
Termodynamika statystyczna. Cele teoriitiger.chem.uw.edu.pl/staff/tania/WykladyBJ/Termo_POL.pdf ·...
49
Termodynamika statystyczna. Cele teorii • Stworzenie pomostu pomi , edzy teori , a mikro´ swiata (teori , a moleku l i ich oddzia lywa´ n) a teori , a zjawisk makroskopowych • Wyja´ snienie (ilo´ sciowe) wlasno´ sciuklad´ ow makroskopowych (np. funkcji termodynamicznych) w oparciu o znajomo´ s´ cwlasno´ sci indywidualnych moleku l (uzyskanych metodami chemii kwantowej lub spektroskopii) • Zrozumienie definicji wielko´ sci termodynamicznych i praw termodynamiki fenomenologicznej w oparciu o prawa mechaniki kwantowej • Wnioskowanie o wlasno´ sciach pojedynczych molekul i o ich oddzia lywaniu na podstawie znajmo´ sciwlasno´ sci uk lad´ow makroskopowych (ma to znaczenie glownie historyczne)
Transcript of Termodynamika statystyczna. Cele teoriitiger.chem.uw.edu.pl/staff/tania/WykladyBJ/Termo_POL.pdf ·...

Termodynamika statystyczna. Cele teorii
• Stworzenie pomostu pomi edzy teori a mikroswiata (teori a moleku li ich oddzia lywan) a teori a zjawisk makroskopowych
•Wyjasnienie (ilosciowe) w lasnosci uk ladow makroskopowych (np. funkcjitermodynamicznych) w oparciu o znajomosc w lasnosci indywidualnychmoleku l (uzyskanych metodami chemii kwantowej lub spektroskopii)
• Zrozumienie definicji wielkosci termodynamicznych i praw termodynamikifenomenologicznej w oparciu o prawa mechaniki kwantowej
•Wnioskowanie o w lasnosciach pojedynczych moleku l i o ich oddzia lywaniuna podstawie znajmosci w lasnosci uk ladow makroskopowych (ma toznaczenie g lownie historyczne)

Przyk lad
Termodynamika fenomenologiczna:
Eenergia swobodna F (T, V )=E−TS.
dF = −S dT − p dV⇓
S = −(∂F
∂T
)V
p = −(∂F
∂V
)T
⇓(∂S
∂V
)T
=
(∂p
∂T
)V
Funkcje S=S(T, V ) i p=p(T, V ) dla dowolnej substancji.
Termodynamika statystyczna:
Energia swobodna F=−kT lnQ, gdzie Q=ΣI e−EI/kT (suma statystyczna)
Dla gazu N atomow o masie m:
p(T, V ) =NkT
V, S(T, V ) = Nk ln
(2πmkT )3/2V
h3N+
5
2Nk

Termodynamika statystyczna. Przedmiot badanNa podstawie znajomosci sta lych uniwersalnych (takich jak k, h, c, e, me) orazparamerow ,,materia lowych” chrakteryzuj acych moleku ly rozwazanej substancji,takich jak:
• masy i spiny j ader atomowych (http://www.nist.gov)
• d lugosci wi azan chemicznych
• k aty pomi edzy wi azaniami
• sta le si lowe
• energie wzbudzen elektronowych
• potencja ly oddzia lywn mi edzymolekularnych
formalizm termodynamki statystycznej pozwala obliczac:
• funkcje termodynamiczne (entropi e, entalpi e swobodn a, pojemnosc ciepln a)
• sta le rownowagi chemicznej
• rownanie stanu
• szybkosci reakcji chemicznych
• w lasnosci elektryczne i magnetyczne substancji
• temperatury i ciep la przejsc fazowych
• parametry charakteryzuj ace zjawiska krytyczne

Termodynamika statystyczna. Podzia l teorii
Termodynamika, a ogolnie mechanika statystyczna, jest bardzo obszernymdzialem nauki scis lych. Mozna j a roznie dzielic, np. na:
− termodynamik e statystyczn a klasyczn a
− termodynamik e statystyczn a kwantow a
lub na:
− termodynamik e statystyczn a stanow rownowagi
− termodynamik e statystyczn a procesow nieodwracalnych
Pod wzgl edem stosowanych technik rachunkowych istotny jest podzia l na:
− teorie wykorzystuj ace metody analityczne
− metody symulacji komputerowych (Monte Carlo, dynamika molekularna).
Polecana literatura uzupe lniej aca:
1. F. Reif Fizyka statystyczna, PWN, 1973. Rozdz. 3, 4, 6.
2. H. Buchowski Elementy termodynamiki statystycznej, WNT, 1998. Rozdz. 1, 3.
3. R. Ho lyst, A. Poniewierski, A. Ciach Termodynamika, WNT, 2005, Rozdz. 13, 16, 17, 18.

Specyficzne w lasnosci uk ladow makroskopowych
Uk lady makroskopowe maj a 3 wazne w lasnosci rozni ace je istotnie od uk ladowmikrosokopowych:
1. W uk ladach makroskopowych zachodz a procesy nieodwracalne prowadz acedo stanow rownowagi, w ktorych w lasnosci uk ladu nie zalez a od czasu.
2. Stany rownowagi charakteryzowane s a przez niewielk a liczb e parametrow,dla uk ladu jednosk ladnikowego wystarcz a 3 takie parametry, np. E, V, i N.
3. W lasnosci uk ladow makroskopowych s a na ogo l zmiennymi losowymio bardzo ma lych fluktuacjach wzgl ednych. Na ogo l fluktuacje te malej az wielkosci a uk ladu jak 1/
√N .
Fluktuacja wzgl edna δ(X) zmiennej losowej X zdefiniowane jest nast epuj aco:
δ(X) =
√σ2(X)
〈X〉=σ(X)
〈X〉
gdzie 〈X〉 jest wartosci a sredni a zmiennej losowej X, a σ2(X) jest wariancj atej zmiennej (kwadratem odchylenia standardowego σ):
σ2(X) = 〈(X − 〈X〉)2〉 = 〈X2〉 − 〈X〉2
W lasnosc 1 jest sprzeczna z tw. Poincare’go o powrocie, ktore mowi, ze uk laddynamiczny o skonczonej energii i obj etosci wraca do swego stanu pocz atkowego.


Proces praktycznie nieodwracalny: rozpr ezanie gazu 200 atomow do prozni

Fluktuacje g estosci

Fluktuacja wzgl edna g estosci
Jako przyk lad obliczmy fluktuacj e liczby cz astek n w elemencie obj etosci v.Jesli w uk ladzie by laby tylko jedna cz astka to:
Pn=1 = v/V ≡ p Pn=0 = 1− v/V ≡ 1− p Pn>1 = 0,
gdzie Pn=k oznacza prawdopodobienstwo, ze w obj etosci v jest k cz astek . St ad
〈n〉 = 〈n2〉 = 0 · (1− p) + 1 · p = p
σ2(n) = 〈n2〉 − 〈n〉2 = p− p2 = p(1− p)
Jesli w uk ladzie jest N cz astek to Pn=k, 〈n〉 oraz σ2(n) mozna obliczyc zrozk ladu Bernoulliego ale nie ma potrzeby. Wystarczy zdefiniowacN zmiennychniezaleznych ni, gdzie ni jest liczb a cz astek o numerze i w obj etosci v:
n =N∑i=1
ni
Wowczas
〈ni〉 = p, σ2(ni) = p(1− p), 〈n〉 = Np, σ2(n) = Np(1− p)
Ostatecznie
δ(n) =
√σ2(n)
〈n〉=
√1− pNp
=
√1− p〈n〉

Postulaty i trzy najwazniejsze rozk lady prawdopodobienstwa
Definicje:
Makrostan jest to stan uk ladu wyznaczony przez niewielka liczb e parametrowmakroskopowych niezb ednych do jego jednoznacznego okreslenia.
Mikrostan jest to konkretny stan kwantowy uk ladu (w kwantowej mechanicestatystycznej) lub ma la komorka w przestrzeni fazowej uk ladu(w klasycznej mechanice statystycznej)
Postulaty:
Postulat 1: W makroskopowym uk ladzie izolowanym spontaniczne procesyprzebiegaj a tak, ze liczba dozwolonych mikrostanow wzrasta
Postulat 2: Jesli uk lad izolowany (o sta lej energii) jest w stanie rownowagi towszystkie dozwolone mikrostany s a jednakowo prawdopodobne
Rozk lady:
1. Mikrokanoniczny. Dla uk ladu izolowanego. Ustalone E, V,N.
2. Kanoniczny Gibbsa. Dla uk ladu termostatowanego. Ustalone T, V,N .
3. Wielki kanoniczny. Dla uk ladu otwartego. Ustalone µ, T, V , gdzie µ jestpotencja lem chemicznym.

Rozk lad mikrokanoniczny
Definicja: Liczba stanow Ω(E, V,N) jest to liczba mikrostanow uk laduo obj etosci V , liczbie cz astek N oraz o okreslonej energii(zawartej w przedziale od E do E+δE, δE=10−30J).
Definicja: Statystyczna (kwantowa) definicja temperatury
1kT =
(∂ ln Ω∂E
)V,N
Definicja: Empiryczna definicja temperatury (Tt.p=273.16 K)
T =1
klimp→0
pV
N
Definicja: Statystyczna (kwantowa) definicja entropii (Boltzmann, Planck)
S = k ln Ω
Definicja: Statystyczna definicja cisnienia
p = kT
(∂ ln Ω
∂V
)E,N

Rozk lad kanoniczny Gibbsa
Uk lad termostatowany o temperaturze T nie ma okreslonej energii. Taki uk ladjest w stanie kwantowym i o energii Ei z prawdopodobienstwem:
Pi = 1Q e−Ei/kT
Q =∑i e−Ei/kT
to suma statystyczna. Znaj ac sum e statystyczn a Q=Q(T, V,N) mozemy latwo obliczyc funkcje termodynamiczne uk ladu:
E = kT 2
(∂ lnQ
∂T
)V
S = k lnQ+E
T
p = kT
(∂ lnQ
∂V
)T
F = −kT lnQ

Rozk lad kanoniczny Gibbsa, c.d.
Jesli zaniedbamy oddzia lywanie mi edzy moleku lami gazu oraz jeslitemperatura T nie jest bardzo niska to to Q ma szczegolnie prost a postac:
Q =qN
N !
gdzie N jest liczb a moleku l w uk ladzie, a q jest sum a statystyczn adla jednej moleku ly, zwan a tez molekularn a funkcj a podzia lu
q =∑i
e−εi/kT
W definicji funkcji podzia lu q sumowanie przebiega po wszystkichstanach kwantowych ψi pojedynczej moleku ly, a εi to energie tych stanow.
Dla sztywnych moleku l q jest w przyblizeniu rowne iloczynowi funkcji:
− translacyjnej qtr
− rotacyjnej qrot
− wibracyjnej qvib
− elektronowej qel
− j adrowej qnucl

Rozk lad kanoniczny Gibbsa. Obliczenie funkcji podzia lu
q = qtr qrot qvib qel qnucl
Faktoryzacja ta jest mozliwa gdy energia εi itego stanu kwantowego moleku lydaje si e przedstawic jako suma energii translacji, rotacji, wibracji, etc.
εi = εtrn1n2n3
+ εrotJKM + εvib
v1...vf+ εel
l + εnuclλ
Funkcje qtr qrot qvib, etc. zdefiniowane s a formalnie tak samo jak q, np.
qtr =∞∑
n1n2n3
exp(−εtrn1n2n3
/kT ),
gdzie energie wzbudzen translacyjnych dane s a wzorem na poziomyenergetyczne cz astki o masie m w pudle o obj etosci V = L3:
εtrn1n2n3
=h2
8mL2(n2
1 + n22 + n2
3)
Sumowanie po n1, n2, n3 mozna sfaktoryzowac i zast apic przez ca lkowanie:
qtr =∞∑
n1n2n3
exp
(−n2
1 + n22 + n2
3
a2
)=
[ ∞∑n
exp
(−n2
a2
)]3
=
[∫ ∞0
exp
(−x2
a2
)dx
]3
=π3/2a3
8
gdzie a=√
8mkT L/h.
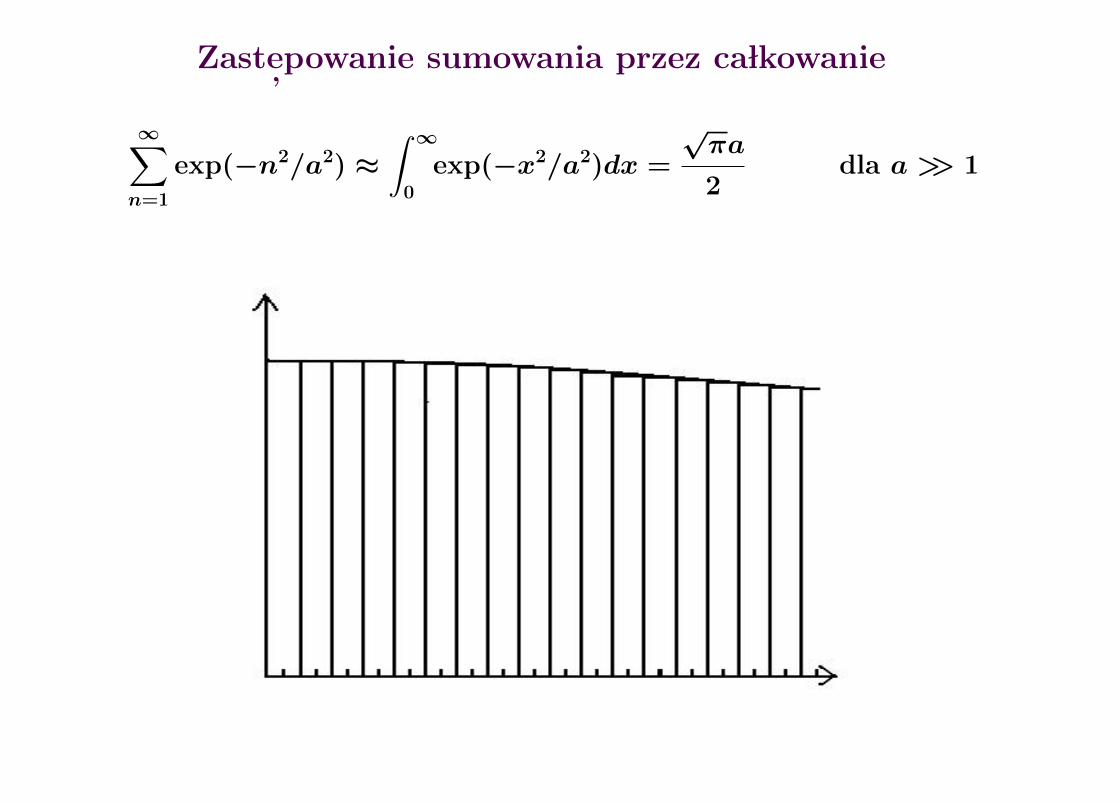
Zast epowanie sumowania przez ca lkowanie
∞∑n=1
exp(−n2/a2) ≈∫ ∞
0
exp(−x2/a2)dx =
√πa
2dla a >> 1

Rozk lad kanoniczny Gibbsa. Obliczenie funkcji podzia lu, c.d.
Po podstawieniu a=√
8mkT L/h otrzymujemy ostatecznie
qtr =(2πmkT )3/2V
h3
lub
qtr =V
λ3B
, gdzie λB =h
√2πmkT
to tzw. termiczna d lugosc fali de Broglie.
Mozemy teraz obliczyc translacyjn a sum e statystyczn a Qtr = (qtr)N/N ! oraz
translacyjn a energi e wewn etrzn a Etr, pojemnosc ciepln a C
trV i entropi e S
tr.
Obliczenie Etr jest szczegolnie proste. Wystarczy zauwazyc, ze Qtr∼T 3N/2.
Otrzymujemy:
Etr =3
2NkT Ctr
V =
(∂E
∂T
)V
=3
2Nk
Str =Nk ln(2πmkT )3/2V
h3N+
5
2Nk = Nk ln
v
λ3B
+5
2Nk,
gdzie v=V/N . Jest to wzor Sackura i Tetrode - stosowalny gdy v> λ3B.
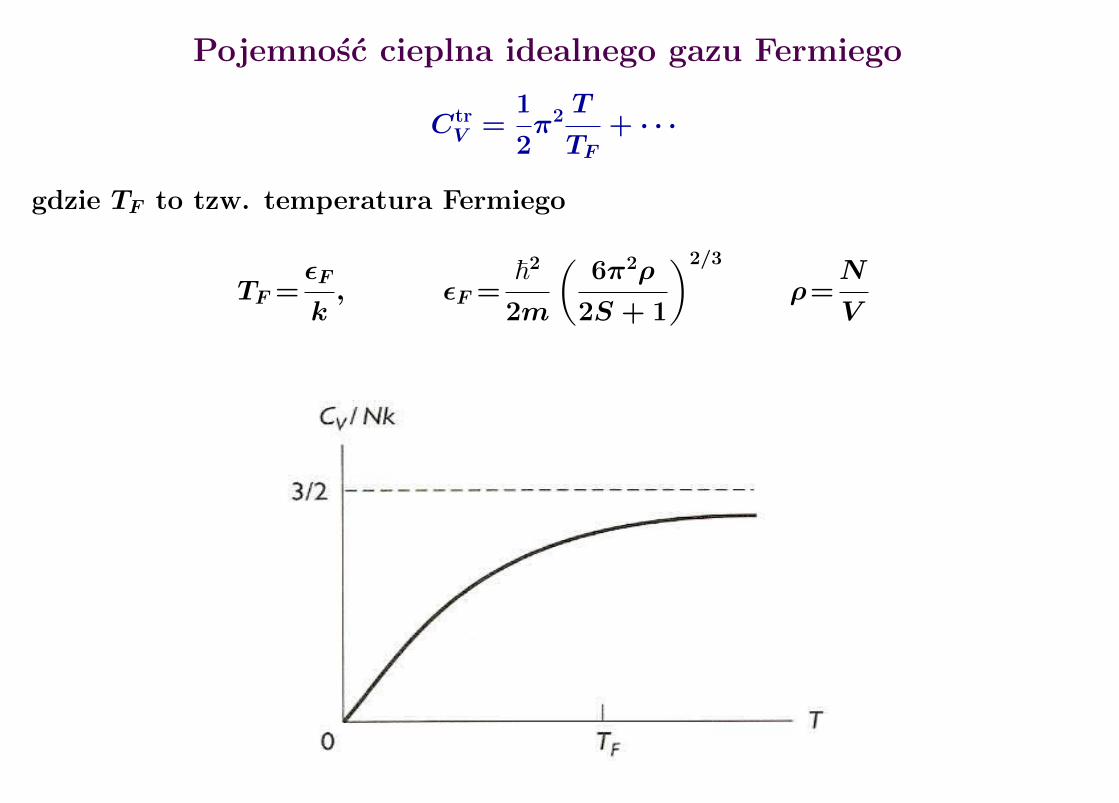
Pojemnosc cieplna idealnego gazu Fermiego
CtrV =
1
2π2 T
TF+ · · ·
gdzie TF to tzw. temperatura Fermiego
TF =εF
k, εF =
~2
2m
(6π2ρ
2S + 1
)2/3
ρ=N
V
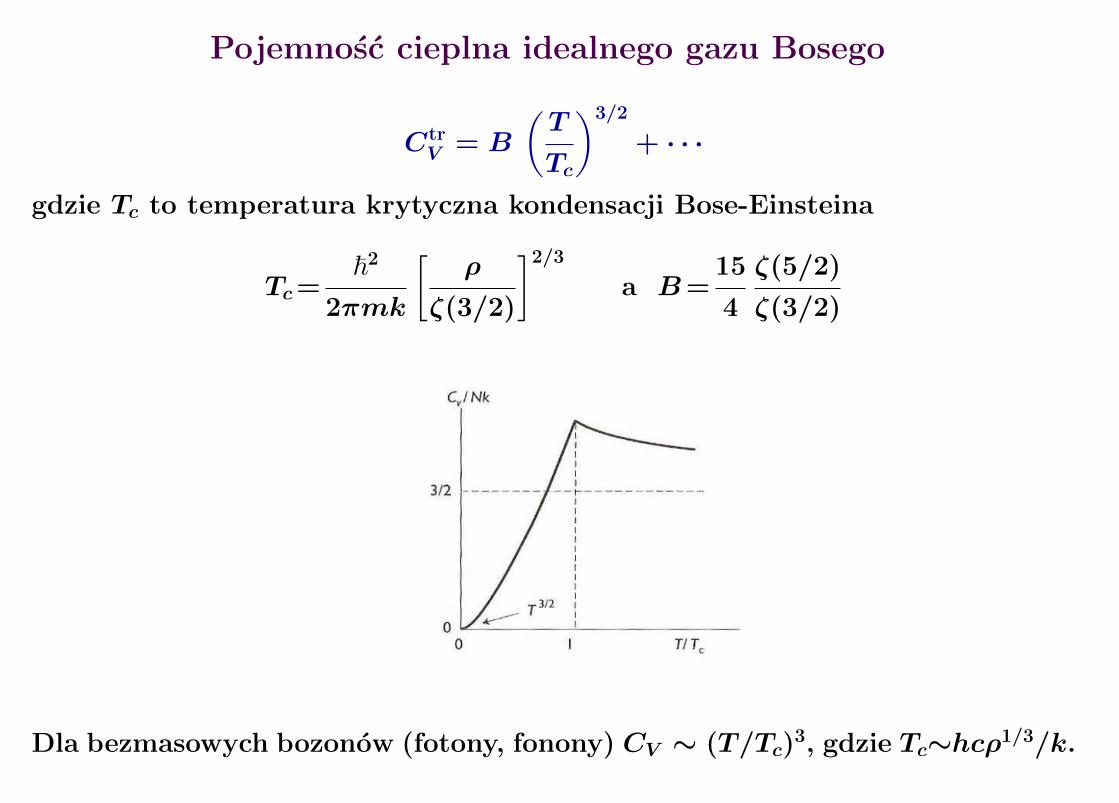
Pojemnosc cieplna idealnego gazu Bosego
CtrV = B
(T
Tc
)3/2
+ · · ·
gdzie Tc to temperatura krytyczna kondensacji Bose-Einsteina
Tc=~2
2πmk
[ρ
ζ(3/2)
]2/3
a B=15
4
ζ(5/2)
ζ(3/2)
Dla bezmasowych bozonow (fotony, fonony) CV ∼ (T/Tc)3, gdzie Tc∼hcρ1/3/k.

Pojemnosc cieplna helu 4
Przejscie lambda dla T=2.17 K (Willem Keesom i Mieczys law Wolfke - 1928).

Rozk lad kanoniczny Gibbsa. Uwzgl ednienie rotacji moleku l
Dla moleku l liniowych energie poziomow rotacyjnych dane s a wzorem:
εrotJ =
~2
2IJ(J + 1)
gdzie I jest momentem bezw ladnosci molek ly (I=µR2 dla dwuatomowych)Rotacyjna funkcja podzia lu dana jest zatem wzorem:
qrot =∞∑J=0
J∑M=−J
e−εrotJ /kT =
∞∑J=0
(2J + 1) e−J(J+1)ΘrotT ≈
T
γΘrot
gdzie Θrot jest charakterystyczn a temperatur a rotacji
Θrot =~2
2Ik
Wzor ten jest s luszny dla moleku l liniowych symetrycznych (γ = 2) i asymet-rycznych (γ = 1) i tylko dla T >> Θrot. Dla T ≤ Θrot sumujemy szereg.Na ogo l T >> Θrot bo Θrot jest rz edu 1 K lub mniejsze. Dla H2, Θrot=85 K.
Erot =NkT CrotV =
(∂E
∂T
)V
=Nk
Srot = Nk lnT
γΘrot
+Nk,
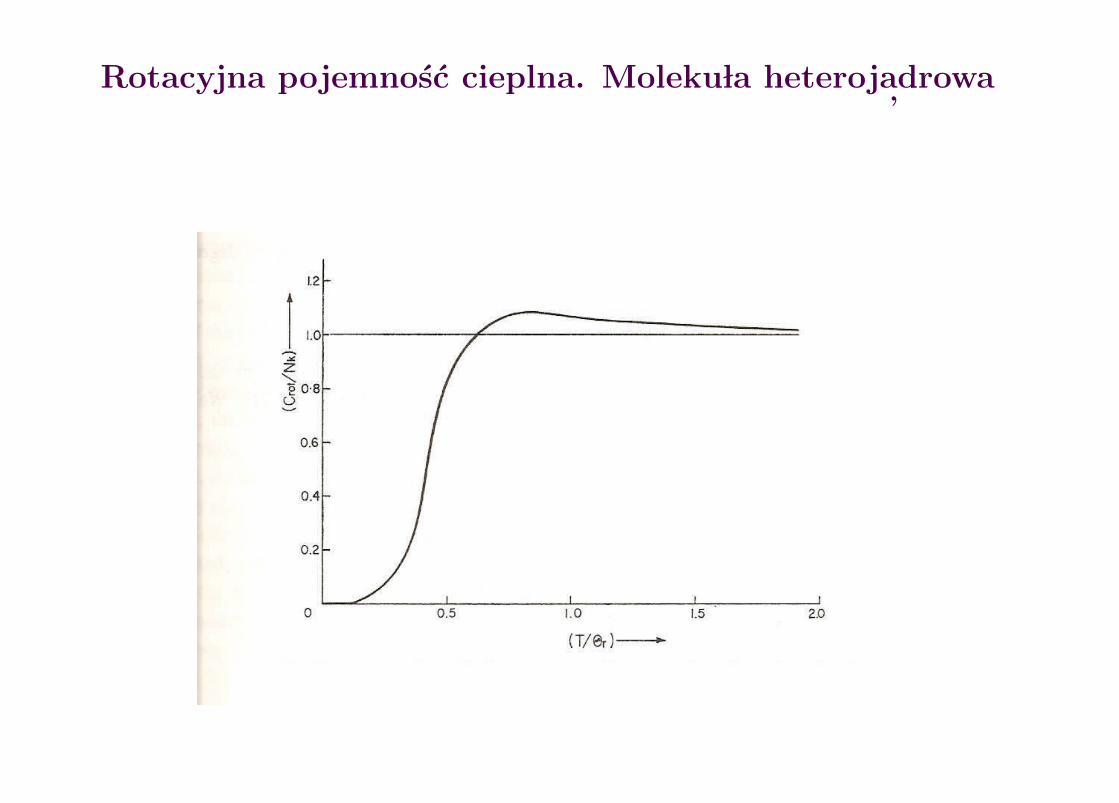
Rotacyjna pojemnosc cieplna. Moleku la heteroj adrowa

Pojemnosc cieplna para (1), orto (2) i mieszaniny 1:3 (3) wodoru
Para wodor Snucl=0, J = 0, 2, 4, 6, . . .Orto wodor Snucl=1, J = 1, 3, 5, 7, . . .

Rozk lad kanoniczny Gibbsa. Uwzgl ednienie wibracji moleku l
Dla moleku l dwuatomowych energia wibracji dana jest wzorm
εvib = nhν
Wibracyjna funkcja podzia lu ma zatem postac
qvib =∞∑n=0
e−nhνkT =
1
1− e−Θv/T,
gdzie Θvib jest charakterystyczn a temperatur a wibracji
Θv =hν
k
Temperatury Θvib s a wysokie, np. Θvib≈6000 K dla H2, Θvib≈3000 K dla CO.
Dla wk ladu wibracji do energii wewn etrznej i pojemnosci cieplnej dostajemy:
Evib =Nhν
ehν/kT − 1⇒ Cvib
V = Nk
(hν
kT
)2 ehν/kT
(ehν/kT − 1)2
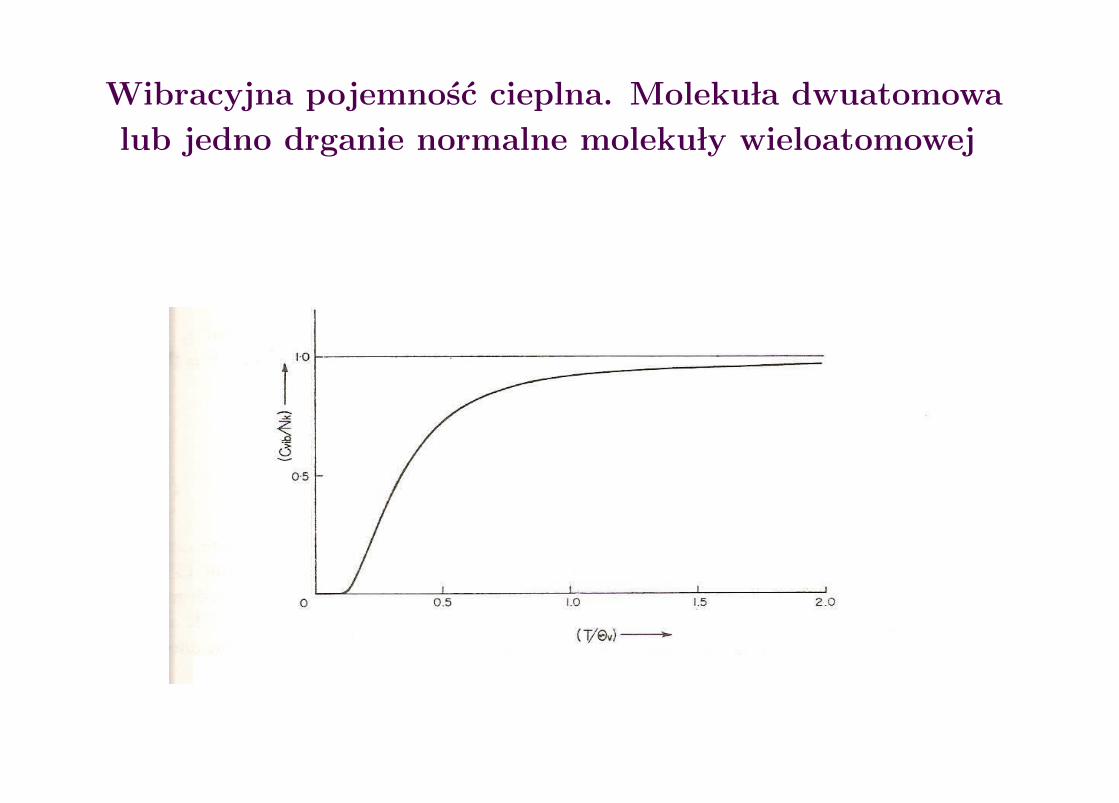
Wibracyjna pojemnosc cieplna. Moleku la dwuatomowa
lub jedno drganie normalne moleku ly wieloatomowej

Rozk lad kanoniczny Gibbsa. Moleku ly wieloatomowe-rotacje
Dla b aka sferycznego, symetrycznego lub asymetrycznego
qrot =
√π
γ
√T 3
ΘAΘBΘC
,
gdzie ΘA, ΘB, i ΘC s a temperaturami charakterystycznymi
ΘA =~2
2IAk, ΘB =
~2
2IBk, ΘC =
~2
2ICk
Wzor ten jest s luszny jedynie gdy T ΘA, T ΘB, i T ΘC.IA, IB i IC to momenty bezw ladnosci moleku ly wzgl edem jej osi g lownycha γ to liczba symetrii rowna liczbie takich permutacji j ader identycznych,ktore mozna uzyskac przez obroty moleku ly: γ=n dla symetrii Cnv, γ=2n dlasymetrii Dnh, γ=12 dla symetrii Td, γ=24 dla symetrii Oh.
Poniewaz Qrot=(qrot)N latwo teraz znalezc funkcje termodynamiczne:
Erot =3
2NkT Crot
V =3
2Nk
Srot = Nk ln
[√π
γ
√T 3
ΘxΘyΘz
]+
3
2Nk,

Rozk lad kanoniczny Gibbsa. Moleku ly wieloatomowe-wibracje
Dla moleku ly wieloatomowej o f drganiach normalnych o cz estosci νj energiawibracji jest sum a energii wszystkich drgan a zatem qvib jest iloczynem:
qvib =
f∏j=1
1
1− e−hνj/kT
Energia wewn etrzna i pojemnosc cieplna na jedn a moleku l e s a sumami wk ladowod poszczegolnych drgan normalnych:
Evib =
f∑j=1
hνj
ehνj/kT − 1
Kryszta l zN atomow to jedna wielka moleku la o f=3N drganiach normalnych.Jesli za lozymy, ze wszystkie cz estosci s e jednakowe i rowne νE to
Evib = 3NhνE
ehνE/kT − 1
Jest to teoria Einsteina pojemnosci cieplnej kryszta low. Niez la dla srednich iwysokich temperatur, s laba dla niskich.

Teoria Debye’a pojemnosci cieplnej kryszta low
Za lozenie, ze wszystkie cz estosci s a rowne jest zbyt drastyczne. Istnieje pewienrozk lad cz estosci drgan w krysztale dany funkcj a g(ν) tak a, ze
g(ν)dν jest liczb a drgan normalnych o cz estosci pomi edzy ν a ν + dν
Wyrazenie na energi e wewn etrzn a ma wowczas postac ca lki:
Evib =
∫ νmax
0
hν g(ν)dν
exp(hν/kT )− 1
Debye za lozy l, ze
g(ν) = 4πV
(1
cl+
2
ct
)ν2 = Aν2 dla ν < νmax
gdzie cl i ct to pod luzna i poprzeczna pr edkosc dzwi eku w krysztale. Wowczas:
Evib = A
∫ νmax
0
hν3dν
exp(hν/kT )− 1= A
k4T 4
h3
∫ ΘD/T
0
x3 dx
ex − 1
gdzie ΘD = hνmax/h to tzw. temperatura Debye’a. Gdy T ΘD wtedy:
Evib ∼ T 4 oraz CV ∼ T 3.
Gdy νmax =∞ otrzymujemy rozk lad Plancka i prawo Stefana-Boltzmanna.

Funkcja spektralna dla fononow w krysztale
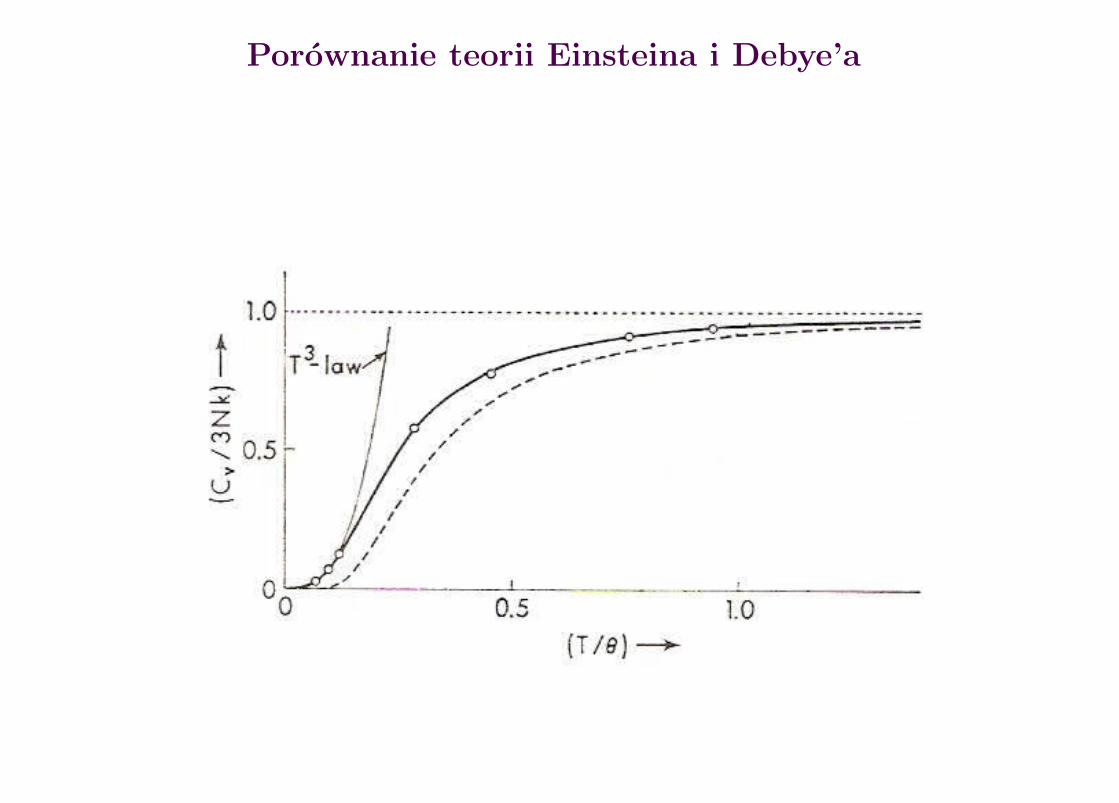
Porownanie teorii Einsteina i Debye’a

Porownanie teorii Debye’a z doswiadczeniem

Widmo promieniowania t la kosmicznego. T=2.73KPorownanie ze wzorem Plancka

Rozk lad kanoniczny Gibbsa. Wzbudzenia elektronowe i j adrowe
Elektronow a funkcj e podzia lu obliczamy bezposrednio z definicji, czyli wg.wzoru (4), wykonuj ac sumowanie po stanach elektronowych moleku ly.
qel =∑i
e−εeli /kT
Jesli j adra maj a spin rozny od zera (np. j adro atomu helu 3 lub j adro atomuazotu) to do iloczynu w rownaniu (5) trzeba jeszcze dopisac j adrow a funkcj epodzia lu
qnucl =∏j
(2sj + 1),
gdzie sj jest spinem j adra j.
Na ogo l wystraczy uwzgl ednic najnizej lez ace stany elektronowe wynikaj acez rozszczepienia spinorbitalnego. Np. dla moleku ly tlenku azotu (NO) stanpodstawowy 2Π rozszczepia si e na dwa stany 2Π3/2 i 2Π1/2 rozni ace si e o energi ewzbudzenia rown a ∆εel/k=178 K. qal ma dla NO postac
qel = 2 + 2e−Θel/T
gdzie Θel =178 K. Taka funkcja podzia lu daje charakterystyczne maksimumna wykresie pojemnosci cieplnej dla T ≈ 178K zwane anomali a Schottkiego.

Rozk lad kanoniczny Gibbsa. Rownowagi chemiczne
Sta l a rownowagi dla reakcji chemicznej
nA A + nB B nC C + nD D
przebiegaj acej w fazie gazowej definiujemy nast epuj aco
Kp(T ) =pnCC p
nDD
pnAA p
nBB
,
gdzie pX jest cisnieniem parcjalnym substancji X, X=A, B, C, D, a nX jejwspo lczynnikiem stechiometrycznym substancji X w rownaniu reakcji. Kp(T )to tzw. sta la cisnieniowa. Rozwaza si e tez sta l a ilosciow a KN(T ), zdefin-iowan a formalnie takim samym wzorem jak Kp(T ), z tym ze cisnienia parcjalnes a zast apione przez liczby moleku l poszczegolnych substancji:
KN(T ) =N
nCC N
nDD
NnAA N
nBB
,
Korzystaj ac z rownania p = NkT/V wi az acego cisnienie parcjalne z liczb amoleku l mozna latwo pokazac, ze sta la ilosciowa i cisnieniowa zwi azane s azaleznosci a
Kp(T ) =
(kT
V
)nC+nD−nA−nB
KN(T ).

Rozk lad kanoniczny Gibbsa. Rownowagi chemiczne
Korzystaj ac z rozk ladu kanonicznego Gibbsa wyprowadza si e nast epuj acy,podstawowy wzor na sta l a ilosciow a:
KN(T ) =qnCC q
nDD
qnAA q
nBB
,
gdzie qX jest funkcj a podzia lu moleku ly rodzaju X zdefiniowan a formalnie tymsamym wzorem co wczesniej
q =∑i
e−εi/kT
z tym, ze energie moleku ly εi obliczane s a teraz wzgl edem energii rozdzielonychatomow a nie wzgl edem energii jej stanu podstwowego E0 . Energia rozdzielonychatomow jest wyzsza od E0 i rozni si e od E0 o energi e atomizacji D0 (energi edysocjacji na atomy). Dlatego
εi = εi −D0
i w konsekwencjiq = eD0/kTq.

Rozk lad kanoniczny Gibbsa. Rownowagi chemiczne
Jesli korzystamy z funkcji podzia lu qX obliczanych wzgl edem energii stanupodstawowego molekul to sta la rownowagi KN(T ) wyraza si e wzorem
KN(T ) = e∆D0/kTqnCC q
nDD
qnAA q
nBB
.
gdzie
∆D0 = nCDC0 + nDD
D0 − nAD
A0 − nBD
B0
a DX0 jest energi a atomizacji moleku ly X. Bior ac pod uwag e, ze DX
0 jest rowneenergii atomow minus enregia stanu podstawowego EX
0 oraz ze liczba atomownie zmienia sie w trakcie rekacji chemicznej widzimy, ze
∆D0 = −∆E0
gdzie
∆E0 = nCEC0 + nDE
D0 − nAE
A0 − nBE
B0
jest energi a reakcji dla zerowej temperatury (∆E0 < 0 dla reakcji egzoter-micznych).

Teoria szybkosci rekacji. Teoria stanu przejsciowego
Jest wiele teorii szybkosci reakcji chemicznych. Scis la teoria wykracza dalekopoza zakres termodynamiki statystycznej stanow rownowagi.
Istnieje prosta teoria stanu przejsciowego (lub teoria kompleksu aktywnego,Eyring), ktora wykorzystuje istotnie termodynamik e statystyczn a.
Rozwazmy reakcj e:
A + BC −→ AB + C
Wg. Eyringa reakcja ta przebiega przez stadium kompleksu przejsciowego X‡
A + BC −→ X‡ −→ AB + C
b ed acego w rownowadze termodynamicznej z substratami A i BC. Koncen-tracja X‡ dana jest wi ec wzorem
[X‡] = K‡ [A] [BC]
gdzie K‡ jest st ezeniow a ([A]=NA/V ) sta l a rownowagi reakcji A + BC X‡
K‡ =[X‡]
[A] [BC]=
N ‡
NANBC
V = e−∆E‡0/kT
q‡
qA qBC
V
∆E‡0 jest tu energi a aktywacji a q‡ funkcj a podzia lu kompleksu aktywnego
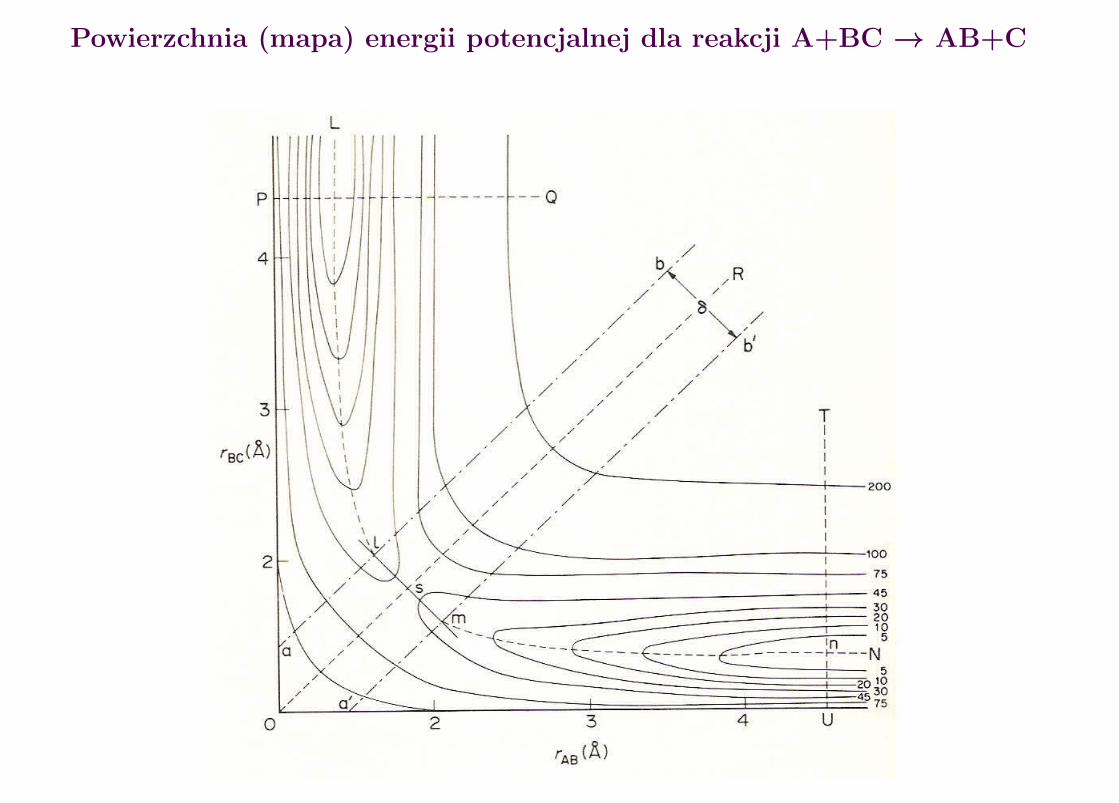
Powierzchnia (mapa) energii potencjalnej dla reakcji A+BC → AB+C

Przekroje powierzchni energii potencjalnej dla reakcji A+BC → AB+C

Sciezka reakcji dla reakcji typu A+BC → AB+C

Teoria szybkosci rekacji. Teoria stanu przejsciowego, c.d.
Szybkosc reakcji jest rowna iloczynowi koncentracji kompleksu [X‡] i szybkosciv‡ jego rozpadu na produkty AB i C
d[C]
dt= v‡ [X‡] = v‡ K‡ [A] [BC]
St ad oczywiscie sta la szybkosci ks dana jest wzorem
ks = v‡ K‡ = v‡ e−∆E‡0/kT
q‡
qA qBC
V
Dok ladne obliczenie v‡ jest trudne ale Eyring pokaza l, ze w przyblizeniu
v‡ =kT
h
1
q∗
gdzie q∗ jest tym czynnikiem w wibracyjnej cz esci q‡, ktory odpowiadazespolonej cz estosci (wspo lrz ednej reakcji). Po podstawieniu, otrzymujemy
ks =kT
he−∆E
‡0/kT
q‡∗qA qBC
V
gdzie q‡∗ jest funkcj a podzia lu kompleksu aktywnego, w ktorej uwzgl edniones a wy l acznie rzeczywiste cz estosci drgan normalnych.

Uk lady otwarte. Wielki rozk lad kanoniczny Gibbsa
Tak jak temperatura decyduje o kierunku przep lywu energii pomi edzyuk ladami, ktore nie s a izolowane adiabatycznie, tak potencja l chemiczny µdecyduje o kierunku przep lywu cz astek pomi edzy uk ladami otwartymi.
Statystyczna definicja potencja lu chemicznego jest nast epuj aca:
µ = −kT(∂ ln Ω∂N
)E,V
µ = −kT(∂ lnQ∂N
)T,V
Dla uk ladu otwartego o sta lych T , µ i V prawdopodobienstwo, ze ma on Ncz astek i znajduje si e w i-tym stanie kwantowym o energii Ei wynosi:
PiN = 1Ξ e
(µN−Ei)/kT
gdzie
Ξ =∑N∑i e
(µN−Ei)/kT
Ξ to wielka suma statystyczna. Znaj ac wielk a sum e statystyczn a Ξ=Ξ(T, µ, V )mozemy znalezc wszystkie funkcje termodynamiczne uk ladu.

Uk lady otwarte. Wielki rozk lad kanoniczny Gibbsa, c.d.
Funkcje termodynamiczne w wielkim rozk ladzie kanonicznym wyrazaj a si enast epuj aco :
N = kT
(∂ ln Ξ
∂µ
)T,V
pV = kT ln Ξ
G = µ N(T,µ,V ) F = G− pV
Fluktuacje
Energii - rozk lad kanoniczny
δ(E) =1
E
√kT 2
(∂E
∂T
)N,V
=1√N
kT
ε
√cV
k
Liczby cz astek - wielki rozk lad kanoniczny (v = V/N)
δ(N) =1
N
√kT
(∂N
∂µ
)T,V
=1√N
√kT
∂p/∂ρ

Statystyki kwantowe Fermiego-Diraca oraz Bosego-Einsteina
Wielki rozk lad kanoniczny mozna zastosowac do pojedynczego spinorbitalu.Mozemy wowczas obliczyc prawdopodobienstwo, ze na tym spinorbitalu bedzieN cz astek a takze sredni a liczb e cz estek Ni na itym spinorbitalu.
Dla fermionow N moze byc rowne tylko 0 lub 1. Zatem
NFi = 0 · P0 + 1 · P1 =
1
Ξe(µ−εi)/kT =
e(µ−εi)/kT
1 + e(µ−εi)/kT
Ostatecznie
NFi =
1
e(εi−µ)/kT + 1
Dla bozonow N moze byc 0,1,2,...,etc. Po prostym obliczeniu Ξ dostajemy
NBi =
1
e(εi−µ)/kT − 1
To s a wzory na statystyki Fermiego-Diraca i Bosego-Einsteina. Gdy NFi >> 1
lub gdy NBi >> 1 to jedynki w mianowniku mozna zaniedbac i dostajemy
statystyk e Maxwella-Boltzmanna stosowaln a gdy λB << r0 = (V/N)1/3:
Ni = e(µ−εi)/kT

Kondensacja Bosego-Einsteina
NBi =
1
e(εi−µ)/kT − 1
NB0 =
1
e−µ/kT − 1≥ 0 −→ eµ/kT ≤ 1 −→ µ ≤ 0
NBi ≤
1
eεi/kT − 1, for i > 0
NBexc ≡
∑i>0
NBi ≤
∑i>0
1
eεi/kT − 1=
∫ ∞0
g(ε)dε
eε/kT − 1≡ Nmax
exc
Kondensacja ma miejsce gdy Nmaxexc < Ntot, Tc ≈ 2 K dla helu

Liczby obsadzen dla bozonow dla bardzo niskich temperatur

Pojemnosc cieplna idealnego gazu Bosego
CtrV = B
(T
Tc
)3/2
+ · · ·
gdzie Tc to temperatura krytyczna kondensacji Bose-Einsteina
Tc=~2
2πmk
[ρ
ζ(3/2)
]2/3
a B=15
4
ζ(5/2)
ζ(3/2)
Dla bezmasowych bozonow (fotony, fonony) CV ∼ (T/Tc)3, gdzie Tc∼hcρ1/3/k.

Pojemnosc cieplna helu 4
Przejscie lambda dla T=2.17 K (Willem Keesom i Mieczys law Wolfke - 1928).

Liczby obsadzen dla fermionow dla bardzo niskich temperatur

Pojemnosc cieplna idealnego gazu Fermiego
CtrV =
1
2π2 T
TF+ · · ·
gdzie TF to tzw. temperatura Fermiego
TF =εF
k, εF =
~2
2m
(6π2ρ
2S + 1
)2/3
ρ=N
V






![Węgiel tania energia i miejsca pracy - giph.com.plukaszczyk.pdf · Węgiel Ropa Gaz ziemny Energia jądrowa Energia odnawialna Inne. TWh. Udział węgla [%] Źródło: Eurostat ...](https://static.fdocuments.pl/doc/165x107/5c785b9f09d3f2fb438b6e21/wegiel-tania-energia-i-miejsca-pracy-giphcompl-wegiel-ropa-gaz-ziemny.jpg)











