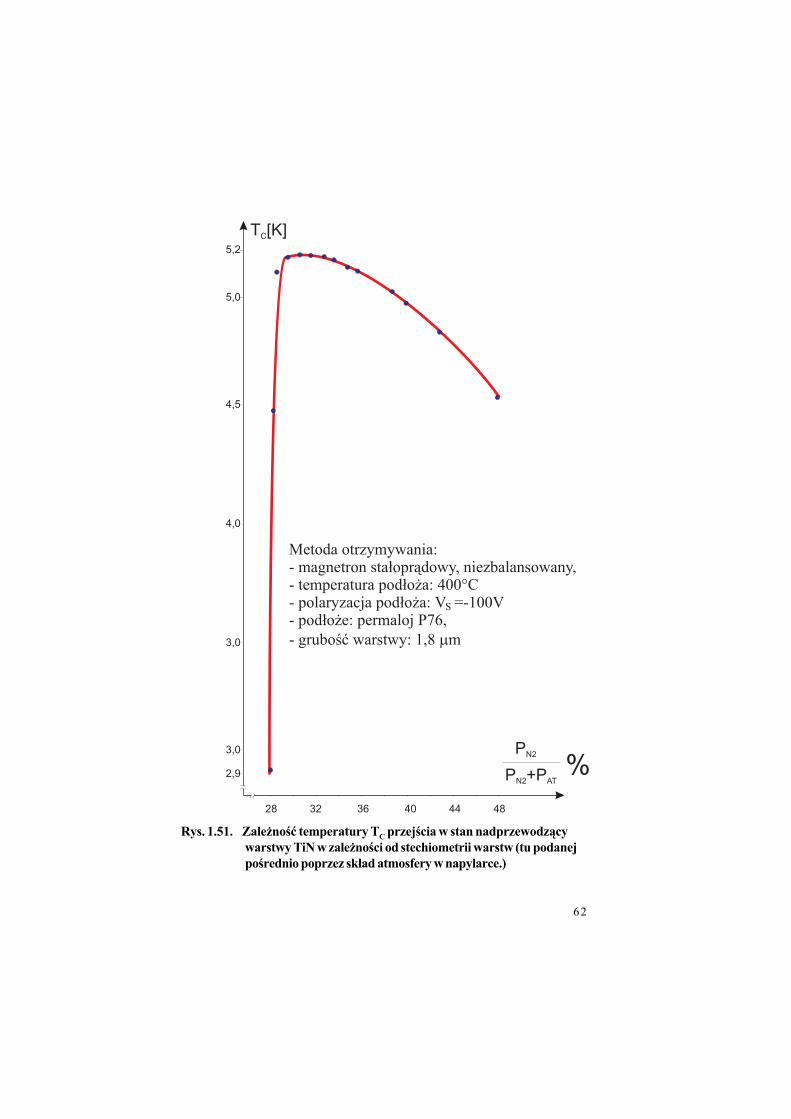
Rys. 1.49. Zale%no"$ oporno"ci w!a"ciwej warstwy TiN od stechiometrii (tu
wyra%onej poprzez stosunek ci"nienia parcjalnego azotu do
ci"nienia ca!kowitego w komorze napylarki).
Rys. 1.50. Zale%no"$ oporno"ci w!a"ciwej kilku warstw stechiometrycznego
TiN od temperatury.
62
Rys. 1.51. Zale%no"$ temperatury TC przej"cia w stan nadprzewodz&cy
warstwy TiN w zale%no"ci od stechiometrii warstw (tu podanej
po"rednio poprzez sk!ad atmosfery w napylarce.)
63
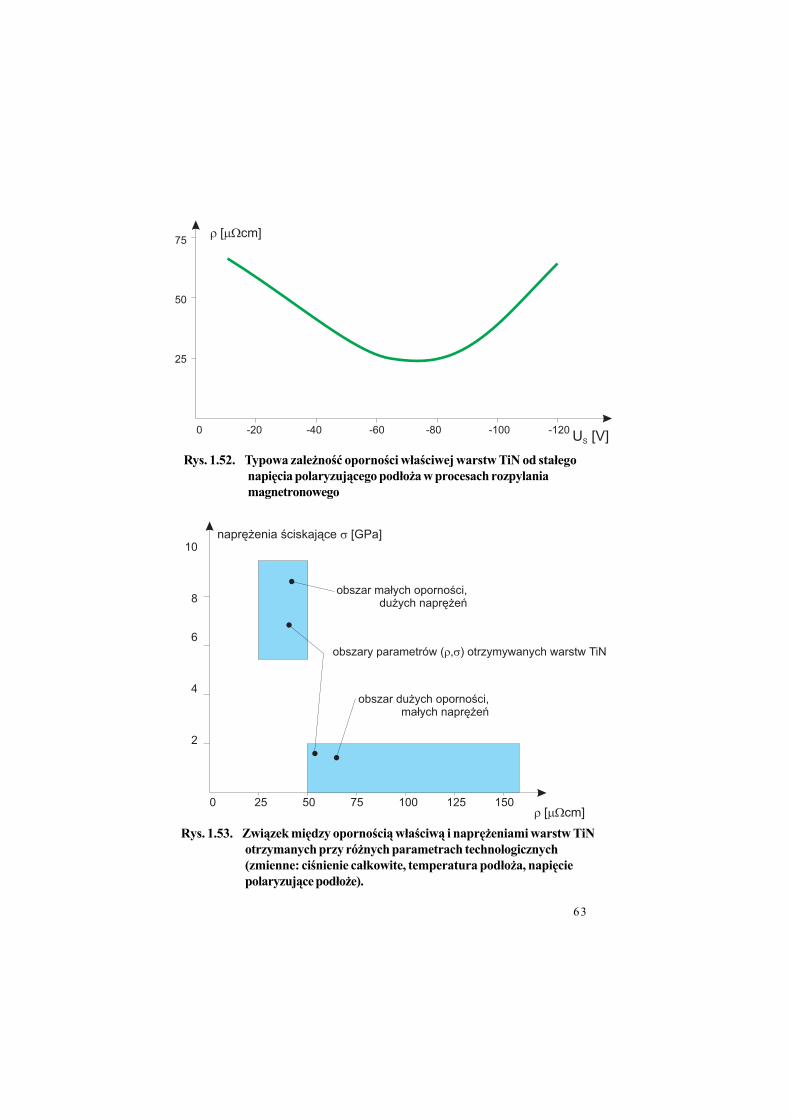
Rys. 1.52. Typowa zale%no"$ oporno"ci w!a"ciwej warstw TiN od sta!ego
napi#cia polaryzuj&cego pod!o%a w procesach rozpylania
magnetronowego
Rys. 1.53. Zwi&zek mi#dzy oporno"ci& w!a"ciw& i napr#%eniami warstw TiN
otrzymanych przy ró%nych parametrach technologicznych
(zmienne: ci"nienie ca!kowite, temperatura pod!o%a, napi#cie
polaryzuj&ce pod!o%e).

64
****Przypis o w!a"ciwo"ciach elektrycznych
Oporno$) w#a$ciw" ρ wprowadza si& poprzez:- tzw. ca#kowite prawo Ohma:
UR
1I =
- tzw. ró!niczkowe prawo Ohma:
Ejρρ
σ=
Oznaczenia: I - nat&!enie pr"du, R - oporno$), U - napi&cie, jρ
- g&sto$)
pr"du, σ - przewodnictwo w#a$ciwe, Eρ
-
nat&!enie pola elektrycznego.
G&sto$) pr"du definiuje si& jako nat&!e-nie pr"du przep#ywaj"cego przez jednost-kow" powierzchni& prostopad#" do kierun-ku przep#ywu. Zatem:
S
Ij =
Ogólniej:
∫ ⋅=S
sdjIρρ
gdzie: $renj υ=ρρ
, n - koncentracja elektronów, $rυρ
- $rednia pr&dko$) elek-
tronów w kierunku pola elektrycznego, e - #adunek elektronu.
Pr&dko$) unoszenia $rυρ
jest proporcjonalna do nat&!enia pola Eρ
(tzw. for-mu#a Langevin`a):
E$r
ρρµ=υ .
'ADUNEK ELEKTRYCZNYe = 1,6×10-19CC - Coulomb
Wspó#czynnik µ nazywany jestRUCHLIWO(CI, NO(NIKÓW 'ADUNKU
⋅
=µsV
cm][
2
Rys. 1.54. Okre"lenie nat#%enia
pr&du elektrycznego

65
Zatem:
µ=ρ
=σ
µ=υ=σ=
en1
EenenEj $r
ρρρρ
Mierz"c ρ mo!na okre$li) iloczyn kon-centracji i ruchliwo$) no$ników #adun-ku.W przypadku regularnej, prostopad#o-$ciennej próbki oporno$) w#a$ciw" ρdefiniuje si& nast&puj"co:
l
S
I
Ul
SR
S
lR
⋅=ρ⇒
⋅=ρ⇒⋅ρ=
Wymiarem ρ jest [Ω·cm]
Do niezale!nego pomiaru kon-centracji mo!na wykorzysta) efektHall'a. Mierzona w tym efekcie tzw.sta#a Hall'a RH jest w modelu elek-tronów swobodnych metalu okre$lo-na formu#":
−=
C
cm
en
1R
3
H .
Zatem mierz"c oporno$) w#a$ciw"oraz efekt Hall'a mo!na znale%) kon-centracj& i ruchliwo$) elektronów:
[ ]
[ ]112H
3
H
sVmR
cmeR
1n
−−
−
ρ=µ
=
Rys. 1.55.
EFEKT HALL'A (odkrycie 1879r.)Je$li próbk& (prostopad#o$cian) przez któ-r" p#ynie pr"d I umie$ci si& w polu magne-
tycznym o indukcji Bρ
, to mi#dzy bokami
próbki prostopad!ymi do Bρ
oraz kierunkuprzep!ywu pr&du powstanie napi#cie UHzwane napi&ciem Hall'a:
L
BIRU HH
⋅=

66
W azotku tytanu dominuj"cym wi"zaniem jest wi"zanie kowalencyjneco manifestuje si& du!" trwa#o$ci" mechaniczn" i wysokim punkcie topnieniazwi"zku. Udzia# wi"zania metalicznego wi"!e si& z utworzeniem przez shy-brydyzowane ostatnie orbitale N i Ti pasma przewodnictwa pokrywaj"cegosi& z pasmem energetycznym elektronów walencyjnych.Jest zatem TiN bardzo dobrym przewodnikiem metalicznym o czym $wiad-czy warto$) jego oporno$ci w temperaturze pokojowej. Charakter metalicz-ny TiN potwierdza temperaturowa zale!no$) ρ. Jest ona bowiem liniowa wzakresie wy!szych temperatur:
ρρρρρ=ρρρρρ0(1+αααααρρρρρ∆∆∆∆∆T)
gdzie: ∆T=T-T0 - ró!nica temperatur pomiaru ρ i ρ0, αρ - temperaturowywspó#czynnik oporu.
Innym argumentem przemawiaj"cym za metalicznym charakteremprzewodnictwa elektrycznego TiN jest sta#o$) koncentracji elektronów wfunkcji temperatury.
Pomiary oporno$ci w#a$ciwej #atwe do przeprowadzenia w porówna-niu z innymi technikami analitycznymi, pozwalaj" szybko okre$li) „jako$)”cienkich warstw TiN. Dodatkowo, ze wzgl&du na istnienie rozbudowanychteorii przewodnictwa elektrycznego cienkich warstw mo!na, wykorzystuj"cgotowe formu#y i posiadane rezultaty pomiaru oporno$ci w#a$ciwej oceni)np. $redni" wielko$) ziaren warstwy, stan powierzchni ziaren itp.Uwaga:
Niekiedy, szczególnie w zastosowaniach optycznych dla scharaktery-zowania w#a$ciwo$ci elektrycznych cienkich warstw u!ywa si& parametrunazywanego oporno$ci"na kwadrat - oznaczenieR.
Metoda pomiarutego parametru przedsta-wiona jest na rysunku1.56.
Na warstw&, nanie-sion" na prostopad#o$cien-ne pod#o!e o szeroko$ci a,nak#ada si& dwie p#askieelektrody (zwykle pastaprzewodz"ca) odleg#e o L.Niech h b&dzie grubo$ci"
Rys. 1.56. Schemat ideowy metody pomiaru
oporno"ci kwadratu R

67
warstwy. Mierzy si& opór warstwy R jako: I
VR = , a nast&pnie dzieli si& uzy-
skan" warto$) przez stosunek d#ugo$ci warstwy L do jej szeroko$ci a. Stosu-
nek a
L okre$la nic innego jak ilo$) kwadratów o boku a na które mo!na po-
dzieli) powierzchni& warstwy.
Zatem: .R
Ra
L = Z drugiej strony: ha
LR
⋅ρ= , st"d:
aL
Rh
R=
ρ= .
Mamy wi&c ostatecznie: hR
Ra
L
ρ== .
Mierz"c niezale!ni grubo$) warstwy mo!na wi&c okre$li) szybko opor-no$) w#a$ciw" warstwy.
****
2.11.Adhezja i tarcie
Silne przyleganie (adhezja) warstw azotku tytanu do pod#o!y jest bo-daj czy nie najwa!niejsz" cech" warunkuj"c" ich wykorzystanie dla celówpraktycznych. Zapewnieniu dobrego przylegania po$wi&ca si& kilka krokóww ka!dej z procedur otrzymywania cienkich warstw TiN.
Niestety pomiar si# wi"!"cych warstw& z pod#o!em jest bardzo trud-ny. Istnieje w tym wzgl&dnie kilka metod, lecz ka!da z nich ma charakterraczej jako$ciowy. Osi"gane rezultaty s" rezultatami porównawczymi i mog"by) rozbie!ne nawet w obr&bie jednej metody, o ile nie u!ywa si& standar-dowych przyrz"dów pomiarowych.
Odnosi si& wra!enie, !e ogóln" akceptacj& je$li chodzi o pomiar adhe-zji warstw TiN, uzyska#a metoda zwana metod" rysy realizowana przy wy-korzystaniu urz"dzenia o nazwie REVETEST produkcji szwajcarskiej. Osi"-gane t" metod" i przy pomocy wymienionego urz"dzenia rezultaty si#y ad-hezji warstw TiN do pod#o!y uznaje si& za porównywalne.
Schemat ideowy urz"dzenia do pomiaru si#y adhezji metod" rysy przed-stawia rysunek 1.57.

68
Przy pomiarze adhezji metod" rysy pod#o!e z warstw" przesuwa si&ruchem jednostajnym ( v = const.) wzgl&dem dotykaj"cego warstwy wg#&b-nika diamentowego typu C Rockwell'a (wg#&bnik hemisferyczny, o k"ciesto!kowym 120° i promieniu 200 µm). Wg#&bnik obci"!ony jest si#" Fn,liniowo narastaj"c" w czasie.
Stosowne czujniki (zwykle piezoelektryczne) mierz" warto$) si#y ob-ci"!enia Fn oraz si#y tarcia Ft w trakcie ruchu warstwy. Oddzielny detektormierzy generowany w tym czasie sygna# akustyczny. Dodatkowo urz"dze-nie wyposa!one jest w mikroskop metalograficzny umo!liwiaj"cy obserwa-cj& powsta#ego $ladu ruchu wg#&bnika po próbce tj. rysy.
Odpowiedni system mechaniczno - elektroniczny s#u!y do sterowa-nia i kontroli urz"dzenia oraz do zbierania, analizy i archiwizacji danych.Dla potrzeb analizy zjawisk zachodz"cych w trakcie testu rysy mo!liwy jestzapis nast&puj"cych zale!no$ci:· si#y nacisku w funkcji czasu lub po#o!enia,· si#y tarcia w funkcji czasu lub po#o!enia,· si#y tarcia w funkcji si#y nacisku,· wspó#czynnika tarcia w funkcji czasu, po#o!enia lub si#y nacisku,· amplitudy sygna#u akustycznego w funkcji czasu lub po#o!enia.Naturalnie, kilka z tych zale!no$ci mo!e by) zapisywane jednocze$nie.
Je$li chodzi o sygna# akustyczny powstaj"cy podczas ruchu wg#&bni-ka, to zapisowi podlega zwykle jego amplituda, b&d"ca ca#k" po charaktery-styce detektora. Istnieje jednak mo!liwo$) przeprowadzania analizy widmo-
Rys. 1.57. Schemat uk!adu do pomiaru adhezji warstw TiN metod& rysy.
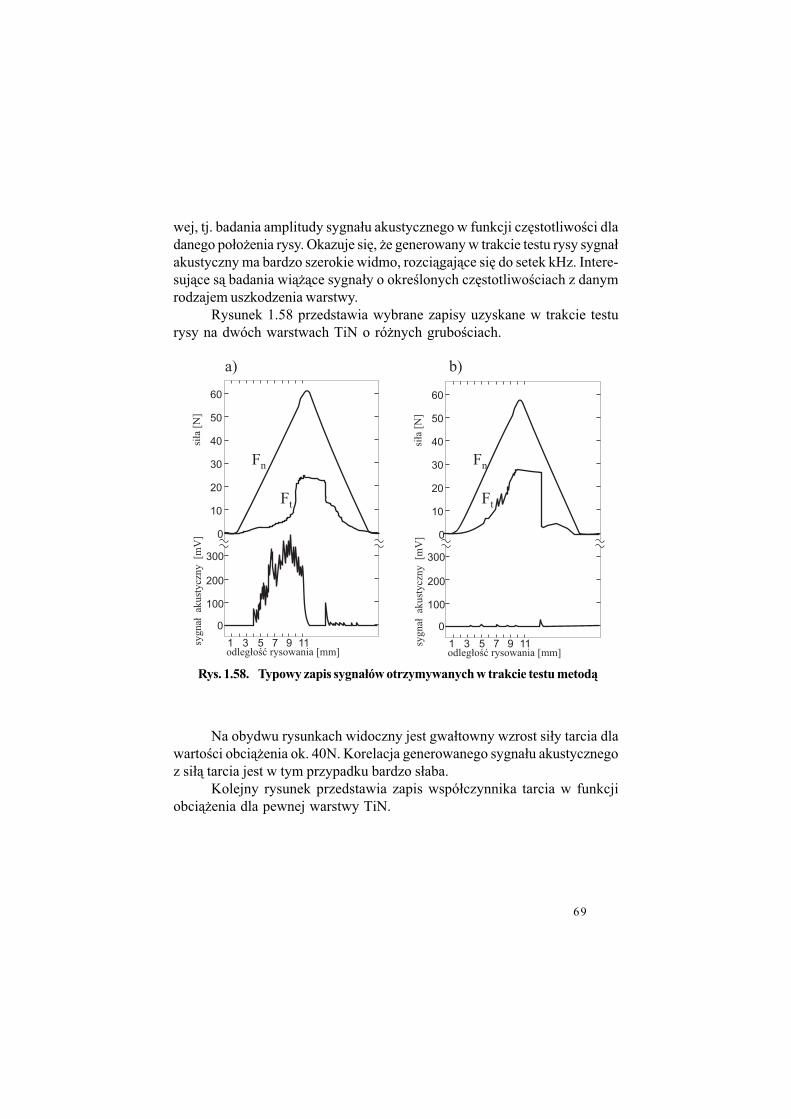
69
wej, tj. badania amplitudy sygna#u akustycznego w funkcji cz&stotliwo$ci dladanego po#o!enia rysy. Okazuje si&, !e generowany w trakcie testu rysy sygna#akustyczny ma bardzo szerokie widmo, rozci"gaj"ce si& do setek kHz. Intere-suj"ce s" badania wi"!"ce sygna#y o okre$lonych cz&stotliwo$ciach z danymrodzajem uszkodzenia warstwy.
Rysunek 1.58 przedstawia wybrane zapisy uzyskane w trakcie testurysy na dwóch warstwach TiN o ró!nych grubo$ciach.
Na obydwu rysunkach widoczny jest gwa#towny wzrost si#y tarcia dlawarto$ci obci"!enia ok. 40N. Korelacja generowanego sygna#u akustycznegoz si#" tarcia jest w tym przypadku bardzo s#aba.
Kolejny rysunek przedstawia zapis wspó#czynnika tarcia w funkcjiobci"!enia dla pewnej warstwy TiN.
Rys. 1.58. Typowy zapis sygna!ów otrzymywanych w trakcie testu metod&

70
Widoczne s" dwie warto$ci ob-ci"!enia wg#&bnika L1 i L2 (stosuje si&te! oznaczenia LC1, LC2) zwane pierw-szym i drugim obci"!eniem krytycz-nym. Zwi"zane s" one ze zmian" si#ytarcia podczas testu. Pierwsze obci"-!enie krytyczne LC1 wi"!e si& z po-cz"tkiem procesu niszczenia warstwy(uszkodzenia kohezyjne), drugie LC2
z odrywaniem warstwy od pod#o!a(tzw. uszkodzenie adhezyjne). Jakwida) z rysunku 1.59 dla badanejwarstwy wynosz" one odpowiednioLC1~16N, LC2=40N.
Warto$) obci"!e* krytycznychmo!na potwierdza) poprzez badanieprzebiegów sygna#u akustycznego lubte! wizualnie poprzez obserwacj& mi-kroskopow" - patrz rysunek 1.60.
Przyjmuje si&, !e warstwy TiNdla zastosowa* praktycznych powin-ny charakteryzowa) si& si#" adhezjiwyra!on" przez drugie obci"!enie kry-tyczne LC2 o warto$ciach 50-80N.
Rys. 1.59. Zapis wspó!czynnika
tarcia w funkcji obci&%e-
nia normalnego dla testu
Rys. 1.60.Obrazy rysy na warstwie TiN:
a) w obszarze pierwszego obci&%enia
krytycznego L1,
b) po przekroczeniu obci&%enia L2.
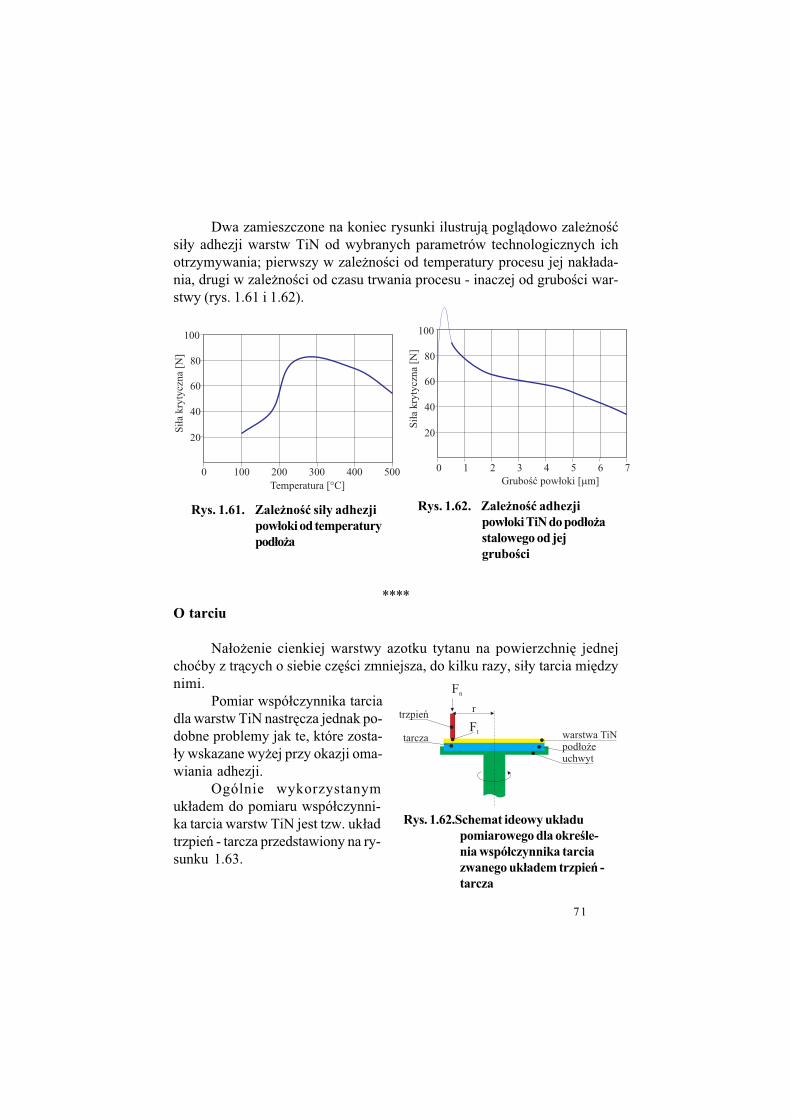
71
Dwa zamieszczone na koniec rysunki ilustruj" pogl"dowo zale!no$)si#y adhezji warstw TiN od wybranych parametrów technologicznych ichotrzymywania; pierwszy w zale!no$ci od temperatury procesu jej nak#ada-nia, drugi w zale!no$ci od czasu trwania procesu - inaczej od grubo$ci war-stwy (rys. 1.61 i 1.62).
****O tarciu
Na#o!enie cienkiej warstwy azotku tytanu na powierzchni& jednejcho)by z tr"cych o siebie cz&$ci zmniejsza, do kilku razy, si#y tarcia mi&dzynimi.
Pomiar wspó#czynnika tarciadla warstw TiN nastr&cza jednak po-dobne problemy jak te, które zosta-#y wskazane wy!ej przy okazji oma-wiania adhezji.
Ogólnie wykorzystanymuk#adem do pomiaru wspó#czynni-ka tarcia warstw TiN jest tzw. uk#adtrzpie* - tarcza przedstawiony na ry-sunku 1.63.
Rys. 1.61. Zale%no"$ si!y adhezji
pow!oki od temperatury
pod!o%a
Rys. 1.62. Zale%no"$ adhezji
pow!oki TiN do pod!o%a
stalowego od jej
grubo"ci
Rys. 1.62.Schemat ideowy uk!adu
pomiarowego dla okre"le-
nia wspó!czynnika tarcia
zwanego uk!adem trzpie' -
tarcza

72
Próbk& w kszta#cie p#askiego walca, która stanowi pod#o!e z naniesion"na* warstw" TiN, mocuje si& w obrotowym uchwycie. Instrumentalnie stanowiona tarcz& urz"dzenia. W odleg#o$ci r od osi tarczy dociskany jest do niejtrzpie* o znanej geometrii wykonany z materia#u wzgl&dem którego zamierzasi& bada) wspó#czynnik tarcia warstwy TiN.
Trzpie* obci"!ony jest zmienn" liniowo w czasie si#" Fn, która wrazze styczn" do pod#o!a si#" tarcia Ft mierzona jest przez czujniki si#y.
Tarcz& wprowadza si& w ruch obrotowy ze sta#" cz&stotliwo$ci" i wtrakcie ruchu mierzony jest wspó#czynnik tarcia jako stosunek Ft do Fn.
Wbrew prostocie w formalnym zdefiniowaniu oraz prostocie urz"dze-nia pomiarowego jednoznaczny pomiar wspó#czynnika jest trudny. Podob-nie trudna jest interpretacja wyników.
Mo!na przyj"), !e wspó#czynnik tarcia warstwy TiN o stale waha si&w granicach: 0,1 ÷ 0,6.
****2.12.Mikrostruktura
Obecnie ogólnie akceptuje si& pogl"d, !e mikrostruktura cienkichwarstw otrzymywanych metodami PVD bardzo silnie wp#ywa na ich w#a-$ciwo$ci fizyczne i chemiczne. Rzeczywi$cie mikrostruktura okre$la przynaj-mniej w cz&$ci, takie w#a$ciwo$ci warstw jak twardo$), wspó#czynnik tarcia,odporno$) na zu!ycie, odporno$) na korozj&, wspó#czynnik za#amania $wiat#aa nawet kolor.
Mikrostruktur& warstw z kolei mo!na zmienia) przez dobór odpo-wiednich parametrów w procesach PVD zwi"zanych, ogólnie mówi"c, zesterowaniem ilo$ci" energii dostarczanej do wzrastaj"cej warstwy. Energiata jest czynnikiem krytycznym dla ostatecznej mikrostruktury warstw, de-cyduje bowiem o ruchliwo$ci padaj"cych na pod#o!e atomów czyli w konse-kwencji o mechanizmach zarodkowania i wzrostu warstw.
Cienka warstwa tworz"ca si&, w procesach PVD z fazy gazowej (pla-zmy), na sta#ym pod#o!u mo!e mie) strukturalnie charakter monokrysta-liczny, polikrystaliczny lub amorficzny.
Wytwarzanie warstw monokrystalicznych (epitaksjalnych) i amorficz-nych (bezpostaciowych) wymaga specjalnych zabiegów - tego rodzaju war-stwy nie b&d" tu omawiane. Standardowo wytwarzane warstwy TiN maj"posta) polikrystaliczn".
Proces kondensacji atomów na pod#o!u mo!na przedstawi) np. w trzechetapach (patrz tak!e rysunek 1.23). Po pierwsze padaj"ce na powierzchni&

73
pod#o!a atomy przekazuj" sieci krystalograficznej pod#o!a swoj" energi& kine-tyczn" i wi"!" si& s#abo, dzi&ki si#om van der Walls'a, z atomami pod#o!a.Dalej, atomy te mog" dyfundowa) po powierzchni wymieniaj"c energi& z sieci",innymi atomami na powierzchni, mog" zosta) zdesorbowane lub te! zlokalizo-wane przez niskoenergetyczne centra na powierzchni - tworzy si& mo!liwo$)zarodkowania. Ostatni etap to dyfuzja obj&to$ciowa atomów w powstaj"cejwarstwie.
W zwi"zku z tym mo!na powiedzie), !e wzrost warstwy zwi"zany jestz nast&puj"cymi podstawowymi procesami:· cieniowaniem (prosta, geometryczna relacja pomi&dzy nierówno$ciami
powierzchni, na której wzrasta warstwa, a k"tami padania atomów),· dyfuzj" powierzchniow",· dyfuzj" obj&to$ciow",· desorpcj".
Procesy te mog" zosta) ilo$ciowo uj&te w ramach parametrów chro-powato$ci, energii aktywacji procesów dyfuzji powierzchniowej i obj&to-$ciowej oraz poprzez energi& sublimacji (desorpcja). Energie te s" proporcjo-nalne do temperatur topnienia (Ttop) materia#u warstwy, zatem nale!y ocze-kiwa), !e w zale!no$ci od stosunku temperatury pod#o!a (T) do temperatu-ry Ttop, zmienia) si& mo!e dominacja wy!ej wymienionych efektów w proce-sie wzrostu warstwy.
Rys. 1.64. Strefowy model mikrostruktury warstw
wg. Movczana - Demcziszina

74
Strefowy model mikrostruktury warstw Movczana - Demcziszina opi-suje rezultaty pró!niowych procesów naparowywania grubych warstw (0,3- 2 mm). Jedynym zmiennym w tym wypadku parametrem jest temperaturapod#o!a T. (rys. 1.64).
Strefa I, dla T/Ttop<0,3, zbudowanajest z pojedynczych, oddzielonych pustkamikrystalitów, których wewn&trzna strukturajest mocno zdeformowana, o du!ej g&sto$cidyslokacji (rys. 1.65). Dla okre$lenia typu mi-krostruktury warstwy w tej strefie u!ywa si&terminu „kolumnowa porowata”. W strefie tejruchliwo$) padaj"cych na powierzchni& ato-mów jest ma#a, atomy przylegaj" do pod#o!aw miejscach na które padn" - podstawowymmechanizmem steruj"cym wzrostem warstwyjest mechanizm cieniowania.
Strefa II (0,3<T/Ttop<0,5) sk#ada si& z kolumnowych ziaren oddzie-lonych przez wyra%ne, g&ste granice mi&dzykrystaliczne. Strefa nazywanajest „kolumnow" - g&st"”. Powierzchnia warstwy jest g#adka, matowa. Dys-lokacje zlokalizowane s" g#ównie na granicach ziaren. Wymiary ziaren wzra-staj" ze wzrostem T/Ttop i mog" rozci"ga) si& na ca#" grubo$) warstwy.Zale!no$) szeroko$ci ziaren od T/Ttop dostarcza warto$ci energii aktywacjidyfuzji powierzchniowej.
Strefa III (0,5<T/Ttop<1) sk#ada si& z du!ych ziaren o jasnych po-wierzchniach. (rednica ziaren wzrasta ze wzrostem stosunku T/Ttop i zale!-no$) ta cechuje si& energi" aktywacji odpowiadaj"c" procesowi dyfuzji obj&-to$ciowej. Struktura i w#a$ciwo$ci warstwy w tej strefie s" podobne do w#a-$ciwo$ci materia#u litego po obróbce cieplnej, tj. po rekrystalizacji i wzro-$cie ziaren.
Strefowy model mikrostruktury warstw Thortona opisuje budow&warstw otrzymywanych metod" rozpylania magnetronowego. Uwzgl&dniaon dodatkowo ci$nienie gazu roboczego - argonu istniej"ce podczas procesu(rys. 1.66). Wyst&puj" w nim trzy strefy podobnie jak w modelu Movczana- Demcziszina.
Rys. 1.65. Mikrostruktura
warstwy w
strefie I

75
Wyra%nie zarysowana strefa I uwarunkowana jest przez chropowato$)powierzchni, wysokie ci$nienie argonu oraz niskok"towe sk#adowe strumieniapadaj"cych na powierzchni& atomów. Pomi&dzy strefami I i II zosta#azidentyfikowana dodatkowa strefa przej"ciowa T
(T od transition - przej$cie) o charakterystycznej w#óknistej, g&stej struktu-rze krystalitów. Struktura ta posiada du!" twardo$) i wytrzyma#o$), wyst&-puj"ce w niej napr&!enia maj" charakter $ciskaj"cy. Powierzchnia warstwyw strefie T jest g#adka. Kolumnowa strefa II posiada bardziej zarysowan"powierzchni& zewn&trzn". Ci$nienie gazu roboczego ma niewielki wp#yw nastruktur& warstwy w strefie w zakresie wysokich temperatur (strefa III). Ba-dania wykaza#y, !e optymalne w#a$ciwo$ci warstw uzyskuje si& dla T/Ttop≈0,3,w warunkach prostopad#ego podawania atomów na pod#o!e.
Kolejny model, Messier'a (rys. 1.67), uwzgl&dnia energi& atomów Ei
padaj"cych na pod#o!e w procesach w których pod#o!e jest dodatkowo ujem-nie polaryzowane. Wskazuje on na fakt poszerzania si& w skali temperaturystrefy T w miar& wzrostu energii padaj"cych na pod#o!e atomów. Strefa ta,w przypadku braku bombardowania pod#o!a przez padaj"ce na* jony i ato-my (jest to równowa!ne wzrostowi ci$nienia roboczego) jest w"ska lub mo!ew ogóle nie istnie). Je!eli energia cz"stek bombarduj"cych wzrasta szero-ko$) strefy T ro$nie kosztem strefy I, przy czym granica strefy II pozostaje wprzybli!eniu bez zmian.
Rys. 1.66. Model strefowy mikrostruktury cienkich warstw wg. Thortona

76
Rysunki 1.68 i 1.69 ilustruj" wp#yw dodatkowego bombardowania powierzchni pod#o!a na zmiany mikrostruktury narastaj"cej warstwy.
Rys. 1.67. Strefowy model budowy warstw Messier`a
Rys. 1.68. Kolumnowy wzrost
warstwy jako
rezultat efektu
cieniowania
Rys. 1.69. Wp!yw bombardowania
jonowego na zmian# charak-
teru mikrostruktury warstwy

77
Gdy nie istnieje bombardowanie powierzchni na której narasta war-stwa przez jony gazu roboczego (rys. 1.68) zarodkowanie warstwy i jej ko-lumnowy wzrost rozpoczynaj" si& na nierówno$ciach pod#o!a (tzw. centrazarodkowania, osobliwo$ci powierzchniowe) prowadz"c do widocznego narysunku efektu cieniowania. Powstaje struktura charakterystyczna dla stre-fy I modelu strefowego mikrostruktury warstw.
Ujemne spolaryzowanie pod#o!a (rys. 1.69) powoduje procesy rozpy-lania (wskazane na rysunku) atomów gromadz"cych si& na nierówno$ciachpowierzchni. W rezultacie prowadzi to do likwidacji efektu cieniowania istwarza mo!liwo$ci powstawania struktury krystalitów charakterystycznejdla stref T lub II modelu strefowego budowy warstw.
Istniej" obecnie bardziej szczegó#owe modele mikrostruktur cienkichwarstw otrzymywanych przy pomocy ró!nych technik PVD wspomaganychjonowo. Bior" one pod uwag&, w superpozycji lub jako niezale!ne nast&pu-j"ce czynniki warunkuj"ce charakter wzrostu i mikrostruktury warstw:• stosunek temperatury pod#o!a do temperatury topnienia materia#u war-
stwy T/Tm(tzw. temperatura homologowana),• ci$nienie gazu w procesie technologicznym,• energi& jonów padaj"cych na pod#o!e (proporcjonalna do ujemnego na-
pi&cia polaryzuj"cego pod#o!e),• stosunek ilo$ci jonów do atomów padaj"cych w jednostce czasu na pod-#o!e.
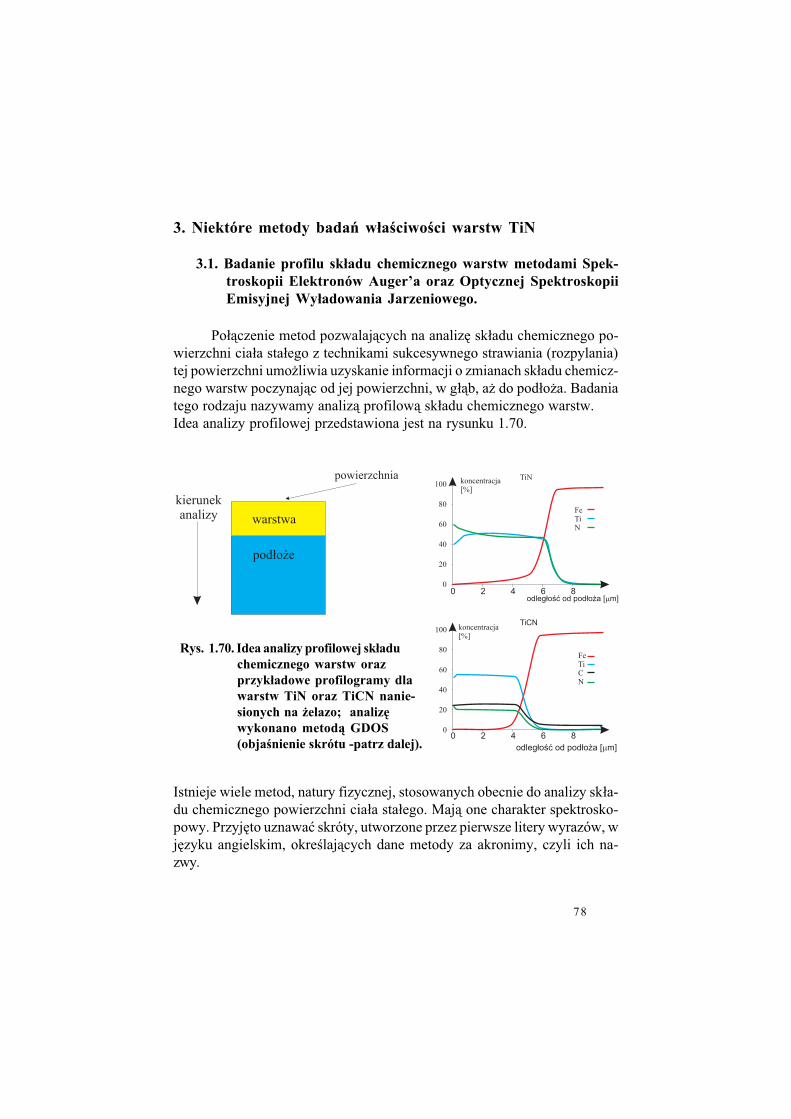
78
3. Niektóre metody bada! w"a#ciwo#ci warstw TiN
3.1. Badanie profilu sk"adu chemicznego warstw metodami Spek-
troskopii Elektronów Auger’a oraz Optycznej Spektroskopii
Emisyjnej Wy"adowania Jarzeniowego.
Po!"czenie metod pozwalaj"cych na analiz# sk!adu chemicznego po-wierzchni cia!a sta!ego z technikami sukcesywnego strawiania (rozpylania) tej powierzchni umo$liwia uzyskanie informacji o zmianach sk!adu chemicz-nego warstw poczynaj"c od jej powierzchni, w g!"b, a$ do pod!o$a. Badania tego rodzaju nazywamy analiz" profilow" sk!adu chemicznego warstw. Idea analizy profilowej przedstawiona jest na rysunku 1.70.
Istnieje wiele metod, natury fizycznej, stosowanych obecnie do analizy sk!a-du chemicznego powierzchni cia!a sta!ego. Maj" one charakter spektrosko-powy. Przyj#to uznawa% skróty, utworzone przez pierwsze litery wyrazów, w j#zyku angielskim, okre&laj"cych dane metody za akronimy, czyli ich na-zwy.
Rys. 1.70. Idea analizy profilowej sk"adu
chemicznego warstw oraz
przyk"adowe profilogramy dla
warstw TiN oraz TiCN nanie-
sionych na $elazo; analiz%wykonano metod& GDOS
(obja#nienie skrótu -patrz dalej).

79
Wymie'my kilka z tych metod:• AES - Auger Electron Spectroscopy - spektroskopia elektronów Auger’a,• SIMS - Secondary Ion Mass Spectroscopy - spektroskopia masowa jo-
nów wtórnych,• SNMS - Secondary Neutrals Mass Spectroscopy - spektroskopia maso-
wa zoboj#tnionych jonów wtórnych,• ESCA - Electron Spectroscopy for Chemical Analysis - spektroskopia
elektronowa dla celów analizy chemicznej. Metoda ta ma dwa warianty,zale$nie od energii fotonów pobudzaj"cych próbk#; mamy wi#c:
• UPS - Ultraviolet Photoelectron Spectroscopy - spektroskopiafotoelektronów wzbudzanych promieniowaniem ultrafioletowym,
• XPS - X-ray Photoelectron Spectroscopy - spektroskopia foto-elektronów wzbudzanych promieniowaniem rentgenowskim,
• RBS - Rutherford Back Scattering - spektroskopia jonów rozpraszanychw geometrii Rutherforda,
• GDOES - Glow Discharge Optical Emission Spectroscopy - optycz-na spektroskopia emisyjna wy!adowania jarzeniowego (niekiedy stoso-wana jest nazwa: GDOS - Glow Discharge Optical Spectroscopy - spek-troskopia optyczna wy!adowania jarzeniowego),
• GDMS - Glow Discharge Mass Spectroscopy - spektroskopia masowawy!adowania jarzeniowego.
Wszystkie z wymienionych metod (oprócz GDOS) pozwalaj" na ana-
liz# próbek na g!#boko&% kilku do kilkudziesi#ciu Ångstromów, tj. kilku dokilkunastu sta!ych sieci. Chc"c dokona% analizy profilowej nale$y sukce-sywnie usuwa% przeanalizowane ju$ warstwy poprzez rozpylanie, zwykle przypomocy dzia!a jonowego.
Ogólna idea wszystkich metod jest taka sama, niezale$nie od wyko-rzystanych do celów analizy zjawisk fizycznych. Powierzchnia próbki pod-dawana jest dzia!aniu strumienia cz"stek (elektronów, jonów, fotonów) ookre&lonych parametrach (energia, nat#$enie), a analizie i detekcji podlega-j" cz"stki wyzwalane z powierzchni (jony, elektrony, fotony). W rezultacieotrzymuje si# zapis nat#$enia emitowanych z powierzchni cz"stek w funkcjienergii, masy lub cz#stotliwo&ci, czyli ich widmo (inaczej spektrum). St"dokre&lenie ich nazw - spektroskopowe.
Inn" wspóln" cech" dla metod analizy powierzchni jest wykorzysty-wanie wysokiej jako&ci systemów pró$niowych w zakresie ultrawysokiej wy-sokiej pró$ni oraz wyrafinowanych elementów techniki i miernictwa pró$-niowego.

80
Dalej zostanieomówiona jedna z wy-mienionych wy$ej me-tod, mianowicieGDOS. Jednak dla zilu-strowania ró$norodno-&ci zjawisk fizycznychwykorzystywanych w
analizie profilowej omówmy najpierw pokrótce najpopularniejsz" chybaz nich tj. Spektroskopi# Elektronów Auger’a.
Schemat ideowy metody AES przedstawiony jest na rysunku 1.71.Na powierzchni# próbki pada wi"zka elektronów Ep o du$ej energii, 5 - 10keV, znacznie przewy$szaj"cej energi# jonizacji atomów próbki z podstawo-wego poziomu energetycznego K. W wyniku oddzia!ywania padaj"cej wi"z-ki elektronów z powierzchni" próbki nast#puje rozpraszanie padaj"cych elek-tronów E
R, wyemitowane zostaj" wybite z poziomów wewn#trznych ato-
mów elektrony EE, wypromieniowana jest fala elektromagnetyczna w za-
kresie rentgenowskim X oraz emitowane s" elektrony o charakterystycz-nym widmie energetycznym, zwane elektronami Auger’a.
Wyja&nijmy mechanizm emisji elektronów Auger’a, wykorzystuj"c w tym celuuproszczony, schematyczny wykres struktury energetycznej elektronów w cielesta!ym, &ci&lej w metalu. Przedstawia go rysunek 1.72.
Auger Pierre Victor ur. 1899 fizyk francuski, prof. Sorbo-ny, i dyr. Dept. Nauk. Przyrodniczych UNESCO; prace zfizyki atomowej i promieniowania kosmicznego.Zjawisko Auger'a - bezradiacyjny sposób przej&cia atomuze wzbudzonego stanu elektronowego do stanu o ni$szejenergii po!"czony z emisj" elektronów (tzw. elektronówAuger'a z zewn#trznej Pow!oki elektronowej)
Rys. 1.71. Schemat ideowy metody AES
EP - wi&zka elektronów jonizuj&cych,
EE - elektrony wybite z próbki w wyniku jonizacji,
ER - elektrony rozproszone,
EA - elektrony Auger’a,

81
Po lewej stronie pokazany jest stan podstawowy uk!adu. Na rysunku&rodkowym padaj"cy elektron o energii E
P>>E
K wybija elektron z pozio-
mu K (jonizacja) wytwarzaj"c na nim dziur#. Dziura ta zape!niana jest przezelektron rekombinuj"cy z poziomu L
1. Wyzwalana jest przy tym energia
(∆E=EK-E
L1, która mo$e zosta% uwolniona poprzez emisj# fotonu o cz#sto-
tliwo&ci
)EE(h
11LK −=ν
lub te$ zosta% przekazana bezpromieni&cie innemu elektronowi natym samym poziomie energetycznym (lub poziomie bardziej p!ytkim) powo-duj"c jego emisj#.
Pierwszy z procesów nazywany jest fluorescencj" rentgenowsk"i wi"$e si# z tzw. charakterystycznym promieniowaniem rentgenowskim,drugi za& jest procesem emisji elektronu Auger’a.
W przyk!adzie na rysunku nadmiar energii zosta! przekazany elektro-nowi na poziomie L2,3, a zatem wyemitowany elektron Augera ma energi#:
∗−−==3,21231 LLKLKLA EEEEE
Rys. 1.72. Schematyczny wykres pasmowej struktury energetycznej metalu
ilustruj&cy mechanizm generacji elektronów Auger’a (w AES
przyjmuje si% zwyczajowo zero energii na poziomie Fermiego).

82
Gwiazdka przy sym-
bolu ∗
3,2LE oznacza, $e war-
to&% energii elektronu natym poziomie mierzona jestw warunkach istnienia dziu-ry na poziomie EL1 , ró$najest zatem od energii EL23.
Naturalnie w ato-mach cia!a sta!ego, któregostruktura energetycznaprzedstawiona jest na rysun-ku mo$liwe s" i realizuj" si#,inne przej&cia np. KL1L1,L1L2,3L2,3. Elektrony bior"ceudzia! w procesie Auger’amog" pochodzi% tak$e z pa-sma walencyjnego. W tymprzypadku przyj#to u$ywa%oznacze' na przyk!ad KL1V- je&li jeden elektron pocho-dzi z pasma walencyjnegolub te$ KVV je&li obydwaelektrony pochodz" z pasmawalencyjnego.
Najistotniejszym jest, $e energie elektronów Auger’a wyra$one for-
mu!" typu3,21LKLE s" funkcj" jedynie atomowych poziomów energetycz-
nych, zatem dla ka$dego pierwiastka uk!adu okresowego istnieje jeden itylko jeden zbiór energii elektronów Auger’a, nie ma bowiem dwóch pier-wiastków o tych warto&ciach energii wi"zania elektronów.
Gwoli &cis!o&ci nale$y wyja&ni% pewn" nie-konsekwencj# oznacze' na rysunku 4.3. Je&li rysu-nek ten ilustruje struktur# energetyczn" elektronóww metalu, to zb#dne jest wydzielanie pasma walen-cyjnego. Z teorii elektronów swobodnych wiadomobowiem, $e w metalach puste pasmo przewodnictwaprzekrywa si# z pasmem elektronów walencyjnychtworz"c kontinuum. W spektroskopii wydziela si# topasmo pragn"c podkre&li%, $e chodzi o elektrony bio-r"ce udzia! w wi"zaniach chemicznych - elektronywalencyjne. Mo$na, id"c na pewnego rodzaju kom-promis, powiedzie%, $e rysunek 4.3. przedstawia sy-tuacj# dla temperatury równej 0°K, w której elektro-ny swobodne zajmuj" wszystkie poziomy energetycz-ne do poziomu Fermiego.ENERGIA FERMIEGO EF - najwy$sza warto&% energii elek-tronów swobodnych w metalu w temperaturze zerabezwzgl#dnego, lub te$, dla T > 0°K, taki poziomenergetyczny w pa&mie przewodnictwa, któregoprawdopodobie'stwo obsadzenia przez elektronywynosi 1/2. Definicje te wynikaj" z funkcji rozk!adustatystycznego energii elektronów swobodnych wmetalu, tzw. funkcji Fermiego - Diraca:
( )1
kT
EEexp
1Ef
F
DF
+
−=−
PRACA WYJ'CIA elektronów z metalu (oznaczenie φ lubw) to warto&% energii, któr" trzeba dostarczy% elek-tronowi przewodnictwa by wyprowadzi% go zwn#trza metalu. Wielko&% ta wprowadzona jest w s!yn-nym wzorze Einstein`a opisuj"cym efekt fotoelek-tryczny (zewn#trzny):
hν=W+Ekinet
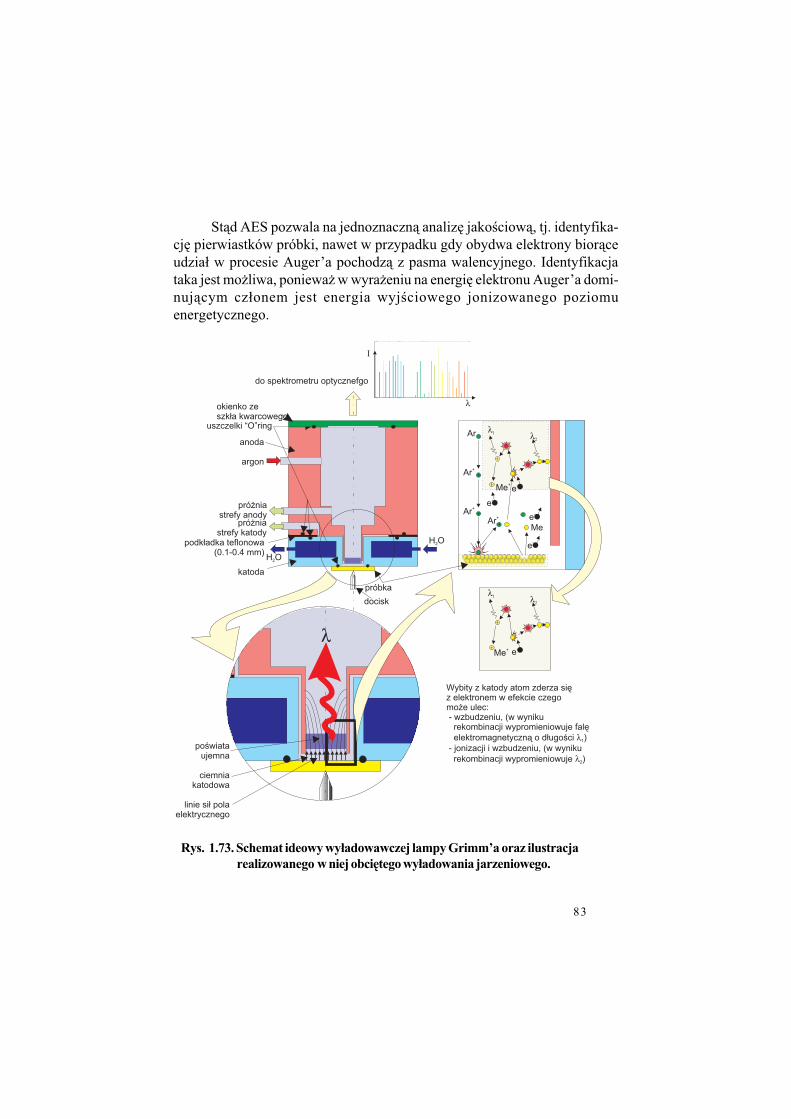
83
St"d AES pozwala na jednoznaczn" analiz# jako&ciow", tj. identyfika-cj# pierwiastków próbki, nawet w przypadku gdy obydwa elektrony bior"ce udzia! w procesie Auger’a pochodz" z pasma walencyjnego. Identyfikacja taka jest mo$liwa, poniewa$ w wyra$eniu na energi# elektronu Auger’a domi-nuj"cym cz!onem jest energia wyj&ciowego jonizowanego poziomu energetycznego.
Rys. 1.73. Schemat ideowy wy"adowawczej lampy Grimm’a oraz ilustracja
realizowanego w niej obci%tego wy"adowania jarzeniowego.

84
Analiza ilo&ciowa przy pomocy AES polega na porównywaniu uzyski-wanych widm z widmami próbek wzorcowych.
Przejd(my do zapowiadanego opisu innej z metod analizy profilowej,GDOS - Optycznej Spektroskopii Emisyjnej Wy!adowania Jarzeniowego.
Do badania profilu sk!adu chemicznego warstw i pod!o$a w GDOSwykorzystuje si# zjawisko wy!adowania jarzeniowego w anormalnej cz#&cijego charakterystyki. Badana próbka (warstwa, lity materia!) stanowi kato-d# komory pomiarowej. Anod# stanowi obudowa.
Metoda GDOS jako narz#dzie analityczne zosta!a wprowadzona w1968 roku przez W. Grimm’a. Grimm poda! te$ rozwi"zanie komory pomia-rowej, która stosowana jest do dzi&. Przedstawiona jest ona na rys. 1.73.
W istocie swej lampa Grimma stanowi napylark# diodow", w którejpod!o$em jest specjalnie skonstruowana anoda (a w!a&ciwie ekran katody),katod# za& stanowi badana próbka. Dzi#ki takiemu rozwi"zaniu realizuj"cesi# lampie Grimma wy!adowanie jarzeniowe zostaje ograniczone do obszaruzorzy (po&wiaty) ujemnej.
W GDOS (ród!em informacji o rodzaju i ilo&ci atomów tworz"cychbadan" próbk# jest powstaj"ce w wy!adowaniu jarzeniowym promieniowa-nie elektromagnetyczne w obszarze ultrafioletu, widzialnym i podczerwieni,
czyli w przybli$eniu w zakresie d!ugo&ci fali od 2000Å do 9000Å .Widmo to mo$e pochodzi% zarówno od atomów jak i jonów wybija-
nych z pod!o$a dzi#ki efektowi rozpylania diodowego i wzbudzanych optycz-nie w aktach zderze' z elektronami.
Jako gaz roboczy stosuje si# zwykle argon o wysokiej czysto&ci, które-go linie emisyjne wyst#puj" oczywi&cie tak$e w badanym widmie optycz-nym stanowi"c niepo$"dane t!o.
Zauwa$my, $e dzi#ki wykorzystaniu wy!adowania jarzeniowego wGDOS uzyskuje si# jednocze&nie: (ród!o informacji o próbce (widmo optycz-ne) oraz efekt ci"g!ego strawiania powierzchni niezb#dnej dla analizy profi-lowej.
Zwró%my teraz uwag# na schemat ideowy spektrometru GDOS(rys. 1.74). Ci&nienie robocze argonu w lampie Grimma mo$na zmienia%typowo w granicach 0,1 - 50 Tor. Za ci&nienie robocze uwa$a si# ci&nienie wstrefie anodowej (patrz rysunek 1.73). Stref# katody, tj. bardzo w"ski obszarmi#dzy katod" a anod", odpompowuje si# do wy$szej pró$ni, tak aby nie-mo$liwe by!o powstanie w niej „fa!szywego” wy!adowania jarzeniowego.Odleg!o&% katoda - anoda w tej strefie musi by% zatem mniejsza od szeroko-&ci ciemni katodowej w ca!ym zakresie stosowanych ci&nie' Ar.

85
Dla uzyskania dok!adno&ci i powtarzalno&ci pomiarów metod" GDOSstabilizuje si# ci&nienie gazu roboczego wykorzystuj"c na przyk!ad sprz#$e-nie pró$niomierza strefy anodowej z elektromagnetycznym zaworem dozu-j"cym ten gaz.
Je$eli przedmiotem bada' s" próbki przewodz"ce (metale, pó!prze-wodniki o ma!ej oporno&ci elektrycznej) do wzbudzenia wy!adowania jarze-niowego stosuje si# regulowane zasilacze sta!opr"dowe stabilizowane b"d(pr"dowo, b"d( napi#ciowo. Typowe warto&ci stosowanych pr"dów i napi#%to:
• pr"d: od kilkunastu do 300 mA,• napi#cie: od kilkuset do 2 kV.
Dla analizy próbek izolacyjnych stosuje si# zasilanie pr"dem prze-miennym wysokiej cz#stotliwo&ci (typowo 13,56 MHz).
W zwi"zku z du$" warto&ci" mocy wydzielanej na katodzie jej korpusch!odzony jest intesywnie wod".
Standardowo &rednica wewn#trzna anody w lampie Grimm’a wynosi4 lub 8 mm, jednak &rednica próbki (w kszta!cie p!askiego walca) musi by%du$o wi#ksza (10 - 300 mm) ze wzgl#du na konieczno&% dobrego kontaktucieplnego z korpusem katody (ch!odzenie).
Rys. 4.5. Schemat ideowy spektrometru GDOS.

86
W torze analizy optycznej u$ywa si# ró$nego rodzaju elementy dysper-syjne i detektory pozwalaj"ce na ci"g!y, w czasie, zapis wybranego obszaruwidma lub tylko wybranych linii emisyjnych.
Typowa, zdolno&% rozdzielcza uk!adów optycznych wynosi od 1Å do 0,1Å.Przy pomocy szybkiego systemu zbierania i obróbki danych zapisuje
si# widmo w wybranym zakresie d!ugo&ci fali, identyfikuje poszczególne li-nie widmowe pierwiastków wchodz"cych w sk!ad próbki, a nast#pnie zapi-suje si# je w funkcji czasu. Przy znanej szybko&ci strawiania próbki transfor-muje si# uzyskane przebiegi czasowe na zale$no&% koncentracji (zwykle wzgl#d-n") okre&lonego pierwiastka od odleg!o&ci od powierzchni. Uzyskuje si# wten sposób obraz profilu sk!adu chemicznego próbki.W metodzie GDOS typowe pr#dko&ci strawiania powierzchni to: 1 - 100nm/s.
Granica detekcji w GDOS wynosi od 1 - 10 ppm.Analiz# ilo&ciow" sk!adu chemicznego przeprowadza si# w oparciu o
wykresy uzyskane dla próbek wzorcowych opieraj"c si# na za!o$eniu, $enat#$enie linii widmowej danego pierwiastka jest proporcjonalne do jegoilo&ci w badanej próbce.
Dodajmy, $e GDOS wyst#puj" typowe dla metod spektrometrycz-nych problemy jak wybór do analizy „czystej” linii emisyjnej nie przekrywa-j"cej si# z liniami od innych pierwiastków, lub jej separacja, czy te$ zagad-nienie zwi#kszania stosunku sygna! u$yteczny - szum.
W GDOS dla celów analitycznych stosuje si# nast#puj"ce parame-try:
• szybko#( rozpylania:
µ
=s
g,
t
mq gdzie: m - ubytek masy katody, t -
czas.• zredukowana szybko#( rozpylania Q, tj. szybko&% rozpylania znorma-
lizowana do pr"du katodowego:
⋅µ
=As
g,
I
• szybko#( penetracji „W” tj. szybko&% rozpylania w jednostkach grubo&%warstwy na czas:
µ
=
⋅
⋅µ⋅ρ
=−−−
s
m
cmcm
g
sg10,
S
q10w
2
3
122
gdzie: q - szybko&% rozpylania,
r - g#sto&% próbki, S - powierzchnia próbki.

87
• zredukowana szybko#( penetracji, tj. szybko&% penetracji znormalizo-
wana do pr"du katodowego:
⋅µ
=As
m,
I
wW
• wydajno#( rozpylania (wydajno&% kwantowa) S, tj. ilo&% wybitych z
katody atomów przez jeden padaj"cy na katod# jon: jon
6
IM
eqN10S
⋅⋅
=−
gdzie:q
- szybko&% rozpylania [(g(s-1], N - liczba Avogadro [mol-1], e - !adunekelektryczny [C], M -ci#$ar atomowy, I
jon - pr"d jonowy
• pr&d jonowy zwi"zany jest z pr"dem ca!kowitym relacj": γ+=
1
II jon ,
gdzie: γ - &rednia ilo&% elektronów wtórnych wybijanych z katody przezjeden jon.
Podstawiaj"c numeryczne dane do wyra$enia na S i wykorzystuj"c definicj#
q oraz Ijon
otrzymuje si# parametr: M
Q0965,0
1
S=
γ+ , który okre&la si# mianem
skorygowanej wydajno&ci kwantowej.Dla celów analizy ilo&ciowej istotna jest znajomo&% szybko&ci rozpyla-
nia danej próbki. Okazuje si#, $e szybko&% rozpylania q spe!nia prost", em-
piryczn" formu!#: ( )0Q UUICq −= gdzie: I, U - pr"d i napi#cie wy!adowa-
nia, CQ,U
0 - sta!e zale$ne od materia!u katody, rodzaju u$ytego gazu robo-
czego i geometrii komory wy!adowawczej, lecz niezale$nie od pr"du i napi#-cia wy!adowania oraz ci&nienia gazu roboczego.
Metoda GDOS znajduje coraz szersze zastosowanie tak w pracachtypowo badawczych jak 2i w kontrolnych laboratoriach przemys!owych zewzgl#du na prostot# konstrukcji i niezawodno&% dzia!ania lampy Grimm’a,wysok" czu!o&%, krótki czas przeprowadzania analiz oraz stosunkowo nie-du$e koszty urz"dzenia.
Inne zalety metody GDOS to:• minimalny wp!yw osnowy (matrycy) materia!u na rezultaty analizy,• liniowo&% krzywych kalibracji dla analizy ilo&ciowej,• mo$liwo&% wykorzystania jednej krzywej kalibracyjnej dla analizy kilku
materia!ów,• mo$liwo&% równoczesnego okre&lenia dominuj"cych, minimalnych i &la-
dowych koncentracji pierwiastków w badanym materiale,• minimalne t!o widmowe.

88
Oprócz zaakcentowanego na pocz"tku zastosowania GDOS do analizy profilu sk!adu chemicznego twardych pokry% typu TiN, metod# t# wykorzy-stuje si# do analizy mi#dzy innymi:• rezultatów azotowania, naw#glania, azotonaw#glania, borowania stali,• sk!adu warstw skorodowanych, utlenionych, spasywowanych,• rezultatów implantacji jonowej,• wszelkiego rodzaju warstw nak!adanych ró$nymi metodami,• analizy ilo&ciowej: $eliwa (bia!ego, szarego, wysokostopowego itd.), stali
wszelkiego rodzaju, niklu i stopów kobaltowych, stopów miedzi (br"zy, mosi"dze), stopy aluminium, stopy tytanu itd.
3.2. Pomiar grubo#ci cienkich warstw metod& Kalotest.
Do pomiaru grubo&ci cienkich warstw wykorzystuje si# wiele metod. W&ród nich znajduj" si# mi#dzy innymi:• metoda grawimetryczna,• metoda profilograficzna,• metoda zwana Kalotest.
Metoda grawimetryczna (rys. 1.75) jest chyba najstarsz" z metod po-miaru grubo&ci warstw. Pro-cedura tej metody sprowadza si# do wa$enia pod!o$a przed i po naniesieniu na nie cien-kiej warstwy.
Je$eli: W - jest ró$nic"w ci#$arze, A - powierzchni"pod!o$a na które naniesiono warstw#, r - ci#$arem w!a&ci-wym materia!u warstwy, to grubo&% warstwy wynosi:
ρ⋅=
A
Wd
Pomijaj"c wszystkie, rzucaj"ce si# w oczy zastrze$enia, które mo$na mie%analizuj"c t# metod# nale$y zauwa$y%, $e jej dok!adno&% jest du$a (~1%) i
$e pozwala zmierzy% grubo&% warstw rz#du kilku Å.
Rys. 1.75. Metoda wagowa pomiaru
grubo#ci warstw

89
W metodzie profilograficznej (rys. 1.76) do okre&lenia grubo&ci warstw wykorzystuje si# standardo-wy profilograf u$ywany zwykle do charakteryzowania chropowato&ci powierzchni. Idea pomiaru przedsta-wiona jest na rysunku. W wyniku za-stosowania tej metody otrzymuje si#profilogram warstwy z pod!o$em, przy czym próbk# wykonuje si# w ten sposób, $e warstwa posiada jeden lub kilka uskoków. Okre&laj"c bezpo-&rednio z profilogramu wysoko&% tego uskoku otrzymuje si# grubo&% war-stwy.
Istnienie uskoków warstwy mo$na zabezpieczy% przed procesem jej nanoszenia poprzez wykonanie odpowiedniego maskowania fragmentów powierzchni pod!o$a lub te$ mo$na je wykona% na naniesionej ju$ warstwie na przyk!ad metodami selektywnego trawienia chemicznego lub jonowego, czy te$ poprzez mechaniczne &cieranie.
Zasada metody Kalotest („calo” - w!. - ubytek, strata na wadze) jest niezwykle prosta. Kula, zwykle stalowa, o znanym promieniu R obraca si#na powierzchni warstwy. W wyniku tarcia nast#puje usuni#cie materia!u warstwy i pod!o$a. Geometrycznie, &lady usuni#tych: warstwy i pod!o-$a maj" kszta!t koncentrycznych kó!. W celu wyznaczenia grubo&ci warstwy wykonuje si# pomiar ich &rednic, zazwyczaj przy pomocy mi-kroskopu metalograficznego.
Kolejny zamieszczony rysunek (rys. 1.77) ilustruje bli$ej t# metod#, nast#pne za& pomocne s" przy wy-prowadzaniu wzorów na grubo&%warstwy.
Rys. 1.76. Metoda profilograficzna
pomiaru grubo#ci warstw
Rys. 1.77. Idea metody Kalotest

90
Dla wyprowadzenia wzoru na grubo&% warstwy mierzonej metod"Kalotest wprowadzimy nast#puj"ce oznaczenia (patrz rysunki 1.78, 1.79,1.80): R - promie' kuli &cieraj"cej warstw#, T - wysoko&% odcinka kuli (tak$e ca!-
kowita g!#boko&% wytarcia), D - &rednica odcinka kuli (tak$e ze-
wn#trzna &rednica wytarcia), d - wewn#trzna &rednica wytarcia
(&rednica wytarcia pod!o$a), S - grubo&% warstwy, t - grubo&% startego pod!o$a. x, y - wielko&ci pomocnicze, przy czym,
jak wida% z rysunku:
2
dDx
+= ,
2
dDy
−= .
Poniewa$: s=T-t ,
a: 2221 DR4RT −−= , 22
21 dR4Rt −−= ,
to grubo&% warstwy S wynosi:
[ ]2 2 2 2 2 1 D R 4 d R 4 S − − − = .
Dla obliczenia s mo$na pos!u$y% si#uproszczon" formu!"; zauwa$my, $e:
d<<R oraz D<<R. Rozwijaj"c, przy tych warunkach, wzór na "S" w szereg Taylora otrzy-mamy:
R
xyS
2= .
B!"d oblicze' S oceniany jest na ≤1%
Rys. 1.78. Oznaczenie parametrów
kuli.
Rys. 1.79. Oznaczenie #ladów wytarcia
w przypadku jednej warstwy

91
Je$eli badana warstwa ma struktur# wielowarstwow" (patrz rysunek 1.80), to do oblicze' gru-bo&ci poszczególnych warstw nale-$y wielokrotnie wykorzysta% wpro-wadzony wzór.
Zwró%my uwag#, $e &lad wy-tarcia warstwy i pod!o$a kulk" sta-low", przy du$ej jej &rednicy - stan-dardem jest ϕ = 10mm - jest w rze-czy samej szlifem uko&nym. Mo$na zatem dodatkowo oceni%:• budow# warstwy (lita, porowata, kompo zytowa itd.),
• struktur# warstwy (jedno- czy wielo-fazowa).Mo$na te$ oceni% jej odporno&% na zu-$ycie poprzez okre&lenie obj#to&ci V
usuni#tej warstwy i pod!o$a w jednostce czasu t (np. 1 minuta).Wprowadza si# w tym celu parametr:
==γ
3tV m
s
V
t1
Obj#to&% odcinka kuli, V wynosi :
( )hR3hV 231 −π=
Na koniec kilka uwag praktycznych:• dla szybkiego uzyskania szlifu stosuje si# drobnoziarnist" past# diamen-
tow",• aby uzyska% wyra(nie zarysowan" granic#: pod!o$e - warstwa mo$na u$y%&rodka trawi"-cego i zaczerniaj"cego pod!o$e, a nie trawi"cego materia!u warstwy. Na przyk!ad dla warstw TiN na stalach wystarczy s!aby roz-twór kwasu azotowego.
• nacisk kulki na pod!o$e mo$na zmienia% przez dobór k"ta pochylenia α Kalotestera (patrz rys. 1.77).
Rys. 1.80. Oznaczenie #ladów
wytarcia w przypadku
trzech warstw
Rys. 1.81. Odcinek kuli.

92
3.3. Pomiar oporno#ci w"a#ciwej i sta"ej Hall'a
Pomiary elektrycznej oporno&ci w!a&ciwej cia! sta!ych wykonuje si#przy pomocy wielu metod pos!uguj"c si# sta!ymi lub zmiennymi polamielektrycznymi. Metody te mo$na generalnie podzieli% na bezkontaktowe ikontaktowe. W przypadku tych ostatnich stosowane s" kontakty stykowe ista!e. Powinny one spe!nia% dwa podstawowe warunki: posiada% jak naj-mniejsz" oporno&% oraz mie% liniow" charakterystyk# U=U(I) - jest to tzw.warunek omowo&ci kontaktu.
W pomiarach oporno&ci w!a&ciwej wyró$nia si# m.in. metody: tech-niczn", jednoson-dow", dwusondow", czterosondow", pier&cieniow" (ma-teria!y izolacyjne).Do pomiarów stosuje si# wysokostabilne regulowane zasilacze pr"dowe orazwoltomierze lub elektrometry o bardzo du$ej oporno&ci wewn#trznej. Po-dobnie, badania efektu Hall'a stanowi" obecnie rozbudowan" ga!"( metro-logii elektrycznej.Istnieje równie$ wiele rozwi"za' uk!adów badawczych do wykonywaniapomiarów w wysokim zakresie temperatur oraz zewn#trznych pól magne-tycznych.Ni$ej zostan" wyprowadzone formu!y na oporno&% w!a&ciw" oraz sta!" Hall'adla najpopularniejszych, przy pomiarach cienkich warstw, metod.
Metoda czterosondowa.
Metoda czterosondowa jest jedn" znajcz#&ciej stosowanych metod pomiaruoporno&ci w!a&ciwej cienkich warstw. Wszczególno&ci pozwala na badanie rozk!a-du oporno&ci w p!aszczy(nie warstwy.
Idea metody jest nast#puj"ca: do po-wierzchni próbki przyciska si# 4 ostrzau!o$one zazwyczaj w jednej linii i w jed-nakowych odst#pach: przez dwa zewn#trz-ne ostrza przepuszcza si# pr"d, wewn#trz-ne s!u$" do pomiaru napi#cia (rys. 1.82).Ostrza wykonuje si# zwykle z drutu wol-
Rys. 1.82. Gruba próbka d>>D,
sondy liniowe

93
framowego, a sto$kowy profil ko'cówki uzyskuje si# w procesach elektroli-tycznych.
Przy za!o$eniu, $e grubo&% próbki d jest znacznie wi#ksza od odleg!o-
&ci D mi#dzy sondami ( 1Dd >> ), oporno&% w!a&ciwa r dana jest wzorem:
I
UDπ=ρ 2 ,
gdzie: D - odleg!o&% mi#dzy sondami, U - napi#cie mi#dzy wewn#trzny-mi ostrzami, I- pr"d p!yn"cy przez próbk#.
Wyprowadzimy ten zwi"zek, wykorzystuj"c za!o$enie, $e pr"d p!yn"-cy przez doln" pó!przestrze', zupe!nie wype!nion" materia!em próbki po-woduje taki sam spadek napi#cia jak pr"d 2I p!yn"cy przez ca!" przestrze'(tak$e zupe!nie wype!nion" materia!em próbki).Zatem w odleg!o&ci r od elektrody wprowadzaj"cej pr"d g#sto&% pr"du wy-
nosi: 22 r
Ij
π= ,
a nat#$enie pola elektrycznego 22)(
r
IjrE
πρ
=ρ= .
Spadek napi#cia U mi#dzy wewn#trznymi ostrzami wynosi wi#c:
''U'UU += ,
gdzie: D
Idr
r
IdrEU
D
D
D
Dπρ
=πρ
−=−= ∫∫ 42'
22
2
jest spadkiem napi#cia wytworzo-
nym przez pr"d I wyp!ywaj"cy z ostrza.
Poniewa$ pr"d odp!ywa przez drug" kra'cowo po!o$on" elektrod#, powo-duje to nast#pny spadek potencja!u na &rodkowym ostrzu:
D
I
D
Idr
r
IU
D
Dπρ
=−π−ρ
=π−ρ
−= ∫ 4)(4
)(
2
)(''
2
2 .
Ca!kowity spadek potencja!u ''U'UU += daje zale$no&%:I
UD2π=ρ .
W wy$ej przytoczonych rachunkach zak!adano, $e pr"d p!ynie naj-pierw z elektrody pierwszej do niesko'czono&ci, a nast#pnie z niesko'czo-no&ci do drugiej elektrody. Wed!ug teorii potencja!u uj#cie takie jest rów-nowa$ne rzeczywistemu przep!ywowi pr"du.

94
Sko'czone wymiary próbek oraz charakter o&rodka otaczaj"cego próbk#(izolator, metal) powoduje wprowadzenie do otrzymanego wzoru poprawek wpostaci sta!ego czynnika. Poprawka ta zale$y równie$ od po!o$enia sondwzgl#dem granic próbki. Odpowiednie funkcje poprawkowe przedstawiones" dalej
W przypadku, gdy grubo&% próbki jest znacznie mniejsza od odleg!o&ci mi#-
dzy sondami ( 1Dd << - rys. 1.83), g#sto&% pr"du wynosi:
rd
Ij
π=
2 .
Wobec tego, przez analogi# z wy$ej przed-stawionym rozumowaniem otrzymujemywyra$enie na napi#cie mi#dzy wewn#trz-nymi sondami
2
''2
2ln
2'
22
UU
d
Idr
rd
IdrEU
D
D
D
D
==π
ρ=
πρ
−=−= ∫∫ .
Zatem w tym przypadku oporno&% w!a&ciwa dla cienkich warstw okre&lonajest wzorem:
I
Ud⋅
π=ρ
2ln, gdzie: d - grubo&% próbki.
Cz#sto ostrza sond u!o$one s" w kwadrat(rys. 1.84).Niech d!ugo&% boku kwadratu wynosi D. Dlagrubych próbek otrzymujemy:
( )D
Idr
r
IdrEU
D
D
D
Dπ
−ρ=
πρ
−=−= ∫∫ 21
2
2
2
1
222 ,
( )2
11−π
=ρI
DU.
Rys. 1.83. Cienka próbka
d<<D.
Rys. 1.84. Sondy w konfigura-
cji kwadratu

95
Dla cienkich próbek mamy za&:
d
Idr
rd
IU
D
Dπ
ρ=
πρ
−= ∫ 2
2ln
22
2
,
2ln
2
I
dUπ=ρ .
Funkcje poprawkowe
Odst#pstwo od za!o$e' co do wymiarów geometrycznych próbki,naturalne w praktyce, poci"gaj" za sob" konieczno&% wprowadzenia pew-nych poprawek do powy$szych wzorów. Poprawki te uwzgl#dnia si# jakoczynnik b#d"cy pewn" funkcj#, gdzie:
l - odleg!o&% sond od granic próbki,D- odleg!o&% mi#dzy sondami.
Ni$ej zostan" przedstawione funkcje poprawkowe dla ró$nych konfiguracjisond (kontaktów) oraz ró$nych ich po!o$e' wzgl#dem granic próbek.
· P"aszczyzna sondy u"o$ona jest prostopadle wzgl%dem kraw%dzi prób-
ki otoczonej przez o#rodek izolacyjny (rys. 1.85).
Skorygowan" warto&% oporno&ci w!a&ciwejobliczamy z zale$no&ci:
( )D
lF1* ⋅ρ=ρ ,
gdzie: I
UDπ=ρ 2 , l - odleg!o&% mi#dzy
kraw#dzi" próbki a najbli$sz" sond", D -
odleg!o&% mi#dzy sondami, ( )D
lF1 - wspó!-
czynnik poprawkowy.
( )
D
l
D
l
D
l
D
l
D
lF
25
1
24
1
22
1
21
1
11
++
+−
+−
+
=.
Rys. 1.85.

96
Niektóre warto&ci wspó!czynnika ( )DlF1 zawiera tabela:
D
l0 0,2 0,5 1,0 2,0 5,0 10,0
! "D
lF1 0,69 0,79 0,882 0,947 0,992 0,996 0,9995
· P"aszczyzna sondy u"o$ona jest równolegle wzgl%dem kraw%dzi próbki
otoczonej przez o#rodek izolacyjny (rys. 1.86).
W tym przypadku: ( )D
lF2* ⋅ρ=ρ ,
gdzie:
( )
( ) ( )222
2
1
1
1
21
1
D
l
D
l
D
lF
+−
++
=.
Niektóre warto&ci wspó!czynnika ( )DlF2
zawiera tabela:
D
l0 0,2 0,5 1,0 2,0 5,0 10,0
! "D
lF2 0,5 0,533 0,658 0,842 0,965 0,997 0,9996
· P"aszczyzna sondy u"o$ona jest prostopadle wzgl%dem kraw%dzi prób-
ki, stanowi&cej granic% z o#rodkiem przewodz&cym (rys. 1.87).
W tym przypadku: ( )Dl
3F* ⋅ρ=ρ ,
gdzie:
( )
Dl
Dl
Dl
Dl
D
lF
2222
3
5
1
4
1
2
1
1
11
1
+−
++
++
+−
=.
Rys. 1.86.
Rys. 1.87.

97
Niektóre warto&ci wspó!czynnika zawiera tabela:
D
l0 0,2 0,5 1,0 2,0 5,0 10,0
! "D
lF3 1,82 1,365 1,182 1,060 0,010 1,004 1,0005
· P"aszczyzna sondy u"o$ona jest równolegle wzgl%dem kraw%dzi próbki
otoczonej przez o#rodek izolacyjny(rys.1.88).
W tym przypadku: ( )D
lF4* ⋅ρ=ρ ,
gdzie:
( )
( ) ( )222
4
1
1
1
21
1
D
l
D
l
D
lF
++
+−
=.
Niektóre warto&ci wspó!czynnika zawiera tabela:
D
l0 0,2 0,5 1,0 2,0 5,0 10,0
! "D
lF4 8,07 2,08 1,232 1,038 1,003 1,0004
Wszystkie przedstawione wy$ej przypadki dotycz" pomiarów w pobli$u kra-w#dzi próbki przy za!o$eniu, $e jej grubo&% jest znacznie wi#ksza od odleg!o-
&ci mi#dzy sondami - 10>D
d.
Je$eli grubo&% próbki jest porównywalna z odst#pem D mi#dzy sondami,wprowadzi% nale$y inne wspó!czynniki poprawkowe.
Rys. 1.88.

98
· Pomiar na cienkiej próbce opartej na pod"o$u izolacyjnym.
Skorygowan" warto&% oporno&ci w!a&ciwej obliczamy z równania:
( )D
dG5
*ρ
=ρ , gdzie: d - grubo&% próbki, a:
( )( ) ( ) ( ) ( )
∑=
=
+−
++=
3
12222
5
22
1
2
141
n
ndD
dD
dD
Dd
nnG .
Niektóre warto&ci zawiera tabela:
D
d0,100 0,141 0,200 0,333 0,500 1,000 1,413 2,000 3,333 5,000 10,000
D
dG5 13,869 9,704 6,931 4,159 2,780 1,504 1,223 1,094 1,0228 1,0070 1,00045
Kiedy grubo&% próbki jest znacznie mniejsza od odleg!o&ci miedzy sondami
tj. 100,0<D
d, równanie opisuj"ce wspó!czynnik poprawkowy ( )
DdG5 przyj-
muje posta%:
( ) 2ln2
5 ⋅=d
DG
D
d.
· Pomiar na cienkiej próbce opartej na pod"o$u przewodz&cym.
W takim przypadku: ( )DdG6
*ρ
=ρ , gdzie:
( ) ( )( ) ( ) ( ) ( )
∑=
=
+−
+−+=
3
122226
22
1
2
1141
n
ndD
dD
n
dD
Dd
nnG .
Wybrane warto&ci zawiera tabela:
D
d0,100 0,141 0,200 0,333 0,500 1,000 1,414 2,000 3,333 5,000 10,000
! "DdG6 0,0000019 0,00018 0,00342 0,0604 0,228 0,683 0,848 0,933 0,983 0,9948 0,9993

99
UWAGA:W przypadkach z!o$onych, gdy pomiaru dokonujemy na próbce któ-
rej wymiary linio-we oraz grubo&% s" porównywalne z wymiarami uk!adusond, uwzgl#dnia% nale$y jednocze&nie odpowiednie wspó!czynniki popraw-kowe, mno$"c je przez siebie.Przyk"ad:
Pomiar w pobli$u kraw#dzi cienkiej p!ytki opartej na pod!o$u izola-cyjnym. P!aszczyzna sond równoleg!a do kraw#dzi próbki. Zgodnie zprzedstawionymi wcze&niej zale$no&ciami, skorygowan" warto&% opor-no&ci w!a&ciwej, dla takiego przypadku, obliczamy na podstawie rów-nania:
( ) ( )D
dD
l
GF
5
2
1* ⋅⋅ρ=ρ .
· W przypadku, gdy sonda czteropunktowa ma konfiguracj% kwadra-
tow& (rys. 1.89), a wymiary próbki s" znacznie wi#ksze od wymiarówuk!adu sond, oporno&% w!a&ciw" ρ opisuje równanie:
I
DU
22
2
−π
=ρ ,
gdzie D - odleg!o&% mi#dzy sondami - jestbokiem kwadratu.Je$eli grubo&% próbki - d jest porównywal-na z odleg!o&ci" D mi#dzy sondami, sko-rygowan" warto&% oporno&ci w!a&ciwej ρ*obliczamy z zale$no&ci:
( )dD
1K*
ρ=ρ .
Wybrane warto&ci wspó!czynnika poprawkowego ( )dD
1K zawiera tabela:
d
D0,1 0,2 0,5 1,0 2,0 5,0 10,0
! "dD
1K 1,0005 1,004 1,057 1,344 2,378 5,916 11,83
Rys. 1.89.

100
Dla próbek o ma!ych grubo&ciach ( 000,10d
D> ) równanie opisuj"ce skorygo-
wan" warto&% oporno&ci w!a&ciwej ρ* przyjmuje posta%:
I2ln
dU2*
⋅π
=ρ .
Metoda van der Pauw'e
Metoda ta pozwala okre&li%oporno&% w!a&ciw" p!askiej p!ytki o do-wolnym kszta!cie. Na brzegach próbkiumieszcza si# w dowolnym po!o$eniu4 styki (rys. 1.90).
Najpierw przepuszcza si# pr"dprzez styki 1 i 2 i mierzy si# spadeknapi#cia na stykach 3 i 4 okre&laj"cpewn" oporno&%:
12
3434,12
I
UR = ,
a nast#pnie przepuszcza si# pr"d przez styki 2 i 3 mierzy napi#cie na kontak-tach 1 i 4 okre&laj"c wielko&%:
23
1441,23
I
UR = .
Te dwa pomiary pozwalaj" okre&li% oporno&% w!a&ciw" próbki. Wynosi ona:
f41,23
34,12
R
R34,1234,12
2
RR
2ln
d*
⋅+
⋅π
=ρ
gdzie: f41,23
34,12
R
R
jest pewn" funkcj" stosunku obydwu oporno&ci.
Rys. 1.90. Próbka w metodzie
van der Pauwe

101
Uzasadnijmy ten wzór:
G#sto&% pr"du p!yn"cego przez stykwynosi:
rd2
Ij
π= .
Uk!ad styków w metodzie Van derPauw'e mo$na przedstawi% jak na ry-sunku 1.92.
Aby zachowa% linie pr"du i linie po-tencja!u ci"g!ymi nale$y uzupe!ni% dol-n" p!aszczyzn# tzn. próbk# przez iden-tyczn" górn". Odpowiada to podwo-jeniu warto&ci pr"du.W takim przypadku:
rd
I
rd2
I2j
π=
π= .
Poniewa$ potencja!: ∫−= EdrV ,
a pole elektryczne:rd
IjE
πρ
=ρ= ,
to otrzymujemy: Crlnd
IV +
πρ
−= .
Stosuj"c to wyra$enie dla obliczenia spadku napi#cia mi#dzy punktami 3 i 4(tzn. V4-V3) otrzymujemy (uwzgl#dniaj"c (ród!o pr"du +I i -I) potencja! wpunkcie 3:
( ) ( ) ( )
21
22123
SS
Sln
d
ISSln
d
ISln
d
IV
+πρ
=+π+ρ
−π
−ρ−= ,
Rys. 1.91. Kontakt pr&dowy
Rys. 1.92. Próbka w metodzie
van der Pauwe

102
potencja! w punkcie 4:
( ) ( ) ( ) ( )
321
32321323
SSS
SSln
d
ISSSln
d
ISSln
d
IV
+++
πρ
=++π+ρ
−+π
−ρ−= .
Wprowadzaj"c oznaczenia:
241,23134,12 RR;RR == ,
mo$emy zapisa%:
( )
( )( )3221
3212341
SSSS
SSSSln
dI
VVR
++++
πρ
−=−
= ,
i analogicznie dla przypadku przep!ywu pr"du mi#dzy stykami 2 i 3:
( )( )3221
31412
SSSS
SSln
dI
VVR
++⋅
πρ
−=−
= .
Przedstawiaj"c te wyra$enia w postaci eksponensjalnej i dodaj"c stronami
otrzymujemy: 1eedRdR 21
=+ ρπ−
ρπ−
.
Je$eli dokonamy nast#puj"cego podstawienia:
( ) ( )[ ]
( ) ( )[ ],2
1
,2
1
21212
21211
RRRRR
RRRRR
−−+=
−++=
to powy$sze równanie mo$na zapisa% w postaci:
2
1
2
RRdcosh 212
RRd
e21
=
ρ
−π⋅ρ
+π−
.
Podstawiaj"c: ( )21
21
R,Rf
2ln
2
RRd =
ρ+
π ,
gdzie: f(R1,R2) - jest pewn" funkcj" zale$n" od R1 i R2, mamy:
e f
2ln
21
21
2
1
f
2ln
RR
RRcosh =
⋅
+−
,

103
lub:
e f
2ln
2
1
2
1
2
1
f
2ln
1R
R
1R
R
cosh =
⋅+
−
.
Zapis wskazuje na jawn" zale$no&% f od stosunku 2
1
R
R.
Równanie powy$sze pozwala okre&li% warto&% funkcji f; dla wybranych war-
to&ci stosunku 2
1
R
R warto&ci te przedstawia tabela:
2
1
R
R1 2 5 10 20 50 100 200 500 1000
f2
1
R
R##$
%&&'
(1 0,95 0,81 0,69 0,59 0,46 0,40 0,34 0,29 0,25
Ostateczne wyra$enie na warto&% oporno&ci w!a&ciwej mierzonej metod"van der Pauw'e ma posta%:
f2
1
R
R21
2
RR
2ln
d
+
⋅π
=ρ .
Standardowy pomiar efektu Hall'a
Je$eli wyd!u$on", prostopad!o&cienn" próbk#, przez któr" p!ynie pr"delektryczny, umie&ci si# w polu magnetycznym prostopad!ym do jednego zjej boków, to w próbce, w kierunku prostopad!ym do kierunku przep!ywupr"du i kierunku pola magnetycznego powstanie dodatkowe pole elektrycz-ne zwane polem Hall'a (rys. 1.93). Odpowiadaj"ce temu polu napi#cie okre-&lone jest wzorem:
a
BIRU HH
⋅⋅=
Za!ó$my model elektronów swobodnych cia!a sta!ego.

104
Przy braku zewn#trzne-go pola magnetycznegono&niki !adunku q poru-szaj" si# w próbce (patrzrysunki) ze &redni" pr#d-
ko&ci" dryfu drvρ
rów-
nolegle do zewn#trznegopola elektrycznego E.Przy!o$enie z zewn"trzpola magnetycznego o in-
dukcji Bρ
powoduje po-wstanie si!y Lorentza:
BvqF drL
ρρρ×= ,
spychaj"cej no&niki !a-dunku do dolnej cz#&cipróbki. Poci"ga to zasob" niedobór !adun-ków w górnej cz#&cipróbki. W rezultacie po-wstaje natychmiast,jako reakcja, przeciwnieskierowane do FL i rów-nowa$"ce j" pole elek-
tryczne HE
ρ zwane po-
lem Hall'a. Ustala si# zatem dynamiczny stan równowagi si!: HL EqF
ρρ= .
Przy za!o$eniu, $e: drvB
ρρ⊥ , otrzymujemy:
Hdrdr qEqn
qjBBqvBvq ===×
ρρ
i dalej ba
BI
qn
1
b
UH
⋅⋅
⋅= ,
EDWIN HERBERT HALL (USA) odkrywa efekt (zwany obec-nie efektem Hall'a) w 1879 roku. badaj"c cienkie warstwyz!ota. Niezwyk!ym jest, $e istnienie elektronu, cz"stki nie-zb#dnej do wyja&nienia tego efektu, odkryto dopiero w1897 r. (J.J. Thomson).
Rys. 4.24. Efekt Hall’a

105
st"d:a
BIRU HH
⋅⋅= ,
gdzie: qn
1R H = .
Wielko&% RH nazwa-na jest sta!" Hall'a. Znak tejsta!ej okre&la rodzaj no&ni-ków !adunku w próbce (wprzypadku pó!przewodni-ków znak no&ników wi#k-szo&ciowych), a jego warto&% s!u$y do wyliczenia koncentracji no&ników.Podkre&lamy, $e tak prost" formu!# na RH otrzymuje si# jedynie przy za!o$e-niu modelu elektronów swobodnych cia!a sta!ego. Przy wzi#ciu pod uwag#z!o$onych mechanizmów rozpraszania no&ników !adunku formu!a na RH
staje si# bardzo z!o$ona.Po przy!o$eniu pola magnetycznego na no&nik !adunku dzia!aj" za-
tem dwa pola: zewn#trzne i Hall'a. St"d wektor wypadkowy pola elektrycz-nego nie jest ju$ równoleg!y do boków próbki.
Z rysunku wida% (rys. 1.93), $e po przy!o$eniu Bρ
, nast#puje pochyle-nie linii ekwipotencjalnych, czego bezpo&redni" konsekwencj" jest powsta-nie napi#cia UH.
****Przypis o pomiarze UH:
1) )"danie by d!ugo&%próbki do pomiaruUH by!a wi#ksza odjej wymiarów po-przecznychwynika zfaktu, $e rozk!ad polaelektrycznego w po-bli$u kontaktów jestzniekszta!cony przezgeometri# kontak-
Rys. 1.94. Prosty uk"ad do pomiaru UH
Je$eli α ≠ 90° to: α⋅
= sina
BIRU HH
Rys. 1.95.

106
tów doprowadzaj"cych pr"d. Zatem kontakty do pomiaru UH musz" by%umieszczone z dala od kontaktów pr"dowych (rys. 1.95).
2) Napi#cie asymetrii. Nie sposób fizycznie wykona% kontaktów do pomia-ru UH, tak, by by!y onepunktowymi i le$a!y do-k!adnie naprzeciw sie-bie (bowiem tylko wte-dy: UH=0 dla B=0)(rys. 4.27). Powodujeto, $e na zaciskach dopomiaru UH powstajenapi#cie bez obecno&cipola magnetycznego.Napi#cie to nazywanejest napi#ciem asymetriiUAS.
Napi#cie asymetrii mo$na skompensowa% zewn#trznymi uk!adamielektronicznymi na wiele sposobów (patrz rys. 1.97). Przy pomiarach napi#-cia Hall'a nale$y pami#ta%, $e mamy cztery wyprowadzenia z czterech punk-tów próbki, z których kazdy znajduje si# na innym potencjale. St"d (ród!opr"dowe i woltomierz musz" by% galwanicznie odizolowane. Jedenz tych przyrz"dów, lecz nie obydwa jednocze&nie, mo$e by% uziemiony.
3) Efekty krzy$oweDok!adny pomiar napi#cia UH jest mo$liwy tylko wtedy, gdy uwzgl#dni
si# fakt, $e napi#cie na zaciskach Hall'a, jest sum" "czystego" napi#cia UH
oraz innych napi#% powsta!ych wskutek zjawisk towarzysz"cych przep!ywo-wi !adunku w skrzy$owanych (st"d nazwa "efekty krzy$owe" - inna nazwa
Rys. 1.97. Uk"ady kompensacji napi%cia asymetrii
Rys. 196.

107
"efekty poprzeczne") polach Eρ
i Bρ
takich jak: przewodnictwo elektryczne,przewodnictwo cieplne, efekty galwanomagnetyczne, termoelektryczne czytermomagnetyczne. Dosy% odnotowa%, $e jak podaje teoria, ilo&% efektówkrzy$owych mo$e si#ga% liczby 560.G!ówne z nich to:• zjawisko asymetrii (patrz wy$ej),• magnetoopór,• efekt Seebeck'a (termoelektryczny),• efekt Ettingshausen'a (poprzeczny galwanotermomagnetyczny),• efekt Nerst'a - Ettingshausen’a (termogalwanomagnetyczny),• efekt Righi - Leduc'a (termomagnetyczny);• efekt Peltier - Nerst’a - Ettingshausena (elektrotermiczny efekt Peltier
zwi"zany z efektem termogalwanomagnetycznym),• efekt Peltier - Righi - Leduc'a (elektrotermiczny efekt Peltier zwi"zany z
efektem termomagnetycznym).Wi#ksza cz#&% z tych efektów ma charakter nieparzysty, tzn. zmienia znak
przy zmianie kierunku zewn#trznego pola Eρ
lub Bρ
. Mo$na je zatem wy-eliminowa% dokonuj"c czterech pomiarów dla czterech konfiguracji kierun-
kowych Eρ
i Bρ
.G!ównym efektem parzystym, nie daj"cym si# wyeliminowa% t" procedur"jest efekt Ettinghausena, którego (ród!em jest ró$nica temperatur mi#dzyp!aszczyznami próbki na których powstaje napi#cie Hall'a. Wspomnianaró$nica temperatur powstaje jako rezultat tworzenia si# pola EH i jest konse-kwencj" rozk!adu pr#dko&ci elektronów w ciele sta!ym. Zatem ró$nica tem-
peratur, a i napi#cie Ettinghausena UE ustala si# po w!"czeniu pola Eρ
lub
Bρ
. St"d je$eli UH ro&nie po w!"czeniu pól, czy te$ nie znika natychmiast poich wy!"czeniu oznacza to, $e wp!yw efektu Ettinghausena jest znaczny.Ostatecznie wi#c mo$na zapisa% wyra$enie na mierzone napi#cie jako:
( ) ( ) ( ) ( )[ ]B,IUB,IUB,IUB,IU4
1UU E
H −−++−+−++++=+
Dodajmy na koniec, $e detektory dzia!aj"ce w oparciu o efekt Hall'a zwanehallotronami lub generatorami Hall'a maj" niezwykle szerokie zastosowa-nie m.in. w: czytnikach kart magnetycznych, czujnikach zbli$eniowych,czujnikach pr#dko&ci obrotowej, miernikach mocy i energii, pomiarach pólmagnetycznych, przetwornikach liniowo - k"towych.
****

108
Pomiar sta"ej Hall'a metod& van der Pauw'e
Oporno&% próbki mierzona na zaciskach 2 i 4 (w metodzie van derPauw'e) wynosi:
13
2424,13
I
UR =
Po przy!o$eniu do próbki jednorodnego pola magnetycznego prostopadledo jej powierzchni warto&% ta zmienia si# o pewn" wielko&% ∆R13,24, któraproporcjonalna jest do sta!ej Halla. Relacja ta wyra$a si# nast#puj"co:
H24,13 Rd
BR =∆
Aby wyja&ni% ten zwi"zek pos!u$my si#rysunkiem (rys. 1.89):W wyniku przy!o$enia do próbki polamagnetycznego przeciwleg!e jej brzegi(2 i 4) !aduj" si# powoduj"c powstaniemi#dzy punktami 2 i 4 napi#cie Halla.Linie pr"du pozostaj" jednak niezmie-
nione. Pole Halla HE
ρ jest prostopad!e
do wektora g#sto&ci pr"du jϖ
. Za!ó$my,
$e punkty 4 i 2' le$" (przed przy!o$e-niem pola B) na jednej linii ekwipoten-
cjalnej, oraz 2 i 2' znajduj" si# na tej samej linii pr"du na brzegach próbki.
Wobec tego dla elementu drogi sdρ wzd!u$ której sdEH
ρρ⊥ s!uszna jest
równo&%:
∫ =⋅=2
'2
H 0sdEUρρ
,
zatem:
∫ ∫ ⋅=⋅='2
4
2
4
H sdEsdEUρρρρ
.
Rys. 4.29. Metoda van der
Pauw'e pomiaru sta"ej
Hall'a

109
Poniewa$ napi#cie Halla mi#dzy punktami 4 i 2' le$"cymi (przed przy!o$eniempola B) na jednej linii ekwipotencjalnej dane jest zale$no&ci":
d
IBRU H
H = ,
wi#c napi#cie to mierzone b#dzie mi#dzy punktami 4 i 2 jako przyrost ∆V24.Wobec tego:
d
BR
I
VR H
13
2424,13 =
∆=∆ .
Ostatecznie zatem otrzymujemy wyra$enie na sta!" Hall’a:
24,13H RB
dR ∆=
Metoda van der Pauw'e jest bardzo szeroko stosowana ze wzgl#du na to, $e(pomijaj"c $"danie sta!o&ci grubo&ci) nie wymaga ona regularnych kszta!-tów próbki.
3.4. Pomiar mikrotwardo#ci metod& Vickers’a
W zwi"zku z niewielk" grubo&ci" warstw TiN pomiar twardo&ci prze-prowadza si# przy pomocy metody Vickers'a (u$ywane jest te$ okre&lenie -mikrotwardo&% Vickers'a).
Metoda ta polega na wciskaniu wg!#bnika, w postaci foremnego, czwo-rok"tnego czworok"tnego ostros!upa diamentowego o k"cie dwu&ciennymα = 136° w p!ask" powierzchni# przedmiotu pod obci"$eniem F prostopa-d!ym do tej powierzchni oraz odczycie (po zdj#ciu obci"$enia) przek"tnejpowsta!ego kwadratu.Popularnie stosuje si# tzw. obiektyw Hannemana z przytwierdzonym cen-tralnie wg!#bnikiem i uk!adem si!ownika spr#$ynowego. Obiektyw ten po-zwala (po zamontowaniu go w mikroskopie metalograficznym) na bezpo-&redni odczyt si!y nacisku. Odczytu warto&ci przek"tnej dokonuje si# przypomocy przeskalowanego okularu pomiarowego.
Twardo&ci" Vickers'a (oznaczan" HV) nazywa si# stosunek obci"$e-nia F (wyra$onego w kG lub N) do pola S (w mm2) powierzchni bocznejotrzymanego odcisku. HV podaje si# te$ w Pascalach (1 Pa = 1N/m2). Wdolnej cz#&ci symbolu HV podaje si# cz#sto warto&% nacisku, przy którym

110
wykonano pomiar. Na przyk!ad HV50 oznacza twardo&% Vickers'a mierzonaprzy obci"$eniu 50 G.Zatem:
[ ]Pamm
N
mm
kG
S
FHV
22
=
Powierzchni# odcinka mo$na wyliczy% ze znajomo&ci przek"tnej kwadrato-wego &ladu wg!#bnika (zwykle mierzy si# obydwie przek"tne i bierze ich&redni" arytmetyczn")
Pole S wynosi: 854,1
dS
2
= ,
g!#boko&% wnikania za&: 95,4
dh = ,
Je$eli wi#c si!# wyrazimy w kG to: 2d
F854,1HV =
2mm
kG.
Dla si!y wyra$onej w niutonach: 2d
F1891,0HV =
2mm
N.
Przy pomiarze mikrotwardo&ci cienkich warstw, gdzie g!#boko&% wnikaniajest z regu!y wi#ksza ni$ grubo&% warstwy nale$y uwzgl#dni% grubo&% war-stwy oraz w!a&ciwo&ci mechaniczne pod!o$a.
Rys. 4.30. Konstrukcje geometryczne pomocne dla obliczenia pola odcisku oraz
g"%boko#ci wnikania wg"ebnika.
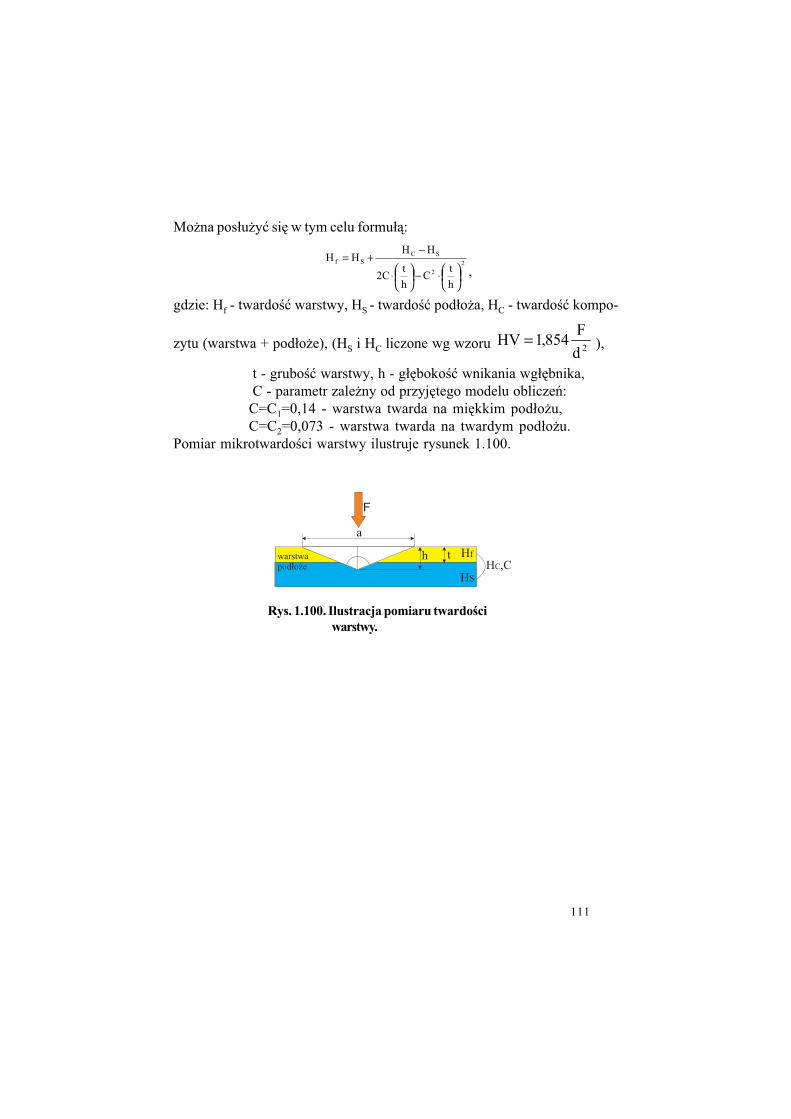
111
Mo$na pos!u$y% si# w tym celu formu!":
2
2
SCSf
h
tC
h
tC2
HHHH
⋅−
⋅
−+=
,
gdzie: Hf - twardo&% warstwy, HS - twardo&% pod!o$a, HC - twardo&% kompo-
zytu (warstwa + pod!o$e), (HS i HC liczone wg wzoru 2d
F854,1HV = ),
t - grubo&% warstwy, h - g!#boko&% wnikania wg!#bnika, C - parametr zale$ny od przyj#tego modelu oblicze':
C=C1=0,14 - warstwa twarda na mi#kkim pod!o$u,C=C2=0,073 - warstwa twarda na twardym pod!o$u.
Pomiar mikrotwardo&ci warstwy ilustruje rysunek 1.100.
Rys. 1.100. Ilustracja pomiaru twardo#ci
warstwy.
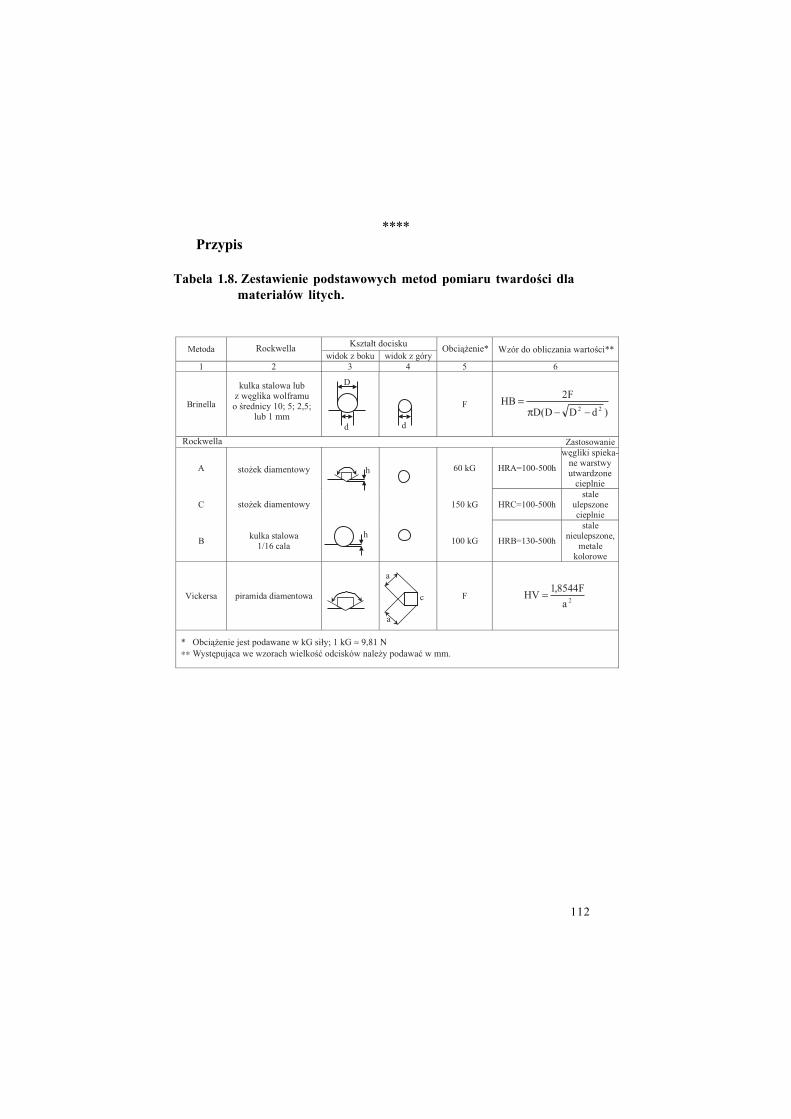
112
****Przypis
Tabela 1.8. Zestawienie podstawowych metod pomiaru twardo#ci dla
materia"ów litych.

113
Rys. 4.32. Porównanie skal twardo#ci okre#lanych sposobami: Mohs’a,
Vickers’a, i Rockwell’a oraz przedzia"y twardo#ci niektórych
materia"ów litych i warstw.

114
II. Technologie warstw azotku tytanu
Dalej opisane zostan! bli"ej niektóre sposoby otrzymywania cienkich
warstw TiN oraz urz!dzenia s#u"!ce do tego celu. Nacisk po#o"ony b$dzie
na metody z grupy PVD, przy czym dok#adniej opisane zostan! dwie z
nich, a mianowicie: metoda sta#opr!dowego magnetronu niezbalansowane-
go oraz metoda #ukowa - te bowiem metody wykorzystujemy w VTT.
1. Metody CVD.
Zasada metod CVD znana by#a ju" w ubieg#ym stuleciu, jednak"edla uzyskania twardych warstw zastosowano j! dopiero w latach czterdzie-
stych tego wieku w USA.
Pierwsze handlowe narz$dzia z w$glików spiekanych pokryte war-
stwami TiC i TiN uzyskanymi tymi metodami pokaza#y si$ na rynku w
drugiej po#owie lat sze%&dziesi!tych.
W metodach CVD lotne sk#adniki maj!ce utworzy& warstw$ mog!reagowa& mi$dzy sob! w fazie gazowej lub na pod#o"u umieszczonym w
komorze reakcyjnej. Uwa"a si$, "e pod#o"e ma katalityczny wp#yw na prze-
bieg reakcji. Je%li reakcja zachodzi w fazie gazowej mówimy o zarodkowaniu
homogenicznym, je"eli za% proces zarodkowania zachodzi wy#!cznie na pod-
#o"u reakcja ma charakter heterogeniczny.
Niezale"nie od rodzaju reakcji, która zachodzi, zdecydowany wp#yw
na jej przebieg ma temperatura. Procesy CVD w których aktywacja reakcji
odbywa si$ wy#!cznie dzi$ki temperaturze nazywane s! termicznymi lub
konwencjonalnymi CVD. Procesy te przeprowadza si$ zwykle przy ci%nie-
niu atmosferycznym gazów lub ma#ym nadci%nieniu. Sygnalizuje si$ tym
samym istnienie niekonwencjonalnych CVD. Rzeczywi%cie mo"liwa jest
inna aktywacja procesów CVD, o czym dalej.
Wyró"nia si$ trzy podstawowe typy reakcji termo-chemicznych wy-
korzystanych w procesach CVD:
1. Dysocjacja
W przypadku tym zachodzi reakcja typu:
2AB ⇔ A + AB2

115
Je%li temperatura procesu spada, sta#a równowagowa reakcji przesuwa si$ wlewo i zachodzi dysocjacja AB na A i AB2. Je"eli temperatura ro%nie zacho-
dzi synteza i powstaje cz!stka AB.
Przyk#adem mo"e by& dysocjacja dwujodku germanu dla wytwarza-
nia epitaksjalnego germanu:
2GeJ2(gazowy) ⇔ Ge(sta#y) + GeJ4(gazowy)
2. Rozk!ad
Tutaj zachodzi reakcja typu:
AB(gaz) → A(sta#y) + B(gazowy)
Pod#o"e, na które ma by& naniesiona warstwa A podgrzewane jest w
atmosferze gazu AB do takiej temperatury, w której zachodzi rozk#ad cz!-steczek AB. Faza A wydziela si$ w postaci sta#ej na pod#o"u, za% sk#adnik B
unoszony jest na zewn!trz komory reakcyjnej poprzez roboczy gaz oboj$t-ny, zwykle argon.
Przyk#ady:
a) proces Mond'a
Ni (CO)4(gaz) → Ni(sta#y) + 4CO(gaz)
b) nak#adanie pyrolitycznego w$gla z w$glowodorów
C2H2 → 2C(sta#y) + H2(gaz)
2. Utlenianie i reakcja chlorków
Jest to bardzo popularny typ reakcji.
Na przyk#ad utlenianie chlorku cyny:
SnCl4(gaz) + 2H2O(gaz) → SnO2(sta#y) + 4HCl(gaz)
(proces ten zwany jest cz$sto pyrohydrolitycznym)
Inny przyk#ad to redukcja chlorku molibdenu:
2MoCl5(gaz) + 5H2(gaz) → 2Mo(sta#y) + 10HCl(gaz)

116
Procesy CVD daj! mo"liwo%& wytwarzania nie tylko warstw metali
czy pó#przewodników - pozwalaj! tak"e na nak#adanie warstw tlenkowych,
azotkowych i w$glikowych, w tym naturalnie azotków i w$glików tytanu.
Podkre%lmy jeszcze, "e CVD mo"na przeprowadzi& w uk#adach otwar-
tych i zamkni$tych. W uk#adach otwartych gazowe substraty reakcji dopro-
wadzane s! z zewn!trz do komory reakcyjnej; równie" uboczne produkty
reakcji odprowadzane s! na zewn!trz. Tego rodzaju procesy wykorzystuje
si$ przemys#owo do nanoszenia twardych warstw.
Procesy CVD w uk#adach zamkni$tych wykorzystuje si$ mi$dzy in-
nymi do monokrystalizacji zwi!zków chemicznych tzw. metod! transportu
chemicznego.
Nie mo"na w tym miejscu oprze& si$ pokusie przedstawienia idei
metody transportu chemicznego ze wzgl$du na jej niezwyk#y przebieg (cho&pozostaje ona bez zwi!zku z metodami technologicznymi stosowanymi dla
otrzymywania warstw azotku tytanu).
Omówimy j! na przyk#adzie monokrystalizacji siarczku cynku (ZnS).
W jednym z ko'ców poziomej, kwarcowej ampu#y, z której odpom-
powano powietrze, umieszcza si$ polikrystaliczny, sproszkowany ZnS oraz
kryszta#ki jodu (J).
Ampu#k$ wprowadza si$ do pieca z odpowiednim rozk#adem tempe-
ratury (rys. 2.1).
Rys. 2.1. Schemat uk!adu do krystalizacji ZnS metod" transportu chemicznego

117
W wysokiej temperaturze ZnS gwa#townie reaguje z jodem:
1. ZnS(sta#y) + J2(gaz) → ZnJ2(gaz) + 1 S2(gaz)
Nast$puje transport gazów do drugiego ko'ca ampu#y, o ni"szej tem-
peraturze i tam wydziela si$ (krystalizuje) ZnS.
2. ZnJ2(gaz) + 1S2 → ZnS(monokryszta#) + J2(gaz)
Jod wraca do strefy o wysokiej temperaturze i powtarza si$ reakcja 1.
Ca#o%& procesu ma charakter cykliczny. Z fizycznego punktu widze-
nia jego si#ami nap$dowymi s! gradienty st$"e' sk#adników powoduj!ce
ich dyfuzj$, z chemicznego za% warto%ci entalpii reakcji w obydwu ko'cach
ampu#y i sta#a równowagowa procesu.
Dla otrzymania monokryszta#ów o kilku milimetrowych wymiarach
potrzeba zwykle kilkudziesi$ciu godzin.
W zale"no%ci od temperatury pod#o"y procesy CVD dzieli si$ nast$puj!co:
1) Wysokotemperaturowe CVD T ∈ 850°C - 1200°C
Przyk#ady reakcji:
TiCl4 + CH4 → TiC + 4HCl
TiCl4 + 2H2 + ½N2 → TiN + 4HCl
2AlCl3 + 3CO2 + 3H2 → Al2O3 + 3CO + 6HCl
2) (redniotemperaturowe CVD T ∈ 700°C - 900°C
Przyk#ad reakcji:
(TiCl4, CH3CN, H2) → (TiCxN1-x, HCl)
3) Niskotemperaturowe CVD T ∈ 300°C - 600°C
Przyk#ad reakcji:
(WF6, C6H6, H2,) → (W2C, HF)
4) Plazmowe CVD T ∈ 3000°C - 6000°C
Przyk#ad reakcji:
(TiCl4, H2, N2, Ar) → (TiN, HCl, NH3, Ar)
Reakcje typu 1 to typowe, konwencjonalne reakcje zachodz!ce w
procesach CVD z aktywacj! wy#!cznie termiczn!. Procesy: 2, 3, i 4 to pro-
cesy niekonwencjonalne. W procesach 2 i 3 obni"ono temperatur$ procesu
poprzez dodanie do substratów gazowych pewnych substancji metaloorga-

118
nicznych u#atwiaj!cych zachodzenie reakcji g#ównej (substancje te nazywane
s! prekursorami).
W procesie 4 obni"enie temperatury procesu jest mo"liwe dzi$ki za-
stosowaniu aktywacji plazmowej.
Aktywacja plazmowa w CVD polega na wzbudzaniu wy#adowania
jarzeniowego w gazowej mieszance reakcyjnej, do której dodaje si$ gaz obo-
j$tny, zwykle argon (Ar).
Technicznie polega to na do#!czeniu wysokiego napi$cia (800 V -
2000 V) mi$dzy pod#o"a (wsad) - katod$, a obudow$ pieca - anod$. Jony
dodatnie i elektrony wytworzone w wy#adowaniu u#atwiaj! rozpad i synte-
z$ cz!stek w fazie gazowej, a tak"e modyfikuj! warunki wzrostu warstwy na
pod#o"ach poprzez ich intensywne bombardowanie jonowe.
Procesy 2 ÷ 4 okre%la si$ tak"e mianem: wspomagane CVD, a dla
procesu 4 rezerwuje si$ okre%lenie PACVD [Plasma Assisted (Activated)
Chemical Vapor Deposition]. Procesy PACVD przeprowadza si$ przy ob-
ni"onych ci%nieniach (0,1 ÷ 20) mbar.
Warto zauwa"y&, "e w przypadku wysokotemperaturowych procesów
CVD otrzymana warstwa zwi!zana jest z pod#o"em dyfuzyjnie. Ten charak-
ter powi!zania warstwy z pod#o"em zmienia si$ w procesach niskotempera-
turowych CVD na adhezyjny.
Procesy CVD przeprowadza si$ przy pomocy urz!dze', których ogól-
ny, ideowy schemat przedstawiony jest na rysunku 2.2.
Uchwyt na którym umieszczone s! pod#o"a znajduje si$ w pró"niosz-
czelnym ko#paku umieszczonym w piecu, zwykle oporowym. Powietrze z
ko#paka usuwa si$ przy pomocy pompy pró"niowej, a nast$pnie doprowa-
dza si$ do niego odpowiednie gazy reakcyjne.
Parametrami decyduj!cymi o charakterze procesu s!:· temperatura i jej rozk#ad w komorze reakcyjnej,
· ci%nienia cz!steczkowe gazów i ci%nienie ca#kowite,
· jednorodno%& rozk#adu g$sto%ci gazu w komorze reakcyjnej,
· szybko%& przep#ywu gazów przez komor$,· czas.
Szybko%& osadzania warstw, w przypadku metali redukowanych z
chlorków przy pomocy wodoru, wynosi kilka mikrometrów na minut$. Przy
innych metodach, w szczególno%ci rozk#adzie termicznym, jest ona wielo-
krotnie mniejsza i waha si$ od 0,01 ÷ 0,1 mm/min.
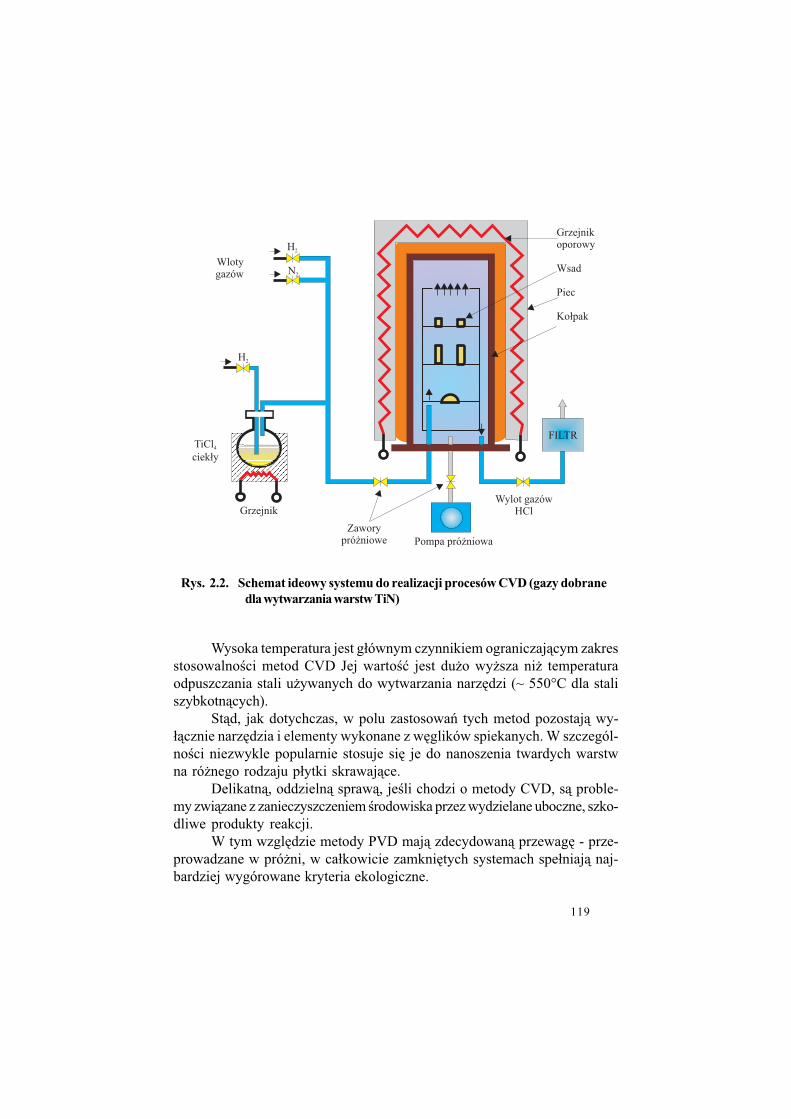
119
Wysoka temperatura jest g#ównym czynnikiem ograniczaj!cym zakres
stosowalno%ci metod CVD Jej warto%& jest du"o wy"sza ni" temperatura
odpuszczania stali u"ywanych do wytwarzania narz$dzi (~ 550°C dla stali
szybkotn!cych).
St!d, jak dotychczas, w polu zastosowa' tych metod pozostaj! wy-
#!cznie narz$dzia i elementy wykonane z w$glików spiekanych. W szczegól-
no%ci niezwykle popularnie stosuje si$ je do nanoszenia twardych warstw
na ró"nego rodzaju p#ytki skrawaj!ce.
Delikatn!, oddzieln! spraw!, je%li chodzi o metody CVD, s! proble-
my zwi!zane z zanieczyszczeniem %rodowiska przez wydzielane uboczne, szko-
dliwe produkty reakcji.
W tym wzgl$dzie metody PVD maj! zdecydowan! przewag$ - prze-
prowadzane w pró"ni, w ca#kowicie zamkni$tych systemach spe#niaj! naj-
bardziej wygórowane kryteria ekologiczne.
Rys. 2.2. Schemat ideowy systemu do realizacji procesów CVD (gazy dobrane
dla wytwarzania warstw TiN)

123
V. Bibliografia
Przedstawione zestawienie literatury jest arbitralne. Zawiera wy cznie ogólnodost pne
pozycje polskoj zyczne, a jedynym jego porz dkiem jest kolejno alfabetyczna nazwisk au-
torów. Nie jest ono zupe ne wobec adnej kategorii przedmiotowej tekstu, nie posiada w nim
tak e odno ników.
Zadaniem poni szej bibliografii jest wskazanie zainteresowanemu czytelnikowi ró-
de , w oparciu o które móg by poszerzy swoj wiedz w zakresie poruszanym w niniejszym
opracowaniu.
Wbrew pozorom jednak bibliografia ta nie jest zestawieniem cz stkowym, czy te
jakimkolwiek "wyborem literatury". Z ubolewaniem bowiem nale y stwierdzi , e pi mien-
nictwo polskie w omawianym przedmiocie jest skromne.
1. Blicharski M. Wst p do in ynierii materia owej. Warszawa, WNT 1998.
2. B a ewski S., Mikoszewski J. Pomiary twardo ci metali. Warszawa, WNT 1981.
3. Bujak J., Miernik K., Rogowska R., Smolik J., Walkowicz J. Przygotowanie powierzchni
narz dzi dla nanoszenia pow ok przeciwzu yciowych metodami PVD.W: Przegl d Mecha-
niczny nr 14, 1994.
4. Burakowski T. Miernik K., Walkowicz J. Technologie wytwarzania, z udzia em plazmy,
cienkich warstw trybologicznych. W: Trybologia nr 4/5, 1993.
5. Burakowski T., Roli ski E., Wierzcho T. In ynieria powierzchni metali Warszawa, Wy-
dawnictwo Politechniki Warszawskiej, 1992.
6. Burakowski T., Wierzcho T. In ynieria powierzchni metali. Warszawa, WNT, 1995.
7. Büttner J. Nowoczesne metody pró niowe osadzania warstw. W: Elektronika nr 7-8, 1986.
8. Celi ski Z. Plazma. Warszawa, PWN 1980.
9. Celi ski Z. Podstawy fizyki plazmy w zastosowaniach technicznych. Warszawa, WNT 1970.
10. Celi ski Z., Miernik K. Plazmowo - chemiczne metody wytwarzania warstw odpornych na
zu ycie. W: Trybologia nr 6, 1991.
11. Czernietski A.W. Wst p do fizyki plazmy. Warszawa, PWN 1971.
12. Groszkowski J. Technika wysokiej pró ni. Warszawa, WNT, 1978.
13. Ha as A., Technologia wysokiej pró ni. Warszawa, PWN 1980.
14. Heinz B. Metoda rozpylania magnetronowego do produkcji cienkich warstw dla elektroni-
ki W: Elektronika nr 9, 1981.
15. Karpi ski T., Kacalak W., ukianowicz Cz., ukianowicz T. wiczenia laboratoryjne
z metrologii mechanicznej. Koszalin, Wydawnictwo Uczelniane Politechniki Koszali skiej
1997.
16. Kirczuk Cz., Stabilizacja gazu roboczego w napylarce pró niowej. W: Elektronika nr 9,
1986.
17. Kirczuk Cz., Markowski J., Prajzner A. Nagrzewanie jonowe pod o y w napylarce pró -
niowej. W: Elektronika nr 9, 1986.
18. Kittel C. Wst p do fizyki cia a sta ego. Warszawa, PWN 1970.
19. Kordus A. Plazma, w a ciwo ci i zastosowania w technice. Warszawa, WNT 1985.
20. Kowtioniuk N.F., Koncewoj A.J. Pomiary parametrów materia ów pó przewodnikowych.
Warszawa PWN, 1973.
21. Krall N.A., Trivelpiece A.W. Fizyka plazmy. Warszawa, PWN 1979.

124
22. Kupczyk M. Jako technologiczna kó z batych naci tych d utakami modu owymi pokry-
tymi warstewk TiNX-Ti w warunkach adchezyjnego zu ycia si ostrzy. Pozna , Wydaw-
nictwo Pozna skiego Towarzystwa Naukowego 1994.
23. Ladmanowski W. Zarys teorii wy adowa w dielektrykach. Warszawa, WNT 1988.
24. Legutko S., Wieczorowski K. Techniki cienkich warstw w zastosowaniach do narz dzi
skrawaj cych. W: Mechanik nr 8-9, 1993.
25. Leja E., Horodyski T., Budzy ski K. Magnetronowa technika wytwarzania cienkich warstw.
W: Elektronika nr 9, 1982.
26. Michalski A. Metody PVD stosowane do nanoszenia warstw materia ów twardych i trud-
notopliwych na narz dzia skrawaj ce. W: Metaloznawstwo. Obróbka Cieplna nr 79 str.
18. 1986.
27. Michalski A., Zdunek K., Soko owska A., Olszyna A. Impulsowo-plazmowa metoda na-
noszenia warstw TiN na narz dziach w temperaturze ni szej ni 500K. W: Przegl d Tech-
niczny nr 15, 1991.
28. Miernik K. Generacja i separacja mikrokropli w metodzie ukowo - pró niowej. W: Prze-
gl d Mechaniczny nr22, 1997.
29. Miernik K. Dzia anie i budowa magnetronowych urz dze rozpylaj cych. Radom, Biblio-
teka Problemów Eksploatacji 1997.
30. Miernik K. Dzia anie i budowa magnetronowych urz dze rozpylaj cych. Radom, Wy-
dawnictwo Instytutu Technologii Eksploatacji, 1997.
31. Mikoszewski J. Twardo ciomierze. Warszawa, Wydawnictwo Normalizacyjne "Alfa" 1985.
32. Münz W.D. Rozpylanie reaktywne azotków i w glików W: Elektronika nr 5, 1986.
33. Muszy ska D., Posadowski W. Badanie w a ciwo ci elektrycznych cienkich warstw do-
mieszkowanych azotem, otrzymywanych metod reaktywnego rozpylania magnetronowe-
go. W: Elektronika nr 3, 1985.
34. Nawrocka A., Bako A. Metalizacja diamentów syntetycznych. W: Przegl d Mechanicz-
nych nr 1, 1988.
35. Ole A. Metody eksperymentalne fizyki cia a sta ego. Warszawa, WNT 1983.
36. Ole A. Metody eksperymentalne fizyki cia a sta ego. Warszawa, WNT 1998
37. Orajewski W.N. Plazma na ziemi i w kosmosie. Warszawa, PWN 1989.
38. Precht W. Nowoczesne technologie plazmowe: Materia y Konferencji w Mielnie z lat 1990-
1998. Koszalin, Wyd. Politechniki Koszali skiej.
39. Redakcja Narz dzia pokrywane azotkiem tytanu. W: Mechanik nr 12, 1983.
40. Redakcja Pokrywanie narz dzi skrawaj cych azotkiem lub w glikiem tytanu. W: Mecha-
nik nr 2, 1984.
41. Redakcja Pokrywanie narz dzi ze stali szybkotn cej. W: Mechanik nr 6, 1984.
42. Roli ski E. Wspó czesna in ynieria powierzchni. W: Przegl d Mechaniczny nr 8, 1987.
43. Soko owska A. Niekonwencjonalne rodki syntezy materia ów. Warszawa, PWN 1991.
44. Wysiecki M. Nowoczesne materia y narz dziowe. Warszawa, WNT, 1997.
45. Zdanowski J. Jonowe metody nanoszenia warstw azotku tytanu jako pokry utwardzaj -
cych, antykorozyjnych i dekoracyjnych. W: Elektronika nr 2, 1988.
46. Zdanowski J. Jonowe trawienie powierzchni cia a sta ego i jego zastosowania. Wroc aw,
Wydawnictwo Politechniki Wroc awskiej, 1976.
47. Zdanowski J. Wy adowania elektryczne w gazach. Wroc aw, Skrypty Politechniki Wro-
c awskiej 1975.


















