
ĆWICZENIE 3: Pasywność metali - Alchemik · metalu cienkiej warstewki produktów reakcji metalu...
12
ĆWICZENIE 3: Pasywność metali WPROWADZENIE Odpornośd korozyjna konstrukcyjnych tworzyw metalowych w wielu wypadkach zależy od ich skłonności do pasywacji. Rozwój badao w tej dziedzinie przyczynił się miedzy innymi do otrzymania szeregu nowych stali i stopów odpornych na korozję. W nowoczesnej technice zabezpieczeo przeciwkorozyjnych zjawisko pasywności jest szeroko wykorzystywane przy doborze odpowiednich tworzyw do agresywnych środowisk przemysłu chemicznego, inhibitorów korozji (pasywatorów), przy stosowaniu ochrony anodowej, itp. 1. Podstawy teoretyczne pasywności metali Termin „pasywność” odnosi się do metali zajmujących w szeregu napięciowym miejsce aktywne, a mimo to korodujących z bardzo małą szybkością. Metal pasywny zachowuje się pod względem elektrochemicznym jak metal szlachetny. Pasywności zawdzięczają swoją odpornośd na działanie większości środowisk naturalnych takie metale jak Al., Cr, Ti i ich stopy, oraz stale odporne na korozję. Niektóre metale lub stopy, normalnie aktywne w danym środowi sku korozyjnym można przeprowadzid w stan pasywny przez ekspozycję w środowisku pasywującym (np. żelazo w roztworach chromianu lub azotynu) lub przez polaryzację anodową dostatecznie dużą gęstością prądu (np. w H 2 SO 4 ). Czyste żelazo nie koroduje w stężonym HNO 3 , zaś w rozcieoczonym HNO 3 ulega szybkiemu rozpuszczaniu. Próbka żelazna zanurzona najpierw w stężonym kwasie azotowym zachowuje odpornośd korozyjną po przeniesieniu do rozcieoczonego kwasu. Jednak nawet nieznaczne uszkodzenie mechaniczne powierzchni próbki powoduje gwałtowną reakcję żelaza z rozcieoczonym kwasem, jak przy bezpośrednim zanurzeniu. Powyższe doświadczenia, przeprowadzone przez Faraday’a w latach 40-tych ubiegłego wieku, dowodzą podobnie jak szereg innych późniejszych eksperymentów, że pasywnośd metali jest ściśle związane z obecnością pewnych warstewek na ich powierzchni. Grubośd warstewek zawierających znaczne ilości wody hydratacyjnej ocenia się na 3 mm lub mniej. Natura warstewek pasywnych, a w konsekwencji podstawowy charakter pasywności pozostaje nadal niewyjaśniony. Spośród wielu teorii dotyczących budowy i powstawaniu warstewek pasywnych, zaproponowanych przez różnych badaczy, dwie uważane są za najważniejsze: warstewkowa i adsorpcyjna. Pierwsza utrzymuje, że pasywnośd jest wynikiem obecności na powierzchni metalu cienkiej warstewki produktów reakcji metalu ze środowiskiem, najczęściej tlenków. Warstewka ma charakter bariery dyfuzyjnej i oddzielając metal od środowiska znacznie zmniejsza szybkośd reakcji. Teoria adsorpcyjna zakłada natomiast istnienie na metalu chemisorpcyjnej warstewki, np. tlenu, wypierającej normalnie adsorbowane cząsteczki H2O, przez co obniżona zostaje szybkośd reakcji rozpuszczania anodowego obejmującej hydratację
Transcript of ĆWICZENIE 3: Pasywność metali - Alchemik · metalu cienkiej warstewki produktów reakcji metalu...

ĆWICZENIE 3: Pasywność metali
WPROWADZENIE
Odpornośd korozyjna konstrukcyjnych tworzyw metalowych w wielu wypadkach
zależy od ich skłonności do pasywacji. Rozwój badao w tej dziedzinie przyczynił się miedzy
innymi do otrzymania szeregu nowych stali i stopów odpornych na korozję.
W nowoczesnej technice zabezpieczeo przeciwkorozyjnych zjawisko pasywności jest
szeroko wykorzystywane przy doborze odpowiednich tworzyw do agresywnych środowisk
przemysłu chemicznego, inhibitorów korozji (pasywatorów), przy stosowaniu ochrony
anodowej, itp.
1. Podstawy teoretyczne pasywności metali
Termin „pasywność” odnosi się do metali zajmujących w szeregu napięciowym
miejsce aktywne, a mimo to korodujących z bardzo małą szybkością. Metal pasywny
zachowuje się pod względem elektrochemicznym jak metal szlachetny. Pasywności
zawdzięczają swoją odpornośd na działanie większości środowisk naturalnych takie metale
jak Al., Cr, Ti i ich stopy, oraz stale odporne na korozję.
Niektóre metale lub stopy, normalnie aktywne w danym środowisku korozyjnym
można przeprowadzid w stan pasywny przez ekspozycję w środowisku pasywującym (np.
żelazo w roztworach chromianu lub azotynu) lub przez polaryzację anodową dostatecznie
dużą gęstością prądu (np. w H2SO4).
Czyste żelazo nie koroduje w stężonym HNO3, zaś w rozcieoczonym HNO3 ulega
szybkiemu rozpuszczaniu. Próbka żelazna zanurzona najpierw w stężonym kwasie azotowym
zachowuje odpornośd korozyjną po przeniesieniu do rozcieoczonego kwasu. Jednak nawet
nieznaczne uszkodzenie mechaniczne powierzchni próbki powoduje gwałtowną reakcję
żelaza z rozcieoczonym kwasem, jak przy bezpośrednim zanurzeniu. Powyższe
doświadczenia, przeprowadzone przez Faraday’a w latach 40-tych ubiegłego wieku, dowodzą
podobnie jak szereg innych późniejszych eksperymentów, że pasywnośd metali jest ściśle
związane z obecnością pewnych warstewek na ich powierzchni. Grubośd warstewek
zawierających znaczne ilości wody hydratacyjnej ocenia się na 3 mm lub mniej. Natura
warstewek pasywnych, a w konsekwencji podstawowy charakter pasywności pozostaje nadal
niewyjaśniony.
Spośród wielu teorii dotyczących budowy i powstawaniu warstewek pasywnych,
zaproponowanych przez różnych badaczy, dwie uważane są za najważniejsze: warstewkowa i
adsorpcyjna. Pierwsza utrzymuje, że pasywnośd jest wynikiem obecności na powierzchni
metalu cienkiej warstewki produktów reakcji metalu ze środowiskiem, najczęściej tlenków.
Warstewka ma charakter bariery dyfuzyjnej i oddzielając metal od środowiska znacznie
zmniejsza szybkośd reakcji. Teoria adsorpcyjna zakłada natomiast istnienie na metalu
chemisorpcyjnej warstewki, np. tlenu, wypierającej normalnie adsorbowane cząsteczki H2O,
przez co obniżona zostaje szybkośd reakcji rozpuszczania anodowego obejmującej hydratację

jonów metalu. Zaadsorbowany tlen zmniejsza gęstośd prądu wymiany (zwiększa nadnapięcie
anodowe) reakcji:
Me Men+ + ne
2. Potencjostatyczne krzywe polaryzacji anodowej
W pomiarach korozyjnych szeroko wykorzystuje się techniki otrzymywania krzywych
polaryzacyjnych. W badaniach zjawiska pasywności metali i stopów najbardziej przydatne
okazało się wyznaczanie krzywych polaryzacji anodowej przy użyciu techniki
potencjostatycznej. Polega ona na wyznaczaniu zależności gęstości zewnętrznego prądu
polaryzacyjnego od potencjału badanej elektrody metalowej względem określonej elektrody
odniesienia , iz = f(E).
Jeżeli potencjał elektrody jest zmieniany skokowo (np. co 10 mV) w odstępach czasu
dostatecznie dużych do ustalenia się wartości prądu polaryzującego, wówczas otrzymana
krzywa iz = f(E) nosi nazwę potencjostatycznej. Jeżeli natomiast zmiana potencjału następuje
w sposób ciągły z określoną szybkością, otrzymuje się krzywą potencjokinetyczną lub
potencjodynamiczną.
Utrzymywanie stałej wartości potencjału elektrody lub odpowiednio
zaprogramowaną zmianę potencjału realizuje się w praktyce za pomocą potencjostatu w
układzie trójelektrodowym jak to przedstawiono na rysunku 1.
Rys. 1. Schemat układu do badao potencjostatycznych: EB – elektroda badana, EO –
elektroda odniesienia, EP – elektroda pomocnicza.
Aktualna wartośd różnicy potencjałów między elektrodą badaną (EB) a elektrodą
odniesienia (EO) jest porównywalna z wartością zadaną prze układ programujący potencjału.

Różnica zostaje wzmocniona przez wzmacniacz różnicowy, powodując przepływ między (EB)
a elektrodą pomocniczą (EP) prądu o takim natężeniu i kierunku, aby różnicę potencjałów
sprowadzid do wartości zadanej. Zadana wartośd potencjału może byd stała lub byd
programowana w dowolny sposób, np. skokowo, liniowo, impulsowo. W tym celu układ
programujący wyposażony jest w odpowiedni system napędzający.
Rysunek 2 przestawia schematycznie anodową krzywą polaryzacji żelaza w 1 M
H2SO4.
Rys. 2. Potencjostatyczna krzywa polaryzacji anodowej żelaza w 1 M H2SO4.
W warunkach bez prądu zewnętrznego na żelazie ustala się potencjał korozyjny
(stacjonarny) Ekor. Przy stopniowym podwyższaniu potencjału żelaza przez przepuszczanie
prądu anodowego rośnie szybkośd rozpuszczania żelaza (na wykresie obrazuje odcinek AB).
Przy wartości potencjału około ±250 mV gęstośd prądu osiąga maksimum wynoszące prawie
20000 A/m2 i utrzymuje się na stałym poziomie aż do wartości potencjału +600 mV (punkt B),
przy której następuje gwałtowny spadek gęstości prądu do wartości 0,07 A/m2 (punkt C).
Przy dalszym wzroście potencjału żelaza do około +1,5 V gęstośd prądu prawie nie ulega
zmianie (odcinek CD). Powyżej wartości potencjału transpasywacji (ET) gęstośd prądu
ponownie rośnie (odcinek DE).
Zakres potencjałów miedzy punktami A i B krzywej polaryzacji nazywa się zakresem
aktywnym. Żelazo rozpuszcza się z tworzeniem jonów Fe2+. Potencjał i gęstośd prądu
odpowiadające punktowi B wykresu noszą nazwę potencjału krytycznego pasywacji (Ekr) i

krytycznej gęstości prądu pasywacji (ikr). Odcinek BC przedstawia przejście w stan pasywny,
a odcinek CD odpowiada stanowi pasywnemu metalu w danym roztworze, (tutaj żelaza w 1
M H2SO4). Gęstośd prądu w tym stanie nazywa się gęstością prądu pasywnego (ip). W
zakresie pasywnym żelazo praktycznie nie koroduje. Zakres potencjałów powyżej wartości
potencjału trans pasywacji ET (odcinek DE) nosi nazwę transpasywnego. Na powierzchni
żelaza wydziela się tlen zgodnie z reakcją:
2 H2O → O2 + 4 H+ + 4 e
Przebieg anodowych krzywych polaryzacyjnych dla innych metali lub stopów
skłonnych do pasywacji oraz różnych środowisk elektrolitycznych może w mniejszym lub
większym stopniu różnid się od przykładowej krzywej z rysunku 2. Trzy podstawowe typy
anodowych krzywych polaryzacyjnych uzyskiwanych za pomocą techniki potencjostatycznej
podaje rysunek 3b. Dla lepszego zrozumienia na rysunku 3a przedstawiono teoretyczne
krzywe reakcji anodowej i katodowej (zachodzące na powierzchni metalu zanurzonego w
roztworze kwasu), którym odpowiadają anodowe krzywe polaryzacyjne z rysunku 3b. Dla
uproszczenia przyjęto, że zachodzi tylko jedna reakcja anodowa (rozpuszczanie metalu) oraz jedna
reakcja katodowa (redukcja jonów H+ w środowisku kwaśnym). Z teorii potencjału mieszanego
wiadomo, że sumaryczne szybkości reakcji anodowych i katodowych na powierzchni tej samej
elektrody (lub odpowiadające im gęstości prądu) musza byd sobie równe. Warunek ten na wykresie
3a spełniony jest w punktach przecięcia krzywych obu reakcji.
W przypadku I (rys. 3) teoretyczne krzywe reakcji anodowej i katodowej przecinają się
w punkcie A, którego rzędna Ekor (potencjał korozyjny) leży w zakresie aktywnym potencjału,
zaś dieta ikor odpowiada szybkości obu reakcji wyrażonej w jednostce natężenia prądu.
Współrzędne punktu A (Ekor, ikor) stanowią punkt wyjścia dla anodowej krzywej
polaryzacyjnej przedstawionej z prawej strony rysunku. Przypadek I odpowiada
omówionemu wcześniej przykładowi, gdy żelazo znajduje się w 1 M H2SO4. W warunkach
stacjonarnych występuje aktywna korozja, zaś przesunięcie potencjału pod wpływem
przyłożonego z zewnątrz prądu o dostatecznie dużej gęstości (ikr) powoduje przejście żelaza
w stan pasywny przy jednoczesnym znacznym zmniejszeniu gęstości prądu do wartości ip.
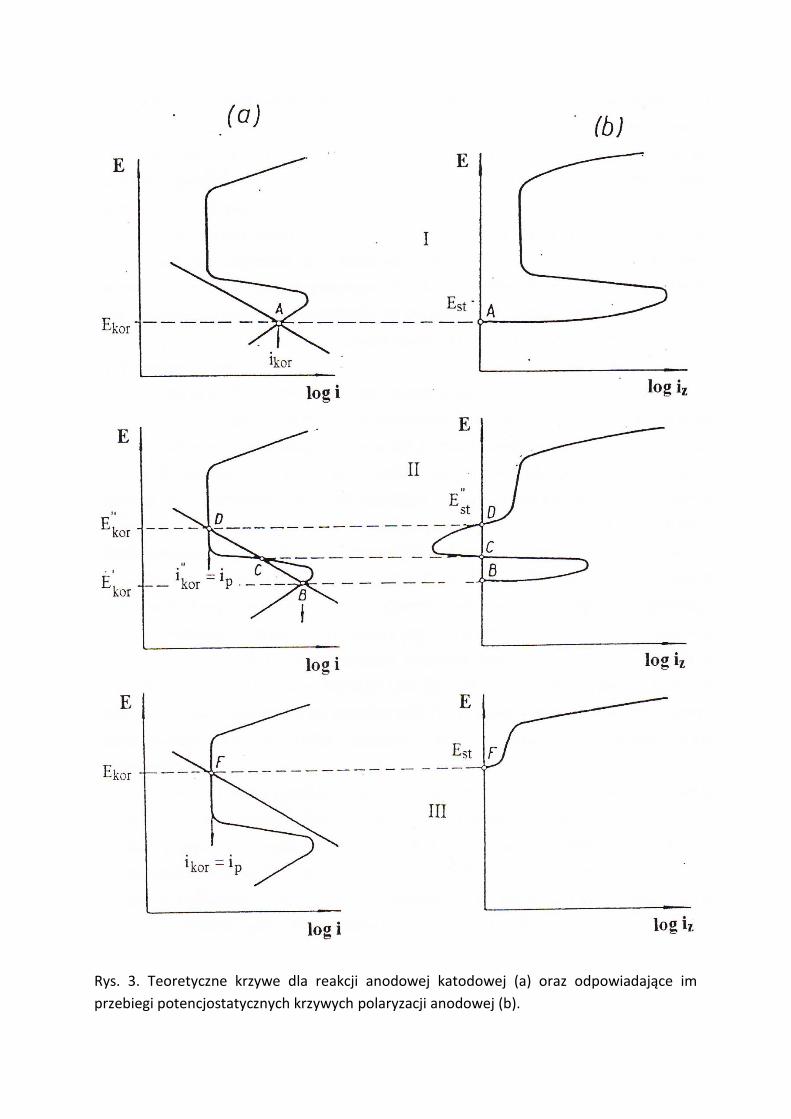
Rys. 3. Teoretyczne krzywe dla reakcji anodowej katodowej (a) oraz odpowiadające im
przebiegi potencjostatycznych krzywych polaryzacji anodowej (b).

Stan pasywny utrzymuje się tak długo, jak długo potencjał metalu utrzymywany jest
w zakresie pasywnym pod działaniem zewnętrznego prądu – wyłączenie prądu spowoduje
przejście metalu ponownie do stanu aktywnego (Ekor, ikor). Możliwośd uzyskania i
utrzymywania pasywności za pomocą zewnętrznego prądu polaryzacyjnego stanowi
podstawę jednej z elektrochemicznych metod ochrony metali przed korozją – ochrony
anodowej.
Przypadek II na rysunku 3 jest ilustracją spotykanych w praktyce możliwości
występowania metalu w identycznych warunkach środowiskowych w dwóch trwałych
stanach: aktywnym i pasywnym. Teoretyczne krzywe reakcji anodowej katodowej maja trzy
punkt wspólne B, C, D z tym, że punkt C jest pomijany jako odpowiadający stanowi
nietrwałemu. Stacjonarny potencjał korozyjny E’kor dla punktu B leży w zakresie aktywnym,
zaś E’’kor dla punktu D – w zakresie pasywnym potencjału. Szybkośd korozji w stanie
pasywnym i’kor = ip jest znacznie mniejsza od szybkości korozji w stanie aktywnym i’’kor.
Zależnie od stanu wyjściowego (punkt B lub D) anodowa krzywa polaryzacyjna może mied
dwojaki przebieg (rys 3b). Charakterystyczne jest występowanie ujemnego zakresu
prądowego w przedziale potencjału między rzędnymi punktów C i D. Przykładem
ilustrującym przypadek II jest wspomniane wcześniej doświadczenie Faradaya: żelazo w
rozcieoczonym HNO3 szybko koroduje (pkt. B), zaś po uprzednim spasywowaniu (w stężonym
kwasie) praktyczni nie koroduje (pkt. D). Przejście z jednego stanu do drugiego może
nastąpid albo na drodze chemicznej albo za pomocą zewnętrznego prądu polaryzującego.
Przypadek III na rysunku 3 przedstawia trwały stan pasywny metalu w rozworze
kwasu (punkt F przecięcia teoretycznych krzywych reakcji anodowej i katodowej leży w
zakresie pasywnym) – metal po zanurzeniu do kwasu ulega samorzutnej pasywacji, tzw.
autopasywacji. Przykładem może byd żelazo w stężonym HNO3 lub stal odporna na korozję w
roztworze kwasu z dodatkiem utleniaczy, np. soli żelazowych. Z korozyjnego punktu widzenia
przypadek III jest najbardziej pożądany w praktyce.
3. Wpływ temperatury i składu środowiska elektrolitycznego
Przejście metalu ze stanu aktywnego w pasywny charakteryzują trzy parametry, które
można wyznaczyd z potencjostatycznych krzywych polaryzacji anodowej: krytyczną gęstośd
prądu pasywacji ikr, krytyczny potencjał pasywacji Ekr oraz gęstośd prądu w stanie pasywnym
ip. Wartości wymienionych parametrów są ściśle związane z rodzajem (składem) metalu lub
stopu oraz środowiska.
Przyjmując, że metal jest dwuwartościowy, proces pasywacji można zapisad
schematycznie:
2 OH- Me + H2O + 2 e
H2O MeO + 2 H+ + 2e
lub:
2 OH- Me/O + H2O + 2 e

H2O Me/O + 2 H+ + 2 e
Symbolem Me/O oznaczono powierzchnię pokrytą warstwę chemisorbowanego
tlenu. Obie formy zapisu są równocenne. Przyjmując, że , dla
czystego żelaza w 25 oC wyrażenie na potencjał początku pasywacji (Ep) ma postad:
A z tego
Podwyższenie pH przesuwa potencjał początku pasywacji w kierunku wartości
ujemnych, czyli ułatwia przejście metalu w stan pasywny.
Wzrost temperatury z reguły wpływa dośd znacznie na zwiększenie wartości ikr i ip,
natomiast w niewielkim stopniu na zmianę wartości Ekr.
Obecnośd w środowisku niektórych aktywnych jonów, np. chlorkowych, może mied
Duzy wpływ na anodowe zachowanie się metali i stopów skłonnych do pasywacji, szczególnie
żelaza i stali odpornych na korozję. Jony chlorkowe adsorbując się w określonych miejscach
warstewki pasywnej powodują jej zniszczenie (przebicie). Przy dostatecznie dużym stężeniu
jonów Cl- warstewka pasywna może wcale się nie utworzyd. Obserwując potencjostatyczne
krzywe polaryzacji anodowej metali w roztworach zawierających dodatek chlorków można
zauważyd zwiększenie krytycznej gęstości prądu pasywacji oraz prądu w stanie pasywnym
przy znacznym zmniejszeniu zakresu pasywnego potencjału, a w granicznym przypadku –
zanik zakresu pasywnego. Na powierzchni próbki spolaryzowanej anodowo powyżej pewnej
wartości potencjału (potencjał przebicia Epp) zaczynają rozwijad się wżery.
Obecnośd utleniaczy (np. azotynów, chromianów, soli żelazowych) w roztworze
elektrolitu sprzyja na ogół przejściu metalu lub stopu w stan pasywny, tzn. powoduje
zmniejszenie krytycznej gęstości prądu pasywacji i rozszerza zakres pasywny. Dodane w
dostatecznej ilości utleniacze mogą spowodowad autopasywację metalu (por. przypadek III
na rys. 3).
4. Zastosowanie potencjostatycznych krzywych polaryzacyjnych.
Potencjostatyczna technika otrzymywania anodowych krzywych polaryzacyjnych
stanowi podstawę badao zjawisk pasywności metalu. Uzyskane wykresy pozwalają określid
korozyjne zachowanie się metalu w danym środowisku, możliwośd przeprowadzenia i
utrzymania danego metalu w stanie pasywnym na drodze zarówno chemicznej jak i
elektrochemicznej, wpływ zmian temperatury i składu środowiska na przebieg procesów
elektrodowych na metalu, itp.
Praktycznym zastosowaniem metod potencjo statycznych, o dużym znaczeniu
przemysłowym, jest dynamicznie rozwijająca się nowoczesna technika zabezpieczeo
przeciwkorozyjnych – ochrona anodowa.

O ile ochrona katodowa stosowana jest szeroko i z dużym powodzeniem dla ochrony
ano konstrukcji podziemnych (rurociągi, zbiorniki) oraz statków i konstrukcji portowych to
nie zdaje egzaminu w agresywnych warunkach przemysłu chemicznego. Tutaj lepsza okazuje
się ochrona anodowa. Np. w 0,3 M H2SO4 ochronna gęstośd prądu dla stali węglowej przy
ochronie katodowej wynosi 3,5·10-2 A/dm2, zaś przy ochronie anodowej gęstośd prądu w
stanie pasywacji wynosi poniżej 10-3 A/dm2.
Zastosowanie ochrony anodowej sprzyja szersze wykorzystanie jako materiału
konstrukcyjnego stali nierdzewnej i tytanu, które dobrze pasywują się w wielu środowiskach i
wykazują szeroki obszar pasywny. Przy zastosowaniu potencjałów skuteczną ochronę można
zapewnid jeśli obszar ten wynosi co najmniej 30 mV. Przykładowe wielkości
charakterystyczne dla obszaru pasywnego chromu, żelaza i stali w 0,5 M H2SO4
przedstawiono w tabeli 1.
Tabela 1. Charakterystyka stanu pasywnego chromu, żelaza i stali austenitycznej w 0,5 M
H2SO4 w 25oC.
Metal ikr
[A/dm2] Ekr [V]
Ep ÷ ET [V]
ip przy E=0,6 V [A/dm2]
Chrom 3,2·10-4 -0,35 od -0,05 do 1,10 5,0·10-10
Stal austeniczna chro-moniklowa (18% Cr, 12% Ni)
1,0·10-6
-0,15
od 0,15 do 0,65
3,0·10-10
żelazo 1,7·10-1 0,16 od 0,50 do 1,90 7,0·10-8
CZĘŚĆ PRAKTYCZNA
Celem dwiczenia jest zapoznanie się z techniką potencjostatyczną oraz jej
wykorzystanie do badania zjawisk pasywności metali i stopów z uwzględnieniem wpływu:
rodzaju tworzywa metalicznego,
rodzaju środowiska elektrolitycznego oraz jego stężenia i temperatury,
obecnośd utleniaczy,
obecnośd jonów niszczących stan pasywny (np. Cl-).
Przyrządy i sprzęt laboratoryjny:
Potencjostat, miliamperomierz, mieszadło magnetyczne, naczyoko pomiarowe, nasycona
elektroda kalomelowa, pipety, naciągarka do pipet, próbki stalowe: St3 szt. 2, OH13,
OH17N12M2T, zlewka poj. 50 dm3
Odczynniki chemiczne: 1 M H2SO4, 10% HCl, 2 M NaCl, aceton.

Wykonanie dwiczenia
1. Przygotowanie roztworu do badao.
2. Napełnienie sporządzonym roztworem układu pomiarowego.
3. Przygotowanie badanej elektrody, tj.: odtłuszczenie w rozpuszczalniku organicznym,
wytrawienie w 10% roztworze HCl, dokładne opłukanie w wodzie bieżącej oraz
destylowanej, osuszenie.
4. Umieszczenie elektrody w naczyniu pomiarowym i przyłączenie do odpowiednich
zacisków potencjostatu.
5. Uruchomienie mieszadła magnetycznego.
6. Odczekanie 15 minut, celem ustalenia się potencjału stacjonarnego.
7. Zmierzenie potencjału stacjonarnego, a następnie uruchomienie właściwe
zaprogramowanego układu sterowania potencjału w potencjostacie.
8. Odczytywanie wielkości natężenia wielkości natężenia prądu po każdej zmianie
potencjału elektrody o 20 mV i zapisywanie w tabelce według wzoru (rys. 4).
9. Obserwowane (wizualne) zachodzących procesów na powierzchni elektrody.
Tabela pomiarów potencjodynamicznych
Rodzaj metalu: …………………………………………………………Elektrolit:………………………………….
Temperatura:…………………………………… Szybkośd zmian potencjału: ………………. mV/min
Powierzchnia probki:……………………………m2
Lp. Potencjał względem NEK [mV]
Natężenie prądu [A]
Gęstośd prądu [A/m2]
Rys. 4. Wzór tabeli pomiarów potencjodynamicznych.
Opracowanie wyników
A. Wykreślenie uzyskanych potencjodynamicznych krzywych polaryzacji anodowej
iz=f(E) oraz log iz=f(E) wg wzoru na rys. 5
B. Zestawienie odczytanych charakterystycznych wielkości z potencjodynamicznych
krzywych polaryzacji anodowej w tabeli wg wzoru na rys. 6. Tabele należy wykonad
dla każdej krzywej oddzielnie i dołączyd do sprawozdania.
C. Określenie wpływu badanych czynników na przebieg potencjodynamicznych
krzywych polaryzacji anodowej oraz korozyjne zachowanie się metalu w badanych
układach.
D. Wyciągniecie wniosków o przebiegu procesów elektrodowych zachodzących na
powierzchni próbek.

Literatura:
1. Shreir L.L., Korozja metali i stopów, „Ochrona przed korozją”, WNT, Warszawa, 1966.
2. Baszkiewicz J., Kamioski M., Korozja materiałów, Oficyna Wydawnicza Politechniki
Warszawskiej, Warszawa, 2006.
3. Uhlig H., H., Korozja i jej zapobieganie WNT, Warszawa, 1976.

Rys. 5. Wyznaczanie charakterystycznych wielkości na potencjodynamicznej krzywej
polaryzacji anodowej.

Tabela pomiarów potencjodynamicznych.
Rodzaj tworzywa
(metal, stop)
Przygotowanie powierzchni:
(np.: sposób szlifowania, odtłuszczania, aktywowania, itp.)
Powierzchnia próbki, [m2]
Elektrolit
Mieszanie
Temperatura, [oC]
Szybkość zmian potencjału,
[mV/min]
Est Potencjał stacjonarny mV Opis wyglądu próbki:
(zmiana barwy próbki, wydzielanie
gazów, zmiana zabarwienia
roztworu, itp.)
Ekr Potencjał krytyczny mV
Ep Potencjał pasywacji mV
ikr Krytyczna gęstość prądu A/m2
ip Gęstość prądu pasywacji mA/m2
ET Potencjał początkowy stanu trans
pasywnego
mV
EW Potencjał wtórnej pasywacji mV
iW Gęstość prądu przy EW A/m2
Eg Potencjał wydzielania tlenu mV
ig Początkowa gęstość prądu
wydzielania tlenu
A/m2
Epp Potencjał przebicia warstewki mV
Ep Zakres stanu pasywnego mV
iu Maksymalna gęstość prądu
katodowego
A/m2
Eu Zakres potencjałów dla prądu
katodowego
mV
E’u Początek zakresu katodowego mV
E“u Koniec zakresu katodowego mV
Eu Potencjał przy maksymalnym
prądzie katodowym
mV
Współczynnik nachylenia krzywej
anodowego rozpuszczania metalu
V/dek
’ Współczynnik nachylenia krzywej
wydzialania tlenu
V/dek
Rys. 6. Zestawienie charakterystycznych parametrów potencjodynamicznej krzywej
polaryzacyjnej.




![WKRĘTY DO METALUMETAL SCREWS INSERT …...STEEL HOOK with slot cut HAK WKRĘCANY z przecięciem METAL SCREWS / WKRĘTY DO METALU METAL SCREWS / WKRĘTY DO METALU Art. d Ø [mm] d](https://static.fdocuments.pl/doc/165x107/5e9d49834c449a4aa949d47c/wkrty-do-metalumetal-screws-insert-steel-hook-with-slot-cut-hak-wkrcany.jpg)













