Politechnika Łódzka Wydział Biotechnologii i Nauk o Żywnościnzbqmub4354qqc... · Niektóre...
10
Politechnika Łódzka Wydział Biotechnologii i Nauk o Żywności Ćwiczenie 1 Polarograficzna metoda oznaczania śladowych ilości Pb(II) w obecności innych jonów metali w próbie badanej
Transcript of Politechnika Łódzka Wydział Biotechnologii i Nauk o Żywnościnzbqmub4354qqc... · Niektóre...

Politechnika Łódzka Wydział Biotechnologii i Nauk o Żywności
Ćwiczenie 1
Polarograficzna metoda oznaczania śladowych ilości Pb(II)
w obecności innych jonów metali w próbie badanej

1.Cel ćwiczenia Celem ćwiczenia jest zapoznanie się z możliwościami użycia technik
woltamperometrycznych i polarograficznych do oznaczania wybranego składnika
tu - jony Pb2+
w próbie: wody, gleby lub materiału roślinnego w obecności innych metali.
2. Wstęp teoretyczny
Polarografia należy do metod woltamperometrycznych, które polegają na realizowaniu
i analizowaniu zależności natężenia prądu od napięcia przykładanego z zewnątrz do elektrod
znajdujących się w naczyńku elektrolitycznym. Jedna z elektrod wchodząca w skład układu
elektrolitycznego ulega polaryzacji (może to być katoda lub anoda). W metodzie tej
wykorzystuje się zjawisko elektrolizy, która nie zachodzi w całej objętości roztworu ale
jedynie w warstwie dyfuzyjnej bezpośrednio przy polaryzowanej elektrodzie. W związku z
tym stężenia składników roztworu w dostatecznie dużej odległości od elektrody pozostają nie
zmienione.
Polarografia jest szczególnym przypadkiem woltamperometrii z zastosowaniem elektrody
rtęciowej. W praktyce stosuje się kilka technik polarograficznych, należą do nich m.in.:
a) polarografia stałoprądowa, w której do kroplowej elektrody rtęciowej jest
przykładany potencjał zmieniający się liniowo w czasie,
b) polarografia zmiennoprądowa, w której liniowa zmiana potencjału elektrody
pracującej jest modulowana napięciem zmiennym (prostokątnym lub sigmoidalnym),
c) polarografia pulsowa, w której na potencjał elektrody pracującej nakładane są impulsy
napięcia prądu pulsującego,
d) oscylopolarografia, w której zmiany przykładanego do elektrod napięcia następują z
szybkością dochodzącą do kilkudziesięciu woltów na sekundę.
Techniki te umożliwiają realizację oznaczenia polarograficznego w wersji najbardziej
odpowiedniej dla danej substancji.
Twórcą metod polarografii był Jaroslav Heyrowski – profesor Uniwersytetu Karola w
Pradze, W 1959 r. otrzymał Nagrodę Nobla jako wynik uznania za odkrycie i
systematyczne ich rozwijanie. Najbardziej rozpowszechnioną w polarografii elektrodą
wskaźnikową (często nazywaną – roboczą) jest klasyczna kapiąca elektroda rtęciowa, ale
coraz częściej stosowana jest stacjonarna wisząca kroplowa elektroda rtęciowa. Mogą być
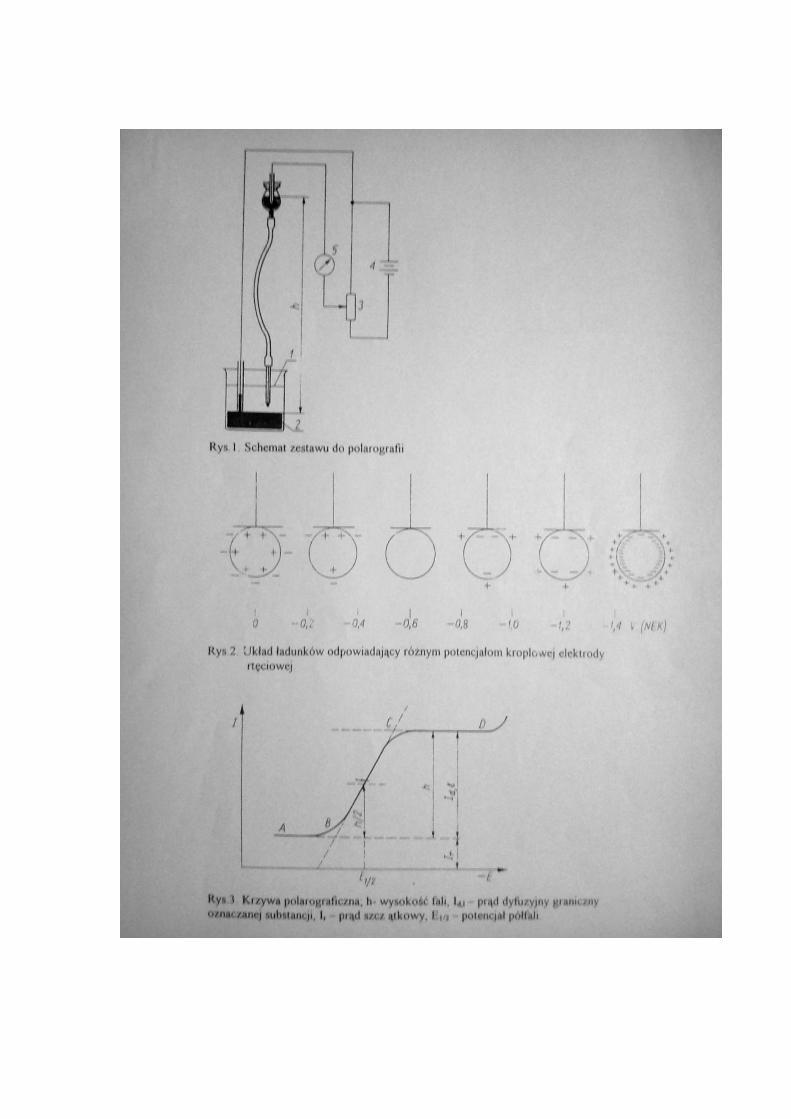
też zastosowane inne elektrody, np. z innych metali bądź z grafitu. Podstawowy schemat
zestawu do polarografii przedstawiono na rys.1. Jedną z elektrod jest rtęć, kapiąca
drobnymi kroplami z kapilary (1) a druga elektroda to warstwa rtęci (2) umieszczona na
dnie naczyńka o dużej powierzchni. Źródłem przykładanego napięcia jest bateria
akumulatorów (4), ujemny biegun baterii połączony jest ze zbiornikiem kroplowej
elektrody, która zwykle pracuje jako katoda (chociaż może też być anodą), napięcie
można w sposób ciągły regulować za pomocą opornicy suwakowej (3). Natężenie prądu
płynącego przez elektrolit mierzy się galwanometrem (5). Roztworu poddanego
elektrolizie w metodzie polarograficznej nie miesza się podczas wykonywania pomiaru.
Natomiast przed wykonaniem pomiaru układ odtlenia się. Ma to na celu usunięcie tlenu
występującego w elektrolicie gdyż bez tego mógłby wchodzić w reakcję elektrolityczną i
w sposób niekontrolowany zmieniać wynik reakcji. Usuwanie tlenu z elektrolitu
przeprowadza się poprzez przepuszczanie gazu obojętnego (argon, azot lub hel) w ściśle
określonym czasie z kontrolowaną szybkością.

Kroplowa elektroda rtęciowa (KER)
Zasadniczym elementem w zestawie do polarografii jest kroplowa elektroda rtęciowa
(KER). Składa się ona z grubościennej kapilary szklanej o długości kilkunastu
centymetrów i średnicy wewnętrznej 0,03-0,05 mm. Kapilara jest połączona za pomocą
węża polietylenowego ze szklanym zbiornikiem rtęci. Prędkość wypływu rtęci zależy od
położenia zbiorniczka (wysokości słupa) oraz długości i średnicy kapilary.
Kroplowa elektroda rtęciowa charakteryzuje się następującymi właściwościami:
- powierzchnia rtęci wypływającej systematycznie w postaci kropel jest stale
odnawiana,
a produkty reakcji elektrodowej są usuwane wraz z odrywającą się kroplą,
- każda narastająca świeża kropla rtęci styka się ze świeżym roztworem,
- mała powierzchnia kropli powoduje, że natężenie płynącego prądu jest bardzo małe,
wobec czego zużycie depolaryzatora jest minimalne w trakcie pomiaru
polarograficznego. Dzięki tym zaniedbywalnym zmianom stężenia możliwe jest
wielokrotne rejestrowanie krzywych dla tego samego roztworu, co ma istotne
znaczenie przy prowadzeniu oznaczeń ilościowych (kontrola powtarzalności
wykonania oznaczenia)
- duże nadnapięcie wodoru na rtęci pozwala na prowadzenie pomiarów w szerokim
zakresie potencjałów
- rtęć jako metal szlachetny nie reaguje chemicznie ze stosowanymi w polarografii
roztworami jako elektrolity podstawowe.
Kroplowa elektroda rtęciowa jest całkowicie polaryzowalna, tzn., że pod wpływem
zewnętrznej siły elektromotorycznej powstaje na KER potencjał, który powoduje
wytworzenie siły elektromotorycznej polaryzacji P skierowanej przeciw przyłożonemu
napięciu równemu co do wielkości. Polaryzacja dąży do zmniejszenia natężenia prądu:
R
PVI
gdzie: I - natężnie prądu
R- opór
P – polaryzacja
Iloczyn IR jest wielkością stałą mniejszą od 1mV, a wynika z tego, że natężenie prądu
jest bardzo małe, rzędu 10-6
A (1mikroamper) i opór R jest niewielki – rzędu 1000 .
W porównaniu z przyłożonym napięciem, które osiąga wartość jednego lub dwóch
woltów iloczyn IR jest więc bardzo mały i można go pominąć. Wówczas zgodnie z
równaniem polaryzacja jest prawie równa zewnętrznemu napięciu przyłożonemu do
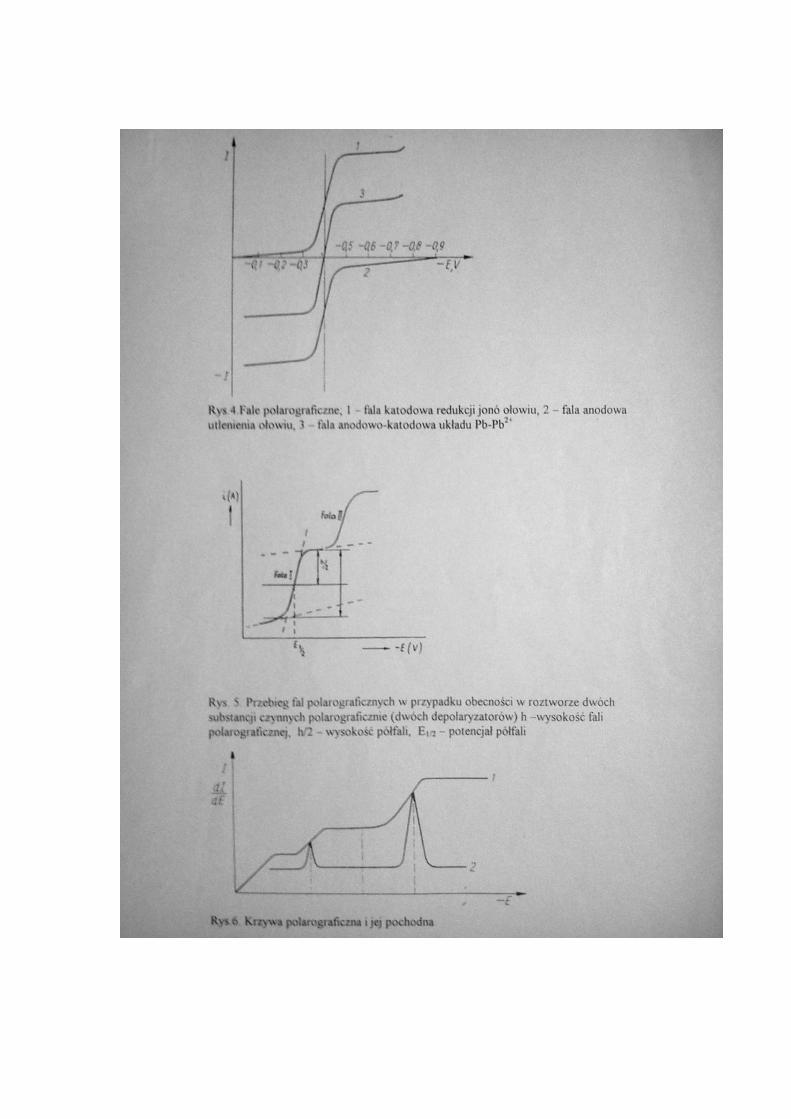
elektrod. Dzięki zjawisku polaryzacji na elektrodzie (rys.2.) pojawia się dokładnie tyle
ładunków ile zostanie przyłożonych z zewnątrz, ale o znaku przeciwnym.
V P
Krzywa polarograficzna
Jeżeli w elektrolitycznym naczyńku znajduje się roztwór elektrolitu nie ulegający
reakcjom elektrochemicznym w zakresie stosowanego napięcia (np. podstawowe sole
sodu, litu lub potasu; np.: NaNO3, NaClO4, Na2SO4, KCl, LiCl i itp.), zawierający małą
ilość kationów zdolnych do reakcji elektrodowych, np.: Zn2+
, Cu2+
, Cd2+
, Pb2+
, Fe3+
, Fe2+
,
Se4+
a także i inne, wówczas zacznie się przykładać napięcie wzrastające w czasie oraz
będzie się mierzyć i rejestrować odpowiadające mu natężenie prądu, to otrzyma się

krzywą zależności I=f(E) rys.3. Na rysunku tym krzywa stanowiąca wykres zależności I
od E składa się z trzech części; AB, BC i CD. Odcinek AB jest prawie równoległy do osi
X i mimo wzrostu napięcia natężenie jest bardzo małe. Prąd ten nazywany jest prądem
szczątkowym, obejmuje on prąd pojemnościowy elektrolitu podstawowego i wszystkie
elektroaktywne zanieczyszczenia obecne w układzie. Ta część krzywej odpowiada
procesowi polaryzacji elektrody rtęciowej. Punkt B odpowiada potencjałowi wydzielania
depolaryzatora czyli w tym przypadku Pb2+
, od tego punktu następuje znaczny wzrost
natężenia prądu, krzywa zagina się. Ten punkt obrazuje początek procesu - normalnej
elektrolizy, jony cynku redukują się na kroplowej elektrodzie rtęciowej i w postaci
amalgamatu rozpuszczają się w rtęci. Ogólnie reakcję zachodzącą na elektrodzie możemy
zapisać następująco:
Mn+
+ ne- M
0(Hg)
Proces dyfuzji
Zredukowany na katodzie metal w postaci amalgamatu jest usuwany z roztworu razem
z odrywającą się kroplą rtęci. W wyniku tego procesu jony ołowiu ubywają z najbliższego
otoczenia elektrody, w rezultacie czego powstaje różnica stężeń jonów ołowiu między
cienką warstwą roztworu przy elektrodzie, a pozostałą częścią roztworu. W wyniku tej
różnicy stężeń następuje dyfuzja jonów ołowiu w kierunku powierzchni elektrody z głębi
roztworu. Płynie prąd zwany prądem dyfuzyjnym, którego wyrazem graficznym jest
stroma część krzywej polarograficznej (BC) zwana falą polarograficzną lub strefą
depolaryzacji elektrody. Dlatego też substancje, które wydzielają się na elektrodzie noszą
nazwę depolaryzatorów. Wolny wzrost napięcia powyżej potencjału wydzielania
powoduje więc wzrost natężenia prądu zgodnie z prawem Ohma, ze względu na to, że
zwiększa się ilość jonów ołowiu redukujących się na KER i zwiększa się w ten sposób
szybkość dyfuzji. Jednak liczba jonów doprowadzonych do powierzchni elektrody
uwarunkowana jest ich dyfuzją z wnętrza roztworu do powierzchni elektrody i nadchodzi
taki moment, że wszystkie jony, które dotrą do elektrody ulegną natychmiast redukcji.
Stężenie jonów ołowiu w warstwie katodowej praktycznie staje się równe zero i różnica
stężeń w warstwie katodowej i reszcie roztworu równa się stężeniu ołowiu w głębi
roztworu. W tych warunkach ustala się stała szybkość dyfuzji, przy czym prąd dyfuzyjny
osiąga wartość maksymalną, na którą nie ma wpływu dalsze zwiększanie napięcia
(odcinek CD). Ta ustalona wartość natężenia prądu nazywa się prądem granicznym.
Fala polarograficzna jest to część krzywej polarograficznej odpowiadająca
przebiegowi określonej reakcji elektrodowej. Potencjał, przy którym natężenie prądu
osiąga wartość równą połowie wartości granicznego prądu dyfuzyjnego nazywa się
potencjałem półfali i oznacza się symbolem E1/2.
Prąd dyfuzyjny – nazwa ta wskazuje, że natężenie prądu zależy wyłącznie od
szybkości dyfuzji depolaryzatora z głębi roztworu do powierzchni elektrody kroplowej.
Ponieważ szybkość ta zależy od stężenia oznaczanej substancji w roztworze, wartość
natężenia prądu dyfuzyjnego płynącego przez roztwór w czasie trwania kropli rtęci można
obliczyć na podstawie równania wyprowadzonego przez Ilkoviča (współpracownik
Heyrowskiego).
Id,l = 607.n
.c
.D
1/2 .m
2/3 .t1/6
gdzie: Id,l – średnie natężenie granicznego prądu dyfuzyjnego
607 – współczynnik obejmuje wielkości stałe w temp.25C

n - liczba elektronów biorących udział w reakcji elektrodowej
c - stężenie substancji oznaczanej w głębi roztworu (mol/l)
D – współczynnik dyfuzji substancji ulegającej redukcji lub utlenieniu
m – wydajność kapilary, czyli masa rtęci wypływająca z kapilary w czasie 1s,
t - czas trwania kropli rtęci
W danych warunkach doświadczalnych wartości - n, D, m i t są wielkościami stałymi
więc równanie można przedstawić w formie uproszczonej
Id,l = k. c
gdzie: k – obejmuje wszystkie współczynniki ustalone w warunkach wykonania analizy.
Ta postać równania wskazuje na prostoliniowy przebieg zależności natężenia prądu
dyfuzyjnego od stężenia elektroaktywnej substancji i jest podstawą polarografii jako
metody analitycznej. Równanie fali polarograficznej (katodowej) zgodnie z równaniem
Nernsta dla roztworów rozcieńczonych przedstawia się następująco:
red
ox
c
c
nEE lg
059,0
Fala anodowa i anodowo-katodowa
Dotychczas była mowa o fali katodowej, która powstaje podczas redukcji zachodzącej
na kroplowej elektrodzie rtęciowej. Przykładem takim może być redukcja jonów Pb2+
,
które zgodnie z równaniem tworząc amalgamat rozpuszczają się w kropli rtęci:
Pb2+
+ 2e- Pb
0(Hg)
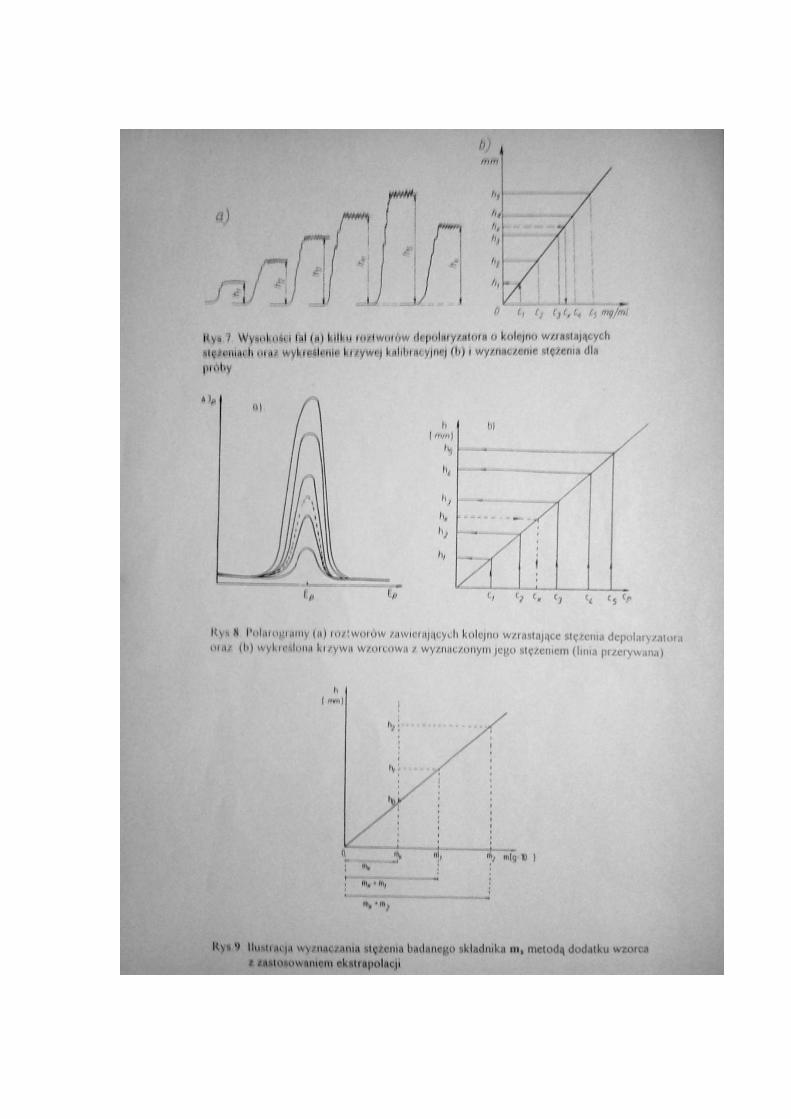
Wykres przebiegu tej reakcji przedstawiony jest na rys.4., jako krzywa . Proces ten można
odwrócić, zastępując czystą rtęć w kroplowej elektrodzie rtęciowej bardzo słabym
amalgamatem ołowiu i wkraplając amalgamat do roztworu elektrolitu pozbawionego
powietrza. Wtedy gdy do elektrod przyłożone jest duże ujemne napięcie (około - 1.0 V),
galwanometr wykazuje tylko przepływ prądu szczątkowego. W miarę stopniowego
przykładania bardziej dodatniego napięcia otrzymuje się położenie, w którym
galwanometr wskazuje szybki wzrost natężenia prądu, ale w przeciwnym kierunku do
natężenia otrzymanego przy redukcji jonów ołowiu na katodzie. Gdy przyłożone ujemne
napięcie dalej się zmniejsza, otrzymuje się natężenie stałe, niezależne od napięcia. Zostaje
więc otrzymany prąd dyfuzyjny, spowodowany rozpuszczeniem ołowiu z amalgamatu:
Pb(Hg) Pb2+
+ 2e-
Otrzymuje się wtedy falę anodową, jak na rys.4., krzywa 2. Jeżeli zaś krople amalgamatu
ołowiu spadają do roztworu elektrolitu zawierającego rozcieńczony roztwór soli ołowiu,
to otrzymuje się falę anodowo-katodową, rys.4., krzywa 3.
Na rys.5. przedstawiono przykład fal polarograficznych dla elektroredukcji dwóch
substancji elektroaktywnych obecnych w tej samej próbie badanej. Przedstawiono też
sposób wyznaczania wysokości fal polarograficznych dla obecnych tam depolaryzatorów.
Niektóre krzywe polarograficzne wykreślone jako fale są trudne do zinterpretowania na

skutek niedostatecznie rozdzielonych fal. Dokładne ustalenie potencjałów półfali jest
jednak możliwe. Na przykład jeżeli zastosuje się rejestrację krzywych polarograficznych
jako pochodne, czyli różniczki podstawowej zależności funkcji
f(E) = di/dE
Przykład takiego wykresu podano na rys.6.
Oznaczenie zawartości jonów Pb(II) w obecności jonów miedzi(II) i kadmu(II)
Oznaczenie kilku substancji obok siebie metodą polarograficzną jest możliwe
wówczas, gdyż potencjały półfali (depolaryzacji) tych pierwiastków różnią się
dostatecznie między sobą. Tylko w takim przypadku dla każdego z badanych składników
mieszaniny otrzymuje się dobrze wykształconą fale lub pik polarograficzny, który nie jest
zakłócony i/lub zniekształcony przez falę sąsiednią. Innym sposobem jest skorzystanie z
wykresu w postaci pochodnej funkcji podstawowej (jak na rys.6.).
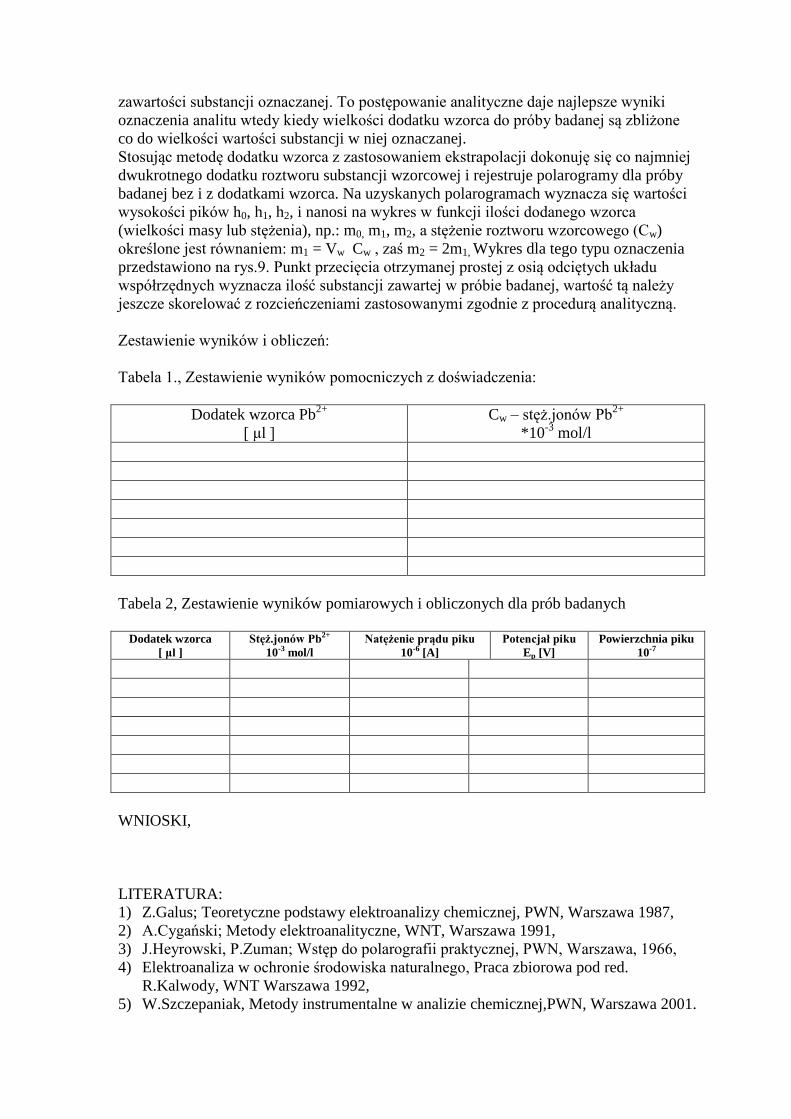
Zasada oznaczenia polega na wykreśleniu krzywych zależności id =f(E) w układzie
KER, Pt i NEK (Nasycona Elektroda Kalomelowa) dla różnych stężeń jonów Pb2+
w
roztworze elektrolitu podstawowego. Wartość prądu id jest liniowo zależna od stężenia
jonów ołowiu w roztworze. Dla próby badanej na zawartość ołowiu wykreśla się w tych
samych warunkach pomiarowych wykres polarograficzny id =f(E) a dla otrzymanej
wartości id tej próby odczytuje się z krzywej kalibracyjnej (rys.7.) wartość stężenia
ołowiu. Ponadto w obliczeniach końcowego jego stężenia należy uwzględnić wszystkie
rozcieńczenia związane z przygotowaniem próby badanej do analizy polarograficznej.
I analogicznie kiedy wykreślamy nie fale a piki polarograficzne (rys.8a.) to dla max
wartości ich prądów i odpowiadających im wartości stężeń analitu (jony Pb2+
) w
roztworze wzorcowym wykreślamy krzywą wzorcową (rys.8b.) z której odczytujemy
wartości stężenia analitu dla próby badanej (na rys.8a i b - linia przerywana) i przeliczamy
odczytaną wartość dla uwzględnienia wszystkich operacji wykonanych na roztworze
badanym podczas przygotowania doświadczalnego zgodnie z procedurą analityczną.
Polarograficzne oznaczenie zawartości jonów Pb2+
w roztworze możemy dokonać
także metodą dodatku wzorca i z użyciem techniki ekstrapolacji. W metodzie tej dokonuje
się rejestracji co najmniej dwóch polarogramów (można też więcej, np.: 4-6). Najpierw
dokonuje się rejestracji polarogramu dla próby badanej (o znanej objętości V1 i
nieznanym stężeniu Cx) dla której odczytuje się wysokość piku h1 = max wartości prądu
tego piku. Następnie do roztworu dodaje się znaną objętość V2 roztworu o znanym
stężeniu C2 oznaczanego składnika – analitu, i wykonuję się drugą rejestrację
polarogramu dla próby badanej z dodatkiem roztworu wzorcowego, i dla niej odczytuje
się wysokość piku h2 = max wartości prądu tego drugiego piku. Stężenie substancji
badanej (jony Pb2+
) w próbie poddanej analizie polarograficznej, określone jako Cx
wyznacza się ze wzoru :
Cx = h1C2
h2−h1 V1V2+h2
Stosując tą metodę można dodać kolejne dodatki wzorca i wówczas jeszcze raz
zarejestrować polarogramy i analogicznie jak poprzednio i wyznaczyć wartość składnika
oznaczanego (wg. wzoru) oraz dokonać obliczenia wyznaczanej doświadczalnie
zawartości substancji oznaczanej. To postępowanie analityczne daje najlepsze wyniki
oznaczenia analitu wtedy kiedy wielkości dodatku wzorca do próby badanej są zbliżone
co do wielkości wartości substancji w niej oznaczanej.
Stosując metodę dodatku wzorca z zastosowaniem ekstrapolacji dokonuję się co najmniej
dwukrotnego dodatku roztworu substancji wzorcowej i rejestruje polarogramy dla próby
badanej bez i z dodatkami wzorca. Na uzyskanych polarogramach wyznacza się wartości
wysokości pików h0, h1, h2, i nanosi na wykres w funkcji ilości dodanego wzorca
(wielkości masy lub stężenia), np.: m0, m1, m2, a stężenie roztworu wzorcowego (Cw)
określone jest równaniem: m1 = Vw Cw , zaś m2 = 2m1, Wykres dla tego typu oznaczenia
przedstawiono na rys.9. Punkt przecięcia otrzymanej prostej z osią odciętych układu
współrzędnych wyznacza ilość substancji zawartej w próbie badanej, wartość tą należy
jeszcze skorelować z rozcieńczeniami zastosowanymi zgodnie z procedurą analityczną.
Zestawienie wyników i obliczeń:
Tabela 1., Zestawienie wyników pomocniczych z doświadczenia:
Dodatek wzorca Pb2+
[ μl ]
Cw – stęż.jonów Pb2+
*10-3
mol/l
Tabela 2, Zestawienie wyników pomiarowych i obliczonych dla prób badanych
Dodatek wzorca
[ μl ]
Stęż.jonów Pb2+
10-3 mol/l
Natężenie prądu piku
10-6 [A]
Potencjał piku
Ep [V]
Powierzchnia piku
10-7
WNIOSKI,
LITERATURA:
1) Z.Galus; Teoretyczne podstawy elektroanalizy chemicznej, PWN, Warszawa 1987,
2) A.Cygański; Metody elektroanalityczne, WNT, Warszawa 1991,
3) J.Heyrowski, P.Zuman; Wstęp do polarografii praktycznej, PWN, Warszawa, 1966,
4) Elektroanaliza w ochronie środowiska naturalnego, Praca zbiorowa pod red.
R.Kalwody, WNT Warszawa 1992,
5) W.Szczepaniak, Metody instrumentalne w analizie chemicznej,PWN, Warszawa 2001.