
New olefin metathesis catalysts bearing polyether clamp in N-heterocyclic carbenes ligands
7
New olefin metathesis catalysts bearing polyether clamp in N-heterocyclic carbenes ligands Agnieszka Hryniewicka a , Iwona Misztalewska a , Dorota Czajkowska-Szczykowska a , Zofia Urba nczyk-Lipkowska b , Jacek W. Morzycki a , Stanis1aw Witkowski a, * a Institute of Chemistry, University of Bialystok, Pilsudskiego 11/4, 15-443 Bialystok, Poland b Institute of Organic Chemistry, Polish Academy of Science, Kasprzaka 44, 01-224 Warsaw, Poland article info Article history: Received 6 May 2014 Received in revised form 3 July 2014 Accepted 15 July 2014 Available online 22 July 2014 Keywords: Catalyst design Olefin metathesis N-Heterocyclic carbenes Ruthenium abstract Synthesis of new type of the HoveydaeGrubbs catalysts containing modified N-heterocyclic carbene ligands is described herein. New catalysts bear different in size polyether clamp embracing N,N 0 -2,4- dimethylphenyl substituents in N-heterocyclic carbene. New complexes were tested in model RCM, enyne and CM reactions. They showed comparable activity to that of commercially available Grubbs second generation and HoveydaeGrubbs second generation complexes. Complex with larger polyether clamp proved Z-stereoselective in a macrocycle formation and yielded more Z isomers than commercial complexes in CM reactions. The catalysts are stable and easy to purify. Ó 2014 Elsevier Ltd. All rights reserved. 1. Introduction Olefin metathesis has emerged as a powerful tool for the for- mation of carbonecarbon double bonds. The success of this methodology has spurred the intense investigation of new catalysts showing a better application profile. 1e3 The development of robust metathesis catalysts, 4e6 that carry out this transformation, has fa- cilitated the adoption of this methodology by various areas of sci- ence including polymer chemistry, 7e9 organic synthesis, 10,11 biochemistry, 11 and green chemistry. 12 Commercially available catalysts 1e3 (Fig. 1) 13e16 have been used in many synthesis, but have disadvantages, including lack of reactivity during the syn- thesis of tetrasubstituted olefins and poor Z/E stereocontrol in some cases. Over the last years considerable efforts have been focused on the NHC modifications in new catalysts, especially for making them more environmentally friendly (immobilization, tagging) 17e20 or improving selectivity. 21 Cavallo et al. tried to theoretically predict the effect of the NHC modification on catalytic activity in olefin metathesis. 22 Nolan and Lujan studied correlation between elec- tronic and steric effects of the NHC ligands attached to diverse Ru metal centers 23 and influence of the ancillary ligand for the E/Z selectivity outcome. 24 Attempts to address the problem of stereoselectivity can gen- erally be divided into two areas, catalyst control and substrate control, with examples of the latter recently achieving notable successes. 25,26 On the other hand, selectivity improvement via catalyst design is much more challenging since the catalysts in majority prefer formation of thermodynamically favored E-olefins. Until recently all attempts to synthesize a Z-selective catalyst failed. The pioneering work of Schrock and Hoveyda groups enabled preparation of highly Z-efficient molybdenum catalysts. 27e30 Fig. 1. Examples of olefin metathesis catalysts. * Corresponding author. Tel.: þ48 85 745 7586; fax: þ48 85 745 7580; e-mail addresses: [email protected], [email protected] (S. Witkowski). Contents lists available at ScienceDirect Tetrahedron journal homepage: www.elsevier.com/locate/tet http://dx.doi.org/10.1016/j.tet.2014.07.056 0040-4020/Ó 2014 Elsevier Ltd. All rights reserved. Tetrahedron 70 (2014) 6810e6816
Transcript of New olefin metathesis catalysts bearing polyether clamp in N-heterocyclic carbenes ligands

lable at ScienceDirect
Tetrahedron 70 (2014) 6810e6816
Contents lists avai
Tetrahedron
journal homepage: www.elsevier .com/locate/ tet
New olefin metathesis catalysts bearing polyether clamp inN-heterocyclic carbenes ligands
Agnieszka Hryniewicka a, Iwona Misztalewska a, Dorota Czajkowska-Szczykowska a,Zofia Urba�nczyk-Lipkowska b, Jacek W. Morzycki a, Stanis1aw Witkowski a,*a Institute of Chemistry, University of Bialystok, Piłsudskiego 11/4, 15-443 Białystok, Polandb Institute of Organic Chemistry, Polish Academy of Science, Kasprzaka 44, 01-224 Warsaw, Poland
a r t i c l e i n f o
Article history:Received 6 May 2014Received in revised form 3 July 2014Accepted 15 July 2014Available online 22 July 2014
Keywords:Catalyst designOlefin metathesisN-Heterocyclic carbenesRuthenium
* Corresponding author. Tel.: þ48 85 745 7586; faaddresses: [email protected], [email protected] (S. Witko
http://dx.doi.org/10.1016/j.tet.2014.07.0560040-4020/� 2014 Elsevier Ltd. All rights reserved.
a b s t r a c t
Synthesis of new type of the HoveydaeGrubbs catalysts containing modified N-heterocyclic carbeneligands is described herein. New catalysts bear different in size polyether clamp embracing N,N0-2,4-dimethylphenyl substituents in N-heterocyclic carbene. New complexes were tested in model RCM,enyne and CM reactions. They showed comparable activity to that of commercially available Grubbssecond generation and HoveydaeGrubbs second generation complexes. Complex with larger polyetherclamp proved Z-stereoselective in a macrocycle formation and yielded more Z isomers than commercialcomplexes in CM reactions. The catalysts are stable and easy to purify.
� 2014 Elsevier Ltd. All rights reserved.
Fig. 1. Examples of olefin metathesis catalysts.
1. Introduction
Olefin metathesis has emerged as a powerful tool for the for-mation of carbonecarbon double bonds. The success of thismethodology has spurred the intense investigation of new catalystsshowing a better application profile.1e3 The development of robustmetathesis catalysts,4e6 that carry out this transformation, has fa-cilitated the adoption of this methodology by various areas of sci-ence including polymer chemistry,7e9 organic synthesis,10,11
biochemistry,11 and green chemistry.12 Commercially availablecatalysts 1e3 (Fig. 1)13e16 have been used in many synthesis, buthave disadvantages, including lack of reactivity during the syn-thesis of tetrasubstituted olefins and poor Z/E stereocontrol in somecases. Over the last years considerable efforts have been focused onthe NHCmodifications in new catalysts, especially for making themmore environmentally friendly (immobilization, tagging)17e20 orimproving selectivity.21 Cavallo et al. tried to theoretically predictthe effect of the NHC modification on catalytic activity in olefinmetathesis.22 Nolan and Lujan studied correlation between elec-tronic and steric effects of the NHC ligands attached to diverse Rumetal centers23 and influence of the ancillary ligand for the E/Zselectivity outcome.24
x: þ48 85 745 7580; e-mailwski).
Attempts to address the problem of stereoselectivity can gen-erally be divided into two areas, catalyst control and substratecontrol, with examples of the latter recently achieving notablesuccesses.25,26 On the other hand, selectivity improvement viacatalyst design is much more challenging since the catalysts inmajority prefer formation of thermodynamically favored E-olefins.Until recently all attempts to synthesize a Z-selective catalyst failed.The pioneering work of Schrock and Hoveyda groups enabledpreparation of highly Z-efficient molybdenum catalysts.27e30

A. Hryniewicka et al. / Tetrahedron 70 (2014) 6810e6816 6811
As a complement to their work, Grubbs et al. developedruthenium-based catalysts showing highly improved Z-stereo-selectivity control. The new complexes contained a RueC bondformed by carboxylate-induced intramolecular CeH activation (4and 5, Fig. 1).31e33 Buchmeiser et al. reported Ru-based metathesiscatalysts that reached high Z-selectivity in ROMP.34 Jensen et al.also reported synthesis of a new catalyst revealing high Z-selec-tivity, but only in low conversion.35 Another recent modification,described by Hoveyda et al., consisting in introduction of a dithio-late ligand, which provided high efficiency and Z-selectivity.36
The stereochemical outcome of metathetic transformation ismostly influenced by spatial arrangement of substituents aroundthe reaction intermediate (the ruthenacyclobutane). We assumedthat the approach of an olefinic substrate to the active 14-electronRu benzylidene complex can be controlled by steric hindrance oftheN-heterocyclic carbene (NHC) ligand.With this premise inmindwe have designed new catalysts bearing polyether clamps linkingN,N0-2,4-dimethylphenyl substituents (6 and 7, Fig. 2). In ouropinion, the polyether macrocyclic ring may interact with the re-active metallic center and influence the reactivity and stereo-chemistry of metathetic transformation.
Fig. 2. New modified ruthenium metathesis catalysts.
The incorporation of ions or neutral molecules into the poly-etheral cavity may be expected. Luning et al. synthesized macro-bicyclic concave salts as precursors for N-heterocyclic carbenes.Their application was not investigated, but authors expectedchemo-, regio- and diastereoselectivity in organocatalysis andmetal catalyzed reactions.37
Scheme 2. Synthesis of new catalysts.
2. Results and discussion
2.1. Synthesis of NHC precursors
New imidazolinium salts were obtained in a simple and efficientsynthetic way (Scheme 1). The reaction of 3,5-dimethyl-2-
Scheme 1. Synthesis of new NHC precursors. Reagents and conditions: a) BrCH2(CH2OCHdioxane, HCOOH, THF, molecular sieves 4 �A, then NaBH3CN; d) NH4Cl, HC(OMe)3, HCOOH;
nitrophenol38 with a,u-dibromoether derivatives prepared fromdi- and triethylene glycols (1,5-dibromo-3-oxapentane and 1,8-dibromo-3,6-dioxaoctane)39 gave 8a and 8b, respectively. The re-duction of nitro groups with hydrazine and Pd/C40 provided pureamines 9a and 9b in 65% and 92% yields, respectively. The standardreaction of 9a and 9b with glyoxal solution led to the formation ofimines only in very poor yields. However, replacement of glyoxalwith 2,3-dihydroxy-1,4-dioxane followed by reduction of theresulting imines with sodium cyanoborohydride provided ethyl-enediamines (10a and 10b) in high yields (60% and 85%, re-spectively). The imidazolinium salts 11a and 11b were obtainedusing a classical method.41 The salts can be deprotonated to thecorresponding NHC by potassium tert-amylate and used immedi-ately to the preparation of catalysts. However, it is advisable totransform them into the stable and easy to handle NHC chloroformadducts 12a and 12b according to the literature.42
2.2. Synthesis of new catalysts
New catalysts 6 and 7 were synthesized in two alternative ways(Scheme 2). The carbene adducts 12a and 12b were subjected tothe ligand exchange with Hoveyda-Grubbs first generation catalystto give 6 and 7 (48% and 73% yield, respectively). In a shorter way,the salts 11a and 11b were deprotonated in situ with t-AmOK andreacted with HoveydaeGrubbs first generation complex, providingthe catalysts in comparable yields. This method is faster and itdoes not involve isolation of free carbene. However, the purity ofsalt is very important. Excess base should be avoided because itdecreases significantly the reaction yield. The alternative methodof the catalyst preparation from chloroform adducts seems to beeasier and more efficient (fewer side products are formed). Thecatalysts can be subjected to purification by silica gel columnchromatography in air using undistilled, reagent-grade solvents.
2)nCH2Br, K2CO3, 18-C-6, DMF; b) NH2NH2 hydrate, Pd/C, EtOH; c) 2,3-dihydroxy-1,4-e) NaOH, CHCl3, toluene.

A. Hryniewicka et al. / Tetrahedron 70 (2014) 6810e68166812
They are storable in a fridge for a few months without significantloss of reactivity.
However, the catalyst 7 is more stable and was formed in betteryield than 6. It should be noticed that yields of every step in thesynthesis of the salts 11 are better in case of products with largerpolyether clamp (compounds named as b). This may be explainedby the bigger steric strain in the structure of compounds namedas a.
An X-ray crystal structure analysis of the catalyst 7 was per-formed to determine the molecular structure and to explain on thisbasis its catalytic stereoselectivity (Fig. 3). Indeed, location of thepolyether linker significantly reduces accessibility of the upper-back whereas two methyl groups in o-position hinder front partof the catalytic site of 7.
Fig. 3. Single crystal X-ray structure of catalyst 7. Thermal ellipsoids are drawn at 30%probability level. Selected interatomic distances: Ru1eC1¼1.973(3), Ru1eC26¼1.825(3);Ru1eCl1¼2.3350(7); Ru1eCl2¼2.3392(6); Ru1eO5¼2.258(2) �A; angles: C1eRu1eO5¼178.17(9); C1eRu1eC26¼102.3(1); O5eRu1eC26¼79.56(9); Cl1eRu1eCl2¼92.65(7),C1eRu1eCl1¼92.65(7); C1eRu1eCl2¼93.23(7); O5eRu1eCl1¼87.02(5); O5eRu1eCl2¼86.52(5), C26eRu1eCl1¼98.43(9); C26eRu1eCl2¼98.40(9)� .
2.3. Testing of new catalysts
The new complexes appeared to be very efficient as RCM cata-lysts, initiating (faster) formation of disubstituted olefins (Table 1,entry 1) than the commercial Grubbs second generation (2) andHoveyda second generation complexes (3). In the more challengingsynthesis of tetrasubstituted double bonds (Table 1, entry 3) theyshowed higher reactivity compared to the complex 3 and lowerreactivity compared to 2. Nevertheless, it is worth emphasized thatthe newcatalysts proved very efficient in an enyne reaction (Table 1,entry 4). All enyne reactionswere carried out at 0 �Cwith a charge of0.5mol % of the catalyst. They gavemuch higher yields compared tothat of the commercial complexes 2 and 3 and similar to that of thenitro-catalyst.43 It should be mentioned that the chromenyl andnitrochromenyl catalysts synthesized earlier in our laboratoryrevealed comparable reactivity in this type of reaction.44,45
The catalysts were also tested in model CM reactions for the de-termination of E/Z stereoselectivity. One can notice that the newcatalysts showed high efficiency in CM reactions comparable to thatof commercially available catalysts (Table 2). No significant increasein Z-selectivity was observed. However, it should be noticed that incase of newcomplexes 6 and 7 contribution of olefinic productswithZ-configuration were slightly higher than that obtained using com-mercial catalysts 2 and 3 (Table 2). In the case of macrocyclization
(RCM) of the 13-membered ring in lactone 33 using new catalysts 6and 7 contribution of Z-isomer exceeded 20% (Table 3, entry 1). Thecomplex 7was also used for the synthesis of molecular rotors basedon steroids.46 The macrocyclization of diene 34 in the presence of 7led to the 34-membered macrocycle 35 with preference to the Z-isomer E/Z 1:2 (Table 3, entry 2). In order to avoid intermolecularreactions high dilution of the reaction mixture was used.
In order to improve selectivity of the obtained catalysts in-corporation of ions or neutral molecules into the macrocyclic cavein NHC is planned in due course.
3. Conclusions
In summary, simple and efficient synthesis of NHC pre-cursorsdsalts 11a and 11b, containing new structural ele-mentdpolyether climbing ring, was described. New catalysts 6 and7 bearingmodified NHC units were synthesized from the respectivesalts by exchange of the phosphinic ligand in Hoveyda first gener-ation complex with NHC fragment obtained directly by deproto-nation of the proper salts or thermal decomposition ofNHCechloroform adducts. The new complexes 6 and 7were testedin model RCM, leading to formation of five-membered and mac-rocyclic rings, as well as enyne and CM reactions. The catalystsshowed activity comparable to that of commercially availableGrubbs second generation and HoveydaeGrubbs second genera-tion complexes. The catalysts showed also slightly better Z-selec-tivity in model CM reactions. The complex 7 appeared to be Z-selective in formation of macrocyclic product 35.
4. Experimental section
4.1. General remarks
All manipulations of organometallic compounds were per-formed using standard Schlenk techniques under an atmosphereof dry argon. CH2Cl2 and DMF were dried by distillation over CaH2,THF over Na/benzophenone, toluene over Na (deoxygenated bybubbling He for 5 min). Melting points were determined ona Kofler apparatus of the Boetius type and were uncorrected. 1Hand 13C NMR spectra were recorded on a Bruker Avance II spec-trometer (400 and 100 MHz, respectively). Spectra are referencedrelative to the chemical shift (d) of TMS. Mass spectra were ob-tained with Micromass LCT TOF and AutoSpec Premier (Waters)spectrometers. IR spectra were recorded on a Nicolet series IIMagna-IR 550 FT-IR spectrometer. Flash chromatography (FC) wasperformed on silica gel 230e400 mesh. Catalysts: 1, 2, 3 andHoveydaeGrubbs first generation complex were commerciallyavailable. 3,5-Dimethyl-2-nitrophenol,38 1,5-dibromo-3-oxapentane and 1,8-dibromo-3,6-dioxaoctane,47 2,3-dihydroxy-1,4-dioxane48 were synthesized according to literature procedures.Substrates for testing catalysts are prepared by etherification ofproper commercial compounds with allyl bromide or 3-chloro-2-methylpropene. Other chemicals are commercially available andused as received.
4.2. Synthesis of NHC precursors
4.2.1. 1,5-Bis(3,5-dimethyl-2-nitrophenoxy)-3-oxapentane (8a). Tothe stirred mixture of K2CO3 (1.32 g, 9.62 mmol) and crown ether18-C-6 (5 mg) in dry DMF (10 mL) a solution of 3,5-dimethyl-2-nitrophenol (1.60 g, 9.6 mmol) in DMF (8 mL) was added and re-action mixture was stirred for 1 h. After that time 1,5-dibromo-3-oxapentane (1.11 g, 4.8 mmol) was added. The solution was stir-red at room temperature for 16 h and 2 h in 80 �C.Water was addedand reaction mixture was extracted with diethyl ether. Organiclayers were washed with brine and water and evaporated to
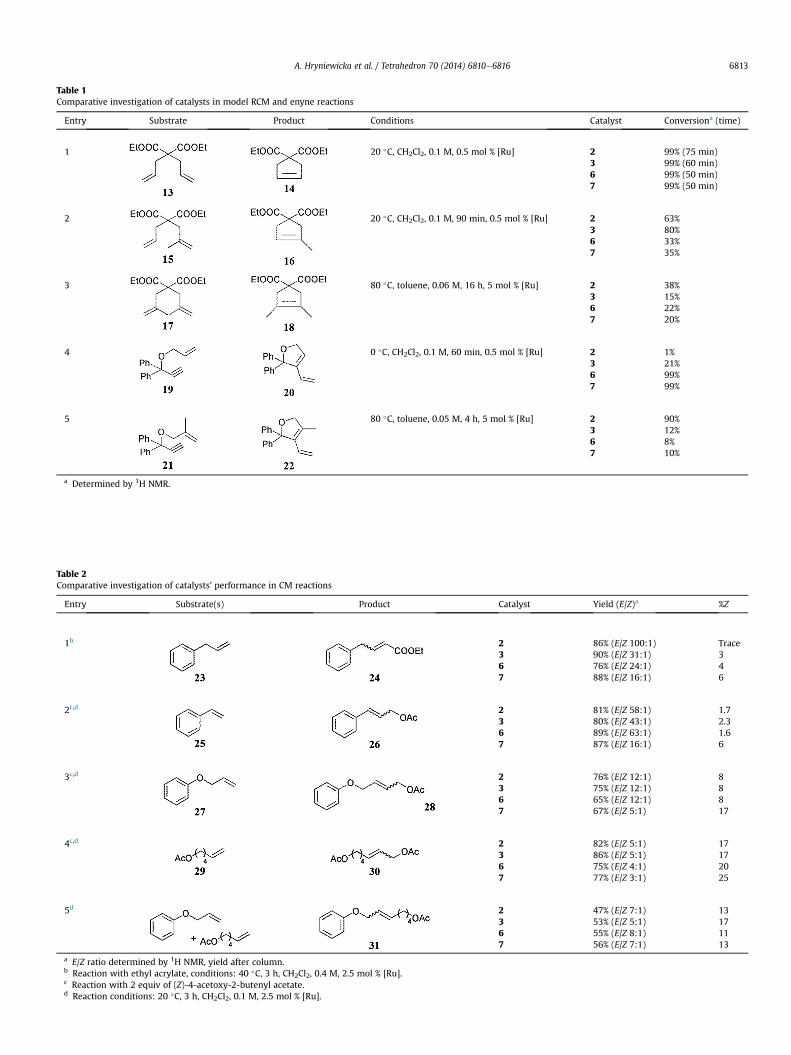
Table 2Comparative investigation of catalysts’ performance in CM reactions
Entry Substrate(s) Product Catalyst Yield (E/Z)a %Z
1b 2 86% (E/Z 100:1) Trace3 90% (E/Z 31:1) 36 76% (E/Z 24:1) 47 88% (E/Z 16:1) 6
2c,d 2 81% (E/Z 58:1) 1.73 80% (E/Z 43:1) 2.36 89% (E/Z 63:1) 1.67 87% (E/Z 16:1) 6
3c,d 2 76% (E/Z 12:1) 83 75% (E/Z 12:1) 86 65% (E/Z 12:1) 87 67% (E/Z 5:1) 17
4c,d 2 82% (E/Z 5:1) 173 86% (E/Z 5:1) 176 75% (E/Z 4:1) 207 77% (E/Z 3:1) 25
5d 2 47% (E/Z 7:1) 133 53% (E/Z 5:1) 176 55% (E/Z 8:1) 117 56% (E/Z 7:1) 13
a E/Z ratio determined by 1H NMR, yield after column.b Reaction with ethyl acrylate, conditions: 40 �C, 3 h, CH2Cl2, 0.4 M, 2.5 mol % [Ru].c Reaction with 2 equiv of (Z)-4-acetoxy-2-butenyl acetate.d Reaction conditions: 20 �C, 3 h, CH2Cl2, 0.1 M, 2.5 mol % [Ru].
Table 1Comparative investigation of catalysts in model RCM and enyne reactions
Entry Substrate Product Conditions Catalyst Conversiona (time)
1 20 �C, CH2Cl2, 0.1 M, 0.5 mol % [Ru] 2 99% (75 min)3 99% (60 min)6 99% (50 min)7 99% (50 min)
2 20 �C, CH2Cl2, 0.1 M, 90 min, 0.5 mol % [Ru] 2 63%3 80%6 33%7 35%
3 80 �C, toluene, 0.06 M, 16 h, 5 mol % [Ru] 2 38%3 15%6 22%7 20%
4 0 �C, CH2Cl2, 0.1 M, 60 min, 0.5 mol % [Ru] 2 1%3 21%6 99%7 99%
5 80 �C, toluene, 0.05 M, 4 h, 5 mol % [Ru] 2 90%3 12%6 8%7 10%
a Determined by 1H NMR.
A. Hryniewicka et al. / Tetrahedron 70 (2014) 6810e6816 6813

Table 3Comparative investigation of catalysts’ performance in RCM formation of macrocycles 33 and 35
Entry Substrate Product Catalyst Yield (E/Z)a %Z
1b 2 38% (E/Z 6:1) 143 39% (E/Z 6:1) 146 35% (E/Z 4:1) 207 36% (E/Z 3.5:1) 22
2c 1 58% (E/Z 1:1) 502 62% (E/Z 2:1) 333 63% (E/Z 1:1) 507 53% (E/Z 1:2) 67
a E/Z ratio determined by 1H NMR, yield after column.b Reaction conditions: 60 �C, 3 h, toluene, 0.001 M, 2.5 mol % [Ru].c Reaction conditions: 80 �C, toluene, 0.00001 M, 15 mol % [Ru].
A. Hryniewicka et al. / Tetrahedron 70 (2014) 6810e68166814
dryness to give white solid (1.76 g, 91%), mp 77e79 �C; nmax (CHCl3)2932, 2873, 1597, 1520, 1449, 1325, 1249, 1093 cm�1; dH (400 MHz,CDCl3) 6.70 (2H, s, HAr), 6.65 (2H, s, HAr), 4.18e4.15 (4H, m,eCH2CH2Oe), 3.85e3.83 (4H, m, eCH2CH2Oe), 2.32 (6H, s, CH3),2.26 (6H, s, CH3); dC (100 MHz, CDCl3) 150.1, 141.4, 140.3, 130.7,123.5, 112.2, 69.6, 69.4, 21.6, 17.0; m/z (ESI) 427 ([MþNa]þ 100%);HRMS (ESI): found 427.1467. [C20H24N2O7Na]þ requires 427.1476.
4.2.2. 1,8-Bis(3,5-dimethyl-2-nitrophenoxy)-3,6-dioxaoctane(8b). The above procedure for the synthesis of the compound 8awas followed using 3,5-dimethyl-2-nitrophenol (1.20 g, 7.2 mmol),1,8-dibromo-3,6-dioxaoctane (0.99 g, 3.6 mmol), K2CO3 (0.99 g,7.16 mmol) and 18-C-6 (5 mg). Reaction time at 80 �C was pro-longed to 4 h. After work-up, white solid was obtained (1.47 g, 91%),mp 143e145 �C; nmax (CHCl3) 2957, 2886, 1599, 1532, 1479, 1313,1249, 1113 cm�1; dH (400 MHz, CDCl3) 6.70 (2H, s, HAr), 6.65 (2H, s,HAr), 4.19e4.16 (4H, m, eCH2CH2Oe), 3.83e3.81 (4H, m,eCH2CH2Oe), 3.68 (4H, s, eCH2CH2Oe), 2.32 (6H, s, CH3), 2.26 (6H,s, CH3); dC (100 MHz, CDCl3) 150.2, 141.3, 140.3, 130.7, 123.4, 112.1,71.0, 69.3, 21.6, 17.0; m/z (ESI) 471 (100% [MþNa]þ); HRMS (ESI):found 471.1724. [C22H28N2O8Na]þ requires 471.1738.
4.2.3. 1,5-Bis(2-amino-3,5-dimethylphenoxy)-3-oxapentane (9a). Tothe stirredmixture of compound 8a (1.76 g, 4.36mmol) and 10% Pd/C (0.29 g) in absolute ethanol (15 mL) hydrazine hydrate was addeddropwise (80%, 10 mL) for 30 min. Reaction mixture was refluxedfor 2 h. Catalyst was filtered and supernatant was cooled to 5 �C.White solid precipitated, which was filtered off and dried to obtaina product (0.97 g, 65%), mp 103e105 �C; nmax (CHCl3) 3408, 2915,1588, 1453, 1299, 1251, 1122 cm�1; dH (400 MHz, CDCl3) 6.58 (4H, s,HAr), 4.19e4.17 (4H, m, eCH2CH2Oe), 3.94e3.91 (4H, m,eCH2CH2Oe), 3.71 (4H, br s, NH2), 2.26 (6H, s, CH3), 2.16 (6H, s,CH3); dC (100 MHz, CDCl3) 145.9, 132.5, 126.8, 123.7, 122.9, 111.7,69.9, 68.6, 20.8, 17.1; m/z (ESI) 367 (100% [MþNa]þ); HRMS (ESI):found 367.1983. [C20H28N2O3Na]þ requires 367.1992.
4.2.4. 1,8-Bis(2-amino-3,5-dimethylphenoxy)-3,6-dioxaoctane(9b). The above procedure for the synthesis of the compound 9awas followed using compound 8b (1.47 g, 3.28mmol), Pd/C (0.22 g),hydrazine hydrate (7.5 mL) to obtain white solid (1.17 g, 92%), mp
92e95 �C; nmax (CHCl3) 3430, 2912, 1589, 1505, 1458, 1304, 1253,1131 cm�1; dH (400 MHz, CDCl3) 6.55 (4H, s, HAr), 4.17e4.12 (4H, m,eCH2CH2Oe), 3.89e3.84 (4H, m, eCH2CH2Oe), 3.76 (4H, s,eCH2CH2Oe), 3.58 (4H, br s, NH2), 2.24 (6H, s, CH3), 2.12 (6H, s,CH3); dC (100 MHz, CD3OD) 148.0, 133.0, 128.9, 125.1, 124.7, 112.6,71.7, 70.9, 69.6, 21.0, 17.3; m/z (ESI) 411 (100% [MþNa]þ); HRMS(ESI): found 411.2242. [C22H32N2O4Na]þ requires 411.2254.
4.2.5. Ethylenediamine 10a. To a solution of amine 9a (500 mg,1.45 mmol) in dry THF (20 mL) 2,3-dihydroxy-1,4-dioxane (171 mg,1.45 mmol), formic acid (3 drops) and molecular sieves 4 �A wereadded. The reaction mixturewas stirred for 48 h. Yellow precipitateappeared. Then sodium cyanoborohydride (183 mg, 2.9 mmol) wasadded and reactionmixturewas stirred for 1 h. The reactionmixturewas quenched with water and extracted with diethyl ether. Organiclayers were washed with brine and water and evaporated to dry-ness. The crudeproductwas purified by FC (hexane/ethyl acetate v/v5:1) to give colorless oil (350mg, 65%); nmax (CHCl3) 2935,1611,1592,1454, 1244, 1099 cm�1; dH (400 MHz, CDCl3) 6.62 (2H, s, HAr), 6.57(2H, s, HAr), 4.21e4.19 (4H, m, eCH2CH2Oe), 3.90e3.88 (4H, m,eCH2CH2Oe), 3.02 (4H, s,eNCH2CH2Ne), 2.28 (6H, s, CH3), 2.27 (6H,s, CH3); dC (100 MHz, CDCl3): 150.8, 135.1, 131.0, 130.8, 125.0, 111.7,69.1, 68.0, 47.8, 21.0, 18.5; m/z (ESI) 393 (100% [MþNa]þ); HRMS(ESI): found 393.2138. [C22H30N2O3Na]þ requires 393.2149.
4.2.6. Ethylenediamine 10b. The above procedure for the synthesisof the compound 10a was followed using compound 9b (500 mg,1.29 mmol), 2,3-dihydroxy-1,4-dioxane (152 mg, 1.29 mmol),HCOOH (3 drops) and sodium cyanoborohydride (162 mg,2.58 mmol) to obtain colorless oil (460 mg, 86%); nmax (CHCl3) 2927,1725, 1613, 1592, 1456, 1101 cm�1; dH (400 MHz, CDCl3) 6.60 (2H, s,HAr), 6.54 (2H, s, HAr), 4.16e4.13 (4H, m, eCH2CH2Oe), 3.90e3.88(4H, m, eCH2CH2Oe), 3.71 (4H, s, eCH2CH2Oe), 3.07 (4H, s,eNCH2CH2Ne), 2.26 (6H, s, CH3), 2.25 (6H, s, CH3); dC (100 MHz,CDCl3) 151.0, 134.7, 131.0, 130.7, 124.4, 111.0, 70.9, 69.7, 68.7, 48.5,21.0, 18.2; m/z (ESI) 437 (100% [MþNa]þ); HRMS (ESI): found437.2399. [C24H34N2O4Na]þ requires 437.2411.
4.2.7. Imidazolinium salt 11a. To the solution of ethylenediamine10a (300 mg, 0.81 mmol) in trimethyl orthoformate (10 mL)

A. Hryniewicka et al. / Tetrahedron 70 (2014) 6810e6816 6815
ammonium chloride (65 mg, 1.22 mmol) and formic acid (2 drops)were added. The reaction mixture was refluxed for 2 h. Methylenechloride was added to reaction mixture, and solids were filtered off.The filtrate was evaporated to dryness, crude product was redis-solved in CH2Cl2 and precipitatedwith ethyl ether. The solid residuewas filtered and dried under vacuum to afford white product(260mg, 77%), mp 243e245 �C; nmax (CHCl3) 2934,1637,1592,1455,1255, 1142, 1104 cm�1; dH (400 MHz, CDCl3) 8.26 (1H, s, eCH]),6.67 (2H, s, HAr), 6.61 (2H, s, HAr), 4.58 (4H, s, eNCH2CH2Ne),4.18e4.02 (4H, m, eCH2CH2Oe), 3.99e3.82 (4H, m, eCH2CH2Oe),2.35 (6H, s, CH3), 2.33 (6H, s, CH3); dC (100 MHz, CDCl3) 161.3, 154.0,142.0, 136.5, 124.0, 120.5, 111.5, 68.9, 67.4, 51.9, 21.7, 17.4; m/z (ESI)381 (100% [M�Cl]þ); HRMS (ESI): found 381.2160. [C23H29N2O3]þ
requires 381.2173.
4.2.8. Imidazolinium salt 11b. The above procedure for the syn-thesis of the compound 11a was followed using compound 10b(300 mg, 0.72 mmol), NH4Cl (58 mg,1.1 mmol), HCOOH (2 drops) intrimethyl orthoformate (10 mL) to obtain of white solid (320 mg,89%), mp 257.0 �C; nmax (CHCl3) 2954, 1638, 1591, 1454, 1259, 1170,1101 cm�1; dH (400 MHz, CDCl3) 8.78 (1H, s, eCH]), 6.68 (2H, s,HAr), 6.59 (2H, s, HAr), 4.55 (4H, s, eNCH2CH2Ne), 4.17e4.15 (4H, m,eCH2CH2Oe), 3.82e3.80 (4H, m, eCH2CH2Oe), 3.63 (4H, s,eCH2CH2Oe), 2.38 (6H, s, CH3), 2.28 (6H, s, CH3); dC (100 MHz,CDCl3) 160.9, 153.4, 141.3, 135.6, 124.2, 120.7, 111.8, 69.7, 68.5, 67.8,51.4, 21.7, 17.6; m/z (ESI) 425 (100% [M�Cl]þ); HRMS (ESI): found425.2421. [C25H33N2O4]þ requires 425.2435.
4.2.9. NHC chloroform adduct 12a. To a stirred suspension ofpowdered (ground in a mortar) sodium hydroxide (688 mg,17.2 mmol) in dry, degassed toluene (10 mL), chloroform (130 mL,1.42 mmol) was added by microsyringe. After 10 min at roomtemperature, salt 11a (200 mg, 0.43 mmol) was added, and thereaction mixture was heated at 60 �C for 2 h. The mixture wasallowed to cool down to room temperature and filtered. The filtratewas concentrated under vacuum to a white solid. This crudeproduct was purified through a silica gel plug (hexane/ethyl acetatev/v 5:1) to yield of product as awhite solid (101mg, 47%), mp 165 �C(decomposition); nmax (CHCl3) 2926, 2958, 1578, 1489, 1317, 1091,808, 625 cm�1; dH (400 MHz, CDCl3) 6.69 (2H, s, HAr), 6.64 (2H, s,HAr), 6.38 (1H, s, HCeCCl3), 4.31e4.30 (4H, m, eCH2CH2Oe),4.22e4.21 (4H, m, eCH2CH2Oe), 3.75e3.74 (2H, m, eNCH2CH2Ne),3.24e3.23 (2H, m, eNCH2CH2Ne), 2.44 (6H, s, CH3), 2.33 (6H, s,CH3); dC (100MHz, CDCl3) 155.1, 139.1, 135.3, 133.1, 124.1, 111.3, 86.2,71.2, 67.8, 52.7, 21.3, 19.1; m/z (ESI) 381 (100% [M�CCl3]þ); HRMS(ESI): found 381.2159. [C23H29N2O3]þ requires 381.2173.
4.2.10. NHC chloroform adduct 12b. The above procedure for thesynthesis of the compound 12awas followed using compound 11b(300 mg; 0.6 mmol), sodium hydroxide (960 mg, 24 mmol), chlo-roform (165 mL, 2 mmol) to obtain a white solid (244 mg, 75%), mp133 �C (decomposition); nmax (CHCl3) 2939, 2910, 1576, 1485, 1310,1074, 801, 627 cm�1; dH (400MHz, CDCl3) 6.74 (2H, s, HAr), 6.68 (2H,s, HAr), 6.22 (1H, s, HCeCCl3), 4.31e4.21 (4H, m, eCH2CH2Oe),4.13e4.08 (2H, m, eCH2CH2Oe), 4.02e3.97 (2H, m, eCH2CH2Oe),3.80 (4H, s, eCH2CH2Oe), 3.76e3.73 (2H, m, eNCH2CH2Ne),3.22e3.18 (2H, m, eNCH2CH2Ne), 2.41 (6H, s, CH3), 2.3 (6H, s, CH3);dC (100 MHz, CDCl3) 156.0, 138.8, 135.2, 134.3, 124.4, 114.2, 87.2,71.3, 70.6, 69.9, 52.5, 21.2, 19.1; m/z (ESI) 425 (100% [M�CCl3]þ);HRMS (ESI): found 425.2419. [C25H33N2O4]þ requires 425.2435.
4.3. Synthesis of catalysts
4.3.1. Catalyst 6. Method (I) The HoveydaeGrubbs first generationcatalyst (120 mg, 0.2 mmol) and adduct 12a (100 mg, 0.2 mmol)were heated at 110 �C in dry deoxygenated toluene (10mL) over 3 h.
The reaction mixture was purified by FC (first CH2Cl2, then hexane/ethyl acetate v/v 1:1) to give green catalyst 6 (67 mg, 48%).
Method (II) To a suspension of 11a (83 mg, 0.2 mmol) in dry tol-uene (10 mL) in a Schlenk flask potassium tert-amylate (118 mL,0.2 mmol) was added and reaction was stirred for 30 min at roomtemperature. Then solution of HoveydaeGrubbs first generationcatalyst (120 mg, 0.2 mmol) in toluene (5 mL) and refluxed reactionmixture for 3 h. After purification catalyst 6 was obtained (63 mg,45%), mp 129e130 �C; nmax (CHCl3) 2929, 1726, 1668, 1589, 1493,1330, 1277, 1114 cm�1; dH (400 MHz, CDCl3) 16.83 (1H, s, Ru]CHe),7.55e7.46 (1H, m, HAr), 6.98e6.96 (1H, m, HAr), 6.88e6.81 (2H, m,HAr), 6.79 (2H, s, HAr), 6.68 (2H, s, HAr), 4.92 (1H, septet, J 6.1 Hz,eOCH(CH3)2), 4.34e4.30 (2H, m, eCH2CH2Ne), 4.29e4.19 (2H, m,eCH2CH2Oe), 4.12e4.05 (4H, m, eCH2CH2Oe), 4.04e4.01 (2H, m,eNCH2CH2Ne), 3.79e3.73 (2H, m, eCH2CH2Oe), 2.51 (6H, s, CH3),2.43 (6H, s, CH3), 1.37 (6H, d, J 6.1 Hz, eOCH(CH3)2); dC (100 MHz,CDCl3) 287.5, 212.9,157.2,152.2,145.1,139.5,139.2,129.2,122.8,122.6,122.2, 112.8, 110.4, 74.6, 66.6, 60.4, 52.2, 21.8, 21.3, 18.8;m/z (EI) 702(33), 700 (35), 586 (46), 381 (52), 296 (29), 212 (56),162 (41),147 (55),131 (36),120 (100), 93 (43), 91 (64), 69 (88), 63 (40), 43 (36%); HRMS(EI): found: 700.1423. [C33H40
35Cl2N2O4102Ru]þ requires 700.1403.
4.3.2. Catalyst 7. The above method (I) for the synthesis of thecatalyst 6 was followed using Hoveyda catalyst first (111 mg,0.18mmol) and compound 12b (100mg; 0.18mmol) to afford greencatalyst 7 (98 mg, 73% yield).
The above method (II) for the synthesis of the catalyst 6 wasfollowed using salt 11b (92 mg, 0.2 mmol), t-AmOK (118 mL,0.2 mmol) and Hoveyda catalyst first (120 mg, 0.2 mmol) to affordcatalyst 7 (97 mg, 65%), mp 120e121 �C; nmax (CHCl3) 2985, 1731,1590, 1475, 1454, 1329, 1262, 1113 cm�1; dH (400 MHz, CDCl3) 16.97(1H, s, Ru]CHe), 7.50e7.46 (1H, m, HAr), 7.00e6.98 (1H, m, HAr),6.88e6.79 (2H, m, HAr), 6.82 (2H, s, HAr), 6.69 (2H, s, HAr), 4.90 (1H,septet, J 6.1 Hz, eOCH(CH3)2), 4.38 (2H, t, J 8.7 Hz, eNCH2CH2Ne),4.25e4.07 (8H,m,eCH2CH2Oe), 4.01 (2H, t, J 8.7 Hz,eNCH2CH2Ne),3.79e3.69 (4H, m, eCH2CH2Oe), 2.48 (6H, s, CH3), 2.42 (6H, s, CH3),1.29 (6H, d, J 6.1 Hz,eOCH(CH3)2); dC (100MHz, CDCl3) 294.8, 210.4,157.4,152.3,145.2,140.5,139.6,129.1,125.9,123.3,122.4,122.2,112.9,110.6, 74.7, 69.5, 69.2, 68.7, 51.3, 21.7, 21.1,19.2 ppm;m/z (EI) 746 (9),744 (10), 630 (2), 499 (2), 425 (7), 422 (9), 296 (21), 212 (29),162 (9),150 (25), 147 (17), 120 (26), 108 (100), 107 (37), 91 (17), 77 (13), 69(24), 63 (11), 43 (25), 41 (11%); HRMS (EI): found 744.1688.[C35H44
35Cl2N2O5102Ru]þ requires 744.1665. Crystals suitable for X-ray
analysis were obtained by slow evaporation of a dichloromethane/n-heptane solution of the catalyst for 10 h in the refrigerator.
4.4. X-ray crystallographic data
C35H44Cl2N2O5Ru, green plates of 0.23�0.14�0.03 mm; mono-clinic space group P21/n; a¼11.5099(8), b¼18.4086(14),c¼16.0400(12) �A; b¼93.772(4)�; V¼3391.2(4) �A3; Z¼4;Dcalcd¼1.459 mG m�3; m(CuKa)¼5.546 mm�1; 27,226 reflectionsmeasured at Bruker APEX-II CCD diffractometer at room tempera-ture; 5496 reflections with I>2s(I) were used for structure solutionand refinement; data were corrected for numerical absorption us-ing program SADABS of the Bruker SHELXTL system: Tmin¼0.3569,Tmax¼0.8425; R¼0.0325; Rw¼0.0860.
CCDC 988535 contains the supplementary crystallographic datafor this compound. These data can be obtained free of charge fromThe Cambridge Crystallographic Data Centre via www.ccdc.cam.a-c.uk/data_request/cif.
Acknowledgements
The authors acknowledge financial support from the PolishNational Science Centre (DEC-2011/02/A/ST5/00459). We also

A. Hryniewicka et al. / Tetrahedron 70 (2014) 6810e68166816
thank Dr. Krzysztof Brzezi�nski for helpful remarks in preparation ofsample for X-ray measurement.
Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.tet.2014.07.056.
References and notes
1. Samoj1owicz, C.; Bieniek, M.; Grela, K. Chem. Rev. 2009, 109, 3708.2. Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2003, 42, 4592.3. Vougioukalakis, G. C.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746.4. Astruc, D. New J. Chem. 2005, 29, 42.5. F€urstner, A. Angew. Chem., Int. Ed. 2000, 39, 3012.6. Trnka, T. M.; Grubbs, R. H. Acc. Chem. Res. 2001, 34, 18.7. Leitgeb, A.; Wappel, J.; Slugovc, C. Polymer 2010, 51, 2927.8. Liu, X.; Basu, A. J. Organomet. Chem. 2006, 691, 5148.9. Sutthasupa, S.; Shiotsuki, M.; Sanda, F. Polym. J. 2010, 42, 905.
10. Cossy, J.; Arseniyadis, S.; Meyer, C. Metathesis in Natural Product Synthesis:Strategies, Substrates, and Catalysts; Wiley-VCH: Weinheim, Germany, 2010.
11. Binder, J. B.; Raines, R. T. Curr. Opin. Chem. Biol. 2008, 12, 767.12. Schrodi, Y.; Ung, T.; Vargas, A.; Mkrtumyan, G.; Lee, C. W.; Champagne, T. M.;
Pederson, R. L.; Hong, S. H. Clean Soil Air Water 2008, 36, 669.13. Schwab, P.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc. 1996, 118, 100.14. Scholl, M.; Ding, S.; Lee, C. W.; Grubbs, R. H. Org. Lett. 1999, 1, 953.15. Garber, S. B.; Kingsbury, J. S.; Gray, B. L.; Hoveyda, A. H. J. Am. Chem. Soc. 2000,
122, 8168.16. Gessler, S.; Randl, S.; Blechert, S. Tetrahedron Lett. 2000, 41, 9973.17. �Sled�z, P.; Mauduit, M.; Grela, K. Chem. Soc. Rev. 2008, 37, 2433.18. Buchmeiser, M. R. Chem. Rev. 2008, 109, 303.19. Ko�snik, W.; Grela, K. Dalton Trans. 2013, 42, 7463.20. Kluciar, M.; Grela, K.; Mauduit, M. Dalton Trans. 2013, 42, 7354.21. Shahane, S.; Bruneau, C.; Fischmeister, C. ChemCatChem 2013, 5, 3436.22. Poater, A.; Falivene, L.; Urbina-Blanco, C. A.; Manzini, S.; Nolan, S. P.; Cavallo, L.
Dalton Trans. 2013, 42, 7433.23. Lujan, C.; Nolan, S. P. J. Organomet. Chem. 2011, 696, 3935.
24. Lujan, C.; Nolan, S. P. Catal. Sci. Technol. 2012, 2, 1027.25. Gallenkamp, D.; F€urstner, A. J. Am. Chem. Soc. 2011, 133, 9232.26. Wang, Y.; Jimenez, M.; Hansen, A. S.; Raiber, E. A.; Schreiber, S. L.; Young, D. W.
J. Am. Chem. Soc. 2011, 133, 9196.27. Jiang, A. J.; Zhao, Y.; Schrock, R. R.; Hoveyda, A. H. J. Am. Chem. Soc. 2009, 131,
16630.28. Marinescu, S. C.; Levine, D. S.; Zhao, Y.; Schrock, R. R.; Hoveyda, A. H. J. Am.
Chem. Soc. 2011, 133, 11512.29. Marinescu, S. C.; Schrock, R. R.; M€uller, P.; Takase, M. K.; Hoveyda, A. H. Or-
ganometallics 2011, 30, 1780.30. Meek, S. J.; O’Brien, R. V.; Llaveria, J.; Schrock, R. R.; Hoveyda, A. H. Nature 2011,
471, 461.31. Endo, K.; Grubbs, R. H. J. Am. Chem. Soc. 2011, 133, 8525.32. Keitz, B. K.; Endo, K.; Herbert, M. B.; Grubbs, R. H. J. Am. Chem. Soc. 2011, 133,
9686.33. Rosebrugh, L. E.; Herbert, M. B.; Marx, V. M.; Keitz, B. K.; Grubbs, R. H. J. Am.
Chem. Soc. 2013, 135, 1276.34. Buchmeiser, M. R.; Ahmad, I.; Gurram, V.; Kumar, P. S. Macromolecules 2011, 44,
4098.35. Occhipinti, G.; Hansen, F. R.; T€ornroos, K. W.; Jensen, V. R. J. Am. Chem. Soc. 2013,
135, 3331.36. Khan, R. K. M.; Torker, S.; Hoveyda, A. H. J. Am. Chem. Soc. 2013, 135, 10258.37. Winkelmann, O.; N€ather, C.; L€uning, U. Eur. J. Org. Chem. 2007, 981.38. Adams, R.; Stewart, H. J. Am. Chem. Soc. 1941, 63, 2859.39. Singh, P.; Verma, R. K.; Singh, M. S. Tetrahedron Lett. 2011, 52, 3818.40. Shargi, H.; Nicknam, K.; Pooyan, M. Tetrahedron 2001, 57, 6057.41. Arduengo, A. J., III; Krafczyk, R.; Schmutzler, R. Tetrahedron 1999, 55, 14523.42. Trnka, T. M.; Morgan, J. P.; Sanford, M. S.; Wilhelm, T. E.; Scholl, M.; Choi, T. L.;
Ding, S.; Day, M. W.; Grubbs, R. H. J. Am. Chem. Soc. 2003, 125, 2546.43. Michrowska, A.; Bujok, R.; Harutyunyan, S.; Sashuk, V.; Dolgonos, G.; Grela, K. J.
Am. Chem. Soc. 2004, 126, 9318.44. Hryniewicka, A.; Koz1owska, A.; Witkowski, S. J. Organomet. Chem. 2012, 701, 87.45. Hryniewicka, A.; Morzycki, J. W.; Witkowski, S. J. Organomet. Chem. 2010, 695,
1265.46. Czajkowska-Szczykowska, D.; Jastrzebska, I.; Santillan, R.; Morzycki, J. W.
Tetrahedron, in press.47. Berube, G.; Rabouin, D.; Perron, V.; N’Zemba, B.; Gaudreault, R.-C.; Parent, S.;
Asselin, E. Steroids 2006, 71, 911.48. Venuti, M. C. Synthesis 1982, 61.












