
Obliczenia metodami DFT niektórych własności i przemian ...juleks.com/download/MGR.pdf ·...
53
Uniwersytet Warszawski Wydzial Chemii Juliusz Stasiewicz Nr albumu: 249054 Obliczenia metodami DFT niektórych własności i przemian fazowych pod wysokim ciśnieniem dla siarczanu srebra(II), AgSO 4 Praca magisterska na kierunku Chemia w zakresie chemia fizyczna Praca wykonana pod kierunkiem dra hab. Wojciecha Grochali Wydział Chemii UW Warszawa, 7 września 2010
Transcript of Obliczenia metodami DFT niektórych własności i przemian ...juleks.com/download/MGR.pdf ·...

Uniwersytet WarszawskiWydział Chemii
Juliusz Stasiewicz
Nr albumu: 249054
Obliczenia metodami DFT niektórych własnościi przemian fazowych pod wysokim ciśnieniem dla
siarczanu srebra(II), AgSO4
Praca magisterskana kierunku Chemia
w zakresie chemia fizyczna
Praca wykonana pod kierunkiemdra hab. Wojciecha Grochali
Wydział Chemii UW
Warszawa, 7 września 2010

Oświadczenie kierującego pracą
Oświadczam, że niniejsza praca została przygotowana pod moim kierunkiem i stwier-dzam, że spełnia ona warunki do przedstawienia jej w postępowaniu o nadanie tytu-łu zawodowego.
Data Podpis kierującego pracą
Oświadczenie autora pracy
Świadom odpowiedzialności prawnej oświadczam, że niniejsza praca dyplomowa zostałanapisana przez mnie samodzielnie i nie zawiera treści uzyskanych w sposób niezgodnyz obowiązującymi przepisami.Oświadczam również, że przedstawiona praca nie była wcześniej przedmiotem procedurzwiązanych z uzyskaniem tytułu zawodowego w wyższej uczelni.Oświadczam ponadto, że niniejsza wersja pracy jest identyczna z załączoną wersją elek-troniczną.
Data Podpis autora pracy

StreszczeniePrzedmiotem niniejszej pracy było przewidzenie niektórych wysokociśnieniowychwłasności AgSO4 na gruncie teorii funkcjonału gęstości (DFT) w ramach LocalDensity Approximation (LDA) i Generalized Gradient Approximation (GGA).
Wyznaczono równanie stanu struktury P1 AgSO4 (w temperaturze bliskiej zerubezwzględnemu) poprzez dopasowanie równania Bircha–Murnaghana do obliczonej
zależności objętości od ciśnienia. Oceniono, że w zakresie 0–20 GPa moduł ściśliwościB0 mieści się w granicach 18–27 GPa. Wyznaczono także, metodą LSDA+U,
zależność przerwy energetycznej AgSO4 od ciśnienia, nie przewidując metalizacjimateriału do 20 GPa. Rozważano także dziewięć wysokociśnieniowych odmian
polimorficznych dla siarczanu srebra(II), z czego osiem staje się termodynamiczniekonkurencyjne wobec fazy eksperymentalnej w miarę kompresji do 20 GPa. Ponadto
wskazano na istotne rozbieżności w wynikach otrzymanych z LDA oraz GGA.
Słowa kluczoweDFT, teoria funkcjonału gęstości, wysokie ciśnienie, AgSO4, Ag(II), przemiany fazowe
Dziedzina pracy (kody wg programu Socrates-Erasmus)13.3 Chemia
Tytuł pracy w języku angielskimDFT calculations of some properties and the pressure-induced phase transitions of
silver(II) sulfate, AgSO4

Spis treści
I Część literaturowa 6
1 Pojęcia chemii kwantowej . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71.1 Notacja . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71.2 Równanie Schrödingera . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
1.2.1 Hamiltonian elektronowy . . . . . . . . . . . . . . . . . . . . . . 81.2.2 Twierdzenie Hellmanna–Feynmana . . . . . . . . . . . . . . . . 9
1.3 Warunki dodatkowe . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 91.3.1 Spin . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 91.3.2 Antysymetria . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101.3.3 Normalizacja . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
2 DFT: Teoria Funkcjonału Gęstości . . . . . . . . . . . . . . . . . . . . . . . . 112.1 Twierdzenia Hohenberga–Kohna . . . . . . . . . . . . . . . . . . . . . . 112.2 Metoda Kohna–Shama . . . . . . . . . . . . . . . . . . . . . . . . . . . 122.3 Funkcjonały wymienno–korelacyjne . . . . . . . . . . . . . . . . . . . . 13
2.3.1 LDA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 132.3.2 GGA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
2.4 Układy otwartopowłokowe . . . . . . . . . . . . . . . . . . . . . . . . . 15
3 Układy periodyczne . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 163.1 Twierdzenie Blocha . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 173.2 Przestrzeń odwrotna . . . . . . . . . . . . . . . . . . . . . . . . . . . . 173.3 Elektronowa struktura pasmowa . . . . . . . . . . . . . . . . . . . . . . 173.4 Układy krystalograficzne i ich symetrie . . . . . . . . . . . . . . . . . . 18
3.4.1 Grupy punktowe i przestrzenne . . . . . . . . . . . . . . . . . . 19
4 Stosowane metody obliczeniowe . . . . . . . . . . . . . . . . . . . . . . . . . 204.1 Warunki brzegowe Borna–von Kármána . . . . . . . . . . . . . . . . . . 204.2 Bazy fal płaskich . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 204.3 Pseudopotencjały i metoda PAW . . . . . . . . . . . . . . . . . . . . . 214.4 Metoda LSDA+U . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
5 Srebro . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 245.1 Informacje ogólne . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 245.2 Siarczan srebra(II) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
4

SPIS TREŚCI 5
II Wyniki własne 28
6 Metodologia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 297 Struktura eksperymentalna pod wysokim ciśnieniem . . . . . . . . . . . . . . 30
7.1 Optymalizacja geometrii komórki elementarnej . . . . . . . . . . . . . . 307.2 Równanie stanu AgSO4 . . . . . . . . . . . . . . . . . . . . . . . . . . . 327.3 Elektronowa struktura pasmowa . . . . . . . . . . . . . . . . . . . . . . 34
7.3.1 Warunki zerowego ciśnienia zewnętrznego . . . . . . . . . . . . . 347.3.2 Obliczenia wysokociśnieniowe . . . . . . . . . . . . . . . . . . . 36
8 Przejścia fazowe AgSO4 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 398.1 Procedura . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 398.2 Potencjalne struktury wysokociśnieniowe . . . . . . . . . . . . . . . . . 408.3 Porównanie entalpii . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
8.3.1 Metoda wspólnej stycznej . . . . . . . . . . . . . . . . . . . . . 458.3.2 Dokładniejsze wyniki . . . . . . . . . . . . . . . . . . . . . . . . 46
9 Podsumowanie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 499.1 Porównanie wyników LDA i GGA . . . . . . . . . . . . . . . . . . . . . 49

Część I
Część literaturowa
6

Rozdział 1
Pojęcia chemii kwantowej
1.1 Notacja
W niniejszej pracy posłużono się następującym zestawem oznaczeń:
δij – delta Kroneckera, równa 1 gdy i = j i 0 w pozostałych przypadkach,∂t – pochodna cząstkowa po wielkości t,
〈a|b〉 – iloczyn skalarny a i b,∫
R3f(r1)dτ1 – całka z funkcji f po trójwymiarowej przestrzeni,
∆ – operator Laplace’a, (∂2x + ∂2y + ∂
2z ),
c – sprzężenie zespolone liczby c,K∗ – przestrzeń dualna (sprzężona algebraicznie) do K,σ – tzw. współrzędna spinowa, przyjmująca wartości ±1
2,
~ – stała równa h2π, gdzie h to stała Plancka,
ε0 – przenikalność elektryczna próżni,a0 – promień Bohra, 4πε0~
2
mee2,
e – ładunek elementarny,me – masa spoczynkowa elektronu,mn – masa spoczynkowa nukleonu (niemal równa dla protonu i neutronu),Ne – liczba elektronów w układzie,Nj – liczba jąder atomowych w układzie,
Wielkości wektorowe oznaczono literami pogrubionymi. Iloczyn skalarny wektorów z R3
przyjęto w następującej postaci:
〈k | r〉 := kxrx + kyry + kzrz. (1.1)
7

Pojęcia chemii kwantowej
Natomiast w przypadku funkcji e, f : D → R i operatora H działającego w przestrzeni
tychże funkcji:
〈e | f〉 :=∫
D
ef, (1.2)
⟨
e∣∣∣H∣∣∣ f⟩
:=∫
D
e(Hf). (1.3)
1.2 Równanie Schrödingera
Jako postulat przyjmowane jest (np. wg [1]), że w problemach niezależnych od
czasu funkcja falowa Ψ, opisująca układ kwantowy, musi spełniać stacjonarne równanie
Schrödingera:
HΨ = EΨ. (1.4)
gdzie operator H, przez analogię do mechaniki klasycznej [2], zwany jest hamiltonianem
układu.
W większości nierelatywistycznych problemów chemii kwantowej operator H ma
postać (1.5), zwaną także hamiltonianem kulombowskim:
H = − ~2
2me
Ne∑
k=1
∆ek −
e2
4πε0
Ne∑
k=1
Nj∑
m=1
Zm|rk −Rm|
+e2
4πε0
Ne∑
k<l
1
|rk − rl|+
−Nj∑
m=1
~2
2mm∆jm +
e2
4πε0
Nj∑
m<n
ZmZn|Rm −Rn|
, (1.5)
gdzie oznaczono:
rk – wektor zwany położeniem k-tego elektronu,Rm – wektor zwany położeniem m-tego jądra,Zm – ładunek m-tego jądra w jednostkach e,mm – masa m-tego jądra,
Operator Laplace’a ∆ek zawiera różniczkowanie po k-tej współrzędnej elektronowej, zaś
∆jm – po m-tej współrzędnej jądrowej, zgodnie z definicją podaną w paragrafie 1.1.1.
1.2.1 Hamiltonian elektronowy
Gdy mn ≫ me, to w przybliżeniu można rozdzielić ruch jąder i elektronów [3].
Prowadzi to do tzw. hamiltonianu elektronowego Hel (ang. clamped nuclei Hamiltonian,
8

Pojęcia chemii kwantowej
wzór (1.6)), w którym położenia jąder są parametrami.
Hel := −~2
2me
Ne∑
k=1
∆ek −
e2
4πε0
Ne∑
k=1
Nj∑
m=1
Zm|rk −Rm|
+e2
4πε0
Ne∑
k<l
1
|rk − rl|+
+e2
4πε0
Nj∑
m<n
ZmZn|Rm −Rn|
. (1.6)
i-tą funkcję własną operatora (1.6) oznaczano odtąd przez Ψiel, a odpowiadającą jej
wartość własną przez Eiel. W szczególności przyjęto, że i = 0 odpowiada stanowi pod-
stawowemu układu (tj. E0el ¬ Eiel).
1.2.2 Twierdzenie Hellmanna–Feynmana
Parametryczna zależność Eiel od zestawu współrzędnych jądrowych Rm nasuwa
interpretację jakoby jądra atomowe były klasycznymi cząstkami, poruszającymi się
w potencjale Eiel. Sensownie jest więc mówić o siłach działających na jądra. Na mocy
twierdzenia Hellmanna–Feynmana [4, 5] siła Fm ≡ (F xm, F ym, F zm)T działająca na m-te
jądro wynosi:
F jm =⟨
Ψiel∣∣∣∂RjmHel
∣∣∣Ψiel
⟩
= ∂RimEiel, j = x, y, z (1.7)
gdzie pochodna cząstkowa operatora jest granicą ciągu operatorów [1]:
∂RjmH ≡ limδRjm→0+
H(Rjm + δRjm)− H(Rjm)δRjm
. (1.8)
Definicja (1.7) będzie potrzebna do oceny zbieżności prowadzonych obliczeń (rozdział 6.).
1.3 Warunki dodatkowe
1.3.1 Spin
Istnieją także dodatkowe warunki nałożone na funkcje falowe, nieujęte w równaniu
Schrödingera, jak na przykład zależność Ψiel od współrzędnych spinowych. W ujęciu
nierelatywistycznym postuluje się bowiem, że elektron posiada wewnętrzny moment
pędu zwany spinem [7]. Eksperymenty (np. [8]) wskazały, że rzut spinu elektronu na
dowolnie wybraną oś (zwaną zwyczajowo z) może przyjmować jedynie dwie wartości: ~
2
i −~
2. Operator z-towej składowej spinu posiada więc dwa ortonormalne stany własne
9

Pojęcia chemii kwantowej
|α〉 i |β〉, będące funkcjami na przestrzeni12,−12
, takie że:
|α〉 : α(1
2
)
= 1, α(
−12
)
= 0 oraz
|β〉 : β(1
2
)
= 0, β(
−12
)
= 1.
(1.9)
Aby przedstawiony obraz był kompletny, funkcję falową należy wzbogacić o nowe stop-
nie swobody: zależność od „współrzędnej spinowej” każdego k-tego elektronu, σk ∈12,−12
.
1.3.2 Antysymetria
Połówkowy spin elektronu niesie daleko idące konsekwencje dla Ne-elektronowej
funkcji falowej [9], które można wyrazić jako warunek antysymetrii:
Ψiel(r1, σ1; . . . ; rk, σk; rl, σl; . . . ; rNe, σNe) = −Ψiel(r1, σ1; . . . ; rl, σl; rk, σk; . . . ; rNe
, σNe).
(1.10)
1.3.3 Normalizacja
Kolejną własnością elektronowej funkcji falowej, nie wynikającą z postaci (1.6), jest
normalizowalność funkcji falowej (całkowalność w kwadracie), którą można zapisać:
∑
σ1
∑
σ2
· · ·∑
σNe
∫
R3Ne
∣∣∣Ψiel(r1, σ1; . . . ; rNe
, σNe)∣∣∣
2dτ1dτ2 . . . dτNe
<∞. (1.11)
Ze względu na interpretację probabilistyczną funkcji falowej [6] przyjmuje się, że (1.11)
wynosi 1.
Przemilczano dotąd problem znajdowania Ψel, czyli rozwiązywania elektronowego
równania Schrödingera w dziedzinie funkcji spełniających (1.10) i (1.11). Następny
rozdział poświęcono jednemu z ujęć tego zagadnienia.
10

Rozdział 2
DFT: Teoria Funkcjonału Gęstości
Teoria Funkcjonału Gęstości (ang. Density Functional Theory, DFT) jest koncepcją
chemii kwantowej opartą na pojęciu gęstości elektronowej. Gęstość (jednoelektrono-
wa) ρi we wszystkich dalszych rozważaniach będzie unormowana do liczby elektronów
w układzie Ne:
ρi(r1) := Ne
∑
σ1
∑
σ2
· · ·∑
σNe
∫
R3
· · ·∫
R3
︸ ︷︷ ︸
Ne−1
∣∣∣Ψiel
∣∣∣
2dτ2dτ3 . . . dτNe
. (2.1)
Wprost z definicji wynikają takie własności ρi jak nieujemność i normalizacja. Oka-
zuje się że prosty obiekt matematyczny i fizyczna obserwabla, jaką jest ρ0, umożliwia
(teoretycznie) pełny opis układu kwantowego.
2.1 Twierdzenia Hohenberga–Kohna
U podstaw DFT leżą dwa twierdzenia sformułowane w 1964 roku [10], od nazwisk
autorów zwane twierdzeniami Hohenberga–Kohna. Pierwsze z nich uzasadnia zastoso-
wanie gęstości elektronowej jako podstawowego narzędzia chemii kwantowej:
Twierdzenie 1. H–K
Gęstość elektronowa niezdegenerowanego stanu podstawowego, ρ0, daje możliwość by
wyznaczyć elektronowy hamiltonian układu, a zatem niesie o nim (teoretycznie) pełną
informację.
Drugie twierdzenie pozwala natomiast na sformułowanie zasady wariacyjnej dla gęstości
elektronowych. Można je wyrazić następująco:
11

DFT: Teoria Funkcjonału Gęstości
Twierdzenie 2. H–K
Istnieje funkcjonał Eel[ρ], który na przestrzeni gęstości elektronowych ρ o ustalonym
Ne przyjmuje wartość minimalną dla ρ0, równą energii stanu podstawowego.
Choć twierdzenie to zapewnia o istnieniu odpowiedniego funkcjonału energii, to nie po-
daje jego dokładnej postaci. Do dziś nie doniesiono o jej odkryciu, choć zaproponowano
wiele użytecznych przybliżeń, które omówione zostaną w najbliższych podrozdziałach.
2.2 Metoda Kohna–Shama
Kohn i Sham w pracy [11] zaproponowali praktyczny algorytm znajdowania ρ0
w oparciu o DFT. Rozważyli oni układ Ne nieoddziałujących wzajemnie elektronów,
znajdujący się w takim potencjale V0, który prowadzi do stanu podstawowego o gęsto-
ści elektronowej identycznej z ρ0. W takim przypadku równanie Schrödingera można
rozdzielić na Ne niezależnych równań jednoelektronowych:(
− ~2
2me
∆+ V0
)
φi = ǫiφi, (2.2)
gdzie ǫi to energia własna unormowanego spinorbitalu φi. Rozwiązawszy równania (2.2)
można obliczyć energię kinetyczną T0 fikcyjnego układu:
T0 = −~2
2me
Ne∑
i=1
〈φi |∆|φi〉 . (2.3)
Korzystając z uzyskanego wyniku, funkcjonał energii można zapisać jako:
Eel[ρ] = T0−e2
4πε0
Nj∑
m=1
∫
R3
ρ(r)Zm|r−Rm|
dτ+e2
8πε0
∫
R3
∫
R3
ρ(r1)ρ(r2)
|r1 − r2|dτ1dτ2+Ejj+Exc[ρ]. (2.4)
Wyraz Ejj to energia oddziaływań jądro–jądro, stała z obecnego punktu widzenia. Na-
tomiast człon Exc[ρ] zwany jest funkcjonałem wymienno–korelacyjnym i z definicji za-
wiera wszystkie poprawki do energii całkowitej nieujęte w pozostałych wyrazach wzoru
(2.4).
Oczywiście, podany algorytm byłby bezwartościowy, gdyby nie towarzyszył mu
przepis na potencjał V0. Można wykazać, że V0 o pożądanych własnościach dane jest
wzorem:
V0(r) = −e2
4πε0
Nj∑
m=1
Zm|r−Rm|
+e2
4πε0
∫
R3
ρ(r1)
|r− r1|dτ1 +
δExc
δρ, (2.5)
12

DFT: Teoria Funkcjonału Gęstości
gdzie pochodna funkcjonalna δExc/δρ jest funkcją (ogólniej: dystrybucją), taką by dla
dowolnej funkcji próbnej f : R3 → R zachodziło:
⟨
δExc
δρ| f⟩
= limǫ→0+
ddǫExc[ρ+ ǫf ]. (2.6)
W praktyce obliczeniowej zakłada się pewną początkową gęstość elektronową ρ
i korzystając z niej wyznacza potencjał V0 (wzór (2.5)). Dalej rozwiązuje się jedno-
elektronowe równania (2.2). Z otrzymanych spinorbitali φi wyznacza się poprawioną
gęstość:
ρ(r) :=Ne∑
i=1
∑
σ
|φi(r, σ)|2 , (2.7)
z niej zaś poprawiony potencjał V0 itd. do uzyskania zadowalającej zbieżności.
2.3 Funkcjonały wymienno–korelacyjne
Spośród wielu znanych przybliżeń dla Exc[ρ] w niniejszej pracy stosowano dwa:
LDA (ang. Local Density Approximation) oraz GGA (ang. Generalized Gradient Ap-
proximation), dobrze ugruntowane teoretycznie i sugerowane już w pierwszych pracach
dotyczących DFT ([11, 10]).
2.3.1 LDA
W ramach LDA zakłada się, że energia wymienno–korelacyjna w obrębie niewielkiej
objętości dτ jest równa energii wymienno–korelacyjnej jednorodnego gazu elektrono-
wego o identycznej gęstości. Zwykle zapisuje się:
ELDAxc [ρ] =
∫
R3
ǫLDAxc (ρ(r))ρ(r)dτ ≡
∫
R3
ǫLDAx (ρ(r))ρ(r)dτ +
∫
R3
ǫLDAc (ρ(r))ρ(r)dτ. (2.8)
Nieznaną funkcję ǫxc często zapisuje się jako sumę członu wymiennego ǫx oraz kore-
lacyjnego ǫc, bowiem dla jednorodnego gazu fermionowego znana jest ścisła wartość
ǫx [12]:
ǫLDAx (ρ(r)) = −3
4
3
√
3ρ(r)
π. (2.9)
13

DFT: Teoria Funkcjonału Gęstości
Poprawka korelacyjna w niniejszej pracy obliczana była według metody VWN (Vosko–
Wilk–Nusair, [13]):
ǫLDAc (ρ(r)) = A
ln
(
y2
y2 + by + c
)
+2b
Qarctan
(
Q
2y + b
)
+ (2.10)
− by0y20 + by0 + c
[
ln
(
(y − y0)2y2 + by + c
)
+2(b+ 2y0)
Qarctan
(
Q
2y + b
)]
,
gdzie: y := 6
√
3/(4πρ(r)), natomiast A, Q, y0, b i c to pewne stałe, dopasowane do
wyników precyzyjnych symulacji Monte Carlo [14].
2.3.2 GGA
Funkcjonał wymienno–korelacyjny (2.8) można uogólnić, rozszerzając ǫxc o zależ-
ność od gradientu ρ:
EGGAxc [ρ] =
∫
R3
ǫGGAxc (ρ(r),∇ρ(r))ρ(r)dτ. (2.11)
W niniejszej pracy korzystano z funkcjonału GGA w wersji PBE (Perdew–Burke–
Ernzerhof, [15]), która w przypadku bez polaryzacji spinowej ma ogólną postać (2.12):
ǫGGAxc (ρ(r)) = ǫ
LDAc (ρ(r))+A ln
[
1 +Bt2(
1 + Ct2
1 + Ct2 + C2t4
)]
+ǫLDAx
(
1 + F − F
1 +Gs2
)
,
(2.12)
gdzie C jest funkcją ǫLDAc (ρ(r)):
C :=B
exp(
− ǫLDAc (ρ(r))A
)
− 1, (2.13)
zaś t i s są tzw. bezwymiarowymi gradientami gęstości:
t(ρ(r)) :=|∇ρ(r)|√πa04ρ(r) 6
√
3π2ρ(r), (2.14)
s(ρ(r)) :=|∇ρ(r)|
2ρ(r) 3√
3π2ρ(r). (2.15)
Pozostałe symbole w (2.12), tj. A, B, F i G to stałe, których wartości można wyznaczyć
numerycznie.
14

DFT: Teoria Funkcjonału Gęstości
2.4 Układy otwartopowłokowe
W niektórych przypadkach wkład do gęstości ρ od elektronów o σ = 12
i σ = −12
nie jest (choćby lokalnie) jednakowy. Wówczas należy uogólnić dotychczasowe rozwa-
żania poprzez wprowadzenie dwóch gęstości ρα i ρβ, opisujących elektrony o przeciwnie
skierowanych spinach. Oczywiście zachodzi:
∀r∈R3 ρ(r) = ρα(r) + ρβ(r). (2.16)
Rozważa się także nową wersję funkcjonału Exc[ρα, ρβ], zależną od obu gęstości spi-
nowych. W szczególności istnieją „otwartopowłokowe” odpowiedniki ǫLDAxc oraz ǫGGA
xc .
Pierwszy z nich, stosowany przy przygotowaniu tej pracy, występuje w literaturze pod
nazwą LSDA lub LSD (ang. Local Spin Density Approximation). Dokładną postać ma-
tematyczną tych przybliżeń znaleźć można w [13].
15

Rozdział 3
Układy periodyczne
W teorii ciała stałego przyjmuje się powszechnie, że rozmiar rozważanego kryszta-
łu jest nieskończony we wszystkich trzech kierunkach przestrzennych, co nadaje sieci
krystalicznej symetrię translacyjną. Istnieją więc takie wektory e1, e2, e3 ∈ R3, że dla
dowolnego r ∈ R3 układ w punkcie:
r′ := r+ n1e1 + n2e2 + n3e3, gdzie: n1, n2, n3 ∈ Z
3 (3.1)
jest identyczny jak w punkcie r. Istnieje nieskończenie wiele możliwości wyboru e1, e2, e3by spełniały powyższy warunek i stanowiły bazę w R
3. W szczególności można zażądać,
by objętość rozpinanego przez nie równoległościanu była minimalna – taki zestaw wek-
torów nazywa się wektorami prymitywnymi. Kombinacje ich całkowitych wielokrotno-
ści wskazują sieć punktów zwaną siecią Bravais. Sieć ta, wraz z otoczeniem atomowym
każdego węzła (bazą atomową) tworzy strukturę krystaliczną.
Równoległościan rozpięty przez wektory prymitywne nazywany jest komórką prymi-
tywną; komórka taka, powielona wektorami prymitywnymi, wypełnia ściśle całą prze-
strzeń. Innym przykładem komórki prymitywnej jest komórka Wignera–Seitza, czyli
zbiór punktów leżących bliżej danego węzła sieci niż któregokolwiek innego. Komórki
Wignera–Seitza można znaleźć za pomocą operacji tesselacji Voronoi sieci Bravais [17].
W praktyce jednak dużo częściej używa się pojęcia komórki elementarnej, czyli ta-
kiego elementu sieci krystalicznej, który poddany operacjom translacji wypełnia całą
przestrzeń, a ponadto posiada możliwie wysoką symetrię. Zagadnienia symetrii zostały
krótko omówione w jednym z następnych podrozdziałów.
16

Układy periodyczne
3.1 Twierdzenie Blocha
Związek (3.1) pociąga za sobą niezmienniczość translacyjną potencjału, w którym
poruszają się elektrony:
V0(r) = V0(r+ n1e1 + n2e2 + n3e3), gdzie: n1, n2, n3 ∈ Z3. (3.2)
Z niezmienniczości translacyjnej potencjału wynika translacyjna niezmienniczość jed-
noelektronowego hamiltonianiu. Wynika stąd, że jego funkcje własne φi spełniają za-
leżność [18]:
∀R=∑
i
nieiφi(r+R, σ) = exp (i 〈k|R〉)φi(r, σ) (3.3)
Kowektor k stanowi element przestrzeni (R3)∗ i jest charakterystyczny dla danej funkcji
falowej φi (oznaczanej odtąd jako φik). Można więc powiedzieć, że jednoelektronowe
funkcje falowe w krysztale są zmodulowane falami płaskimi o wektorze falowym k.
3.2 Przestrzeń odwrotna
Spostrzeżenie wyrażone wzorem (3.3) daje podstawy by sformułować pojęcie prze-
strzeni odwrotnej, czyli przestrzeni wektorów k. Stanowi ona sprzężenie algebraiczne
przestrzeni rzeczywistej, rozpiętej przez wektory sieciowe. Choć obie przestrzenie są
wzajemnie izomorficzne, to zwyczajowo jako bazę przestrzeni odwrotnej przyjmuje się
tzw. bazę biortogonalną do e1, e2, e3, czyli bazę f1, f2, f3 taką, że:
〈fi|ej〉 = 2πδij. (3.4)
Komórkę Wignera–Seitza w sieci odwrotnej nazywa się pierwszą strefą Brillouina (PSB).
Można wykazać, że wektory należące do PSB wystarczają do opisu własności trans-
lacyjnych wszystkich funkcji falowych φik. Ponadto, każdy wektor spoza tej strefy
posiada swój odpowiednik wewnątrz PSB, nadający funkcjom falowym identyczne wła-
sności translacyjne [1].
3.3 Elektronowa struktura pasmowa
Do określania funkcji elektronowych w krysztale stosuje się wskaźnik k zamiast ze-
stawu liczb kwantowych n, l,m, używanych np. w fizyce atomowej. Ponieważ (w krysz-
17

Układy periodyczne
tale nieskończonym) k może zmieniać się w sposób ciągły, zależność energii i-tego
spinorbitalu od wektora falowego, ǫik, nazywa się zwyczajowo pasmem energetycznym.
Przerwę energetyczną Eg definiuje się jako różnicę energii między najniższym nie-
obsadzonym (lowest unoccupied, ǫLUCOk1
), a najwyższym obsadzonym (highest occupied,
ǫHOCOk2
) orbitalem krystalicznym (crystal orbital) w stanie podstawowym:
Eg := mink1,k2
(
ǫLUCOk1
− ǫHOCOk2
)
. (3.5)
Jeśli obu energiom odpowiadają równe wektory falowe k1 = k2 to mówi się o przerwie
energetycznej prostej, w przeciwieństwie do przerwy energetycznej skośnej. Wartość
Eg decyduje o przynależności związku do metali (Eg = 0), bądź półprzewodników
i izolatorów (Eg > 0).
Kolejną definicją jest poziom Fermiego ǫF:
ǫF :=1
2
(
ǫLUCOk1
+ ǫHOCOk2
)
. (3.6)
3.4 Układy krystalograficzne i ich symetrie
Definiuje się sześć (lub siedem, jeśli wyróżnić układ trygonalny) różnych układów
[19], opisujących kształty komórek elementarnych. Wyróżnia się:
• regularny (α = β = γ = 90, a = b = c),
• tetragonalny (α = β = γ = 90, a = b 6= c),
• heksagonalny (α = β = 90, γ = 120, a = b 6= c),
• rombowy (α = β = γ = 90, a 6= b 6= c),
• jednoskośny (α = γ = 90 6= β, a 6= b 6= c),
• trójskośny (α 6= β 6= γ; α, β, γ 6= 90; a 6= b 6= c),
gdzie a, b, c to stałe sieciowe, zaś α, β, γ – kąty między parami krawędzi (kolejno bc,
ac i ab) w komórce elementarnej. By określić rozmieszczenie węzłów w sieci Bravais
należy podać także sposób centrowania (prymitywne, na podstawach, na ścianach lub
przestrzenne). Łącznie występuje 14 różnych typów sieci Bravais.
18

Układy periodyczne
3.4.1 Grupy punktowe i przestrzenne
Obok translacji istnieje szereg innych operacji przekształcających sieć krystaliczną
w samą siebie. Wyróżnia się następujące typy przekształceń:
• obrót wokół osi o kąt 2πn
(tzw. n-krotna oś obrotu),
• odbicie w płaszczyźnie,
• inwersja (zamiana r, mierzonego względem pewnego punktu, na −r),
• obrót inwersyjny (złożenie obrotu wokół osi i inwersji)
• oś śrubowa (złożenie translacji z obrotem wokół kierunku translacji),
• płaszczyzna poślizgu (złożenie translacji z odbiciem w płaszczyźnie).
Daje to podstawy, by zdefiniować 230 grup symetrii przestrzennej. Szczegółowe zasady
ich tworzenia i oznaczania można znaleźć w [19].
19

Rozdział 4
Stosowane metody obliczeniowe
4.1 Warunki brzegowe Borna–von Kármána
Do celów obliczeniowych wygodnie jest rozważać kryształ złożony z Lj komórek pry-
mitywnych w każdym j-tym kierunku, w którym dodatkowo utożsamiono przeciwległe
ściany, tj.:
φik(r, σ) = φik(r+ Ljej, σ) gdzie: j = 1, 2, 3. (4.1)
Warunek (4.1) nakłada ograniczenia na rozkład punków w przestrzeni odwrotnej. Przy
przyjęciu warunków brzegowych Borna–von Kármána dozwolone są tylko takie k, które
spełniają:
Lj 〈k|ej〉 = 2πnj gdzie: nj ∈ Z oraz: j = 1, 2, 3. (4.2)
4.2 Bazy fal płaskich
Dogodną bazą do opisu jednoelektronowych funkcji falowych w układach perio-
dycznych jest zbiór fal płaskich. Mając na uwadze (3.3), dany spinorbital φik można
przybliżyć przez φik postaci:
φik(r, σ) =∑
~2
2(K+k)2¬Ecut
ck,K(σ)ei〈k+K|r〉. (4.3)
Górne ograniczenie liczb falowych K zwykło podawać się jako tzw. energię odcięcia
Ecut (ang. cut-off energy).
Za stosowaniem fal płaskich przemawia kilka argumentów. Korzystny jest fakt, że
tworzą one zbiór ortogonalny:⟨
ei〈k1+K1|r〉 | ei〈k2+K2|r〉⟩
= 0 ⇔ (k1 +K1 6= k2 +K2). (4.4)
20

Stosowane metody obliczeniowe
Ponadto istnieją bardzo wydajne procedury rozkładu danej funkcji φk na fale płaskie
(FFT, ang. Fast Fourier Transform). Wiele całek zawierających fale płaskie ma także
znaną postać analityczną.
4.3 Pseudopotencjały i metoda PAW
Rozwiązanie równań Kohna–Shama (2.2) przy podanej postaci potencjału V0 (2.5)
wywołuje trudności dwojakiego rodzaju. Po pierwsze, funkcje falowe w pobliżu jąder
wykazują szybkie oscylacje, toteż do ich opisu potrzebne byłyby fale płaskie o bardzo
dużych wartościach |k|. Po drugie, większość czasu zajmowałoby symulowanie elektro-
nów związanych blisko jąder, mimo że z chemicznego punktu widzenia są one nieinte-
resujące.
Trudności te stały się motywacją do wynalezienia pseudopotencjałów. Pseudopoten-
cjały to abstrakcyjne pola, które – poglądowo mówiąc – opisują energię oddziaływania
elektronów walencyjnych nie tylko z jądrem atomowym, ale też z elektronami rdzenia.
Istnieje kilka strategii takiego opisu, między innymi można wyróżnić:
• pseudopotencjały zachowujące normę (ang norm-conserving pseudopotentials).
Rozwiązania z ich wykorzystaniem spełniają z definicji cztery warunki:
1. rzeczywista funkcja falowa oraz „pseudofunkcja” falowa muszą być tożsamo-
ściowo równe od pewnej odległości rc od jądra atomowego.
2. Energia własna pseudofunkcji w „pseudohamiltonianie” jest równa energii
jej rzeczywistej odpowiedniczki.
3. Warunkiem zszycia na brzegu kuli (tj. w rc) jest równość pochodnych loga-
rytmicznych funkcji i pseudofunkcji.
4. Pseudofunkcja falowa musi prawidłowo odtwarzać ładunek rdzenia atomo-
wego (zachowanie normy).
Okazuje się jednak, że podejście takie nie upraszcza obliczeń dla atomów o kon-
figuracji powłoki walencyjnej 2p i 3d [20], co wynika z faktu, że orbitale te są
niemal identyczne w ujęciu „pseudo” jak w ujęciu tradycyjnym.
• Pseudopotencjały Vanderbilta. Vanderbilt spostrzegł [21], że zrezygnowanie z wa-
runku zachowania normy pozwala uzyskać lepsze wyniki dla wspomnianych ato-
21

Stosowane metody obliczeniowe
mów. Tak zdefiniowane pseudopotencjały nazwał „ultramiękkimi” (ang. ultrasoft).
• Metoda PAW (ang. projector augumented wave). Choć formalnie wyprowadzona
[22] z metody LAPW (ang. linearized augmented-plane-wave), okazała się nie-
mal tożsama matematycznie z podejściem Vanderbilta [23]. W ramach metody
tej zakłada się, że w obszarze kuli o promieniu rcut wokół m–tego jądra każ-
da prawdziwa funkcja falowa φmi zadana jest liniowym przekształceniem pewnej
wolnozmiennej funkcji pomocniczej φmi :
φmi = (1 + Tm)φmi . (4.5)
Operator Tm poza kulą, tj. w obszarze r : |r−Rm| > rcut jest tożsamościowo
równy zeru. Dodatkowym warunkiem stawianym Tm jest by funkcje φmi i φmigładko łączyły się na brzegu kuli.
Przy takim podejściu rozważane są wszystkie elektrony. Niewielkim modyfikacjom
ulega jednak równanie na energię całkowitą w DFT (2.4), a także na gęstość
elektronową (2.7), co zostało szerzej omówione w [24]. Należy także dodać, że
odpowiednio modyfikując funkcje falowe w pobliżu jąder można uwzględnić część
efektów relatywistycznych, mimo użycia nierelatywistycznego hamiltonianiu.
Każda z wyżej wymienionych strategii ma wiele sposobów realizacji. W niniejszej
pracy korzystano z ostatniej z nich, czyli metody PAW, w wersji dostarczonej z pro-
gramem VASP 4.6.
4.4 Metoda LSDA+U
Obliczenia L(S)DA okazują się mało precyzyjne, gdy dotyczą układów o silnej ko-
relacji elektronowej. Najbardziej spektakularnym przykładem ich niedoskonałości są
izolatory Mott’a, takie jak tlenek kobaltu(II) czy tlenek żelaza(II). W ramach LSDA
obydwa związki powinny być metalami, choć w rzeczywistości są izolatorami. Okazuje
się jednak, że dokładność takich obliczeń można znacznie zwiększyć poprzez wzboga-
cenie hamiltonianu elektronowego o człon opisujący oddziaływania Hubbarda:
HHub =Nm∑
j=1
∑
X=s,p,d
UX,jnX,j,αnX,j,β − J∑
<ij>,σ
(a†i,σaj,σ + a†j,σai,σ), (4.6)
22

Stosowane metody obliczeniowe
gdzie nX,j,α to operator liczby elektronów typu X o spinie α w otoczeniu j–tego jądra.
Natomiast a†i,σ i ai,σ to odpowiednio operatory kreacji i anihilacji elektronu o współrzęd-
nej spinowej σ w otoczeniu i-tego atomu. Zapis 〈ij〉 oznacza parę sąsiadujących ato-
mów. Parametr U można interpretować jako energię związaną z umieszczeniem dwóch
elektronów typu X w otoczeniu jednego atomu:
UX := E(Xn+1) + E(Xn−1)− 2E(Xn). (4.7)
Uzupełnienie hamiltonianiu o człon Hubbard, zastosowane do przybliżenia LSDA, wy-
stępuje w literaturze pod nazwą LSDA+U. Metoda ta daje znakomite wyniki w prze-
widywaniu własności elektrycznych i magnetycznych różnych materiałów [25]. Niestety
odbywa się to przy znacznym zwiększeniu kosztochłonności obliczeń, toteż LSDA+U
stosowane jest przeważnie do obliczeń typu single-point, rzadziej do optymalizacji.
23

Rozdział 5
Srebro
5.1 Informacje ogólne
Srebro jest srebrzystoszarym metalem przejściowym, leży w 11. grupie układu okre-
sowego pierwiastków. Jego liczba atomowa wynosi 47, zaś średnia masa atomowa
107.8682 u. Konfiguracja elektronowa atomu Ag to [Kr] 4d105s1. Elektroujemność w ska-
li Paulinga jest równa 1.93. Znane są związki srebra na trzech stopniach utlenienia: +1,
+2 oraz +3. Istnieją także obliczenia wskazujące na stabilność srebra(V) (w anionie
Ag[F6]−) [27].
Czyste srebro w warunkach normalnych jest ciałem stałym, o temperaturze top-
nienia 1234.93 K i temperaturze wrzenia 2435 K. Jego gęstość wynosi 10490 kg/m3.
Wykazuje właściwości diamagnetyczne. W przyrodzie występują dwa stabilne izotopy:107Ag (51.84%) i 109Ag (48.16%), w większości w postaci związków (tworzy minerały
takie jak argentyt, prustyt czy pirargyryt), choć także w stanie metalicznym.
Srebro nie reaguje z powietrzem wolnym od zanieczyszczeń, jednak matowieje pod
wpływem ozonu i związków siarki. Halogenki srebra rozkładają się na świetle. Tlenki
wykazują właściwości amfoteryczne. W szeregu napięciowym srebro znajduje się za
wodorem (EAg+/Ag0 = 0.80 V względem normalnej elektrody wodorowej).
Srebro stosuje się głównie w jubilerstwie, a także do wyrobu instrumentów muzycz-
nych i luster. W medycynie związki srebra(I) używane są jako środki dezynfekujące
ze względu na toksyczność dla grzybów, bakterii i wirusów. Halogenki i azotan(V)
srebra(I) wykorzystywane są w fotografii.
24

Srebro
W układach elektronicznych znalazło ono zastosowanie jako doskonały przewodnik
elektryczności (przewodność właściwa 6.3 · 105cm−1Ω−1). W chemii stosuje się je ja-
ko katalizator w niektórych reakcjach utleniania.
5.2 Siarczan srebra(II)
Znanych jest około 100 związków srebra(II), z czego niemal wszystkie zawierają
aniony fluorkowe lub aniony pochodne silnych kwasów Lewisa, takie jak BF−4 , SbF−6 ,
czy SO3F−. W warunkach normalnych są to przeważnie paramagnetyki. Jedynym ko-
mercyjnie dostępnym związkiem Ag2+ jest brązowy, światłoczuły AgF2.
Możliwość istnienia „bezfluorowych” wyjątków w tej grupie była z początku przed-
miotem badań teoretycznych metodami DFT. Wskazano m.in. na możliwość istnie-
nia nadchloranu, azotanu, metafosforanu oraz pewnych perfluorowanych organicznych
związków srebra(II) [29]. Wykazano także, że w fazie gazowej anion siarczanowy mo-
że ulec utlenieniu przez Ag2+ do postaci rodnikoanionu [29]. Późniejsze eksperymenty
udowodniły, że aniony siarczanowe, podobnie jak chromianowe, azotanowe, nadchlora-
nowe i nadmanganianowe, w fazie stałej, są względnie odporne na utleniające własności
srebra(II) [30].
Siarczan(VI) srebra(II) w fazie stałej także był przedmiotem badań kwantowoche-
micznych. Przy użyciu metod DFT próbowano stwierdzić stabilność kryształu AgSO4i przewidzieć jego strukturę [31, 32]. Jak się później okazało, prawidłowo przewidziano
płaską, niemal kwadratową sferę koordynacyjną srebra, a także przybliżone odległości
Ag–O. Oprócz tego poprawnie przewidziano tetraedryczną budowę jonu siarczanowego,
bez terminalnych atomów tlenu, a zatem bez krótkich wiązań podwójnych S=O. Pro-
duktem ubocznym tych poszukiwań było kilkanaście innych, metastabilnych struktur,
które posłużyły w dalszej części niniejszej pracy jako potencjalne formy wysokociśnie-
niowe.
Pierwsze wiarygodne doniesienie o udanej syntezie AgSO4 pojawiło się w styczniu
2010 r. [33]. Otrzymano go w reakcji:
AgF2 + H2SO4 −→ AgSO4 (↓) + 2 HF (↑).
Autorzy stwierdzili, że jest to czarna krystaliczna substancja o własnościach antyfer-
romagnetycznych. Przejście fazowe antyferromagnetyk–paramagnetyk nie następuje aż
25

Srebro
do rozkładu związku z wydzieleniem tlenu (ok. 400 K). Przerwę energetyczną osza-
cowano wówczas na 0.18 eV na podstawie elektronowego widma absorpcji w bliskiej
i średniej podczerwieni. Późniejsze pomiary widma impedancji [34] wskazały jednak na
znacznie większą wartość 1.12 eV (w temperaturze -50C) i 1.24 eV (w temp. +33.5C).
Liniowa ekstrapolacja tych wyników prowadzi do wartości 0.80 eV w 0 K. Należy tu
zaznaczyć, że pomiary dyfrakcji neutronów [28] nie wykazały przejścia fazowego AgSO4przy chłodzeniu do temperatury 2 K.
Pomiary XRPD (rentgenowskiej dyfrakcji proszkowej) wykazały że siarczan sre-
bra(II) krystalizuje w układzie trójskośnym, o symetrii P1. Na komórkę elementarną
przypadają dwie jednostki stechiometryczne AgSO4. W Tab. 5.1 zestawiono wybrane
parametry tej struktury:
a, b, c [Å] 8.0125(2) 4.7535(1) 4.6923(1)α, β, γ [] 76.478(1) 118.078(1) 103.403(1)Z 2ρ [g cm−3]∗ 4.46
Tabela 5.1: Parametry struktury krystalicznej AgSO4 pod ciśnieniem atmosferycznym(na podstawie [33]). *Wartość obliczona z parametrów sieci.
Rysunek 5.1: Komórka elementarna AgSO4 o symetrii P1 (Z = 2)na podstawie pomiarów XRPD (za [33]). Kolorami wyróżniono atomy różnych pierwiastków,
odpowiednio: Ag, S, O.
W strukturze tej wyróżnić można dwa nierównocenne typy atomów srebra, połączo-
ne poprzez ciąg wiązań -O–S–O- w trójwymiarową sieć. Sfery koordynacyjne obu jonów
26

Srebro
Ag2+ przypominają kwadraty. Mimo iż atomy srebra są nierównocenne, to różnice w ich
koordynacji są raczej niewielkie, co – wsparte pomiarami podatności magnetycznej –
pozwala przypuszczać, że otrzymano rzeczywiście siarczan srebra(II), nie zaś związek
o mieszanej wartościowości (I + III).
Warto wspomnieć, że jest to struktura jakościowo różna od struktury CuSO4, w któ-
rej kationy miedzi połączone są mostkami tlenkowymi Cu–O–Cu, a także od AuSO4,
w której zaobserwować można wiązanie Au–Au.
Obliczenia własności magnetycznych metodą GGA+U prowadzą do rozwiązań ferri-
magnetycznych (o momentach -0.39 µB i +0.44 µB na atomach srebra [33]). Rozwiązanie
ferromagnetyczne leży wyżej na skali energii o 3.1 meV na „cząsteczkę” AgSO4. Ponadto
obliczenia te wskazują, że oddziaływania magnetyczne przenoszone są nie przez „most-
ki” -O–S–O-, ale w głównej mierze bezpośrednio między atomami tlenu -O· · ·O-, na co
wskazuje mała gęstość spinowa na atomach siarki.
27

Część II
Wyniki własne
28

Rozdział 6
Metodologia
Do obliczeń wykorzystano program VASP (Vienna Ab-initio Simulation Package,
[35]) w wersji 4.6. Program ten realizuje metody DFT, ogólnie rzecz biorąc, w sposób
opisany w części literaturowej. Przy jego użyciu można wyznaczyć m.in. strukturę pa-
smową, gęstość stanów i rozkład ładunku dla zadanej struktury krystalicznej pod usta-
lonym ciśnieniem. Ponadto pozwala on na optymalizację struktur, także pod zewnętrz-
nym ciśnieniem. Rozszerzeniem programu VASP jest Phonon, pozwalający wyznaczać
widmo dyspersji fononów i funkcje termodynamiczne [36]. Manipulacji strukturami oraz
wizualizacji dokonywano przy użyciu programów Materials Studio i MedeA. Obliczenia
wykonywano w centrum Komputerów Dużej Mocy (KDM) Interdyscyplinarnego Cen-
trum Modelowania Matematycznego i Komputerowego Uniwersytetu Warszawskiego
(ICM UW).
Algorytm Kohna–Shama iterowano dopóki kolejne poprawki do energii były większe
niż 10−6 eV/atom. Optymalizację geometrii prowadzono tak długo, jak siły działające
na jony przekraczały 5 · 10−4 eV/Å/atom. Obliczenia prowadzono przy Ecut = 600 eV,
na siatce w przestrzeni odwrotnej o gęstości 0.5 Å−1.
Obliczenia struktury pasmowej prowadzono metodą LSDA+U, przy nieco ostrzej-
szym kryterium zbieżności dla procedury Kohna–Shama (10−7 eV/atom). Zgodnie
z [37] przyjęto następujące współczynniki: U(Ag4d)=4 eV oraz, dla wszystkich ato-
mów, J=1 eV. Atomom tlenu przypisano typową wartość U(O2p)=4 eV [38], natomiast
atomom siarki, która jest cięższym tlenowcem, U(S3p)=2 eV. Pozostałe parametry były
identyczne jak przy optymalizacji geometrii.
29

Rozdział 7
Struktura eksperymentalna pod
wysokim ciśnieniem
Jednym z celów, dla którego dokonano syntezy AgSO4, było poszukiwanie nadprze-
wodników wśród związków srebra(II). Choć pomiary podatności magnetycznej AgSO4(pod ciśnieniem atmosferycznym) [33] nie wykazały obecności efektu Meissnera [43],
to istnieje szansa, że przejście fazowe do stanu nadprzewodzącego uda się osiągnąć po-
przez zwiększenie ciśnienia. Znanych jest wiele materiałów, w których udało się tego
dokonać; jako przykłady można wymienić O, S, Se, Te [39]; CsI [40], czy CeRhIn5 [41].
Stanowi to także istotną motywację dla niniejszej pracy.
7.1 Optymalizacja geometrii komórki elementarnej
Strukturę eksperymentalną (Rys. 5.1) o parametrach z Tab. 5.1 poddano optyma-
lizacji geometrii w ramach LDA oraz GGA pod zewnętrznym, izotropowym ciśnieniem
z zakresu 0 – 20 GPa. Na Rys. 7.1 i 7.2 przedstawiono stałe sieci oraz kąty między nimi
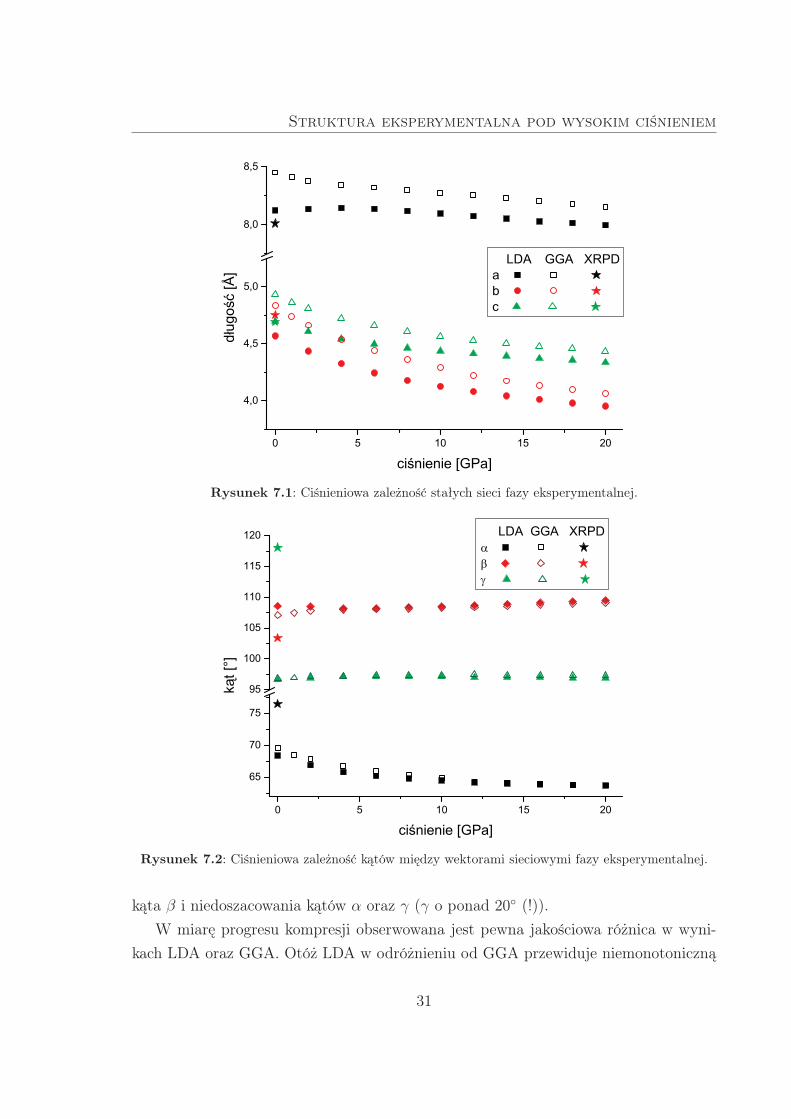
w funkcji ciśnienia.
Niepokój budzą rozbieżności między wynikami obliczeń i danymi eksperymentalny-
mi, obserwowane już przy nieobecności zewnętrznego ciśnienia. Różnice te zestawiono
w Tab. 7.1. Wszystkie stałe sieciowe obliczone w ramach GGA są przeszacowane, z błę-
dami na poziomie 2–5%, co prowadzi do znacznego przeszacowania objętości komórki
elementarnej (o ok. +18%). Natomiast obliczenia w LDA przeszacowują stałą siecio-
wą a i niedoszacowują b, tak że poprawnie odtwarzają objętość komórki elementarnej
(z błędem zaledwie +0.5%). Obydwa przybliżenia zgodnie prowadzą do przeszacowania
30
Struktura eksperymentalna pod wysokim ciśnieniem
0 5 10 15 20
4,0
4,5
5,0
8,0
8,5
LDA GGA XRPDa b c
dug
o [Å
]
ci nienie [GPa]
Rysunek 7.1: Ciśnieniowa zależność stałych sieci fazy eksperymentalnej.
0 5 10 15 20
65
70
75
95
100
105
110
115
120 LDA GGA XRPD
kt [
°]
ci nienie [GPa]
Rysunek 7.2: Ciśnieniowa zależność kątów między wektorami sieciowymi fazy eksperymentalnej.
kąta β i niedoszacowania kątów α oraz γ (γ o ponad 20 (!)).
W miarę progresu kompresji obserwowana jest pewna jakościowa różnica w wyni-
kach LDA oraz GGA. Otóż LDA w odróżnieniu od GGA przewiduje niemonotoniczną
31

Struktura eksperymentalna pod wysokim ciśnieniem
LDA GGA XRPD [33]a [Å] 8.126 8.452 8.0125(2)b [Å] 4.570 4.836 4.7535(1)c [Å] 4.697 4.933 4.6923(1)α [] 68.4 69.6 76.478(1)β [] 108.6 107.1 103.403(1)γ [] 96.6 96.8 118.078(1)V [Å3] 152.561 179.379 151.761(6)
Tabela 7.1: Zestawienie różnic między obliczonymi parametrami sieci a danymi eksperymentalnymi.Wielkości niedoszacowane wyróżniono kolorem niebieskim, zaś przeszacowane — czerwonym.
zależność a(p), z lokalnym maksimum w okolicach 4 GPa. Ponadto stałe sieciowe z GGA
są o kilka procent wyższe od tych z LDA, w całym rozważanym zakresie ciśnień. Z ko-
lei wartości kątów, przewidywane w ramach obu przybliżeń, wykazują tym większą
wzajemną zgodność im wyższy jest stopień kompresji.
7.2 Równanie stanu AgSO4
Istotną informacją dla badań wysokociśnieniowych jest równanie stanu rozważanej sub-stancji. Do opisu kryształu AgSO4 zastosowano często używane równanie trzeciegorzędu Bircha–Murnaghana [42]:
p(V ) =3
2B0
(V0V
) 73
−(V0V
) 53
1 +3
4(B′0 − 4)
(V0V
) 23
− 1
. (7.1)
Oznaczono: p – ciśnienie, V – objętość komórki elementarnej, V0=V (p=0); B0, B′0 –
współczynniki charakterystyczne dla danego materiału. W szczególności B0 posiada
interpretację modułu ściśliwości (współczynnika sprężystości objętościowej, ang. bulk
modulus):
B0 = −V(
∂p
∂V
)
T
. (7.2)
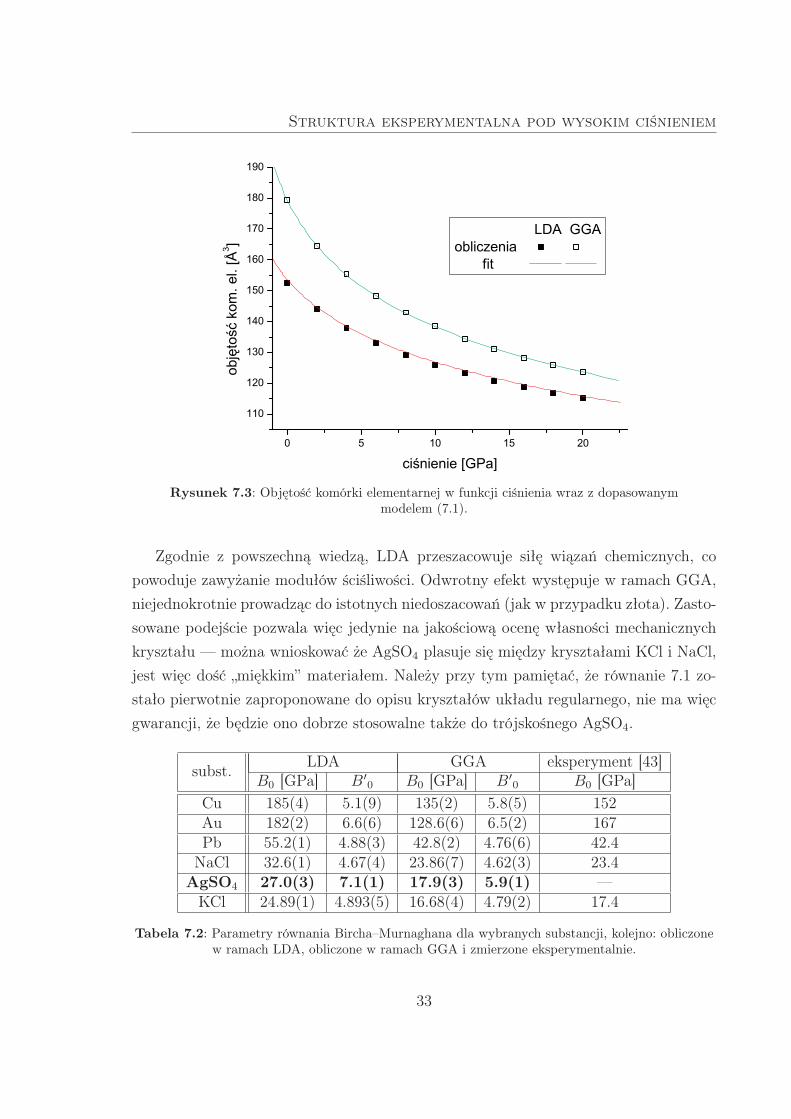
Dla każdej rozważanej wartości ciśnienia wyznaczono objętość komórki elementarnej.
Następnie do uzyskanych wyników dopasowano równanie (7.1), czego rezultat przed-
stawiono na Rys. 7.3. Odczytano także współczynniki dopasowania: B0 oraz B′0. Aby
sprawdzić poprawność stosowanego podejścia, wyznaczono B0 dla kilku substancji
o znanych modułach ściśliwości. Uzyskane wyniki zebrano w Tab. 7.2.
32
Struktura eksperymentalna pod wysokim ciśnieniem
0 5 10 15 20
110
120
130
140
150
160
170
180
190
LDA GGAobliczenia fit
obj
to k
om. e
l. [Å
3 ]
ci nienie [GPa]
Rysunek 7.3: Objętość komórki elementarnej w funkcji ciśnienia wraz z dopasowanymmodelem (7.1).
Zgodnie z powszechną wiedzą, LDA przeszacowuje siłę wiązań chemicznych, co
powoduje zawyżanie modułów ściśliwości. Odwrotny efekt występuje w ramach GGA,
niejednokrotnie prowadząc do istotnych niedoszacowań (jak w przypadku złota). Zasto-
sowane podejście pozwala więc jedynie na jakościową ocenę własności mechanicznych
kryształu — można wnioskować że AgSO4 plasuje się między kryształami KCl i NaCl,
jest więc dość „miękkim” materiałem. Należy przy tym pamiętać, że równanie 7.1 zo-
stało pierwotnie zaproponowane do opisu kryształów układu regularnego, nie ma więc
gwarancji, że będzie ono dobrze stosowalne także do trójskośnego AgSO4.
subst.LDA GGA eksperyment [43]
B0 [GPa] B′0 B0 [GPa] B′0 B0 [GPa]Cu 185(4) 5.1(9) 135(2) 5.8(5) 152Au 182(2) 6.6(6) 128.6(6) 6.5(2) 167Pb 55.2(1) 4.88(3) 42.8(2) 4.76(6) 42.4
NaCl 32.6(1) 4.67(4) 23.86(7) 4.62(3) 23.4AgSO4 27.0(3) 7.1(1) 17.9(3) 5.9(1) —
KCl 24.89(1) 4.893(5) 16.68(4) 4.79(2) 17.4
Tabela 7.2: Parametry równania Bircha–Murnaghana dla wybranych substancji, kolejno: obliczonew ramach LDA, obliczone w ramach GGA i zmierzone eksperymentalnie.
33

Struktura eksperymentalna pod wysokim ciśnieniem
7.3 Elektronowa struktura pasmowa
7.3.1 Warunki zerowego ciśnienia zewnętrznego
L(S)DA nie może być użyte do wyznaczania struktury pasmowej, gdyż daje jako-
ściowo błędne wyniki, kwalifikując AgSO4 jako metal. Wspomnianej wady nie posiada
metoda L(S)DA+U, dlatego to ją zastosowano dalej, by wyznaczyć struktury pasmowe
siarczanu srebra(II). Na Rys. 7.4 dokonano porownania struktur pasmowych AgSO4obliczonych przy różnych doborach parametrów U . W Tab. 7.3 porównano wartości
przerwy energetycznej, odczytane z tychże struktur pasmowych.
wybór ∆Egap [eV]U(Ag) = U(S) = U(O) = 0 eV 0
U(Ag) = 4 eV, U(S) = U(O) = 0 eV 0.70U(Ag) = U(O) = 4 eV, U(S) = 0 eV 0.80U(Ag) = U(O) = 4 eV, U(S) = 2 eV 0.82
Tabela 7.3: Wartości przerwy energetycznej AgSO4 w zależności od wyboru zestawu parametrów U .
Jak widać, wprowadzenie odpychania on-site elektronów na atomach srebra powo-
duje otworzenie przerwy na poziomie Fermiego. Wielkość przerwy energetycznej tylko
nieznacznie się powiększa po dodaniu korekt „U” na atomach tlenu i siarki. Wartość
przerwy otrzymana dla zestawu parametrów U(Ag) = U(O) = 4 eV, U(S) = 2 eV,
wynosi 0.82 eV i tylko nieznacznie odbiega od eksperymentalnej wartości przerwy wy-
ekstrapolowanej do 0 K (0.80 eV).
34
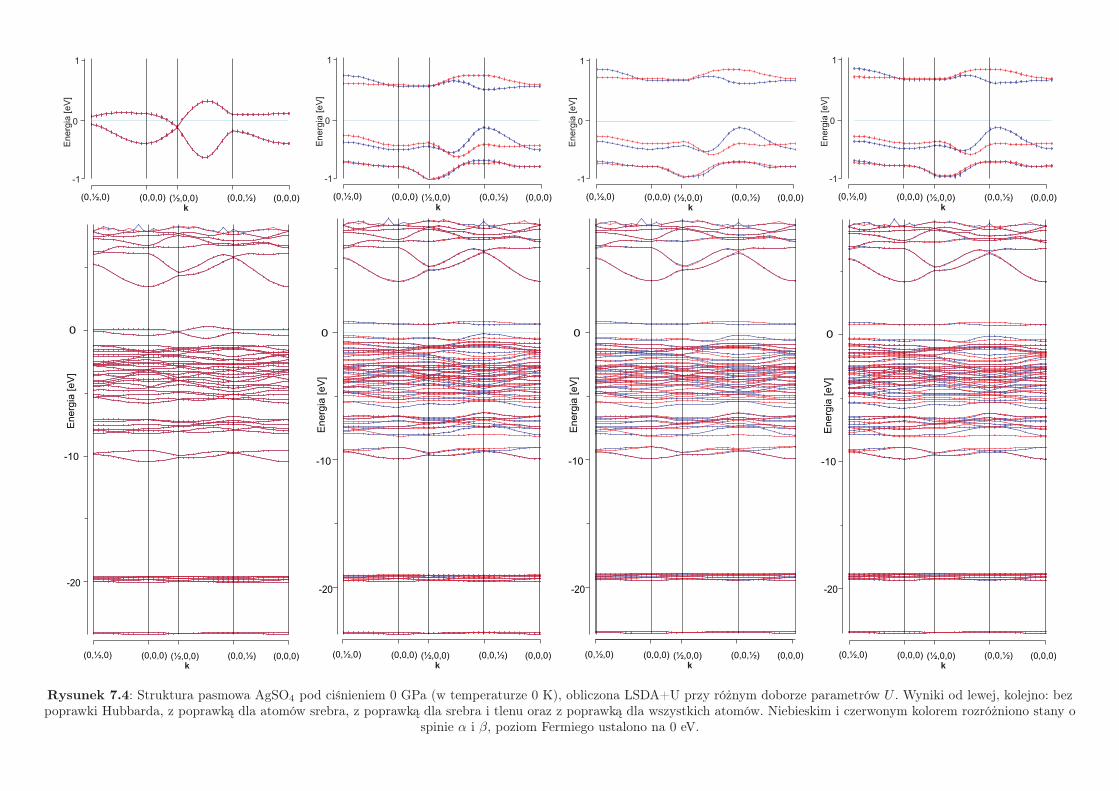
Rysunek 7.4: Struktura pasmowa AgSO4 pod ciśnieniem 0 GPa (w temperaturze 0 K), obliczona LSDA+U przy różnym doborze parametrów U . Wyniki od lewej, kolejno: bezpoprawki Hubbarda, z poprawką dla atomów srebra, z poprawką dla srebra i tlenu oraz z poprawką dla wszystkich atomów. Niebieskim i czerwonym kolorem rozróżniono stany o
spinie α i β, poziom Fermiego ustalono na 0 eV.

Struktura eksperymentalna pod wysokim ciśnieniem
7.3.2 Obliczenia wysokociśnieniowe
Dalsze obliczenia struktury pasmowej przeprowadzono na komórkach fazy ekspery-
mentalnej, zoptymalizowanych LDA w zakresie ciśnień 0–20 GPa. Następnie z otrzyma-
nych związków dyspersyjnych odczytano wartości przerw energetycznych. Celem tych
działań było przewidzenie możliwości metalizacji kryształu AgSO4 pod wpływem ze-
wnętrznego ciśnienia. Podobne obliczenia przeprowadzono równolegle dla krzemu, by
zilustrować poprawność stosowanego podejścia, choć w tym przypadku poprawka „U”
oraz uwzględnienie polaryzacji spinowej były oczywiście niepotrzebne. Należy wspo-
mnieć, że w ogólności L(S)DA+U nie daje ilościowo poprawnych wyników [25], dlatego
bardziej poważnie należy traktować przewidywane przezeń tendencje aniżeli bezwzględ-
ne rezultaty liczbowe. Przykładowo, prawidłowa wartość przerwy energetycznej krzemu
przy p = 0 GPa to ok. 1.11 eV [43] (nie 0.44 eV, jak obliczono tutaj).
Dwie przykładowe struktury pasmowe AgSO4 (P1) zestawiono na Rys. 7.5. Odczy-
tane wartości przerwy energetycznej pokazano zaś na Rys. 7.8. Dla krzemu zaobser-
wowano zawężanie przerwy energetycznej w miarę kompresji, co jest typowym wyni-
kiem, związanym ze zwiększonym mieszaniem orbitali atomowych i – w konsekwencji
– efektywniejszym oddziaływaniem pasm obsadzonych oraz nieobsadzonych [45]. Choć
pomiary przewodnictwa kryształów Si [46] wskazują, że metalizacja powinna nastąpić
już poniżej 13 GPa, to uzyskany wynik (metalizacja powyżej 20 GPa) można uznać
za jakościowo poprawny. Dużo mniej zrozumiała jest zależność przerwy energetycznej
od ciśnienia dla AgSO4. Z początku (do 4 GPa) obserwowany jest niewielki wzrost,
a dalej monotoniczny spadek do wartości ok. 0.71 eV przy 20 GPa. Zachowanie to moż-
na określić jako anomalne, mimo iż znane są przypadki materiałów, których przerwa
energetyczna rośnie z ciśnieniem (jak MgO, NaCl czy diament [45]). W badanym za-
kresie ciśnień zmiany ∆Egap w AgSO4 są niewielkie (ok. 0.15 eV), co oznacza, że nie
przewiduje się jego metalizacji pod ciśnieniem p ¬ 20 GPa.
36
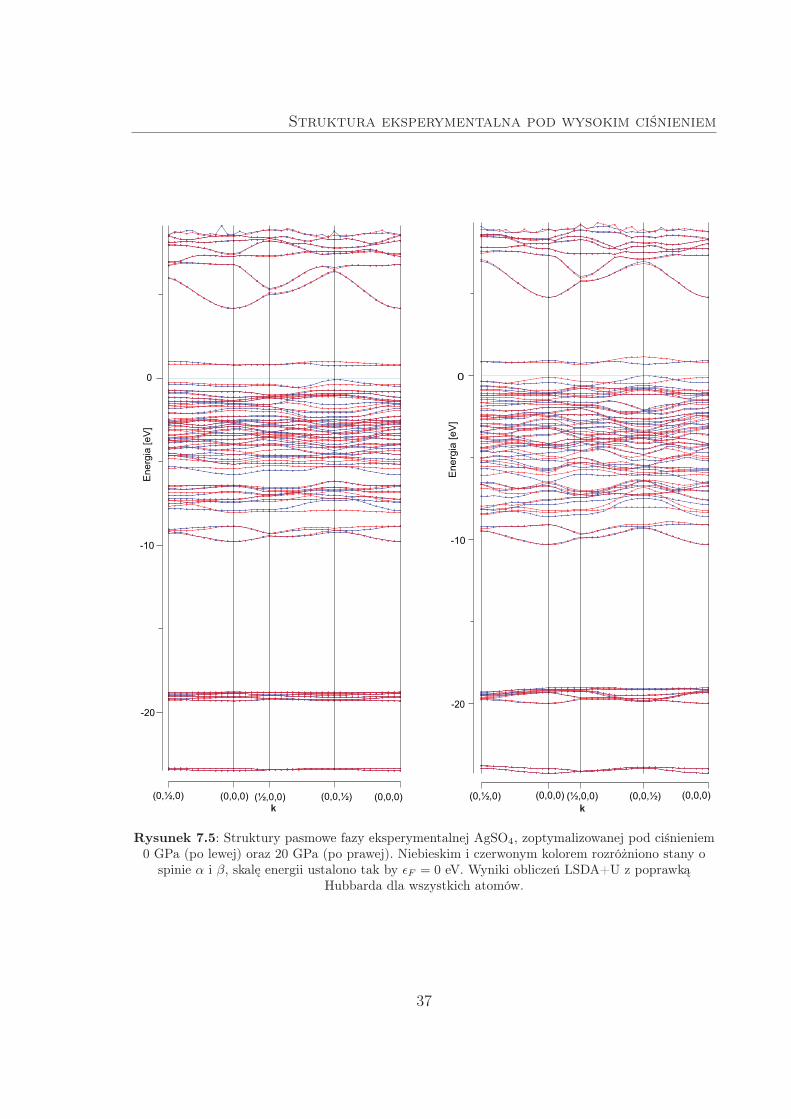
Struktura eksperymentalna pod wysokim ciśnieniem
Rysunek 7.5: Struktury pasmowe fazy eksperymentalnej AgSO4, zoptymalizowanej pod ciśnieniem0 GPa (po lewej) oraz 20 GPa (po prawej). Niebieskim i czerwonym kolorem rozróżniono stany o
spinie α i β, skalę energii ustalono tak by ǫF = 0 eV. Wyniki obliczeń LSDA+U z poprawkąHubbarda dla wszystkich atomów.
37
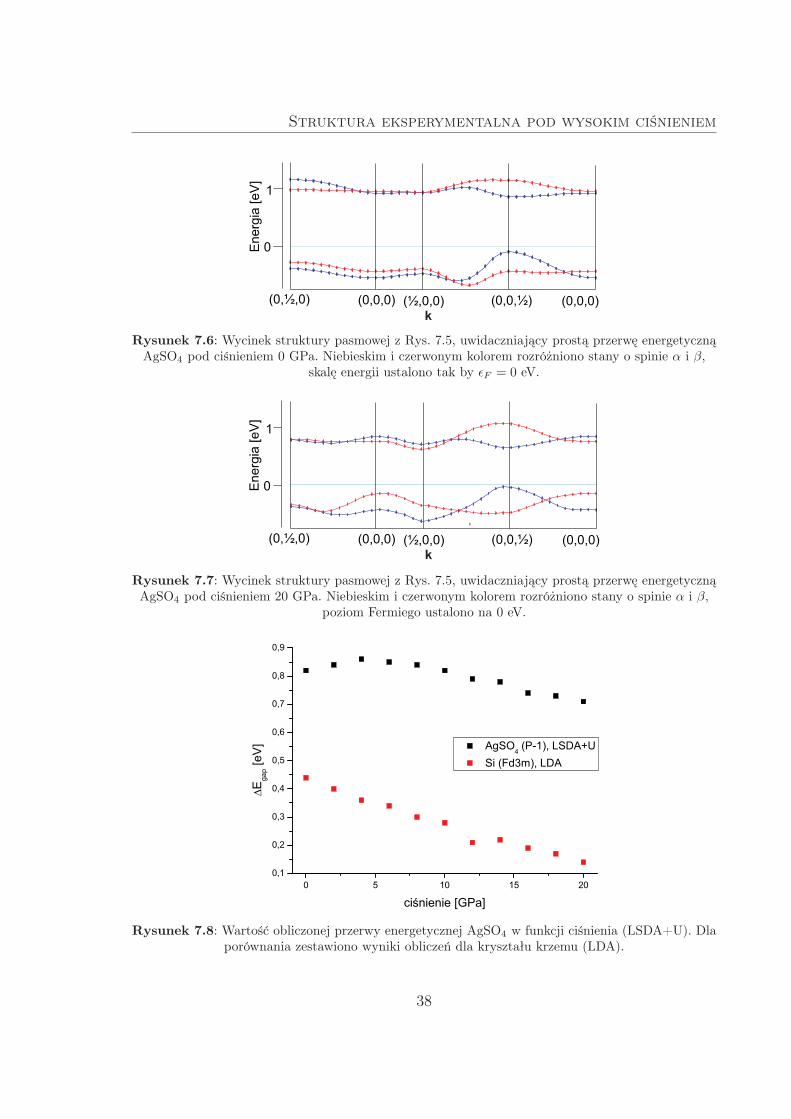
Struktura eksperymentalna pod wysokim ciśnieniem
Rysunek 7.6: Wycinek struktury pasmowej z Rys. 7.5, uwidaczniający prostą przerwę energetycznąAgSO4 pod ciśnieniem 0 GPa. Niebieskim i czerwonym kolorem rozróżniono stany o spinie α i β,
skalę energii ustalono tak by ǫF = 0 eV.
Rysunek 7.7: Wycinek struktury pasmowej z Rys. 7.5, uwidaczniający prostą przerwę energetycznąAgSO4 pod ciśnieniem 20 GPa. Niebieskim i czerwonym kolorem rozróżniono stany o spinie α i β,
poziom Fermiego ustalono na 0 eV.
0 5 10 15 200,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
AgSO4 (P-1), LSDA+U
Si (Fd3m), LDA
Ega
p [eV
]
ci nienie [GPa]
Rysunek 7.8: Wartość obliczonej przerwy energetycznej AgSO4 w funkcji ciśnienia (LSDA+U). Dlaporównania zestawiono wyniki obliczeń dla kryształu krzemu (LDA).
38

Rozdział 8
Przejścia fazowe AgSO4
Omówione do tej pory obliczenia przeprowadzono przy założeniu, że w zakresie 0–
20 GPa nie nastąpi przemiana fazowa AgSO4. Takiej ewentualności nie można jednak
wykluczyć. W niniejszym rozdziale próbowano odpowiedzieć na pytanie czy w ściśnię-
tym AgSO4 nastąpi przejście fazowe, a jeśli tak, to jaką strukturę będzie miała forma
wysokociśnieniowa.
Skuteczne przewidzenie przemiany fazowej jest istotne dla badań nad potencjalnym
nadprzewodnictwem, gdyż wysokociśnieniowe odmiany polimorficzne AgSO4 mogą cha-
rakteryzować się odmiennymi własnościami elektronowymi.
8.1 Procedura
Przewidywanie przemian polimorficznych rozpoczęto od wytypowania prawdopo-
dobnych struktur wysokociśnieniowych. Następnie wyznaczono molową entalpię h każ-
dej z nich w zakresie ciśnień 0–20 GPa:
h := u+ pv. (8.1)
Oznaczono: u – molowa energia wewnętrzna, v – molowa objętość pod ciśnieniem p.
Fazę o najniższej entalpii interpretowano jako najtrwalszą termodynamicznie pod da-
nym ciśnieniem. Przyjęto że obliczenia odpowiadają eksperymentom w temperaturze
bliskiej zeru bezwzględnemu, co uzasadnia stosowanie entalpii zamiast entalpii swo-
bodnej Gibbsa.
Poprawki na drgania zerowe zaniedbano, gdyż – jak wykazano w [32] – są one bardzo
zbliżone dla wszystkich rozważonych dotąd odmian polimorficznych AgSO4 (różnice nie
przekraczają 2 kJ/mol).
39

Przejścia fazowe AgSO4
8.2 Potencjalne struktury wysokociśnieniowe
Ogólna reguła mówi, że pod wysokim ciśnieniem przeważają fazy o dużej gęstości
[45]. Kierując się tą prostą zasadą, wytypowano kilka potencjalnych struktur wysokoci-
śnieniowych (Tab. 8.1). Powstały one z właściwych struktur macierzystych (wybranych
za [44] i [32]), poprzez odpowiednią zamianę atomów i optymalizację DFT.
typ strukt.gęstość [g cm−3]LDA GGA
Ag2O2 4.280 3.685AgSO4 4.408 3.754CuSO4 4.441 3.912CoSO4 4.563 3.923CrVO4 4.575 4.060ZrSiO4 4.736 4.046KReO4 4.756 3.830BaWO4 4.926 3.720LaPO4 4.955 3.644CaWO4 5.041 4.373
Tabela 8.1: Obliczone gęstości hipotetycznych odmian polimorficznych oraz fazy eksperymentalnejAgSO4. Wyniki dla zerowego ciśnienia zewnętrznego.
W strukturze typu CuSO4 (Rys. 8.1) sfera koordynacyjna atomu srebra przypomi-
na wydłużony, lekko zdeformowany oktaedr. Można wyróżnić nieskończone łańcuchy
[-Ag–O-]∞, zarówno jak [-Ag–O–S–O-]∞. Z kolei w strukturze typu Ag2O2 (Rys. 8.2)
atomy srebra mają sfery koordynacyjne w postaci kwadratów. Ponieważ komórka ele-
mentarna tej fazy zawiera aż 16 jednostek stechiometrycznych AgSO4, do obliczeń
użyto dwukrotnie mniejszej komórki prymitywnej.
W fazie typu KReO4 (Rys. 8.3) jony srebra połączone są jedynie przez mostki
[-Ag–O–S–O-]∞. Każdy z jonów srebra otoczony jest przez cztery atomy tlenu, ułożone
w narożach lekko zdeformowanego kwadratu.
W strukturze typu BaWO4 (Rys. 8.4) każdy atom srebra otoczony jest przez sześć
atomów tlenu, rozmieszczonych w wierzchołkach wygiętego oktedru. W strukturze ty-
pu CaWO4 (Rys. 8.5) sfera koordynacyjna srebra jest bardzo nietypowa i przypomina
mocno spłaszczony tetraedr. Obydwie struktury są wyjątkowe, ze względu na war-
stwową budowę. W pierwszej z nich warstwy zawierają łańcuchy [-Ag–O–S–O-]∞ oraz
[-Ag–O-]∞; w drugiej wyłącznie [-Ag–O–S–O-]∞.
40
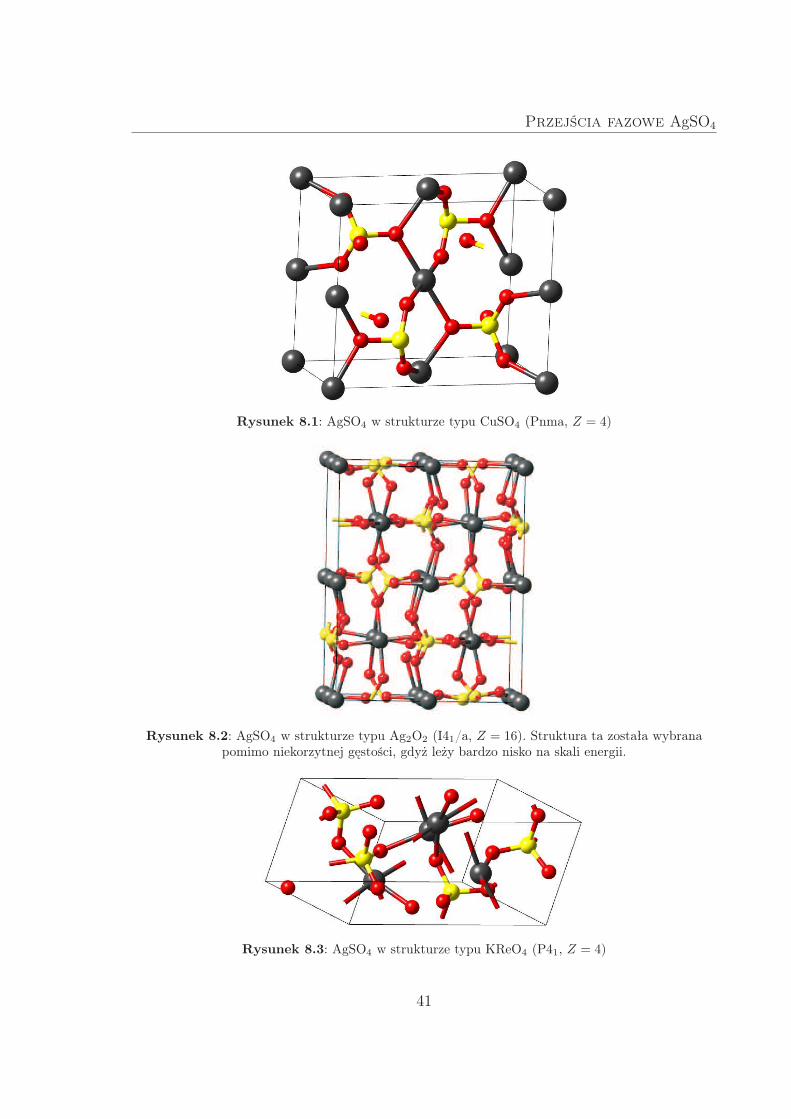
Przejścia fazowe AgSO4
Rysunek 8.1: AgSO4 w strukturze typu CuSO4 (Pnma, Z = 4)
Rysunek 8.2: AgSO4 w strukturze typu Ag2O2 (I41/a, Z = 16). Struktura ta została wybranapomimo niekorzytnej gęstości, gdyż leży bardzo nisko na skali energii.
Rysunek 8.3: AgSO4 w strukturze typu KReO4 (P41, Z = 4)
41
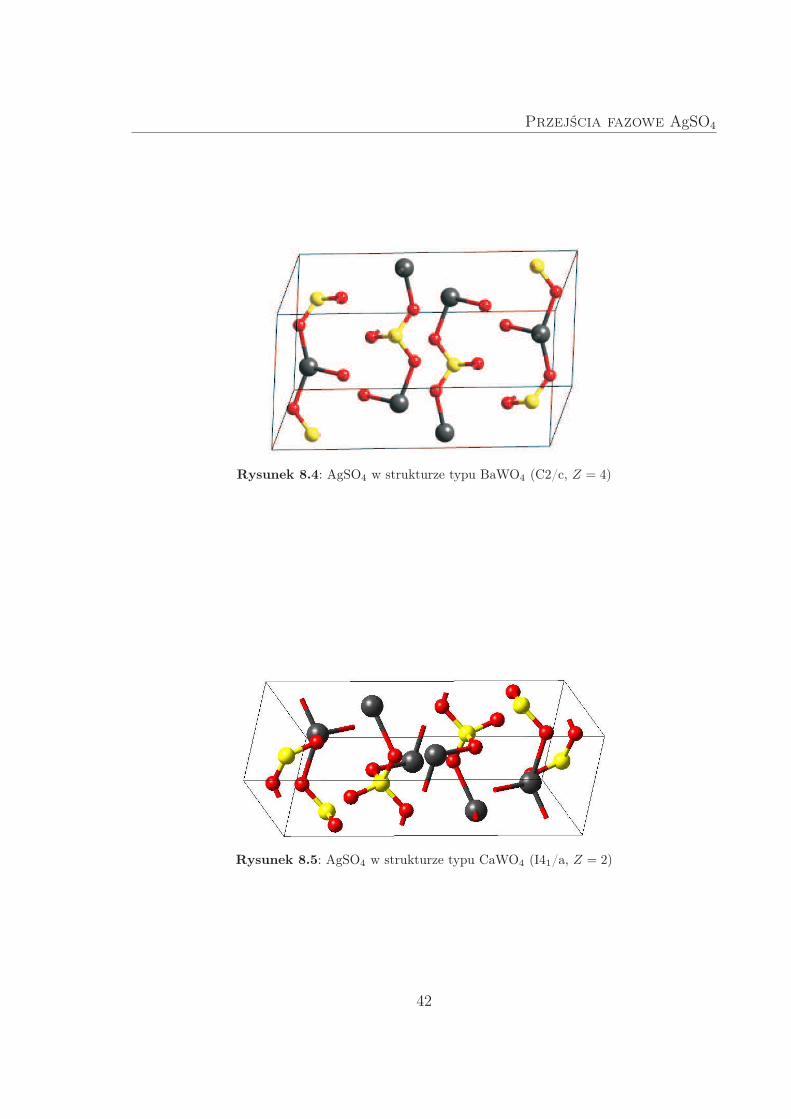
Przejścia fazowe AgSO4
Rysunek 8.4: AgSO4 w strukturze typu BaWO4 (C2/c, Z = 4)
Rysunek 8.5: AgSO4 w strukturze typu CaWO4 (I41/a, Z = 2)
42
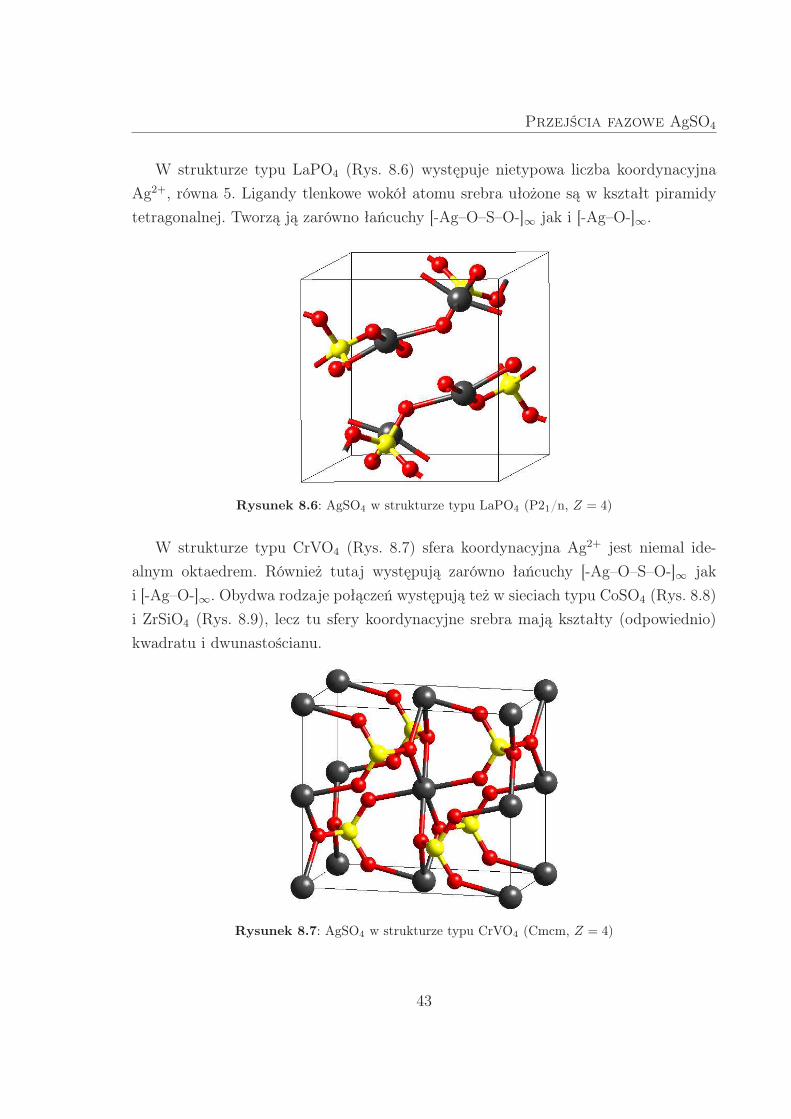
Przejścia fazowe AgSO4
W strukturze typu LaPO4 (Rys. 8.6) występuje nietypowa liczba koordynacyjna
Ag2+, równa 5. Ligandy tlenkowe wokół atomu srebra ułożone są w kształt piramidy
tetragonalnej. Tworzą ją zarówno łańcuchy [-Ag–O–S–O-]∞ jak i [-Ag–O-]∞.
Rysunek 8.6: AgSO4 w strukturze typu LaPO4 (P21/n, Z = 4)
W strukturze typu CrVO4 (Rys. 8.7) sfera koordynacyjna Ag2+ jest niemal ide-
alnym oktaedrem. Również tutaj występują zarówno łańcuchy [-Ag–O–S–O-]∞ jak
i [-Ag–O-]∞. Obydwa rodzaje połączeń występują też w sieciach typu CoSO4 (Rys. 8.8)
i ZrSiO4 (Rys. 8.9), lecz tu sfery koordynacyjne srebra mają kształty (odpowiednio)
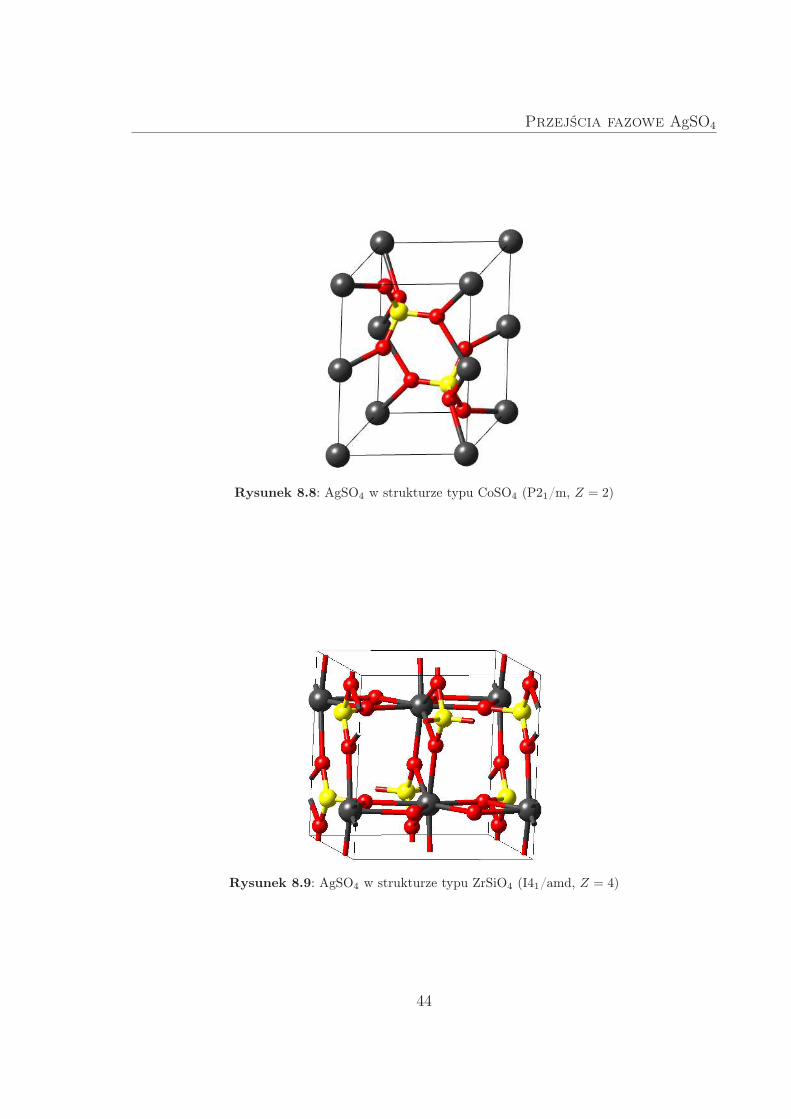
kwadratu i dwunastościanu.
Rysunek 8.7: AgSO4 w strukturze typu CrVO4 (Cmcm, Z = 4)
43
Przejścia fazowe AgSO4
Rysunek 8.8: AgSO4 w strukturze typu CoSO4 (P21/m, Z = 2)
Rysunek 8.9: AgSO4 w strukturze typu ZrSiO4 (I41/amd, Z = 4)
44

Przejścia fazowe AgSO4
8.3 Porównanie entalpii
8.3.1 Metoda wspólnej stycznej
Dotychczasowe rozważania na temat gęstości z pewnością nie są wystarczające, by
prawidłowo wskazać wysokociśnieniowe odmiany polimorficzne. Nieco dokładniejszy,
choć nadal przybliżony, obraz można uzyskać dzięki metodzie wspólnej stycznej (ang.
common tangent).
Niech hX oznacza entalpię molową fazy X, zaś h0 – entalpię molową fazy ekspery-
mentalnej (lub dowolnej innej, którą przyjmuje się za poziom odniesienia). W najprost-
szym ujęciu można założyć, że objętości i energie rozważanych faz jednakowo zmieniają
się pod wpływem ciśnienia, tj.:
h0(p) = u0 +∆u(p) + p (v0 +∆v(p))
hX(p) = uX +∆u(p) + p (vX +∆v(p)) (8.2)
gdzie ∆u i ∆v są pewnymi funkcjami ciśnienia. Po odjęciu obu równań stronami otrzy-
muje się wyrażenie na względną entalpię fazy X:
hX(p)− h0(p) = uX − u0 + p (vX − v0) . (8.3)
Z postaci równania (8.3) wynika, że wykresy (hX−h0)(p) są liniami prostymi, o współ-
czynniku kierunkowym (vX − v0) charakterystycznym dla każdej ze struktur. Metoda
common tangent pozwala więc oszacować zmiany entalpii jedynie na podstawie wyni-
ków obliczeń energii i objętości w p = 0 GPa.
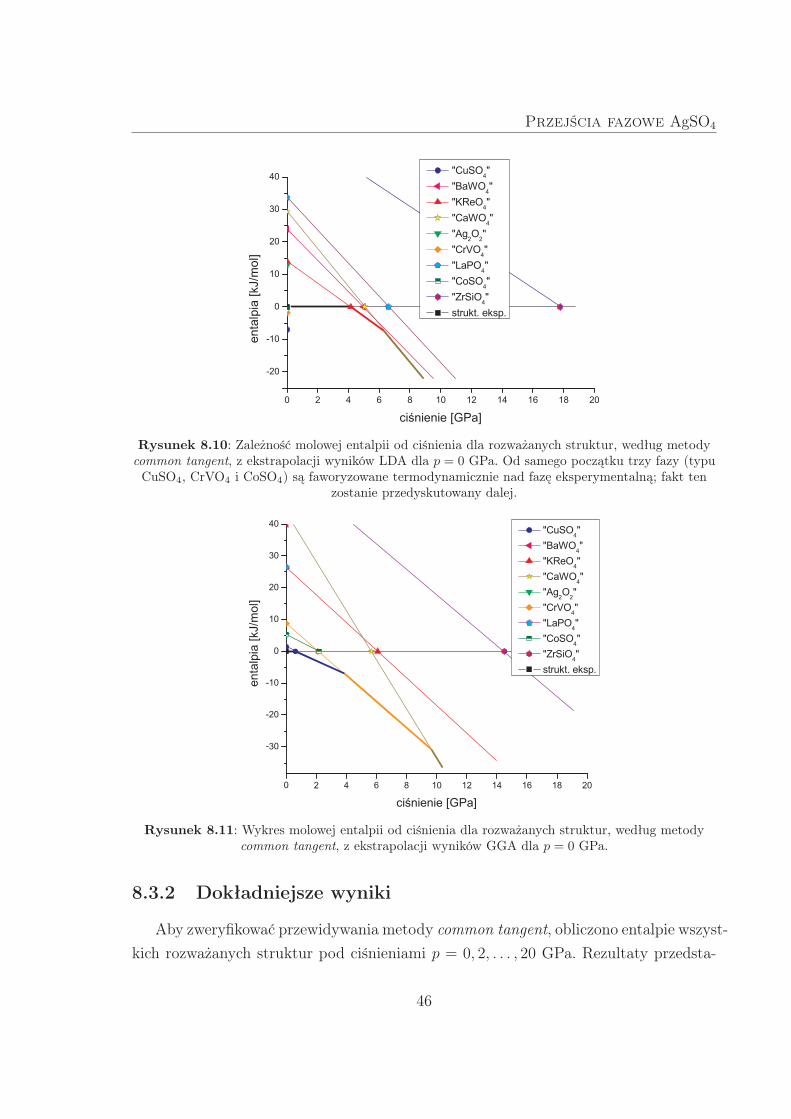
Wykresy h(p) dla rozważanych struktur przedstawiono na Rys. 8.10 (LDA) i Rys. 8.11
(GGA). Liniami pogrubionymi oznaczono ewolucję struktur o najniższej entalpii w stro-
nę wysokich ciśnień. Część rozważanych faz jest niewidoczna na wykresach, gdyż do-
piero w formalnie ujemnych ciśnieniach stają się one konkurencyjne z fazą eksperymen-
talną.
Zgodnie z przewidywaniami metody common tangent dla wyników LDA, dla p =
4 GPa spodziewane jest przejście fazowe do struktury typu KReO4. Następnie, między
6–7 GPa, powinna nastąpić kolejna przemiana, do struktury typu CaWO4. Ekstrapo-
lacja wyników GGA prowadzi do nieco innego scenariusza: między 0–1 GPa przewiduje
się przejście fazowe do strukury typu CuSO4, dalej (ok. 4 GPa) do CrVO4, a następ-
nie (ok. 10 GPa) do CaWO4. Struktura typu CaWO4 jest zatem najstabilniejszą dla
p 7 GPa (LDA) i p 10 GPa (GGA).
45
Przejścia fazowe AgSO4
Rysunek 8.10: Zależność molowej entalpii od ciśnienia dla rozważanych struktur, według metodycommon tangent, z ekstrapolacji wyników LDA dla p = 0 GPa. Od samego początku trzy fazy (typuCuSO4, CrVO4 i CoSO4) są faworyzowane termodynamicznie nad fazę eksperymentalną; fakt ten
zostanie przedyskutowany dalej.
Rysunek 8.11: Wykres molowej entalpii od ciśnienia dla rozważanych struktur, według metodycommon tangent, z ekstrapolacji wyników GGA dla p = 0 GPa.
8.3.2 Dokładniejsze wyniki
Aby zweryfikować przewidywania metody common tangent, obliczono entalpie wszyst-
kich rozważanych struktur pod ciśnieniami p = 0, 2, . . . , 20 GPa. Rezultaty przedsta-
46

Przejścia fazowe AgSO4
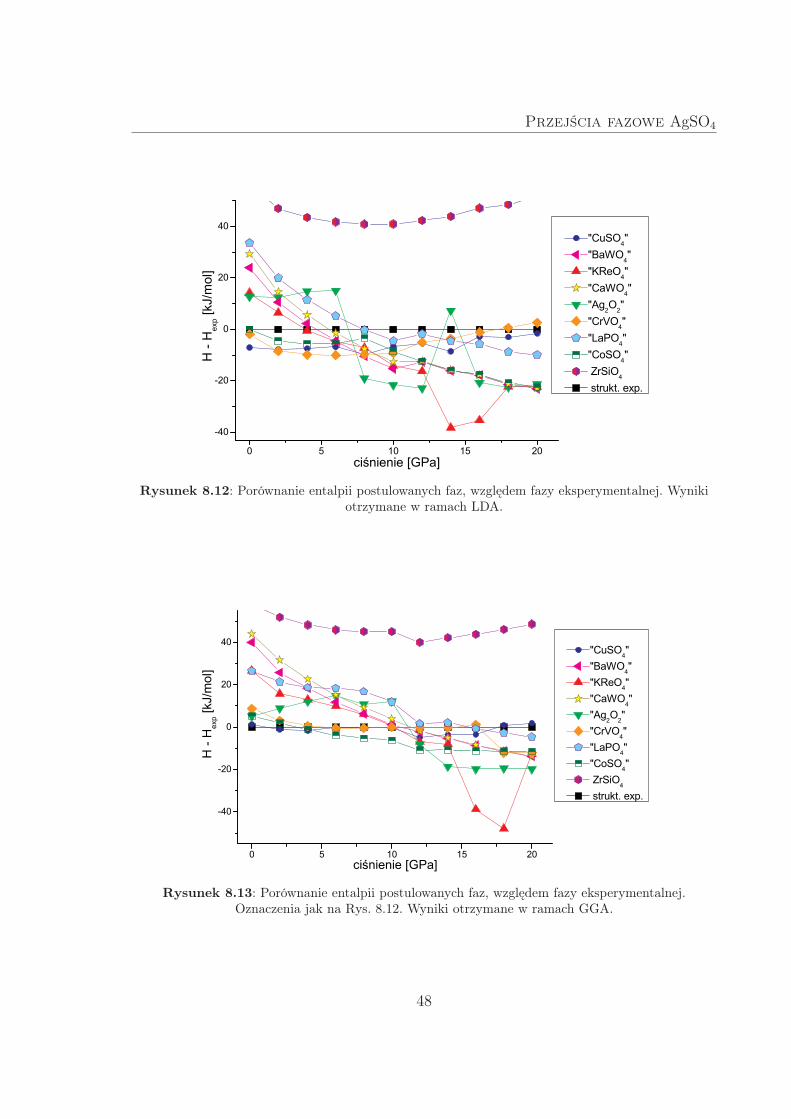
wiono na Rys. 8.12 oraz 8.13. Wyniki te dowodzą, że większość zaproponowanych struk-
tur wysokociśnieniowych została trafnie wytypowana – wszystkie z wyjątkiem ZrSiO4rzeczywiście są termodynamicznie konkurencyjne wobec fazy eksperymentalnej w roz-
maitych zakresach ciśnień.
Z wyników LDA można wnioskować, że istnieją co najmniej trzy fazy (typu CoSO4,
CuSO4 i CrVO4), które już pod ciśnieniem atmosferycznym są faworyzowane termo-
dynamicznie nad fazę eksperymentalną. Wynik ten jest prawdopodobnie artefaktem
metody obliczeniowej, gdyż GGA, które w ogólności lepiej odtwarza parametry ter-
modynamiczne, nie przewiduje podobnego zjawiska. Uwagę zwraca też entalpowa „bi-
stabilność” struktury typu Ag2O2, obserwowana jedynie w ramach LDA, a także dość
nietypowa zmienność entalpii struktury typu KReO4 w funkcji ciśnienia dla p > 12 GPa
(zarówno LDA jak i GGA przewidują spadek, a następnie wzrost entalpii).
Obydwa przybliżenia wskazują na możliwość przejścia fazowego, między 4–8 GPa
(LDA) lub 10–16 GPa (GGA). Niestety, trudno jest jednoznacznie wskazać, która ze
struktur będzie wysokociśnieniową odmianą polimorficzną, gdyż – podobnie jak wska-
zywała metoda commont tangent – w różnych zakresach ciśnień dominują różne fazy,
a różnice entalpii między nimi są bardzo małe i często w granicach błędu metody.
47
Przejścia fazowe AgSO4
0 5 10 15 20
-40
-20
0
20
40
H -
Hex
p [kJ/
mol
]
ci nienie [GPa]
"CuSO4""BaWO4""KReO4""CaWO4""Ag2O2""CrVO4""LaPO4""CoSO4" ZrSiO4
strukt. exp.
Rysunek 8.12: Porównanie entalpii postulowanych faz, względem fazy eksperymentalnej. Wynikiotrzymane w ramach LDA.
0 5 10 15 20
-40
-20
0
20
40"CuSO4""BaWO4""KReO4""CaWO4""Ag2O2""CrVO4""LaPO4""CoSO4" ZrSiO4
strukt. exp.
H -
Hex
p [kJ/
mol
]
ci nienie [GPa]
Rysunek 8.13: Porównanie entalpii postulowanych faz, względem fazy eksperymentalnej.Oznaczenia jak na Rys. 8.12. Wyniki otrzymane w ramach GGA.
48

Rozdział 9
Podsumowanie
Przedmiotem niniejszej pracy było zbadanie wysokociśnieniowych własności AgSO4na gruncie dwóch przybliżeń DFT: LDA oraz GGA. Własności mechaniczne kryształu
AgSO4 z powodzeniem opisano równaniem Bircha–Murnaghana, z modułem ściśliwości
równym 27.0 GPa (LDA) lub 17.9 GPa (GGA). Zbadano także ciśnieniową zależność
przerwy energetycznej, nie przewidując metalizacji materiału do ciśnienia 20 GPa.
Zaproponowano także kilka nowych struktur dla AgSO4. Większość z nich okazała
się dobrymi kandydatami na wysokociśnieniowe odmiany polimorficzne, co wykazały
obliczenia entalpii w funkcji ciśnienia przy założeniu niskiej temperatury. Przewidzia-
no przejścia fazowe między 4–16 GPa, jednak, z powodu rozbieżności wyników LDA
i GGA, nie udało się jednoznacznie ustalić, która struktura (typu CuSO4, Ag2O2,
KReO4, BaWO4, CaWO4, CrVO4, CoSO4 czy LaPO4) będzie najlepszym kandydatem
na formę wysokociśnieniową.
9.1 Porównanie wyników LDA i GGA
Na osobny komentarz zasługuje porównanie wyników LDA i GGA, które otrzymano
w niniejszej pracy. Struktura eksperymentalna została odtworzona przez obie metody
ze słabą dokładnością. Rozbieżności między obliczonymi parametrami sieci i danymi
eksperymentalnymi są miejscami znacząco wyższe niż standardowe 2–3%. Prawidłowe
odtworzenie objętości komórki elementarnej przez LDA może więc być zupełnie przy-
padkowym wynikiem; nie ma podstaw by wnioskować o wyższości LDA nad GGA,
mimo że takie przypadki bywały odnotowywane w literaturze [47].
Kolejnym interesującym wynikiem, kilkukrotnie sygnalizowanym w tekście niniej-
49

Podsumowanie
szej pracy, jest przewidywanie istnienia trzech struktur o energii niższej niż struktura
eksperymentalna. Oczywiście, ten wynik LDA może być błędny i wynikać np. z przesza-
cowania energii wiązań przy danej geometrii komórki. Taki scenariusz jest dość prawdo-
podobny, gdyż LDA w ogólności nie jest zalecane do obliczeń termodynamicznych [12].
Mimo to autor uważa, że nie należy z góry przekreślać hipotezy o metastabilności fazy
P1 – wszak ogromna większość związków chemicznych otrzymana została w postaci
metatrwałej.
Oczywiście nie wszystkie rozbieżności w wynikach obliczeń są niepokojące. Zawyża-
nie modułu ściśliwości w ramach LDA i jego zaniżanie w GGA oraz zaniżanie gęstości
w LDA i jej zawyżanie w GGA to typowe efekty, opisane dla wielu układów, choćby
takich jak azotki metali przejściowych [48].
Reasumując: wyniki przeprowadzonych obliczeń pozostawiają jak dotąd więcej py-
tań niż odpowiedzi. Można być pewnym, że modelowanie AgSO4 sprawia duże kłopoty
standardowym metodom DFT i dopiero porównanie obliczeń z wynikami eksperymen-
tów pozwoli ocenić czy zastosowane podejście było prawidłowe. Eksperymenty wyso-
kociśnieniowe są aktualnie w toku [49].
50

Bibliografia
[1] L. Piela „Idee chemii kwantowej” PWN, Warszawa (2002)
[2] E. Schrödinger, Phys. Rev. 28:1049 (1926)
[3] M. Born, J. R. Oppenheimer, Ann. Physik 84:457 (1927)
[4] H. Hellmann „Einführung in die Quantenchemie” Franz Deuticke, Leipzig (1937)
[5] R. P. Feynman, Phys. Rev. 56:340 (1939)
[6] M. Born, wykład noblowski, 11 grudnia 1954
(http://nobelprize.org/nobel_prizes/physics/laureates/1954/born-lecture.html)
[7] G. E. Uhlenbeck, S. Goudsmit, Naturwissenschaften 13:953 (1925)
[8] W. Gerlach, O. Stern, Z. Phys. A – Hadron Nucl. 9:353 (1922)
[9] W. Pauli, Phys. Rev. 58:716 (1944)
[10] P. Hohenberg, W. Kohn, Phys. Rev. 136:864 (1964)
[11] W. Kohn, L. J. Sham, Phys. Rev. 140:1133 (1965)
[12] W. Koch, M. C. Holthausen „A Chemist’s Guide to Density Functional Theory”
2nd ed. Wiley VCH, Weinheim (2001)
[13] S. H. Vosko, L. Wilk, M. Nusair, Can. J. Phys. 58:1200 (1980)
[14] D. M. Ceperley, B. J. Alder, Phys. Rev. Lett. 45:566 (1980)
[15] J. P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77:3865 (1996)
[16] J. P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 78:1396 (1996)
51

Bibliografia
[17] „Generalized Voronoi Diagram: A Geometry-Based Approach to Computationa”
red. M. L Gavrilova, Springer–Verlag Berlin Heidelberg (2008)
[18] F. Bloch, Z. Physik 52:555 (1928)
[19] Z. Trzaska Durski, H. Trzaska Durska „Podstawy krystalografii” WPW, Warszawa
(2003)
[20] D. R. Hamann et al., Phys. Rev. Lett. 43:1494 (1979)
[21] D. Vanderbilt, Phys. Rev. B 41:7892 (1985)
[22] P. E. Blöchl, Phys. Rev. B 50:17953 (1994)
[23] G. Kresse, D. Joubert, Phys. Rev. B 59:1758 (1999)
[24] C. Rostgaard, arXiv:0910.1921v2 (2009)
[25] V. I. Anisimov et al.. Phys. Rev. B 44:943 (1991)
[26] „Tablice Chemiczne” red. W. Mizerski, Adamantan, Warszawa (2000)
[27] S. Riedel, M. Kaupp, Coord. Chem. Rev. 253:606 (2009)
[28] M. Derzsi, W. Grochala, wyniki nieopublikowane (2010)
[29] W. Grochala, Inorg. Chem. Comm. 11:155 (2008)
[30] P. Malinowski et al., Z. Anorg. Allg. Chem. 634:2608 (2008)
[31] M. Derzsi, K. Dymkowski, W. Grochala, Inorg. Chem. 49:2735 (2010)
[32] K. Dymkowski, praca magisterska, Wydział Chemii UW, Warszawa (2009)
[33] P. J. Malinowski et al., Angew. Chem. Int. Ed. Engl. 49:1683 (2010)
[34] R. Jurczakowski, W. Grochala, wyniki nieopublikowane (2010)
[35] G. Kresse, W. Furthmüller, Phys. Rev. B 54:11169 (1996)
[36] K. Parliński et al.. Phys. Rev. Lett. 78:4063 (1997)
[37] Z. Mazej et al.. Cryst. Eng. Comm. 11:1702 (2009)
52

Bibliografia
[38] K. Fijałkowski, W Grochala, Dalton Trans. 40:5447 (2008)
[39] K. Amaya et al.. J. Phys.: Condens. Matter 10:11179 (1998)
[40] M. I. Eremets et al.. J. Phys.: Condens. Matter 10:11519 (1998)
[41] H. Hegger et al.. Phys. Rev. Lett. 84:4986 (2000)
[42] F. Birch, Phys. Rev. 71:809 (1947)
[43] „Tablice fizyczno-astronomiczne” red. W. Mizerski, Adamantan, Warszawa (2002)
[44] O. Fukunaga, S. Yamaoka, Phys. Chem. Minerals 5:167 (1979)
[45] W. Grochala et al.. Angew. Chem. Int. Ed. 46:3620 (2007)
[46] S. D. Gilev, A. M. Trubachev, Phys. Stat. Sol. (b) 211:379 (1999)
[47] G. D. Barrera et al., Chem. Phys. 317:119 (2005)
[48] C. Stampfl, Phys. Rev. B 63:155106 (2001)
[49] V. V. Struzkhin, A. Budzianowski, W. Grochala, wyniki nieopublikowane (2010)
53







![A 3)1, î> · teorii funkcjonałów gęstości, określanej skrótem DFT (od nazwy angielskiej: Density Functional Theory) [4-10]. W tej teorii gęstość elektronowa, q (r), zdefiniowana](https://static.fdocuments.pl/doc/165x107/6071f34cf5588609b16972fe/a-31-teorii-funkcjonaw-gstoci-okrelanej-skrtem-dft-od-nazwy.jpg)










