
Uniwersytet Warszawski - ichf.edu.plichf.edu.pl/Praca_W3_Zofia_Tomasiewicz.pdf · Dzięki temu, nie...
46
1 Uniwersytet Warszawski Wydział Chemii Zofia Tomasiewicz Nr albumu: 292743 Synteza analogów kapu zawierających linkery aminokarboksylowe w obrębie reszt rybozy - substratów do znakowania fluorescencyjnego i funkcjonalizacji nanomateriałów. Projekt licencjacki na kierunku Chemia w ramach Kolegium Międzywydziałowych Indywidualnych Studiów Matematyczno - Przyrodniczych w zakresie Chemii Organicznej Praca wykonana pod kierunkiem dr Jacka Jemielitego Zakład Biofizyki IFD, Wydziału Fizyki UW Warszawa, czerwiec 2012
Transcript of Uniwersytet Warszawski - ichf.edu.plichf.edu.pl/Praca_W3_Zofia_Tomasiewicz.pdf · Dzięki temu, nie...

1
Uniwersytet Warszawski
Wydział Chemii
Zofia Tomasiewicz
Nr albumu: 292743
Synteza analogów kapu zawierających
linkery aminokarboksylowe w obrębie reszt rybozy
- substratów do znakowania fluorescencyjnego
i funkcjonalizacji nanomateriałów.
Projekt licencjacki
na kierunku Chemia
w ramach Kolegium Międzywydziałowych
Indywidualnych Studiów Matematyczno - Przyrodniczych
w zakresie Chemii Organicznej
Praca wykonana pod kierunkiem dr Jacka Jemielitego
Zakład Biofizyki IFD, Wydziału Fizyki UW
Warszawa, czerwiec 2012

2

3
Oświadczenie kierującego pracą
Oświadczam, że niniejsza praca została przygotowana pod moim kierunkiem i stwierdzam, że spełnia ona warunki do przedstawienia jej w postępowaniu o nadanie tytułu zawodowego.
Data Podpis kierującego pracą
Oświadczenie autora pracy
Świadoma odpowiedzialności prawnej oświadczam, że niniejsza praca dyplomowa została napisana przez mnie samodzielnie i nie zawiera treści uzyskanych w sposób niezgodny z obowiązującymi przepisami.
Oświadczam również, że przedstawiona praca nie była wcześniej przedmiotem procedur związanych z uzyskaniem tytułu zawodowego w wyższej uczelni.
Oświadczam ponadto, że niniejsza wersja pracy jest identyczna z załączoną wersją elektroniczną.
Data Podpis autora pracy .

4

5
Streszczenie
Celem niniejszego projektu była synteza analogów końca 5’ mRNA zawierających linkery
w resztach rybozy 7-metyloguanozyny, pozwalające na przyłączenie znaczników
fluorescencyjnych, bądź funkcjonalizację nanocząstek. Otrzymano serię analogów kapu z trzema
różnymi linkerami aminokarboksylowymi w resztach 2’ oraz 3’ rybozy.
Słowa kluczowe
mRNA, 5’-kap, linkery, znakowanie fluorescencyjne, funkcjonalizacja nanocząstek, eIF4E, Dcp1/Dcp2
Dziedzina pracy (kody wg programu Socrates-Erasmus)
13.3 Chemia

6

7
Składam serdeczne podziękowania
dr Jackowi Jemielitemu
za przekazaną wiedzę, poświęcony czas
oraz okazaną serdeczność.

8

9

10

11
Spis treści
1. Ekspresja genu ............................................................................................................................ 13
1.1. Transkrypcja .......................................................................................................................... 13
1.2. Translacja ............................................................................................................................... 15
1.3. Degradacja mRNA .................................................................................................................. 16
2. Modyfikacje chemiczne struktury kapu .................................................................................... 17
2.1. Budowa i właściwości 7-metyloguanozyny ........................................................................... 17
2.2. Struktura kapu ....................................................................................................................... 17
2.3. Biologiczne funkcje kapu ....................................................................................................... 18
2.4. Modyfikacje w obrębie rybozy 7-metyloguanozyny ............................................................. 18
2.5. Cele syntezy analogów kapu zawierających linker w obrębie reszt rybozy .......................... 18
2.5.1. Analogi kapu w terapii przeciwnowotworowej ..................................................................... 18
2.5.2. Znakowanie fluorescencyjne ................................................................................................. 19
2.5.3. Koniungaty z nanocząstkami ................................................................................................. 20
3. Synteza chemiczna analogów kapu ........................................................................................... 21
3.1. Tworzenie wiązania pirofosforanowego ............................................................................... 21
3.1.1. Aktywacja nukleotydów ........................................................................................................ 21
3.1.1.1. Synteza p-imidazolidów Hashimoto .............................................................................. 21
3.1.1.2. Aktywacja fosforanu N,N-karbonylodiimidazolem........................................................ 22
3.2. Metylowanie w pozycji N7 guanozyny ................................................................................... 22
3.3. Otrzymywanie dinukleotydów .............................................................................................. 23
4. Badania własne ........................................................................................................................... 25
4.1. Cel pracy ................................................................................................................................ 25
4.2. Strategia syntezy analogów kapu zawierających linkery aminokarboksylowe ..................... 25
4.3. Badania NMR ......................................................................................................................... 28
4.4. Podsumowanie ...................................................................................................................... 30
5. Część eksperymentalna .............................................................................................................. 31
5.1. Informacje ogólne ................................................................................................................. 31
Rozpuszczalniki.................................................................................................................................. 31
Kolumnowa chromatografia jonowymienna .................................................................................... 31
Wysokociśnieniowa chromatografia kolumnowa (HPLC) ................................................................. 31
Spektrometria mas i NMR ................................................................................................................. 31
5.2. Syntezy chemiczne ................................................................................................................ 32
5’-O-(β-cyjanoetylo)difosforan guanozyny (4) .................................................................................. 32
5’-difosforan 2’-O/3’-O-(2-karboksyetylo)-karbamoilo-guanozyny (5)............................................. 32
5’-difosforan 2’-O/3’-O-(2-karboksyetylo)-karbamoilo-7-metyloguanozyny (8) (BAPA-CO-m7GDP) 33
BAPA-CO-m7GpppG (1) ..................................................................................................................... 33
5’-difosforan 2’-O/3’-O-(3-karboksybutylo)-karbamoilo-guanozyny (6) .......................................... 34
5’-difosforan 2’-O/3’-O-(3-karboksybutylo)-karbamoilo-7-metyloguanozyny (9) (GABA-CO-m7GDP)
34
GABA-CO-m7GpppG (2) ..................................................................................................................... 35

12
5’-difosforan 2’-O/3’-O-(6-karboksyheksylo)-karbamoilo-guanozyny (7) ........................................ 36
5’-difosforan 2’-O/3’-O-(2-karboksyheksylo)-karbamoilo-7-metyloguanozyny (10) (EACA-CO-
m7GDP) .............................................................................................................................................. 36
EACA-CO-m7GpppG (3)...................................................................................................................... 37
5.3. Uzupełnienie .......................................................................................................................... 38
5.3.1. BAPA-CO-m7GpppG ............................................................................................................... 38
5.3.2. GABA-CO-m7GpppG ............................................................................................................... 40
5.3.3. EACA-CO-m7GpppG ............................................................................................................... 42
Bibliografia ........................................................................................................................................... 45

13
1. Ekspresja genu
Ekspresja genu jest to proces odczytywania informacji genetycznej i wykorzystania jej
do syntezy odpowiednich makromolekuł [1]. W procesie tym powstają białka, jak również
funkcjonalne kwasy rybonukleinowe. Na mechanizm ekspresji genu składa się kilka podstawowych
etapów. Pierwszym z nich jest transkrypcja podczas której syntetyzowany jest matrycowy
(informacyjny) RNA (mRNA) o sekwencji komplementarnej do matrycowego odcinka DNA. Pierwotny
transkrypt jest modyfikowany na końcach 5’, 3’ a następnie poddawany jest tzw. procesowi składania
(ang. splicing), czyli wycinaniu fragmentów niekodujących – intronów. Dojrzały mRNA
transportowany jest z jądra do cytoplazmy, gdzie podlega translacji, w której bierze udział
wiele makrocząsteczek – czynniki białkowe, kwasy rybonukleinowe oraz rybosomy. Gotowy łańcuch
polipeptydowy poddawany jest modyfikacjom posttranslacyjnym oraz ulega zwijaniu (ang. folding).
Powstałe w ten sposób białko transportowane jest do odpowiedniego miejsca w organizmie.
Na poniższym schemacie została przedstawiona typowa droga przepływu informacji
genetycznej w komórce. Jej poszczególne etapy omówiono bardziej szczegółowo w kolejnych
podrozdziałach .
Rys. 1.1. Schemat ekspresji genów u eukariontów.
1.1. Transkrypcja
Transkrypcja u organizmów eukariotycznych zachodzi w jądrze komórkowym. Na matrycy
przechowywanego w nim DNA syntetyzowane są cząsteczki RNA o sekwencji zasad komplementarnej
do odcinka antysensownej nici DNA. Proces jest katalizowany przez zależną od DNA polimerazę RNA,
która najpierw wiąże się z promotorowym rejonem DNA i rozplątuje fragment nici DNA, a następnie
przyłącza kolejne trójfosforany rybonukleozydów obecne w nukleoplazmie, wydłużając łańcuch
mRNA. Obecność enzymu pirofosfatazy umożliwia hydrolizę powstających w wyniku kondensacji
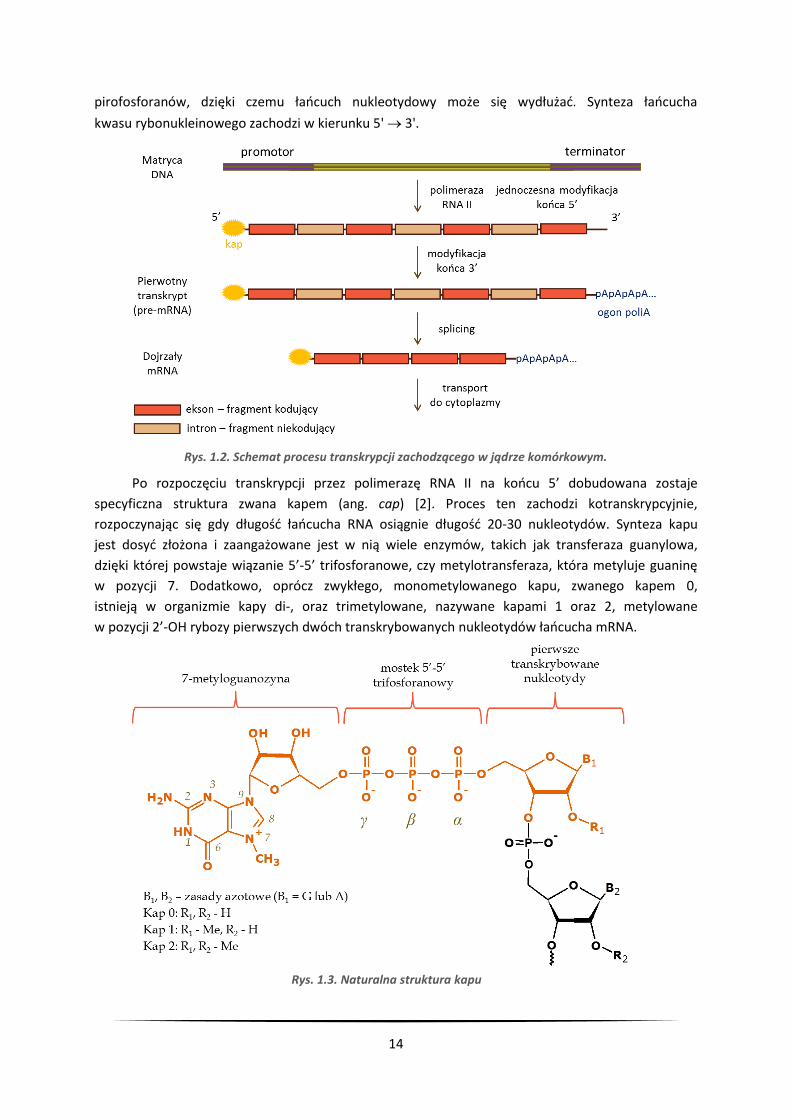
14
pirofosforanów, dzięki czemu łańcuch nukleotydowy może się wydłużać. Synteza łańcucha
kwasu rybonukleinowego zachodzi w kierunku 5' 3'.
Po rozpoczęciu transkrypcji przez polimerazę RNA II na końcu 5’ dobudowana zostaje
specyficzna struktura zwana kapem (ang. cap) [2]. Proces ten zachodzi kotranskrypcyjnie,
rozpoczynając się gdy długość łańcucha RNA osiągnie długość 20-30 nukleotydów. Synteza kapu
jest dosyć złożona i zaangażowane jest w nią wiele enzymów, takich jak transferaza guanylowa,
dzięki której powstaje wiązanie 5’-5’ trifosforanowe, czy metylotransferaza, która metyluje guaninę
w pozycji 7. Dodatkowo, oprócz zwykłego, monometylowanego kapu, zwanego kapem 0,
istnieją w organizmie kapy di-, oraz trimetylowane, nazywane kapami 1 oraz 2, metylowane
w pozycji 2’-OH rybozy pierwszych dwóch transkrybowanych nukleotydów łańcucha mRNA.
Rys. 1.2. Schemat procesu transkrypcji zachodzącego w jądrze komórkowym.
Rys. 1.3. Naturalna struktura kapu

15
Po zakończeniu syntezy łańcucha, na końcu 3’, dobudowywany jest ciąg nukleotydów
adeninowych, zwany ogonem poli-A. Przeciętnie składa się on z 50 – 250 nukleotydów.
Modyfikacja ta między innymi zwiększa odporność łańcucha mRNA na hydrolizę enzymatyczną.
Jest również istotna z punktu widzenia translacji i innych procesów w których uczestniczy RNA
(np. represja translacji za pośrednictwem mikroRNA) [3].
Powstały w ten sposób pierwotny transkrypt mRNA (pre mRNA), ulega składaniu
(ang. splicing). Polega to na wycięciu niekodujących sekwencji nukleotydowych - intronów
i połączeniu fragmentów kodujących – egzonów. Utworzony w ten sposób dojrzały matrycowy RNA
transportowany jest do cytoplazmy.
1.2. Translacja
Proces ten u eukariontów ma miejsce w cytoplazmie bądź na powierzchni retikulum
endoplazmatycznego szorstkiego i składa się z trzech etapów: inicjacji, elongacji oraz terminacji.
W translację, oprócz mRNA, które tworzy matrycę do biosyntezy białka, zaangażowane są także
dwa inne typy kwasu rybonukleinowego: tRNA (transportowy RNA) i rRNA (rybosomalny RNA).
Inicjacja polega na przyłączeniu małej podjednostki rybosomu do końca 5’ mRNA przy pomocy
czynników inicjujących translację. Sygnałem do rozpoczęcia translacji jest związanie struktury kapu
przez białko eIF4E [2]. Etap ten limituje przebieg całego procesu translacji [4].
Pod koniec procesu inicjacji translacji, po przyłączeniu dużej podjednostki rybosomu,
odnalezieniu kodonu AUG oraz zakotwiczeniu pierwszego aminokwasu (metioniny) rozpoczyna się
proces elongacji łańcucha peptydowego. Gdy tylko rybosom napotka kodon stop następuje
terminacja transkrypcji i uwolnienie zsyntetyzowanego polipeptydu.
Rys. 1.4. Schemat procesu translacji zachodzącego w cytoplazmie.

16
Gotowy łańcuch polipeptydowy poddawany jest modyfikacjom posttranslacyjnym
oraz ulega zwijaniu. Powstałe w ten sposób białko transportowane jest do odpowiedniego miejsca
w organizmie.
1.3. Degradacja mRNA
Na stabilność struktury łańcucha mRNA wpływa struktura kapu na końcu 5’ oraz ogona poli-A
na końcu 3’ [5, 6]. W związku z tym degradacja łańcucha mRNA może przebiegać na dwa sposoby
rozróżniane w zależności od kierunku działania nukleaz [7, 8]. Zarówno degradacja 5’->3’ jak i 3’-> 5’
wymaga wcześniejszej deadenylacji [9].
Dostęp do matrycy mRNA od końca 5’ jest niemożliwy dla egzonukleaz gdy na 5’ końcu
znajduje się osłaniająca transkrypt struktura kapu, jednak gdy tylko kap zostanie zhydrolizowany
egzonukleazy natychmiast rozpoczynają degradację kwasu nukleinowego w kierunku jego 3’ końca.
Utrata kapu następuje pod wpływem kompleksu enzymatycznego Dcp1/Dcp2. Degradacja od strony
3’ przeprowadzana jest przez egzosom. Powstałe krótkie oligonukleotydowe fragmenty zawierające
kap są degradowane przez enzym DcpS (Decapping Scavenger).Enzym DcpS odcina kap hydrolizując
wiązanie pomiędzy β i γ fosforanem w przeciwieństwie do Dcp1/Dcp2, który hydrolizuje wiązanie
pomiędzy α i β fosforanem [10, 11].
Rys. 1.5. Degradacja mRNA.
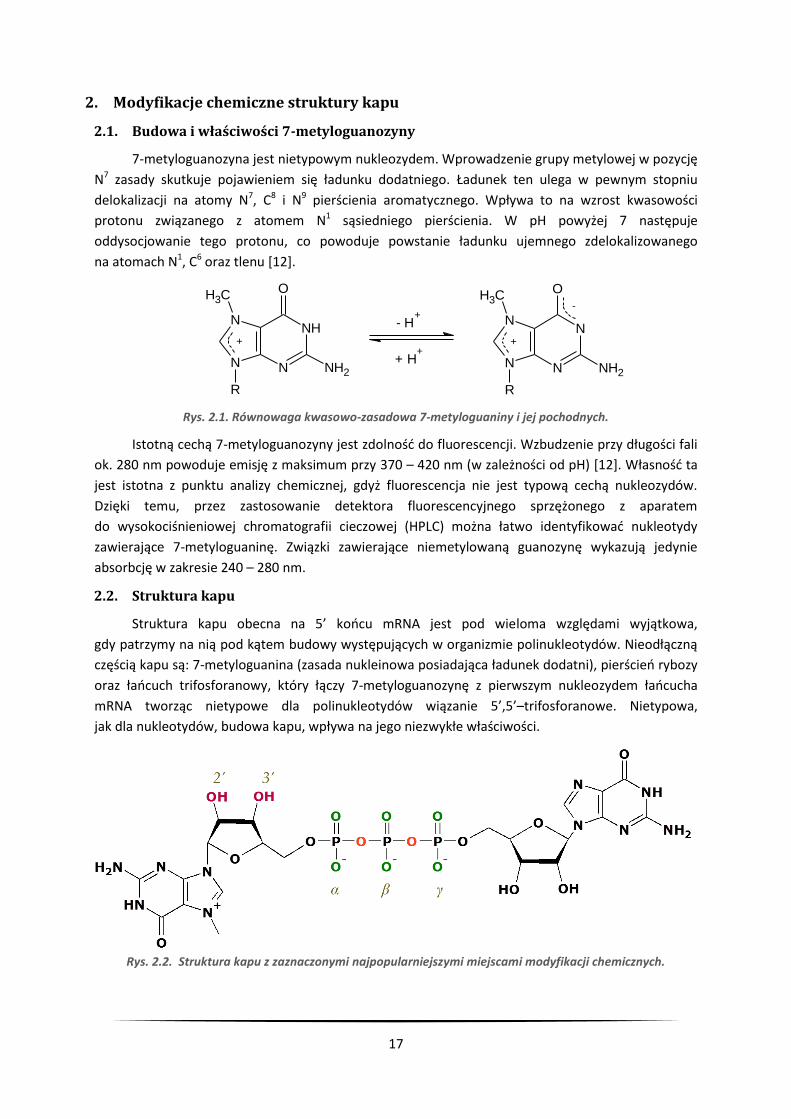
17
2. Modyfikacje chemiczne struktury kapu
2.1. Budowa i właściwości 7-metyloguanozyny
7-metyloguanozyna jest nietypowym nukleozydem. Wprowadzenie grupy metylowej w pozycję
N7 zasady skutkuje pojawieniem się ładunku dodatniego. Ładunek ten ulega w pewnym stopniu
delokalizacji na atomy N7, C8 i N9 pierścienia aromatycznego. Wpływa to na wzrost kwasowości
protonu związanego z atomem N1 sąsiedniego pierścienia. W pH powyżej 7 następuje
oddysocjowanie tego protonu, co powoduje powstanie ładunku ujemnego zdelokalizowanego
na atomach N1, C6 oraz tlenu [12].
N
NH
N
N
NH2
O
R
CH3
+
- H+
+ H+
N
N
N
N
NH2
O
R
CH3
+
-
Rys. 2.1. Równowaga kwasowo-zasadowa 7-metyloguaniny i jej pochodnych.
Istotną cechą 7-metyloguanozyny jest zdolność do fluorescencji. Wzbudzenie przy długości fali
ok. 280 nm powoduje emisję z maksimum przy 370 – 420 nm (w zależności od pH) [12]. Własność ta
jest istotna z punktu analizy chemicznej, gdyż fluorescencja nie jest typową cechą nukleozydów.
Dzięki temu, przez zastosowanie detektora fluorescencyjnego sprzężonego z aparatem
do wysokociśnieniowej chromatografii cieczowej (HPLC) można łatwo identyfikować nukleotydy
zawierające 7-metyloguaninę. Związki zawierające niemetylowaną guanozynę wykazują jedynie
absorbcję w zakresie 240 – 280 nm.
2.2. Struktura kapu
Struktura kapu obecna na 5’ końcu mRNA jest pod wieloma względami wyjątkowa,
gdy patrzymy na nią pod kątem budowy występujących w organizmie polinukleotydów. Nieodłączną
częścią kapu są: 7-metyloguanina (zasada nukleinowa posiadająca ładunek dodatni), pierścień rybozy
oraz łańcuch trifosforanowy, który łączy 7-metyloguanozynę z pierwszym nukleozydem łańcucha
mRNA tworząc nietypowe dla polinukleotydów wiązanie 5’,5’–trifosforanowe. Nietypowa,
jak dla nukleotydów, budowa kapu, wpływa na jego niezwykłe właściwości.
Rys. 2.2. Struktura kapu z zaznaczonymi najpopularniejszymi miejscami modyfikacji chemicznych.

18
Ze względu na istotne funkcje pełnione w organizmie, struktura kapu jest interesującym obiektem do
badań. Wprowadzane do niej modyfikacje mogą znacząco wpływać na jej właściwości biologiczne,
takie jak powinowactwo do białek wiążących kap, czy odporność na hydrolizę z udziałem enzymów
degradujących kap [13].
Modyfikacje w mostku trifosforanowym polegają na podstawieniu atomu tlenu innym atomem
lub ugrupowaniem, mogą one być w pozycjach mostkowych – pomiędzy atomami fosforu, bądź
w pozycjach niemostkowych. Znacząco wpływają one na podatność kapu na degradację enzymami
hydrolitycznymi, oraz na powinowactwo kapu do białek wiążących kap.
Możliwe są również modyfikacje w obrębie rybozy 7-metyloguanozyny. Dzięki nim można badać
oddziaływania między kapem, a wiążącymi go białkami.
2.3. Biologiczne funkcje kapu
Struktura kapu obecna na końcu 5’ mRNA jest znakiem rozpoznawczym dla białek pełniących
istotne funkcje w procesie ekspresji genu. Już w jądrze komórkowym, kap obecny na końcu 5’
pre-mRNA oddziałuje z białkiem CBP (Cap Binding Protein), które to umożliwia splicing
oraz poliadenylację końca 3’ mRNA [14]. CBP bierze również udział w transporcie dojrzałego
transkryptu z jądra do cytoplazmy [15]. Bez obecności kapu na końcu 5’ mRNA żaden z tych procesów
nie mógłby być prawidłowo przeprowadzony.
Kap pełni również kluczową rolę w procesie inicjacji translacji. Oddziaływanie kapu
z eukariotycznym czynnikiem translacji eIF4E jest etapem limitującym szybkość syntezy białek [16].
Bez obecności kapu na końcu 5’ mRNA nie byłoby możliwe utworzenie kompleksu inicjującego
translację, a więc i wydajne powstawanie białek.
Niezwykle ważną funkcją kapu jest również ochrona mRNA przed degradacją.
Mostek 5’-5’-trifosforanowy stanowi skuteczną ochronę przed hydrolizą enzymatyczną, wydłużając
dzięki temu czas życia mRNA w komórce.
2.4. Modyfikacje w obrębie rybozy 7-metyloguanozyny
Jak wykazały badania krystalograficzne, modyfikacje w obrębie rybozy 7-metyloguanozyny
nie mają specjalnego wpływu na powinowactwo struktury kapu do czynnika inicjującego.
Grupy –OH w pozycjach 2’, 3’ rybozy 7-metyloguanozyny znajdują się poza centrum aktywnym białka,
a więc najprawdopodobniej słabo z nim oddziałują. Dzięki temu, nie tracąc na sile powinowactwa
można w te pozycje wprowadzać różne modyfikacje chemiczne (np. znaczniki fluorescencyjne),
nie zmieniając ich właściwości biologicznych [17].
Dodatkowo wprowadzenie modyfikacji w resztach 2’,3’ rybozy 7-metyloguanozyny zapewnia
prawidłowe wbudowywanie się kapu do łańcucha mRNA – z 7-metyloguanozyną na końcu 5’.
Kap z wolną grupą 3’-OH może zostać wbudowany odwrotnie, tzn. z 7-metyloguanozyną bliżej
3’ końca łańcuch mRNA. Transkrypt taki jest bezużyteczny.
2.5. Cele syntezy analogów kapu zawierających linker w obrębie reszt rybozy
2.5.1. Analogi kapu w terapii przeciwnowotworowej
W wielu komórkach nowotworowych stwierdzono podwyższony poziom ekspresji czynnika
inicjującego translację eIF4E [18,19]. Przy prawidłowym poziomie tego białka w komórkach translacji
ulegają tylko tzw. silne mRNA, wysycające obecne w komórce eIF4E, natomiast tzw. słabe mRNA,

19
kodujące zazwyczaj czynniki onkogenne, pozostają nieaktywne. Przy podwyższonym poziomie
białka eIF4E możliwe jest również wiązanie się słabych mRNA, a przez to translacja onkoprotein [20].
Redukcja poziomu eIF4E w komórce uniemożliwiłaby translację czynników związanych
z kancerogenezą, a więc rozwój nowotworu. Analogi kapu, dzięki wysoce specyficznemu wiązaniu
z eIF4E, mogłyby zmniejszyć pulę białka dostępnego w procesie translacji i zahamować
proces nowotworzenia [21, 22].
Innym potencjalnym zastosowaniem mRNA jest wykorzystanie go w terapii genowej [23].
Do tej pory w terapii genowej najczęściej stosowano DNA jednak, w porównaniu do zastosowania
mRNA, ma ono sporo wad. RNA jest znacznie bezpieczniejsze w użyciu, nie wymaga ingerencji
w genom pacjenta, więc zmniejsza ryzyko wprowadzania uszkodzeń w istotne fragmenty genomu,
co może prowadzić do nowotworzeni i innych groźnych chorób. Nie wprowadza nieodwracalnych
zmian. Jest również mniej immunogenne. Działa w cytoplazmie przez co nie jest konieczne
wprowadzanie do jądra komórkowego [24]. Jednak istnieje też pewne ograniczenie w stosowalności
RNA – naturalne mRNA jest dosyć nietrwałe w warunkach komórkowych. Przez wprowadzenie
odpowiedniej modyfikacji chemicznej w mostku trifosforanowym struktury kapu, możliwe jest jednak
znaczne wydłużenie jego czasu życia [25]. Wykazano, że mRNA wstrzyknięte do węzła chłonnego
zostaje spontanicznie wchłonięte przez niedojrzałe komórki dendrytyczne, gdzie ulega translacji,
a powstałe białko prezentowane jest na powierzchni komórek dendrytycznych i bierze udział
w specyficznej aktywacji układu immunologicznego [26, 27].
2.5.2. Znakowanie fluorescencyjne
Analogi kapu wprowadzone do komórki stwarzają wiele trudności badawczych.
Szczególnym problemem jest ocena ich lokalizacji oraz stężenia w komórce. Przyłączenie
do analogu kapu znacznika fluorescencyjnego umożliwiłoby wyznaczenie tych parametrów.
Stworzyłoby to nowe możliwości w badaniu procesów posttranskrypcyjnych, takich jak degradacja
mRNA przez enzym Dcp1/Dcp2. Pozwoliłoby też na lepsze poznanie mechanizmu działania analogów
kapu jako inhibitorów translacji poprzez badania na liniach komórkowych
oraz modelowych organizmach zwierzęcych. Znakowane fluorescencyjnie analogi kapu umożliwiłyby
również zbadanie mechanizmu pochłaniania mRNA przez komórki dendrytyczne oraz mechanizm
jego translacji podczas terapii genowej z wykorzystaniem RNA.
Dzięki zastosowaniu analogów kapu z odpowiednio dobranym znacznikiem fluorescencyjnym,
będzie można prowadzić badania wprowadzonych do komórki analogów kapu nawet przy niskich
stężeniach (rzędu dziesiątek nanomoli na litr). Dotychczas prowadzenie badań przy stężeniach
poniżej 1 M stanowiło nierozwiązany problem.
Ze względu na niską wydajność kwantową fluorescencji naturalnej struktury kapu pewne
techniki fluorescencyjne, jak np. FRET, czy badania oparte na zaniku anizotropii fluorescencji,
są niemożliwe do zastosowania. Zastosowanie znakowanych analogów umożliwiłoby wykorzystanie
tych metod.
Z drugiej strony, mimo niskiej wydajności kwantowej, fluorescencja 7-metyloguanozyny
stwarza trudności w badaniach oddziaływań analogów kapu z białkami. Przy dużych stężeniach kapu,
znacznie komplikuje się interpretacja danych eksperymentalnych, gdyż widmo emisji
7-metyloguanozyny pokrywa się z widmem emisji reszt tryptofanowych białek. Zastosowanie

20
analogów znakowanych fluorescencyjnie umożliwiłoby wygaszenie fluorescencji 7-metyloguanozyny
na zasadzie wewnątrzcząsteczkowego transferu energii (FRET).
2.5.3. Koniungaty z nanocząstkami
Analogi kapu, jak wszystkie nukleotydy, nie przenikają przez błony komórkowe. Przyłączenie
analogów kapu o znaczeniu terapeutycznym do nanocząstek stanowi jeden z pomysłów
na rozwiązanie tego problemu. Enkapsulacja analogów kapu wewnątrz nanocząstek przedłużyłoby
również ich żywotność, ponieważ krócej przebywałyby w środowisku fizjologicznym, gdzie
są narażone na działanie enzymów hydrolizujących kap. Połączenie z nanocząstkami magnetycznymi
umożliwiłyby natomiast ich kierowanie za pomocą pola magnetycznego i precyzyjne ulokowanie
w wybranych miejscach organizmu pacjenta.
Koniungaty kapu z nanocząstkami można również wykorzystać do tworzenia złóż
powinowactwa. Jest to bardzo prosty i wydajny sposób na oczyszczenie białek wiążących kap.
Znacznie ułatwiałoby to otrzymanie wysoko oczyszczonych preparatów białkowych niezbędnych
do badań biologicznych. Złoża powinowactwa na nośniku magnetycznym mogłyby również znaleźć
zastosowanie w poszukiwaniu nowych białek wiążących kap oraz do ich wstępnej charakterystyki.
Analogi kapu z przyłączonymi linkerami mogą posłużyć również do tworzenia biosensorów
do diagnostyki medycznej. Selektywność nadana sensorowi poprzez dołączenie analogu kapu
umożliwiłaby ilościowe oznaczenie białek wiążących kap nawet w próbkach o skomplikowanym
składzie bez konieczności ich oczyszczania. Przykładem mogą być elektrody grafenowe
z przyłączonymi analogami kapu – w zależności od tego, czy struktura kapu związana jest
przez czynnik eIF4E, zmienia się natężenie przepływającego prądu.

21
3. Synteza chemiczna analogów kapu
3.1. Tworzenie wiązania pirofosforanowego
Wytworzenie wiązania pirofosforanowego wymaga zwiększenia elektrofilowości jednego
z atomów fosforu. Aktywny P-imidazolid reagując z anionem fosforanowym daje odpowiedni
5’-difosforan nukleozydu.
N
NH
N
N
NH2
O
OO
OH OH
P
O-
O
NN
N
NH
N
N
NH2
O
OO
OH OH
P
O-
O
O-
O
O
O-
PZnCl2, DMF
PO4
3- sól TEAH
+
Rys. 3.1. Tworzenie wiązania pirofosforanowego.
W reakcji tej istotną rolę odgrywają sole metali
dwuwartościowych np. chlorek cynku lub magnezu [28]. Ich rola
w przebiegu reakcji jest dość złożona. Kationy metalu kompleksując
atomy tlenu w reszcie fosforanowej oraz P-imidazolidu, a także
oddziałując również z atomem azotu pierścienia imidazolu zwiększają
elektrofilowość atomu fosforu, stabilizując stan przejściowy reakcji.
Prawdopodobną strukturę stanu przejściowego przedstawiono
na rys. 3.2. Chlorki metali dwuwartościowych w istotny sposób
zwiększają też rozpuszczalność nukleotydów w wykorzystywanych
rozpuszczalnikach.
3.1.1. Aktywacja nukleotydów
Opracowano wiele metod chemicznych aktywacji grupy fosforanowej, jednak
w mojej pracy stosowane są pochodne, w której aktywną formą fosforanu są P-imidazolidy,
zaproponowane przez Hoarda i Otta [29]. W literaturze można znaleźć kilka sposobów otrzymywania
imidazolowych pochodnych nukleotydów, jednak przedstawię tylko dwie z nich, najważniejsze
z punktu widzenia niniejszej pracy.
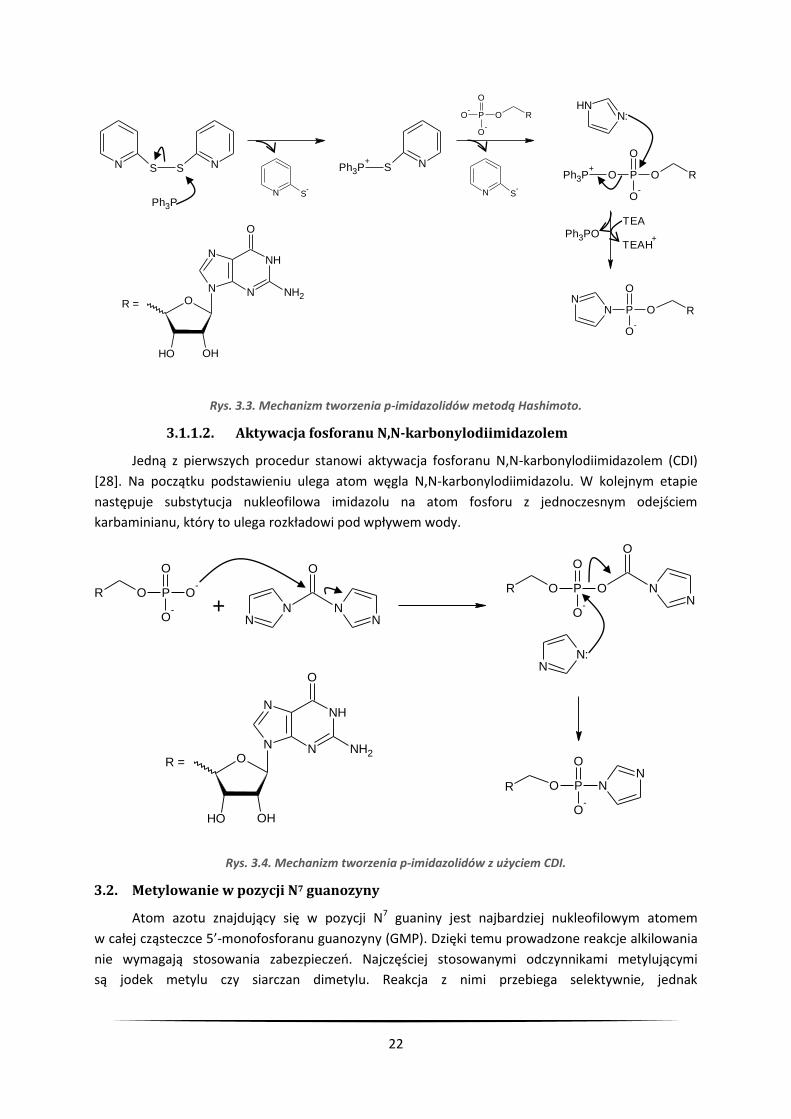
3.1.1.1. Synteza p-imidazolidów Hashimoto
Reakcja Mukaiyamy-Hashimoto [30] przebiega w obecności trifenylofosfiny
i 2',2'-ditiodipirydyny, które stanowią układ aktywujący. W pierwszym etapie trifenylofosfina atakuje
atom siarki 2,2-ditiodipirydyny z utworzeniem soli fosfoniowej i anionu tiolanowego o żółtym
zabarwieniu. Taka sól fosfoniowa reaguje z grupą fosforanową aktywując ją, dzięki czemu ulega ona
substytucji nukleofilowej, tworząc p-imidazolid. Podstawienie atomu fosforu imidazolem możliwe
jest dzięki wytworzeniu bardzo trwałego wiązania P=O w odchodzącej trifenylofosfinie, co stanowi
siłę napędową reakcji.
Zn
ClCl
NN
ZnCl
Cl
P
OR
Nu
O-
O-
Rys. 3.2. Prawdopodobny mechanizm
działania ZnCl2. Nu oznacza centrum
nukleofilowe, natomiast R pozostałą
część nukleotydu.
22
N S NS
N S-
Ph3P+ NS
N S-
P
O
O
O-
O-
R
P
O
O
O-
O RPh3P+
NHN:
TEA
TEAH+Ph3PO
ROP
O
O-
NN
N
NH
N
N
NH2
O
O
OHOH
R =
Ph3P
Rys. 3.3. Mechanizm tworzenia p-imidazolidów metodą Hashimoto.
3.1.1.2. Aktywacja fosforanu N,N-karbonylodiimidazolem
Jedną z pierwszych procedur stanowi aktywacja fosforanu N,N-karbonylodiimidazolem (CDI)
[28]. Na początku podstawieniu ulega atom węgla N,N-karbonylodiimidazolu. W kolejnym etapie
następuje substytucja nukleofilowa imidazolu na atom fosforu z jednoczesnym odejściem
karbaminianu, który to ulega rozkładowi pod wpływem wody.
R O P
O
O-
NN
N
NH
N
N
NH2
O
O
OHOH
R =
P
O
O
O-
O-
R
NN
NN
O
+P
O
O
O-
OR NN
O
N:N
Rys. 3.4. Mechanizm tworzenia p-imidazolidów z użyciem CDI.
3.2. Metylowanie w pozycji N7 guanozyny
Atom azotu znajdujący się w pozycji N7 guaniny jest najbardziej nukleofilowym atomem
w całej cząsteczce 5’-monofosforanu guanozyny (GMP). Dzięki temu prowadzone reakcje alkilowania
nie wymagają stosowania zabezpieczeń. Najczęściej stosowanymi odczynnikami metylującymi
są jodek metylu czy siarczan dimetylu. Reakcja z nimi przebiega selektywnie, jednak
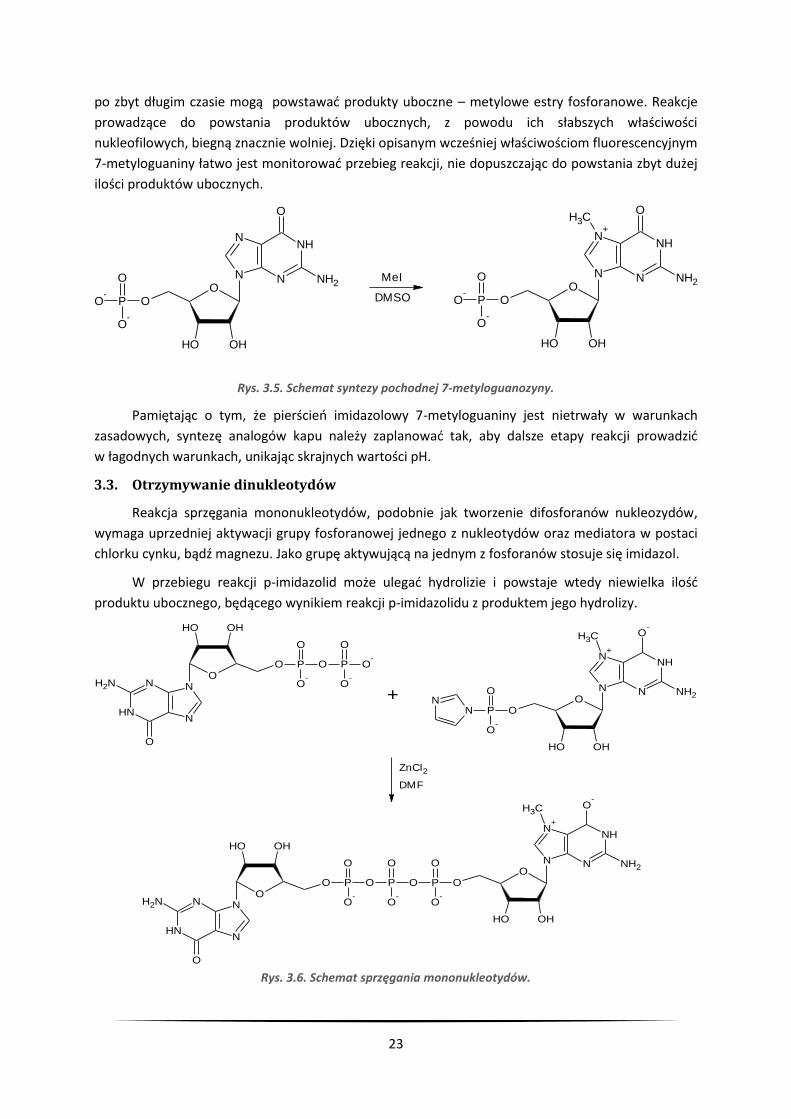
23
po zbyt długim czasie mogą powstawać produkty uboczne – metylowe estry fosforanowe. Reakcje
prowadzące do powstania produktów ubocznych, z powodu ich słabszych właściwości
nukleofilowych, biegną znacznie wolniej. Dzięki opisanym wcześniej właściwościom fluorescencyjnym
7-metyloguaniny łatwo jest monitorować przebieg reakcji, nie dopuszczając do powstania zbyt dużej
ilości produktów ubocznych.
N
NH
N
N
NH2
O
OO
OHOH
O-
O
PO-
MeI
DMSO
N
NH
N
N+
NH2
O
OO
OHOH
O-
O
PO-
CH3
Rys. 3.5. Schemat syntezy pochodnej 7-metyloguanozyny.
Pamiętając o tym, że pierścień imidazolowy 7-metyloguaniny jest nietrwały w warunkach
zasadowych, syntezę analogów kapu należy zaplanować tak, aby dalsze etapy reakcji prowadzić
w łagodnych warunkach, unikając skrajnych wartości pH.
3.3. Otrzymywanie dinukleotydów
Reakcja sprzęgania mononukleotydów, podobnie jak tworzenie difosforanów nukleozydów,
wymaga uprzedniej aktywacji grupy fosforanowej jednego z nukleotydów oraz mediatora w postaci
chlorku cynku, bądź magnezu. Jako grupę aktywującą na jednym z fosforanów stosuje się imidazol.
W przebiegu reakcji p-imidazolid może ulegać hydrolizie i powstaje wtedy niewielka ilość
produktu ubocznego, będącego wynikiem reakcji p-imidazolidu z produktem jego hydrolizy.
N
NH
N
N
NH2
O
OO
OH OH
P
O-
O
O-
O
O
O-
P
N
NH
N
N+
NH2
O-
OO
OHOH
P
O
O-
N
CH3
N
N
NH
N
N
NH2
O
OO
OH OH
P
O-
O
O
O
O
O-
P
N
NH
N
N+
NH2
O-
OO
OHOH
P
O
O-
CH3
+
ZnCl2
DMF
Rys. 3.6. Schemat sprzęgania mononukleotydów.

24

25
4. Badania własne
4.1. Cel pracy
Celem niniejszej pracy było zaprojektowanie ścieżki syntezy i otrzymanie analogów
końca 5’ mRNA zawierających linkery aminokarboksylowe w resztach 2’ oraz 3’ rybozy
7-metyloguanozyny, umożliwiających znakowanie fluorescencyjne, bądź funkcjonalizację
nanocząstek. Kluczowym etapem było opracowanie przyłączania linkera aminokarboksylowego
do reszty cukrowej poprzez ugrupowanie karbaminianowe.
Zaplanowano syntezę trzech dinukleotydowych analogów kapu z następującymi
linkerami: β-alaninowym, γ-aminomasłowym oraz -kapronowym.
W przyszłości mogą one znaleźć zastosowanie między innymi w opracowywaniu terapii
genowej w oparciu o RNA (tzw. szczepionek RNA), czy też w diagnostyce medycznej.
4.2. Strategia syntezy analogów kapu zawierających linkery aminokarboksylowe
Związkiem wyjściowym do syntezy analogów kapu z linkerami aminokarboksylowymi
w obrębie 2’/3’ reszt rybozy 7-metyloguanozyny sól trietyloamoniowa GMP. Strategię syntezy
ilustrują poniższe schematy.
N
NH
N
N
NH2
O
O
O
OHOH
O-
O
PO-
P
O
N
OH
ON
N
ZnCl2, DMF
N
NH
N
N
NH2
O
O
O
OHOH
O-
O
POP
O
O-
O
N
N
NH
N
N
NH2
O
O
O
OHOH
O-
O
POP
O
O-
O-
OH(CH2)nNHO
O
1) CDI / DMF
2) linker, DBU
MeI
DMSO
N
N
N
N+
NH2
O-
O
O
OHOH
O-
O
POP
O
O-
O-
CH3
OH(CH2)nNHO
O
N
N
N
N+
NH2
O-
O
O
OHOH
O-
O
POP
O
O-
O
CH3
N
NH
N
N
NH2
O
O
O
OH OH
O
O-
P
OH(CH2)nNHO
O
GMP-Im, ZnCl 2
DMF
n = 2, 3, 5
1, 2 lub 3
5, 6 lub 7 8, 9 lub 10
4
Rys. 4.1. Strategia syntezy analogów kapu zawierających linker aminokarboksylowy w obrębie reszt rybozy.
Pierwszym etapem było zabezpieczenie grupy fosforanowej w pozycji 5’-OH pierścienia rybozy.
Aby wprowadzić linker na reszty rybozy, niezbędne jest wcześniejsze zabezpieczenie grupy
fosforanowej na końcu 5’-OH rybozy. Substratami do reakcji są p-imidazolid fosforanu

26
β-cyjanoetylowego oraz monofosforan nukleozydu. Mechanizm reakcji jest analogiczny,
jak w opisanym tworzeniu wiązania pirofosforanowego.
N
NH
N
N
NH2
O
OO
OHOH
O-
O
PO-
N
NH
N
N
NH2
O
OO
OHOH
O-
O
PO
O-
O
PO
N
P
O
N
OH
ON
N
ZnCl 2, DMF
Rys. 4.2. Schemat reakcji zabezpieczania grupy fosforanowej.
Zabezpieczenie fosforanu grupą cyjanoetylową jest niezbędne, gdyż karbonylodiimidazol,
jak było opisane wcześniej może aktywować fosforan, co w przypadku prowadzonej przeze mnie
syntezy było niepożądane (powstałyby dinukleotydy połączone mostkiem tetrafosforowym).
W przeprowadzonej przeze mnie syntezie CDI jest wykorzystany do utworzenia reaktywnego
węglanu na resztach 2’/3’-OH rybozy, w celu wprowadzenia tam linkera aminokarboksylowego.
N
NH
N
N
NH2
O
OO
OHOH
P
O
O-
O
O-
O
O
P
N GO
O
OO
P
O
O-
O
O-
O
O
P
N
O
NN
NN
O
DMF
H3N+(CH2)nCOO
-
DBU (1 eqv.)
OH (CH2)nNH2
O
GO
O
OO
P
O
O-
O
O-
O
O
P
N
O
GO
O
OO
P
O
O-
O
O-
O
O
P
N
O-
N+
OH (CH2)n
O H
H
OH (CH2)nNH
O
GO
O
OOH
P
O
O-
O
O-
O
O
P
N
O
n = 2, 3, 5
Rys. 4.3. Schemat wprowadzania linkera aminokarboksylowego w pozycje 2’/3’ rybozy.
N,N-karbonylodiimidazol przyłącza się do pierścienie rybozy z utworzeniem cyklicznego
węglanu. Następnie grupa aminowa linkera (zdeprotonowana przez DBU) atakuje karbonylowy atom
węgla i następuje addycja nukleofilowa z przejściem pary elektronowej na atom tlenu. Następnie
rozrywa się jedno z wiązań C-O i następnie przeskok protonu z grupy aminowej z jednoczesnym
utworzeniem wiązania O-H, odtwarza się również podwójne wiązanie C=O.
Otrzymany 5’-cyjanoetylodifosforan 2’/3’-karbamoiloguanozyny z przyłączonym linkerem
poddano in situ reakcji odbezpieczania. Zabezpieczenie cyjanoetylowe zdejmuje się w obecności
DBU, który odrywa proton od węgla w pozycji α, potem następuje β-eliminacja, a produktami
są difosforan nukleozydu oraz akrylonitryl. Etap ten jest odwracalny, w związku z czym reakcję

27
prowadzi się pod zmniejszonym ciśnieniem w temperaturze 40-50oC w celu usunięcia ze środowiska
reakcji powstającego akrylonitrylu (Twrz. akrylonitrylu wynosi 78oC).
GO
O
OHOH
P
O
O-
O
O-
O
O
P
N
n = 2, 3, 5
DBU
OH (CH2)nNH
O
O
GO
O
OHOH
P
O
O-
O-
O-
O
O
P
OH (CH2)nNH
O
O
Rys. 4.4. Schemat reakcji odbezpieczania fosforanu.
Dinukleotyd z linkerem w resztach 2’/3’ rybozy poddano metylowaniu za pomocą jodku metylu
otrzymując jego metylową pochodną. Wydzielony produkt poddano reakcji sprzęgania
z P-imidazolidem monofosforanu guanozyny.
N
NH
N
N
NH2
O
O
O
OHOH
O-
O
PNN
N
N
N
N+
NH2
O-
O
O
OHOH
O-
O
POP
O
O-
O-
CH3
OH(CH2)nNHO
O
N
N
N
N+
NH2
O-
O
O
OHOH
O-
O
POP
O
O-
O
CH3
N
NH
N
N
NH2
O
O
O
OH OH
O
O-
P
OH(CH2)nNHO
O
ZnCl 2
DMF
n = 2, 3, 5
+
Rys. 4.5. Schemat otrzymywania dinukleotydów.
Powyższe syntezy przeprowadzono kolejno dla każdego z linkerów. W ten sposób otrzymano
trzy pary izomerów, każdy z innym linkerem karboksylowym – odpowiednio były to β-alanina (BAPA),
kwas γ-aminomasłowy (GABA) oraz kwas -aminokapronowy (EACA).

28
4.3. Badania NMR
W celu zidentyfikowania struktury otrzymanych związków wykonano ich widma 1H NMR
oraz 31P NMR. Widma fosforowe pozwalają na potwierdzenie struktury otrzymanego mostka
5’,5’-trifosforanowego. We wszystkich widmach 31P NMR widać dublet dubletów przy δ= -22/-23 ppm
pochodzący od fosforu w pozycji β oraz nałożone dublety przy ok. -10/-11 ppm odpowiadający
jądrom fosforu w pozycji α oraz γ. Ze schematu rozszczepień wynika, że są one niemal równocenne,
a ich stałe sprzężenia z fosforem β są prawie identyczne (J=19,5 Hz dla związku 2 oraz J= 18,1 Hz
dla związku 3b).
Widma 1H NMR otrzymanych analogów kapu wykazują pewne podobieństwo. Wszystkie
składają się z trzech części zawierających sygnały od protonów guaniny w zakresie 9-8 ppm
oraz singlet przy ok. 4 ppm pochodzący od 3 protonów grupy metylowej, protonów rybozy
od 6 do 4 ppm oraz protonów linkera. Tylko część zawierająca sygnały od linkera jest znacząco inna
dla każdego z linkerów. Pozostałe dwie są bardzo podobne do siebie.
W widmach protonowych analogów z linkerem BAPA oraz GABA nie widać piku od protonu H8
m7-guaniny. Prawdopodobnie spowodowane jest to wymianą na deuter – proton ten jest lekko
kwaśny, w związku z czym, po pewnym czasie w wodzie ciężkiej, ulega on wymianie.
W widmie protonowym 2’-EACA-CO-m7GpppG (3a) w części obejmującej piki pochodzące
od protonów linkera multiplet o przesunięciu chemicznym 3,08 ppm pochodzi od protonów grupy
CH2. Według przewidywanego schematu rozszczepień powinien to być tryplet (sprzężenie z dwoma
równocennymi protonami grupy δCH2), jednak tak naprawdę protony przy kolejnych węglach linkera
są diastereotopowe (cząsteczka kapu jest chiralna). Jako, że protony od grupy CH2 znajdują się
najbliżej chiralnego centrum cząsteczki, to ma ono na nie największy wpływ, dlatego schematu
rozszczepienia nie da się tak łatwo przewidzieć. Zjawiska tego nie zaobserwowano w pozostałych
widmach.
Aby zidentyfikować miejsce przyłączenia linkera – reszta 2’/ 3’ rybozy, dla analogu kapu
zawierającego jako linker kwas 6-aminoheksanowy wykonano widma odprzęgania. Uprzednio
rozdzielono oba izomery na HPLC preparatywnym. Tzw. decoupling jest to metoda używana
w spektroskopii NMR, w której na próbkę działamy określoną częstością, aby wyeliminować efekt
sprzęgania pomiędzy określonymi jądrami.
Porównując widmo protonowe zarejestrowane normalnie oraz widmo z odprzęganiem
można określić, do której reszty rybozy przyłączony jest linker. Widma odprzęgania zostały
zarejestrowane dla analogów kapu z linkerem -aminokapronowym (EACA) a izomery zostały
rozdzielone za pomocą preparatywnej chromatografii HPLC. Co ciekawe, okazało się, że oczyszczone
produkty nie izomeryzują, a przynajmniej zjawisko to nie zostało zaobserwowane. Można to dosyć
łatwo wyjaśnić – aby zaszła izomeryzacja, musiałby powstać zatłoczony stan przejściowy
z czwartorzędowym atomem węgla, otoczonym przez cztery elektroujemne atomy (trzy tleny i azot).
Poniżej przedstawione jest widmo odprzęgania dla związku 3a, o krótszym czasie retencji
na HPLC. W ramkach ujęte są różnicujące fragmenty widma protonowego. Odprzęgnięty został
proton H2’ pochodzący od 7-metyloguanozyny, co oznacza, że zniknęły sprzężenia pomiędzy
protonami H1’m7G–H2’m7G oraz H2’m7G–H3’m7G. W ten sposób bardzo łatwo jest zidentyfikować
sygnały pochodzące od protonów H1’m7G, H2’m7G oraz H3’m7G. Pik przy δ=5,97 w widmie
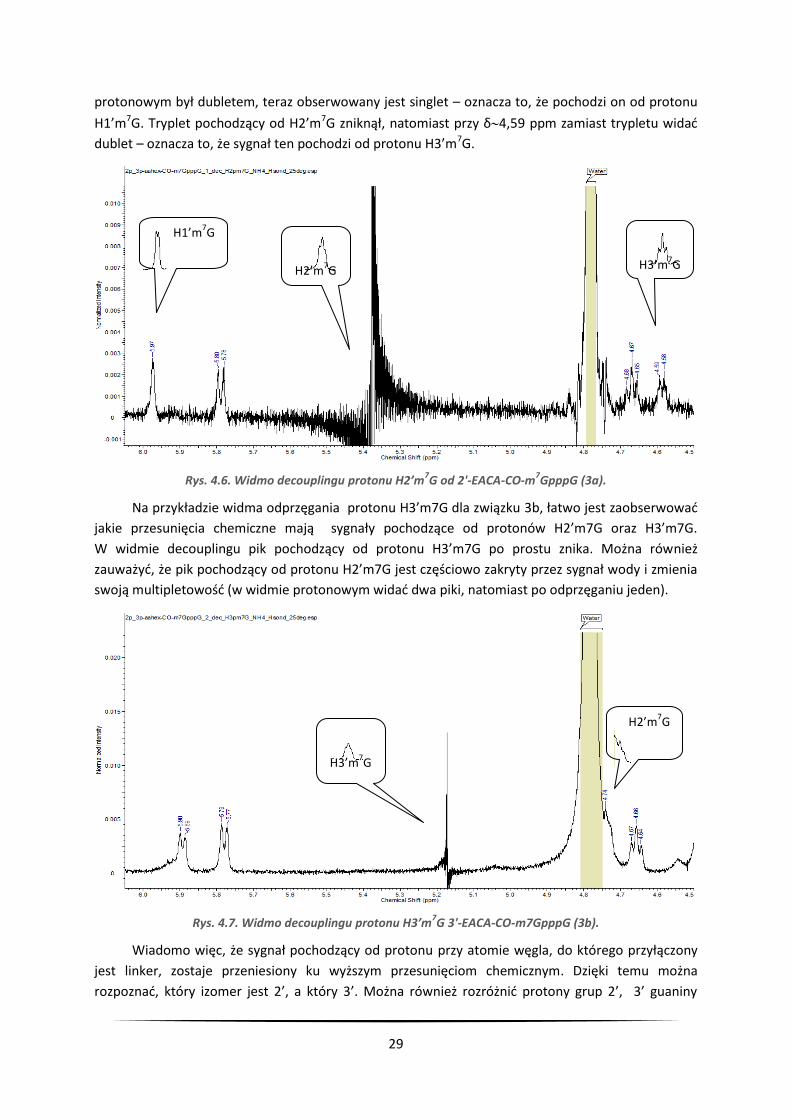
29
protonowym był dubletem, teraz obserwowany jest singlet – oznacza to, że pochodzi on od protonu
H1’m7G. Tryplet pochodzący od H2’m7G zniknął, natomiast przy δ4,59 ppm zamiast trypletu widać
dublet – oznacza to, że sygnał ten pochodzi od protonu H3’m7G.
Rys. 4.6. Widmo decouplingu protonu H2’m7G od 2'-EACA-CO-m
7GpppG (3a).
Na przykładzie widma odprzęgania protonu H3’m7G dla związku 3b, łatwo jest zaobserwować
jakie przesunięcia chemiczne mają sygnały pochodzące od protonów H2’m7G oraz H3’m7G.
W widmie decouplingu pik pochodzący od protonu H3’m7G po prostu znika. Można również
zauważyć, że pik pochodzący od protonu H2’m7G jest częściowo zakryty przez sygnał wody i zmienia
swoją multipletowość (w widmie protonowym widać dwa piki, natomiast po odprzęganiu jeden).
Rys. 4.7. Widmo decouplingu protonu H3’m7G 3'-EACA-CO-m7GpppG (3b).
Wiadomo więc, że sygnał pochodzący od protonu przy atomie węgla, do którego przyłączony
jest linker, zostaje przeniesiony ku wyższym przesunięciom chemicznym. Dzięki temu można
rozpoznać, który izomer jest 2’, a który 3’. Można również rozróżnić protony grup 2’, 3’ guaniny
H2’m7G
H3’m7G
H1’m7G
H3’m7G
H2’m7G

30
od 7-metyloguaniny. Izomer 2’ ma krótszy czas retencji na HPLC, dzięki czemu wiadomo, że izomer 3’
jest bardziej preferowaną formą – w przypadku każdej syntezy, izomeru 3’ powstawało trochę więcej.
4.4. Podsumowanie
W ciągu pracy nad moim projektem licencjackim opracowano metodę wprowadzania linkera
w reszty 2’/ 3’-OH rybozy. Jako grupę aktywującą wybrano karbonyl, otrzymywany w wyniku reakcji
reszt 2’,3’-OH rybozy z karbonylodiimidazolem. Konieczne było więc zabezpieczenie fosforanu,
co osiągnięto przez wprowadzenie grupy cyjanoetylowej. Dobrano również odpowiednie warunki
zdejmowania zabezpieczenia z reszty fosforanowej, nie powodujące rozpadu wiązania między
węglem karbonylowym, a azotem z grupy aminowej linkera.
W rezultacie zsyntetyzowałam trzy analogi kapu zawierające linker w obrębie reszt rybozy
7-metyloguanozyny. Badania spektroskopowe potwierdziły strukturę otrzymanych przeze mnie
związków.
Rys. 4.8. Struktury otrzymanych analogów kapu.
W przyszłości mogą one posłużyć do badań nad procesami posttranslacyjnymi,
jak też do badania mechanizmów wchłaniania oraz translacji analogów kapu o znaczeniu
terapeutycznym. Mogą również być wykorzystane w diagnostyce nowotworów.

31
5. Część eksperymentalna
5.1. Informacje ogólne
Rozpuszczalniki
Do wszystkich eksperymentów używano wody miliporowej (MQ) dejonizowanej
za pomocą aparatury MiliQ Milipore. W syntezach wykorzystano handlowo dostępne rozpuszczalniki
o czystości cz.d.a. – DMF, DMSO, (CH3O)3PO i przechowywano je nad sitami molekularnymi 4Å.
Kolumnowa chromatografia jonowymienna
Chromatografię kolumnową przeprowadzono na złożu jonowymiennym DEAE-Sephadex A-25
w formie HCO3- (anionit). Do elucji stosowano liniowy gradient buforu wodorowęglanu
trietyloamoniowego w wodzie (TEAB, pH 7,8). Bufor otrzymywano przepuszczając gazowy CO2
przez roztwór wodny trietyloaminy, do uzyskania odpowiedniego pH. Zebrane frakcje badano
przez pomiar absorbancji przy 260 nm oraz RP HPLC. Frakcje zawierające oczyszczone związki
odparowywano pod zmniejszonym ciśnieniem z dodatkiem etanolu lub bezwodnego etanolu
w celu usunięcia buforu TEAB.
Wysokociśnieniowa chromatografia kolumnowa (HPLC)
Analityczna chromatografia HPLC, przeprowadzana była na kolumnie RP (z odwróconym
układem faz) Supelcosil LC-18-T RP column (wymiary kolumny: 4,6 x 250 mm, śr. 5μm;
przepływ 1,3 ml/min). Detekcja odbywała się przy pomocy detektora UV przy długości fali 260 nm
oraz detektora fluorescencyjnego (wzbudzenie falą o długości 280 nm, detekcja przy 370 nm).
Stosowano elucję gradientową przy użyciu dwóch układów: A – bufor 0,05 M CH3COONH4/CH3COOH
o pH 5,9; B – mieszaninę 1:1 (v/v) buforu A i metanolu. Rozdział prowadzono w liniowym gradiencie
buforu B w buforze A od 0 do 50% w czasie 15 minut.
Rozdział preparatywny prowadzono na kolumnie RP Discovery RP Amide C-16 (wymiary kolumny:
21.2 x 250 mm, śr. 5μm; przepływ 5,0 ml/min), wykorzystując bufor A (tak jak w przypadku
chromatografii analitycznej) oraz bufor C – 7:3 (v/v) buforu A oraz acetonitrylu. Zastosowano
gradient liniowy od 0 do 50% buforu C w 120 minut.
Spektrometria mas i NMR
Widma masowe otrzymanych związków zarejestrowane zostały na aparacie Micromass QToF
1 MS spectrometer, z jonizacją elektrosprej w trybie jonów ujemnych. Widma 1H NMR i 31P NMR
zarejestrowano przy pomocy aparatu Varian Inova 400 odpowiednio przy częstościach 399.94 MHz
i 161.90 MHz. Do rejestracji widm 1H NMR wykorzystano wzorzec wewnętrzny – TMSP
(2,2,3,3 – tetradeutero–3–(trimetylosililo)–propionian sodu), natomiast do widm 31P NMR
zastosowano wzorzec zewnętrzny – 85% roztwór kwasu ortofosforowego H3PO4 w D2O.
Z racji na niezbyt wysoką rozdzielczość zarejestrowanych widm, nie jesteśmy w stanie
zaobserwować multipletowości niektórych pików – zostały one oznaczone jako bs (ang. broad signal).

32
5.2. Syntezy chemiczne
5’-O-(β-cyjanoetylo)difosforan guanozyny (4)
Do zawiesiny 3,994 g (7,78 mmol) guanozyny w 60,0 ml DMF dodano 1,1 eqv. (8,55 mmol)
p-imidazolidu fosforanu β-cyjanoetylowego w formie soli trietyloamoniowej oraz 16 eqv.
(124,5 mmol) chlorku magnezu. Zawartość kolby mieszano za pomocą mieszadła magnetycznego
przez około 5 godzin. Reakcję zakończono dodając 600 ml wody i rozdzielono za pomocą
chromatografii jonowymiennej (bufor: od 0 do 0,7 M TEAB).
Otrzymano 2,421 g (3,47 mmol) soli trietyloamoniowej 5’-O-(-cyjanoetylo)difosforanu guanozyny.
Wydajność całkowita 45%. RT 4,1 min, ESI M(-) m/z 495.
5’-difosforan 2’-O/3’-O-(2-karboksyetylo)-karbamoilo-guanozyny (5)
W kolbie rozpuszczono 170,0 mg (0,244 mmol)cyjanoetylodifosforanu GDP (4) oraz 50 eqv. (12,2
mmol) karbonylodiimidazolu w 9,74 ml DMSO. Po 10 minutach, gdy całe GDP-ETCN przereagowało,
do mieszaniny reakcyjnej dodano 95 eqv. (23,1 mmol) wody w celu rozłożenia pozostałego CDI.
Następnie do kolby dodano równomolową mieszaninę (40 eqv.; 9,74 mmol) β-alaniny
oraz diazabicykloundecenu. Po 2 godzinach, gdy aminokwas przyłączył się do nukleotydu, kolbę
umieszczono na wyparce pod zmniejszonym ciśnieniem w temperaturze 40oC aby zdjąć z fosforanu
zabezpieczenie cyjanoetylowe. Po 5 godzinach reakcję zakończono przez 10-krotne rozcieńczenie
wodą i wyekstrachowano octanem etylu, aby pozbyć się DBU. Pozostałości octanu etylu odpędzono
na wyparce. Mieszaninę poreakcyjną rozdzielono za pomocą chromatografii jonowymiennej
(bufor: od 0 do 1,1 M TEAB).
Otrzymano 150,9 mg (0,229 mmol) soli trietyloamoniowej 5’- difosforanu 2’-O/3’-O-
-(2-karboksyetylo)-karbamoilo-guanozyny (5). Wydajność całkowita 94%. RT1 5,9 min, RT
2 6,2 min,
ESI M(-) m/z 557.

33
5’-difosforan 2’-O/3’-O-(2-karboksyetylo)-karbamoilo-7-metyloguanozyny (8)
(BAPA-CO-m7GDP)
Odparowane BAPA-CO-GDP (5) (150,9 mg; 0,229 mmol) zawieszono w 3,0 ml DMSO. Do roztworu
dodano 8 eqv. (1,83 mmol) jodku metylu i pozostawiono na 7 godzin mieszając na mieszadle
magnetycznym. W miarę zachodzenia reakcji roztwór zmieniał barwę z bezbarwnej na brązową
pod wpływem uwalniającego się jodu. W celu zakończenia reakcji mieszaninę rozcieńczono
10-krotnie wodą i wyekstrahowano eterem , w celu pozbycia się jodu z warstwy wodnej.
Resztek jodu pozbyto się przez dodanie odrobiny Na2S2O5. Pozostałości eteru odparowano
na wyparce. Mieszaninę poreakcyjną rozdzielono za pomocą chromatografii jonowymiennej
(bufor: od 0 do 0,9 M TEAB).
Otrzymano 69,6 mg (0,092 mmol) soli trietyloamoniowej 5’- difosforanu 2’-O/3’-O-(2-karboksyetylo)-
-karbamoilo-7-metyloguanozyny (8). Wydajność całkowita 40%. RT1 5,3 min, RT
2 5,5 min,
ESI M(-) m/z 571.
BAPA-CO-m7GpppG (1)
Do zawiesiny 69,6 mg (0,092 mmol) BAPA-CO-m7GDP (8) w 1,8 ml DMF dodano 1,25 eqv.
(0,115 mmol) p-imidazolidu (M=760,00 g/mol) oraz 16 eqv. (1,46 mmol) chlorku cynku. Zawartość
kolby mieszano za pomocą mieszadła magnetycznego przez 2,5 godziny. Reakcję zakończono
roztworem EDTA/NaHCO3 w wodzie i rozdzielono za pomocą chromatografii jonowymiennej
(bufor: od 0 do 1,1 M TEAB).
Otrzymano 49,2 mg (0,048 mmol) soli trietyloamoniowej BAPA-CO-m7GpppG (1). Wydajność
całkowita 42%. RT1a 5,9 min, RT
1b 6,2 min, ESI M(-) m/z 916. 1H NMR (399,94 MHz, D2O, TMSP),
δ(ppm): 8,11 (nałożone s, 2H, 2H8G); 5,97 (d, 3J(H1’2’-BAPA-CO-m7G–H2’2’-BAPA-CO-m7G)=3,2 Hz, 1H, H1’2’-BAPA-
CO-m7G); 5,88 (d, 3J(H1’3’-BAPA-CO-m7G–H2’3’-BAPA-CO-m7G)=5,5 Hz, 1H, H1’3’-BAPA-CO-m7G); 5,79 (d, 3J(H1’G
–H2’G)=5,2 Hz, 2H, 2H1’G); 5,36 (dd, 3J(H1’2’-BAPA-CO-m7G–H2’2’-BAPA-CO-m7G)=3,2 Hz, 3J(H2’2’-BAPA-CO-m7G–H3’2’-
BAPA-CO-m7G)=5,0 Hz, 1H, H2’2’-BAPA-CO-m7G); 5,14 (dd, 3J(H2’3’-BAPA-CO-m7G–H3’3’-BAPA-CO-m7G)=4 Hz, 3J(H3’3’-BAPA-

34
CO-m7G–H4’3’-BAPA-CO-m7G)=4 Hz, 1H, H3’3’-BAPA-CO-m7G); 4,70 (dd, 3J(H1’3’-BAPA-CO-m7G–H2’3’-BAPA-CO-m7G)=5,5 Hz, 3J(H2’3’-BAPA-CO-m7G–H3’3’-BAPA-CO-m7G)=4 Hz, 1H, H2’3’-BAPA-CO-m7G); 4,60 (dd, 3J(H1’G–H2’G)=5,2 Hz, 3J(H2’G
–H3’G)=4,7 Hz, 2H, 2H2’G); 4,53 (dd, 3J(H2’2’-BAPA-CO-m7G–H3’2’-BAPA-CO-m7G)=5,0 Hz, 3J(H3’2’-BAPA-CO-m7G
–H4’2’-BAPA-CO-m7G)=6,2 Hz, 1H, H3’2’-BAPA-CO-m7G); 4,45-4,15 (m, 14H, 2H3’G, 2H4’BAPA-CO-m7G, 2H4’G,
4H5’BAPA-CO-m7G, 4H5’G); 4,04 (nałożone s, 6H, m7); 3,34 (nałożone t, 3J(αCH2– βCH2)=6,8 Hz, 3J(αCH2
– βCH2)=6,1 Hz, 4H, 2βCH2); 2,39 (nałożone t, 3J(αCH2– βCH2)=6,1 Hz, 3J(αCH2– βCH2)=6,8 Hz, 4H,
2αCH2); 31P NMR (161,90 MHz, D2O, H3PO4), δ(ppm): -11,12 (bs, 4P, 2Pα, 2Pγ); -22,76 (bs, 2P, 2Pβ).
5’-difosforan 2’-O/3’-O-(3-karboksybutylo)-karbamoilo-guanozyny (6)
W kolbie rozpuszczono 59,5 mg (0,085 mmol) cyjanoetylodifosforanu GDP (4) oraz 50 eqv. (4,26
mmol) karbonylodiimidazolu w 3,41 ml DMSO. Po 20 minutach, gdy całe GDP-ETCN przereagowało,
do mieszaniny reakcyjnej dodano 95 eqv. (8,10 mmol) wody w celu rozłożenia pozostałego CDI.
Następnie do kolby dodano równomolową mieszaninę (40 eqv.; 3,41 mmol) kwasu
γ-aminomasłowego oraz diazabicykloundecenu. Po 4 godzinach, gdy aminokwas przyłączył się
do nukleotydu, kolbę umieszczono na wyparce pod zmniejszonym ciśnieniem w temperaturze 40oC
aby zdjąć z fosforanu zabezpieczenie cyjanoetylowe. Dodano również kolejne 20 eqv. (1,70 mmol)
DBU. Po 7 godzinach reakcję zakończono przez 10-krotne rozcieńczenie wodą i wyekstrahowano
octanem etylu, aby pozbyć się DBU. Pozostałości octanu etylu odpędzono na wyparce. Mieszaninę
poreakcyjną rozdzielono za pomocą chromatografii jonowymiennej (bufor: od 0 do 1,1 M TEAB).
Otrzymano 52,5 mg (0,073 mmol) soli trietyloamoniowej 5’- difosforanu 2’-O/3’-O-
-(3-karboksybutylo)-karbamoilo-guanozyny (6). Wydajność całkowita 85%. RT1 6,1 min, RT
2 6,3 min,
ESI M(-) m/z 571.
5’-difosforan 2’-O/3’-O-(3-karboksybutylo)-karbamoilo-7-metyloguanozyny (9)
(GABA-CO-m7GDP)

35
Odparowane GABA-CO-GDP (6) (52,5 mg; 0,073 mmol) zawieszono w 1,1 ml DMSO. Do roztworu
dodano 8 eqv. (0,582 mmol) jodku metylu i pozostawiono na 5 godzin mieszając na mieszadle
magnetycznym. W miarę zachodzenia reakcji roztwór zmieniał barwę z bezbarwnej na brązową
pod wpływem uwalniającego się jodu. W celu zakończenia reakcji mieszaninę rozcieńczono
10-krotnie wodą i wyekstrahowano eterem , w celu pozbycia się jodu z warstwy wodnej.
Resztek jodu pozbyto się przez dodanie odrobiny Na2S2O5. Pozostałości eteru odparowano
na wyparce. Mieszaninę poreakcyjną rozdzielono za pomocą chromatografii jonowymiennej
(bufor: od 0 do 0,9 M TEAB).
Otrzymano 17,9 mg (0,024 mmol) soli trietyloamoniowej 5’- difosforanu 2’-O/3’-O-(2-karboksyetylo)-
-karbamoilo-7-metyloguanozyny (9). Wydajność całkowita 33%. RT1 6,2 min, RT
2 6,8 min,
ESI M(-) m/z 585.
GABA-CO-m7GpppG (2)
Do zawiesiny 17,9 mg (0,024 mmol) GABA-CO-m7GDP (9) w 1,8 ml DMF dodano 1,25 eqv.
(0,030 mmol) p-imidazolidu oraz 16 eqv. (0,390 mmol) chlorku cynku. Zawartość kolby mieszano
za pomocą mieszadła magnetycznego przez 3,5 godziny. Reakcję zakończono roztworem
EDTA/NaHCO3 w wodzie i rozdzielono za pomocą chromatografii jonowymiennej
(bufor: od 0 do 1,1 M TEAB).
Otrzymano 14,6 mg (0,014 mmol) soli trietyloamoniowej GABA-CO-m7GpppG (2). Wydajność
całkowita 47%. RT2a 7,2 min, RT
2b 7,4 min, ESI M(-) m/z 930. 1H NMR (399,94 MHz, D2O, TMSP),
δ(ppm): 9,07 (nałożone s, 2H, 2H8G); 5,98 (bs, 1H, H1’2’-GABA-CO-m7G); 5,90 (d, 3J(H1’3’-GABA-CO-m7G
–H2’3’-GABA-CO-m7G) =3,7 Hz, 1H, H1’3’-GABA-CO-m7G); 5,81 (d, 3J(H1’G–H2’G)=4,2 Hz, 2H, 2H1’G); 5,40 (bs, 1H,
H2’2’-GABA-CO-m7G); 5,17 (bs, 1H, H3’3’-GABA-CO-m7G); 4,74 (m, 1H, H2’3’-GABA-CO-m7G); 4,66 (nałożone t, 3J(H1’G
–H2’G)=4,7 Hz, 3J(H1’G–H2’G)=4,2 Hz, 2H, 2H2’G); 4,59 (dd, 3J(H2’2’-GABA-CO-m7G–H3’2’-GABA-CO-m7G)= 3J(H3’2’-
GABA-CO-m7G–H4’2’-GABA-CO-m7G)=4,2 Hz, 1H, H3’2’-GABA-CO-m7G); 4,55-4,17 (m, 14H, 2H3’G, 2H4’GABA-CO-m7G,
2H4’G, 4H5’GABA-CO-m7G, 4H5’G); 4,08 ( nałożone s, 6H, m7);3,14 (m, częściowo nakłada się z sygnałem
TEA-CH2, 4H, 2γCH2); 2,36 (nałożone t, 3J=7 Hz, 3J=7,5 Hz, 4H, 2αCH2); 1,78 (m, 4H, 2βCH2); 31P NMR
(161,90 MHz, D2O, H3PO4), δ(ppm): -10,68 (nałożone d, J(Pα–Pβ)= J(Pβ–Pγ)=19,5 Hz, 4P, 2Pα, 2Pγ);
-22,29 (dd, 2P, 2Pβ).

36
5’-difosforan 2’-O/3’-O-(6-karboksyheksylo)-karbamoilo-guanozyny (7)
W kolbie rozpuszczono 68,0 mg (0,097 mmol) cyjanoetylodifosforanu GDP oraz 50 eqv. (4,87 mmol)
karbonylodiimidazolu w 3,89 ml DMSO. Po 10 minutach, gdy całe GDP-ETCN przereagowało,
do mieszaniny reakcyjnej dodano 95 eqv. (9,25 mmol) wody w celu rozłożenia pozostałego CDI.
Następnie do kolby dodano równomolową mieszaninę (40 eqv.; 3,90 mmol) kwasu
-aminokapronowego oraz DBU. Po 2 godzinach, gdy aminokwas nie przyłączał się do nukleotydu,
kolbę umieszczono na wyparce pod zmniejszonym ciśnieniem w temperaturze 40oC. Po 7 godzinach
reakcję zakończono przez 10-krotne rozcieńczenie wodą i wyekstrahowano octanem etylu,
aby pozbyć się DBU. Pozostałości octanu etylu odpędzono na wyparce. Mieszaninę poreakcyjną
rozdzielono za pomocą chromatografii jonowymiennej (bufor: od 0 do 1,1 M TEAB).
Otrzymano 47,3 mg (0,063 mmol) soli trietyloamoniowej 5’- difosforanu 2’-O/3’-O-
-(6-karboksyheksylo)-karbamoilo-guanozyny (7). Wydajność całkowita 65%. RT1 10,1 min,
RT2 10,6 min, ESI M(-) m/z 599.
5’-difosforan 2’-O/3’-O-(2-karboksyheksylo)-karbamoilo-7-metyloguanozyny (10)
(EACA-CO-m7GDP)
Odparowane EACA-CO-GDP (7) (47,3 mg; 0,063 mmol) zawieszono w 0,94 ml DMSO. Do roztworu
dodano 8 eqv. (0,506 mmol) jodku metylu i pozostawiono na 3,5 godziny mieszając
na mieszadle magnetycznym. W celu zakończenia reakcji mieszaninę rozcieńczono 10-krotnie wodą
i wyekstrahowano eterem , w celu pozbycia się jodu z warstwy wodnej. Resztek jodu pozbyto się
przez dodanie odrobiny Na2S2O5. Pozostałości eteru odparowano na wyparce. Mieszaninę
poreakcyjną rozdzielono za pomocą chromatografii jonowymiennej (bufor: od 0 do 0,9 M TEAB).
Otrzymano 10,6 mg (0,014 mmol) soli trietyloamoniowej 5’- difosforanu 2’-O/3’-O-(2-karboksyetylo)-
-karbamoilo-7-metyloguanozyny (10). Wydajność całkowita 22%. RT1 10,9 min, RT
2 11,7 min,
ESI M(-) m/z 613.

37
EACA-CO-m7GpppG (3)
Do zawiesiny 10,6 mg (0,014 mmol) EACA-CO-m7GDP (10) w 0,83 ml DMF dodano 1,25 eqv.
(0,017 mmol) p-imidazolidu oraz 16 eqv. (0,222 mmol) chlorku cynku. Zawartość kolby mieszano
za pomocą mieszadła magnetycznego przez 5 godzin. Reakcję zakończono roztworem EDTA/NaHCO3
w wodzie i rozdzielono za pomocą chromatografii jonowymiennej (bufor: od 0 do 1,2 M TEAB).
Otrzymano 16,3 mg (0,014 mmol) zanieczyszczonej soli trietyloamoniowej EACA-CO-m7GpppG (3).
Wydajność całkowita 78%. RT3a 15,6 min, RT
3b 16,3 min, ESI M(-) m/z 958.
Produkt oczyszczono za pomocą preparatywnego HPLC. Frakcje zawierające te same izomery
połączono, odparowano bufor i zliofilizowano. Otrzymano 4,9 mg (0,0046 mmol) izomeru 2’ oraz
3,4 mg (0,0032 mmol) izomeru 3’. Wydajności całkowite syntezy wyniosły odpowiednio 33 % i 23 %.
2’-EACA-CO-m7GpppG (3a): RT(HPLC RP) 15,6 min, RT
(HPLC prep.) 53,7 min, 1H NMR (399,94 MHz, D2O,
TMSP), δ(ppm): 9,04 (s, 1H, H8EACA-CO-m7G); 8,01 (s, 1H, H8G); 5,98 (d, 3J(H1’EACA-CO-m7G–H2’EACA-CO-m7G)=3
Hz, 1H, H1’EACA-CO-m7G); 5,79 (d, 3J(H1’G–H2’G)=5,8 Hz, 1H, H1’G); 5,38 (dd, 3J(H1’EACA-CO-m7G–H2’EACA-CO-
m7G)=3 Hz, 3J(H2’EACA-CO-m7G–H3’EACA-CO-m7G)=5,4 Hz, 1H, H2’EACA-CO-m7G); 4,67 (dd, 3J(H1’G–H2’G)=5,8 Hz, 3J(H2’G–H3’G)=5,3 Hz, 1H, H2’G); 4,59 (dd, 3J(H2’EACA-CO-m7G–H3’EACA-CO-m7G)=5,4 Hz, 3J(H3’EACA-CO-m7G
–H4’EACA-CO-m7G)=5,2 Hz, 1H, H3’EACA-CO-m7G); 4,47 (dd, 3J(H2’G–H3’G)=5,3 Hz, 3J(H3’G–H4’G)=3,3 Hz, 1H,
H3’G); 4,44-4,20 (m, 6H, H4’EACA-CO-m7G, H4’G, 2H5’EACA-CO-m7G, 2H5’G); 4,07 (s, 3H, m7); 3,08 (m, 2H,
CH2); 2,26 (t, 3J(αCH2–βCH2)=7,5 Hz, 2H, αCH2); 1,57 (tt, 3J(δCH2–CH2)=7,5 Hz, 3J(γCH2–δCH2)=7,5 Hz,
2H, δCH2); 1,46 (tt, 3J(αCH2–βCH2)=7,5 Hz, 3J(βCH2–γCH2)=6,8 Hz, 2H, βCH2); 1,31 (tt, 3J(γCH2
–δCH2)=7,5 Hz, 3J(βCH2–γCH2)=6,8 Hz, 2H, γCH2); 31P NMR (161,90 MHz, D2O, H3PO4), δ(ppm):
-10,72 (nałożone d, 2P, Pα, Pγ); -22,29 (dd, 1P, Pβ).
3’-EACA-CO-m7GpppG (3b): RT(HPLC RP) 15,6 min, RT
(HPLC prep.) 56,0 min, 1H NMR (399,94 MHz, D2O,
TMSP), δ(ppm): 9,08 (s, 1H, H8EACA-CO-m7G); 7,98 (s, 1H, H8G); 5,90 (d, 3J(H1’EACA-CO-m7G–H2’EACA-CO-
m7G)=5,5 Hz, 1H, H1’EACA-CO-m7G); 5,78 (d, 3J(H1’G–H2’G)=5,7 Hz, 1H, H1’G); 5,17 (dd, 3J(H2’EACA-CO-m7G
–H3’EACA-CO-m7G)=5,2 Hz, 3J(H3’EACA-CO-m7G–H4’EACA-CO-m7G)=4,3 Hz, 1H, H3’EACA-CO-m7G); 4,73 (dd, 3J(H1’EACA-CO-
m7G–H2’EACA-CO-m7G)=5,5 Hz, 3J(H2’EACA-CO-m7G–H3’EACA-CO-m7G)=5,2 Hz, 1H, H2’EACA-CO-m7G); 4,66 (dd, 3J(H1’G
–H2’G)=5,7 Hz, 3J(H2’G–H3’G)=5,5 Hz, 1H, H2’G); 4,45 (dd, 3J(H2’G–H3’G)=5,5 Hz, 3J(H3’G–H4’G)=3,4 Hz,
1H, H3’G); 4,40- 4,18 (m, 6H, H4’EACA-CO-m7G, H4’G, 2H5’EACA-CO-m7G, 2H5’G); 4,08 (s, 3H, m7); 3,13
(t, 3J(δCH2–CH2)=6,6 Hz, 2H, CH2); 2,29 (t, 3J(αCH2–βCH2)=7,3 Hz, 2H, αCH2); 1,59 (tt, 3J(δCH2
–CH2)=6,6 Hz, 3J(γCH2–δCH2)=7,5 Hz, 2H, δCH2); 1,52 (tt, 3J(αCH2–βCH2)=7,3 Hz, 3J(βCH2
–γCH2)=7,0 Hz, 2H, βCH2); 1,35 (tt, 3J(γCH2–δCH2)=7,5 Hz, 3J(βCH2–γCH2)=7,0 Hz, 2H, γCH2); 31P NMR
(161,90 MHz, D2O, H3PO4), δ(ppm): -10,65 (nałożone d, J(Pα–Pβ)= J(Pβ–Pγ)=18,1 Hz, 2P, Pα, Pγ);
-22,27 (dd, 1P, Pβ)
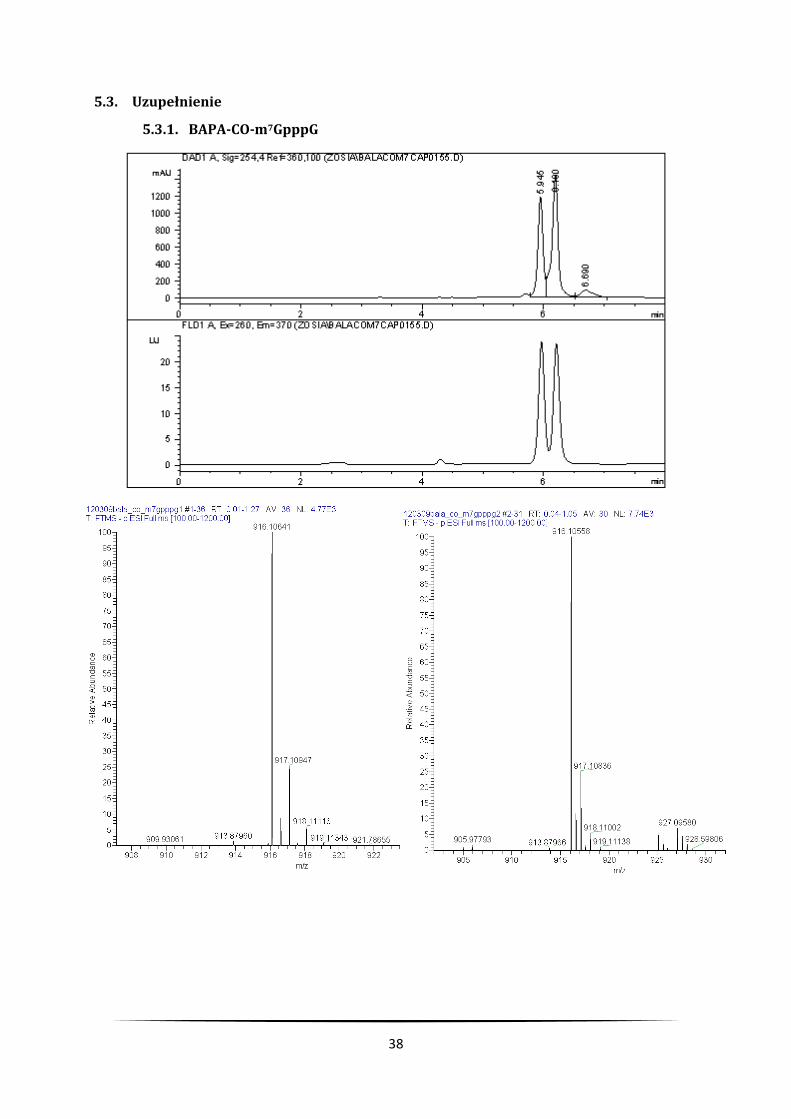
38
5.3. Uzupełnienie
5.3.1. BAPA-CO-m7GpppG
39

40
5.3.2. GABA-CO-m7GpppG
41
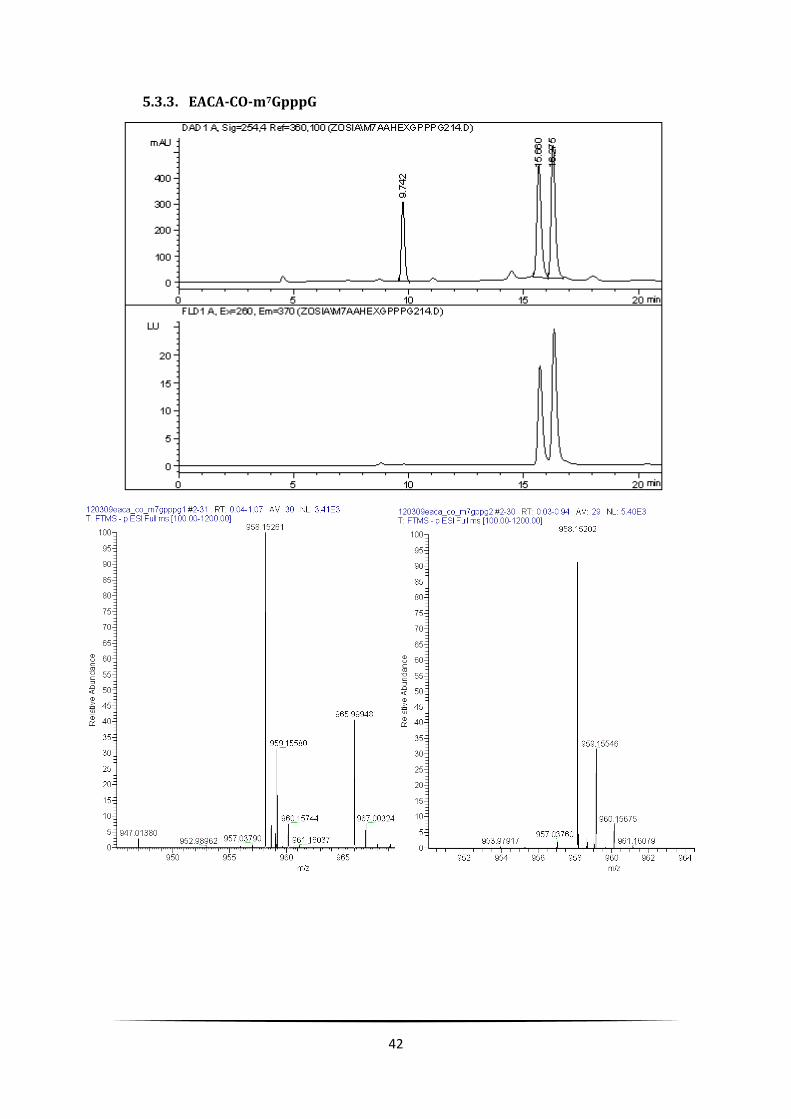
42
5.3.3. EACA-CO-m7GpppG
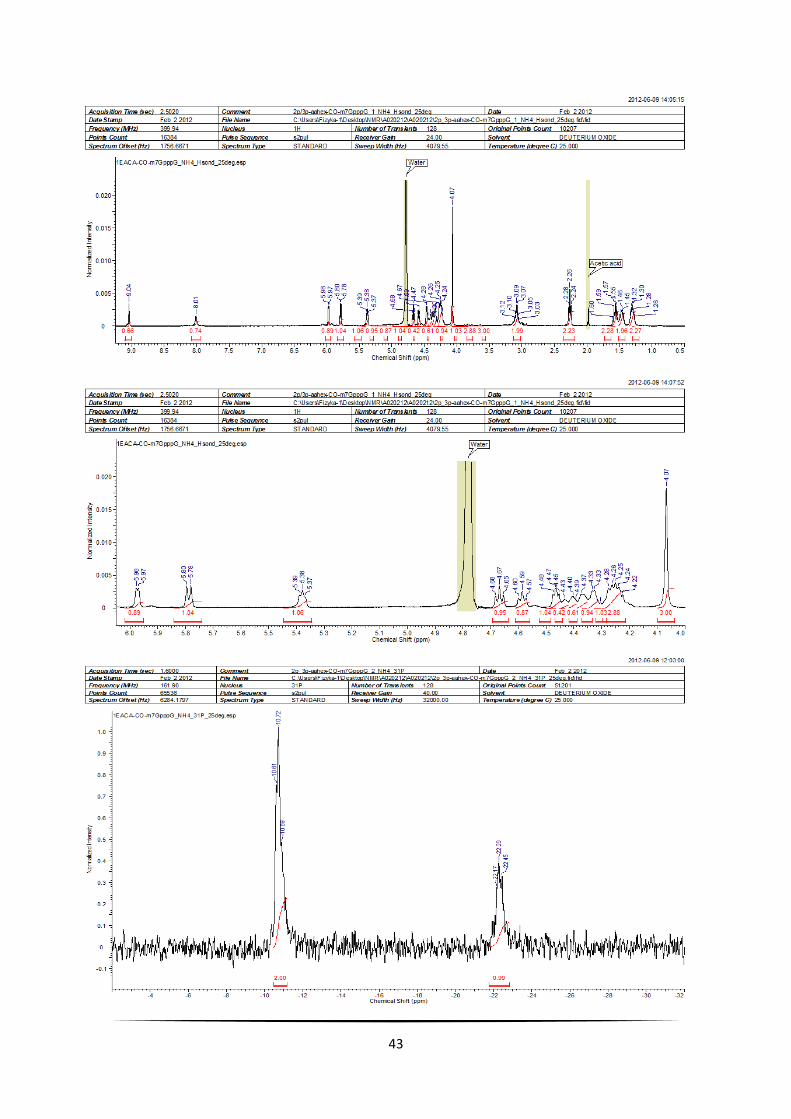
43
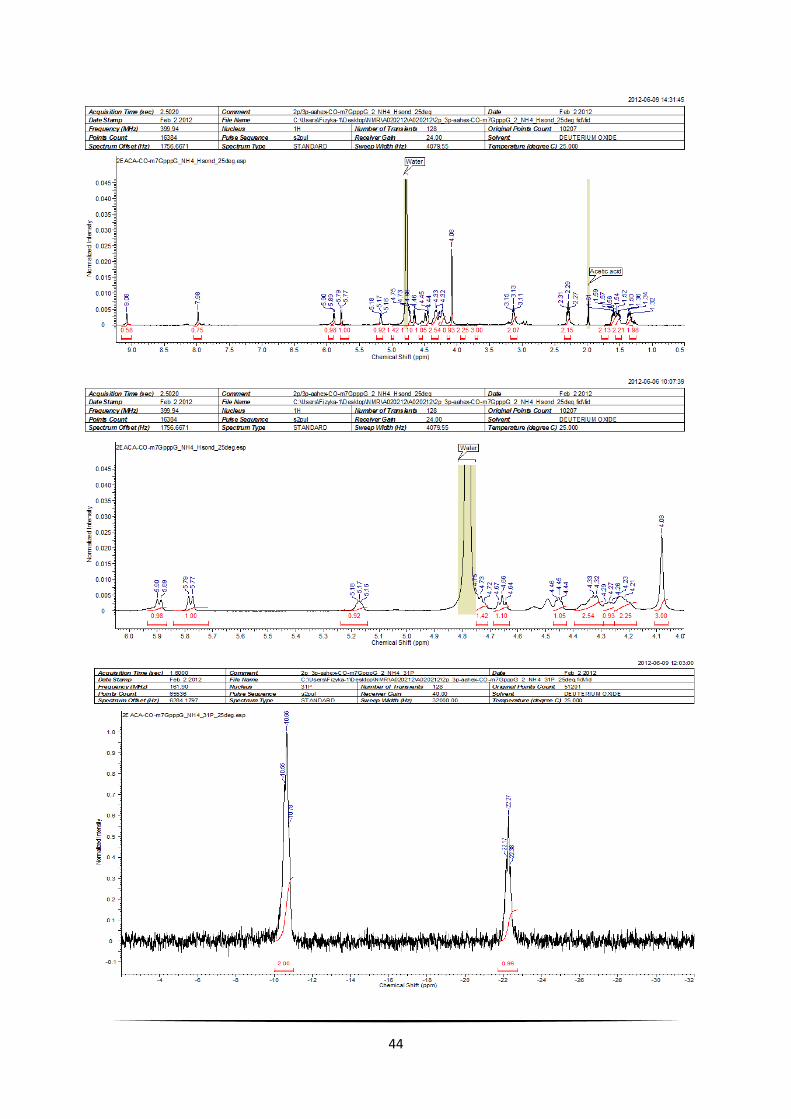
44

45
Bibliografia
1. Lodish H. Berk, Zipursky S.L, Metsudaira P, Baltimore, Darnell J; Molecular Cell Biology,
4th edition. New York: W. H. Freeman (2000).
2. Shatkin AJ; mRNA cap binding proteins: essential factors for initiating trnslation; Cell, 40, 223-234,
(1985).
3. Mathonnet G, Fabian M.R, Svitkin Y.V, Parsyan A, Huck L, Jaramillo M, Murata T, Biffo S, Merrick
W.C, Darzynkiewicz E, Pillai R.S, Filipowicz W, Duchaine T.F, Sonenberg N; microRNA function
in vitro: inhibition of translational initiation by targeting eIF4F; Science, 317, 1764-1767 (2007).
4. Darnbrough C, Legon S, Hunt T, Jackson RJ; Initiation of protein synthesis: evidence
for messenger RNA-independent binding of methionyl-transfer RNA to the 40 S ribosomal
subunit.; J Mol Biol., 76, 379-403 (1973).
5. Coller J, Parker R, Annu. Rev. Biochem., 73, 861-890 (2004).
6. Parker R, Sheth U; P Bodies and the Control of mRNA Translation and Degradation; Mol.Cell., 25,
635–646 (2007).
7. Sachs AB; Messenger RNA degradation in eukaryotes; Cell, 74, 413-421 (1993).
8. Parker R, Song H; The enzymes and control of eukaryotic mRNA turnover; Nat Struct Mol Biol., 11,
121-127 (2004).
9. Couttet P, Fromont-Racine M, Steel D, Pictet R, Grange T; Messenger RNA deadenylylation
precedes decapping in mammalian cells; Proc Natl Acad Sci U S A., 94, 5628-5633 (1997).
10. Muhlrad D, Decker CJ, Parker R; Turnover mechanisms of the stable yeast PGK1 mRNA;
Mol Cell Biol., 15, 2145-2156 (1995).
11. Liu H, Rodgers ND, Jiao X, Kiledjian M; The scavenger mRNA decapping enzyme DcpS is a member
of the HIT family of pyrophosphatases; EMBO J., 21, 4699-4708 (2002).
12. Nishimura Y, Takahashi S.I, Yamamoto T, Tsuboi M, Hattori M, Miura K.I, Yamaguchi K, Ohtani S,
Hata T; On the base-stacking in the S'-terminal cap structure of mRNA: a fluorescence study;
Nucl. Acids Res., 8, 1107-1119 (1980).
13. Kałek M, Jemielity J, Darzynkiewicz Z.M, Bojarska E, Stępiński J, Stolarski R, Davis RE,
Darzynkiewicz E; Enzymatically stable 5' mRNA cap analogs: synthesis and binding studies with
human DcpS decapping enzyme; Bioorg Med Chem., 14, 3223-3230 (2006).
14. Hodel A.E, Gershon P.D, Shi X, Wang S.M, Quiocho F.A; Specific protein recognition of an mRNA
cap through its alkylated base; Nat. Struct. Biol., 4, 350-354 (1997).
15. Visa N, Izaurralde E, Ferreira J, Daneholt B, Mattaj I.W; A nuclear cap-binding complex binds
Balbiani ring pre-mRNA cotranscriptionally and accompanies the ribonucleoprotein particle during
nuclear export; J Cell Biol., 133, 5-14 (1996).
16. Kozak M; Initiation of translation in prokaryotes and eukaryotes; Gene, 234, 187-208 (1999).

46
17. Niedźwiecka A, Marcotrigiano J, Stępiński J., Jankowska-Anyszka M, Wysłouch-Cieszyńska A,
Dadlez M, Gingras A.C, Mak P, Darżynkiewicz P, Sonenberg N, Burley S.K, Stolarski R;
Biophysical studies of eIF4E cap-binding protein: recognition of mRNA 5' cap structure
and synthetic fragments of eIF4G and 4E-BP1 proteins; J. Mol. Biol., 319, 615-635 (2002).
18. Herbert T.P, Fshraeus, Prescott A, Lane D.P, Proud C.G; Rapid induction of apoptosis mediated
by peptides that bind initiation factor eIF4E; Curr. Biol., 10, 793 (2000).
19. Moerke N.J, Aktas H, Chen H, Cantel S, Reibarkh M.Y, Fahmy A, Gross J.A, Degterev A, Yuan J,
Chorev M; Small-molecule inhibition of the interaction between the translation initiation factors
eIF4E and eIF4G; Cell, 28, 257 (2007).
20. Mikkola S, Salomaki S, Zhang Z, Maki E, Lonnberg H; Preparation and properties of mRNA 5 '-cap
structure; Curr. Org. Chem., 9, 999-1022 (2005).
21. Żuberek J, Jemielity J, Jabłonowska A, Stępiński J, Dadlez M, Stolarski R, Darzynkiewicz E; Influence
of electric charge variation at residues 209 and 159 on the interaction of eIF4E with the mRNA
5' terminus; Biochemistry, 43, 5370-5379 (2004).
22. Jemielity J, Fowler T, Żuberek J, Stępiński J, Lewdorowicz M, Niedźwiecka A, Stolarski R,
Darzynkiewicz E, Rhoads R.E; Novel 'anti-reverse' cap analogues with superior translational
properties RNA; RNA, 9, 1108 (2003).
23. Mitchell D.A, Nair S.K; RNA transfected dendritic cells as cancer vaccines; Curr. Opin. Mol. Ther., 2,
176-181 (2000).
24. Yamamoto A, Kormann M, Rosenecker J, Rudolph C; Eur. J. Pharm. Biopharm., 71, 484-489 (2009)
25. Kowalska J, Lewdorowicz M, Żuberek J, Grudzień-Nogalska E, Bojarska E, Stępiński J, Rhoads R.E,
Darzynkiewicz E, Davis R.E, Jemielity J; RNA, 14, 1119-1132 (2008).
26. Mitchell D. A, Nair S. K; RNA-transfected dendritic cells in cancer immunotherapy; J. Clin Invest., 9,
1065-1069 (2000).
27. Kuhn A.N, Diken M, Kreiter S, Selmi A, Kowalska J, Jemielity J, Darzynkiewicz E, Huber C, Tureci O,
Sahin U; Phosphorothioate cap analogs increase stability and translational efficiency of RNA
vaccines in immature dendritic cells and induce superior immune responses in vivo; Gene
Therapy, 17, 961-971 (2010).
28. Kadokura M, Wada T, Urashima C, Sekine M; Efficient synthesis of gamma-methyl-capped
guanosine 5'-triphosphate as a 5'-terminal unique structure of U6 RNA via a new triphosphate
bond formation involving activation of methyl phosphorimidazolidate using ZnCl2 as a catalyst
in DMF under anhydrous co; Tet. Lett., 38, 8359-8362 (1997).
29. Hoard D.E, Ott D.G; Conversion of Mono- and Oligodeoxyribonucleotides to 5'-Triphosphates;
J. Am. Chem. Soc., 87, 1785–1788 (1965).
30. Mukaiyama T, Hashimoto M; Synthesis of oligothymidylates and nucleoside cyclic phosphates
by oxidation-reduction condensation; J. Am. Soc., 94, 8528-8532 (1972).


















